

Abstract
RAS genes are the most frequently mutated oncogenes and play critical roles in the development and progression of malignancies. The mutation, isoform (KRAS, HRAS, and NRAS), position, and type of substitution vary depending on the tissue types. Despite decades of developing RAS‐targeted therapies, only small subsets of these inhibitors are clinically effective, such as the allele‐specific inhibitors against KRASG12C. Targeting the remaining RAS mutants would require further experimental elucidation of RAS signal transduction, RAS‐altered metabolism, and the associated immune microenvironment. This study reviews the mechanisms and efficacy of novel targeted therapies for different RAS mutants, including KRAS allele‐specific inhibitors, combination therapies, immunotherapies, and metabolism‐associated therapies.
Keywords: RAS mutation, Signal transduction, RAS‐targeted therapy, Combination therapy, Immunotherapy, Cancer metabolism
Abbreviations
- GEF
guanine nucleotide exchange‐factor
- GAP
GTPase‐activating proteins
- SOS
Son of Sevenless
- ERK
extracellular signal regulated kinase
- PI3K
phosphoinositide 3‐kinase
- mTOR
mammalian target of rapamycin
- NF1
neuro‐fibromin 1
- SHP2
Src homology‐2 domain‐containing protein tyrosine phosphatase 2
- PDAC
pancreatic ductal adenocarcinoma
- CRC
colorectal cancer
- NSCLC
non‐small cell lung cancer
- G12Ci
inhibitors targeting the KRAS G12C mutant
- C12
cysteine‐12
- EDTA
ethylenediaminetetraacetic acid
- PK
pharmacokinetics
- PD‐1
programmed cell death protein 1
- RTK
receptor tyrosine kinase
- STK11
serine/threonine kinase 11
- LKB1
liver kinase B1
- KEAP1
kelch‐like ECH‐associated protein 1
- PD‐L1
programmed cell death ligand 1
- FDA
Food and Drug Administration
- ADME
absorption, distribution, metabolism, and excretion
- TME
tumor microenvironment
- IFN
interferon
- MAPK
mitogen‐activated protein kinase
- DUSP
dual‐specificity phosphatase
- SPRY
sprouty
- EGFR
epidermal growth factor receptor
- LUAD
lung adenocarcinoma
- cfDNA
cell‐free DNA
- PROTACs
proteolysis‐targeting chimeras
- RC‐U
(RBD+CRD)CRAF‐U‐Box
- DARPins
designed ankyrin repeat proteins
- WT
wild type
- CRBN
cereblon
- VHL
von Hippel‐Lindau
- siRNA
small interfering RNA
- ASO
antisense oligonucleotide
- PR
partial response
- SD
stable disease
- mAbs
monoclonal antibodies
- HSPG
heparan sulfate proteoglycan
- PTP
protein tyrosine phosphatase
- PTPN11
protein tyrosine phosphatase non‐receptor type 11
- GEMM
genetically engineered mouse model
- PI3KCA
PI3K catalytic subunit alpha
- SPRED1
Sprouty‐related protein with an EVH1 domain
- FOXO
forkhead box, class O
- IRS‐1
insulin receptor substrate 1
- S6K
S6 kinase
- PTEN
phosphatase and tensin homolog
- MAGI1
membrane‐associated guanylate kinase, WW and PDZ domain‐containing protein 1
- PIP3
phosphatidylinositol 3,4,5‐trisphosphate
- TSC
tuberous sclerosis complex
- Rheb
Ras homologue enriched in brain
- IGF1R
insulin‐like growth factor1 receptor
- mFOLFOX
modified FOLFOX
- KSR1
kinase suppressor of Ras‐1
- GAB1
Grb2‐associated binder‐1
- RBD
RAS‐binding domain
- PDX
patient‐derived xenograft
- MTD
maximum tolerated dose
- MLD
multiple low dose
- CTLA‐4
cytotoxic T‐lymphocyte‐associated protein 4
- ICI
immune checkpoint inhibitor
- TIL
tumor‐infiltrating lymphocytes
- TTP
tristetraprolin
- IRF2
interferon regulatory factor 2
- GM‐CSF
granulocyte macrophage colony‐stimulating factor
- IL‐6
interleukin 6
- AP1
activator protein 1
- TMB
tumor mutational burden
- Treg
regulatory T cell
- MDSC
myeloid derived suppressive cell
- HLA
human leukocyte antigen
- TCR
T cell receptor
- CAR
chimeric antigen receptor
- MSLN
mesothelin
- LNP
lipid nanoparticles
- HK1
hexokinase 1
- HK2
hexokinase 2
- PFK1
phosphofructokinase 1
- LDHA
lactate dehydrogenase A
- TCA
tricarboxylic
- GLUT
glucose transporter
- ATR
rad3‐related protein
- CHK1
checkpoint kinase 1
- DHA
dehydroascorbate
- ROS
reactive oxygen species
- GAPDH
glyceraldehyde 3‐phosphate dehydrogenase
- NEAA
non‐essential amino acid
- SLC
solute carrier
- GLS
glutaminase
- α‐KG
α‐ketoglutarate
- GLUD
glutamate dehydrogenase
- GOT
glutamate oxaloacetate transaminase
- GSH
glutathione
- ME1
malic enzyme 1
- NRF2
nuclear factor erythroid 2–related factor 2
- GSK3α/β
glycogen synthase kinase 3 α/β
- CDK4/6
cyclin‐dependent kinase 4 and 6
- PARP
poly (ADP‐ribose) polymerase
- SNAT2
serotonin N‐acetyltransferase 2
- LAT1
L‐type amino acid transporter
- AMPK
AMP‐activated protein kinase
- ULK1
Unc‐51‐like kinase 1
- EIPA
5‐(N‐ethyl‐N‐ isopropyl) amiloride
- HCQ
hydroxychloroquine
- PPT1
palmitoyl‐protein thioesterase 1
1. BACKGROUND
RAS mutations are detected in ∼21% of all cancers and occur in three isoforms, namely KRAS, HRAS, and NRAS [1, 2, 3, 4], all of which encode 21 kDa guanine nucleotide‐binding proteins that cycle between GTP‐ and GDP‐bound states. This binary switch is regulated by guanine nucleotide exchange factors (GEFs) and GTPase‐activating proteins (GAPs). Without upstream stimuli, the RAS protein maintains a GDP‐bound state due to its intrinsic GTPase activity and is unable to engage in downstream signal transduction. When activated by upstream receptors, GEFs, such as son of sevenless (SOS), promote the change from GDP to GTP. GTP‐bound RAS undergoes conformational changes in the switch I and II regions, which recruit various downstream effector molecules, including RAF‐MEK‐extracellular‐signal‐regulated kinase (ERK), phosphoinositide 3‐kinase (PI3K)‐Akt‐mammalian target of rapamycin (mTOR), and other non‐canonical downstream cascades (Figure 1). GAPs, such as neuro‐fibromin 1 (NF1), stimulate the GTP hydrolysis to return RAS to the inactive state [5, 6, 7].
FIGURE 1.
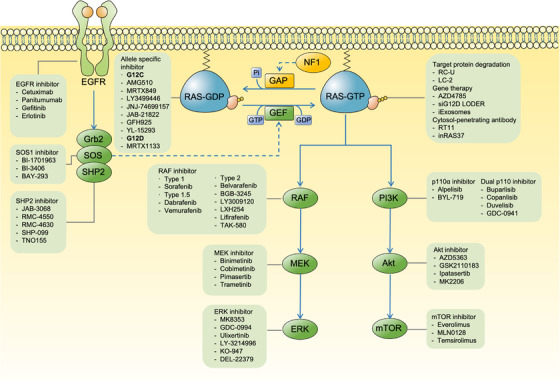
The signal transduction network of RAS and corresponding inhibitors targeting RAS itself and related molecules. External stimuli lead to the activation of RTKs, such as EGFR which in turn activate the SOS, increasing the level of GTP‐bound activated RAS. Blockade of upstream RTKs, SOS and SHP2 potentially down‐regulates the exchange of GDP for GTP in RAS. Inhibitors directly targeting RAS proteins themselves have been developed, such as KRAS G12C allele‐specific inhibitors that lock KRAS in the GDP‐bound inactivated state. Activated RAS orchestrates a large spectrum of biological behaviors of cancers by activating downstream effectors, including RAF‐MEK‐ERK and PI3K‐Akt‐mTOR. Thus, multiple inhibitors have been developed to targeting these cascades at different levels. Abbreviations: RTKs, receptor tyrosine kinase; EGFR, epidermal growth factor receptor; SOS, Son of Sevenless; GEF, guanine nucleotide exchange‐factor; GAP, GTPase‐activating proteins; NF1, neuro‐fibromin 1; SHP2, Src homology‐2 domain‐containing protein tyrosine phosphatase‐2; ERK, extracellular signal regulated kinase; PI3K, phosphoinositide 3‐kinase; mTOR, mammalian target of rapamycin; Grb2, growth factor receptor‐bound protein 2; RC‐U, (RBD+CRD)CRAF‐U‐Box
Single missense mutations at codons 12, 13, and 61 around the nucleotide‐binding site of RAS have been found in multiple human cancers [8]. Early studies indicated that these single amino acid substitutions disrupted both intrinsic and GAP‐stimulated GTPase activity, favoring the accumulation of GTP‐bound RAS proteins [9]. GTP‐bound RAS activates downstream signaling pathways, leading to a spectrum of biological behaviors in cancer [10].
Fifty years since the identification of RAS in rat sarcoma virus, several inhibitors have been developed (Figure 1). However, only the KRASG12C inhibitor has been approved for clinical use. The targeting of RAS is complicated by several factors. First, the lack of a drug‐binding pocket makes it difficult to develop inhibitors with high affinity and selectivity. Second, the efficacy of inhibitors targeting key molecules of the RAS signaling pathway is limited by feedback inhibition and compensatory circuits within the signaling network. Third, inhibitors that do not discriminate between wild‐type and mutated RAS proteins show poor results in clinical trials.
Allele‐specific RAS inhibitors, targeted protein degradation, and gene therapy have been applied in the direct targeting of RAS. However, these inhibitors are effective only for a small subset of RAS mutants. The application of RAS upstream inhibitors (i.e., SOS1 and Src homology‐2 domain‐containing protein tyrosine phosphatase 2 [SHP2]) and concurrent inhibition of RAS downstream effectors is considered an effective indirect RAS‐targeted therapy. Moreover, there is growing evidence for the regulation of the immune microenvironment and cellular metabolism by RAS signaling—a connection that has been innovatively exploited in drug discovery and therapy. This review describes the mechanisms and anti‐tumor efficacy of novel RAS‐targeted therapies and provides perspectives on the development of well‐tolerated and effective inhibitors for all RAS mutants.
2. RAS MUTATIONS AND VARIANTS
The mutation, isoform (i.e., KRAS, HRAS, and NRAS), position, and type of substitutions in RAS vary depending on tissue type (data from GENIE 10.1 [11]) (Figure 2). In terms of isoform variability, KRAS mutations occur in 14.3% of all malignancies, 76.5% of pancreatic ductal adenocarcinoma (PDAC), 42.9% of colorectal cancer (CRC), and 27.4% of non‐small cell lung cancer (NSCLC) cases. NRAS mutations have been detected in 2.5% of malignancies, especially in melanoma (23.0%). HRAS mutations (0.8%) occur less frequently and have been detected in a subset of bladder (4.06%) and head and neck cancers (4.21%). Regarding the position of greatest variability, ∼78.3% of KRAS mutations occur at G12, while 58.2% and 29.1% of NRAS and HRAS mutations occur at Q61, respectively. Regarding the associated type of substitution, the KRAS G12D mutation is common in PDAC, while KRAS G12C is the predominant mutation in NSCLC. These distinct RAS mutant distribution patterns may be due to the selective environmental pressures during tumor initiation and progression [12].
FIGURE 2.
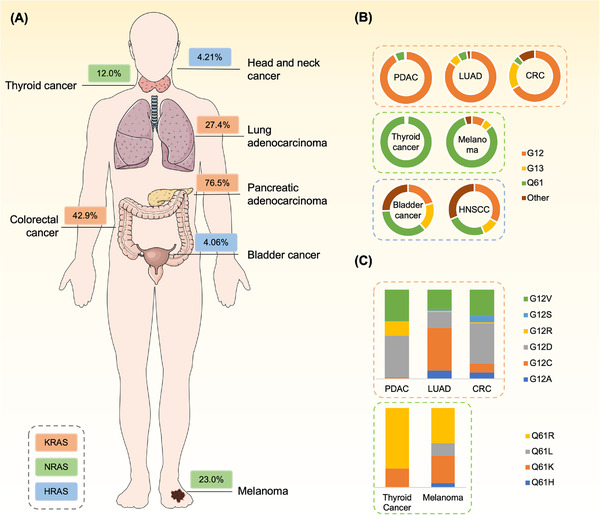
The frequency of RAS mutations in human malignancies. The isoforms, position and type of substitution of RAS mutations vary by tissue types in clinical observations. (A) The frequency of the most common isoforms of RAS mutations across tumor types. (B) Distribution of different position of substitution of KRAS, NRAS and HRAS mutations across tumor types. (C) Distribution of KRAS mutations that are in G12 for pancreatic, lung and colorectal cancers and that of NRAS mutations that are in Q61 for melanoma and thyroid cancer. Data are acquired from GENIE (Version 10.1). Abbreviations: PDAC, pancreatic ductal adenocarcinoma; LUAD, lung adenocarcinoma; CRC, colorectal cancer. HNSCC, head and neck squamous cell cancer
Notably, KRAS is unique in that it encodes for two variants (KRAS4A and KRAS4B) via alternative splicing. These variants have different C‐terminal membrane‐targeting regions owing to the alternative splicing of exon 4 and thus have different post‐translational modifications and membrane localizations [13]. Previous studies showed that KRAS4A was not necessary in embryo development [14] and its relative mRNA level was lower than those of KRAS4B in human cancer [15, 16]. However, a recent study indicated that KRAS4A protein levels were equal to or higher than that of KRAS4B in several cancer cell lines [13], and KRAS4B was dispensable at high levels of KRAS4A [15]. Furthermore, while both isoforms were required in KRAS‐mutated tumor initiation [17], they had distinct functions in stress resistance [17] and metabolism [18]. Thus, approaches targeting RAS‐driven cancers vary depending on the specific isoform and codon mutations as these determine biological and clinical behaviors.
3. THERAPEUTIC STRATEGIES TO TARGET RAS DIRECTLY
RAS proteins are difficult to target given the lack of a pocket for allosteric inhibitor binding. Moreover, the high binding affinity of RAS to nucleotides hinders the efficacy of inhibitors by increasing competition for the GTP‐binding domain. Below we discuss novel covalent inhibitors that can directly target RAS.
3.1. Allele‐specific inhibitors
3.1.1. KRASG12C allele‐specific inhibitors
Pioneering studies adopted a structure‐guided disulfide fragment‐based screening of inhibitors targeting the KRAS G12C mutant (hereafter G12Ci) and identified a novel allosteric switch II pocket [19]. The crystal structure of the compound‐bound KRASG12C mutant indicated that G12Ci bound to cysteine‐12 (C12) and extended to occupy the pocket, allosterically inhibiting interaction with effectors and regulatory proteins, thus restricting activation and signal transduction [19, 20]. Further experiments confirmed that G12Ci attenuated the SOS1‐ and ethylenediaminetetraacetic acid (EDTA)‐catalyzed nucleotide exchange of KRASG12C and the phosphorylation level of downstream kinases [19, 20, 21, 22]. However, it is puzzling how the G12Ci that preferentially interacts with GDP‐bound KRAS mutant can decrease KRAS‐GTP levels. Indeed, the impairment of GTPase activity and guanine nucleotide exchange rate vary between RAS isoforms, mutation positions and substitution types. GTPase activity is maintained in the G12C mutation [9], which allows the KRASG12C mutant to cycle between the GTP‐ and GDP‐bound states, making it susceptible to inhibitors. Introducing co‐mutations (e.g., A59G, Q61L, and Y64A) to KRASG12C inhibited the intrinsic hydrolysis level and decreased the inhibitory effect of G12Ci on KRASG12C‐GTP level [21], indicating that G12Ci required intact GTPase activity to trap KRASG12C in the inactive state.
Although in vitro experiments showed a decrease in KRASG12C‐GTP levels after treatment with inhibitors, many early G12Ci (e.g., compound 12) showed poor KRASG12C engagement [20]. As a result, studies attempted to optimize the in vivo pharmacokinetics (PK) and binding affinity of G12Ci to KRASG12C proteins. Structure‐based drug screening identified a more potent compound, ARS‐853, with a 600‐fold faster engagement rate than compound 12. Although it showed profound anti‐proliferative effects in a panel of KRAS G12C cell lines, ARS‐853 and its analogs showed reduced metabolic stability and bioavailability in a mouse model [22]. Moreover, narrow structure‐activity relationships limited the optimization of ARS‐853. Janes et al. [22] redesigned the drug scaffold by shortening the metabolically active 2‐amino‐1‐(piperazin‐1‐yl)ethan‐1‐one linker and adopting a rigid bicyclic quinazoline core, thus establishing ARS‐1620. Crystal structure analysis of drug‐bound KRASG12C suggested that, compared to ARS‐853, ARS‐1620 could fit into the region between the switch II and α3 helices, enabling additional interaction with H95 that supported a more rigid conformation for covalent binding to C12. Besides, the fluorophenol warhead of ARS‐1620 extended into the hydrophobic region of the switch II pocket and forms several hydrogen bonds, further enhancing the binding potency. Biochemical assay showed that the covalent binding of ARS‐1620 with KRASG12C occurred 10‐fold faster than for ARS‐853, thus exhibiting selective therapeutic effects in cell line‐ and patient‐derived xenografts with the KRAS G12C mutant [22]. ARS‐3248 (JNJ‐74699157) is the latest derivate of ARS‐1620, with improved potency. Its clinical efficacy remains unknown as the phase I clinical trial (NCT04006301) was incomplete.
Amgen (Thousand Oaks, CA, USA) in collaboration with Carmot Therapeutics (Berkeley, CA, USA) identified and optimized a set of compounds with potent anti‐KRASG12C activity through their custom electrophile drug screening platform [23]. They assessed the crystallographic structure of the most selective compounds during each screening iteration and found that the additional occupancy of the previously unexploited Y96/H95/Q99 groove, in which H95 adopted an alternative orientation, could enhance the potency. This led to the identification of AMG 510 (sotorasib)—a clinically approved G12Ci with improved PK properties and 10‐fold greater potency compared to ARS‐1620, according to a nucleotide exchange assay [24]. In the preclinical experiments, AMG 510 exhibited profound allele‐specific anti‐tumor efficacy and synergized with chemotherapy and targeted therapy in a KRAS G12C‐mutated cancers. in vivo tumor inhibition studies of CT26‐KRAS G12C xenografts indicated that AMG 510 may have immunomodulatory effects because it showed lower therapeutic efficacy in immune‐deficient than in immune‐competent mice. In the same disease model, AMG 510 acted synergistically with anti‐programmed cell death protein 1 (PD‐1) and improved survival rates compared with the individual agents. The underlying mechanism of immunomodulation has been attributed to a pro‐inflammatory microenvironment with enhanced long‐term tumor‐specific T‐cell responses [24]. A phase I clinical trial of AMG 510 (NCT03600883) in the treatment of advanced KRAS G12C‐mutated solid tumors demonstrated excellent safety and anti‐tumor activity [25]. No dose‐limiting toxic effects or treatment‐related deaths were observed, 19 of 59 patients (32.2%) with NSCLC had a confirmed response, compared to 3 of 31 patients (7.1%) with CRC. In a phase II trial of AMG 510, Amgen found that 46 of 124 (37.1%) NSCLC patients with KRAS G12C achieved a confirmed response, of which four had a complete response [26]. However, in the phase II trial for KRAS G12C‐mutated CRC, only 6 of 62 patients (9.7%) had objective response [27]. Differences in disease biology and adaptive signaling responses to AMG 510 might underlie the differences in the clinical activity of AMG 510 observed between NSCLC and CRC [28]. For example, it was reported that CRC with KRAS G12C had higher basal receptor tyrosine kinase (RTK) activation and stronger response to growth factor stimulation than NSCLC. In this setting, KRASG12C inhibition induced higher phospho‐ERK rebound in CRC, leading to less profound and more transient inhibition of KRAS downstream signaling in CRC [29]. Subgroup analysis based on molecular biomarkers was conducted to explore the relationships between drug response, mutations, and immune profiles. AMG 510 elicited clinical responses from a wide spectrum of patients with co‐mutations, such as serine/threonine kinase 11 (STK11)/liver kinase B1 (LKB1) and kelch‐like ECH‐associated protein 1 (KEAP1), which were generally associated with a poor prognosis for patients with NSCLC. AMG 510 also induced a long‐term therapeutic effect in all programmed cell death ligand 1 (PD‐L1) expression subgroups, most of whom did not respond to immunotherapy with ramucirumab and docetaxel [26]. The US Food and Drug Administration (FDA) has approved AMG 510 for the treatment of locally advanced or metastatic KRAS G12C‐mutated NSCLC, and a phase III trial comparing sotorasib with docetaxel in end‐stage NSCLC patients with the KRASG12C mutant is underway (NCT04303780).
