

Abstract
Enhancing the recellularization of a decellularized heart valve in situ may lead to an improved or ideal heart valve replacement. A promising approach is leveraging the immune response for inflammation-mediated recellularization. However, this mechanism has not been previously demonstrated in vitro. This study investigated loading the chemokine MCP-1 into decellularized porcine heart valve tissue and measured the migration of human peripheral blood mononuclear cells (PBMCs) and mesenchymal stem cells (MSCs) toward the chemokine loaded valve tissue. The results of this study demonstrate that MCP-1-loaded tissues increase PBMC migration compared to non-loaded tissues. Additionally, we demonstrate MCP-1-loaded tissues that have recruited PBMCs lead to increased migration of MSCs compared to decellularized tissue alone. The results of this study provide evidence for the inflammation-mediated recellularization mechanism. Furthermore, the results support the use of such an approach for enhancing the recellularization of a decellularized heart valve.
Keywords: Tissue engineering, monocyte chemoattractant protein, recellularization
Introduction
Decellularized heart valves have demonstrated great potential as a standalone heart valve replacement and as a scaffold for heart valve tissue engineering.32 Early attempts using xenogeneic decellularized heart valves in clinical trials were unsuccessful and demonstrated the critical need for effective decellularization and antigen removal.25 However, more recent clinical studies using allogeneic decellularized valves have demonstrated increased durability over cryopreserved (non-decellularized) counterparts, the current gold standard for pediatric patients.3 This is encouraging since the primary mode of failure for the cryopreserved allograft is a lack of durability as the tissue degrades, leading to valve stenosis or regurgitation. While decellularized allografts have demonstrated increased freedom from reintervention, the results are only marginally improved.3, 16 Further improving the durability of a decellularized valve will require tissue engineering strategies to encourage recellularization of the decellularized tissue by the patient’s cells, creating living tissue and allowing for continual growth and remodeling after implantation.32 To this end, decellularized heart valves are well suited due to their inherent physiologic architecture and acellular nature. Indeed, both clinical and animal trials have demonstrated partial autologous recellularization of decellularized heart valves post-implantation.1, 7, 10, 21 However, the whole valve does not recellularize and incomplete recellularization, particularly of the distal edge of the leaflet tissue, is thought to ultimately lead to valve failure.16 Therefore, enhancing the recellularization of decellularized heart valves could lead to an improved or even ideal heart valve replacement.
A promising approach is to enhance recellularization by taking advantage of the body’s own paracrine signaling using in vivo tissue engineering. Recent studies have identified a substantial role of inflammation and monocyte migration in natural wound healing and have leveraged these pathways for regenerative therapies, such as cardiovascular tissue engineering.5, 15, 17, 28 Seeding cardiovascular constructs with mononuclear cells has been shown to aid in recellularization via cytokine signaling by the seeded cells.24, 29, 33 The inflammatory chemokine monocyte chemoattractant protein-1 (MCP-1; chemokine ligand-2, CCL2) has been identified as one of the primary recruiters of monocytes in vivo. MCP-1 acts via its receptor CCR2, which is constitutively expressed by monocytes and contingently expressed by lymphocytes.9 Many human cells are capable of producing MCP-1, which functions by creating a concentration gradient leading to migration and recruitment of circulating monocytes.23 Additionally, increased systemic levels of MCP-1 has been shown to increase systemic circulating monocyte production from the bone marrow.23 A previous study by Roh et al. demonstrated that seeding mononuclear cells onto a polymeric vascular graft in mice led to recellularization by native vascular cells.22 Furthermore, when MCP-1 was included into the polymer vascular grafts, they observed similar recellularization outcomes. As a result, the group proposed a mechanism of inflammation-mediated recellularization wherein MCP-1 recruits native monocytes, which release further signaling cytokines leading to the subsequent recruitment of vascular cells. However, this mechanism has not been definitely demonstrated and the processes leading to repopulation by cardiovascular cells are poorly understood.
The goal of this study was to demonstrate the mechanism of chemokine induced monocyte migration leading to the subsequent migration of a re-populating cell type. Additionally, this study aimed to investigate the feasibility of using such a mechanism to enhance the recellularization of a decellularized heart valve. For the purposes of this study, mesenchymal stem cells (MSCs) were considered an appropriate re-populating cell type because of their similar phenotype to valve interstitial cells (VICs), their ability to differentiate into VICs, and their presence in circulating blood, albeit at a low concentration.17, 26 To demonstrate the mechanism, we first evaluated the loading and release of MCP-1 onto decellularized valve tissues and then used Boyden chamber migration assays to measure the migration of PBMCs and MSCs toward the chemokine-loaded tissues.
Materials and Methods
Tissues and Tissue Processing
Tissue samples were prepared from decellularized porcine pulmonary valves. Whole porcine hearts were obtained fresh from a commercial source (Sierra for Medical Science) and the pulmonary valves were dissected upon arrival. Following removal from the heart, the valves were washed in PBS and 1% Anti-Anti overnight at 4 °C and then stored frozen at −80 °C until decellularization. Decellularization was performed on the whole valve following a previously established protocol.31 Briefly, the pulmonary valves were subjected to reciprocating osmotic shock, detergent washes (Triton X-100 and sodium-lauroyl sarcosinate), and enzymatic (Benzonase) removal of nuclear material. Then, extraction of organic material was performed using ion exchange resins in water. Following decellularization, the valves were dissected and tissue samples were obtained using a sterile 7mm punch from the belly of the leaflet region and the sinus of the conduit region. The tissue samples were again frozen at −80 °C until further use.
Cells and Cell Characterization
Human peripheral blood mononuclear cells (PBMCs) were obtained from research designated Plateletpheresis Leukoreduction Filters (LRS Chambers) purchased from a blood donation center (Vitalent). The isolation of PBMCs from the LRS chambers was performed as described by others.18 Briefly, the contents of each LRS chamber was drained and further purified via density gradient centrifugation using Ficoll-Paque media. The buffy coat layer was removed following density gradient centrifugation and washed in serum free cell media (RPMI; Gibco). Finally, the cells were counted and resuspended in freeze media (RPMI supplemented with 20% FBS and 10% dimethyl sulfoxide) to be cryopreserved in liquid nitrogen until further use.
