

Key Points
Question
Does 50 mg of fluvoxamine twice daily for 10 days, compared with placebo, shorten symptom duration among adult (aged ≥30 years) outpatients with symptomatic mild to moderate COVID-19?
Findings
In this platform randomized clinical trial conducted during a period of predominance for the Delta and Omicron variants in the US, including 1288 adult outpatients with COVID-19 treated with fluvoxamine vs placebo, the hazard ratio was 0.96 (95% credible interval, 0.86-1.06) for time to sustained recovery with a posterior P = .21 for the probability of improvement. This did not meet the prespecified threshold of greater than 0.95 for posterior probability.
Meaning
These findings do not support the use of fluvoxamine at this dose and duration in patients with mild to moderate COVID-19.
Abstract
Importance
The effectiveness of fluvoxamine to shorten symptom duration or prevent hospitalization among outpatients with mild to moderate symptomatic COVID-19 is unclear.
Objective
To evaluate the efficacy of low-dose fluvoxamine (50 mg twice daily) for 10 days compared with placebo for the treatment of mild to moderate COVID-19 in the US.
Design, Setting, and Participants
The ongoing Accelerating COVID-19 Therapeutic Interventions and Vaccines (ACTIV-6) platform randomized clinical trial was designed to test repurposed medications in outpatients with mild to moderate COVID-19. A total of 1288 participants aged 30 years or older with test-confirmed SARS-CoV-2 infection and experiencing 2 or more symptoms of acute COVID-19 for 7 days or less were enrolled between August 6, 2021, and May 27, 2022, at 91 sites in the US.
Interventions
Participants were randomized to receive 50 mg of fluvoxamine twice daily for 10 days or placebo.
Main Outcomes and Measures
The primary outcome was time to sustained recovery (defined as the third day of 3 consecutive days without symptoms). There were 7 secondary outcomes, including a composite outcome of hospitalization, urgent care visit, emergency department visit, or death through day 28.
Results
Among 1331 participants who were randomized (median age, 47 years [IQR, 38-57 years]; 57% were women; and 67% reported receiving ≥2 doses of a SARS-CoV-2 vaccine), 1288 completed the trial (674 in the fluvoxamine group and 614 in the placebo group). The median time to sustained recovery was 12 days (IQR, 11-14 days) in the fluvoxamine group and 13 days (IQR, 12-13 days) in the placebo group (hazard ratio [HR], 0.96 [95% credible interval, 0.86-1.06], posterior P = .21 for the probability of benefit [determined by an HR >1]). For the composite outcome, 26 participants (3.9%) in the fluvoxamine group were hospitalized, had an urgent care visit, had an emergency department visit, or died compared with 23 participants (3.8%) in the placebo group (HR, 1.1 [95% credible interval, 0.5-1.8], posterior P = .35 for the probability of benefit [determined by an HR <1]). One participant in the fluvoxamine group and 2 participants in the placebo group were hospitalized; no deaths occurred in either group. Adverse events were uncommon in both groups.
Conclusions and Relevance
Among outpatients with mild to moderate COVID-19, treatment with 50 mg of fluvoxamine twice daily for 10 days, compared with placebo, did not improve time to sustained recovery. These findings do not support the use of fluvoxamine at this dose and duration in patients with mild to moderate COVID-19.
Trial Registration
ClinicalTrials.gov Identifier: NCT04885530
This randomized, placebo-controlled platform trial compares the use of low-dose fluvoxamine (50 mg twice daily) for 10 days compared with placebo in outpatients with mild to moderate COVID-19.
Introduction
There remains a need for oral therapies to prevent progression to severe COVID-19.1 Novel oral antivirals have demonstrated a clinical benefit in unvaccinated persons; however, the efficacy of the currently recommended therapies for mild to moderate COVID-19 among vaccinated patients is unclear and may be lower than what was reported among unvaccinated populations.2,3 Repurposing approved drugs developed for other conditions presents an attractive strategy to identify new treatment options and expand access to potentially lifesaving care.4,5
Fluvoxamine is a selective serotonin reuptake inhibitor approved by the US Food and Drug Administration in 1994 for the treatment of obsessive-compulsive disorder and is now used to treat a variety of psychiatric conditions, including social anxiety disorder and depression.6 Fluvoxamine also has been noted to activate the σ-1 receptor, which may decrease inflammation by reducing endoplasmic reticulum stress and downregulating the expression of inflammatory genes. Early COVID-19 studies reported improved clinical outcomes in participants receiving fluvoxamine.7,8,9
A placebo-controlled, randomized, adaptive platform trial (TOGETHER trial)10 of 1497 symptomatic adults in Brazil who had confirmed SARS-CoV-2 and a known risk factor for progression to severe COVID-19 found treatment with 100 mg of fluvoxamine twice daily for 10 days reduced the need for hospitalization (defined as retention in an emergency setting or transfer to a tertiary hospital). However, tolerability has been identified as a potential limiting factor for use of this dose of fluvoxamine, with 74% of participants in the fluvoxamine group reporting completion of more than 80% of possible doses.