MRTX849 (adagrasib), developed by Mirati Therapeutics (San Diego, CA, USA), is another leading G12Ci currently under clinical evaluation (NCT03785249). From the Array BioPharma covalent fragment collection, Fell et al. [30] identified a series of tetrahydropyridopyrimidines as irreversible covalent inhibitors of KRASG12C in vivo, though these compounds required further optimization owing to their rapid clearance and poor bioavailability. Subsequent structure‐based analyses of metabolite liabilities associated with the unsatisfactory PK properties led to the synthesis of analogs with enhanced potency, absorption, distribution, metabolism, and excretion (ADME) [31]. Among these, MRTX849 is the most promising candidate with significantly optimized PK properties and robust anti‐tumor efficacy [31]. MRTX849 and anti‐PD‐1 acted synergistically in vivo; however, this effect was significantly reduced in an immune‐deficient KRASG12C mouse model. Therefore, the underlying mechanism is related to tumor microenvironment (TME) remodeling by MRTX849, which increases the antigen presentation and the infiltration of anti‐tumor immune cells, while decreasing immunosuppressive components [32]. Preliminary clinical data from phase I/Ib and II trials showed that 23 of 51 (45%) NSCLC patients and 3 of 18 (17%) CRC patients with the KRAS G12C mutant showed a confirmed response to MRTX849 [33, 34]. KRAS G12C patients harboring an STK11 co‐mutation showed a better response (9 of 14) to MRTX849 than those with wild‐type STK11 [33]. At the transcriptional level, the STK11 co‐mutation conferred a cold immune phenotype to the tumor, with low expression of immune‐related signatures, such as CD4 and CD8. MRTX849 upregulated the levels of immune transcripts and interferon (IFN) signatures in 2 of 3 patients with STK11 co‐mutations, possibly through the recruitment of cytotoxic T cells to the TME, thus reversing the STK11‐mediated immune suppression [33].
Although preclinical experiments and clinical trials showed promising therapeutic results for G12Ci, some patients inevitably developed resistance to monotherapies. Several cell line‐ and patient‐based models were used to identify the potential mechanisms of resistance, which can be divided into two categories. First, the drug‐induced signal reprogramming bypasses the inhibitory effect. For instance, MRTX849 induced molecular changes in five KRAS G12C xenograft‐bearing mice and, although G12Ci rapidly inhibit KRAS‐associated signaling (conferring their anti‐tumor effects), downregulated a set of genes involved in the negative feedback of mitogen‐activated protein kinase (MAPK) pathway, such as dual‐specificity phosphatase (DUSP) and sprouty(SPRY), allowing the reactivation of ERK‐dependent signaling [35]. Compensatory activation of upstream RTKs occurred after G12Ci treatment [35, 36, 37]. The active RTKs shifted KRASG12C to a G12Ci‐insensitive GTP‐bound state, thus simultaneously promoting the activation of wild‐type KRAS‐MAPK/PI3K cascades [35, 36, 37, 38]. Single‐cell transcriptional profiling of KRAS G12C‐mutated lung cancer cells revealed an adaptive cell state in response to G12Ci. The subsets of adaptive cancer cells can circumvent the effects of GDP‐bound state‐specific G12Ci as they produced new KRASG12C cells undergoing epidermal growth factor receptor (EGFR)‐ and aurora kinase‐promoted GTP exchange [39]. Second, inhibition by G12Ci is reduced by the secondary mutation of KRAS G12C or de novo mutation of RAS‐associated signaling molecules. Multiple de novo activating mutations in the RAS‐MAPK cascade (KRAS G12D , KRAS G12V, NRAS, and BRAF) can drive oncogenesis in a KRASG12C‐independent manner. A previously undescribed allele KRAS Y96D potentially conferred “survival advantages” under the selective pressure of G12Ci. Further in vitro structural and functional analyses indicated that the Y96 residue was located at the switch II pocket, where the substitution of single amino acid can disrupt the hydrogen bond between the Y96 and G12Ci and promoted the resistance [40]. Similarly, acquired RAS‐associated signaling mutations, including substitution, amplification, and fusion of oncogenes, were detected in 17 of 38 MRTX849‐refractory patients with RTK/RAS/MAPK/PI3K cascades. Similar secondary KRAS mutations were observed in the switch II pocket of 4 patients, including Y96C, H95Q, H95R, H95D, and R68S [41]. A positive‐selection screening of underlying G12Ci‐resistant mutations in a KRAS G12C Ba/F3 cell population harboring secondary lentivirus‐ or chemical‐induced mutations also suggested similar alterations [41, 42]. Mapping these residues onto the crystallographic structures of the G12Ci‐bound KRASG12C protein indicated that these mutations disrupted the binding of drugs non‐covalently. This differential binding mechanism caused drug‐specific resistance mutations, because exogenous expression of several H95 mutants in KRAS G12C Ba/F3 cells confer resistance to MRTX849 but not AMG 510. However, the R68S and Y96C could mediate the resistance to both AMG 510 and MRTX849. Notably, multiple concurrent resistance mutations appear to be more commonly detected in CRC than in NSCLC, which explains different clinical responses to the G12Ci.
In conclusion, novel KRASG12C allele‐specific inhibitors have shown promising results in preclinical models and clinical trials (Table 1). G12Ci showed better clinical response in lung adenocarcinoma (LUAD) than in other cancers. However, the emergence of drug resistance generally limits the therapeutic effect. Combination therapies co‐targeting both KRASG12C proteins and reactivating pathways and novel compounds with different inhibitory mechanisms may effectively overcome these limitations.
TABLE 1.
Therapeutic strategies to target RAS directly in clinical trials
| Drug | Study phase | Disease setting | ClinicalTrials.gov identifier | Status |
|---|---|---|---|---|
| KRAS G12C allele‐specific inhibitors | ||||
| AMG 510 | I/II | Advanced solid tumors | NCT03600883 | Recruiting |
| II | NSCLC | NCT04625647 | Recruiting | |
| III | NSCLC | NCT04303780 | Active, not recruiting | |
| MRTX849 | I/II | Advanced solid tumors | NCT03785249 | Recruiting |
| III | NSCLC | NCT04685135 | Recruiting | |
| JAB‐21822 | I/II | Advanced solid tumors | NCT05002270 | Recruiting |
| JNJ‐74699157 | I | Advanced solid tumors | NCT04006301 | Completed |
| GFH925 | I/II | Advanced solid tumors | NCT05005234 | Not yet recruiting |
| YL‐15293 | I/II | Advanced solid tumors | NCT05173805 | Not yet recruiting |
| JDQ‐443 | I/II | Advanced solid tumors | NCT04699188 | Recruiting |
| III | NSCLC | NCT05132075 | Not yet recruiting | |
| GDC‐6036 | I | Advanced solid tumors | NCT04449874 | Recruiting |
| LY3499446 | I/II | Advanced solid tumors | NCT04165031 | Terminated |
| LY3537982 | I | Advanced solid tumors | NCT04956640 | Recruiting |
| BI 1823911 | I | Advanced solid tumors | NCT04973163 | Recruiting |
| D‐1553 | I/II | Advanced solid tumors | NCT04585035 | Recruiting |
| Other direct‐targeting RAS inhibitors | ||||
| AZD4785 | I | Advanced solid tumors | NCT03101839 | Completed |
| siG12D LODER | I | PDAC | NCT01188785 | Completed |
| II | PDAC | NCT01676259 | Recruiting | |
| iExosomes | I | PDAC | NCT03608631 | Recruiting |
Abbreviations: NSCLC, non‐small cell lung carcinoma; PDAC, pancreatic ductal adenocarcinoma
3.1.2. KRASG12D allele‐specific inhibitors
The KRAS G12C mutation predominantly occurs in LUAD, whereas KRAS G12D is the most frequently detected mutation in RAS‐mutated cancers. The inhibitory effects of G12Ci are dependent on the warhead forming covalent bond with C12 and extending to occupy the switch II pocket. Unlike KRASG12C, the KRASG12D protein lacks a reactive residue adjacent to the switch II pocket and necessitates novel targeting strategies. Several attempts were made to target a pocket in the RAS between switch I and II [43, 44]. Inhibitors, such as BI‐2852, bind to the switch I/II pocket in both the GTP‐ and GDP‐bound state of KRASG12D, preventing the interaction between RAS and SOS1 as well as downstream effectors. A binding site near proline 110 in KRAS (P110 site) was used to develop a series of inhibitors involving KAL‐21404358 [45]. However, owing to their weak affinity, these inhibitors showed limited cellular activity. MRTX1133 is a potent selective non‐covalent G12Di generated by structure‐based optimization of the binding affinity to the mutant D12 side chain. MRTX1133 demonstrated nanomolar activity in the cellular assays and anti‐tumor efficacy in the tumor models bearing the KRAS G12D mutation [46]. TH‐Z835, which was generated using a similar strategy as that for MRTX1133, effectively disrupted MAPK signaling and reduced tumor volume in KRAS G12D‐mutated PDAC by forming a salt‐bridge bond with D12 [47]. In a mouse model, it appeared to act synergistically with anti‐PD‐1 antibodies. Despite these promising results, more comprehensive pharmacological characterization and preclinical testing are required before applying G12Di in clinical trials.
3.2. Other direct‐targeting strategies
As previously stated, KRAS G12C mutations account for a small subset of cancers, and drug resistance inevitably impedes the therapeutic effects of existing G12Ci. Moreover, the anti‐tumor efficacy of G12Di needs further testing in clinical settings. To address these shortcomings, researchers are developing complementary direct‐targeting strategies for RAS‐mutated malignancies.
3.2.1. Targeted protein degradation
Targeted protein degradation techniques allow for the direct removal of pathogenic proteins. Proteolysis‐targeting chimeras (PROTACs) and macromolecules (e.g., macrodrug degraders) are synthesized to act as a “bridge” between proteins of interest and E3 ligases. The recruitment of E3 ligases results in ubiquitination of the protein, which serves as a marker for degradation by the ubiquitin‐proteasome system. Because the clearance of target proteins ablates all of their associated functions, direct protein degradation is assumed to be superior to traditional treatments involving small‐molecule inhibitors [48, 49]. Given that RAS proteins participate in a complex and redundant signal network and rely heavily on protein‐protein interaction, degradation is a powerful strategy for the sequestration of oncogenic RAS. Therefore, efforts have been made to develop KRAS‐specific degrader molecules.
Initial efforts focused on using the RAS‐binding domain along with the cystine‐rich domain of CRAF and incorporating them into the U‐Box‐based ubiquitin ligase to create chimeric proteins known as (RBD+CRD)CRAF‐U‐Box (RC‐U) [50]. Transfection of RC‐U into pancreatic cancer cell lines significantly reduced the KRAS protein levels in a ubiquitin‐dependent proteasome‐mediated manner. Similarly, lowering the KRAS protein level inhibited pancreatic cancer proliferation both in vitro and in vivo. However, RC‐U also reduced the protein levels of HRAS and NRAS. Nonselective RAS degradation may cause severe on‐target toxicity, preventing the use of this approach in clinical practice. Bery et al. [51] engineered KRAS‐specific designed ankyrin repeat proteins (DARPins) with E3 ligases to specifically target KRAS and generate macrodrug degraders. This KRAS‐specific degrader selectively inhibited RAS‐downstream signaling and proliferation of KRAS‐mutated cancers in vivo and in vitro. While the degrader reduced wild‐type KRAS levels, no obvious decrease in proliferation was observed in RAS wild‐type cells, including untransformed cells, which supported a favorable safety profile. Nevertheless, the clinical applications of these macrodrugs are limited by a lack of an efficient delivery method for macromolecules into RAS‐mutated cells. More viable options include cell‐permeable small‐molecule PROTACs or small‐molecule degraders converted from G12Ci. Zeng et al. [52] designed a PROTAC with a quinazoline scaffold of ARS‐1620 as a binder and thalidomide derivatives as E3 ligase ligand. The lead compound successfully engaged in cereblon (CRBN) and enabled CRBN/KRASG12C dimerization, resulting in the efficient reduction of exogenously expressed GFP‐KRASG12C in reporter cells. However, it did not degrade endogenous KRAS in lung or pancreatic cancer cells. Subsequent experiments revealed that the insufficient polyubiquitination of KRASG12C and the ineffective recruitment of CRLCRBN to membrane‐anchored exogenous KRAS significantly reduced the degradation activity. LC‐2, an endogenous KRASG12C degrader, was generated using a similar strategy [53]. This compound, which combined MRTX849 with a von Hippel‐Lindau (VHL) E3 ligase ligand, caused rapid and sustained degradation in cancer cell lines with homozygous or heterozygous KRAS G12C alleles. Despite its potential in vitro, LC‐2 was unable to participate in catalytic rounds of degradation, a critical procedure that allowed for maximal degradation in vivo [54]. These PROTACs have a similar warhead to that of G12Ci, implying that they are susceptible to the second mutations in the switch II pocket. Further research is needed to determine whether these compounds can overcome the reactivation caused by signal reprogramming and acquired resistance. In summary, both PROTACs and macrodrug degraders induced robust degradation of exogenously expressed RAS in vitro, though several bottlenecks limited future clinical applications. Further research is required to improve in vivo delivery efficiency and mutant‐allele targeting specificity.
3.2.2. Gene therapy
Gene therapy has seen early success in treating monogenic disorders, such as immunodeficiencies and storage disorders. Gene therapy, as opposed to small‐molecule inhibitors that require an ideal binding pocket and PK properties, provides a tailored treatment that specifically targets pathogenic genes with few off‐target toxicities. Antisense oligonucleotide (ASO) or RAS‐specific small interfering RNA (siRNA)‐containing vectors have been designed to facilitate RAS knockdown in oncogenic RAS mutations. AZD4785, a high‐affinity constrained ethyl‐containing therapeutic ASO, was designed to selectively reduce cellular KRAS mRNA levels and showed significant therapeutic effects in KRAS‐mutated cancer models both in vitro and in vivo [55, 56]. Notably, AZD4785‐induced inhibition of KRAS was not hampered by feedback reactivation of the RAS pathway. However, following phase I study (NCT03101839), the clinical trial of AZD4785 was halted owing to insufficient knockdown of KRAS. Moreover, AZD4785 did not distinguish between wild‐type and mutated KRAS, which may contribute to its on‐target toxicity in normal cells. Another group of researchers used a biodegradable polymer matrix to load anti‐KRAS G12D siRNA, known as siG12D LODER [57], and found a significant decrease in KRAS expression and tumor growth in vitro and in vivo. In addition, siG12D LODER combined with chemotherapy was evaluated in a phase I/IIa clinical trial (NCT01188785), achieving 2 partial responses (PR) and 10 stable diseases (SD) out of 12 patients with locally advanced pancreatic cancer [58]. In addition to polymers, nanoparticles, such as liposomes, were used to target KRAS or downstream effectors by RNA interference and are superior to viral‐based systems. However, low tumor‐targeting specificity and rapid clearance hamper the systematic delivery of these nanoparticles. Unlike nanoparticles, exosomes contain membrane proteins, such as CD47, which act as “don't eat me” signals and promote circulation retention [59, 60]. Furthermore, KRAS mutations confer cancer cells with constitutive macropinocytosis, which increases exosome uptake. iExosomes—derived from fibroblast‐like mesenchymal cells and loaded with siRNA specifically targeting the KRAS G12D mutation—reduced pancreatic tumor growth and metastasis in a mouse model, whereas regular liposomes containing the same siRNA had a much smaller effect [61]. A phase I clinical trial evaluating iExosomes for the treatment of patients with pancreatic cancer harboring the KRAS G12D mutation has recently begun participant recruitment (NCT03608631). Gene therapy using various vectors to deliver oligonucleotides targeting RAS has demonstrated promising therapeutic effects, though the safety profile of these nanoparticles requires further evaluation.
3.2.3. Cytosol‐penetrating antibody
Although several monoclonal antibodies (mAbs) against membrane proteins have been approved for clinical use, they are ineffective against cytosolic proteins because of their inability to penetrate the cytosolic region. Earlier studies used retroviruses or transgenic techniques to express immunoglobulin heavy chain variable domain fragments against the RAS protein and confirmed that antibody‐mediated blockade of RAS‐effector interactions could effectively inhibit the growth of RAS‐mutated cancer cells [62, 63]. Consequently, RT11 cytosol‐penetrating antibodies were developed to target intracellular GTP‐bound oncogenic RAS [64]. RT11 entered the cytosol via clathrin‐mediated endocytosis, which used heparan sulfate proteoglycan (HSPG) as a receptor, disrupting the interaction between RAS and its effectors. However, HSPG is ubiquitously expressed in epithelial cells, which may result in on‐target toxicity. The RGD10 cyclic peptide, which bound to tumor‐associated integrins, was linked to produce RT11‐i with a high tumor specificity. RT11‐i demonstrated profound anti‐tumor efficacy and bioavailability in mice bearing RAS‐mutated xenografts. A second generation of iMab, known as inRAS37, demonstrated improved anti‐tumor activity, PK properties and tumor accumulation [65]. Intrinsic or acquired PI3K or β‐catenin mutations drove resistance to inRAS37, which could be overcome by combinatorial treatments with corresponding inhibitors [65]. Despite promising preclinical results, no clinical trials of cytosol‐penetrating antibodies have been conducted.
4. THERAPEUTIC STRATEGIES TO TARGET RAS INDIRECTLY
Despite progress in the direct inhibition of KRAS G12C‐mutated cancers, therapeutic strategies targeting other RAS isoforms and mutant alleles remain elusive. Considerable efforts have been expended to indirectly target proteins involved in RAS nucleotide exchange, processing, membrane anchoring, and upstream and downstream signal transduction.
4.1. Inhibitors disrupting RAS nucleotide exchange
Following the failure of compounds that interfered with RAS processing and membrane anchoring in clinical trials [66, 67], the focus shifted to disrupting RAS nucleotide exchange. Although point mutations impair the intrinsic GTPase activity of RAS proteins, RAS can still undergo nucleotide exchange between the GTP‐ and GDP‐bound states. In this scenario, hyperactivation of RAS is caused by the GEF‐catalyzed GTP upload of RAS. Thus, targeting RAS nucleotide exchange is now considered a potential therapeutic strategy for RAS‐mutated malignancies.
4.1.1. SOS1 inhibitors
SOS1 is one of the most important GEFs of RAS and mediates the allosteric cross‐activation of wild‐type RAS by the KRAS mutant, in addition to increasing the level of GTP‐bound RAS [68]. SOS1 gene ablation and GEF function silencing reduced tumor growth in RAS‐mutated cancers but not RAS‐wild‐type cells [68]. Several early attempts to target SOS1 have been made. Fragment‐based screening was used to identify the first series of small molecule inhibitors that bound KRAS between the switch I and switch II regions, disrupting its interaction with SOS1 [69]. However, these compounds had a low affinity for KRAS. Researchers also developed peptides that mimic the SOS helix, thereby inhibiting the RAS‐SOS interaction. Even with nanomolar affinity, the cellular inhibitory effects of these peptides were relatively low [70, 71], and some of these inhibitors paradoxically activated RAS nucleotide exchange and exhibited biphasic modulation of RAS‐GTP levels via negative feedback [72]. Hillig et al. [73] recently developed potent small‐molecule SOS1 inhibitors using a structure‐guided design, which combined fragment‐based and high‐throughput screening. The final compound, BAY‐293, demonstrated sub‐micromolar anti‐tumor activity against various tumor cell lines. However, BAY‐293 inhibited proliferation and the MAPK pathway more in RAS wild‐type cells than in RAS‐mutated cells, raising concerns regarding intolerant toxicity in vivo. BI‐3406—the first orally administered SOS1‐KRAS interaction inhibitor—reduced the level of GTP‐bound RAS and cell proliferation in most RAS‐driven cancers while sparing RAS wild‐type cells in vitro and in vivo [74]. Tumors harboring RAS Q61H and RAS G12R mutant showed resistance to BI‐3406. The RAS Q61H mutant severely impaired intrinsic GTPase activity [9], requiring fewer upstream signal to maintain RAS‐GTP levels, whereas the RAS G12R mutant abolished the interaction with the allosteric binding site of SOS1, resulting in KRAS‐independent survival [75]. BI‐3406 acted synergistically with the MEK inhibitor trametinib and the KRAS G12C inhibitor AMG 510 in the treatment of RAS‐mutated cancers, as it reduced MAPK reactivation caused by inhibitor‐induced relief of negative feedback. BI 1701963, a clinical candidate derived from BI‐3406, has progressed to phase I clinical trials in combination with MEK inhibitors and G12Ci (NCT04111458). However, a recent study found that KRASG13D had a higher affinity for SOS than the other isoforms and tended to form ternary complexes with SOS, in which KRASG13D‐GTP allosterically modulated SOS activity to accelerate the nucleotide exchange rate of wild‐type KRAS [76]. Surprisingly, RAS‐SOS inhibitors had little effect on oncogenic RAS‐induced SOS‐mediated transactivation of wild‐type RAS. Further experiments revealed that G12Ci ARS‐1640 could not block KRASG12C‐GDP within its ternary complex with SOS. In general, these findings highlighted the critical need for novel RAS‐SOS inhibitors capable of disassociating ternary complexes.