Human PBMCs were obtained from three LRS Chambers from three separate donors and were independently characterized by flow cytometry. For flow analysis, the PBMCs were quickly thawed from cryopreservation and resuspended in complete cell media (RPMI supplemented with 10% FBS and 1% Pen-Strep) at a concentration of 1x106 cells ml−1. The cells were then stained with the following conjugated antibodies : anti-CD45 (FITC; BD Biosciences), anti-CD34 (PE; BD Biosciences), anti-CD14 (PE-Cy7; BD Biosciences), anti-CD4 (APC; BD Biosciences), anti-CD8 (Alexa fluor 700; BD Biosciences), anti-CD192 (APC-Cy7; Biolegend) and anti-CD16 (BV510; BD Biosciences). Cell viability was assessed with DAPI. The labelled cells were washed and counted using a benchtop flow cytometer (Gallios 561). Gating and data analysis were performed with the Kaluza software (Representative gating strategy shown in Supplemental Figure 1).
Human mesenchymal stem cells (MSCs) were purchased from a commercial source (Fisher Scientific) and cultured on tissue culture plastic in DMEM/F12 supplemented with 10% FBS and 1% Pen-Strep. Upon reaching 70-80% confluency, the MSCs were passaged with Accutase (Stemcell Technologies) and culture continued until a sufficient number were available for storage. The MSCs were then resuspended in freeze media and cryopreserved in liquid nitrogen until further use. When needed, the MSCs were quickly thawed and resuspended in complete DMEM/F12. The cells were then re-plated on tissue culture plastic and cultured until use.
MCP-1 Loading and Release
Decellularized pulmonary valve leaflet and conduit samples were loaded with MCP-1 to analyze the release profile and for use in the chemotactic assays. MCP-1 loading of the decellularized tissue was performed by soaking the samples in a solution of PBS containing 100 ng ml−1 of MCP-1 and 0.1% BSA as a carrier protein for 24 hours on an orbital shaker. Following MCP-1 loading, the release rate of MCP-1 from the decellularized tissues was measured. MCP-1-loaded tissue sections (n=3 leaflet and n=3 conduit) were placed into new tubes of PBS with 0.1% BSA and placed on an orbital shaker. Samples of the release solution were taken at 0, 0.5, 1, 4, 24, and 48 hours. The concentration of MCP-1 in the release solutions was detected using an enzyme linked immunosorbent assay (ELISA) kit (DuoSet ELISA) following the manufacturers protocol. The resulting absorbances were measured at 450 nm using a microplate reader (VersaMax).
MCP-1 distribution within the tissue
MCP-1-loaded tissue samples were processed for histology to visualize the distribution of MCP-1 within the tissue. The tissues were fixed in formalin and embedded in paraffin before being sectioned onto glass slides. The tissue sections were blocked in 10% FBS and stained using a rabbit anti-MCP-1 primary antibody (Abcam; 1:100 dilution) and goat anti-rabbit secondary antibody (ThermoFisher Scientific; 1:200 dilution). The sections were counterstained with DAPI to visualize the scaffolds and confirm the absence of cell nuclei in the decellularized tissues. Images were captured on a Zeiss microscope.
Boyden Chamber Chemotaxis Assays
The chemotactic migration of cells was measured using a variety of Boyden chamber migration assays, depending on the experiment performed. First, the migration toward an MCP-1 concentration by the monocytes from the PBMC populations was evaluated. PBMCs were quickly thawed from cryopreservation and resuspended in complete cell media at a concentration of 1x106 cells ml−1. PBMCs (100,000 cells) were placed into the top chamber of a Boyden chamber in a 24 well plate using a PET filter with 5 μM pores (Corning). The bottom chamber was then filled with complete cell media supplemented with 0, 5 10, 25, 50, 100, 250, or 500 ng ml−1 of MCP-1 (n=3 wells per concentration). Following 4 hours of incubation at 37 °C and 5% CO2, the filters were removed and fixed in 70% ethanol for 10 minutes. The insides of the filter was then wiped with a cotton swab to remove cells that did not migrate and the filter was stained with hematoxylin for 10 minutes to visualize migrated cells. The filter was then washed in water, cut from the frame, and mounted onto a glass slide. The number of migrated cells were counted with a high power field (20x, Zeiss microscope) from 5 regions of each filter and the average migrated cells per area was calculated. Three filters were tested for each MCP-1 concentration and the entire experiment was performed with each of the three PBMC lines to confirm similar results between individuals. In all cell migration experiments the number of migrated cells was counted from bottom of the filter. This was to provide consistency between experiments, since future experiments contain tissue samples and upon observation there were very few cells suspended in the media.
Next, the migration of PBMCs toward MCP-1-loaded or non-loaded tissue was measured. MCP-1 loading into decellularized leaflet and conduit sections was performed as described earlier (n=5 per tissue type). Control, non-loaded leaflet and conduit sections were also included and were incubated for 24 hours in a solution of PBS with 0.1% BSA prior to testing (n=5 per tissue type). The MCP-1-loaded or non-loaded tissue sections were then used in a Boyden chamber assay using 5 μm pore filters as described previously. The tissue samples were placed into the bottom chamber along with complete cell media. A control group with only complete cell media in the bottom chamber was also included (n=4). PBMCs were prepared as previously described and placed in the top chamber (100,000 total cells). The plate was then incubated for 4 hours at 37 °C and 5% CO2. Following incubation, the insides of the filters were similarly wiped with a cotton swab and the whole filter was then fixed, stained, washed, cut from the frame, and mounted onto glass slides. Again, the number of migrated cells were counted with a high power field (20x) from 5 regions of each filter and the average migrated cells per area was calculated.
Finally, the successive migration of MSCs following PBMC migration was evaluated. Conduit sections of decellularized pulmonary valve were loaded with MCP-1 in a 100 ng/mL loading solution as described earlier. Only conduit sections were used due to the small amount of leaflet tissue available from each valve. PBMCs were thawed, resuspended at a concentration of 1x106 cells ml−1, and labeled with DiO cell labeling solution (5 μL/mL of media; ThermoFisher Scientific) for 20 minutes. MSCs were cultured until sufficient numbers were available and were labeled with DiI cell labeling solution (5 μL/mL; ThermoFisher Scientific) for 20 minutes. The MSCs were then washed and resuspended in complete cell media at a concentration of 1x105 cells ml−1. The MSC migration study included six groups, the test group and five controls (Figure 1). In the test group, MCP-1-loaded tissue sections were placed in the bottom chamber of a Boyden chamber assay using a 5 μm filter and the top chamber was filled with labeled PBMCs (100,000 cells). After four hours incubation, the PBMC filter was removed and an 8 μM pore filter was placed in the well with labeled MSCs (10,000 cells) in the top chamber. This group tested the subsequent migration of MSCs following PBMC migration (n=5). Additional groups (n=5 per group) included only MCP-1-loaded tissue in the bottom chamber, labeled PBMCs (100,000 cells) placed directly in the bottom chamber, and a negative control media only group. A positive control group was also included using the known stem cell chemoattractant stromal cell-derived factor 1 (SDF1) at a concentration of 100 ng/mL. Lastly, since FBS is a known chemoattractant, an additional negative control media only group without FBS was included using insulin-transferrin-sodium selenite supplement (Sigma Aldrich). For all groups, MSC migration occurred for 8 hours in an incubator (37 °C and 5% CO2) using 8 μM pore filters. After incubation, the insides of the filters were wiped with a cotton swab and the filters were fixed in formalin, washed in PBS, cut from the frame, and mounted onto glass slides with a DAPI fluorescent mounting media. The whole filter was imaged using tiled images at 5x. Since the cotton swab could not wipe the outside edges of the filter, they were excluded from cell counting. The area of the counting region and number of migrated MSCs were calculated using Zeiss software.