The STOP COVID 2 trial,11 in which participants were randomized to receive 100 mg of fluvoxamine twice daily or placebo for 15 days, was stopped for futility in May 2021 after an interim analysis found that the low event rate (for hypoxia or hospitalization due to COVID-19) seen in the trial with the original sample size was associated with a conditional probability of less than 10% for demonstrating efficacy of fluvoxamine. A subsequent meta-analysis7 of 3 clinical trials found a high probability (94.1%-98.6%) that fluvoxamine was associated with a reduced risk of hospitalization for COVID-19 with a risk ratio of 0.75 (95% CI, 0.58-0.97). Given these conflicting results, regulatory authorities and guideline committees have not recommended fluvoxamine as an early treatment for COVID-19. In addition, uncontrolled observational data suggested a lower 50-mg dose would be more tolerable.12
The ongoing Accelerating COVID-19 Therapeutic Interventions and Vaccines (ACTIV-6) platform randomized clinical trial is investigating the use of repurposed drugs (a 50-mg, twice-daily dose of fluvoxamine for 10 days vs placebo in this report) among nonhospitalized adults with mild to moderate COVID-19.
Methods
Trial Design and Oversight
This ongoing double-blind, placebo-controlled platform randomized clinical trial is being conducted using a decentralized approach, allowing for use in a wide range of settings within health care systems and the community. The study has enrolled outpatients with mild to moderate COVID-19 and a confirmed positive polymerase chain reaction or antigen test for SARS-CoV-2 infection, including positive results from home-based testing. The trial protocol and the statistical analysis plan appear in Supplement 1.
The trial protocol was approved by each site’s institutional review board. Informed consent was obtained from each participant either via written or electronic consent. An independent data and safety monitoring committee oversaw participant safety, efficacy, and trial conduct.
Participants
Recruitment into the platform trial opened on June 11, 2021, and is ongoing. Participants were enrolled into the fluvoxamine group or the placebo group between August 6, 2021, and May 27, 2022, at 91 sites in the US. The 2 groups (fluvoxamine and matching placebo) were closed after meeting the prespecified accrual goal. Participants were identified by investigators at trial sites or were self-identified by contacting a central study telephone hotline. Participants without a local site were managed via interactions with the coordinating call center or website.
The trial sites verified eligibility criteria, including (1) age of 30 years or older, (2) confirmed SARS-CoV-2 infection for 10 days or less, and (3) whether experiencing 2 or more COVID-19 symptoms for 7 days or less from the time of consent (the full eligibility criteria appear in Supplement 1). Symptoms included the following: fatigue, dyspnea, fever, cough, nausea, vomiting, diarrhea, body aches, chills, headache, sore throat, nasal symptoms, and loss of sense of taste or smell. Receipt of a SARS-CoV-2 vaccine was not an exclusion criterion. Use of standard COVID-19 therapies available under US Food and Drug Administration approval or emergency use authorization was permissible.
Exclusion criteria included hospitalization, known allergies or contraindications (including to prohibited concomitant medications), and use of fluvoxamine within 14 days of enrollment.
Randomization
Within the platform trial, drugs could be added or removed according to the adaptive design, emerging evidence, or both. During the period that randomization to the fluvoxamine group was open, participants were assigned using a random number generator to 1 of the drugs actively enrolling, which included 50 mg of fluvoxamine twice daily for 10 days, 400 μg/kg/d of oral ivermectin for 3 days, and 200 μg of inhaled fluticasone furoate once daily for 14 days, or to a matching placebo. Because multiple study drugs were available, randomization occurred based on the appropriateness of each drug for the participant as determined by the eligibility criteria (Figure 1). Participants could choose to opt out of being randomized to specific drugs during the consent process.
Figure 1. Flow of Patients in the ACTIV-6 Platform Trial Investigating Repurposed Drugs for Nonhospitalized Persons With Mild to Moderate COVID-19.

ACTIV-6 indicates Accelerating COVID-19 Therapeutic Interventions and Vaccines.
aOne patient met more than 1 of the exclusion criteria.
bIn this platform trial with multiple study drugs, participants were able to choose which agents they were willing to potentially take. At the first step of randomization, participants were assigned to receive either placebo or active drug in a ratio of 1:m, where m is the number of study drug groups for which participants were eligible. The more study groups for which a participant was eligible, the greater the chance of receiving the active study drug. Subsequently, participants were randomized among the m study groups with equal probability.
At the first step of randomization, participants were assigned to receive either placebo or active drug in a ratio of 1:m, where m is the number of study drug groups for which participants were eligible. The more study groups for which a participant was eligible, the greater the chance of receiving the active study drug. Subsequently, participants were randomized among the m study groups with equal probability. The trial was designed to share information and participants who were randomized to receive placebo contributed data to all of the placebo groups for which eligibility to the matching study drug was met (Figure 1).
Interventions
Participants received a 10-day supply of either fluvoxamine or placebo matching fluvoxamine in bottles provided by the manufacturer (Apotex) via direct home delivery from a central pharmacy. Participants self-administered 50 mg of fluvoxamine or matching placebo twice daily for 10 consecutive days. For the alternative active drugs (ivermectin or inhaled fluticasone furoate), packaging for the matching placebo was identical to that of the associated drug.
