

Abbreviations
- CCLE
Cancer Cell Line Encyclopedia
- CRC
colorectal cancer
- GSEA
Gene Set Enrichment Analysis
- HCC
hepatocellular cancer
- IF
immunofluorescence
- IHC
Immunohistochemistry
- KD
knockdown
- KO
knockout
- LAMP1
lysosomal associated membrane protein 1
- LAMTOR1‐5
late endosomal/lysosomal adaptor, MAPK, and mTOR activator 1‐5
- LAMTOR4
late endosomal/lysosomal adaptor, MAPK, and mTOR activator 4
- LysoIP‐MS
lysosome immunoprecipitation‐mass spectrometry
- MMTV‐PyMT
The mammary‐specific polyomavirus middle T antigen overexpression mouse model
- mTOR
mammalian target of rapamycin
- mTORi
mTOR inhibitors
- p‐PRAS40
phosphorylated proline‐rich Akt substrate of 40 kDa
- TMEM9
transmembrane protein 9
- v‐ATPase
vacuolar ATPase
Dear Editor,
Accumulating evidence suggests that dysregulated lysosomal membrane proteins, including vacuolar ATPase (v‐ATPase) and the mammalian target of rapamycin (mTOR), are involved in tumorigenesis [1]. Therefore, lysosomal proteins were proposed as potential therapeutic targets in cancer [1]. As one of the lysosome‐related signaling pathways, mTOR signaling regulates cell proliferation, survival, motility, and metabolism [2]. Since mTOR signaling activation promotes tumorigenesis, mTOR inhibitors (mTORi), AZD8055 [3], MLN0128 [4], and Rapalink‐1 (the latest third‐generation mTORi) [4], have been applied to several cancers. However, the limitations of mTORi include drug resistance and the lack of biomarkers.
Recently, we identified transmembrane protein 9 (TMEM9) as a positive regulator of Wnt/β‐catenin signaling in colorectal [5] and hepatocellular cancers [6]. TMEM9 facilitated the assembly of v‐ATPase, which induced vesicular acidification and subsequent hyperactivation of β‐catenin‐mediated gene expression [5]. Given the vital role of TMEM9 in vesicular acidification and lysosomal dysfunction, we sought to determine the impact of TMEM9‐dysregulated lysosomes on tumorigenesis. Detailed methods can be found in the Supplementary file.
To assess the pathologic relevance of TMEM9 to cancer, we first analyzed the TMEM9 gene alteration and expression. Among human cancers, TMEM9 gene amplification and upregulation were frequently observed in breast cancer tissues and cell lines (Supplementary Figure S1A‐G; Supplementary Tables S1‐S2). Furthermore, high expression of TMEM9 was associated with the reduced relapse‐free survival of breast cancer patients as compared to low expression of TMEM9 (Supplementary Figure S1H). These results led us to hypothesize that TMEM9 plays oncogenic roles in mammary tumorigenesis. In MDA‐MB‐231 and MDA‐MB‐453 breast cancer cells, TMEM9‐knockdown (KD) cells showed reduced proliferation compared to control cells (Supplementary Figure S2A‐C). Conversely, ectopic expression of TMEM9 promoted the proliferation of breast cancer cells (Supplementary Figure S2D‐E).
Next, we determined in vivo roles of TMEM9 in mammary tumorigenesis using a mammary‐specific polyomavirus middle T antigen overexpression (MMTV‐PyMT) mouse model. We confirmed that Tmem9 was highly expressed in mammary tumors of MMTV‐PyMT mice (Supplementary Figure S3A‐B). Utilizing Tmem9‐knockout (KO) mice previously established [5], we generated an MMTV‐PyMT:Tmem9‐KO compound strain (Supplementary Figure S3C‐D). While MMTV‐PyMT:Tmem9 wild‐type (WT) mice developed early mammary hyperplasia at the age of 10 weeks, MMTV‐PyMT:Tmem9‐KO mice did not show any lesions in the mammary glands (Supplementary Figure S3E‐G). Moreover, at the age of 150 days, MMTV‐PyMT:Tmem9‐KO mice exhibited reduced tumor weight and number compared to MMTV‐PyMT:Tmem9‐WT mice (Figure 1A; Supplementary Figure S3H‐I). Additionally, MMTV‐PyMT:Tmem9‐KO mice showed extended tumor‐free survival compared to MMTV‐PyMT:Tmem9‐WT mice (Figure 1B). Microscopic analyses confirmed the decreased tumor cell proliferation, validated by Ki‐67 immunostaining (Figure 1C‐D; Supplementary Figure S3J). These data suggest that TMEM9 plays an oncogenic role in mammary tumorigenesis.