Figure 1:

Groups included for the MSC migration assay. (+)MCP-1 & Migrated PBMCs used MCP-1-loaded tissue to recruit PBMCs for 4 hours before the filter was switched and the top chamber was filled with MSCs. A (+)MCP-1 tissue control group used MCP-1-loaded tissue directly with MSC migration. A PBMC control group included PMBCs placed directly in the bottom chamber. A media only control used only media in the bottom chamber.
Cell Seeding on MCP-1 Tissue Samples
PBMCs or PBMCs plus MSCs were seeded onto MCP-1-loaded and non-loaded tissue samples to investigate cell-matrix and cell-cell interactions. Decellularized pulmonary valve leaflet and conduit samples were taken using a 7 mm punch tool. MCP-1 loading of the tissue samples was performed as described previously. Loaded and non-loaded tissue samples were then seeded with 200 μL of media containing 2x106 PBMCs or a combination of 2x106 PBMCs and 5x104 MSCs. Following 24h of culture, the samples were fixed and processed for histology using H&E staining and immunohistochemistry (IHC). For IHC staining, the sections were blocked in 10% FBS and stained using mouse anti-CD68 (Invitrogen; 1:100 dilution) and rabbit anti-CD206 (Proteintech; 1:100 dilution) primary antibodies. Goat anti-mouse and anti-rabbit secondary antibodies (ThermoFisher Scientific; 1:200 dilution) were then applied and the sections were counterstained with DAPI. Images were captured on a Zeiss microscope.
Statistical Analysis
Statistical analysis was performed using Prism software (Graphpad). Differences between groups were detected by analysis of variance (ANOVA), and post-hoc comparisons were made using the Tukey test. Differences between groups were considered statistically significant at p < 0.05.
Results
LRS chambers provide an easily obtainable PBMC source
PBMC isolation from the LRS chambers was easy and efficient. Each chamber provided approximately 500 million viable cells. Flow cytometry analysis of the PBMC populations determined the cell phenotypes are similar to previously reported results and are considered a normal distribution of PBMCs from a healthy adult (Table 1).18 Human monocytes are comprised of three subtypes which can be distinguished by their CD16 and CD14 expression: Mon1 (CD14+/CD16−), Mon2 (CD14+/CD16+), and Mon3 (CD14+/CD16+). 27 It is the Mon1 and Mon2 subtypes, those that express CD14+, that have also been shown to express CCR2 (CD192). The CD192+ monocytes are the cells that are targeted for recruitment with the MCP-1-loaded tissue scaffolds. Our gating strategy did not account for the Mon3 population, but because those cells do not express CD192 they are of little interest for this study. Other cell types that may express CD192 include Th cells, B cells, and dendritic cells.
Table 1:
Cell phenotypes of PBMC populations isolated from the LRS chambers and analyzed by flow cytometry. Data is presented as the mean percentage +/− standard deviation of the total cell population (n=3). Numbers in parentheses are the mean percentage from the gating population shown in parentheses. The ‘Other’ cell population is the remaining CD45 + cells after the lymphocytes, monocytes, and endothelial progenitor cells (EPCs) were identified and contains cells such as natural killer cells, neutrophils, and dendritic cells.
| Cell Type | |
|---|---|
| Live | 87.94% +/− 9.41% |
| CD45+ | 95.13% +/− 6.00% |
| Lymphocytes | 52.18% +/− 5.80% |
| CD4+ (% of Lymphs) | 41.35% +/− 6.05% (79.27% +/−7.99%) |
| CD8+ (% of Lymphs) | 10.69% +/− 4.10% (20.44% +/− 7.83%) |
| DP+ T Cells (CD4+,CD8+) (% of Lymphs) | 0.15% +/− 0.08% (0.29% +/− 0.17%) |
| Monocytes | 7.83% +/− 3.69% |
| Mon1 (CD14+) (% of Monos) | 7.21% +/− 3.85% (90.12% +/− 6.41%) |
| Mon 2 (CD14+CD16+) (% of Monos) | 0.61% +/− 017% (9.68% +/− 6.48%) |
| CD192+ | 15.18% +/− 5.82% |
| EPCs | 0.10% +/− 0.03% |
| Other | 39.88% +/− 2.95% |
| Cell Type | |
|---|---|
| Live | 87.94% +/− 9.41% |
| CD45+ | 95.13% +/− 6.00% |
| Lymphocytes | 52.18% +/− 5.80% |
| CD4+ (% of Lymphs) | 41.35% +/− 6.05% (79.27% +/−7.99%) |
| CD8+ (% of Lymphs) | 10.69% +/− 4.10% (20.44% +/− 7.83%) |
| DP+ T Cells (CD4+,CD8+) (% of Lymphs) | 0.15% +/− 0.08% (0.29% +/− 0.17%) |
| Monocytes | 7.83% +/− 3.69% |
| Mon1 (CD14+) (% of Monos) | 7.21% +/− 3.85% (90.12% +/− 6.41%) |
| Mon 2 (CD14+CD16+) (% of Monos) | 0.61% +/− 017% (9.68% +/− 6.48%) |
| CD192+ | 15.18% +/− 5.82% |
| Other (Tcell neg, CD16/CD14−, & CD192−) | 39.88% +/− 2.95% |
| EPCs | 0.10% +/− 0.03% |
MCP-1 Loading & Release from Decellularized Tissue
Immunohistochemistry staining of the MCP-1-loaded tissue samples demonstrated that MCP-1 was present throughout the thickness of tissue sections (Figure 2A). Notably, there is also an absence of cell nuclei, confirming removal of nuclear material by decellularization.
