

Key Points
Question
Are features of predicate medical devices referenced in 510(k) regulatory submissions associated with product recalls?
Findings
In the exploratory cross-sectional analysis of 35 176 medical devices cleared by the US Food and Drug Administration between 2003 and 2018 via the 510(k) regulatory submission pathway, referencing predicate medical devices with 3 or more ongoing recalls was significantly associated with a 9.31–percentage-point increase in recall probability. A 1-SD increase in the total number of predicate medical devices cited by the applicant device was significantly associated with a 1.25–percentage-point increase in recall probability, and a 1-SD increase in age of the newest predicate medical device cited by the applicant device was significantly associated a 0.78–percentage-point decrease in recall probability.
Meaning
This study demonstrated significant associations between submission characteristics for 510(k) medical devices and medical device recalls; further research is needed to understand the implications of these associations.
Abstract
Importance
Most regulated medical devices enter the US market via the 510(k) regulatory submission pathway, wherein manufacturers demonstrate that applicant devices are “substantially equivalent” to 1 or more “predicate” devices (legally marketed medical devices with similar intended use). Most recalled medical devices are 510(k) devices.
Objective
To examine the association between characteristics of predicate medical devices and recall probability for 510(k) devices.
Design, Setting, and Participants
In this exploratory cross-sectional analysis of medical devices cleared by the US Food and Drug Administration (FDA) between 2003 and 2018 via the 510(k) regulatory submission pathway, linear probability models were used to examine associations between a 510(k) device’s recall status and characteristics of its predicate medical devices. Public documents for the 510(k) medical devices were collected using FDA databases. A text extraction algorithm was applied to identify predicate medical devices cited in 510(k) regulatory submissions. Algorithm-derived metadata were combined with 2003-2020 FDA recall data.
Exposures
Citation of predicate medical devices with certain characteristics in 510(k) regulatory submissions, including the total number of predicate medical devices cited by the applicant device, the age of the predicate medical devices, the lack of similarity of the predicate medical devices to the applicant device, and the recall status of the predicate medical devices.
Main Outcomes and Measures
Class I or class II recall of a 510(k) medical device between its FDA regulatory clearance date and December 31, 2020.
Results
The sample included 35 176 medical devices, of which 4007 (11.4%) were recalled. The applicant devices cited a mean of 2.6 predicate medical devices, with mean ages of 3.6 years and 7.4 years for the newest and oldest, respectively, predicate medical devices. Of the applicant devices, 93.9% cited predicate medical devices with no ongoing recalls, 4.3% cited predicate medical devices with 1 ongoing class I or class II recall, 1.0% cited predicate medical devices with 2 ongoing recalls, and 0.8% cited predicate medical devices with 3 or more ongoing recalls. Applicant devices citing predicate medical devices with 3 or more ongoing recalls were significantly associated with a 9.31–percentage-point increase (95% CI, 2.84-15.77 percentage points) in recall probability compared with devices without ongoing recalls of predicate medical devices, or an 81.2% increase in recall probability relative to the mean recall probability. A 1-SD increase in the total number of predicate medical devices cited by the applicant device was significantly associated with a 1.25–percentage-point increase (95% CI, 0.62-1.87 percentage points) in recall probability, or an 11.0% increase in recall probability relative to the mean recall probability. A 1-SD increase in the newest age of a predicate medical device was significantly associated with a 0.78–percentage-point decrease (95% CI, 1.29-0.30 percentage points) in recall probability, or a 6.8% decrease in recall probability relative to the mean recall probability.
Conclusions and Relevance
This exploratory cross-sectional study of 510(k) medical devices cleared by the FDA between 2003 and 2018 demonstrated significant associations between 510(k) submission characteristics and recalls of medical devices. Further research is needed to understand the implications of these associations.
This study uses US Food and Drug Administration data and examines the association between characteristics of predicate medical devices and recall probability for applicant medical devices cleared for use via the 510(k) regulatory submission pathway.
Introduction
In the US, most moderate-risk medical devices enter the market through the US Food and Drug Administration (FDA) 510(k) clearance pathway because they are “substantially equivalent” to 1 or more “predicate” devices (legally marketed devices with similar intended use).1,2 The FDA often evaluates substantial equivalence via similarities in the composition and design between applicant medical devices and predicate medical devices, and human clinical data for the applicant device or the predicate device are not typically required.3 Manufacturers may cite competitors’ devices or recalled devices as predicate medical devices as long as the recalled devices were voluntarily recalled by their manufacturers.4,5
Even though the 510(k) pathway facilitates expedient iterative device development,4 hundreds of 510(k) devices are recalled annually due to potential patient harm,6 with more than 300 recalls occurring in 2020 alone. Recalls of 510(k) devices, including transvaginal surgical meshes7,8 and reperfusion catheters,9 have raised concerns that substantial equivalence may be a poor marker of safety and efficacy.
In 2011, the Institute of Medicine asserted that substantial equivalence to predicate medical devices is insufficient for demonstrating safety and efficacy.4 In 2018, the FDA called for modernizing the 510(k) pathway and proposed actions to decrease reliance on “outdated” predicate medical devices, including publicly identifying applicant devices that cite predicate medical devices that are more than 1 decade old.10
Few studies7,11,12 have systematically examined the relationship between predicate medical devices and applicant device safety. An unadjusted analysis by Maisel12 at the FDA of 2003-2009 FDA data found that applicant 510(k) devices that cited newer and higher numbers of predicate medical devices experienced a greater number of recalls. Understanding how regulatory submissions of 510(k) medical devices relate to their safety could inform evidence-based improvements of the 510(k) pathway.
The FDA does not publish analysis-ready data on 510(k) predicate medical devices.13 A text extraction algorithm was used in this study to construct a database of 510(k) predicate medical devices. In this exploratory cross-sectional analysis of 510(k) devices cleared by the FDA between 2003 and 2018, the newly created database was used to estimate the associations between characteristics of the predicate medical devices and the probability of the applicant device experiencing a significant product recall.
Methods
Study Sample
This exploratory cross-sectional analysis of FDA-cleared 510(k) medical devices was performed using public FDA databases describing clearances, recalls, and adverse events.13,14,15 The FDA databases were linked to a new predicate medical device database derived by applying an automated text extraction algorithm to public evidence summary documents for 510(k) devices.13 The study was determined to be exempt from review by an institutional review board at the University of Minnesota.
The study sample included all 2003-2018 FDA-cleared 510(k) devices with at least 1 predicate medical device identified by the algorithm. An FDA committee assigns all medical devices to granular product type codes (>1300 in total) that determine which of the 19 FDA medical specialty review panels will review them (eg, products with the code “BSZ” for “gas-machine, anesthesia” are reviewed by the “anesthesiology” review panel)16; product codes with only 1 FDA clearance were excluded to facilitate product-type, fixed-effects analyses. Additional study design details appear below and in eAppendices 1-2 in Supplement 1.
Predicate Medical Device Identification
The 510(k) predicate medical devices are typically only identifiable in device-specific public evidence summary documents. For all 2003-2018 FDA-cleared 510(k) devices, evidence summary documents were downloaded from the FDA’s website.13 An automated text extraction algorithm was then applied17 that identified unique predicate medical device identifiers. After removing likely erroneous predicate medical devices (eg, those that received regulatory clearance prior to their applicant device), the performance of the automated algorithm was compared with a manually coded random sample of 1800 evidence summaries. Compared with the manually coded sample, the algorithm identified predicate medical devices with 95% sensitivity and 96% specificity. In addition, averages of predicate medical device characteristics that were identified with the algorithm were almost identical to and statistically indistinguishable from averages of manually identified predicate medical device characteristics. Additional details appear in eAppendix 1 (eFigures 1-3 and eTables 1-3) in Supplement 1.