FIGURE 1.
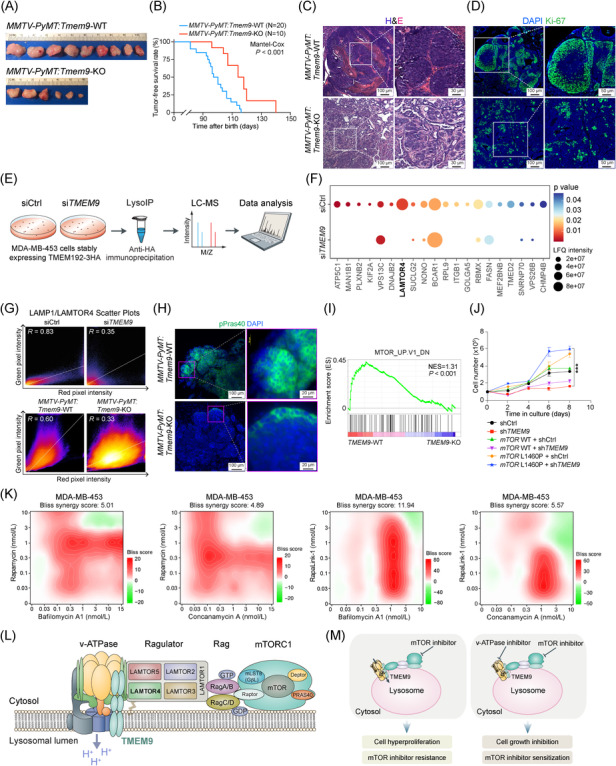
TMEM9 is required for mammary tumorigenesis. (A‐B) Tmem9 KO suppressed mammary tumorigenesis. All tumors were harvested at 150 days to evaluate tumor weight and the number of mammary glands with the tumor. Representative images of mammary tumors isolated from mice (A). Tumor‐free survival (TFS) analysis of MMTV‐PyMT:Tmem9‐WT (n = 20, median TFS = 97 days) and MMTV‐PyMT:Tmem9‐KO mice (n = 10, median TFS = 116 days) (B). (C) H&E‐stained sections of mammary tumors isolated from MMTV‐PyMT:Tmem9‐WT and MMTV‐PyMT:Tmem9‐KO mice. (D) Decreased cell proliferation by Tmem9 KO. Ki‐67‐stained sections of mammary tumors isolated from MMTV‐PyMT:Tmem9‐WT and MMTV‐PyMT:Tmem9‐KO mice. (E) Illustration of the workflow for the LysoIP method. Control and siTMEM9 cells were prepared from the parental cells (MDA‐MB‐453) stably expressing 3 × HA‐tagged TMEM192. Each condition contained three biological replicates. (F) Top 20 differentially expressed proteins (fold change > 1.5 over control, P < 0.05) in the dot plot. The colors of the dots represent the adjusted P value, and the sizes of the nodes represent the enrichment of the proteins that are differentially expressed to the total proteins. (G) Co‐localization analysis of LAMTOR4 and LAMP1 using FIJI/ImageJ software. A scatterplot of red (LAMP1) and green (LAMTOR4) pixel intensities represents the upper panel of siCtrl and siTMEM9 cells, the lower panel of MMTV‐PyMT:Tmem9‐WT and MMTV‐PyMT:Tmem9‐KO mice shown in Supplementary Figure S4E & G. (H) Hypo‐phosphorylation of Pras40 by Tmem9 KO. IF of Tmem9‐KO mammary tumors (MMTV‐PyMT:Tmem9‐WT and MMTV‐PyMT:Tmem9‐KO; age, 10 weeks) for phosphor‐Pras40 (pPras40). (I) Gene set enrichment analysis of the mTOR signaling in RNA‐sequencing data from TMEM9‐WT and TMEM9‐KD MDA‐MB‐453 cells. (J) Growth curves of MDA‐MB‐453 cells with indicated treatments. mTOR L1460P is an active mTOR mutant. The mean and SD of three independent experiments are shown. ***, P < 0.001. (K) Dose‐response matrix as heatmaps of Bliss synergy scores for drug combinations (from left to right: rapamycin and bafilomycin A1; rapamycin and concanamycin A; Rapalink‐1 and bafilomycin A1; Rapalink‐1 and concanamycin A) in MDA‐MB‐453 cells. (L) Illustration of the functional interaction between the TMEM9‐v‐ATPase axis and mTOR signaling. TMEM9‐facilitated v‐ATPase assembly promotes localization of LAMTOR4 on the lysosomal membrane to activate mTORC1 via Ragulator. (M) A schematic model of the tumorigenic role of TMEM9 in promoting breast cancer cell proliferation and mTORi resistance. The experiments were carried out in triplicate. Representative images are shown; Student's t‐test; error bars: SEM.