Figure 2:

(A) MCP-1 staining of decellularized pulmonary valve leaflet and conduit samples demonstrates distribution of MCP-1 throughout the tissue. Secondary Only group did not include an anti-MCP-1 antibody in order to evaluate background staining. Scale bars represent 100 μm. (B) MCP-1 release from leaflet and conduit tissue as measured by ELISA. Leaflet tissue exhibited a burst release profile within the first hour and slowly increased out to 24 hours. Conduit tissue exhibited a slower release with MCP-1 release continuing up to 48 hours. Data points represent the mean concentration with dotted lines representing standard deviation (n=3).
The release of MCP-1 from the loaded tissue scaffolds followed a burst release profile from both the leaflet and conduit tissue (Figure 2B), though the leaflet exhibited a faster burst release. Most of the MCP-1 was released from the leaflet tissue in the first hour and all of MCP-1 appears to be released within 24 hours. The conduit tissue exhibited a more prolonged release profile, with most MCP-1 being released within the first 4 hours but continuing to release for 48 hours. However, the total amount of MCP-1 released per tissue weight appears similar between tissue types. The difference in release profiles is likely due to the increased thickness of the conduit tissue compared to the leaflet tissue, since MCP-1 adsorbed within the conduit will release slower. Additionally, the conduit samples had a greater mass than the leaflet samples. The MCP-1 is likely loaded through adsorption to the surface of the extracellular matrix proteins and therefore the conduit samples had a greater amount MCP-1 loaded. Leaflet samples released an average of 1.3 +/− 0.3 ng of MCP-1 and conduit samples released an average of 2.9 +/− 0.4 ng of MCP-1. The slower release of the conduit samples may also be due to decreased diffusion kinetics as a greater overall amount of MCP-1 was released from the conduit. It is also worth noting the amount of MCP-1 released represents 0.5% and 1.1% of the total MCP-1 in the loading solutions for the leaflet and conduit samples, respectively.
MCP-1-Loaded Tissue Increases PBMC Migration
The initial chemotaxis assays using direct concentrations of MCP-1 were used to evaluate the degree of monocyte migration to MCP-1 from the PBMC populations that were obtained from the LRS chambers. As expected, MCP-1 increases cell migration and increased concentrations of MCP-1 have an increased effect (Figure 3). We found that any tested amount of MCP-1 significantly increased the amount of cell migration compared to a baseline media without MCP-1. It was also demonstrated that the higher concentrations of 250 and 500 ng ml−1 demonstrated significantly greater migration than the lower concentrations of 5 and 10 ng ml−1.
Figure 3:
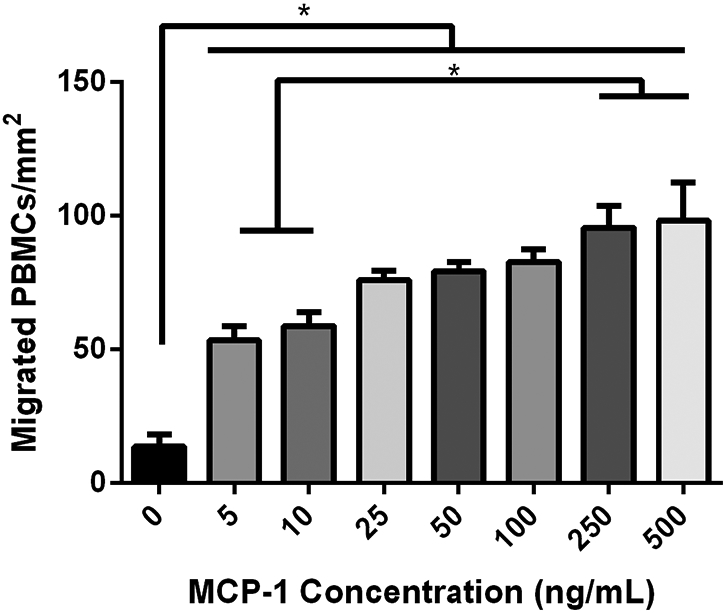
PBMC migration toward various MCP-1 concentrations. Any tested amount of MCP-1 significantly increased PBMC migration and increased concentrations had an increased effect. Data represents the mean ± SEM of the three PBMC cell lines (n=3 for each cell line). * indicates a statistically significant difference between groups (p<0.05).
Next, the PBMC migration toward decellularized valve tissues either loaded with MCP-1 or without were evaluated with chemotaxis assays (Figure 4). These experiments measured significantly increased cell migration in both the MCP-1-loaded conduit and leaflet tissues compared to the non-loaded conduit and leaflet tissues and compared to media only (p<0.001). In fact, more than four times increased migration was observed in the MCP-1-loaded tissues than in the non-loaded tissues. No significant differences were measured between the non-loaded tissues and the media only group. However, the media only group does show a slight increase in cell migration over the non-loaded tissues. While not compared statistically, it is also worth noting the magnitude of migration by the MCP-1-loaded tissues, using a loading solution of 100 ng ml−1 of MCP-1, was comparable to the migration observed in the prior assay at a 100 ng ml−1 MCP-1 concentration. Histology of the tissue samples used in the migration assays revealed only isolated cells present on the tissue surface and no cellular infiltration into any of the tissues (data not shown). Very few cells were suspended in the cell media. Cells were observed on the bottom of the well plates but were not counted since some samples contained tissues.
Figure 4:

PBMC migration toward MCP-1-loaded and non-loaded leaflet and conduit tissues (n=5). A media only group was included with no tissue in the bottom chamber (n=4). MCP-1-loaded tissues led to significantly increased PBMC migration over the non-loaded tissues and more than cells only. **** indicates a statistically significant difference between groups ( p<0.0001).