In instances in which the text extraction algorithm could not identify at least 1 predicate medical device, the characteristics of the predicate medical device were by definition not identifiable and thus missing (necessitating the exclusion of applicant devices without algorithm-identifiable predicate medical devices). When at least 1 predicate medical device was identifiable, the devices could be linked to the FDA’s census of medical device clearances (ie, no missing characteristics).
Exposures
The exposures of interest were characteristics of predicate medical devices referenced in 510 (k) regulatory submissions. Several predicate medical device characteristics were examined, including the total number of predicate medical devices cited by the applicant device, the age of the predicate medical devices, the lack of similarity of the predicate medical devices to the applicant device, and the recall status of the predicate medical devices.
The age of the predicate medical devices was defined in 3 ways. First, corresponding to the FDA’s 2018 statement10 discouraging the referencing of predicate medical devices more than 1 decade old, a binary indicator was created to determine whether an applicant device cited at least 1 predicate medical device aged 10 years or older (age was defined as years between predicate device clearance and applicant device clearance). Second, the mean age of all predicate medical devices cited in a 510(k) submission was calculated. Third, the age of the newest (ie, most recently cleared) predicate medical device and the age of the oldest predicate medical device were measured, similar to prior FDA analyses of predicate medical device age.12
The lack of similarity of the predicate medical device to the applicant device was measured in 2 complementary ways. First, the unique number of product types for the predicate medical devices that did not match the product type of the applicant device was counted. Second, the unique number of medical specialties for the predicate medical devices (as determined by the FDA medical specialty review panel assigned to the predicate medical devices) that did not match the medical specialty of the applicant device was counted.
Predicate medical device recall status was measured using the count of class I or class II recalls for predicate medical devices that were initiated and classified by the FDA but not yet terminated (resolved) at the time of the applicant device’s clearance (“ongoing” recalls). Class I recalls occur when products may cause “serious health problems or death.” Class II recalls occur when products may cause “temporary or reversible” health problems or when there is a “slight chance” that they “will cause serious health problems or death.”18 Class III recalls were excluded because they do not pose patient safety concerns. Recalls for predicate medical devices that were initiated and terminated prior to clearance of an applicant device were not considered as an exposure. The FDA has not consistently reported recalls initiated before 2002,14 therefore, terminated recalls of predicate medical devices would be disproportionately undercounted among applicant devices cleared earlier during the sample period (additional details appear in eFigure 2 in Supplement 1).
Ongoing recalls for predicate medical devices were attributed to 8 problem categories: component or device design, labeling, materials or composition, regulatory application, packaging, software, manufacturing environment, and miscellaneous (eTable 3 in Supplement 1). The miscellaneous category primarily included cases when the recall cause was listed as “other” or was still under investigation by the manufacturer.
Outcomes
The primary outcome was whether a device experienced at least 1 class I or class II recall (a recall with potential for patient harm) between its FDA clearance date and December 31, 2020 (ie, follow-up ranges from 2-17 years because devices in the sample were cleared between 2003 and 2018).
In addition to recalls, device-related patient deaths were identified as an outcome using the FDA’s Manufacturer and User Facility Device Experience (MAUDE) database. The MAUDE database collects reports of deaths, serious injuries, and malfunctions that reporters suspect to be associated with the medical device. The MAUDE database collects reports from both mandatory reporters (eg, device manufacturers and device user facilities) and voluntary reporters (eg, health care professionals, patients, and consumers). The FDA does not verify whether deaths or injuries reported in the MAUDE database were directly caused by the listed medical devices.15
Statistical Analysis
Linear probability models were used to assess whether a 510(k) device experienced a class I or class II recall after its regulatory clearance as a function of its predicate medical devices’ characteristics. Linear probability models were chosen to facilitate fixed-effects analyses (described below).19,20,21 In sensitivity analyses, logistic and accelerated failure time models were estimated as well as models that only considered recalls occurring within 2 years of a device’s clearance (eAppendix 2 [eTables 4-9] in Supplement 1).
The models included 1 of 3 definitions for the age of the predicate medical device (mean age, newest and oldest age, or aged ≥10 years), the number of predicate medical devices cited, the number of nonmatching product types, the number of nonmatching medical specialties, the number of ongoing recalls for predicate medical devices, and binary indicator variables for the year of FDA clearance for the applicant device as covariates. To adjust for differences in manufacturers’ regulatory experiences over time, the models controlled for the number of 510(k) devices by the manufacturer cleared by the FDA prior to the applicant device’s clearance (ie, overall experience) and the number of prior devices by the manufacturer cleared by the FDA with the same product type as the applicant device (ie, product-specific experience).
The adjusted models also included binary indicator variables for each unique manufacturer and product type to account for time-invariant manufacturer and product-type characteristics that could be spuriously correlated with recall probability (manufacturer and product-type fixed effects).22 For example, some product types likely had differences in their underlying recall probability, independent of which predicate medical devices were referenced. Including product-type indicators allowed for the examination of how recall probability varied by the characteristics of the predicate medical devices within the same product type (eg, an applicant semiconstrained cemented metal and polymer hip prosthesis that cited a 5-year-old predicate medical device vs a semiconstrained cemented metal and polymer hip prosthesis that cited a 10-year-old predicate medical device), thus preventing correlations between the characteristics of the predicate medical devices and unobserved time-invariant characteristics of the specific product types from biasing the results.
Heterogeneity across medical specialties (and their accompanying review panels) was examined by estimating separate models for each medical specialty. To describe which types of predicate medical device recalls were most relevant to applicant recalls, the applicant device recalls were modeled as a function of binary indicators for cause-specific ongoing predicate medical device recalls. Cause-specific predicate medical device recalls were not mutually exclusive; an applicant could cite one predicate medical device recalled due to labeling issues and another recalled due to software concerns.
The SEs used in computing the 95% CIs for the coefficient estimates were clustered at the manufacturer level. All tests of statistical significance were performed using 2-sided tests and P<.05 was considered statistically significant. All data were analyzed using Stata MP version 16 (StataCorp).
Results
Study Sample
Among the 48 747 medical devices cleared by the FDA via the 510(k) submission pathway between 2003 and 2018, 4361 were excluded from the sample because a public evidence summary was unavailable, 4248 were excluded because their predicate medical devices were not identifiable, and 4962 were excluded because fixed-effects analyses were not feasible.23 The final sample of 35 176 devices represented 72.2% of the devices cleared by the FDA via the 510(k) submission pathway between 2003 and 2018 (eFigure 3 in Supplement 1).
Device Characteristics
The mean age of the predicate medical devices was 5.4 years (Table 1). The mean age of the newest predicate medical device was 3.6 years and the mean age of the oldest predicate medical device was 7.4 years. The applicant devices in the sample cited a mean of 2.6 predicate medical devices. Of the applicant devices, 27.0% cited at least 1 predicate medical device aged 10 years or older. Of the applicant devices, 93.9% cited predicate medical devices with no ongoing recalls, 4.3% cited predicate medical devices with 1 ongoing class I or class II recall, 1.0% cited predicate medical devices with 2 ongoing recalls, and 0.8% cited predicate medical devices with 3 or more ongoing recalls. The age of the predicate medical devices, the number of predicate medical devices, and the number of ongoing recalls for the predicate medical devices increased slightly over time. The lack of similarity between the applicant device and the predicate medical device remained relatively constant (as measured by nonmatching product types and nonmatching medical specialties with the predicate medical device; Figure). Of 35 176 devices, 4007 (11.4%) experienced at least 1 class I or class II recall after receiving 510(k) regulatory clearance from the FDA (Table 1).