Abbreviations: v‐ATPase, vacuolar ATPase; mTOR, mammalian target of rapamycin; TMEM9, transmembrane protein 9; KD, knockdown; KO, knockout; LAMTOR4, late endosomal/lysosomal adaptor, MAPK, and mTOR activator 4; LAMTOR1‐5, late endosomal/lysosomal adaptor, MAPK, and mTOR activator 1‐5; p‐PRAS40, phosphorylated proline‐rich AKT1 substrate 1; mTORi, mTOR inhibitors; CRC, colorectal cancer; HCC, hepatocellular cancer; CCLE, Cancer Cell Line Encyclopedia; MMTV‐PyMT, The mammary‐specific polyomavirus middle T antigen overexpression mouse model; LysoIP‐MS, lysosome immunoprecipitation‐mass spectrometry; GSEA, Gene Set Enrichment Analysis; IHC, Immunohistochemistry; IF, immunofluorescence; LAMP1, lysosomal associated membrane protein 1
Next, we sought to dissect the mechanism by which TMEM9 contributes to mammary tumorigenesis. Since TMEM9 binds to and activates v‐ATPase for lysosomal vesicular acidification [5, 6], we examined the impact of TMEM9 on lysosomal proteins. We isolated the lysosomes from TMEM9‐WT and TMEM9‐KD MDA‐MB‐453 cells and performed lysosome immunoprecipitation‐mass spectrometry (LysoIP‐MS) (Figure 1E; Supplementary Figure S4A‐C). Approximately 1,200 lysosome‐associated proteins were identified (Supplementary Figure S4D; Supplementary Table S3). Intriguingly, among the top 20 lysosomal proteins differentially detected, LAMTOR4, a component of the LAMTOR complex, became undetectable in TMEM9‐KD cells (Figure 1F; Supplementary Table S4). Next, we examined the effect of TMEM9 on lysosomal LAMTOR4 by immunofluorescence staining for LAMTOR4 and lysosomal associated membrane protein 1 (LAMP1), a lysosome marker, in TMEM9‐KD ZR‐75‐1 cells (Supplementary Figure S4E‐F). Co‐localization analysis showed the reduced co‐localization between LAMTOR4 (green) and LAMP1 (red) in TMEM9‐KD cells (Supplementary Figure S4G). The scatter dot plot also exhibited decreased co‐localization of LAMTOR4 with LAMP1 by TMEM9 KD (Figure 1G). Consistently, MMTV‐PyMT:Tmem9‐KO tumors also showed a decrease in co‐localization between LAMTOR4 and LAMP1, compared to MMTV‐PyMT:Tmem9‐WT tumors (Figure 1G; Supplementary Figure S4H‐I). These results suggest that TMEM9 is required for the lysosomal localization of LAMTOR4.