4.1.2. SHP2 inhibitors
SHP2 is a non‐receptor ubiquitous protein tyrosine phosphatase (PTP) involved in RAS‐associated signaling and is encoded by the protein tyrosine phosphatase non‐receptor type 11 (PTPN11) gene. SHP2 modulates RAS signaling in two ways. The canonical model showed that SHP2 acted as a scaffold protein by binding Grb2 and SOS1 and integrating signals from upstream RTKs, thus increasing RAS‐GTP levels [77, 78]. On the other hand, Src were found to disrupt RAS binding to downstream effectors as well as guanine nucleotide exchange by phosphorylation of RAS tyrosine. SHP2 can dephosphorylate tyrosine in RAS, thus restoring the downstream signaling and GTPase cycling [79, 80]. SHP2 inhibition is a promising strategy for the treatment of RAS‐mutated cancers, particularly those that rely on RAS nucleotide cycling. Genetic ablation of PTPN11 inhibited tumor initiation and delayed the tumor development in genetically engineered mouse model (GEMM) harboring KRAS G12D‐mutated lung and pancreatic cancers [81]. However, orthosteric SHP2 inhibitors that showed promising effects in preclinical models were unsuccessful in clinical trials owing to their low potency and lack of selectivity for SHP2 [82]. SHP099 is the first allosteric inhibitor that concurrently binds to the three domains of SHP2 and stabilizes SHP2 in an auto‐inhibitory conformation. SHP099 was highly sensitive to SHP2 because it had no affinity for a panel of 21 phosphatases and 66 kinases [83]. While SHP099 did not affect the cell growth and proliferation of RAS‐mutated cell lines in two‐dimensional culture systems [83], experiments using three‐dimensional spheroids revealed the sensitivity of SHP2 in KRAS‐mutated cancers [84]. Eight SHP2 inhibitors have entered the clinical development (Table 2). In addition to its direct action on tumor cells, SHP2 is involved in the modulation of other components of the TME. PD‐1 plays an important role in the T cell exhaustion [85]. When activated by its ligands, PD‐1 recruits SHP2 to its immune‐inhibitory motifs; activated SHP2 can dephosphorylated nearby signal molecules, such as CD28, resulting in decreased cytokine secretion and T cell proliferation [86, 87]. Inhibition of SHP2 increased CD8+ T cell infiltration in tumors, while decreasing the amount of pro‐tumorigenic M2 macrophages via attenuation of CSF1 receptor signaling [88]. The synergistic anti‐tumor effect of SHP2 inhibitor and anti‐PD‐1 antibody was demonstrated in the KRAS‐mutated immune‐competent mouse model [89]. Consequently, a phase Ib trial of TNO155 in combination with the PD‐1 antibody spartalizumab is now actively recruiting participants (NCT04000529).
TABLE 2.
Therapeutic strategies to target RAS indirectly in clinical trials
| Drug | Study phase | Disease setting | ClinicalTrials.gov identifier | Status |
|---|---|---|---|---|
| SOS1 inhibitors | ||||
| BI‐1701963 | I | Advanced solid tumors | NCT04111458 | Active, not recruiting |
| SHP2 inhibitors | ||||
| TNO155 | I | Advanced solid tumors | NCT03114319 | Recruiting |
| RMC‐4630 | I | Advanced solid tumors | NCT03634982 | Recruiting |
| JAB‐3068 | I/II | Advanced solid tumors | NCT04721223 | Recruiting |
| JAB‐3312 | I | Advanced solid tumors | NCT04045496 | Recruiting |
| BBP‐398 | I | Advanced solid tumors | NCT04528836 | Recruiting |
| ERAS‐601 | I | Advanced solid tumors | NCT04670679 | Recruiting |
| RLY‐1971 | I | Advanced solid tumors | NCT04252339 | Recruiting |
| SH3809 | I | Advanced solid tumors | NCT04843033 | Recruiting |
| RAF inhibitors | ||||
| Belvarafenib | I | Melanoma | NCT04835805 | Recruiting |
| LXH‐254 | I | Advanced solid tumors | NCT02607813 | Active, not recruiting |
| Lifirafenib | I/II | Advanced solid tumors | NCT03905148 | Recruiting |
| BGB‐3245 | I | Advanced solid tumors | NCT04249843 | Recruiting |
| TAK‐580 | I | Melanoma | NCT02723006 | Terminated |
| MEK inhibitors | ||||
| Binimetinib | III | Melanoma | NCT01763164 | Completed |
| Pimasertib | II | Melanoma | NCT01693068 | Completed |
| ERK inhibitors | ||||
| LY‐3214996 | I | Advanced solid tumors | NCT02857270 | Active, not recruiting |
| KO‐947 | I | Advanced solid tumors | NCT03051035 | Terminated |
Abbreviations: SOS1, son of sevenless 1; SHP2, Src homology‐2 domain‐containing protein tyrosine phosphatase 2; ERK, extracellular signal regulated kinase
4.2. Inhibitors of RAS upstream signaling
Upstream stimuli are considered dispensable for constitutively active RAS mutants to transduce oncogenic signals to downstream, and early clinical trials showed that RAS‐mutated malignancies were unresponsive to EGFR inhibition [90, 91, 92]. Moreover, the emergence and amplification of KRAS mutation was observed in the CRC patients who had become refractory to anti‐EGFR mAbs [93, 94], supporting the idea that RAS mutations generate aberrant proteins with constitutively active catalytic activity conferring independence from upstream signaling. However, two pooled retrospective analyses of patients with chemotherapy‐refractory CRC revealed that those with the KRAS G13D mutant had a better prognosis when treated with cetuximab than those with other KRAS mutations [95, 96]. Two further retrospective analyses and a prospective‐cohort study (ICECREAM) showed no statistically significant difference between the two subgroups in terms of the clinical benefits of EGFR‐targeted therapies [97, 98, 99]. Nevertheless, recent experimental and clinical data revealed an unexpected role for an overactivated upstream signal in RAS‐induced tumors. An oncogenic RAS mutant was found to upregulate EGFR expression early in tumorigenesis, and deletion of the egfr gene in the GEMM significantly reduced the development of Kras G12D‐induced PDAC [100, 101]. In addition to EGFR, other members of the ERBB family (originally named because of their homology to the erythroblastoma viral gene product, v‐erbB) transduce upstream signals to promote tumorigenesis in an autochthonous Kras G12D lung cancer mouse model. The pan‐ERBB inhibitors afatinib and neratinib have demonstrated promising therapeutic effects in preclinical models [102, 103, 104]. It is clear from these controversial findings that EGFR‐targeted therapies cannot yet be used in patients with RAS‐mutated tumors, including those with KRAS G13D mutations.
4.3. Inhibitors of RAS downstream signaling
Disrupting the aberrant binding of RAS proteins to downstream effectors could eliminate their oncogenic function as the signal is propagated via downstream interactions. However, with more than ten distinct downstream effectors, it remains difficult to determine the appropriate cascade to block. Recent efforts have yielded multiple inhibitors targeting the MAPK and PI3K pathways (Table 2). Although conceptually straightforward and simple, the limitations of RAS downstream effector inhibitors were revealed in preclinical models and clinical trials of RAS‐mutated malignancies.
4.3.1. MAPK pathway inhibitors
For decades, treatment strategies for RAS‐mutated malignancies have prioritized the development of inhibitors that block kinases in the MAPK pathway, which is thought to be the primary oncogenic effector molecules. However, these inhibitors provide only limited therapeutic benefits for tumors harboring RAS mutant.
RAF inhibitors. Sorafenib, a first‐generation RAF inhibitor, has been approved by the US FDA for advanced renal cell carcinoma, hepatocellular carcinoma, and thyroid cancer. Its anti‐tumor efficacy is related to the inhibition of multi‐tyrosine kinases involved in tumor progression and angiogenesis [105], though the clinical results for RAS‐mutated malignancies were disappointing [106], probably due to the insufficient suppression of ERK‐driven cancer growth [107]. Next‐generation RAF inhibitors (type 1.5), such as vemurafenib and dabrafenib, induced significant responses in BRAF V600 metastatic melanoma [108, 109, 110, 111]. However, in RAS‐mutated cells, these type 1.5 inhibitors were found to paradoxically activate ERK [112, 113, 114]. The “paradox breaker” second‐generation RAF inhibitors (type 2), such as LY3009120, LXH254 and lifirafenib, showed therapeutic effects in the preclinical models of RAS‐mutated and BRAF‐mutated tumors [115, 116, 117]. However, none of these inhibitors have been approved for clinical uses in RAS‐mutated cancers owing to the limited efficacy in the clinical trials [118, 119, 120, 121].
MEK inhibitors. Three MEK inhibitors (trametinib, cobimetinib, and binimetinib) have been approved by the US FDA for clinical use in patients with BRAFV600‐mutated melanoma [122]. No clinical benefit was observed for these inhibitors in the RAS‐mutated tumors [123, 124, 125, 126, 127]. Regarding constitutively active RAS, MEK inhibitors relieve the ERK‐mediated feedback inhibition of RAF, which increases the feedback phosphorylation of MEK and reduce inhibitor binding affinity. However, patients with NRAS‐mutated melanoma who received pimasertib, an oral selective MEK inhibitor, showed longer progression‐free survival than those who received dacarbazine in a phase II clinical trial [128].
ERK inhibitors. Overactivation of ERK is the central mechanism of RAS signaling in cancer. Moreover, various mechanisms of RAF and MEK inhibitor resistance are mediated by recovery of ERK activation. In preclinical studies, ERK inhibitors demonstrated effective anti‐tumor activity in RAS‐mutated cancers [129]. SCH772984, an allosteric and ATP‐competitive ERK inhibitor, rapidly inhibited the activation of ERK in a panel of KRAS‐ and NRAS‐mutated tumors [130]. However, MK8353, a modified compound of SCH772984 with improved PK properties, failed to improve clinical results in RAS‐mutated cancer (there was no response among the five evaluable patients with RAS‐mutated cancer [131]). GDC‐0994 demonstrated promising single‐agent activity in multiple in vivo models, including KRAS‐mutated tumor‐bearing mice [132, 133]. However, it showed unsatisfactory anti‐tumor efficacy among RAS‐mutated cancers in a phase I clinical trial [134]. It was reported that incomplete RAF‐MEK‐ERK cascade suppression may resulted in adaptive resistance that circumvented ERK inhibition mediated by upstream signaling molecules, which emphasized the need for a combination of MEK, BRAF, and EGFR inhibitors to maintain a durable MAPK inhibition [134]. Other ERK inhibitors, including LY‐3214996 and KO‐947, are currently being tested in phase I clinical trials. In addition to directly targeting ERK catalytic subunits, disruption of ERK dimerization was found to suppress tumor development [135]. DEL‐22379 bound to the dimerization interface and prevented the interaction of two ERK isoforms, resulting in significant pro‐apoptotic effects in RAS‐mutated cell line xenografts [136]. Furthermore, DEL‐22379 had milder adverse effects than those that directly inhibit the catalytic activity. To date, no clinical trials of DEL‐22379 have been initiated.
4.3.2. PI3K pathway inhibitors
Aberrant flux through the RAS‐PI3K cascade plays a critical role in the development and maintenance of RAS‐induced tumors. The induced mutation in pi3k catalytic subunit alpha (pi3kca) which encodes the p110α catalytic subunit, is unable to interact with RAS and attenuates tumor formation, progression and metastasis in a mouse model [137, 138, 139]. These findings largely promoted the development of pharmacological compounds targeting the PI3K pathway in recent years.
Wortmannin is the first PI3K inhibitor that non‐selectively inhibits all isoforms of class I PI3K. Recently, several synthetic pan‐PI3K inhibitors were developed. These compounds demonstrated broad‐spectrum anti‐tumor effects in various preclinical models via multiple molecular targets. The US FDA has only approved four class I PI3K inhibitors thus far. However, these compounds showed limited anti‐tumor activity in RAS‐transformed cell lines and mouse models [140, 141, 142, 143]. A recent computational simulation of PI3Kα activation revealed a new drug‐binding site between the lipid substrate‐binding and the ATP‐binding pockets of active PI3Kα, which provided a novel active state‐specific drug design principle for PI3K inhibitors [144].
This field also attempted to develop inhibitors that target Akt, the main downstream effector of PI3K. Similarly, Akt inhibitors alone demonstrated no significant anti‐tumor effects in KRAS‐mutated xenografts [145, 146], and a significant level of adverse effects was observed in multiple clinical trials, primarily due to on‐target toxicities to systemic metabolism and off‐target toxicities. Subsequent dose‐limiting effect severely limited the clinical efficacy of these compounds [147]. In addition, relief of the negative feedback of the PI3K cascade led to the pathway reactivation and abolished the long‐term inhibitory capacity of the inhibitors.
4.4. Regulatory circuits and feedback inside the RAS signaling network
As a single agent, inhibitors of RAS downstream effectors have only promoted tumor regression and survival in a small subset of patients. This unresponsiveness could be attributed to the redundancy and adaptive reprogramming of the RAS signaling network. Instead of being a simple linear and unidirectional pathway, the RAS downstream pathways are interconnected, redundant, and regulated by multiple negative feedbacks, ensuring stable signal outputs under natural conditions.
ERK‐mediated negative‐feedback circuits have been extensively studied. ERK interacts with and phosphorylates a wide range of substrates, including key components of the RTK‐MAPK cascade (Figure 3A). For instance, ERK inhibits EGF‐induced EGFR kinase activity by phosphorylating T669 [148]; SOS1 can be phosphorylated by ERK, thereby preventing RAS activation [149, 150]; ERK phosphorylates multiple S/TP sites in BRAF and CRAF, disrupting their upstream interactions and heterodimerization [151, 152]; and ERK phosphorylation at the T292 residue of MEK1 may reduce the catalytic activity of the MEK1/MEK2 dimer [153]. In addition to the catalytic components of the MAPK cascade, some protein scaffolds are associated with signaling. The kinase suppressor of Ras‐1 (KSR1) forms a ternary complex with BRAF and MEK, thereby increasing MEK activation. However, the binding of active ERK to KSR1 allows for the phosphorylation of KSR1, resulting in its dissociation and release from the plasma membrane, thus weakening signal transduction [154]. Furthermore, ERK translocation to the nucleus promotes the expression of numerous genes, some of which encode proteins that regulate the activation of the MAPK cascade, known as “transcriptional‐dependent feedback”. ERK‐mediated expression of DUSPs attenuates ERK activity via dephosphorylation [155, 156]. Similarly, ERK activates the expression of SPRY proteins, which are then translocated to the plasma membrane [157] and phosphorylated at a specific N‐terminal tyrosine residue. Evidence suggested that SPRY proteins uncoupled multiple upstream growth factor signals and repressed the pathway activation of RAS [158]. Moreover, Sprouty‐related protein with an EVH1 domain (SPRED1), a member of the SPRY superfamily that is also transcriptionally induced by ERK, may induce the plasma membrane localization of NF1, thereby decreasing RAS‐GTP levels [159].
FIGURE 3.
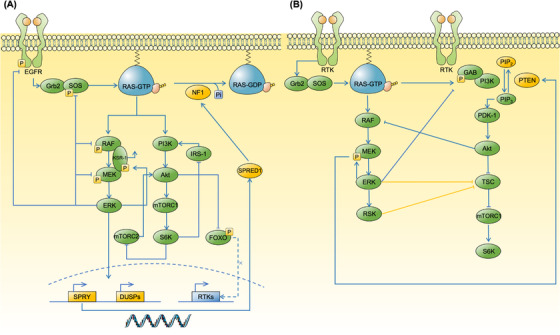
Regulatory circuits and feedbacks inside the RAS signaling network. Regulatory circuits and feedbacks inside the RAS pathways are activated following extracellular stimuli to ensure the stable signal outputs. These pro‐homeostatic mechanisms could cause pathway reactivation in response to target therapies against RAS‐associated cascades. (A) Vertical inhibitory feedbacks involving upstream molecules, downstream MAPK and PI3K pathways. The activation of ERK feeds back to negatively regulate the MAPK pathway by phosphorylation of SOS1, RAF and MEK, as well as promoting the transcription of SPRY and DUSPs, which increase the activity of NF1 and subsequently downregulate the level of RAS‐GTP. Likewise, the activation of Akt prevents the nuclear localization of FOXO, decreasing the transcription of multiple RTKs thus reducing the upstream stimuli. Activated S6K could reduce PI3K signaling by impeding the activity of mTORC2 and IRS‐1. (B) Horizontal circuits include cross‐inhibition (blue line) and cross‐activation (yellow line) between MAPK and PI3K pathways. The activation of ERK leads to the phosphorylation of GAB1, attenuating the GAB1‐mediated recruitment of PI3K to the EGFR. In addition, activated ERK phosphorylates MEK, which in turn promotes the membrane recruitment of PTEN to inhibit the activation of PI3K cascade. Akt‐mediated phosphorylation of RAF de‐couples its interaction of upstream activators or downstream substrates, thus halting the MAPK pathways. Except cross‐inhibition, both ERK and RSK participate in the cross‐activation of PI3K pathways by phosphorylating TSC, impeding its inhibition effect on Rheb which in turn activates the mTROC1. Abbreviations: MAPK, mitogen‐activated protein kinase; PI3K, phosphoinositide 3‐kinase; ERK, extracellular signal regulated kinase; SOS, Son of Sevenless; DUSP, dual‐specificity phosphatase; SPRY, Sprouty; NF1, neuro‐fibromin 1; FOXO, forkhead box, class O; RTK, receptor tyrosine kinase; S6K, S6 kinase; EGFR, epidermal growth factor receptor; PTEN, phosphatase and tensin homolog; IRS‐1, insulin receptor substrate‐1; KSR‐1, kinase suppressor of Ras‐1; GAB1, Grb2‐associated binder‐1; PDK‐1, phosphoinositide‐dependent kinase‐1; TSC, tuberous sclerosis complex; SPRED1, Sprouty Related EVH1 Domain Containing 1;
Similar negative regulatory mechanisms may apply to the PI3K pathway, which have mostly been attributed to the forkhead box, class O (FOXO)‐dependent activation of RTK expression and mTORC1‐mediated insulin receptor substrate‐1 (IRS‐1) inhibition (Figure 3A). Akt‐mediated phosphorylation of FOXO prevents its nuclear localization, which inhibits the transcriptional activation of RTKs [160]. The activation of mTORC1 and downstream S6 kinase (S6K) decreases the expression and stability of IRS‐1, an adaptor protein that connects the growth factor signal to the PI3K‐Akt pathway [161, 162, 163], thereby attenuating pathway activation. S6K activation results in the phosphorylation of mTORC2, which inhibits the maximal activation of Akt [164].
In addition to vertical inhibitory circuits, some effector molecules with broad‐spectrum catalytic activity in both MAPK and PI3K pathways (e.g., ERK, RSK, AKT, and S6K) are linked to the output signal [160, 165] (Figure 3B). Notably, the MAPK and PI3K cascades can repress each other's activity, so called cross‐inhibition. For instance, the activation of ERK leads to the phosphorylation of Grb2‐associated binder‐1 (GAB1) at six serine/threonine residues, four of which are adjacent to the SH2‐binding motif specific to the PI3K p85 subunit; thus, the GAB1‐mediated recruitment of PI3K to EGFR is attenuated, as is the subsequent Akt activation [166]. Moreover, ERK‐mediated phosphorylation of MEK1 at the Y292 residue contributes to the membrane recruitment of phosphatase and tensin homolog (PTEN) as part of a ternary complex containing the adaptor protein membrane‐associated guanylate kinase, WW and PDZ domain‐containing protein 1 (MAGI1), decreasing the phosphatidylinositol 3,4,5‐trisphosphate (PIP3) concentration and Akt activation [167, 168]. Akt may also dampen MAPK activation by phosphorylating BRAF and CRAF at specific serine residues, disrupting RAF coupling to upstream activators or the downstream substrate MEK [169]. In addition to cross‐inhibition, extensive research shows that excessive activation of the MAPK cascade can also promote mTORC1 activation. Both ERK and RSK can phosphorylate tuberous sclerosis complex (TSC) at different serine residues, preventing its inhibition of Ras homologue enriched in brain (Rheb), which activates the mTORC1 [170, 171, 172, 173].
However, tumors may use these pro‐homeostatic mechanisms to avoid downstream RAS inhibitors. Treatment with a single‐agent inhibitor reduces negative feedback and cross‐inhibition of the RAS downstream pathway, preventing long‐term pathway repression and causing pathway reactivation.
4.5. Vertical and horizontal inhibition
Signal reprogramming through reactivation of upstream RTKs and downstream effectors of the MAPK and PI3K cascades mediates resistance to RAS‐targeted therapy. Therefore, a single drug is insufficient in silencing upstream oncoproteins, and concurrent inhibition at multiple levels should be conducted to improve its efficacy (Table 3).