Table 1. Characteristics of the Medical Devices That Received 510(k) Clearance From the US Food and Drug Administration (FDA) by Recall Status.
| Recall status of medical devices that received 510(k) clearance | ||
|---|---|---|
| Recalleda | Not recalled | |
| No. of devices | 4007 | 31 169 |
| No. of unique manufacturersb | 994 | 4120 |
| No. of unique product typesc | 676 | 1326 |
| No. of deaths in MAUDE database | 24 918 | 4923 |
| Age of predicate medical devices, yd | ||
| Mean (SD) | 5.1 (4.2) | 5.4 (4.7) |
| Median (IQR) | 4.0 (2.1-6.9) | 4.1 (2.1-7.3) |
| Age of newest predicate medical devices, y | ||
| Mean (SD) | 3.1 (3.7) | 3.7 (4.4) |
| Median (IQR) | 1.8 (0.9-3.8) | 2.1 (1.0-4.7) |
| Age of oldest predicate medical devices, y | ||
| Mean (SD) | 7.4 (6.3) | 7.4 (6.3) |
| Median (IQR) | 5.8 (2.6-10.6) | 5.7 (2.6-10.4) |
| No. of predicate medical devices | ||
| Mean (SD) | 2.9 (3.4) | 2.5 (2.4) |
| Median (IQR) | 2 (1-3) | 2 (1-3) |
| No. of unique nonmatching product typese | ||
| Mean (SD) | 0.8 (1.2) | 0.6 (1.0) |
| Median (IQR) | 0 (0-1) | 0 (0-1) |
| No. of unique nonmatching medical specialtiese,f | ||
| Mean (SD) | 0.2 (0.5) | 0.2 (0.5) |
| Median (IQR) | 0 (0-0) | 0 (0-0) |
| No. of prior 510(k) clearances from the manufacturers | ||
| Mean (SD) | 74.4 (118.4) | 47.8 (98.5) |
| Median (IQR) | 21 (4-85) | 8 (2-40) |
| No. of prior 510(k) clearances from the manufacturers for the same product types | ||
| Mean (SD) | 5.4 (9.9) | 4.5 (9.4) |
| Median (IQR) | 2 (0-6) | 1 (0-4) |
| ≥1 Predicate medical devices received FDA clearance ≥10 y prior to FDA decision for applicant device, No. (%) | 1117 (27.9) | 8375 (26.9) |
| No. of ongoing class I or class II recalls for predicate medical devices, No. (%)g | ||
| 0 | 3439 (85.8) | 29 589 (94.9) |
| 1 | 346 (8.6) | 1180 (3.8) |
| 2 | 109 (2.7) | 249 (0.8) |
| ≥3 | 113 (2.8) | 151 (0.5) |
| ≥1 Ongoing class I or class II recalls for predicate medical devices by reason, No. (%)h | ||
| Miscellaneous | 236 (5.9) | 533 (1.7) |
| Software | 179 (4.5) | 326 (1.0) |
| Device design or component | 144 (3.6) | 352 (1.1) |
| Materials or composition | 87 (2.2) | 213 (0.7) |
| Manufacturing environment | 66 (1.6) | 230 (0.7) |
| Labeling | 41 (1.0) | 149 (0.5) |
| Packaging | 20 (0.5) | 84 (0.3) |
| Application | 3 (0.1) | 9 (<0.1) |
| Medical specialty of FDA review panel for applicant device, No. (%)i | ||
| Radiology | 777 (19.4) | 3678 (11.8) |
| Orthopedic | 759 (18.9) | 5996 (19.2) |
| Cardiovascular | 605 (15.1) | 3952 (12.7) |
| General and plastic surgery | 338 (8.4) | 3475 (11.1) |
| General hospital | 285 (7.1) | 2238 (7.2) |
| Clinical chemistry | 237 (5.9) | 1500 (4.8) |
| Gastroenterology and urology | 212 (5.3) | 1533 (4.9) |
| Anesthesiology and respiratory therapy | 209 (5.2) | 1490 (4.8) |
| Neurology | 150 (3.7) | 1238 (4.0) |
| Dental | 83 (2.1) | 2281 (7.3) |
| Microbiology | 67 (1.7) | 660 (2.1) |
| Hematology | 64 (1.6) | 310 (1.0) |
| Ophthalmic | 66 (1.6) | 632 (2.0) |
| Immunology | 38 (0.9) | 282 (0.9) |
| Obstetrics and gynecology | 37 (0.9) | 654 (2.1) |
| Ear, nose, and throat | 24 (0.6) | 350 (1.1) |
| Toxicology | 25 (0.6) | 277 (0.9) |
| Physical medicine | 20 (0.5) | 572 (1.8) |
| Pathology | 11 (0.3) | 51 (0.2) |
Abbreviation: MAUDE, Manufacturer and User Facility Device Experience.
Indicates whether a device experienced 1 or more class I or class II recalls between its FDA regulatory clearance date and December 31, 2020. Recalls are classified as class I when there is potential for serious patient harm or death and class II when there is potential for temporary or reversible patient harm or a slight chance of serious patient harm or death.
The variations on similar manufacturer names present in the FDA databases were standardized into a single manufacturer name (additional information appears in eAppendix 1 in Supplement 1).
These are FDA-assigned identifiers describing the generic function of a medical device (additional information appears in the study sample subsection of the Methods section).
Defined as the number of years between FDA clearance of a predicate device and clearance of an applicant device.
The data in these rows reflect dissimilarity between the applicant device and the predicate device.
There are 19 FDA medical specialty review panels that are responsible for reviewing a medical device. The medical specialty review panels are assigned based on a medical device’s product type.
Investigation has not yet terminated (resolved) for the predicate device at the time of FDA clearance for the applicant device.
Recalls were manually assigned to these categories based on the reason for recall (additional information appears in eTable 3 in Supplement 1).
The categories in this section are the specialty descriptions used by the FDA review panels.
Figure. Characteristics of Predicate Medical Devices Over Time.
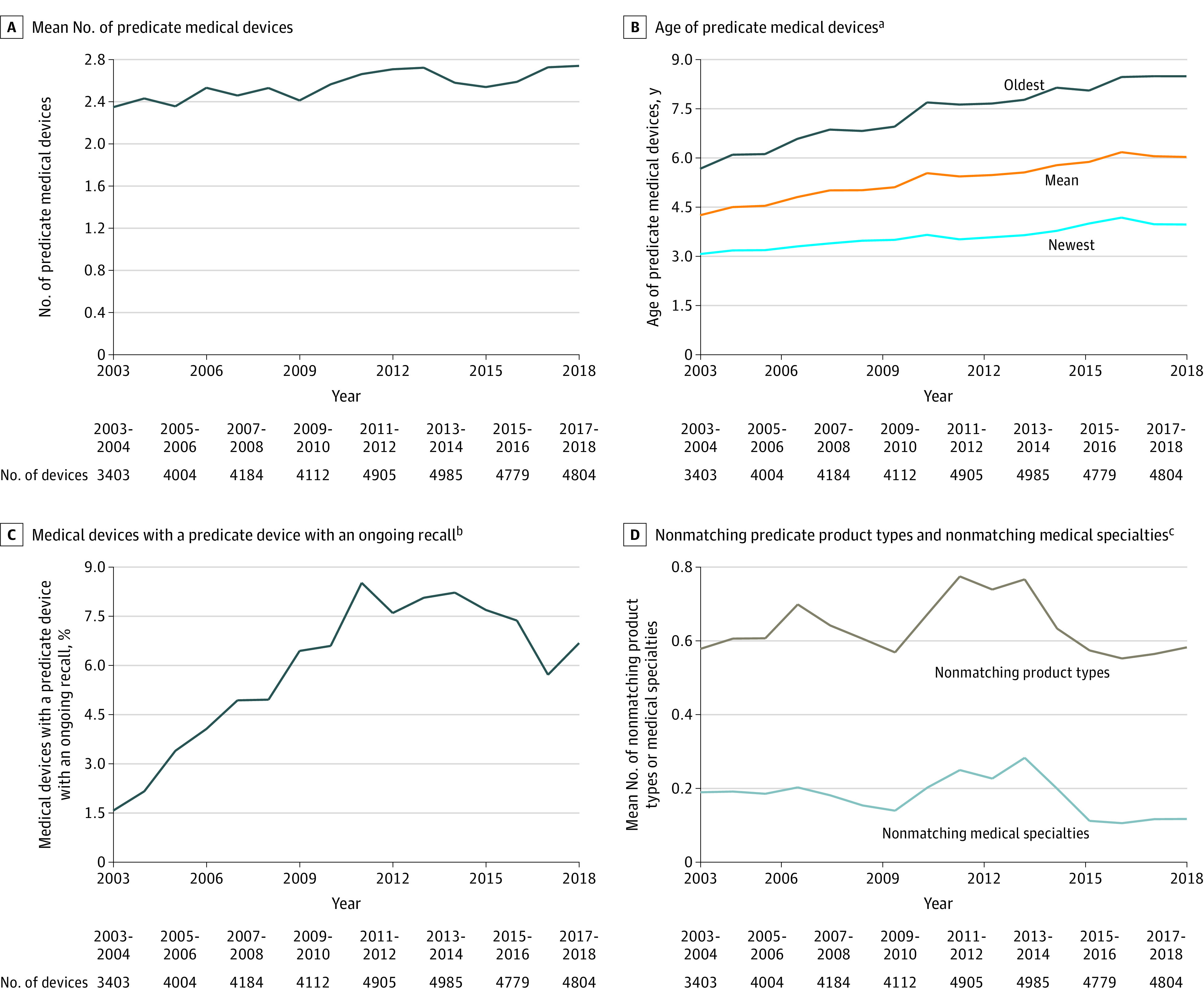
aDefined as the number of years between US Food and Drug Administration (FDA) clearance of a predicate medical device and FDA clearance of the applicant device.