The lysosomal v‐ATPase‐Ragulator complex activates the mTORC1 signaling [7]. As a member of the Ragulator complex, LAMTOR4 modulates Ragulator assembly and contributes to anchoring mTOR to the lysosomal membrane [7]. Therefore, decreased levels of LAMTOR4 in the lysosome by TMEM9 KD may disrupt mTOR signaling activity. Indeed, mTOR and mTOR downstream effector phosphorylated proline‐rich Akt substrate of 40 kDa (p‐PRAS40) were downregulated in Tmem9‐KO mammary tumors (Figure 1H; Supplementary Figure S5A‐C). Additionally, RNA‐sequencing‐based Gene Set Enrichment Analysis (GSEA) of MDA‐MB‐453 cells (TMEM9‐WT vs. TMEM9‐KD) showed that the geneset modulated by mTOR signaling was suppressed by TMEM9 KD (Figure 1I; Supplementary Figure S5D; Supplementary Table S5). We also observed that knockdown of TMEM9 in TMEM9high breast cancer cells (MDA‐MB‐453 and MCF7) decreased the activity of mTOR downstream effectors, while ectopic expression of TMEM9 in TMEM9low cells (MDA‐MB‐231) increased the activity of mTOR effectors (Supplementary Figure S5E‐F). Additionally, we observed the interaction of TMEM9 with LAMTOR4 (Supplementary Figure S5G‐H). To further determine the impact of TMEM9‐regulated mTOR signaling on cell proliferation, we performed a rescue experiment by lentiviral overexpression of wild‐type or constitutively active mutant of mTOR (L1460P) [8] in TMEM9‐KD cells. mTOR L1460P rescued the cell proliferation inhibition induced by TMEM9 KD, while mTOR WT failed to do (Figure 1J). These results suggest that TMEM9‐activated mTOR signaling is required for breast cancer cell proliferation.
Although several mTORi have been introduced for cancer therapy, mTORi resistance is a recurring issue [9]. To overcome mTORi resistance, combinatorial therapies have been used [10]. Given the TMEM9 gene amplification in breast cancer and TMEM9‐activated mTOR signaling, it is plausible that mTORi resistance might be due to TMEM9 upregulation in breast cancer. It should be noted that mTOR inhibition did not upregulate TMEM9 expression (Supplementary Figure S6A). To determine whether the TMEM9‐v‐ATPase axis can be a therapeutic target to overcome mTORi resistance, we treated breast cancer cells with v‐ATPase inhibitors (bafilomycin A1 and concanamycin A) in combination with mTORi (rapamycin and Rapalink‐1). It should be noted that TMEM9high breast cancer cells (MDA‐MB‐453) were more sensitive to v‐ATPase inhibitors than TMEM9low breast cancer cells (BT‐474) (Supplementary Figure S6B). We assessed the impact of pharmacological blockade of the TMEM9‐v‐ATPase axis on mTORi response. The bliss synergy scoring analysis showed that rapamycin or Rapalink‐1 combined with bafilomycin A1 or concanamycin A synergistically inhibited breast cancer cell viability across multiple concentrations (Figure 1K; Supplementary Figure S6C‐D). Additionally, TMEM9‐KD cells showed a similar response to v‐ATPase and mTORC1 co‐inhibition in the context of cell viability (Supplementary Figure S6E). These results suggest that the pharmacological blockade of the TMEM9‐v‐ATPase axis sensitizes tumor cells to mTORi.
Herein our results identified a new role of the TMEM9‐v‐ATPase axis in modulating mTOR signaling via LAMTOR4. In breast cancer cells harboring the TMEM9 gene amplification, TMEM9‐activated v‐ATPase aberrantly regulates lysosome‐associated signaling, including the mTOR pathway. Upon TMEM9 upregulation, TMEM9‐induced v‐ATPase assembly and activation likely stabilize the LAMTOR4‐containing Ragulator complex assembly in the lysosomal membrane, contributing to mTOR signaling hyperactivation (Figure 1L‐M). The detailed molecular mechanisms of mTOR downregulation and mislocalization of LAMTOR4 by TMEM9 depletion need to be elucidated in future studies. The efficacy of further combinatorial therapies remains to be tested using in vivo preclinical models.
Together, our results unveiled that the lysosomal TMEM9‐v‐ATPase‐Ragulator‐Rag axis is indispensable for mTOR signaling integrity and breast cancer cell proliferation and that TMEM9 may serve as a biomarker and a molecular target, which may overcome mTORi resistance and offer a viable therapeutic strategy for TMEM9‐associated breast cancer.
DECLARATIONS
AUTHOR CONTRIBUTIONS
Shengzhe Zhang, Sung Ho Lee, and Jae‐Il Park conceived the experiments. Shengzhe Zhang, Sung Ho Lee, Litong Nie, Yuanjian Huang, Gengyi Zou, Youn‐Sang Jung, Sohee Jun, Jie Zhang, Esther M. Lien, and Jae‐Il Park performed the experiments. Shengzhe Zhang, Sung Ho Lee, Junjie Chen, and Jae‐Il Park analyzed the data. Shengzhe Zhang and Jae‐Il Park wrote the manuscript.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable.