Outcome Measures
The primary outcome was time to sustained recovery (defined as the third day of 3 consecutive days without symptoms). This was selected a priori from among the 2 coprimary outcomes that remained available to the other drugs (additional information about the other drugs in this platform trial appears in Supplement 1). By definition, participants who died did not recover regardless of reported freedom from symptoms.
There were 7 secondary outcomes, including hospitalization or death by day 28; the mean amount of time spent feeling unwell; COVID Clinical Progression Scale scores on days 7, 14, and 28; mortality through day 28; and the composite outcome of hospitalization, urgent care visit, emergency department visit, or death through day 28. The final secondary outcome, the PROMIS-29 (Patient-Reported Outcomes Measurement Information System 29) profile, was to be assessed through day 90 and is not reported here because of the longer follow-up.
Trial Procedures
The study was designed as a decentralized trial and all study visits were planned as remote. Screening and eligibility were self-reported by the participants and confirmed by the investigators at the study sites. A positive SARS-CoV-2 test result was verified by the investigators at the study sites prior to randomization. For the screening visit, participants self-reported demographic information, eligibility criteria, medical history, concomitant medications, COVID-19 symptoms, and completed quality-of-life questionnaires.
A central investigational pharmacy distributed the drugs (either active or placebo) to residential addresses provided by the participants, and shipping and delivery were tracked. Confirmation that the drugs were delivered to the participant’s address was required for the participant to be included in the analysis. Receipt of the study drug was defined as day 1.
Participants were asked to complete assessments and report safety events daily through the first 14 days. From days 15 to 28, participants continued to report if they had symptoms until they had experienced 3 consecutive days without symptoms. Follow-up visits occurred at days 28 and 90. At each study assessment, participants self-reported symptoms and severity, health care visits, and whether they were taking any new medications. Additional details appear in Supplement 2.
Statistical Analysis
This platform trial was designed to be analyzed with the possibility of adding and dropping drug groups as the trial progresses. Regression modeling was the general analytical approach used. Proportional hazard regression was used for the time-to-event analysis. Cumulative probability ordinal regression models were used for the ordinal outcomes. The mean time spent unwell was estimated using a longitudinal ordinal regression model as a quantification of benefit (additional information appears in the statistical analysis plan in Supplement 1).
The planned primary outcome analysis was a Bayesian proportional hazards model. The primary inferential, decision-making quantity was the posterior distribution for the treatment assignment hazard ratio (HR), with an HR greater than 1 indicating benefit. If the posterior probability of benefit exceeded 0.95 at any of the interim or final analyses, the trial would conclude efficacy of the intervention. To preserve a type I error rate of less than 0.05, the prior used for the treatment effect parameter on the loge relative hazard scale was a normal distribution centered at 0 and scaled to an SD of 0.1. All other parameter priors were noninformative using the software default of 2.5 times the ratio of the SD of the outcome divided by the SD of the predictor variable. The study design was estimated to have 80% power to detect an HR of 1.2 for the primary outcome.
In addition to randomization assignment, the primary outcome model included the following predictor variables: age (as a restricted cubic spline), sex, duration of symptoms prior to receipt of study drug, calendar time (as a restricted cubic spline), vaccination status, geographic region (Midwest, Northeast, South, and West), call center indicator, and baseline symptom severity. The proportional hazards assumption of the primary outcome was evaluated by generating visual diagnostics such as the log-log plot and plots of time-dependent regression coefficients for each predictor in the model (a diagnostic that indicates deviations from proportionality if the time-dependent coefficients are not constant in time).
The secondary outcomes were analyzed with Bayesian regression models (either proportional hazards or proportional odds). Noninformative priors were used for all parameters. The secondary outcomes were not used for formal decision-making and no decision threshold was selected. The findings for the analyses of the secondary outcomes should be interpreted as exploratory because of the potential for type I error due to multiple comparisons. The same set of covariates used in the primary outcome model was used in the analysis of the secondary outcomes, provided the outcome accrued enough events to be analyzed with covariate adjustment.
To achieve the sample size in this ongoing platform trial, once 1200 participants had been randomized to the drug group or matching placebo and had received the drug, the choice for additional participants in the drug group was removed from the platform. Some participants had already consented to participate but had not yet been randomized or received the drug at the time of the drug group closure, and these participants were allowed to continue in the trial. As a platform trial, the primary analysis is implemented separately for each drug group. The placebo group consists of all randomized participants who meet the eligibility criteria for the study drug and includes those who received the fluvoxamine-matched placebo and those who received the placebo matching an alternative drug (ivermectin or inhaled fluticasone furoate).
Based on the experience of other remotely conducted trials,13,14 it was recognized that medication delivery (placebo or drug) may not always occur (eg, delivery failure, participant withdrawal, or hospitalization). For this trial, the full analysis set for the primary outcome analysis included all participants who received fluvoxamine or placebo and the participants were analyzed as randomized. All available data were used to compare each active study drug vs placebo control, regardless of adherence to the trial protocol after randomization. In both the analyses for the primary and secondary outcomes, missing data among covariates were addressed with conditional mean imputation because the amount of missing covariate data was small (<5%).