MSC Migration is Increased toward Recruited PBMCs
Finally, the chemotaxis assays were used to measure the migration of MSCs following the recruitment of PBMCs to the MCP-1-loaded valve tissue. Only conduit tissue was used in this assay due to the small amount of leaflet tissue available from each valve. Additionally, the previous tissue chemotaxis assays determined that MCP-1-loaded conduit and leaflet tissues induced similar PBMC migration. Imaging of the filters revealed MSC migration occurred within all groups, clearly evidenced by the DiI cell labeling (Figure 5). PBMCs labeled with DiO were observed occasionally on the filters from groups containing PBMCs, but were not present in other groups, as expected (not shown). It is obvious in Figure 5 where the outside edge of the filters could not be wiped with the cotton swab and the light blue circles on each filter denote the region of the filter where migrated cells were counted (Figure 5). From figure 5, it can be seen the SDF1 group and the group containing PBMCs recruited by MCP-1-loaded tissue appeared to have more cells in the middle region of the filter compared to other groups. Results from the migration assay revealed that recruited PBMCs increased the amount of MSC migration toward decellularized valve tissue (Figure 6). The MCP-1-loaded tissue & recruited PBMC group demonstrated significantly more migrated MSCs than the MCP-1-loaded tissue only group. The positive control group, SDF1, also demonstrated significantly more migrated MSCs than the MCP-1-loaded tissue only or the PBMCs only groups. Finally, the MCP-1-loaded tissue & recruited PBMC group and the SDF1 group demonstrated significantly more MSC migration than the media only group without FBS. In the media only group with FBS one of the samples was not included since it was a major outlier at 10x the difference of the first and third quartiles. It is worth noting, there was not a significant difference between the tissue & migrated PBMC group and the media only group with FBS. Since FBS is a known chemoattractant it is difficult to separate the effect of the recruited PBMCs compared to FBS. However, since the ultimate goal is recellularization of the valve tissue, and the tissue & migrated PBMC group showed significantly more MSC recruitment than the tissue only group, both of which contained FBS, these results are highly encouraging.
Figure 5:

Representative filter images from the MSC migration assays. MSCs are labeled with DiI (red), PBMCs are labeled with DiO (green; not visible at scale), and cell nuclei are counter stained with DAPI (blue). White dotted lines represent filter edges and the blue circles represent the region used to measure cell migration. Scale bars represent 1mm.
Figure 6:
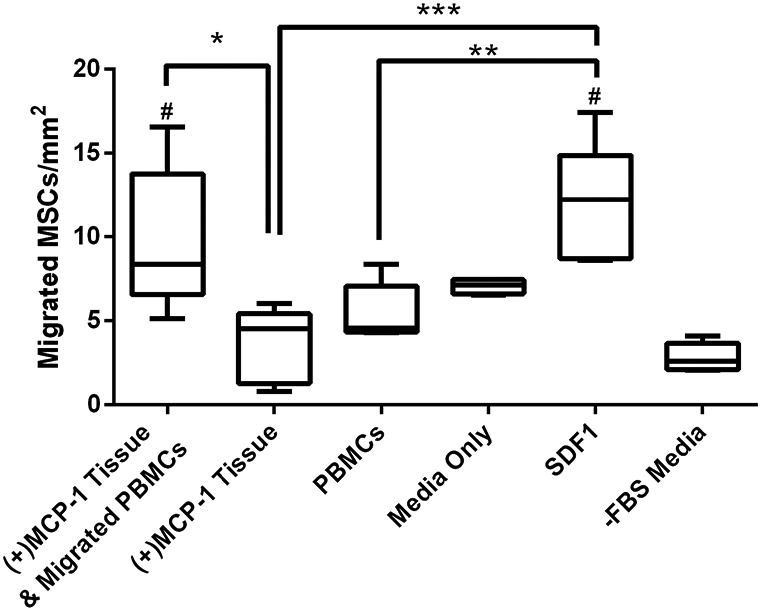
MSC migration toward recruited PBMCs and MCP-1-loaded tissue samples (n=5 per group; n=4 for Media Only group). (+)MCP-1 Tissue & migrated PBMCs allowed PBMC migration to the tissue before the filter was switched for MSC migration. (+)MCP-1 Tissue group included only MCP-1-loaded tissue in the bottom chamber. The PBMC group included PBMCs directly placed in the bottom chamber. A media only group with FBS and a media only group without FBS were used as negative controls. The SDF1 group was included as a positive control. Pre-recruited PBMCs and MCP-1-loaded tissue significantly increased MSC migration compared to only tissue in the bottom well. # indicates a significant difference form the -FBS Media only group and * indicates a statistically significant difference between groups (p<0.05).
Cell Seeding on Tissue Samples
PBMCs or PBMCs and MSCs were seeded onto MCP-1 loaded and non-loaded tissue samples to investigate cell-matrix and cell-cell interactions (Figure 7). Among all groups there was very little cellular infiltration into the decellularized tissue, with only the leaflet samples demonstrating minor cell infiltration. There was very little CD68 expression in any samples, yet CD206, a commonly used marker for alternative macrophage polarization, was expressed in all samples suggesting the monocytes were starting toward a M2 polarization. Seeding the tissue samples with both PBMCs and MSCs appeared to increase CD206 expression, which supports the immunomodulatory properties of MSCs described by others. Therefore, the two cell types may enhance each other such that PBMCs increase the MSC migration and MSCs can then influence the M2 monocyte/macrophage polarization.
Figure 7:

Representative images from seeding PBMCs or PBMCs and MSCs onto MCP-1-loaded (A, B, E, F, I, J, M, N) and non-loaded (C, D, G, H, K, L, O, P) tissue samples. H&E staining (A-H) revealed cells were only on the surface of conduit samples (A-D) and only occasional cells infiltrated into leaflet samples (E-H). CD68 (green) and CD206 (red) staining (I-P) revealed very few cells expressed CD68 but all samples had cells expressing CD206. Samples seeded with both PBMCs and MSCs demonstrated a greater expression of CD206 (J, N, L, P).
MCP-1 loading of the tissue samples did not appear to affect cell-matrix interactions, since similar amounts of cells were present on the tissue surface and there was not an observed increase of infiltration into the tissue. While increased cellular infiltration would be encouraging, the lack of infiltration in the MCP-1-loaded groups was not unexpected since static culture conditions were used. We have demonstrated in previous studies that cells seeded on whole valves do not infiltrate under static conditions, but infiltration increases after bioreactor conditioning.4, 30 Since implanted decellularized valves have also demonstrated partial cellular repopulation in human and animal studies, the lack of cellular infiltration in this study does not discount this approach. Finally, based on the CD68/CD206 staining it also appears that MCP-1 did not have an effect on the cell phenotype or macrophage polarization.
Discussion
Previous clinical studies and animal models have demonstrated that tissue engineered cardiovascular grafts can be recellularized by seeding constructs with mononuclear cells or by directly including chemokines into the construct, such as MCP-1.22, 33 This led to the theory of inflammation-mediated recellularization, wherein chemokines attract increased inflammatory cells, which release further signaling molecules leading to the recruitment of a cardiovascular cell type.22 However, this mechanism had not been definitively shown prior to this study. Our results collectively demonstrate that MCP-1 can increase the migration of PBMCs toward an acellular scaffold, which subsequently increases the migration of MSCs. Furthermore, our results indicate such an approach may be applied to heart valve tissue engineering using decellularized scaffolds. A prior study by Hopper et al. also demonstrated that the secretome of PBMCs led to increased MSC migration.13 However, our study is the first to demonstrate the use of MCP-1 to first recruit PBMCs followed by MSCs and to demonstrate the migration toward heart valve tissue.