bDefined as a class I or a class II ongoing recall for the predicate medical devices classified by the FDA but not yet terminated (resolved) at the time of FDA clearance for the applicant device. A class I recall indicates there is potential for serious patient harm or death. A class II recall indicates there is potential for temporary or reversible patient harm or a slight chance of serious patient harm or death.
cNonmatching product types refer to the number of unique product types of the predicate medical device that do not match the product types of the applicant device. The product types are FDA-assigned identifiers to describe the generic function of a medical device (additional information appears in the study sample subsection of the Methods section). Nonmatching medical specialties refer to the number of unique medical specialties of the predicate medical device that do not match the medical specialties of the applicant device. There are 19 FDA medical specialty review panels that are responsible for reviewing a medical device. The medical specialty review panels are assigned based on a medical device’s product type.
Differences in Outcomes
Compared with devices without recalls, recalled devices cited newer predicate medical devices, a higher number of predicate medical devices, predicate medical devices with more nonmatching product types and nonmatching medical specialties, and predicate medical devices with a higher number of ongoing recalls. The recalls were disproportionately concentrated among devices used in the fields of pathology, radiology, hematology, clinical chemistry, and cardiovascular. Recalled devices were associated with 24 918 deaths in the MAUDE database between 2003 and 2020, whereas devices without recalls were associated with 4923 deaths during the same period (Table 1).
After adjusting for manufacturer and product type binary indicators (fixed effects), a 1-SD increase in mean age of the predicate medical devices (4.6 for the entire sample) was significantly associated with an adjusted 0.46–percentage-point decrease (95% CI, 0.92-0.05 percentage points) in the probability of experiencing a class I or class II recall, or a 4.1% decrease in recall probability relative to the mean recall probability (the per-unit changes appear in Table 2). Increasing the age of the newest predicate medical device by 1 SD (4.3 for the entire sample) was significantly associated with a 0.78–percentage-point decrease (95% CI, 1.29-0.30 percentage points) in recall probability, or a 6.8% decrease in recall probability relative to the mean recall probability. There was no significant association between increasing the age of the oldest predicate medical device and recall probability. Citing a predicate medical device aged 10 years or older was not significantly associated with recall probability.
Table 2. Change in Probability of Class I or Class II Recalls for 35 176 Medical Devices That Received US Food and Drug Administration (FDA) 510(k) Clearance by the Characteristics and the Number of Ongoing Recalls for the Predicate Medical Devicesa.
| Probability of class I or class II recalls of 510(k) medical devices, percentage point change (95% CI)b | ||||||
|---|---|---|---|---|---|---|
| By mean age of predicate medical devices without adjustment for product type and manufacturer fixed effectsc | By newest and oldest age of predicate medical devices without adjustment for product type and manufacturer fixed effectsc | By age ≥10 y for predicate medical devices without adjustment for product type and manufacturer fixed effectsc | By mean age of predicate medical devices adjusted for product type and manufacturer fixed effectsc | By newest and oldest age of predicate medical devices adjusted for product type and manufacturer fixed effectsc | By age ≥10 y for predicate medical devices adjusted for product type and manufacturer fixed effectsc | |
| Characteristics | ||||||
| Mean age of predicate medical devices, y | –0.14 (–0.23 to –0.04) | –0.10 (–0.20 to –0.01) | ||||
| Age of newest predicate medical devices, y | –0.26 (–0.36 to –0.15) | –0.18 (–0.30 to –0.07) | ||||
| Age of oldest predicate medical devices, y | 0.06 (–0.04 to 0.16) | 0.04 (–0.06 to 0.14) | ||||
| ≥1 Predicate medical devices aged ≥10 y | –0.02 (–0.98 to 0.95) | 0.38 (–0.68 to 1.45) | ||||
| No. of predicate medical devices | 0.37 (0.13 to 0.61) | 0.24 (–0.03 to 0.51) | 0.36 (0.12 to 0.60) | 0.55 (0.34 to 0.77) | 0.48 (0.24 to 0.72) | 0.53 (0.31 to 0.75) |
| No. of unique nonmatching product typesd,e | –0.29 (–1.35 to 0.77) | –0.34 (–1.39 to 0.72) | –0.27 (–1.33 to 0.79) | –0.19 (–1.17 to 0.78) | –0.24 (–1.21 to 0.74) | –0.15 (–1.13 to 0.82) |
| No. of unique nonmatching medical specialtiese,f | 0.52 (–0.08 to 1.12) | 0.40 (–0.19 to 0.99) | 0.47 (–0.13 to 1.07) | –0.07 (–0.78 to 0.63) | –0.16 (–0.87 to 0.55) | –0.14 (–0.86 to 0.58) |
| No. of prior 510(k) clearances from the manufacturer | 0.03 (0.01 to 0.05) | 0.03 (0.01 to 0.05) | 0.03 (0.01 to 0.04) | –0.01 (–0.02 to 0) | –0.01 (–0.02 to 0) | –0.01 (–0.02 to 0) |
| No. of prior 510(k) clearances from the manufacturer for the same product type | –0.03 (–0.10 to 0.04) | –0.03 (–0.10 to 0.04) | –0.02 (–0.09 to 0.06) | –0.08 (–0.14 to –0.01) | –0.08 (–0.14 to –0.02) | –0.07 (–0.14 to –0.01) |
| Ongoing recalls g | ||||||
| No. of class I or class II recalls of predicate medical devices | ||||||
| 0 | 0 [Reference] | 0 [Reference] | 0 [Reference] | 0 [Reference] | 0 [Reference] | 0 [Reference] |
| 1 | 11.39 (8.78 to 14.01) | 11.33 (8.71 to 13.95) | 11.36 (8.75 to 13.96) | 1.81 (–0.96 to 4.59) | 1.78 (–1.00 to 4.56) | 1.77 (–1.00 to 4.53) |
| 2 | 18.37 (13.42 to 23.31) | 18.25 (13.31 to 23.18) | 18.35 (13.42 to 23.28) | 2.20 (–1.81 to 6.20) | 2.13 (–1.87 to 6.13) | 2.16 (–1.83 to 6.14) |
| ≥3 | 30.70 (23.42 to 37.97) | 30.56 (23.27 to 37.86) | 30.66 (23.39 to 37.93) | 9.41 (2.98 to 15.84) | 9.31 (2.84 to 15.77) | 9.31 (2.85 to 15.77) |
The final sample of 35 176 devices represented 72.2% of the devices cleared by the FDA between 2003 and 2018.