A prespecified analysis tested for differential treatment effects as a function of preexisting participant characteristics. The analysis of heterogeneity for the treatment effect included age, the number of days with symptoms, body mass index (BMI), symptom severity on day 1, calendar time (surrogate for the circulating SARS-CoV-2 Delta or Omicron variant), sex, and vaccinations status. The continuous variables were modeled as such without creating subgroups.
The analyses were performed using R version 4.1 (R Foundation for Statistical Computing) and the following primary packages: rstanarm, rmsb, and survival.15 Additional details appear in Supplement 2.
Results
Of the 1331 participants who were randomized, 1288 completed the trial (674 in the fluvoxamine group and 614 in the placebo group; Figure 1). Of the 614 participants receiving placebo, 326 (53%) received placebo matching fluvoxamine and 288 (47%) received placebo matching an alternative drug (ivermectin or inhaled fluticasone furoate).
The median age of the participants was 47 years (IQR, 38-57 years) and 42% were aged 50 years or older (Table 1). The population was 57% female, 6.4% identified as Asian, 7.5% identified as Black or African American, 17% reported being of Hispanic or Latino ethnicity, and 81% identified as White. Although not required for enrollment, high-risk comorbidities were prevalent, including a BMI (calculated as weight in kilograms divided by height in meters squared) greater than 30 (36.4%), diabetes (9.2%), hypertension (24.4%), and asthma (13.2%). Overall, 67% of participants reported receiving at least 2 doses of a SARS-CoV-2 vaccine.
Table 1. Baseline Characteristics of Adults in the ACTIV-6 Platform Trial.
| Fluvoxamine (n = 674) | Placebo (n = 614) | |
|---|---|---|
| Age, median (IQR), y | 47.0 (37.0-56.8) | 48.0 (39.0-58.0) |
| Age group, No. (%) | ||
| <50 y | 395 (58.6) | 350 (57.0) |
| ≥50 y | 279 (41.4) | 264 (43.0) |
| Sex, No. (%) | ||
| Female | 387 (57.4) | 347 (56.5) |
| Male | 286 (42.4) | 265 (43.2) |
| Prefer not to answera | 1 (0.2) | 2 (0.3) |
| Race, No. (%)b | ||
| American Indian or Alaska Native | 6 (0.9) | 10 (1.6) |
| Asian | 45 (6.7) | 37 (6.0) |
| Black or African American | 47 (7.0) | 49 (8.0) |
| Middle Eastern or North African | 6 (0.9) | 5 (0.8) |
| Native Hawaiian or Other Pacific Islander | 2 (0.3) | 2 (0.3) |
| White | 542 (80.4) | 496 (80.8) |
| None of the above | 27 (4.0) | 15 (2.4) |
| Prefer not to answer | 9 (1.3) | 11 (1.8) |
| Ethnicity, No. (%) | ||
| Hispanic or Latino | 119 (17.7) | 102 (16.6) |
| Not Hispanic or Latino | 555 (82.3) | 512 (83.4) |
| Region, No. (%) | ||
| Midwestc | 146 (21.7) | 137 (22.3) |
| Northeastd | 56 (8.3) | 42 (6.8) |
| Southe | 341 (50.6) | 316 (51.5) |
| Westf | 131 (19.4) | 119 (19.4) |
| Recruited via call center, No. (%)g | 78 (11.6) | 92 (15.0) |
| Body mass indexh | ||
| Median (IQR) | 27.8 (24.5-32.1) | 28.1 (24.4-32.4) |
| >30, No./total (%) | 246/674 (36.5) | 223/613 (36.4) |
| Weight, median (IQR), kg | 81.6 (69.5-96.2) | 81.6 (68.0-97.5) |
| Weight >88 kg, No. (%) | 258 (38.3) | 238 (38.8) |
| Medical history, No./total (%)i | ||
| Hypertension | 153/658 (23.3) | 151/588 (25.7) |
| Asthma | 89/658 (13.5) | 75/587 (12.8) |
| Smoked within past year | 88/658 (13.4) | 76/588 (12.9) |
| Diabetes | 59/658 (9.0) | 56/588 (9.5) |
| Heart disease | 23/658 (3.5) | 30/587 (5.1) |
| Malignant cancer | 18/658 (2.7) | 24/588 (4.1) |
| Chronic obstructive pulmonary disease | 16/658 (2.4) | 14/588 (2.4) |
| Chronic kidney disease | 2/658 (0.3) | 6/588 (1.0) |
| COVID-19 vaccine status, No./total (%) | ||
| Not vaccinated | 209/670 (31.2) | 194/607 (32.0) |
| Received 1 dose of vaccine | 6/670 (0.9) | 7/607 (1.2) |
| Received ≥2 doses of vaccine | 455/670 (67.9) | 406/607 (66.9) |
| Time from symptom onset to receipt of study drug, median (IQR), d | 5 (4-7) | 5 (4-7) |
| Time from symptom onset to enrollment, median (IQR), d | 4 (2-5) | 4 (2-5) |
| Symptom burden on study day 1, No./total (%) | ||
| None | 36/622 (5.8) | 37/569 (6.5) |
| Mild | 396/622 (63.7) | 353/569 (62.0) |
| Moderate | 176/622 (28.3) | 166/569 (29.2) |
| Severe | 14/622 (2.3) | 13/569 (2.3) |
| Allowable COVID-19 medications, No. (%) | ||
| Monoclonal antibodies | 19 (2.8) | 18 (2.9) |
| Ritonavir-boosted nirmatrelvir | 8 (1.2) | 5 (0.8) |
| Remdesivir | 0 | 1 (0.2) |
Abbreviation: ACTIV-6, Accelerating COVID-19 Therapeutic Interventions and Vaccines.