As mentioned, there has been increasing interest in the role of the immune system for regenerative therapies and modulating such pathways to enhance outcomes.12 For example, while it was originally believed cell therapy for infarcted myocardium acts via integration of seeded cells, it is now been shown to act via paracrine signaling and recruitment of autologous cells.36 In fact, mononuclear cells injected into a mouse heart post-myocardial infarction were shown to increase recruitment of CCR2+ and CX3CR1+ monocytes, leading to increased heart function.29 While the recent surge of interest into leveraging inflammation for regeneration is promising, we must continue to exercise caution when provoking inflammatory pathways since they are often elaborately connected, with a single cytokine capable of binding many receptors and eliciting a myriad of effects.23 Many of the inflammatory signaling pathways are not fully understood and inducing a chronic inflammatory response would be antagonistic to tissue repair and cause further harm. For example, while we demonstrated a mechanism using MCP-1 to increase MSC migration, MCP-1 is also a known mediator in many chronic inflammatory pathologies, such as atherosclerosis.9 While MCP-1 is implicated in various roles of both inflammatory healing and chronic inflammation, its primary role appears to simply direct the movement of monocytes to a site of inflammation.23 The monocytes may then activate into macrophages in accordance to the environment they were recruited to (i.e. classical vs alternative macrophage activation). If true, this would be encouraging for our application using decellularized heart valves since they cause a minimal classical inflammatory response and are capable of partial autologous recellularization, as mentioned previously.1, 7, 10, 21 Our data supports this theory since the MCP-1 loaded tissues demonstrated a similar CD68/CD206 expression compared to non-loaded tissues, suggesting that MCP-1 alone may not initiate a negative inflammatory response.
After Roh et al. exhibited that MCP-1 could induce inflammation-mediated recellularization of vascular grafts, other studies have investigated the mechanisms of monocyte recruitment in vitro. Smits et al. used a flow chamber to evaluate the recruitment of monocytes to MCP-1-loaded polymer scaffolds under the physiologic flow conditions of a small diameter artery.27 They found MCP-1-loaded scaffolds led to increased monocyte recruitment only within the first 30 minutes of being exposed to flow. After 30 minutes, the non-loaded scaffolds and MCP-1-loaded scaffolds demonstrated similar degrees of monocyte recruitment out to 24 hours. They attributed the long-term similarity to shear stress becoming the dominant factor in recruitment, while the chemotactic effects of MCP-1 only lasted during the initial burst release in the short-term. They proposed that a controlled release of MCP-1 may lead to better outcomes. Other groups have delivered MCP-1 and VEGF in a controlled release manner from hydrogel vascular grafts and found MCP-1 release over 24 hours led to increased smooth muscle cell recruitment.14 In this study, MCP-1 was also released over a period of 24 hours. It is unknown how shear stresses will affect MCP-1 release and monocyte recruitment in heart valves, but it is worth consideration in future studies. It has been shown that MCP-1 functions in situ by creating concentration gradients through binding with GAGs present in the tissue, which is encouraging for our use of a decellularized tissue scaffold.20 It is also worth noting Smits et al. used a significantly lower MCP-1 concentration in their scaffolds than the Roh et al. study or this present study.
Within the present study we compared MSC migration for the test group of MCP-1-loaded tissue and recruited PBMCs to control groups of media only with and without FBS and a positive control using SDF1. While our test group showed significantly increased migration compared to the media only group without FBS, there was not a statistical difference compared to the media only group with FBS. Furthermore, there was a significant difference between the test group and the group containing MCP-1-loaded tissue only. While not definitive, these results suggest that using tissue in our migration assays may have actually had a slightly negative effect on migration. This was surprising, since it has been suggested that decellularized tissue may have some chemotactic effects to increase cell recruitment.6 However, because the ultimate goal is the recellularization of the decellularized tissue, the results are encouraging since they demonstrate MCP-1-loaded tissue and pre-recruited PBMCs enhances MSC recruitment compared to tissue alone. While the mechanism leading to enhanced MSC migration is not completely understood, Roh et al. proposed in their study that the recruited PBMCs release further signaling cytokines to cause recruitment autologous vascular cells.22 Our results support this theory since the MSCs were separated from the recruited PBMCs in the Boyden chamber and therefore the recruitment of MSCs is likely modulated through chemokine signaling by the PBMCs. The exact cytokines produced by the PBMCs remain to be determined and will be the focus of future studies, though SDF1 is a promising option since our results show the magnitude of MSC recruitment in the test group was similar to the positive control SDF1 group.
While this is the first study to consider using the specific combination of MCP-1-loaded decellularized valve tissue, it is not the first to consider in vivo tissue engineering approaches for heart valves.32 Other approaches have been considered with moderate success. SDF1, the positive control chemoattractant we used in this study, has been applied directly to decellularized heart valves, either in combination with fibronectin or in a hydrogel coating.8, 11 Flameng et al. coated decellularized ovine pulmonary valves in fibronectin and SDF1α before implantation in sheep for five months. They observed a significantly decreased immune response, characterized by CD45+ cell infiltration, and non-significantly increased interstitial cell infiltration compared to normal decellularized valves.11 Others have also used the stem cell antibodies CD133 or CD90 conjugated onto decellularized valves to recruit circulating stem cells.34, 35 Williams et al. demonstrated that CD133 conjugation onto decellularized valves before implantation in the right ventricular outflow tract of sheep led to recellularization similar to native tissue.34 Finally, the XELTIS valve is a polymeric valve scaffold that takes advantage of material properties to encourage inflammatory pathways similar to the current study of inflammation-mediated recellularization. The XELTIS valve utilizes a unique polymer fiber and pore size to encourage the infiltration of inflammatory cells, leading to subsequent recellularization and tissue formation as the polymer scaffold degrades. Preliminary studies and early clinical results are encouraging;2, 19 however, it is worth noting that the use of a polymer scaffolds necessitate complete scaffold remodeling at the same rate as neotissue formation, which could be disastrous if they don’t occur synchronously.