TABLE 3.
Combination therapy for RAS‐mutated malignancies in clinical trials
| Drug | Study phase | Disease setting | ClinicalTrials.gov identifier | Status |
|---|---|---|---|---|
| Combinations of KRAS G12C allele‐specific inhibitors | ||||
| AMG 510 trametinib | I/II | Advanced solid tumors | NCT04185883 | Recruiting |
| AMG 510 TNO155 | ||||
| AMG 510 RMC‐4630 | ||||
| AMG 510 afatinib | ||||
| AMG 510 palbociclib | ||||
| AMG 510 everolimus | ||||
| AMG 510 anti‐PD‐1/PD‐L1 antibodies | ||||
| MRTX849 afatinib | I/II | Advanced solid tumors | NCT03785249 | Recruiting |
| MRTX849 TNO155 | I/II | Advanced solid tumors | NCT04330664 | Active, not recruiting |
| MTRX849 anti‐PD‐1 antibody | II | NSCLC | NCT04613596 | Recruiting |
| MRTX849 cetuximab | III | CRC | NCT04793958 | Recruiting |
| MTRX849 BI‐1701963 | I | Advanced solid tumors | NCT04975256 | Recruiting |
| MRTX849 palbociclib | I | Advanced solid tumors | NCT05178888 | Recruiting |
| GDC‐6036 cetuximab | I | Advanced solid tumors | NCT04449874 | Recruiting |
| GDC‐6036 bevacizumab | ||||
| GDC‐6036 erlotinib | ||||
| GDC‐6036 anti‐PD‐L1 antibody | ||||
| Combinations of MAPK pathway inhibitors | ||||
| Lifirafenib mirdametinib | I/II | Advanced solid tumors | NCT03905148 | Recruiting |
| Cobimetinib GDC‐0994 | I | Advanced solid tumors | NCT02457793 | Completed |
| Combinations of MAPK and PI3K pathway inhibitors | ||||
| Selumetinib MK2206 | I | PDAC | NCT01658943 | Completed |
| Trametinib GSK2141795 | II | Melanoma | NCT01941927 | Completed |
| Trametinib everolimus | I | Advanced solid tumors | NCT00955773 | Completed |
| Pimasertib voxtalisib | I | Advanced solid tumors | NCT01390818 | Completed |
Abbreviations: PD‐1, programmed cell death protein 1; PD‐L1, programmed cell death protein‐ligand 1; PDAC, pancreatic ductal adenocarcinoma; CRC, colorectal cancer; NSCLC, non‐small cell lung cancer; MAPK, mitogen‐activated protein kinase; PI3K, phosphoinositide 3‐kinases
4.5.1. Vertical inhibition
Combination with upstream RTKs and associated molecules. Direct blocking of KRAS and downstream MAPK/PI3K cascades alleviates the negative‐feedback inhibition of RTKs, which in turn reactivates the downstream signaling [102]. RTK expression is intrinsically heterogeneous and sensitive to stimulation by growth factors in a subset of cancers, including CRC, rendering it naturally resistant to inhibitors targeting RAS. Therefore, a combination of RAS and RTK inhibitors could improve treatment efficacy. For instance, Egfr knockout only delayed tumor initiation in Kras/Trp53‐driven PDAC GEMM, whereas the concurrent ablation of Egfr and Raf1 gene led to rapid tumor regression. Similar results were obtained with pharmacological inhibition of EGFR and RAF1 knockdown in patient‐derived PDAC tumor models bearing KRAS mutations [174]. MEK inhibitors acted synergistically with EGFR inhibitors and pan‐ERBB inhibitors to improve anti‐tumor effects in KRAS‐mutated CRC and NSCLC cell lines and prolonged the survival of mice bearing KRAS G12D‐mutated NSCLC [102, 104]. Furthermore, inhibition of KRASG12C leaded to the reactivation of RTKs thus activating wild‐type RAS (NRAS and HRAS) which was unresponsive to G12Ci. Moreover, basal high‐activity or feedback‐reactivated RTKs promote the guanine nucleotide exchange, thus upregulating the expression level of GTP‐bound KRAS and abrogating its affinity to G12Ci. Vertical inhibition using a combination of G12Ci and RTK inhibitors showed some therapeutic effects. For example, findings from in vitro experiments using matrices of G12Ci and inhibitors of HER kinases (i.e., afatinib) indicated a synergetic cytotoxic effect among KRAS G12C cell lines [24, 35]. However, results from in vivo xenograft models bearing KRAS G12C indicated that the combination of G12Ci with individual RTK inhibitors did not effectively overcome the adaptive feedback reactivation in different cancer models, because of their variation in RTK dependency [37]. Alternatively, both SHP2 and SOS1 inhibitors are promising candidates for combination therapies with G12Ci as they can abrogate the reactivation of multiple RTK‐mediated RAS pathways [24, 35, 37]. Several clinical trials evaluating the combination of G12Ci with inhibitors of HER kinase, SHP2, and SOS1 are under active recruiting (NCT04185883, NCT04793958, NCT04975256).
Combination with MAPK effectors. Monotherapies with MAPK inhibitors have shown low efficacy in the treatment of RAS‐mutated malignancies, which is mainly attributed to inhibitor‐induced ERK reactivation. A combination of MAPK inhibitors may act to repress this pathway to a greater extant. For example, drug‐induced ARAF mutant dimers conferred a potential mechanism of acquired resistance to belvarafenib, while a combination with MEK inhibitors could delay the ARAF‐mediated resistance [175]. Co‐targeting of RAF and MEK is significantly effective in BRAF V600E melanoma by abrogating pathway reactivation [176, 177]. A preclinical study indicated that the RAF inhibitor lifirafenib acted synergistically with the MEK inhibitor mirdametinib to alleviate drug‐induced RAF‐dependent MEK reactivation, leading to the sustained blockage of MAPK signaling [178]. An ongoing phase Ib/II clinical trial was recently designed to evaluate the clinical efficacy of the combination of lifirafenib and mirdametinib in patients with KRAS mutations or other MAPK pathway aberrations (NCT03905148). ERK acts as a central node in mediating MAPK inhibitor resistance. Therefore, the combination of ERK and MAPK inhibitors could potentially improve their individual activities. For example, the ERK inhibitor VTX‐11e re‐sensitized KRAS‐mutated cell lines to the MEK inhibitor mirdametinib [179]. In addition, VTX‐11e exhibited synergistic effects with the MEK inhibitors selumetinib and trametinib in NRAS‐mutated melanoma with enhanced anti‐tumor effects and suppression of pathway reactivation [180]. The effect of the combination of GDC‐0994, another ERK inhibitor, with the MEK inhibitor cobimetinib, was evaluated in KRAS‐mutated xenografts and GEMM models. The findings showed that the combination suppressed MAPK pathway and significantly attenuated tumor growth compared to their individual effects [181]. However, a phase Ib trial testing the combination of GDC‐0994 and cobimetinib was terminated because of intolerable overlapping and cumulative toxicity [182]. Synergistic effects were observed between G12Ci and MAPK inhibitors in several KRAS G12C mouse models. Compared with the individual agents, the minimal effective dose of a combination of AMG 510 and the MEK inhibitor mirdametinib induced an enhanced anti‐tumor effect in mice bearing KRAS G12C‐mutated xenografts [24].
4.5.2. Horizontal inhibition
The MAPK and PI3K cascades are key effectors of RAS signaling, and they are interconnected. Repression of one cascade can relieve feedback inhibition of the other cascades, leading to pathway reactivation. Several preclinical studies reported significant therapeutic effects of a combination of various therapies concurrently inhibiting the MAPK and PI3K pathways in RAS‐mutated malignancies [183, 184, 185]. However, negligible therapeutic effects were observed in clinical trials owing to the limited long‐term tolerability of the co‐inhibition of MEK and PI3K [186, 187, 188]. The ability of KRAS to directly activate PI3K cascades in lung cancer mainly relies on a coordinate signal from insulin‐like growth factor1 receptor (IGF1R), and a combination of IGF1R and MEK inhibitors resulted in selective inhibition of KRAS‐mutated LUAD [189]. Notably, the addition of mTOR inhibitors with G12Ci instead of MEK inhibitor showed further improved efficacy [190]. A phase I trial of the MEK inhibitor selumetinib and Akt inhibitor MK‐2206 showed a partial response in a subset of patients with KRAS‐mutated NSCLC and ovarian cancers. However, the overlapping toxicities of these inhibitors limited the dosage [145]. A phase II trial of this combination showed no effect compared with the use of modified FOLFOX (mFOLFOX) in KRAS‐mutated end‐stage pancreatic cancer. The lack of clinical response was mainly attributed to the high frequency (45%) of toxicity‐related treatment delays and dose reductions [191]. Dual‐inhibition of MEK and Akt was not effective for the treatment of NRAS‐mutated melanoma [192]. The combinations of MEK inhibitors and mTOR inhibitors were evaluated, and the findings show that they were limited by poor tolerability [193, 194, 195]. In addition, RAS activates downstream RAF and PI3K by interacting with the well‐conserved RAS‐binding domain (RBD). Rigosertib is a RAS‐mimetic inhibitors that interacts with the RBDs of RAF and PI3K, resulting in their inability to bind to RAS [196]. Rigosertib effectively inhibited RAF‐MEK‐ERK and PI3K‐Akt signaling and showed promising cytotoxic effect in KRAS‐mutated CRC cells and patient‐derived xenografts (PDX) [197, 198].
Combination therapies against RAS‐associated effectors showed promising therapeutic effects among various preclinical tumor models as they effectively abrogated pathway reactivation. However, the narrow in vivo therapeutic window limited their clinical application. Traditional combinations are administered at the maximum tolerated dose (MTD), which is accompanied by significant overlapping toxicities. Multiple low‐dose (MLD) therapy is a promising alternative that, unlike traditional combinations, comprises multiple inhibitors at low doses to reduce toxicities in normal cells. A low concentration of each drug decreases the selective pressure on each node of the pathway, thereby reducing the possibility of acquired resistance. For instance, vertical inhibition of RAF and ERK at low doses showed long‐term pathway repression and favorable safety profiles in organoids and tumor‐bearing mice with KRAS‐mutated cancers [199]. The therapeutic efficacy of the MLD strategy has yet to be evaluated in clinical trials.
5. IMMUNOTHERAPY
Tremendous progress has been made in understanding the crosstalk between tumor cells and TME, as well as development of immunotherapy. Tumor cells can exhaust immune cells and reshape a pro‐tumorigenic immune microenvironment through direct contact or secretion of signal molecules such as cytokines [200]. Recent findings indicated that oncogenic RAS, in addition to its function in promoting proliferation, played an important role in modulating tumor immune microenvironment.
5.1. Immune checkpoint inhibitors (ICIs)
Immune checkpoint pathways comprising PD‐1, PD‐L1, and cytotoxic T‐lymphocyte‐associated protein 4 (CTLA‐4) downregulate immune function, thus maintaining homeostasis. Tumor cells can utilize immune checkpoints to escape the immune response [201]. For instance, tumors expressing PD‐L1 on the cell membrane can directly repress the immune activity of the microenvironment by interacting with PD‐1 on T cells, resulting in tumor progression.
Seven mAbs targeting the immune checkpoints showing favorable benefits were approved for clinical use. However, significant tumor regression was only observed in a small subset of patients [202, 203]. Therefore, it is imperative to identify the populations that may benefit from ICIs. Tumor PD‐L1 expression, CD8+ tumor‐infiltrating lymphocytes (TILs) are well established biomarkers of response to ICIs [204, 205]. Studies reported that RAS mutation was linked to the upregulation of PD‐L1 [206, 207]. Mechanically, oncogenic RAS promoted MEK signaling‐mediated phosphorylation and inhibition of the AU‐rich element‐binding protein tristetraprolin (TTP), which in turn stabilized PD‐L1 mRNA [208]. However, the association between RAS mutation and CD8+ TILs varies between different cancer types. A recent immunological characterization of Chinese NSCLC patients indicated that KRAS mutation was associated with the increased infiltration of CD8+ T cells [209]. On the contrary, oncogenic KRAS promoted migration of myeloid‐derived suppressive cells to the TME via downregulating the expression of interferon regulatory factor 2 (IRF2) in the KRAS‐mutated CRC [210] and induced immune suppressive TME in the PDAC via BRAF and MYC [211]. Also, KRAS mutant induced high levels of granulocyte macrophage colony‐stimulating factor (GM‐CSF) and interleukin 6 (IL6) secretion that converted T cells to the immunosuppressive regulatory T cells (Treg) via the MEK‐ERK‐activator protein 1 (AP1) pathway [212]. MEK inhibition was reported to reshape the TME and promote the CD8+ T cell infiltration in the breast cancer and CRC mouse model [213]. Increased immune‐suppressive cells in the TME greatly hampered the infiltration and anti‐tumor function of the cytotoxic T cells. Thus, the clinical response to ICIs may discriminate between different tumors and genomic backgrounds.
A retrospective study of patients with advanced NSCLC treated with ICIs reported no differences between patients with KRAS‐mutated and other types of NSCLC [214]. A systematic review and meta‐analysis of patients with advanced NSCLC treated with ICIs showed increased overall survival in the KRAS‐mutated subgroup, but the treatment‐KRAS mutation interaction was not significant [215]. Several confounding factors such as co‐mutations may have resulted in the superior therapeutic effects in KRAS‐mutated NSCLC. ICIs were particularly effective in a subgroup of NSCLC patients harboring concurrent mutations in TP53 and KRAS, as indicated by the high expression levels of PD‐L1 and mutation burden [216]. In contrast, mutations in STK11/LKB1 resulted in resistance to ICIs [217]. Therefore, RAS mutation status alone is not a reliable biomarker for predicting the response to ICIs. Further subgroup analyses should be conducted to identify the populations benefiting from ICI treatments to elucidate the associated mechanisms.
5.2. Adoptive cell therapy and cancer vaccine
Gene mutations in cancer cells lead to the generation of neoantigens—peptides that are processed and presented on cell surfaces. These foreign antigens can be recognized by the immune system and induce the activation of neoantigen‐reactive CD8+ cytotoxic T cells, which infiltrate and kill cancer cells. However, most solid tumors, also known as “cold tumors”, do not present with infiltration of cytotoxic T cells, which is associated with poor prognosis and lack of response to ICIs [204, 218, 219]. Therapies are currently being developed to trigger immune cell infiltration in “cold tumors”. Adoptive cell therapy transfuses ex vivo expanded autologous lymphocytes that recognize tumor cells and exert anti‐tumor effects [220]. Initial attempts to transfuse TILs isolated from metastatic melanoma patients were proved to induce regression of tumor [221]. With regard to RAS‐mutated malignancies, studies indicated that RAS mutations were immunogenic and can be recognized by T cells in a human leukocyte antigen (HLA)‐dependent manner [222]. In addition, CD4+ T cells recognizing the KRAS G12V mutant were reported in patients with NSCLC [223]. However, these therapies rely on personalized isolation and autologous transfusion of TILs from specific patients, and transfusion to different patients remains challenging.
Recent advances in our understanding of tumor antigen recognition and viral vector design enabled the integration of synthetic genes into T cells. This, along with the conserved mutational profile of RAS, allows for the large‐scale generation of T cells that target specific tumor antigens, including engineered T‐cell receptor (TCR) and chimeric antigen receptor (CAR)‐T cell therapies [224]. These techniques overcome the reliance on personalized endogenous TCR‐mediated anti‐tumor functions and significantly improve the efficacy and generalizability of adoptive cell therapy. Murine T cells were highly reactive to KRASG12V protein after the administration of RAS mutant peptides to transgenic mice expressing HLA‐A*11:01, and the G12D mutant can be isolated [225]. TCR can then be identified and genetically integrated into peripheral blood lymphocytes, which selectively exert cytotoxic effects in KRAS‐mutated cancer in vitro and in vivo [225]. Several KRAS mutant epitopes, such as HLA‐A*03:01 and HLA‐A*11:01 in KRAS G12V mutant, were recently identified [226]. The adoptive transfer of T cells carrying TCR specific for these epitopes led to significant tumor regression in a xenograft model of metastatic lung cancer [227]. Transfusion of CD8+ T cells expressing HLA‐C*08:02‐restricted TCRs and targeting KRASG12D antigen in TILs (initially identified in a patient with metastatic CRC) showed a 9‐month partial response and regression in lung metastatic lesions [228]. Recently, similar autologous T cells engineered to express two allogeneic HLA‐C*08:02‐restricted KRASG12D‐reactive TCRs were used to treat a patient with metastatic pancreatic cancer. The patient had regression of visceral metastases, and the response was ongoing at 6 months [229]. However, tumor cells with downregulated expression of class I HLA that do not present and process peptides can escape engineered TCR T cell therapy, which is restricted to patients with compatible HLA [220]. In contrast, CAR‐T cells can recognize tumor cells in an HLA‐independent manner; however, the antigen must be located on the cell membrane. Mesothelin (MSLN), a tumor‐associated antigen, is highly expressed in KRAS‐mutated LUAD [230]. MSLN‐targeted CAR T cells specifically recognized and killed MSLN‐high LUAD both in vitro and in mouse models, with no off‐target toxicity [231]. These results provide a promising basis for clinical trials of CAR‐T cells targeting MSLN in KRAS‐mutated LUAD.
In addition to direct transfection of T cells, peptides from mutated RAS proteins can be synthesized as a vaccine to induce a T‐cell response in RAS‐mutated tumors. KRAS‐peptide vaccines were tested as a single agent in clinical trials where they induced transient T‐cell responses but no clinical effects [232, 233]. Adjuvants, such as GM‐CSF, have been combined with RAS‐mutated peptides to effectively augment the immune response. Co‐administration of TG01, an injectable cancer vaccine, with GM‐CSF induced long‐term immunological memory against the KRAS mutation and promoted the survival of patients with PDAC [234]. A phase I/II trial of TG01/GM‐CSF plus gemcitabine in KRAS‐mutated PDAC patients showed increased immune activation as well as disease‐free and overall survival rates compared to the adjuvant gemcitabine alone [235]. The clinical efficacy of another RAS‐mutated peptide TG02/GM‐CSF as a single agent or combined with PD‐1 mAbs was assessed in the treatment of KRAS‐mutated CRC. However, the trial was terminated because of changing priorities. Lipid nanoparticle (LNP)‐based delivery of mRNA encoding neo‐epitopes of RAS mutations can be used for vaccination to circumvent the limitation of directly injecting peptides into circulation in the TME. The mRNA can be efficiently absorbed by antigen‐presenting cells and translated into peptides, which are subsequently presented on the cell surface to elicit a T‐cell response. mRNA 5671 is an mRNA vaccine comprising four common KRAS mutations. A clinical trial testing the effects of mRNA‐5671/V941 as monotherapy and in combination with pembrolizumab is ongoing (NCT03948763).
6. TUMOR METABOLISM
In addition to activating downstream RAF‐MEK‐ERK and PI3K‐Akt cascades, aberrant RAS signaling modulates tumor metabolism. RAS‐induced metabolic reprogramming results in enhanced glycolysis, glutaminolysis, autophagy and macropinocytosis [236], which support non‐homeostatic proliferation—a hallmark of cancer [237, 238]. RAS‐mutated cells rely heavily on the metabolic adaption to sustain proliferation, which provides rationale for targeting metabolism of RAS‐mutated malignancies (Figure 4).
FIGURE 4.