Linear probability models were used to assess whether the applicant device would experience a class I or class II recall between its FDA regulatory clearance date and December 31, 2020, per unit change in the characteristics of the predicate medical devices. A class I recall indicates there is potential for serious patient harm or death. A class II recall indicates there is potential for temporary or reversible patient harm or a slight chance of serious patient harm or death. The models adjusted for listed predicate and manufacturer characteristics, year fixed effects, and product type and manufacturer fixed effects in columns 5-7. The SEs used in computing the 95% CIs were clustered at the manufacturer level.
The age of the predicate medical device was defined as the number of years between FDA clearance of the predicate device and FDA clearance of the applicant device. See definition for product type in footnote d. Variations on similar manufacturer names in the FDA database were standardized into a single manufacturer name (additional information appears in eAppendix 1 in Supplement 1).
Nonmatching product types refer to the number of unique product types of the predicate medical device that do not match the product types of the applicant device. The product types are FDA-assigned identifiers to describe the generic function of a medical device (additional information appears in the study sample subsection of the Methods section).
The data in these rows reflect dissimilarity between the applicant device and the predicate device.
Nonmatching medical specialties refer to the number of unique medical specialties of the predicate medical device that do not match the medical specialties of the applicant device. There are 19 FDA medical specialty review panels that are responsible for reviewing a medical device. The medical specialty review panels are assigned based on a medical device’s product type.
Investigation has not yet terminated (resolved) for the predicate device at the time of FDA clearance for the applicant device.
In the models with newest and oldest age of the predicate medical devices, a 1-SD increase in the total number of predicate medical devices cited by the applicant device (2.6 for the entire sample) was significantly associated with a 1.25–percentage-point increase (95% CI, 0.62-1.87 percentage points) in recall probability, or an 11.0% increase in recall probability relative to the mean recall probability (the per-unit changes appear in Table 2).
Applicant devices citing predicate medical devices with either 1 or 2 ongoing recalls were not significantly associated with recall probability. Applicant devices citing predicate medical devices with 3 or more ongoing recalls were significantly associated with a 9.31–percentage-point increase (95% CI, 2.84-15.77 percentage points) in recall probability compared with devices without ongoing recalls of predicate medical devices, or an 81.2% increase in recall probability relative to the mean recall probability. Associations were larger in the models that did not adjust for manufacturer and product-type fixed effects (Table 2). The results were similar in the logistic and accelerated failure time models and in the models only considering recalls that occurred within 2 years of a device’s regulatory clearance date (eTables 5-6 in Supplement 1).
There were no significant associations between recall probability and predicate medical device dissimilarity as measured by nonmatching product types and nonmatching medical specialties. When examining heterogeneity across medical specialties, the results were consistent with the overall estimates but less precise (the 3 medical specialties with the most devices appear in Table 3 and all medical specialties appear in eTables 5-6 in Supplement 1).
Table 3. Change in Probability of Class I or Class II Recalls of Medical Devices That Received US Food and Drug Administration (FDA) 510(k) Clearance for the 3 Medical Specialties With the Most Devices by the Characteristics and the Number of Ongoing Recalls for the Predicate Medical Devices.
| Probability of class I or class II recalls of 510(k) medical devices, percentage point change (95% CI)a | ||||||
|---|---|---|---|---|---|---|
| By newest and oldest age of predicate medical devices for orthopedic specialty without adjustment for product type and manufacturer fixed effectsb | By newest and oldest age of predicate medical devices for cardiovascular specialty without adjustment for product type and manufacturer fixed effectsb | By newest and oldest age of predicate medical devices for radiology specialty without adjustment for product type and manufacturer fixed effectsb | By newest and oldest age of predicate medical devices for orthopedic specialty adjusted for product type and manufacturer fixed effectsb | By newest and oldest age of predicate medical devices for cardiovascular specialty adjusted for product type and manufacturer fixed effectsb | By newest and oldest age of predicate medical devices for radiology specialty adjusted for product type and manufacturer fixed effectsb | |
| No. of devices | 6696 | 4440 | 4368 | 6696 | 4440 | 4368 |
| Characteristics | ||||||
| Age of newest predicate medical devices, y | 0.07 (–0.24 to 0.38) | –0.51 (–0.82 to –0.21) | –0.45 (–0.98 to 0.08) | 0 (–0.33 to 0.33) | –0.55 (–0.86 to –0.23) | –0.33 (–0.86 to 0.20) |
| Age of oldest predicate medical devices, y | 0.19 (–0.07 to 0.45) | 0.29 (0.04 to 0.55) | –0.21 (–0.53 to 0.11) | 0.07 (–0.17 to 0.30) | 0.26 (–0.03 to 0.54) | –0.15 (–0.55 to 0.25) |
| No. of predicate medical devices | 0.30 (–0.10 to 0.70) | 0.53 (–0.30 to 1.35) | 0.92 (–0.10 to 1.95) | 0.46 (0.12 to 0.80) | 0.60 (–0.15 to 1.35) | 1.02 (–0.05 to 2.10) |
| No. of unique nonmatching product typesc,d | 0.41 (–1.60 to 2.43) | –1.81 (–4.60 to 0.98) | –0.64 (–5.07 to 3.79) | 0.92 (–1.11 to 2.95) | –1.51 (–4.40 to 1.39) | 1.65 (–2.06 to 5.35) |
| No. of unique nonmatching medical specialtiesd,e | 0.39 (–0.50 to 1.29) | –0.56 (–2.67 to 1.55) | –1.08 (–2.96 to 0.81) | 0.22 (–0.67 to 1.11) | –1.72 (–4.13 to 0.68) | –2.14 (–4.50 to 0.21) |
| No. of prior 510(k) clearances from the manufacturer | 0.02 (–0.01 to 0.05) | 0.02 (0.01 to 0.04) | 0.05 (0.04 to 0.07) | 0 (–0.03 to 0.02) | 0 (–0.04 to 0.03) | –0.01 (–0.03 to 0.01) |
| No. of prior 510(k) clearances from the manufacturer for the same product type | –0.20 (–0.33 to –0.06) | 0.09 (–0.15 to 0.32) | –0.21 (–0.37 to –0.04) | –0.18 (–0.32 to –0.03) | 0.21 (–0.06 to 0.48) | –0.12 (–0.24 to –0.01) |
| Ongoing recalls f | ||||||
| No. of class I or class II recalls of predicate medical devices | ||||||
| 0 | 0 [Reference] | 0 [Reference] | 0 [Reference] | 0 [Reference] | 0 [Reference] | 0 [Reference] |
| 1 | 2.25 (–1.51 to 6.02) | 12.30 (4.45 to 20.15) | 14.08 (8.33 to 19.83) | –1.98 (–5.59 to 1.63) | 0.96 (–6.58 to 8.51) | 7.19 (1.17 to 13.21) |
| 2 | 6.76 (–2.67 to 16.20) | 23.45 (12.85 to 34.05) | 16.31 (6.68 to 25.94) | 1.34 (–8.50 to 11.19) | 7.07 (–5.05 to 19.19) | 3.13 (–4.24 to 10.49) |
| ≥3 | 25.44 (0.81 to 50.07) | 21.27 (7.13 to 35.41) | 26.58 (14.99 to 38.16) | 17.36 (–8.12 to 42.84) | –5.09 (–14.49 to 4.32) | 11.03 (–0.62 to 22.68) |
Linear probability models were used to assess whether the applicant device would experience a class I or class II recall between its FDA regulatory clearance date and December 31, 2020, per unit change in the characteristics of the predicate medical devices. A class I recall indicates there is potential for serious patient harm or death. A class II recall indicates there is potential for temporary or reversible patient harm or a slight chance of serious patient harm or death. The models adjusted for listed predicate and manufacturer characteristics, year fixed effects, and product type and manufacturer fixed effects in columns 5-7. The SEs used in computing the 95% CIs were clustered at the manufacturer level. Three medical specialties (orthopedic, cardiovascular, and radiology) comprise 44.8% of the devices in the sample. The results for the remaining medical specialties appear in eTables 5-6 in Supplement 1.