This study was not without limitations. The method of analysis for determining migration by counting cells adhered to the bottom side of the filter does not account for the total migrated cells. Any cells that migrated and entered suspension or adhered to the bottom of the plate were not measured, which certainly occurred since the entire filter was exchanged during the MSC migration experiments using MCP-1-loaded tissues and PBMCs. We chose to use this method however, because it is consistent between experiments since some groups contained tissue samples and others (media only) did not, which would make counting all migrated cells difficult. Therefore, the actual magnitude of cell migration may be greater than what is reported here. It is unlikely the counting method affected the observed differences and mechanism presented in this study, and in fact if all cells were counted the observed differences between groups may be greater. This study also assumes migrating cells are monocytes from the PBMC population, yet we did not analyze the phenotype of the migrated cells. However, we did demonstrate that the Mon1 and Mon2 monocyte subtypes express CCR2. This observation, in combination with the findings by Smits et al. that did determine most of the migrating cells are monocytes, suggests it is correct to assume the migrated PBMCs are primarily monocytes.27 It is also worth noting the tissue samples were frozen between decellularization and their use in experiments. In practice or future in vivo studies, the valves would be used fresh since freezing causes structural damage to the extracellular matrix. However, it is unlikely that freezing had a major effect on the results of this study since we only investigated the chemotactic properties.
Finally, the application of this approach for a tissue engineered heart valve poses many future questions that were not addressed in the present study and will be the focus of further research. For example, while increasing MCP-1 concentration leads to increased PBMC migration, it remains to be determined if an increased MCP-1 concentration can increase subsequent MSC recruitment. For this study, 100 ng/mL was chosen as a proof of concept with the financial consideration of loading an entire heart valve in the future.
Furthermore, the effectiveness of the loading and release methods of MCP-1 remains to be determined. Only 1% of the MCP-1 in the loading solution is transferred into the tissue and as stated, a controlled release of MCP-1 may have a greater effect in vivo. Lastly, the actual in vivo response needs to be investigated to determine if MCP-1-loaded valve grafts will initiate unforeseen inflammatory pathways or lead to detrimental systemic effects, though previous in vivo experiments by others suggest otherwise.(citations) It is unknown what effect the physiologic valve environment will play on cell recruitment including circulating cells, cyclic shear stress, and mechanical stresses. However, the purpose of this study was a proof of concept to demonstrate the mechanism for inflammation-mediated recellularization by the subsequent recruitment of MSCs by inflammatory cells and suggests future studies to investigate its application for heart valve tissue engineering.
Supplementary Material
Acknowledgements
The authors appreciate the contribution to this research made by E. Erin Smith, HTL(ASCP)CM QIHC, Allison Quador, HTL(ASCP)CM and Jessica Arnold HTL(ASCP)CM of the University of Colorado Denver Tissue Biobanking and Histology Shared Resource. The authors also appreciate the contribution to this research made by the Flow Cytometry Shared Resource. These resources are supported in part by the National Cancer Institute through the Cancer Center Support Grant (P30CA046934). Contents are the authors sole responsibility.
Funding
Research reported in this publication was supported by the National Institutes of Health grant number R01HL130436 (awarded to JGJ), and the American Heart Association grant number 19POST34380541 (awarded to MCV).
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
References
- 1.Baraki H, Tudorache I, Braun M, Hoffler K, Gorier A, Lichtenberg A, Bara C, Calistru A, Brandes G, Hewicker-Trautwein M, Hilfiker A, Haverich A and Cebotari S. Orthotopic replacement of the aortic valve with decellularized allograft in a sheep model. Biomaterials 30: 6240–6246, 2009. [DOI] [PubMed] [Google Scholar]
- 2.Bennink G, Torii S, Brugmans M, Cox M, Svanidze O, Ladich E, Carrel T and Virmani R. A novel restorative pulmonary valved conduit in a chronic sheep model: Mid-term hemodynamic function and histologic assessment. J Thorac Cardiovasc Surg 155: 2591–2601 e2593, 2018. [DOI] [PubMed] [Google Scholar]
- 3.Bibevski S, Ruzmetov M, Fortuna RS, Turrentine MW, Brown JW and Ohye RG. Performance of SynerGraft Decellularized Pulmonary Allografts Compared With Standard Cryopreserved Allografts: Results From Multiinstitutional Data. Ann Thorac Surg 103: 869–874, 2017. [DOI] [PubMed] [Google Scholar]
- 4.Converse GL, Buse EE, Neill KR, McFall CR, Lewis HN, VeDepo MC, Quinn RW and Hopkins RA. Design and efficacy of a single-use bioreactor for heart valve tissue engineering. J Biomed Mater Res B Appl Biomater 105: 249–259, 2015. [DOI] [PubMed] [Google Scholar]
- 5.Cooke JP Inflammation and Its Role in Regeneration and Repair. Circ Res 124: 1166–1168, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Crapo PM, Gilbert TW and Badylak SF. An overview of tissue and whole organ decellularization processes. Biomaterials 32: 3233–3243, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.da Costa FD, Costa AC, Prestes R, Domanski AC, Balbi EM, Ferreira AD and Lopes SV. The early and midterm function of decellularized aortic valve allografts. Ann Thorac Surg 90: 1854–1860, 2010. [DOI] [PubMed] [Google Scholar]
- 8.Dai J, Qiao W, Shi J, Liu C, Hu X and Dong N. Modifying decellularized aortic valve scaffolds with stromal cell-derived factor-1alpha loaded proteolytically degradable hydrogel for recellularization and remodeling. Acta Biomater 88: 280–292, 2019. [DOI] [PubMed] [Google Scholar]
- 9.Deshmane SL, Kremlev S, Amini S and Sawaya BE. Monocyte chemoattractant protein-1 (MCP-1): an overview. J Interferon Cytokine Res 29: 313–326, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dohmen PM, Hauptmann S, Terytze A and Konertz WF. In-vivo repopularization of a tissue-engineered heart valve in a human subject. J Heart Valve Dis 16: 447–449, 2007. [PubMed] [Google Scholar]