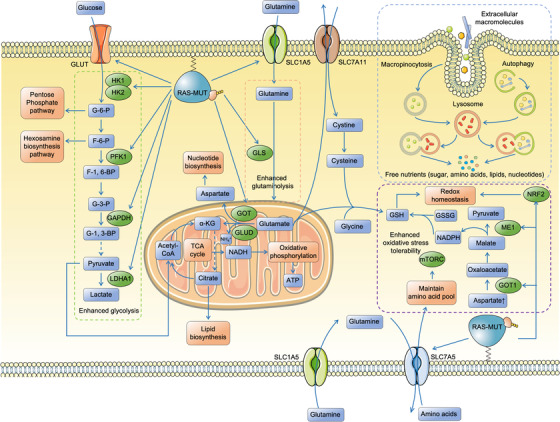
RAS mutations rewire tumor metabolism. RAS‐induced metabolic reprogramming involving enhanced glycolysis, glutaminolysis, autophagy and macropinocytosis supports the tumor proliferation and survival. Mutated RAS dysregulates tumor metabolism by enhancing the expression of transporters and rate‐limiting enzymes of both glucose and glutamine metabolism. The intermediates of glycolysis and glutaminolysis fuel the TCA cycle that provides materials for the biosynthesis of lipids and nucleotides, as well as the production of ATP through oxidative phosphorylation. Glutamine metabolism is critical in maintaining redox homeostasis, as it acts as nitrogen donor and provides NADPH for the production of GSH. Besides, oncogenic RAS activates NRF2 to transcriptionally modulates redox balance of tumor. RAS‐mutated tumor cells exhibit elevated levels of autophagy and macropinocytosis, which digest extra‐ and intra‐cellular macromolecules into free nutrients to support survival and proliferation under nutrient‐depleting condition. Abbreviations: HK1, hexokinase 1; HK2, hexokinase 2; G‐6‐P, glucose 6‐phosphate; F‐6‐P, fructose 6‐phosphate, F‐1,6‐BP, fructose 1,6‐bisphosphate; G‐3‐P, glyceraldehyde 3‐phosphate; G‐1,3‐BP, glycerate 1,3‐bisphosphate; PFK1, phosphofructokinase‐1, GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; LDHA1, lactate dehydrogenase A1; SLC, solute carrier; GLUT, glucose transporter; GOT, glutamate oxaloacetate transaminase; GLS, glutaminase; GLUD, glutamate dehydrogenase; α‐KG, α‐ketoglutarate; TCA, tricarboxylic; GSH, glutathione; GSSG, glutathione disulfide; ME1, malic enzyme 1; NADPH, nicotinamide adenine dinucleotide phosphate hydrogen; NADH, nicotinamide adenine dinucleotide hydrogen; NRF2, nuclear factor erythroid 2‐related factor 2
6.1. Targeting glucose metabolism
Dysregulation of glucose metabolism, particularly enhanced glucose uptake and glycolysis, occurs in various cancers. In addition to ATP, the intermediate products are metabolized to generate amino acids and lipids for cell proliferation and survival. Oncogenes, such as mutated RAS, modulate glucose metabolism in several ways. Transcriptome and metabolomic analyses using an inducible Kras G12D PDAC mouse model revealed that withdrawal of Kras G12D expression decreased glucose uptake and lactate production, as well as the expression of glucose transporters and rate‐limiting enzymes of glycolysis, including hexokinase 1 (Hk1), hexokinase 2 (Hk2), and phosphofructokinase 1 (Pfk1), as well as lactate dehydrogenase A (Ldha) [239]. Moreover, Kras G12D inactivation attenuated the channeling of glucose intermediates into the hexosamine biosynthesis and pentose phosphate pathways [239], which were implicated in the formation of lipids, glycosylation of proteins and synthesis of nucleic acid. In addition to transcriptionally modulating metabolic enzyme expression, KRAS4A can directly interact with the HK1, abrogating the allosteric inhibition of HK1, and enhancing glycolytic flux [18]. A previous study indicated that, although a single endogenous Kras mutant allele was sufficient for tumor initiation, tumor progression required a gain in mutated Kras copy number, as observed in advanced lung tumors of Kras G12D/+;p53null mice [240]. Mechanistically, an increase in the copy number of KRAS resulted in metabolic rewiring with increased glycolysis and channeling of glucose metabolism to the tricarboxylic (TCA) cycle and glutathione (GSH) synthesis. Although RAS mutant‐induced alteration in glucose metabolism supports cell proliferation and survival, it also increases the susceptibility of tumor cells to glucose depletion, thus providing a potential target in RAS‐driven malignancies. Several studies explored the development of inhibitors that blocked glucose transporters (GLUT), the first rate‐limiting step of glycolysis. Fourteen isoforms of GLUT have been identified [241], with GLUT1 being the most widely studied in oncotherapy owing to its overexpression in most cancers [242, 243]. BAY‐867 is a highly selective GLUT1 inhibitor that blocks basal‐ and stress‐induced glycolysis in tumor cells. BAY‐867 impeded the growth of a subset of ovarian and triple‐negative breast cancers with increased glycolysis flux [244, 245, 246]. However, the anti‐tumor efficacy of BAY‐867 has yet to be evaluated in RAS‐mutated models. WZB117 is a selective GLUT1 inhibitor that exhibits synergistic effects with cell cycle checkpoint kinase ataxia telangiectasia and rad3‐related protein (ATR)/ checkpoint kinase 1 (CHK1) inhibitors in the treatment of KRAS‐mutated cancers. This combination treatment exhibited enhanced S‐phase arrest and promoted the accumulation of genotoxic damage and apoptosis in KRAS‐mutated cancer cells [247, 248]. In addition to restricting glucose uptake, the targeting of key glycolytic enzymes provides a promising strategy for cancer treatment. Dehydroascorbate (DHA), the oxidized form of vitamin C, is mainly transported into cells by GLUTs. Elevated level of intracellular DHA decreases the pool of reduced GSH and promotes accumulation of reactive oxygen species (ROS), ultimately causing the inactivation of glyceraldehyde 3‐phosphate dehydrogenase (GAPDH). KRAS‐mutated cells with upregulated GLUT expression and glycolysis flux are highly susceptible to high doses of vitamin C [249]. The clinical application of glucose metabolism inhibitors is limited by their high toxicity, as most targets are ubiquitously expressed in different tissues and cell types. For instance, GLUT1 is highly expressed in erythrocytes and brain epithelial cells [250], and the inhibition of GLUT1 can simultaneously affect blood glucose homeostasis and brain glucose supply. Therefore, further validation should be conducted to evaluate the efficacy and safety profiles of potent inhibitors targeting RAS‐mutated tumor glucose metabolism.
6.2. Targeting glutamine metabolism
Glutamine is a non‐essential amino acid (NEAA) that occurs predominantly in human sera and supports the growth of dividing cells. Many tumors present a “glutamine addiction” or, in other words, an increased demand for glutamine compared to normal cells. RAS‐mutated malignancies exploit metabolic reprogramming that modulates multiple nodes in the glutamine metabolism to supply energy and biosynthetic intermediates for the proliferation and survival [251]. The KRAS mutation is associated with the upregulation of solute carrier (SLC) 1A5, which serves as a key transporter for glutamine uptake, the first rate‐limiting step of glutamine metabolism [252]. In addition, RAS stabilizes the transcriptional factor MYC [253], which promotes the expression of a wide spectrum of key enzymes of glutamine metabolism, including glutaminase (GLS) [254, 255]. The GLS‐mediated deamination of glutamine generates glutamate, which participates in both catabolic and anabolic processes. Glutamate can further be converted in mitochondria to α‐ketoglutarate (α‐KG)—through a process catalyzed by glutamate dehydrogenase (GLUD) or transaminase—and act as a carbon source for the TCA cycle. The TCA cycle is linked to oxidative phosphorylation for ATP generation [256] and provides the precursors for synthesis of nucleotides, amino acids, and fatty acids. Glutamate can donate nitrogen to generate other NEAAs (e.g., aspartate) through the action of transaminase enzymes to produce proteins and nucleotides [257]. The KRAS mutant upregulates the expression of glutamate oxaloacetate transaminase 1 (GOT1) and GOT2 in PDAC cells, which mediate the conversion of glutamate to aspartate for nucleotide production [258]. Furthermore, glutamate plays a key role in sustaining the intracellular amino acid pool, as it can be exchanged for the essential amino acid leucine through SLC7A5/SLC3A2 antiport system to maintain activation of the mTOR pathway [259]. A recent study using GEMM revealed that slc7a5 was indispensable for the growth of Kras G12D‐mutated CRC. Deletion of slc7a5 dysregulated the steady state of intracellular amino acid level and repressed transcription [260]. Glutamine is also implicated in the modulation of cellular redox homeostasis. GSH is a major antioxidant substance generated from glutamate, cysteine and glycine. The level of glutamate substrate limits the synthesis of glutathione as it serves as the substrate and is also involved in the uptake of cysteine and glycine [261]. Glutamine also supports NADPH production, which serves as a reducing equivalent for GSH production [258, 262].
Mutated RAS plays an important role in maintaining redox balance in cancer cells. KRAS‐mutated PDAC cells rely on glutamine as nitrogen donor and exhibit increased aspartate synthesis. Aspartate is transported to the cytosol, where it is converted to malate and utilized to generate NADPH through the action of malic enzyme 1 (ME1) [258]. In addition, the KRAS mutant improves antioxidant effects by activating nuclear factor erythroid 2–related factor 2 (NRF2)—a key regulator of cellular redox status—by modulating GSH metabolism [263]. Loss of the KEAP1 gene was shown to hyperactivate NRF2 and promoted tumorigenesis in a glutaminolysis‐dependent manner through a combination of CRISPR‐Cas9‐based screening and metabolic analyses in the KRAS‐driven LUAD GEMM [264]. Notably, the aberrant KEAP/NRF2 pathway compensated for the redox stress in tumors with loss of LKB1, a common mutation co‐occurring with KRAS, conferring oxidative adaption and tumor progression [265]. Metabolic reprogramming can increase the reliance of tumor cells to glutamine and, combined with glutamine deprivation, can inhibit the growth of KRAS‐mutated malignancies [258, 266].
CB‐839 (telaglenastat) is an allosteric GLS1 inhibitor that effectively blocks the conversion of glutamine to glutamate, leading to significant anti‐tumor effects in multiple preclinical breast cancer [267], ovarian cancer [268] and leukemia models [269]. LUAD with concurrent KRAS/KEAP1 or LKB1 mutations was highly sensitive to CB‐839 [264, 265]. Suppression of glycolysis by chronic mTOR inhibitors in autochthonous Kras G12D;Lkb1 −/− lung squamous cell carcinoma GEMM induced resistance, which was mediated by upregulation of the glycogen synthase kinase 3 α/β (GSK3α/β) signaling pathway that enhanced glutaminolysis to induce the TCA cycle. Therefore, CB‐839 can effectively attenuated GLS activity and overcomed the adaptive resistance against the mTOR inhibitor MLN0128 [270]. A clinical trial evaluating the efficacy of CB‐839 and MLN0128 on advanced‐stage NSCLC (harboring KRAS, KEAP1, and LKB1 mutations) is under active recruitment. CB‐839 can decrease nucleotide levels in tumors by inhibiting the synthesis of glutamine‐derived aspartate required for nucleotide biosynthesis. Consequently, CB‐839 delays the progression of cancer cells to the S‐phase and could potentially act synergistically with other agents inducing cell cycle arrest. Notably, CB‐839 significantly enhanced the anti‐proliferative effects of the cyclin‐dependent kinase 4 and 6 (CDK4/6) inhibitor palbociclib and poly (ADP‐ribose) polymerase (PARP) inhibitors niraparib and talazoparib in KRAS‐mutated CRC xenografts [271]. Clinical trials evaluating the potential of a combination of CB‐839 with CDK4/6 or PARP inhibitors in the treatment of KRAS‐mutated solid tumors are underway (NCT04265534, NCT03875313).
V‐9302 is a small molecule specific antagonist of SLC1A5 that effectively blocks glutamine uptake in multiple tumor cell lines and exhibits a 100‐fold higher potency than other related inhibitors [272]. V‐9302 reduced the viability of more than half of the tumor cell lines, with the most sensitive cell lines harboring alterations in KRAS or downstream effectors. The inhibition of SLC1A5‐mediated glutamine uptake decreased the level of phosphorylated (p)‐S6 and p‐ERK. In addition, deprivation of glutamine by V‐9302 inhibited the synthesis of GSH and increased intracellular ROS levels. A recent study reported that sensitivity to V‐9302 in SLC1A5‐knockout 143B cell line was similar to that of the parental cells, indicating potential off‐target effects [273]. Further experiments demonstrated that the anti‐tumor effect of V‐9302 was mediated by the concurrent inhibition of serotonin N‐acetyltransferase 2 (SNAT2) and L‐type amino acid transporter (LAT1), which disrupted intracellular amino acid homeostasis. Therefore, the exact target of V‐9302 and its therapeutic effects in the treatment of RAS‐mutated malignancies require further investigation.
Inhibition of glutamine metabolism showed promising in vitro and in vivo anti‐tumor activity in KRAS‐mutated LUAD. However, a recent study reported that GLS inhibition was not a viable therapeutic strategy for PDAC [274]. Although exposure to GLS inhibitors, such as BPTES and CB‐839, inhibited proliferation in vitro, these inhibitors did not inhibit tumor growth in vivo. Integration of proteomics and metabolomic analyses revealed that dysregulation of metabolism in cancer cells resulted in drug resistance. Long‐term treatment led to the upregulation of multiple compensatory pathways, such as fatty acid oxidation, and some GLS‐independent glutamate‐producing enzymes, which restored the levels of glutamine, α‐KG and GSH. Although KRAS‐mutated LUAD cells grown in culture relied on glutamine to replenish the TCA cycle, glutamine dependency was lost when these cells were used to induce tumor formation in mice [275], likely because glucose metabolism was the main source of carbon intermediates in the TCA cycle. These findings indicated that multiple factors determined requirement of glutamine in RAS‐induced tumorigenesis [276]. Further experiments are required to explore glutamine‐dependent malignancies.
6.3. Targeting autophagy and nutrient scavenging
Tumor cells require a large amount of nutrient to meet the high demand for biosynthesis and sustain the non‐homeostatic proliferation [238]. Despite the upregulation of membrane transporters conferring higher uptake ability, tumor cells often experience glucose, amino acid, and lipid deprivation, which is attributed to the abnormal vasculature and high interstitial fluid pressure in the TME [277]. A previous study also reported significantly lower levels of various nutrients in tumor region compared to the adjacent region [278]. Tumor cells bypass the limited nutrient supply by alternative methods, including autophagy and nutrient scavenging. Autophagy is a process involving the formation of double‐membrane vesicles that sequester specific organelles and proteins and target them for lysosomal degradation [279]. Scavenging involves uptake of nutrients in the form of less desirable extracellular macromolecules [277]. The oncogenic RAS pathway is associated with enhanced basal autophagic flux and constitutive macropinocytosis (i.e., non‐selective scavenging of extracellular fluid and macromolecules through large vesicles), which promotes the survival and proliferation of tumor cells under low nutrient levels [280, 281]. KRAS suppression or ERK inhibition decreases glycolytic and mitochondrial functions, thus, tumor cells are more reliant on autophagy [282]. Notably, blockade of the MAPK pathway alleviates LKB1 inhibition, which in turn activates the AMP‐activated protein kinase (AMPK)‐Unc‐51‐like kinase 1 (ULK1) pathway to upregulate protective autophagy. Therefore, co‐targeting of the MAPK pathway and autophagy is a promising strategy for the treatment of RAS‐mutated malignancies [282, 283, 284]. A recent study indicated that macropinocytosis of necrotic cell debris (necrocytosis) in TME directly supplied tumor cells with amino acids, sugars, fatty acids and nucleotides. These processes conferred resistance of tumor cells to nutrient stress‐inducing therapies such as gemcitabine, 5‐fluorouracil, doxorubicin and radiotherapy. Resistance can be alleviated by administration of macropinocytosis inhibitors such as 5‐(N‐ethyl‐N‐isopropyl) amiloride (EIPA) [285]. Given that autophagy and nutrient scavenging share the same “digest center” lysosome, lysosome inhibitors could potentially inhibit these alternative metabolic pathways. Recent clinical trials indicated that the lysosome inhibitor hydroxychloroquine sulfate (HCQ), combined with standard chemotherapy, did not improve overall survival but induced a higher overall response rate in patients with metastatic CRC and PDAC [286, 287]. This effect can be attributed to the weak inhibition of autophagy in vivo, which is not sufficient to starve tumor cells [288]. Chloroquine derivatives targeting palmitoyl‐protein thioesterase 1 (PPT1) were recently explored, and the findings showed a dozen‐fold higher potency in lysosome inhibition than HCQ [289, 290]. Future studies should evaluate whether these novel lysosome inhibitors affect the growth and proliferation of RAS‐mutated malignancies and whether their effects are sustained in vivo.
7. CONCLUSIONS AND FURTHER STUDIES
Although past failures created a perception of the RAS protein being undruggable, recent breakthroughs in the development of KRAS G12C allele‐specific inhibitors have greatly contributed to the targeting of RAS mutants in malignancies. Both AMG 510 and MRTX849 have been approved for the treatment of refractory KRAS G12C NSCLC. Although not all mutated RAS proteins have been successfully targeted, the most urgent topics for future research are clear.
First, most KRAS G12C mutants are detected in patients with LUAD; thus, an urgent need exists for inhibitors targeting more common alleles, such as KRAS G13D and KRAS G12V in CRC and PDAC. Preliminary clinical data indicated that monotherapy with allele‐specific inhibitors inevitably induced acquired drug resistance through pathway reactivation and secondary mutations. Combination therapies that reduce the negative feedback inhibition of tumor cells can overcome drug resistance to allele‐specific and RAS‐downstream effector inhibitors. However, this may result in the overlapping of on‐target toxicities. MLD strategies are particularly promising given their high safety profiles and have already shown therapeutic effects in KRAS‐mutated malignancy models.
Second, substantial progress has been made in understanding RAS‐altered metabolism and immunotherapy outcomes. Although these studies provide new therapeutic targets for other RAS mutants, the safety profiles of related therapies are unknown, and selectively targeting key metabolic processes and immune microenvironment components remains challenging.
Third, studies should investigate the context dependency of RAS mutant‐associated phenotypes according to the isoform and codon mutation. For instance, KRAS G12C inhibitors have demonstrated significant clinical responses in LUAD compared to those in CRC. Therefore, further description of the context and complexity related to each malignancy is important for the development of effective therapeutic strategies targeting RAS.
In summary, although the results of targeting RAS over the past few decades have been tortuous, recent advances in direct and indirect targeting provide reasons for optimism.
DECLARATIONS
AUTHOR CONTRIBUTIONS
HY, KD and QX performed and conceived the review. HY and XZ wrote the manuscript. HY, DF, CL prepared all the figures. JW, QZ, XL helped improved the quality of language. YY, KD and QX helped supply financial support. All the authors read and approved the final manuscript.
CONFLICT OF INTERESTS
The authors declare that they have no competing interests.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable.
CONSENT FOR PUBLICATION
Not applicable.
ACKNOWLEDGMENTS
This study was funded by grants from the National Natural Science Foundation of China (No. 82072360 to Q Xiao, No. 82102704 to X Zhou, No. 82072624 to K Ding and No. 81872481 to Y Yuan) and the Natural Science Foundation of Zhejiang Province (LBY20H160002 to Q Xiao)
Yang H, Zhou X, Fu D, Le C, Wang J, Zhou Q, et al. Targeting RAS mutants in malignancies: successes, failures, and reasons for hope. Cancer Commun. 2023;43:42–74. 10.1002/cac2.12377
Contributor Information
Kefeng Ding, Email: dingkefeng@zju.edu.cn.
Qian Xiao, Email: qxiao3@zju.edu.cn.
DATA AVAILABILITY STATEMENT
Not applicable.