The age of the predicate medical device was defined as the number of years between FDA clearance of the predicate device and FDA clearance of the applicant device. See definition for product type in footnote c. Variations on similar manufacturer names in the FDA database were standardized into a single manufacturer name (additional information appears in eAppendix 1 in Supplement 1).
Nonmatching product types refer to the number of unique product types of the predicate medical device that do not match the product types of the applicant device. The product types are FDA-assigned identifiers to describe the generic function of a medical device (additional information appears in the study sample subsection of the Methods section).
The data in these rows reflect dissimilarity between the applicant device and the predicate device.
Nonmatching medical specialties refer to the number of unique medical specialties of the predicate medical device that do not match the medical specialties of the applicant device. There are 19 FDA medical specialty review panels that are responsible for reviewing a medical device. The medical specialty review panels are assigned based on a medical device’s product type.
Investigation has not yet terminated (resolved) for the predicate device at the time of FDA clearance for the applicant device.
The most relevant recalls of predicate medical devices were related to software and those classified as miscellaneous (Table 4). Applicant devices citing predicate medical devices with at least 1 ongoing recall due to software were significantly associated with a 5.92–percentage-point increase (95% CI, 1.97-9.87 percentage points) in recall probability compared with devices without ongoing recalls of predicate medical devices, or a 51.9% increase in recall probability relative to the mean recall probability. Applicant devices citing predicate medical devices with at least 1 ongoing miscellaneous recall were similarly associated with a 5.14–percentage-point increase (95% CI, 1.45-8.81 percentage points) in recall probability, or a 45.1% increase in recall probability relative to the mean recall probability.
Table 4. Change in Probability of Class I or Class II Recalls for the 35 176 Medical Devices That Received US Food and Drug Administration (FDA) 510(k) Clearance by the Characteristics and the Cause of Ongoing Recalls for the Predicate Medical Devicesa.
| Probability of class I or II recalls of 510(k) medical devices, percentage point change (95% CI)b | ||||||
|---|---|---|---|---|---|---|
| By mean age of predicate medical devices without adjustment for product type and manufacturer fixed effectsc | By newest and oldest age of predicate medical devices without adjustment for product type and manufacturer fixed effectsc | By age ≥10 y for predicate medical devices without adjustment for product type and manufacturer fixed effectsc | By mean age of predicate medical devices adjusted for product type and manufacturer fixed effectsc | By newest and oldest age of predicate medical devices adjusted for product type and manufacturer fixed effectsc | By age ≥10 y for predicate medical devices adjusted for product type and manufacturer fixed effectsc | |
| Characteristics | ||||||
| Mean age of predicate medical devices, y | –0.13 (–0.23 to –0.04) | –0.10 (–0.20 to –0.01) | ||||
| Age of newest predicate medical devices, y | –0.26 (–0.37 to –0.15) | –0.18 (–0.30 to –0.07) | ||||
| Age of oldest predicate medical devices, y | 0.07 (–0.03 to 0.16) | 0.04 (–0.06 to 0.14) | ||||
| ≥1 Predicate medical devices aged ≥10 y | 0.06 (–0.89 to 1.01) | 0.40 (–0.67 to 1.46) | ||||
| No. of predicate medical devices | 0.38 (0.14 to 0.61) | 0.25 (–0.02 to 0.52) | 0.37 (0.13 to 0.60) | 0.55 (0.34 to 0.77) | 0.47 (0.24 to 0.71) | 0.53 (0.31 to 0.74) |
| No. of unique nonmatching product typesd,e | –0.27 (–1.33 to 0.79) | –0.32 (–1.38 to 0.73) | –0.25 (–1.31 to 0.80) | –0.17 (–1.16 to 0.81) | –0.22 (–1.20 to 0.76) | –0.13 (–1.11 to 0.84) |
| No. of unique nonmatching medical specialtiese,f | 0.48 (–0.12 to 1.08) | 0.35 (–0.24 to 0.95) | 0.43 (–0.17 to 1.03) | –0.09 (–0.80 to 0.62) | –0.18 (–0.89 to 0.54) | –0.16 (–0.88 to 0.56) |
| No. of prior 510(k) clearances from manufacturer | 0.03 (0.01 to 0.04) | 0.03 (0.01 to 0.04) | 0.03 (0.01 to 0.04) | –0.01 (–0.02 to 0) | –0.01 (–0.02 to 0) | –0.01 (–0.02 to 0) |
| No. of prior 510(k) clearances from manufacturer for same product type | –0.03 (–0.10 to 0.04) | –0.03 (–0.10 to 0.04) | –0.02 (–0.09 to 0.05) | –0.08 (–0.14 to –0.02) | –0.08 (–0.14 to –0.02) | –0.07 (–0.14 to –0.01) |
| Ongoing recalls g | ||||||
| ≥1 Class I or class II recalls of predicate medical devices by causeh | ||||||
| Miscellaneous | 14.50 (10.85 to 18.14) | 14.45 (10.80 to 18.09) | 14.43 (10.79 to 18.06) | 5.17 (1.50 to 8.85) | 5.14 (1.46 to 8.81) | 5.11 (1.45 to 8.78) |
| Software | 16.67 (12.19 to 21.14) | 16.74 (12.28 to 21.20) | 16.79 (12.32 to 21.26) | 5.90 (1.95 to 9.84) | 5.92 (1.97 to 9.87) | 5.92 (1.98 to 9.86) |
| Device design or component | 9.99 (5.57 to 14.41) | 9.84 (5.39 to 14.28) | 9.96 (5.54 to 14.38) | –1.26 (–4.87 to 2.35) | –1.36 (–5.00 to 2.27) | –1.31 (–4.93 to 2.31) |
| Materials or composition | 9.78 (5.04 to 14.52) | 9.63 (4.89 to 14.38) | 9.69 (4.97 to 14.41) | 3.59 (–0.79 to 7.96) | 3.50 (–0.87 to 7.88) | 3.53 (–0.83 to 7.88) |
| Manufacturing environment | 3.69 (–1.11 to 8.49) | 3.62 (–1.19 to 8.44) | 3.65 (–1.15 to 8.45) | –3.41 (–8.01 to 1.19) | –3.42 (–8.03 to 1.19) | –3.40 (–7.98 to 1.19) |
| Labeling | 6.66 (1.29 to 12.02) | 6.57 (1.18 to 11.95) | 6.66 (1.29 to 12.04) | –0.01 (–5.08 to 5.07) | –0.03 (–5.11 to 5.06) | 0 (–5.09 to 5.09) |
| Packaging | 6.78 (–0.08 to 13.63) | 6.77 (–0.08 to 13.63) | 6.42 (–0.41 to 13.24) | 3.37 (–4.04 to 10.78) | 3.44 (–3.96 to 10.84) | 3.15 (–4.24 to 10.55) |
| Application | 9.05 (–17.61 to 35.70) | 8.69 (–18.08 to 35.46) | 8.96 (–17.81 to 35.72) | 12.06 (–9.90 to 34.02) | 11.81 (–10.24 to 33.86) | 11.86 (–10.11 to 33.82) |
The final sample of 35 176 devices represented 72.2% of the devices cleared by the FDA between 2003 and 2018.
Linear probability models were used to assess whether the applicant device would experience a class I or class II recall between its FDA regulatory clearance date and December 31, 2020, per unit change in the characteristics of the predicate medical devices. A class I recall indicates there is potential for serious patient harm or death. A class II recall indicates there is potential for temporary or reversible patient harm or a slight chance of serious patient harm or death. The models adjusted for listed predicate and manufacturer characteristics, year fixed effects, and product type and manufacturer fixed effects in columns 5-7. The SEs used in computing the 95% CIs were clustered at the manufacturer level.