Could have also selected “undifferentiated” or “unknown”; however, neither was selected by any of the participants.
May have selected any combination of the descriptors, therefore, the sum does not match the column total.
Includes Illinois, Indiana, Iowa, Kansas, Michigan, Minnesota, Missouri, Nebraska, North Dakota, Ohio, South Dakota, and Wisconsin.
Includes Connecticut, Maine, Massachusetts, New Hampshire, New Jersey, New York, Pennsylvania, Rhode Island, and Vermont.
Includes Alabama, Arkansas, Delaware, District of Columbia, Florida, Georgia, Kentucky, Louisiana, Maryland, Mississippi, North Carolina, Oklahoma, South Carolina, Tennessee, Texas, Virginia, and West Virginia.
Includes Alaska, Arizona, California, Colorado, Hawaii, Idaho, Montana, Nevada, New Mexico, Oregon, Utah, Washington, and Wyoming.
May have been recruited at local clinical sites.
Calculated as weight in kilograms divided by height in meters squared.
Self-reported based on responses to the following questions: “Has a doctor told you that you have any of the following?” and “Have you ever experienced any of the following (select all that apply)” and “Have you ever smoked tobacco products?”
The median time from symptom onset to receipt of study drug was 5 days (IQR, 4-7 days); 80% received fluvoxamine within 7 days of symptom onset and 77% received placebo (eFigure 1 in Supplement 2). Baseline symptom prevalence and severity are described in eTable 1 in Supplement 2. Although allowable per the trial protocol (Supplement 1), COVID-19 therapeutics approved or authorized for emergency use by the US Food and Drug Administration were uncommonly used (remdesivir by 0.08%, monoclonal antibodies by 2.9%, and ritonavir-boosted nirmatrelvir by 1%; Table 1).
Primary Outcome
The median time to sustained recovery was 12 days (IQR, 11-14 days) in the fluvoxamine group and 13 days (IQR, 12-13 days) in the placebo group (HR, 0.96 [95% credible interval, 0.86-1.06], posterior P = .21 for the probability of benefit [determined by an HR >1]) (Table 2 and Figure 2A). The posterior probability of the primary outcome was below the prespecified threshold of 0.95. No statistical evidence of a treatment benefit remained when using a noninformative prior, no prior, and various approaches for imputing missing symptom data (Table 2 and Figure 3).
Table 2. Primary and Secondary Outcomes.
| No./total (%)a | Adjusted estimate (95% CrI)b | Posterior P value for probability of benefit | ||
|---|---|---|---|---|
| Fluvoxamine (n = 674) | Placebo (n = 614) | |||
| Primary outcome: time to sustained recovery c | ||||
| Skeptical prior (primary analysis) | HR, 0.96 (0.86 to 1.06)d | .21 | ||
| Noninformative prior (sensitivity analysis) | HR, 0.94 (0.82 to 1.06)d | .16 | ||
| No prior (sensitivity analysis) | HR, 0.94 (0.83 to 1.07)d | |||
| Secondary outcomes e | ||||
| Composite outcome of hospitalization, urgent care visit, ED visit, or death through 28 d | 26/670 (3.9) | 23/607 (3.8) | HR, 1.1 (0.5 to 1.8)f | .35 |
| Mortality at 28 d | 0/662 | 0/599 | Not estimatedg | Not estimated |
| Hospitalization or death through 28 d | 1/670 (0.2) | 2/607 (0.3) | HR, 0.45 (0.05 to 4.99)h | Not estimated |
| COVID Clinical Progression Scalei | ||||
| At 7 d (n = 1244) | OR, 1.28 (0.72 to 1.91) | .19 | ||
| At 14 d (n = 1128) | OR, 1.17 (0.47 to 1.95) | .39 | ||
| At 28 d (n = 1126) | OR, 1.47 (0.78 to 2.3) | .10 | ||
| Amount of time spent feeling unwell, mean (95% CrI), d | 11.22 (11.00 to 11.41) | 11.27 (11.08 to 11.47) | Difference, 0.06 (−0.33 to 0.43)j | .61 |
Abbreviations: CrI, credible interval; ED, emergency department; HR, hazard ratio; OR, odds ratio.
Unless otherwise indicated.
The adjustment variables used (unless otherwise indicated) were age (as a restricted cubic spline), sex, duration of symptoms prior to receipt of the study drug, calendar time (as a restricted cubic spline), vaccination status, geographic region (Midwest, Northeast, South, and West), call center indicator, and baseline symptom severity.
Defined as the third day of 3 consecutive days without symptoms.
Estimated from receipt of study drug to time achieving sustained recovery. An HR greater than 1 is favorable for faster recovery with fluvoxamine vs placebo.