REFERENCES
- 1. Muzny DM, Bainbridge MN, Chang K, Dinh HH, Drummond JA, Fowler G, et al. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487(7407):330–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Collisson EA, Campbell JD, Brooks AN, Berger AH, Lee W, Chmielecki J, et al. Comprehensive molecular profiling of lung adenocarcinoma: The cancer genome atlas research network. Nature. 2014;511(7511):543–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Akbani R, Akdemir KC, Aksoy BA, Albert M, Ally A, Amin SB, et al. Genomic Classification of Cutaneous Melanoma. Cell. 2015;161(7):1681–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Raphael BJ, Hruban RH, Aguirre AJ, Moffitt RA, Yeh JJ, Stewart C, et al. Integrated genomic characterization of pancreatic ductal adenocarcinoma. Cancer Cell. 2017;32(2):185–203.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Buday L, Downward J. Epidermal growth factor regulates p21ras through the formation of a complex of receptor, Grb2 adapter protein, and Sos nucleotide exchange factor. Cell. 1993;73(3):611–20. [DOI] [PubMed] [Google Scholar]
- 6. Ebinu JO, Bottorff DA, Chan EY, Stang SL, Dunn RJ, Stone JC. RasGRP, a Ras guanyl nucleotide‐ releasing protein with calcium‐ and diacylglycerol‐binding motifs. Science. 1998;280(5366):1082–6. [DOI] [PubMed] [Google Scholar]
- 7. Xu G, O'Connell P, Viskochil D, Cawthon R, Robertson M, Culver M, et al. The neurofibromatosis type 1 gene encodes a protein related to GAP. Cell. 1990;62(3):599–608. [DOI] [PubMed] [Google Scholar]
- 8. Prior IA, Hood FE, Hartley JL. The Frequency of Ras Mutations in Cancer. Cancer Res. 2020;80(14):2969–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hunter JC, Manandhar A, Carrasco MA, Gurbani D, Gondi S, Westover KD. Biochemical and structural analysis of common cancer‐associated KRAS mutations. Mol Cancer Res. 2015;13(9):1325–35. [DOI] [PubMed] [Google Scholar]
- 10. Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ. Drugging the undruggable RAS: Mission Possible? Nat Rev Drug Discov. 2014;13(11):828–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Project GENIE Consortium AACR. AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov. 2017;7(8):818–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Li S, Balmain A, Counter CM. A model for RAS mutation patterns in cancers: finding the sweet spot. Nat Rev Cancer. 2018;18(12):767–77. [DOI] [PubMed] [Google Scholar]
- 13. Tsai FD, Lopes MS, Zhou M, Court H, Ponce O, Fiordalisi JJ, et al. K‐Ras4A splice variant is widely expressed in cancer and uses a hybrid membrane‐targeting motif. Proc Natl Acad Sci U S A. 2015;112(3):779–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Plowman SJ, Williamson DJ, O'Sullivan MJ, Doig J, Ritchie A‐M, Harrison DJ, et al. While K‐ras is essential for mouse development, expression of the K‐ras 4A splice variant is dispensable. Mol Cell Biol. 2003;23(24):9245–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pells S, Divjak M, Romanowski P, Impey H, Hawkins NJ, Clarke AR, et al. Developmentally‐regulated expression of murine K‐ras isoforms. Oncogene. 1997;15(15):1781–86. [DOI] [PubMed] [Google Scholar]
- 16. Plowman SJ, Berry RL, Bader SA, Luo F, Arends MJ, Harrison DJ, et al. K‐ras 4A and 4B are co‐expressed widely in human tissues, and their ratio is altered in sporadic colorectal cancer. J Exp Clin Cancer Res. 2006;25(2):259–67. [PubMed] [Google Scholar]
- 17. Chen W‐C, To MD, Westcott PMK, Delrosario R, Kim I‐J, Philips M, et al. Targeting KRAS4A splicing through the RBM39/DCAF15 pathway inhibits cancer stem cells. Nat Commun. 2021;12(1):4288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Amendola CR, Mahaffey JP, Parker SJ, Ahearn IM, Chen WC, Zhou M, et al. KRAS4A directly regulates hexokinase 1. Nature. 2019;576(7787):482–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. K‐Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature. 2013;503(7477):548–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Patricelli MP, Janes MR, Li L‐S, Hansen R, Peters U, Kessler LV, et al. Selective Inhibition of Oncogenic KRAS Output with Small Molecules Targeting the Inactive State. Cancer Discov. 2016;6(3):316–29. [DOI] [PubMed] [Google Scholar]
- 21. Lito P, Solomon M, Li L‐S, Hansen R, Rosen N. Allele‐specific inhibitors inactivate mutant KRAS G12C by a trapping mechanism. Science. 2016;351(6273):604–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Janes MR, Zhang J, Li LS, Hansen R, Peters U, Guo X, et al. Targeting KRAS Mutant Cancers with a Covalent G12C‐Specific Inhibitor. Cell. 2018;172(3):578–89.e17. [DOI] [PubMed] [Google Scholar]
- 23. Shin Y, Jeong JW, Wurz RP, Achanta P, Arvedson T, Bartberger MD, et al. Discovery of N‐(1‐Acryloylazetidin‐3‐yl)‐2‐(1 H‐indol‐1‐yl)acetamides as Covalent Inhibitors of KRASG12C. ACS Med Chem Lett. 2019;10(9):1302–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Canon J, Rex K, Saiki AY, Mohr C, Cooke K, Bagal D, et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti‐tumour immunity. Nature. 2019;575(7781):217–23. [DOI] [PubMed] [Google Scholar]
- 25. Hong DS, Fakih MG, Strickler JH, Desai J, Durm GA, Shapiro GI, et al. KRAS G12C Inhibition with Sotorasib in Advanced Solid Tumors. N Engl J Med. 2020;383(13):1207–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Skoulidis F, Li BT, Dy GK, Price TJ, Falchook GS, Wolf J, et al. Sotorasib for Lung Cancers with KRAS p.G12C Mutation. N Engl J Med. 2021;384(25):2371–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fakih MG, Kopetz S, Kuboki Y, Kim TW, Munster PN, Krauss JC, et al. Sotorasib for previously treated colorectal cancers with KRASG12C mutation (CodeBreaK100): a prespecified analysis of a single‐arm, phase 2 trial. Lancet Oncol. 2022;23(1):115–24. [DOI] [PubMed] [Google Scholar]
- 28. Hong DS, Fakih MG, Strickler JH, Desai J, Durm GA, Shapiro GI, et al. KRASG12C Inhibition with Sotorasib in Advanced Solid Tumors. N Engl J Med. 2020;383(13):1207–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Amodio V, Yaeger R, Arcella P, Cancelliere C, Lamba S, Lorenzato A, et al. EGFR Blockade Reverts Resistance to KRASG12C Inhibition in Colorectal Cancer. Cancer Discov. 2020;10(8):1129–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fell JB, Fischer JP, Baer BR, Ballard J, Blake JF, Bouhana K, et al. Discovery of Tetrahydropyridopyrimidines as Irreversible Covalent Inhibitors of KRAS‐G12C with in Vivo Activity. ACS Med Chem Lett. 2018;9(12):1230–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fell JB, Fischer JP, Baer BR, Blake JF, Bouhana K, Briere DM, et al. Identification of the Clinical Development Candidate MRTX849, a Covalent KRASG12CInhibitor for the Treatment of Cancer. J Med Chem. 2020;63(13):6679–93. [DOI] [PubMed] [Google Scholar]
- 32. Briere DM, Li S, Calinisan A, Sudhakar N, Aranda R, Hargis L, et al. The KRASG12C inhibitor MRTX849 reconditions the tumor immune microenvironment and sensitizes tumors to checkpoint inhibitor therapy. Mol Cancer Ther. 2021;20(6):975–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Riely GJ, Ou S‐HI, Rybkin I, Spira A, Papadopoulos K, Sabari JK, et al. 99O_PR KRYSTAL‐1: Activity and preliminary pharmacodynamic (PD) analysis of adagrasib (MRTX849) in patients (Pts) with advanced non–small cell lung cancer (NSCLC) harboring KRASG12C mutation. J Thorac Oncol. 2021;16(4):S751–2. [Google Scholar]
- 34. Johnson ML, Ou SHI, Barve M, Rybkin II, Papadopoulos KP, Leal TA, et al. KRYSTAL‐1: Activity and Safety of Adagrasib (MRTX849) in Patients with Colorectal Cancer (CRC) and Other Solid Tumors Harboring a KRAS G12C Mutation. Eur J Cancer. 2020;138:S2. [Google Scholar]
- 35. Hallin J, Engstrom LD, Hargi L, Calinisan A, Aranda R, Briere DM, et al. The KRASG12C inhibitor MRTX849 provides insight toward therapeutic susceptibility of KRAS‐mutant cancers in mouse models and patients. Cancer Discov. 2020;10(1):54–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Amodio V, Yaeger R, Arcella P, Cancelliere C, Lamba S, Lorenzato A, et al. EGFR Blockade Reverts Resistance to KRASG12C Inhibition in Colorectal Cancer. Cancer Discov. 2020;10(8):1129–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ryan MB, de la Cruz FF, Phat S, Myers DT, Wong E, Shahzade HA, et al. Vertical pathway inhibition overcomes adaptive feedback resistance to KrasG12C inhibition. Clin Cancer Res. 2020;26(7):1617–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Misale S, Fatherree JP, Cortez E, Li C, Bilton S, Timonina D, et al. KRAS G12C NSCLC models are sensitive to direct targeting of KRAS in combination with PI3K inhibition. Clin Cancer Res. 2019;25(2):796–807. [DOI] [PubMed] [Google Scholar]
- 39. Xue JY, Zhao Y, Aronowitz J, Mai TT, Vides A, Qeriqi B, et al. Rapid non‐uniform adaptation to conformation‐specific KRAS(G12C) inhibition. Nature. 2020;577(7790):421–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tanaka N, Lin JJ, Li C, Ryan MB, Zhang J, Kiedrowski LA, et al. Clinical Acquired Resistance to KRASG12C Inhibition through a Novel KRAS Switch‐II Pocket Mutation and Polyclonal Alterations Converging on RAS‐MAPK Reactivation. Cancer Discov. 2021;11(8):1913–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Awad MM, Liu S, Rybkin II, Arbour KC, Dilly J, Zhu VW, et al. Acquired Resistance to KRASG12C Inhibition in Cancer. N Engl J Med. 2021;384(25):2382–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Koga T, Suda K, Fujino T, Ohara S, Hamada A, Nishino M, et al. KRAS Secondary Mutations That Confer Acquired Resistance to KRAS G12C Inhibitors, Sotorasib and Adagrasib, and Overcoming Strategies: Insights From the In Vitro Experiments. J Thorac Oncol. 2021;16(8):1321–32. [DOI] [PubMed] [Google Scholar]
- 43. Maurer T, Garrenton LS, Oh A, Pitts K, Anderson DJ, Skelton NJ, et al. Small‐molecule ligands bind to a distinct pocket in Ras and inhibit SOS‐mediated nucleotide exchange activity. Proc Natl Acad Sci. 2012;109(14):5299–5304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kessler D, Gmachl M, Mantoulidis A, Martin LJ, Zoephel A, Mayer M, et al. Drugging an undruggable pocket on KRAS. Proc Natl Acad Sci. 2019;116(32):15823–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Feng H, Zhang Y, Bos PH, Chambers JM, Dupont MM, Stockwell BR. K‐Ras G12D Has a Potential Allosteric Small Molecule Binding Site. Biochemistry. 2019;58(21):2542–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wang X, Allen S, Blake JF, Bowcut V, Briere DM, Calinisan A, et al. Identification of MRTX1133, a Noncovalent, Potent, and Selective KRASG12D Inhibitor. J Med Chem. 2022;65(4):3123–33. [DOI] [PubMed] [Google Scholar]
- 47. Mao Z, Xiao H, Shen P, Yang Y, Xue J, Yang Y, et al. KRAS(G12D) can be targeted by potent inhibitors via formation of salt bridge. Cell Discov. 2022;8(1):5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chamberlain PP, Hamann LG. Development of targeted protein degradation therapeutics. Nat Chem Biol. 2019;15(10):937–44. [DOI] [PubMed] [Google Scholar]
- 49. Schapira M, Calabrese MF, Bullock AN, Crews CM. Targeted protein degradation: expanding the toolbox. Nat Rev Drug Discov. 2019;18(12):949–63. [DOI] [PubMed] [Google Scholar]
- 50. Ma Y, Gu Y, Zhang Q, Han Y, Yu S, Lu Z, et al. Targeted degradation of KRAS by an engineered ubiquitin ligase suppresses pancreatic cancer cell growth in vitro and in vivo. Mol Cancer Ther. 2013;12(3):286–94. [DOI] [PubMed] [Google Scholar]
- 51. Bery N, Miller A, Rabbitts T. A potent KRAS macromolecule degrader specifically targeting tumours with mutant KRAS. Nat Commun. 2020;11(1):1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zeng M, Xiong Y, Safaee N, Nowak RP, Donovan KA, Yuan CJ, et al. Exploring Targeted Degradation Strategy for Oncogenic KRASG12C. Cell Chem Biol. 2020;27(1):19–31.e6. [DOI] [PubMed] [Google Scholar]
- 53. Bond MJ, Chu L, Nalawansha DA, Li K, Crews CM. Targeted Degradation of Oncogenic KRASG12Cby VHL‐Recruiting PROTACs. ACS Cent Sci. 2020;6(8):1367–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bondeson DP, Mares A, Smith IED, Ko E, Campos S, Miah AH, et al. Catalytic in vivo protein knockdown by small‐molecule PROTACs. Nat Chem Biol. 2015;11(8):611–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ross SJ, Revenko AS, Hanson LL, Ellston R, Staniszewska A, Whalley N, et al. Targeting KRAS‐dependent tumors with AZD4785, a high‐affinity therapeutic antisense oligonucleotide inhibitor of KRAS. Sci Transl Med. 2017;9(394):1–14. [DOI] [PubMed] [Google Scholar]
- 56. Sacco A, Federico C, Todoerti K, Ziccheddu B, Giacomini A, Ravelli C, et al. Specific Targeting of KRAS Using a Novel High‐Affinity KRAS Antisense Oligonucleotide in Multiple Myeloma. Blood. 2019;134(Supplement_1):3104–3104. [Google Scholar]
- 57. Khvalevsky EZ, Gabai R, Rachmut IH, Horwitz E, Brunschwig Z, Orbach A, et al. Mutant KRAS is a druggable target for pancreatic cancer. Proc Natl Acad Sci U S A. 2013;110(51):20723–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Golan T, Khvalevsky EZ, Hubert A, Gabai RM, Hen N, Segal A, et al. RNAi therapy targeting KRAS in combination with chemotherapy for locally advanced pancreatic cancer patients. Oncotarget. 2015;6(27):24560–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Jaiswal S, Jamieson CHM, Pang WW, Park CY, Chao MP, Majeti R, et al. CD47 Is Upregulated on Circulating Hematopoietic Stem Cells and Leukemia Cells to Avoid Phagocytosis. Cell. 2009;138(2):271–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Vader P, Mol EA, Pasterkamp G, Schiffelers RM. Extracellular vesicles for drug delivery. Adv Drug Deliv Rev. 2016;106:148–56. [DOI] [PubMed] [Google Scholar]
- 61. Kamerkar S, Lebleu VS, Sugimoto H, Yang S, Ruivo CF, Melo SA, et al. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature. 2017;546(7659):498–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Tanaka T, Williams RL, Rabbitts TH. Tumour prevention by a single antibody domain targeting the interaction of signal transduction proteins with RAS. EMBO J. 2007;26(13):3250–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Tanaka T, Rabbitts TH. Interfering with RAS–effector protein interactions prevent RAS‐dependent tumour initiation and causes stop–start control of cancer growth. Oncogene. 2010;29(45):6064–70. [DOI] [PubMed] [Google Scholar]
- 64. Shin S‐M, Choi D‐K, Jung K, Bae J, Kim J, Park S‐W, et al. Antibody targeting intracellular oncogenic Ras mutants exerts anti‐tumour effects after systemic administration. Nat Commun. 2017;8(1):15090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Shin S‐M, Kim J‐S, Park S‐W, Jun S‐Y, Kweon H‐J, Choi D‐K, et al. Direct targeting of oncogenic RAS mutants with a tumor‐specific cytosol‐penetrating antibody inhibits RAS mutant‐driven tumor growth. Sci Adv. 2020;6(3):eaay2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Sharma S, Kemeny N, Kelsen DP, Ilson D, O'Reilly E, Zaknoen S, et al. A phase II trial of farnesyl protein transferase inhibitor SCH 66336, given by twice‐daily oral administration, in patients with metastatic colorectal cancer refractory to 5‐fluorouracil and irinotecan. Ann Oncol. 2002;13(7):1067–71. [DOI] [PubMed] [Google Scholar]
- 67. Van Cutsem E, van de Velde H, Karasek P, Oettle H, Vervenne WL, Szawlowski A, et al. Phase III Trial of Gemcitabine Plus Tipifarnib Compared With Gemcitabine Plus Placebo in Advanced Pancreatic Cancer. J Clin Oncol. 2004;22(8):1430–8. [DOI] [PubMed] [Google Scholar]
- 68. Jeng H‐H, Taylor LJ, Bar‐Sagi D. Sos‐mediated cross‐activation of wild‐type Ras by oncogenic Ras is essential for tumorigenesis. Nat Commun. 2012;3(1):1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Sun Q, Burke JP, Phan J, Burns MC, Olejniczak ET, Waterson AG, et al. Discovery of Small Molecules that Bind to K‐Ras and Inhibit Sos‐Mediated Activation. Angew Chemie Int Ed. 2012;51(25):6140–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Patgiri A, Yadav KK, Arora PS, Bar‐Sagi D. An orthosteric inhibitor of the Ras‐Sos interaction. Nat Chem Biol. 2011;7(9):585–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Leshchiner ES, Parkhitko A, Bird GH, Luccarelli J, Bellairs JA, Escudero S, et al. Direct inhibition of oncogenic KRAS by hydrocarbon‐stapled SOS1 helices. Proc Natl Acad Sci U S A. 2015;112(6):1761–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Howes JE, Akan DT, Burns MC, Rossanese OW, Waterson AG, Fesik SW. Small Molecule‐Mediated Activation of RAS Elicits Biphasic Modulation of Phospho‐ERK Levels that Are Regulated through Negative Feedback on SOS1. Mol Cancer Ther. 2018;17(5):1051–60. [DOI] [PubMed] [Google Scholar]
- 73. Hillig RC, Sautier B, Schroeder J, Moosmayer D, Hilpmann A, Stegmann CM, et al. Discovery of potent SOS1 inhibitors that block RAS activation via disruption of the RAS–SOS1 interaction. Proc Natl Acad Sci U S A. 2019;116(7):2551–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Hofmann MH, Gmachl M, Ramharter J, Savarese F, Gerlach D, Marszalek JR, et al. Bi‐3406, a potent and selective sos1–kras interaction inhibitor, is effective in kras‐driven cancers through combined mek inhibition. Cancer Discov. 2021;11(1):142–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Hobbs GA, Baker NM, Miermont AM, Thurman RD, Pierobon M, Tran TH, et al. Atypical KRASG12R Mutant Is Impaired in PI3K Signaling and Macropinocytosis in Pancreatic Cancer. Cancer Discov. 2020;10(1):104–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Moghadamchargari Z, Shirzadeh M, Liu C, Schrecke S, Packianathan C, Russell DH, et al. Molecular assemblies of the catalytic domain of SOS with KRas and oncogenic mutants. Proc Natl Acad Sci U S A. 2021;118(12):e2022403118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Hadari YR, Kouhara H, Lax I, Schlessinger J. Binding of Shp2 Tyrosine Phosphatase to FRS2 Is Essential for Fibroblast Growth Factor‐Induced PC12 Cell Differentiation. Mol Cell Biol. 1998;18(7):3966–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Dance M, Montagner A, Salles JP, Yart A, Raynal P. The molecular functions of Shp2 in the Ras/Mitogen‐activated protein kinase (ERK1/2) pathway. Cell Signal. 2008;20(3):453–9. [DOI] [PubMed] [Google Scholar]
- 79. Bunda S, Burrell K, Heir P, Zeng L, Alamsahebpour A, Kano Y, et al. Inhibition of SHP2‐mediated dephosphorylation of Ras suppresses oncogenesis. Nat Commun. 2015;6(1):8859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Kano Y, Gebregiworgis T, Marshall CB, Radulovich N, Poon BPK, St‐Germain J, et al. Tyrosyl phosphorylation of KRAS stalls GTPase cycle via alteration of switch I and II conformation. Nat Commun. 2019;10(1):224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Ruess DA, Heynen GJ, Ciecielski KJ, Ai J, Berninger A, Kabacaoglu D, et al. Mutant KRAS‐driven cancers depend on PTPN11/SHP2 phosphatase. Nat Med. 2018;24(7):954–60. [DOI] [PubMed] [Google Scholar]
- 82. Vainonen JP, Momeny M, Westermarck J. Druggable cancer phosphatases. Sci Transl Med. 2021;13(588):1–14. [DOI] [PubMed] [Google Scholar]
- 83. Lamarche MJ, Chan HM, Fekkes P, Garcia‐fortanet J, Acker MG, Antonakos B, et al. Allosteric inhibition of SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature. 2016;535(7610):148–52. [DOI] [PubMed] [Google Scholar]