The age of the predicate medical device was defined as the number of years between FDA clearance of the predicate device and FDA clearance of the applicant device. See definition for product type in footnote d. Variations on similar manufacturer names in the FDA database were standardized into a single manufacturer name (additional information appears in eAppendix 1 in Supplement 1).
Nonmatching product types refer to the number of unique product types of the predicate medical device that do not match the product types of the applicant device. The product types are FDA-assigned identifiers to describe the generic function of a medical device (additional information appears in the study sample subsection of the Methods section).
The data in these rows reflect dissimilarity between the applicant device and the predicate device.
Nonmatching medical specialties refer to the number of unique medical specialties of the predicate medical device that do not match the medical specialties of the applicant device. There are 19 FDA medical specialty review panels that are responsible for reviewing a medical device. The medical specialty review panels are assigned based on a medical device’s product type.
Investigation has not yet terminated (resolved) for the predicate device at the time of FDA clearance for the applicant device.
Manually assigned to categories based on the reason for the recall (eTable 3 in Supplement 1).
Discussion
This exploratory cross-sectional study of 510(k) medical devices cleared by the FDA between 2003 and 2018 demonstrated significant associations between recalls of 510(k) medical devices and the regulatory submission characteristics of the predicate medical devices. Recalled devices were attributed to more patient deaths than devices without recalls.
The significant associations between recall probability and predicate medical device characteristics are consistent with prior concerns that substantial equivalence may be insufficient in certain circumstances for assessing the safety profile of moderate-risk medical devices, particularly when the safety profiles of predicate medical devices are not well understood or when applicant devices differ in some meaningful way from their predicate medical devices.4,9 For example, the relationship between device safety and the age of the predicate medical device is ambiguous. It could be that older predicate medical devices represent less modern technology, suggesting a higher-risk profile. However, this study found that the age of the predicate medical device was inversely associated with recall probability. This finding may reflect that market learning occurs over time. If a manufacturer compares their applicant device with a predicate device that has only been on the market for a short period, there may be underlying safety issues about both the applicant device and the predicate device not known to the manufacturer, potentially necessitating a recall. Conversely, a predicate medical device that has been on the market for several years may have stood the test of time. A manufacturer citing an older predicate medical device may have a more complete understanding of safety issues with the predicate medical device and thus the applicant device, such that recalls of the applicant device are less likely.
The association between the number of predicate medical devices cited and the recall probability may work through a similar mechanism. If a manufacturer only cited 1 predicate medical device in their 510(k) submission and effectively created an exact copy of an existing device, then it may be that the applicant device will not have additional safety concerns beyond what is already known about the predicate device, meaning a recall would be unlikely. In contrast, if a manufacturer combined different features from multiple predicate medical devices, those features could interact in unexpected ways, potentially resulting in a recall.
Citing predicate medical devices with nonmatching product types or nonmatching medical specialties was not significantly associated with recall probability despite device dissimilarity contributing to high-profile recalls of devices such as metal-on-metal hip implants.24 This lack of a significant association could reflect a true null relationship between dissimilarity with the predicate medical device and recall probability, or it may be that the relevant differences between applicant devices and predicate medical devices are concentrated in more granular design features not captured in this study’s measures of nonmatching product types and nonmatching medical specialties.
The results from this study must be viewed in a broader context and by themselves do not suggest that either the continued use or prohibition of certain types of predicate medical devices is necessarily appropriate. For example, the finding that applicant devices that cite older predicate medical devices experience fewer recalls appears to be in conflict with the FDA’s stated intent to “retire outdated predicates”10; however, there may be other reasons to cite newer predicate medical devices, potentially related to differences in effectiveness or indication. Regulators would need to weigh multiple factors, potentially including the associations between predicate medical device characteristics and recall probability demonstrated in this study, when deciding whether to change the requirements about which predicate medical devices can be cited in 510(k) submissions and how substantial equivalence is determined.
However, these results suggest that the FDA may benefit from collecting additional information for 510(k) submissions. In particular, there is substantially higher recall probability across multiple medical specialties among devices that cite predicate medical devices with several ongoing recalls. It may be valuable to ask manufacturers to disclose to the FDA the recall status of predicate medical devices cited in their 510(k) submissions (manufacturers currently are not required to do this),5 and explicitly request that manufacturers describe whether applicant devices are subject to the same concerns that led to the recalls of the predicate medical devices.
Additional information may be especially relevant in instances when the reasons for the predicate medical device recalls are unclear. Recalls of predicate medical devices classified as “miscellaneous,” which included cases classified as “other” or “under investigation by firm,” and software-related recalls were significantly associated with the 2 largest increases in recall probability among the studied reasons for recalls of predicate medical devices. Given the wide variety of potential problems within these categories (along with the FDA’s ongoing reevaluation of how it regulates software in the context of medical devices25), the FDA may benefit from receiving and reviewing more information about the applicant devices and their predicate medical devices during the 510(k) review process.
Limitations
This study has several limitations. First, this exploratory analysis could not provide a causal explanation why predicate medical device characteristics were associated with recall probability. It could be that the examined predicate medical device characteristics were correlated with unobserved device characteristics that ultimately caused recalls. For example, if some devices in the sample assessed substantial equivalence via bench testing and others assessed substantial equivalence via human clinical data, and the evidence type used affected recall risk, the results in this study could be confounded by the effects of the evidence type. The use of product-type fixed effects likely mitigated the extent of this confounding, given that some premarket evidence requirements for 510(k) devices are set at the product-type level.26,27 However, the results of this study would still be confounded to the extent that evidence type and other relevant, but unobserved, device characteristics varied within product types.
Second, the MAUDE database, which was used to attribute deaths to 510(k) devices, has known limitations. The FDA does not verify reported deaths in the MAUDE database and relies on passive collection of reports from both mandatory and voluntary reporters. Total death counts, as well as the relative frequency of deaths among recalled devices vs devices without any recalls, may be reported with errors in the MAUDE database.
Third, the predicate medical devices cited in some 510(k) submissions were not identifiable, and this study excluded 510(k) devices without identifiable predicate medical devices. If the relationship between the characteristics of predicate medical devices and recall probability differed based on whether predicate medical devices were identifiable, then the associations presented in this study would not be representative of all 510(k) medical devices. However, the results of this study were consistent with prior unadjusted analyses performed directly by the FDA.12
Fourth, due to a lack of publicly available recall data prior to 2002, the association between already terminated recalls of predicate medical devices and applicant recalls could not be assessed. Future studies could explore this relationship if older recall data become available.
Fifth, this study was only able to identify broad categories of reasons for predicate medical device recalls. Future analyses using more granular classification of predicate medical device recalls, perhaps derived though text analysis of open-response fields within FDA databases, could offer deeper insights into the relationship between predicate medical device recalls and applicant device recalls.
Sixth, this study did not describe the clinical value of the included medical devices. It may be that using certain types of predicate medical devices in 510(k) submissions confer unique clinical benefits, such that the benefits of certain citation practices may outweigh the potential risk of recalls. As such, a comprehensive assessment of the 510(k) pathway is beyond the scope of this study.