- 11.Flameng W, De Visscher G, Mesure L, Hermans H, Jashari R and Meuris B. Coating with fibronectin and stromal cell-derived factor-1alpha of decellularized homografts used for right ventricular outflow tract reconstruction eliminates immune response-related degeneration. J Thorac Cardiovasc Surg 147: 1398–1404 e1392, 2014. [DOI] [PubMed] [Google Scholar]
- 12.Garash R, Bajpai A, Marcinkiewicz BM and Spiller KL. Drug delivery strategies to control macrophages for tissue repair and regeneration. Exp Biol Med (Maywood) 241: 1054–1063, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hopper N, Wardale J, Howard D, Brooks R, Rushton N and Henson F. Peripheral blood derived mononuclear cells enhance the migration and chondrogenic differentiation of multipotent mesenchymal stromal cells. Stem Cells Int 2015: 323454, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jay SM, Shepherd BR, Andrejecsk JW, Kyriakides TR, Pober JS and Saltzman WM. Dual delivery of VEGF and MCP-1 to support endothelial cell transplantation for therapeutic vascularization. Biomaterials 31: 3054–3062, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Julier Z, Park AJ, Briquez PS and Martino MM. Promoting tissue regeneration by modulating the immune system. Acta Biomater 53: 13–28, 2017. [DOI] [PubMed] [Google Scholar]
- 16.Konuma T, Devaney EJ, Bove EL, Gelehrter S, Hirsch JC, Tavakkol Z and Ohye RG. Performance of CryoValve SG decellularized pulmonary allografts compared with standard cryopreserved allografts. Ann Thorac Surg 88: 849–854; discussion 554-845, 2009. [DOI] [PubMed] [Google Scholar]
- 17.Mahler GJ and Butcher JT. Inflammatory regulation of valvular remodeling: the good(?), the bad, and the ugly. Int J Inflam 2011: 721419, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Neron S, Thibault L, Dussault N, Cote G, Ducas E, Pineault N and Roy A. Characterization of mononuclear cells remaining in the leukoreduction system chambers of apheresis instruments after routine platelet collection: a new source of viable human blood cells. Transfusion 47: 1042–1049, 2007. [DOI] [PubMed] [Google Scholar]
- 19.Xeltis Prodan Z. 1-year in vivo data of a novel bioabsorbable pulmonary heart valved conduit. In: 30th Annual Meeting of the European Association of Cardio-Thoracic Surgery (EACTS). Barcelona, Spain: 2016. [Google Scholar]
- 20.Proudfoot AE, Handel TM, Johnson Z, Lau EK, LiWang P, Clark-Lewis I, Borlat F, Wells TN and Kosco-Vilbois MH. Glycosaminoglycan binding and oligomerization are essential for the in vivo activity of certain chemokines. Proc Natl Acad Sci U S A 100: 1885–1890, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Quinn RW, Bert AA, Converse GL, Buse EE, Hilbert SL, Drake WB and Hopkins RA. Performance of Allogeneic Bioengineered Replacement Pulmonary Valves in Rapidly Growing Young Lambs. J Thorac Cardiovasc Surg 2016. [DOI] [PubMed] [Google Scholar]
- 22.Roh JD, Sawh-Martinez R, Brennan MP, Jay SM, Devine L, Rao DA, Yi T, Mirensky TL, Nalbandian A, Udelsman B, Hibino N, Shinoka T, Saltzman WM, Snyder E, Kyriakides TR, Pober JS and Breuer CK. Tissue-engineered vascular grafts transform into mature blood vessels via an inflammation-mediated process of vascular remodeling. Proc Natl Acad Sci U S A 107: 4669–4674, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shi C and Pamer EG. Monocyte recruitment during infection and inflammation. Nat Rev Immunol 11: 762–774, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Simionescu A, Schulte JB, Fercana G and Simionescu DT. Inflammation in cardiovascular tissue engineering: the challenge to a promise: a minireview. Int J Inflam 2011:958247, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Simon P, Kasimir MT, Seebacher G, Weigel G, Ullrich R, Salzer-Muhar U, Rieder E and Wolner E. Early failure of the tissue engineered porcine heart valve SYNERGRAFT in pediatric patients. Eur J Cardiothorac Surg 23: 1002–1006; discussion 1006, 2003. [DOI] [PubMed] [Google Scholar]
- 26.Singer NG and Caplan AI. Mesenchymal stem cells: mechanisms of inflammation. Annu Rev Pathol 6: 457–478, 2011. [DOI] [PubMed] [Google Scholar]
- 27.Smits AI, Ballotta V, Driessen-Mol A, Bouten CV and Baaijens FP. Shear flow affects selective monocyte recruitment into MCP-1-loaded scaffolds. J Cell Mol Med 18: 2176–2188, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Spiller KL and Koh TJ. Macrophage-based therapeutic strategies in regenerative medicine. Adv Drug Deliv Rev 122: 74–83, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vagnozzi RJ, Maillet M, Sargent MA, Khalil H, Johansen AKZ, Schwanekamp JA, York AJ, Huang V, Nahrendorf M, Sadayappan S and Molkentin JD. An acute immune response underlies the benefit of cardiac stem cell therapy. Nature 577: 405–409, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.VeDepo MC, Buse EE, Quinn RW, Hopkins RA and Converse GL. Extended bioreactor conditioning of mononuclear cell-seeded heart valve scaffolds. Journal of Tissue Engineering 9: 2041731418767216, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.VeDepo MC, Buse EE, Quinn RW, Williams T, Detamore MS, Hopkins RA and Converse GL. Species-Specific Effects of Aortic Valve Decellularization. Acta Biomater 50: 249–258, 2017. [DOI] [PubMed] [Google Scholar]
- 32.VeDepo MC, Detamore M, Hopkins RA and Converse GL. Recellularization of decellularized heart valves: Progress toward the tissue-engineered heart valve. Journal of Tissue Engineering 8: 2041731417726327, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Weber B, Scherman J, Emmert MY, Gruenenfelder J, Verbeek R, Bracher M, Black M, Kortsmit J, Franz T, Schoenauer R, Baumgartner L, Brokopp C, Agarkova I, Wolint P, Zund G, Falk V, Zilla P and Hoerstrup SP. Injectable living marrow stromal cell-based autologous tissue engineered heart valves: first experiences with a one-step intervention in primates. Eur Heart J 32: 2830–2840, 2011. [DOI] [PubMed] [Google Scholar]
- 34.Williams JK, Miller ES, Lane MR, Atala A, Yoo JJ and Jordan JE. Characterization of CD133 Antibody-Directed Recellularized Heart Valves. J Cardiovasc Transl Res 8: 411–420, 2015. [DOI] [PubMed] [Google Scholar]
- 35.Ye X, Zhao Q, Sun X and Li H. Enhancement of mesenchymal stem cell attachment to decellularized porcine aortic valve scaffold by in vitro coating with antibody against CD90: a preliminary study on antibody-modified tissue-engineered heart valve. Tissue Eng Part A 15: 1–11, 2009. [DOI] [PubMed] [Google Scholar]
- 36.Zwetsloot PP, Vegh AM, Jansen of Lorkeers SJ, van Hout GP, Currie GL, Sena ES, Gremmels H, Buikema JW, Goumans MJ, Macleod MR, Doevendans PA, Chamuleau SA and Sluijter JP. Cardiac Stem Cell Treatment in Myocardial Infarction: A Systematic Review and Meta-Analysis of Preclinical Studies. Circ Res 118: 1223–1232, 2016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.