- 84. Lu H, Liu C, Velazquez R, Wang H, Dunkl LM, Kazic‐Legueux M, et al. SHP2 Inhibition Overcomes RTK‐Mediated Pathway Reactivation in KRAS‐Mutant Tumors Treated with MEK Inhibitors. Mol Cancer Ther. 2019;18(7):1323–34. [DOI] [PubMed] [Google Scholar]
- 85. Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. 2015;15(8):486–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Wallweber HA, et al. T cell costimulatory receptor CD28 is a primary target for PD‐1‐mediated inhibition. Science. 2017;355(6332):1428–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Marasco M, Berteotti A, Weyershaeuser J, Thorausch N, Sikorska J, Krausze J, et al. Molecular mechanism of SHP2 activation by PD‐1 stimulation. Sci Adv. 2020;6(5):eaay4458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Quintana E, Schulze CJ, Myers DR, Choy TJ, Mordec K, Wildes D, et al. Allosteric Inhibition of SHP2 Stimulates Antitumor Immunity by Transforming the Immunosuppressive Environment. Cancer Res. 2020;80(13):2889–2902. [DOI] [PubMed] [Google Scholar]
- 89. Zhao M, Guo W, Wu Y, Yang C, Zhong L, Deng G, et al. SHP2 inhibition triggers anti‐tumor immunity and synergizes with PD‐1 blockade. Acta Pharm Sin B. 2019;9(2):304–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Karapetis CS, Khambata‐Ford S, Jonker DJ, O'Callaghan CJ, Tu D, Tebbutt NC, et al. K‐ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med. 2008;359(17):1757–65. [DOI] [PubMed] [Google Scholar]
- 91. Linardou H, Dahabreh IJ, Kanaloupiti D, Siannis F, Bafaloukos D, Kosmidis P, et al. Assessment of somatic k‐RAS mutations as a mechanism associated with resistance to EGFR‐targeted agents: a systematic review and meta‐analysis of studies in advanced non‐small‐cell lung cancer and metastatic colorectal cancer. Lancet Oncol. 2008;9(10):962–72. [DOI] [PubMed] [Google Scholar]
- 92. Amado RG, Wolf M, Peeters M, Van Cutsem E, Siena S, Freeman DJ, et al. Wild‐Type KRAS Is Required for Panitumumab Efficacy in Patients With Metastatic Colorectal Cancer. J Clin Oncol. 2008;26(10):1626–34. [DOI] [PubMed] [Google Scholar]
- 93. Misale S, Yaeger R, Hobor S, Scala E, Janakiraman M, Liska D, et al. Emergence of KRAS mutations and acquired resistance to anti‐EGFR therapy in colorectal cancer. Nature. 2012;486(7404):532–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Siravegna G, Mussolin B, Buscarino M, Corti G, Cassingena A, Crisafulli G, et al. Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat Med. 2015;21(7):795–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. De Roock W, Jonker DJ, Di Nicolantonio F, Sartore‐Bianchi A, Tu D, Siena S, et al. Association of KRAS p.G13D mutation with outcome in patients with chemotherapy‐refractory metastatic colorectal cancer treated with cetuximab. JAMA. 2010;304(16):1812–20. [DOI] [PubMed] [Google Scholar]
- 96. Tejpar S, Celik I, Schlichting M, Sartorius U, Bokemeyer C, Van Cutsem E. Association of KRAS G13D Tumor Mutations With Outcome in Patients With Metastatic Colorectal Cancer Treated With First‐Line Chemotherapy With or Without Cetuximab. J Clin Oncol. 2012;30(29):3570–7. [DOI] [PubMed] [Google Scholar]
- 97. Peeters M, Douillard J‐Y, Van Cutsem E, Siena S, Zhang K, Williams R, et al. Mutant KRAS Codon 12 and 13 Alleles in Patients With Metastatic Colorectal Cancer: Assessment As Prognostic and Predictive Biomarkers of Response to Panitumumab. J Clin Oncol. 2013;31(6):759–65. [DOI] [PubMed] [Google Scholar]
- 98. Rowland A, Dias MM, Wiese MD, Kichenadasse G, McKinnon RA, Karapetis CS, et al. Meta‐analysis comparing the efficacy of anti‐EGFR monoclonal antibody therapy between KRAS G13D and other KRAS mutant metastatic colorectal cancer tumours. Eur J Cancer. 2016;55:122–30. [DOI] [PubMed] [Google Scholar]
- 99. Segelov E, Thavaneswaran S, Waring PM, Desai J, Robledo KP, Gebski VJ, et al. Response to Cetuximab With or Without Irinotecan in Patients With Refractory Metastatic Colorectal Cancer Harboring the KRAS G13D Mutation: Australasian Gastro‐Intestinal Trials Group ICECREAM Study. J Clin Oncol. 2016;34(19):2258–64. [DOI] [PubMed] [Google Scholar]
- 100. Ardito CM, Grüner BM, Takeuchi KK, Lubeseder‐Martellato C, Teichmann N, Mazur PK, et al. EGF Receptor Is Required for KRAS‐Induced Pancreatic Tumorigenesis. Cancer Cell. 2012;22(3):304–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Navas C, Hernández‐Porras I, Schuhmacher AJ, Sibilia M, Guerra C, Barbacid M. EGF Receptor Signaling Is Essential for K‐Ras Oncogene‐Driven Pancreatic Ductal Adenocarcinoma. Cancer Cell. 2012;22(3):318–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Sun C, Hobor S, Bertotti A, Zecchin D, Huang S, Galimi F, et al. Intrinsic resistance to MEK inhibition in kras mutant lung and colon cancer through transcriptional induction of ERBB3. Cell Rep. 2014;7(1):86–93. [DOI] [PubMed] [Google Scholar]
- 103. Moll HP, Pranz K, Musteanu M, Grabner B, Hruschka N, Mohrherr J, et al. Afatinib restrains K‐RAS‐driven lung tumorigenesis. Sci Transl Med. 2018;10(44):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Kruspig B, Monteverde T, Neidler S, Hock A, Kerr E, Nixon C, et al. The ERBB network facilitates KRAS‐driven lung tumorigenesis. Sci Transl Med. 2018;10(446):eaao2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Wilhelm SM, Carter C, Tang L, Wilkie D, McNabola A, Rong H, et al. BAY 43‐9006 Exhibits Broad Spectrum Oral Antitumor Activity and Targets the RAF/MEK/ERK Pathway and Receptor Tyrosine Kinases Involved in Tumor Progression and Angiogenesis. Cancer Res. 2004;64(19):7099–109. [DOI] [PubMed] [Google Scholar]
- 106. Gonçalves A, Gilabert M, François E, Dahan L, Perrier H, Lamy R, et al. BAYPAN study: a double‐blind phase III randomized trial comparing gemcitabine plus sorafenib and gemcitabine plus placebo in patients with advanced pancreatic cancer. Ann Oncol. 2012;23(11):2799–805. [DOI] [PubMed] [Google Scholar]
- 107. Ryan MB, Der CJ, Wang‐Gillam A, Cox AD. Targeting RAS‐mutant Cancers: Is ERK the Key? Trends in Cancer. 2015;1(3):183–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, et al. Inhibition of Mutated, Activated BRAF in Metastatic Melanoma. N Engl J Med. 2010;363(9):809–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. Improved Survival with Vemurafenib in Melanoma with BRAF V600E Mutation. N Engl J Med. 2011;364(26):2507–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Hauschild A, Grob J‐J, Demidov L V, Jouary T, Gutzmer R, Millward M, et al. Dabrafenib in BRAF‐mutated metastatic melanoma: a multicentre, open‐label, phase 3 randomised controlled trial. Lancet. 2012;380(9839):358–65. [DOI] [PubMed] [Google Scholar]
- 111. Sosman JA, Kim KB, Schuchter L, Gonzalez R, Pavlick AC, Weber JS, et al. Survival in BRAF V600–Mutant Advanced Melanoma Treated with Vemurafenib. N Engl J Med. 2012;366(8):707–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Hatzivassiliou G, Song K, Yen I, Brandhuber BJ, Anderson DJ, Alvarado R, et al. RAF inhibitors prime wild‐type RAF to activate the MAPK pathway and enhance growth. Nature. 2010;464(7287):431–5. [DOI] [PubMed] [Google Scholar]
- 113. Heidorn SJ, Milagre C, Whittaker S, Nourry A, Niculescu‐Duvas I, Dhomen N, et al. Kinase‐Dead BRAF and Oncogenic RAS Cooperate to Drive Tumor Progression through CRAF. Cell. 2010;140(2):209–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Poulikakos PI, Persaud Y, Janakiraman M, Kong X, Ng C, Moriceau G, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature. 2011;480(7377):387–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Bin Peng S, JR Henry, Kaufman MD, Lu WP, Smith BD, Vogeti S, et al. Inhibition of RAF Isoforms and Active Dimers by LY3009120 Leads to Anti‐tumor Activities in RAS or BRAF Mutant Cancers. Cancer Cell. 2015;28(3):384–98. [DOI] [PubMed] [Google Scholar]
- 116. Monaco KA, Delach S, Yuan J, Mishina Y, Fordjour P, Labrot E, et al. LXH254, a potent and selective ARAF‐Sparing inhibitor of BRAF and CRAF for the treatment of MAPK‐Driven tumors. Clin Cancer Res. 2021;27(7):2061–73. [DOI] [PubMed] [Google Scholar]
- 117. Yuan X, Tang Z, Du R, Yao Z, Cheung S, Zhang X, et al. RAF dimer inhibition enhances the antitumor activity of MEK inhibitors in K‐RAS mutant tumors. Mol Oncol. 2020;14(8):1833–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Sullivan RJ, Hollebecque A, Flaherty KT, Shapiro GI, Rodon Ahnert J, Millward MJ, et al. A Phase I Study of LY3009120, a Pan‐RAF Inhibitor, in Patients with Advanced or Metastatic Cancer. Mol Cancer Ther. 2020;19(2):460–67. [DOI] [PubMed] [Google Scholar]
- 119. Desai J, Gan H, Barrow C, Jameson M, Atkinson V, Haydon A, et al. Phase I, Open‐Label, Dose‐Escalation/Dose‐Expansion Study of Lifirafenib (BGB‐283), an RAF Family Kinase Inhibitor, in Patients With Solid Tumors. J Clin Oncol. 2020;38(19):2140–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Harris SJ, de Miguel Luken MJ, Roda Perez D, Perez Lopez R, Parmar M, Prathapan V, et al. Updated efficacy and safety results from the phase I study of intermittent dosing of the dual MEK/RAF inhibitor, RO5126766 in patients (pts) with RAS/RAF mutated advanced solid tumours. J Clin Oncol. 2016;34(15_suppl):2582–82. [Google Scholar]
- 121. Kim TW, Lee J, Shin SJ, Kim J‐S, Kim YJ, Han HS, et al. Belvarafenib, a novel pan‐RAF inhibitor, in solid tumor patients harboring BRAF, KRAS, or NRAS mutations: Phase I study. J Clin Oncol. 2019;37(15_suppl):3000–3000.31557067 [Google Scholar]
- 122. Flaherty KT, Robert C, Hersey P, Nathan P, Garbe C, Milhem M, et al. Improved survival with MEK inhibition in BRAF‐mutated melanoma. N Engl J Med. 2012;367(2):107–14. [DOI] [PubMed] [Google Scholar]
- 123. Bennouna J, Lang I, Valladares‐Ayerbes M, Boer K, Adenis A, Escudero P, et al. A Phase II, open‐label, randomised study to assess the efficacy and safety of the MEK1/2 inhibitor AZD6244 (ARRY‐142886) versus capecitabine monotherapy in patients with colorectal cancer who have failed one or two prior chemotherapeutic regimens. Invest New Drugs. 2011;29(5):1021–8. [DOI] [PubMed] [Google Scholar]
- 124. Bodoky G, Timcheva C, Spigel DR, La Stella PJ, Ciuleanu TE, Pover G, et al. A phase II open‐label randomized study to assess the efficacy and safety of selumetinib (AZD6244 [ARRY‐142886]) versus capecitabine in patients with advanced or metastatic pancreatic cancer who have failed first‐line gemcitabine therapy. Invest New Drugs. 2012;30(3):1216–23. [DOI] [PubMed] [Google Scholar]
- 125. Blumenschein GR, Smit EF, Planchard D, Kim D‐W, Cadranel J, De Pas T, et al. A randomized phase II study of the MEK1/MEK2 inhibitor trametinib (GSK1120212) compared with docetaxel in KRAS‐mutant advanced non‐small‐cell lung cancer (NSCLC). Ann Oncol. 2015;26(5):894–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Planchard D, Besse B, Groen HJM, Souquet P‐J, Quoix E, Baik CS, et al. Dabrafenib plus trametinib in patients with previously treated BRAFV600E‐mutant metastatic non‐small cell lung cancer: an open‐label, multicentre phase 2 trial. Lancet Oncol. 2016;17(7):984–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Jänne PA, van den Heuvel MM, Barlesi F, Cobo M, Mazieres J, Crinò L, et al. Selumetinib Plus Docetaxel Compared With Docetaxel Alone and Progression‐Free Survival in Patients With KRAS‐Mutant Advanced Non–Small Cell Lung Cancer: The SELECT‐1 Randomized Clinical Trial. JAMA. 2017;317(18):1844–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Lebbe C, Dutriaux C, Lesimple T, Kruit W, Kerger J, Thomas L, et al. Pimasertib (PIM) versus dacarbazine (DTIC) in patients (pts) with cutaneous NRAS melanoma: a controlled, open‐label phase II trial with crossover. Ann Oncol. 2016;27:vi389. [Google Scholar]
- 129. Hatzivassiliou G, Liu B, O'Brien C, Spoerke JM, Hoeflich KP, Haverty PM, et al. ERK inhibition overcomes acquired resistance to MEK Inhibitors. Mol Cancer Ther. 2012;11(5):1143–54. [DOI] [PubMed] [Google Scholar]
- 130. Morris EJ, Jha S, Restaino CR, Dayananth P, Zhu H, Cooper A, et al. Discovery of a novel ERK inhibitor with activity in models of acquired resistance to BRAF and MEK inhibitors. Cancer Discov. 2013;3(7):742–50. [DOI] [PubMed] [Google Scholar]
- 131. Moschos SJ, Sullivan RJ, Hwu W‐J, Ramanathan RK, Adjei AA, Fong PC, et al. Development of MK‐8353, an orally administered ERK1/2 inhibitor, in patients with advanced solid tumors. JCI insight. 2018;3(4):e92352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Robarge K, Schwarz J, Blake J, Burkard M, Chan J, Chen H, et al. Abstract DDT02‐03: Discovery of GDC‐0994, a potent and selective ERK1/2 inhibitor in early clinical development. Cancer Res. 2014;74(19_Supplement):DDT02‐03‐DDT02‐03. [Google Scholar]
- 133. Blake JF, Burkard M, Chan J, Chen H, Chou K‐J, Diaz D, et al. Discovery of (S)‐1‐(1‐(4‐Chloro‐3‐fluorophenyl)‐2‐hydroxyethyl)‐4‐(2‐((1‐methyl‐1 H ‐pyrazol‐5‐yl)amino)pyrimidin‐4‐yl)pyridin‐2(1 H)‐one (GDC‐0994), an Extracellular Signal‐Regulated Kinase 1/2 (ERK1/2) Inhibitor in Early Clinical Development. J Med Chem. 2016;59(12):5650–60. [DOI] [PubMed] [Google Scholar]
- 134. Varga A, Soria JC, Hollebecque A, LoRusso P, Bendell J, Huang SMA, et al. A first‐in‐human phase I study to evaluate the ERK1/2 inhibitor GDC‐0994 in patients with advanced solid tumors. Clin Cancer Res. 2020;26(6):1229–36. [DOI] [PubMed] [Google Scholar]
- 135. Casar B, Pinto A, Crespo P. Essential role of ERK dimers in the activation of cytoplasmic but not nuclear substrates by ERK‐scaffold complexes. Mol Cell. 2008;31(5):708–21. [DOI] [PubMed] [Google Scholar]
- 136. Herrero A, Pinto A, Colón‐Bolea P, Casar B, Jones M, Agudo‐Ibáñez L, et al. Small Molecule Inhibition of ERK Dimerization Prevents Tumorigenesis by RAS‐ERK Pathway Oncogenes. Cancer Cell. 2015;28(2):170–82. [DOI] [PubMed] [Google Scholar]
- 137. Gupta S, Ramjaun AR, Haiko P, Wang Y, Warne PH, Nicke B, et al. Binding of Ras to Phosphoinositide 3‐Kinase p110α Is Required for Ras‐ Driven Tumorigenesis in Mice. Cell. 2007;129(5):957–68. [DOI] [PubMed] [Google Scholar]
- 138. Castellano E, Sheridan C, Thin MZ, Nye E, Spencer‐Dene B, Diefenbacher ME, et al. Requirement for Interaction of PI3‐Kinase p110α with RAS in Lung Tumor Maintenance. Cancer Cell. 2013;24(5):617–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Murillo MM, Zelenay S, Nye E, Castellano E, Lassailly F, Stamp G, et al. RAS interaction with PI3K p110α is required for tumor‐induced angiogenesis. J Clin Invest. 2014;124(8):3601–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Engelman JA, Chen L, Tan X, Crosby K, Guimaraes AR, Upadhyay R, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med. 2008;14(12):1351–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Ihle NT, Lemos R, Wipf P, Yacoub A, Mitchell C, Siwak D, et al. Mutations in the Phosphatidylinositol‐3‐Kinase Pathway Predict for Antitumor Activity of the Inhibitor PX‐866 whereas Oncogenic Ras Is a Dominant Predictor for Resistance. Cancer Res. 2009;69(1):143–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Sos ML, Fischer S, Ullrich R, Peifer M, Heuckmann JM, Koker M, et al. Identifying genotype‐dependent efficacy of single and combined PI3K‐ and MAPK‐pathway inhibition in cancer. Proc Natl Acad Sci. 2009;106(43):18351–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Dan S, Okamura M, Seki M, Yamazaki K, Sugita H, Okui M, et al. Correlating Phosphatidylinositol 3‐Kinase Inhibitor Efficacy with Signaling Pathway Status: In silico and Biological Evaluations. Cancer Res. 2010;70(12):4982–94. [DOI] [PubMed] [Google Scholar]
- 144. Zhang M, Jang H, Nussinov R. The mechanism of PI3Kα activation at the atomic level. Chem Sci. 2019;10(12):3671–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Tolcher AW, Khan K, Ong M, Banerji U, Papadimitrakopoulou V, Gandara DR, et al. Antitumor activity in ras‐driven tumors by blocking akt and mek. Clin Cancer Res. 2015;21(4):739–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Weisner J, Landel I, Reintjes C, Uhlenbrock N, Trajkovic‐Arsic M, Dienstbier N, et al. Preclinical efficacy of covalent‐allosteric AKT inhibitor borussertib in combination with trametinib in KRAS‐mutant pancreatic and colorectal cancer. Cancer Res. 2019;79(9):2367–78. [DOI] [PubMed] [Google Scholar]
- 147. Janku F, Yap TA, Meric‐Bernstam F. Targeting the PI3K pathway in cancer: Are we making headway? Nat Rev Clin Oncol. 2018;15(5):273–91. [DOI] [PubMed] [Google Scholar]
- 148. Li X, Huang Y, Jiang J, Frank SJ. ERK‐dependent threonine phosphorylation of EGF receptor modulates receptor downregulation and signaling. Cell Signal. 2008;20(11):2145–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Corbalan‐Garcia S, Yang SS, Degenhardt KR, Bar‐Sagi D. Identification of the mitogen‐activated protein kinase phosphorylation sites on human Sos1 that regulate interaction with Grb2. Mol Cell Biol. 1996;16(10):5674–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Chen D, Waters SB, Holt KH, Pessin JE. SOS phosphorylation and disassociation of the Grb2‐SOS complex by the ERK and JNK signaling pathways. J Biol Chem. 1996;271(11):6328–32. [DOI] [PubMed] [Google Scholar]
- 151. Dougherty MK, Müller J, Ritt DA, Zhou M, Zhou XZ, Copeland TD, et al. Regulation of Raf‐1 by direct feedback phosphorylation. Mol Cell. 2005;17(2):215–24. [DOI] [PubMed] [Google Scholar]
- 152. Ritt DA, Monson DM, Specht SI, Morrison DK. Impact of Feedback Phosphorylation and Raf Heterodimerization on Normal and Mutant B‐Raf Signaling. Mol Cell Biol. 2010;30(3):806–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Catalanotti F, Reyes G, Jesenberger V, Galabova‐Kovacs G, De Matos Simoes R, Carugo O, et al. A Mek1‐Mek2 heterodimer determines the strength and duration of the Erk signal. Nat Struct Mol Biol. 2009;16(3):294–303. [DOI] [PubMed] [Google Scholar]
- 154. McKay MM, Ritt DA, Morrison DK. Signaling dynamics of the KSR1 scaffold complex. Proc Natl Acad Sci U S A. 2009;106(27):11022–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Brondello JM, Brunet A, Pouysségur J, McKenzie FR. The dual specificity mitogen‐activated protein kinase phosphatase‐1 and ‐2 are induced by the p42/p44(MAPK) cascade. J Biol Chem. 1997;272(2):1368–76. [DOI] [PubMed] [Google Scholar]
- 156. Amit I, Citri A, Shay T, Lu Y, Katz M, Zhang F, et al. A module of negative feedback regulators defines growth factor signaling. Nat Genet. 2007;39(4):503–12. [DOI] [PubMed] [Google Scholar]
- 157. Impagnatiello MA, Weitzer S, Gannon G, Compagni A, Cotten M, Christofori G. Mammalian sprouty‐1 and ‐2 are membrane‐anchored phosphoprotein inhibitors of growth factor signaling in endothelial cells. J Cell Biol. 2001;152(5):1087–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Hanafusa H, Torii S, Yasunaga T, Nishida E. Sprouty1 and Sprouty2 provide a control mechanism for the Ras/MAPK signalling pathway. Nat Cell Biol. 2002;4(11):850–58. [DOI] [PubMed] [Google Scholar]
- 159. Stowe IB, Mercado EL, Stowe TR, Bell EL, Oses‐Prieto JA, Hernández H, et al. A shared molecular mechanism underlies the human rasopathies Legius syndrome and Neurofibromatosis‐1. Genes Dev. 2012;26(13):1421–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Greer EL, Brunet A. FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene. 2005;24(50):7410–25. [DOI] [PubMed] [Google Scholar]