Conclusions
This exploratory cross-sectional study of 510(k) medical devices cleared by the FDA between 2003 and 2018 demonstrated significant associations between 510(k) submission characteristics and recalls of medical devices. Further research is needed to understand the implications of these associations.
eAppendix 1. Data collection and sample construction
eAppendix 2. Statistical analysis and additional results
eFigure 1. Flow diagram of automated text extraction steps
eFigure 2. Mean predicate recalls by date of recall termination relative to applicant device clearance
eFigure 3. Sample construction steps
eTable 1. Accuracy of automated text extraction algorithm
eTable 2. Comparison of predicate characteristics between predicates identified via text extraction and predicates identified via manual search
eTable 3. Manual assignment of FDA recall cause problem categories
eTable 4. Condition number and variance inflation factors for predicate characteristics of interest
eTable 5. Change in unadjusted probability of class I or II recalls of 510(k) medical devices for different medical specialties by predicate characteristics
eTable 6. Change in adjusted probability of class I/II recalls of 510(k) medical devices for different medical specialties by predicate characteristics
eTable 7. Characteristics of 510(k) medical devices by recall status
eTable 8. Change in probability of class I or II recalls of 510(k) medical devices by predicate characteristics
eTable 9. Change in adjusted probability of class I or II recalls of 510(k) medical devices by predicate characteristics as estimated by linear probability models, logistic regression
eReferences
Data sharing statement
References
- 1.US Food and Drug Administration . Premarket notification 510(k). Published March 13, 2020. Accessed October 2, 2022. https://www.fda.gov/medical-devices/premarket-submissions-selecting-and-preparing-correct-submission/premarket-notification-510k
- 2.Darrow JJ, Avorn J, Kesselheim AS. FDA regulation and approval of medical devices: 1976-2020. JAMA. 2021;326(5):420-432. doi: 10.1001/jama.2021.11171 [DOI] [PubMed] [Google Scholar]
- 3.Zuckerman D, Brown P, Das A. Lack of publicly available scientific evidence on the safety and effectiveness of implanted medical devices. JAMA Intern Med. 2014;174(11):1781-1787. doi: 10.1001/jamainternmed.2014.4193 [DOI] [PubMed] [Google Scholar]
- 4.Institute of Medicine . Medical Devices and the Public’s Health: The FDA 510(k) Clearance Process at 35 Years. National Academies Press; 2011. doi: 10.17226/13150 [DOI] [Google Scholar]
- 5.US Food and Drug Administration . The 510(k) program: evaluating substantial equivalence in premarket notifications [510(k)]: guidance for industry and Food and Drug Administration staff. Published July 2014. Accessed May 15, 2022. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/510k-program-evaluating-substantial-equivalence-premarket-notifications-510k
- 6.Dubin JR, Simon SD, Norrell K, et al. Risk of recall among medical devices undergoing US Food and Drug Administration 510(k) clearance and premarket approval, 2008-2017. JAMA Netw Open. 2021;4(5):e217274-e217274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zargar N, Carr A. The regulatory ancestral network of surgical meshes. PLoS One. 2018;13(6):e0197883. doi: 10.1371/journal.pone.0197883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rosh J, Bell CM, Urbach DR. The 510(k) ancestry of transvaginal mesh. JAMA Surg. 2021;156(8):701-702. [DOI] [PubMed] [Google Scholar]
- 9.Kadakia KT, Beckman AL, Ross JS, Krumholz HM. Renewing the call for reforms to medical device safety—the case of penumbra. JAMA Intern Med. 2022;182(1):59-65. [DOI] [PubMed] [Google Scholar]
- 10.US Food and Drug Administration . Statement from FDA Commissioner Scott Gottlieb, MD, and Jeff Shuren, MD, Director of the Center for Devices and Radiological Health, on transformative new steps to modernize FDA’s 510(k) program to advance the review of the safety and effectiveness of medical devices. Published November 26, 2018. Accessed January 28, 2020. https://www.fda.gov/news-events/press-announcements/statement-fda-commissioner-scott-gottlieb-md-and-jeff-shuren-md-director-center-devices-and?utm_campaign=11262018_Statement_FDA
- 11.Pai DB. Mapping the genealogy of medical device predicates in the United States. PLoS One. 2021;16(10):e0258153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maisel WH. C: 510(k) premarket notification analysis of FDA recall data. In: Public Health Effectiveness of the 510(k) Clearance Process: Measuring Postmarket Performance and Other Selected Topics: Workshop Report. National Academies Press; 2011. https://www.ncbi.nlm.nih.gov/books/NBK209655/ [PubMed] [Google Scholar]
- 13.US Food and Drug Administration . Downloadable 510(k) files. Accessed August 11, 2022. https://www.fda.gov/medical-devices/510k-clearances/downloadable-510k-files
- 14.US Food and Drug Administration . Medical device recalls. Accessed January 22, 2021. https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfRES/res.cfm
- 15.US Food and Drug Administration . MAUDE—Manufacturer and User Facility Device Experience. Accessed October 29, 2022. https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfmaude/search.cfm
- 16.US Food and Drug Administration . Product classification. Accessed July 19, 2022. https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPCD/classification.cfm
- 17.Zhu Y, Everhart A, Karaca-Mandic P, Sen S. Using NLP to extract predicate history from medical device approvals. Accessed December 12, 2022. https://experts.umn.edu/en/publications/using-nlp-to-extract-predicate-history-from-medical-device-approv
- 18.US Food and Drug Administration . What is a medical device recall? Accessed July 19, 2022. https://www.fda.gov/medical-devices/medical-device-recalls/what-medical-device-recall
- 19.Katz E. Bias in conditional and unconditional fixed effects logit estimation. Polit Anal. 2001;9(4):379-384. doi: 10.1093/oxfordjournals.pan.a004876 [DOI] [Google Scholar]
- 20.Greene W. The behaviour of the maximum likelihood estimator of limited dependent variable models in the presence of fixed effects. Econom J. 2004;7(1):98-119. doi: 10.1111/j.1368-423X.2004.00123.x [DOI] [Google Scholar]
- 21.Coupé T. Bias in conditional and unconditional fixed effects logit estimation: a correction. Polit Anal. 2005;13(3):292-295. doi: 10.1093/pan/mpi019 [DOI] [Google Scholar]
- 22.Huntington-Klein N. The Effect: An Introduction to Research Design and Causality. Chapman & Hall; 2022. [Google Scholar]
- 23.Correia S. Singletons, cluster-robust standard errors and fixed effects: a bad mix. Accessed November 3, 2022. http://scorreia.com/research/singletons.pdf
- 24.Ardaugh BM, Graves SE, Redberg RF. The 510(k) ancestry of a metal-on-metal hip implant. N Engl J Med. 2013;368(2):97-100. [DOI] [PubMed] [Google Scholar]
- 25.US Food and Drug Administration . Digital health software precertification (pre-cert) pilot program. Accessed November 3, 2022. https://www.fda.gov/medical-devices/digital-health-center-excellence/digital-health-software-precertification-pre-cert-pilot-program
- 26.US Food and Drug Administration . Regulatory controls. Accessed October 27, 2022. https://www.fda.gov/medical-devices/overview-device-regulation/regulatory-controls
- 27.US Food and Drug Administration . Class II special controls documents. Accessed October 27, 2022. https://www.fda.gov/medical-devices/guidance-documents-medical-devices-and-radiation-emitting-products/class-ii-special-controls-documents
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
eAppendix 1. Data collection and sample construction
eAppendix 2. Statistical analysis and additional results
eFigure 1. Flow diagram of automated text extraction steps
eFigure 2. Mean predicate recalls by date of recall termination relative to applicant device clearance
eFigure 3. Sample construction steps
eTable 1. Accuracy of automated text extraction algorithm
eTable 2. Comparison of predicate characteristics between predicates identified via text extraction and predicates identified via manual search
eTable 3. Manual assignment of FDA recall cause problem categories
eTable 4. Condition number and variance inflation factors for predicate characteristics of interest
eTable 5. Change in unadjusted probability of class I or II recalls of 510(k) medical devices for different medical specialties by predicate characteristics
eTable 6. Change in adjusted probability of class I/II recalls of 510(k) medical devices for different medical specialties by predicate characteristics
eTable 7. Characteristics of 510(k) medical devices by recall status
eTable 8. Change in probability of class I or II recalls of 510(k) medical devices by predicate characteristics
eTable 9. Change in adjusted probability of class I or II recalls of 510(k) medical devices by predicate characteristics as estimated by linear probability models, logistic regression
eReferences
Data sharing statement