CONSENT FOR PUBLICATION
Not applicable.
Supporting information
Supporting information
Supporting information
ACKNOWLEDGMENTS
We thank Pierre D. McCrea, Malgorzata Kloc, and Rachel Miller for the discussion and Ann Sutton (Research Medical Library, MD Anderson) for editing the manuscript. This work was supported by the Cancer Prevention and Research Institute of Texas (RP200315 to J.‐I.P.), the National Cancer Institute (CA193297 and CA256207 to J.‐I.P.), an Institutional Research Grant (MD Anderson to J.‐I.P.), a Specialized Program of Research Excellence (SPORE) grant in endometrial cancer (P50 CA83639 to J.‐I.P.), and Radiation Oncology Research Initiatives. The core facilities used (DNA sequencing, Genetically Engineered Mouse Facility, and FACS Facility) were supported by Cancer Center Support Grant (P30 CA016672) from the National Cancer Institute to MD Anderson.
Contributor Information
Shengzhe Zhang, Email: szhang23@mdanderson.org.
Jae‐Il Park, Email: jaeil@mdanderson.org.
DATA AVAILABILITY STATEMENT
Data used in this study are maintained by the MD Anderson Cancer Center (https://www.mdanderson.org/). Data requests are subject to approval by the review committee.
REFERENCES
- 1. Towers CG, Thorburn A. Targeting the Lysosome for Cancer Therapy. Cancer Discov. 2017;7(11):1218‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Popova NV, Jucker M. The Role of mTOR Signaling as a Therapeutic Target in Cancer. Int J Mol Sci. 2021;22(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chresta CM, Davies BR, Hickson I, Harding T, Cosulich S, Critchlow SE, et al. AZD8055 is a potent, selective, and orally bioavailable ATP‐competitive mammalian target of rapamycin kinase inhibitor with in vitro and in vivo antitumor activity. Cancer research. 2010;70(1):288‐98. [DOI] [PubMed] [Google Scholar]
- 4. Kuroshima K, Yoshino H, Okamura S, Tsuruda M, Osako Y, Sakaguchi T, et al. Potential new therapy of Rapalink‐1, a new generation mammalian target of rapamycin inhibitor, against sunitinib‐resistant renal cell carcinoma. Cancer Science. 2020;111(5):1607‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jung YS, Jun S, Kim MJ, Lee SH, Suh HN, Lien EM, et al. TMEM9 promotes intestinal tumorigenesis through vacuolar‐ATPase‐activated Wnt/beta‐catenin signalling. Nat Cell Biol. 2018;20(12):1421‐33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jung YS, Stratton SA, Lee SH, Kim MJ, Jun S, Zhang J, et al. TMEM9‐v‐ATPase Activates Wnt/beta‐Catenin Signaling Via APC Lysosomal Degradation for Liver Regeneration and Tumorigenesis. Hepatology. 2021;73(2):776‐94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhang C‐S, Jiang B, Li M, Zhu M, Peng Y, Zhang Y‐L, et al. The lysosomal v‐ATPase‐Ragulator complex is a common activator for AMPK and mTORC1, acting as a switch between catabolism and anabolism. Cell metabolism. 2014;20(3):526‐40. [DOI] [PubMed] [Google Scholar]
- 8. Grabiner BC, Nardi V, Birsoy K, Possemato R, Shen K, Sinha S, et al. A diverse array of cancer‐associated MTOR mutations are hyperactivating and can predict rapamycin sensitivity. Cancer discovery. 2014;4(5):554‐63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Caro‐Vegas C, Bailey A, Bigi R, Damania B, Dittmer DP, Shenk T, et al. Targeting mTOR with MLN0128 Overcomes Rapamycin and Chemoresistant Primary Effusion Lymphoma. mBio. 2019;10(1):e02871‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zou Z, Tao T, Li H, Zhu X. mTOR signaling pathway and mTOR inhibitors in cancer: progress and challenges. Cell Biosci. 2020;10:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information
Supporting information
Data Availability Statement
Data used in this study are maintained by the MD Anderson Cancer Center (https://www.mdanderson.org/). Data requests are subject to approval by the review committee.