An HR less than 1, an OR less than 1, and a difference less than 0 is favorable for fluvoxamine vs placebo.
An HR greater than 1 favors placebo.
The event rate of 0 precluded analysis.
The low event rate precluded covariate adjustment.
An OR less than 1 is favorable for less progression with fluvoxamine vs placebo. The proportional odds were not evaluated because most participants were at home either with or without limitations, resulting in a model that is approximately a logistic regression. The description of the 8 levels of the scale appears in Supplement 2.
The adjustment variables used were age and calendar time.
Figure 2. Posterior Distribution Effects.
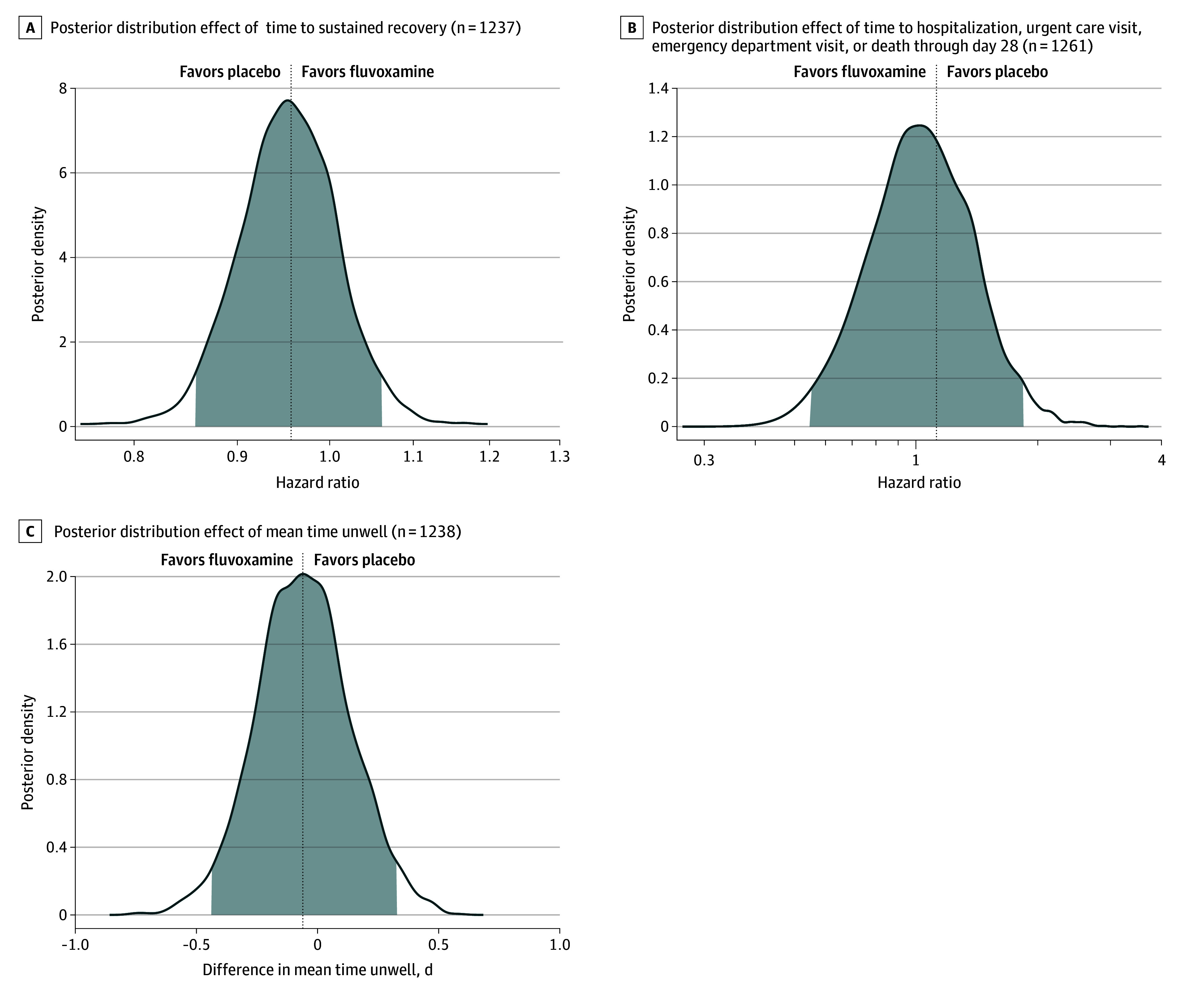
The vertical lines represent the estimated mean of the posterior distribution. Posterior density is the relative likelihood of posterior probability distribution. Outcomes with higher posterior density are more likely than outcomes with lower posterior density.
Figure 3. Primary Outcome of Time to Sustained Recovery.

Sustained recovery was defined as the third day of 3 consecutive days without symptoms. Fifty-two participants were censored for nonresponse and all others were followed up until sustained recovery, death, or the end of 28-day follow-up. The median time to sustained recovery was 12 days (IQR, 11-14 days) in the fluvoxamine group and 13 days (IQR, 12-13 days) in the placebo group.
Secondary Outcomes
For the composite outcome, 26 participants (3.9%) in the fluvoxamine group were hospitalized, had an urgent care visit, had an emergency department visit, or died compared with 23 participants (3.8%) in the placebo group (HR, 1.1 [95% credible interval, 0.5-1.8], posterior P = .35 for the probability of benefit [determined by an HR <1]; Table 2). There was only 1 hospitalization in the fluvoxamine group and there were 2 hospitalizations in the placebo group; there were no deaths in either group (Table 2 and eFigures 2A and 2B in Supplement 2). Thus, the HR on this composite outcome was uninformative.
The mean difference in the amount of time spent feeling unwell was 0.06 days (95% CrI, −0.33 to 0.43 days) in favor of placebo (Table 2 and Figure 2C). The posterior probability of any benefit observed with the COVID Clinical Progression Scale at days 7, 14, and 28 also did not meet prespecified thresholds for beneficial treatment effect (Table 2 and Supplement 2). For example, by day 7, 92.3% (542/587) of the fluvoxamine group and 93.4% (512/548) of the placebo group were not hospitalized and did not report any limitation of activities (eFigure 3 in Supplement 2).
Heterogeneity of Treatment Effect Analyses
There was no evidence of a treatment effect with fluvoxamine compared with placebo for vaccine status, timing of symptom onset, severity of symptoms, age, sex, or calendar time. There was evidence for possible differential treatment effect on time to recovery for BMI (P = .01 for heterogeneity of treatment effect), with a suggestion of increasing treatment benefit with increasing BMI (eFigure 4 in Supplement 2).
Adverse Events
Among participants who reported taking either fluvoxamine or placebo at least once, adverse events were uncommon and similar in both groups (4.7% [29/615] with fluvoxamine vs 5.3% [30/565] with placebo) (eTable 2 in Supplement 2).
Discussion
Among outpatients with mild to moderate COVID-19, treatment with 50 mg of fluvoxamine twice daily for 10 days, compared with placebo, did not improve time to sustained recovery in this large trial of 1288 participants. There was no treatment effect for the secondary outcomes, including for the composite outcome of hospitalization, urgent care visit, emergency department visit, or death through day 28. Hospitalizations and deaths were uncommon in this largely vaccinated study population. These findings do not support the use of fluvoxamine at this dose and duration in patients with mild to moderate COVID-19.
There are numerous conflicting trials for the use of fluvoxamine, and some of the differences may be attributable to dosage.7,8,10,16 Compared with the largest published trial to date (the TOGETHER trial10), the current trial has some similarities and differences. The TOGETHER trial10 used a higher dose of fluvoxamine (100 mg twice daily) compared with 50 mg twice daily in the current trial. The timing of the trial also was different. The TOGETHER trial10 reported randomization to fluvoxamine between January 20, 2021, and August 5, 2021, predating the arrival of the SARS-CoV-2 Omicron variant and the associated subvariants. By contrast, the current trial was conducted during the Delta and Omicron variant surges. In addition, the 2 trials enrolled different populations. The participants in the fluvoxamine group in the TOGETHER trial10 were unvaccinated, high-risk, symptomatic adults living in Brazil with a known risk factor for clinical progression to severe COVID-19. The current trial enrolled adults in the US regardless of COVID-19 risk factors or vaccination status. In fact, the majority (67%) of patients enrolled in this trial reported receiving at least 2 doses of a SARS-CoV-2 vaccine. Similar null results were observed in the COVID-OUT trial,16 which also tested 50 mg of fluvoxamine twice daily vs placebo in a population with the majority of participants who were vaccinated and who were overweight or obese.
This trial has several strengths. As a nationwide trial in the US, it is generalizable for all adults aged 30 years or older with COVID-19. This trial enrolled patients rapidly during the Delta and Omicron variant surges and included individuals who had been vaccinated, thus remaining a highly relevant population for the present time.
Limitations
This trial has several limitations. First, due to the broadly inclusive study population, few clinical events occurred, resulting in an inability to study the treatment effect on clinical outcomes such as hospitalization.
Second, due to the remote nature of the trial, the median time from symptom onset to receipt of study drug was 5 days, which is at the upper limit of the recommend start of antiviral medicines (≤5 days). However, significant interactions with respect to the time from symptom onset to study drug receipt were not observed, consistent with other trials.10,16
Conclusions
Among outpatients with mild to moderate COVID-19, treatment with 50 mg of fluvoxamine twice daily for 10 days, compared with placebo, did not improve time to sustained recovery. These findings do not support the use of fluvoxamine at this dose and duration in patients with mild to moderate COVID-19.
Trial protocol and statistical analysis plan
Executive committee, protocol oversight committee, clinical trial team, independent data monitoring committee, clinical events classification committee, data coordinating center
Primary site investigators and study coordinators
eMethods
COVID-19 ordinal outcome scale
eFigure 1. Time from symptom onset to receipt of drug
eFigure 2A. All-cause hospitalization or death for fluvoxamine versus placebo
eFigure 2B. All-cause hospitalization, urgent care visit, emergency department visit, or death for fluvoxamine versus placebo
eFigure 3. Participants’ clinical status at day 7, 14, and 28
eFigure 4. Heterogeneity of treatment effect between fluvoxamine and placebo for time to recovery
eTable 1. Baseline symptom prevalence and severity
eTable 2. Adverse events
Nonauthor collaborators
Data sharing statement
References
- 1.Ahmad FB, Cisewski JA, Anderson RN. Provisional mortality data—United States, 2021. MMWR Morb Mortal Wkly Rep. 2022;71(17):597-600. doi: 10.15585/mmwr.mm7117e1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hammond J, Leister-Tebbe H, Gardner A, et al. ; EPIC-HR Investigators . Oral nirmatrelvir for high-risk, nonhospitalized adults with Covid-19. N Engl J Med. 2022;386(15):1397-1408. doi: 10.1056/NEJMoa2118542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jayk Bernal A, Gomes da Silva MM, Musungaie DB, et al. ; MOVe-OUT Study Group . Molnupiravir for oral treatment of Covid-19 in nonhospitalized patients. N Engl J Med. 2022;386(6):509-520. doi: 10.1056/NEJMoa2116044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guy RK, DiPaola RS, Romanelli F, Dutch RE. Rapid repurposing of drugs for COVID-19. Science. 2020;368(6493):829-830. doi: 10.1126/science.abb9332 [DOI] [PubMed] [Google Scholar]
- 5.Wiltz JL, Feehan AK, Molinari NM, et al. Racial and ethnic disparities in receipt of medications for treatment of COVID-19—United States, March 2020-August 2021. MMWR Morb Mortal Wkly Rep. 2022;71(3):96-102. doi: 10.15585/mmwr.mm7103e1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sukhatme VP, Reiersen AM, Vayttaden SJ, Sukhatme VV. Fluvoxamine: a review of its mechanism of action and its role in COVID-19. Front Pharmacol. 2021;12:652688. doi: 10.3389/fphar.2021.652688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee TC, Vigod S, Bortolussi-Courval É, et al. Fluvoxamine for outpatient management of COVID-19 to prevent hospitalization: a systematic review and meta-analysis. JAMA Netw Open. 2022;5(4):e226269. doi: 10.1001/jamanetworkopen.2022.6269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lenze EJ, Mattar C, Zorumski CF, et al. Fluvoxamine vs placebo and clinical deterioration in outpatients with symptomatic COVID-19: a randomized clinical trial. JAMA. 2020;324(22):2292-2300. doi: 10.1001/jama.2020.22760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rosen DA, Seki SM, Fernández-Castañeda A, et al. Modulation of the sigma-1 receptor-IRE1 pathway is beneficial in preclinical models of inflammation and sepsis. Sci Transl Med. 2019;11(478):eaau5266. doi: 10.1126/scitranslmed.aau5266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reis G, Dos Santos Moreira-Silva EA, Silva DCM, et al. ; TOGETHER investigators . Effect of early treatment with fluvoxamine on risk of emergency care and hospitalisation among patients with COVID-19: the TOGETHER randomised, platform clinical trial. Lancet Glob Health. 2022;10(1):e42-e51. doi: 10.1016/S2214-109X(21)00448-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lenze E. Fluvoxamine for Early Treatment of Covid-19 (STOP COVID 2). Accessed December 29, 2022. https://clinicaltrials.gov/ct2/show/NCT04668950
- 12.Seftel D, Boulware DR. Prospective cohort of fluvoxamine for early treatment of coronavirus disease 19. Open Forum Infect Dis. 2021;8(2):ofab050. doi: 10.1093/ofid/ofab050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boulware DR, Pullen MF, Bangdiwala AS, et al. A randomized trial of hydroxychloroquine as postexposure prophylaxis for Covid-19. N Engl J Med. 2020;383(6):517-525. doi: 10.1056/NEJMoa2016638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Skipper CP, Pastick KA, Engen NW, et al. Hydroxychloroquine in nonhospitalized adults with early COVID-19: a randomized trial. Ann Intern Med. 2020;173(8):623-631. doi: 10.7326/M20-4207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.R Core Team . R: A language and environment for statistical computing. R Foundation for Statistical Computing. Accessed October 25, 2022. http://www.R-project.org/
- 16.Bramante CT, Huling JD, Tignanelli CJ, et al. ; COVID-OUT Trial Team . Randomized trial of metformin, ivermectin, and fluvoxamine for Covid-19. N Engl J Med. 2022;387(7):599-610. doi: 10.1056/NEJMoa2201662 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Trial protocol and statistical analysis plan
Executive committee, protocol oversight committee, clinical trial team, independent data monitoring committee, clinical events classification committee, data coordinating center
Primary site investigators and study coordinators
eMethods
COVID-19 ordinal outcome scale
eFigure 1. Time from symptom onset to receipt of drug
eFigure 2A. All-cause hospitalization or death for fluvoxamine versus placebo
eFigure 2B. All-cause hospitalization, urgent care visit, emergency department visit, or death for fluvoxamine versus placebo
eFigure 3. Participants’ clinical status at day 7, 14, and 28
eFigure 4. Heterogeneity of treatment effect between fluvoxamine and placebo for time to recovery
eTable 1. Baseline symptom prevalence and severity
eTable 2. Adverse events
Nonauthor collaborators
Data sharing statement