- 161. Harrington LS, Findlay GM, Gray A, Tolkacheva T, Wigfield S, Rebholz H, et al. The TSC1‐2 tumor suppressor controls insulin‐PI3K signaling via regulation of IRS proteins. J Cell Biol. 2004;166(2):213–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Shah OJ, Hunter T. Turnover of the Active Fraction of IRS1 Involves Raptor‐mTOR‐ and S6K1‐Dependent Serine Phosphorylation in Cell Culture Models of Tuberous Sclerosis. Mol Cell Biol. 2006;26(17):6425–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Tzatsos A, Kandror KV. Nutrients Suppress Phosphatidylinositol 3‐Kinase/Akt Signaling via Raptor‐Dependent mTOR‐Mediated Insulin Receptor Substrate 1 Phosphorylation. Mol Cell Biol. 2006;26(1):63–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Dibble CC, Asara JM, Manning BD. Characterization of Rictor Phosphorylation Sites Reveals Direct Regulation of mTOR Complex 2 by S6K1. Mol Cell Biol. 2009;29(21):5657–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Eijkelenboom A, Burgering BMT. FOXOs: Signalling integrators for homeostasis maintenance. Nat Rev Mol Cell Biol. 2013;14(2):83–97. [DOI] [PubMed] [Google Scholar]
- 166. Mendoza MC, Er EE, Blenis J. The Ras‐ERK and PI3K‐mTOR pathways: Cross‐talk and compensation. Trends Biochem Sci. 2011;36(6):320–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Yu CF, Liu ZX, Cantley LG. ERK negatively regulates the epidermal growth factor‐mediated interaction of Gab1 and the phosphatidylinositol 3‐kinase. J Biol Chem. 2002;277(22):19382–8. [DOI] [PubMed] [Google Scholar]
- 168. Lehr S, Kotzka J, Avci H, Sickmann A, Meyer HE, Herkner A, et al. Identification of major ERK‐related phosphorylation sites in Gab1. Biochemistry. 2004;43(38):12133–40. [DOI] [PubMed] [Google Scholar]
- 169. Zmajkovicova K, Jesenberger V, Catalanotti F, Baumgartner C, Reyes G, Baccarini M. MEK1 Is Required for PTEN Membrane Recruitment, AKT Regulation, and the Maintenance of Peripheral Tolerance. Mol Cell. 2013;50(1):43–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Zimmermann S, Moelling K. Phosphorylation and regulation of Raf by Akt (protein kinase B). Science. 1999;286(5445):1741–4. [DOI] [PubMed] [Google Scholar]
- 171. Moelling K, Schad K, Bosse M, Zimmermann S, Schweneker M. Regulation of Raf‐Akt cross‐talk. J Biol Chem. 2002;277(34):31099–106. [DOI] [PubMed] [Google Scholar]
- 172. Dhillon AS, Meikle S, Yazici Z, Eulitz M, Kolch W. Regulation of Raf‐1 activation and signalling by dephosphorylation. EMBO J. 2002;21(1‐2):64–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Guan K‐L, Figueroa C, Brtva TR, Zhu T, Taylor J, Barber TD, et al. Negative Regulation of the Serine/Threonine Kinase B‐Raf by Akt. J Biol Chem. 2000;275(35):27354–9. [DOI] [PubMed] [Google Scholar]
- 174. Blasco MT, Navas C, Martín‐Serrano G, Graña‐Castro O, Lechuga CG, Martín‐Díaz L, et al. Complete Regression of Advanced Pancreatic Ductal Adenocarcinomas upon Combined Inhibition of EGFR and C‐RAF. Cancer Cell. 2019;35(4):573–87.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Yen I, Shanahan F, Lee J, Hong YS, Shin SJ, Moore AR, et al. ARAF mutations confer resistance to the RAF inhibitor belvarafenib in melanoma. Nature. 2021;594(7863):418–23. [DOI] [PubMed] [Google Scholar]
- 176. Flaherty KT, Robert C, Hersey P, Nathan P, Garbe C, Milhem M, et al. Improved survival with MEK inhibition in BRAF‐mutated melanoma. N Engl J Med. 2012;367(2):107–14. [DOI] [PubMed] [Google Scholar]
- 177. Long G V, Stroyakovskiy D, Gogas H, Levchenko E, de Braud F, Larkin J, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med. 2014;371(20):1877–88. [DOI] [PubMed] [Google Scholar]
- 178. Yuan X, Zhang X, Du R, Cheung S‐H, Wei M, Zhou C, et al. Abstract 6415: RAF dimer inhibitor lifirafenib enhances the antitumor activity of MEK inhibitor mirdametinib in RAS mutant tumors. Cancer Res. 2020;80(16_Supplement):6415–5. [Google Scholar]
- 179. Hatzivassiliou G, Liu B, O'Brien C, Spoerke JM, Hoeflich KP, Haverty PM, et al. ERK Inhibition Overcomes Acquired Resistance to MEK Inhibitors. Mol Cancer Ther. 2012;11(5):1143–54. [DOI] [PubMed] [Google Scholar]
- 180. Rebecca VW, Alicea GM, Paraiso KHT, Lawrence H, Gibney GT, Smalley KSM. Vertical inhibition of the MAPK pathway enhances therapeutic responses in NRAS ‐mutant melanoma. Pigment Cell Melanoma Res. 2014;27(6):1154–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Merchant M, Moffat J, Schaefer G, Chan J, Wang X, Orr C, et al. Combined MEK and ERK inhibition overcomes therapy‐mediated pathway reactivation in RAS mutant tumors. PLoS One. 2017;12(10):e0185862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Weekes C, Lockhart A, LoRusso P, Murray E, Park E, Tagen M, et al. A Phase Ib Study to Evaluate the MEK Inhibitor Cobimetinib in Combination with the ERK1/2 Inhibitor GDC‐0994 in Patients with Advanced Solid Tumors. Oncologist. 2020;25(10):833–e1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Engelman JA, Chen L, Tan X, Crosby K, Guimaraes AR, Upadhyay R, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med. 2008;14(12):1351–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Hoeflich KP, Merchant M, Orr C, Chan J, Den Otter D, Berry L, et al. Intermittent Administration of MEK Inhibitor GDC‐0973 plus PI3K Inhibitor GDC‐0941 Triggers Robust Apoptosis and Tumor Growth Inhibition. Cancer Res. 2012;72(1):210–9. [DOI] [PubMed] [Google Scholar]
- 185. Alagesan B, Contino G, Guimaraes AR, Corcoran RB, Deshpande V, Rwojtkiewicz G, et al. Combined MEK and PI3K inhibition in a mouse model of pancreatic cancer. Clin Cancer Res. 2015;21(2):396–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Juric D, Soria J‐C, Sharma S, Banerji U, Azaro A, Desai J, et al. A phase 1b dose‐escalation study of BYL719 plus binimetinib (MEK162) in patients with selected advanced solid tumors. J Clin Oncol. 2014;32(15_suppl):9051–1. [Google Scholar]
- 187. Bedard PL, Tabernero J, Janku F, Wainberg ZA, Paz‐Ares L, Vansteenkiste J, et al. A Phase Ib Dose‐Escalation Study of the Oral Pan‐PI3K Inhibitor Buparlisib (BKM120) in Combination with the Oral MEK1/2 Inhibitor Trametinib (GSK1120212) in Patients with Selected Advanced Solid Tumors. Clin Cancer Res. 2015;21(4):730–8. [DOI] [PubMed] [Google Scholar]
- 188. Shapiro GI, LoRusso P, Kwak E, Pandya S, Rudin CM, Kurkjian C, et al. Phase Ib study of the MEK inhibitor cobimetinib (GDC‐0973) in combination with the PI3K inhibitor pictilisib (GDC‐0941) in patients with advanced solid tumors. Invest New Drugs. 2020;38(2):419–32. [DOI] [PubMed] [Google Scholar]
- 189. Molina‐Arcas M, Hancock DC, Sheridan C, Kumar MS, Downward J. Coordinate direct input of both KRAS and IGF1 receptor to activation of PI3 kinase in KRAS ‐mutant lung cancer. Cancer Discov. 2013;3(5):548–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Molina‐Arcas M, Moore C, Rana S, van Maldegem F, Mugarza E, Romero‐Clavijo P, et al. Development of combination therapies to maximize the impact of KRAS‐G12C inhibitors in lung cancer. Sci Transl Med. 2019;11(510):eaaw7999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Chung V, McDonough S, Philip PA, Cardin D, Wang‐Gillam A, Hui L, et al. Effect of selumetinib and MK‐2206 vs oxaliplatin and fluorouracil in patients with metastatic pancreatic cancer after prior therapy: SWOG S1115 study randomized clinical trial. JAMA Oncol. 2017;3(4):516–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Algazi AP, Esteve‐Puig R, Nosrati A, Hinds B, Hobbs‐Muthukumar A, Nandoskar P, et al. Dual MEK/AKT inhibition with trametinib and GSK2141795 does not yield clinical benefit in metastatic NRAS‐mutant and wild‐type melanoma. Pigment Cell Melanoma Res. 2018;31(1):110–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Tolcher AW, Bendell JC, Papadopoulos KP, Burris HA, Patnaik A, Jones SF, et al. A phase IB trial of the oral MEK inhibitor trametinib (GSK1120212) in combination with everolimus in patients with advanced solid tumors. Ann Oncol. 2015;26(1):58–64. [DOI] [PubMed] [Google Scholar]
- 194. Mita M, Fu S, Piha‐Paul SA, Janku F, Mita A, Natale R, et al. Phase I trial of MEK 1/2 inhibitor pimasertib combined with mTOR inhibitor temsirolimus in patients with advanced solid tumors. Invest New Drugs. 2017;35(5):616–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Schram AM, Gandhi L, Mita MM, Damstrup L, Campana F, Hidalgo M, et al. A phase Ib dose‐escalation and expansion study of the oral MEK inhibitor pimasertib and PI3K/MTOR inhibitor voxtalisib in patients with advanced solid tumours. Br J Cancer. 2018;119(12):1471–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Athuluri‐Divakar SK, Vasquez‐Del Carpio R, Dutta K, Baker SJ, Cosenza SC, Basu I, et al. A Small Molecule RAS‐Mimetic Disrupts RAS Association with Effector Proteins to Block Signaling. Cell. 2016;165(3):643–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Rahmani F, Hashemzehi M, Avan A, Barneh F, Asgharzadeh F, Moradi Marjaneh R, et al. Rigosertib elicits potent anti‐tumor responses in colorectal cancer by inhibiting Ras signaling pathway. Cell Signal. 2021;85:110069. [DOI] [PubMed] [Google Scholar]
- 198. Zhou X, Xiao Q, Fu D, Zhang H, Tang Y, He J, et al. Efficacy of rigosertib, a small molecular RAS signaling disrupter for the treatment of KRAS‐mutant colorectal cancer. Cancer Biol Med. 2021;19(2):213–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Ozkan‐Dagliyan I, Diehl JN, George SD, Schaefer A, Papke B, Klotz‐Noack K, et al. Low‐Dose Vertical Inhibition of the RAF‐MEK‐ERK Cascade Causes Apoptotic Death of KRAS Mutant Cancers. Cell Rep. 2020;31(11):107764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Gajewski TF, Schreiber H, Fu Y‐X. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol. 2013;14(10):1014–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Garon EB, Hellmann MD, Rizvi NA, Carcereny E, Leighl NB, Ahn M‐J, et al. Five‐Year Overall Survival for Patients With Advanced Non‒Small‐Cell Lung Cancer Treated With Pembrolizumab: Results From the Phase I KEYNOTE‐001 Study. J Clin Oncol. 2019;37(28):2518–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, et al. Five‐year survival outcomes for patients with advanced melanoma treated with pembrolizumab in KEYNOTE‐001. Ann Oncol. 2019;30(4):582–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Yi M, Jiao D, Xu H, Liu Q, Zhao W, Han X, et al. Biomarkers for predicting efficacy of PD‐1/PD‐L1 inhibitors. Mol Cancer. 2018;17(1):129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Doroshow DB, Bhalla S, Beasley MB, Sholl LM, Kerr KM, Gnjatic S, et al. PD‐L1 as a biomarker of response to immune‐checkpoint inhibitors. Nat Rev Clin Oncol. 2021;18(6):345–62. [DOI] [PubMed] [Google Scholar]
- 206. Albitar M, Sudarsanam S, Ma W, Jiang S, Chen W, Funari VA, et al. Expression of PD‐L1 in colorectal cancer that lack mutations in RAS or TP53 genes. J Clin Oncol. 2017;35(15_suppl):e14500–e14500. [Google Scholar]
- 207. Schoenfeld AJ, Rizvi H, Bandlamudi C, Sauter JL, Travis WD, Rekhtman N, et al. Clinical and molecular correlates of PD‐L1 expression in patients with lung adenocarcinomas. Ann Oncol. 2020;31(5):599–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Coelho MA, de Carné Trécesson S, Rana S, Zecchin D, Moore C, Molina‐Arcas M, et al. Oncogenic RAS Signaling Promotes Tumor Immunoresistance by Stabilizing PD‐L1 mRNA. Immunity. 2017;47(6):1083–99.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Zhang XC, Wang J, Shao GG, Wang Q, Qu X, Wang B, et al. Comprehensive genomic and immunological characterization of Chinese non‐small cell lung cancer patients. Nat Commun. 2019;10(1):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Liao W, Overman MJ, Boutin AT, Shang X, Zhao D, Dey P, et al. KRAS‐IRF2 Axis Drives Immune Suppression and Immune Therapy Resistance in Colorectal Cancer. Cancer Cell. 2019;35(4):559–72.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Ischenko I, D'Amico S, Rao M, Li J, Hayman MJ, Powers S, et al. KRAS drives immune evasion in a genetic model of pancreatic cancer. Nat Commun. 2021;12(1):1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Zdanov S, Mandapathil M, Eid RA, Adamson‐Fadeyi S, Wilson W, Qian J, et al. Mutant KRAS conversion of conventional T cells into regulatory T cells. Cancer Immunol Res. 2016;4(4):354–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Yang B, Li X, Fu Y, Guo E, Ye Y, Li F, et al. MEK inhibition remodels the immune landscape of mutant kras tumors to overcome resistance to PARP and immune checkpoint inhibitors. Cancer Res. 2021;81(10):2714–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Jeanson A, Tomasini P, Souquet‐Bressand M, Brandone N, Boucekine M, Grangeon M, et al. Efficacy of Immune Checkpoint Inhibitors in KRAS‐Mutant Non‐Small Cell Lung Cancer (NSCLC). J Thorac Oncol. 2019;14(6):1095–101. [DOI] [PubMed] [Google Scholar]
- 215. Lee CK, Man J, Lord S, Cooper W, Links M, Gebski V, et al. Clinical and molecular characteristics associated with survival among patients treated with checkpoint inhibitors for advanced non‐small cell lung carcinoma: A systematic review and meta‐analysis. JAMA Oncol. 2018;4(2):210–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Dong ZY, Zhong WZ, Zhang XC, Su J, Xie Z, Liu SY, et al. Potential predictive value of TP53 and KRAS mutation status for response to PD‐1 blockade immunotherapy in lung adenocarcinoma. Clin Cancer Res. 2017;23(12):3012–24. [DOI] [PubMed] [Google Scholar]
- 217. Skoulidis F, Goldberg ME, Greenawalt DM, Hellmann MD, Awad MM, Gainor JF, et al. STK11/LKB1 Mutations and PD‐1 Inhibitor Resistance in KRAS ‐Mutant Lung Adenocarcinoma. Cancer Discov. 2018;8(7):822–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Fu Q, Chen N, Ge C, Li R, Li Z, Zeng B, et al. Prognostic value of tumor‐infiltrating lymphocytes in melanoma: a systematic review and meta‐analysis. Oncoimmunology. 8(7):1593806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Loi S, Drubay D, Adams S, Pruneri G, Francis PA, Lacroix‐Triki M, et al. Tumor‐Infiltrating Lymphocytes and Prognosis: A Pooled Individual Patient Analysis of Early‐Stage Triple‐Negative Breast Cancers. J Clin Oncol. 2019;37(7):559–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Houot R, Schultz LM, Marabelle A, Kohrt H. T‐cell‐based immunotherapy: Adoptive cell transfer and checkpoint inhibition. Cancer Immunol Res. 2015;3(10):1115–1122. [DOI] [PubMed] [Google Scholar]
- 221. Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwartzentruber DJ, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298(5594):850–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Tran E, Ahmadzadeh M, Lu Y‐C, Gros A, Turcotte S, Robbins PF, et al. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science. 2015;350(6266):1387–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Veatch JR, Jesernig BL, Kargl J, Fitzgibbon M, Lee SM, Baik C, et al. Endogenous CD4 + T Cells Recognize Neoantigens in Lung Cancer Patients, Including Recurrent Oncogenic KRAS and ERBB2 (Her2) Driver Mutations. Cancer Immunol Res. 2019;7(6):910–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. June CH, O'Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. Science. 2018;359(6382):1361–5. [DOI] [PubMed] [Google Scholar]
- 225. Wang QJ, Yu Z, Griffith K, Hanada KI, Restifo NP, Yang JC. Identification of T‐cell receptors targeting KRAS‐mutated human tumors. Cancer Immunol Res. 2016;4(3):204–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Mou N, Yu Y, Yuan J. Abstract 1499: Targeting Ras mutation with TCR therapy. Cancer Res. 2021;81(13_Supplement):1499–9. [Google Scholar]
- 227. Bear AS, Blanchard T, Cesare J, Ford MJ, Richman LP, Xu C, et al. Biochemical and functional characterization of mutant KRAS epitopes validates this oncoprotein for immunological targeting. Nat Commun. 2021;12(1):1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Tran E, Robbins PF, Lu Y‐C, Prickett TD, Gartner JJ, Jia L, et al. T‐Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N Engl J Med. 2016;375(23):2255–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Leidner R, Sanjuan Silva N, Huang H, Sprott D, Zheng C, Shih Y‐P, et al. Neoantigen T‐Cell Receptor Gene Therapy in Pancreatic Cancer. N Engl J Med. 2022;386(22):2112–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Thomas A, Chen Y, Steinberg SM, Luo J, Pack S, Raffeld M, et al. High mesothelin expression in advanced lung adenocarcinoma is associated with KRAS mutations and a poor prognosis. Oncotarget. 2015;6(13):11694–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Minehart J, Eguchi T, Morello A, Adusumilli P. OA14.03 Clinical Rationale and Preclinical Evidence for Chimeric Antigen Receptor (CAR) T Cell Therapy Clinical Trial in KRAS‐Mutant Lung Cancer. J Thorac Oncol. 2019;14(10):S244. [Google Scholar]
- 232. Gjertsen MK, Bakka A, Breivik J, Saeterdal I, Gedde‐Dahl T, Stokke KT, et al. Ex vivo ras peptide vaccination in patients with advanced pancreatic cancer: Results of a phase I/II study. Int J Cancer. 1996;65(4):450–3. [DOI] [PubMed] [Google Scholar]
- 233. Khleif SN, Abrams SI, Hamilton JM, Bergmann‐Leitner E, Chen A, Bastian A, et al. A phase I vaccine trial with peptides reflecting ras oncogene mutations of solid tumors. J Immunother. 1999;22(2):155–65. [DOI] [PubMed] [Google Scholar]
- 234. Gjertsen MK, Buanes T, Rosseland AR, Bakka A, Gladhaug I, Søreide O, et al. Intradermal ras peptide vaccination with granulocyte‐macrophage colony‐stimulating factor as adjuvant: Clinical and immunological responses in patients with pancreatic adenocarcinoma. Int J cancer. 2001;92(3):441–50. [DOI] [PubMed] [Google Scholar]
- 235. Palmer DH, Valle JW, Ma YT, Faluyi O, Neoptolemos JP, Jensen Gjertsen T, et al. TG01/GM‐CSF and adjuvant gemcitabine in patients with resected RAS‐mutant adenocarcinoma of the pancreas (CT TG01‐01): a single‐arm, phase 1/2 trial. Br J Cancer. 2020;122(7):971–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Mukhopadhyay S, Vander Heiden MG, McCormick F. The metabolic landscape of RAS‐driven cancers from biology to therapy. Nat Cancer. 2021;2(3):271–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell. 2011;144(5):646–74. [DOI] [PubMed] [Google Scholar]
- 238. Pavlova NN, Thompson CB. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016;23(1):27–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher‐Sananikone E, et al. Oncogenic kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell. 2012;149(3):656–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. Kerr EM, Gaude E, Turrell FK, Frezza C, Martins CP. Mutant Kras copy number defines metabolic reprogramming and therapeutic susceptibilities. Nature. 2016;531(7592):110–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. Ancey P, Contat C, Meylan E. Glucose transporters in cancer – from tumor cells to the tumor microenvironment. FEBS J. 2018;285(16):2926–43. [DOI] [PubMed] [Google Scholar]
- 242. Yamamoto T, Seino Y, Fukumoto H, Koh G, Yano H, Inagaki N, et al. Over‐expression of facilitative glucose transporter genes in human cancer. Biochem Biophys Res Commun. 1990;170(1):223–30. [DOI] [PubMed] [Google Scholar]
- 243. Brown RS, Wahl RL. Overexpression of Glut‐1 glucose transporter in human breast cancer. An immunohistochemical study. Cancer. 1993;72(10):2979–85. [DOI] [PubMed] [Google Scholar]
- 244. Siebeneicher H, Cleve A, Rehwinkel H, Neuhaus R, Heisler I, Müller T, et al. Identification and Optimization of the First Highly Selective GLUT1 Inhibitor BAY‐876. ChemMedChem. 2016;11(20):2261–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245. Ma Y, Wang W, Idowu M, Oh U, Wang X‐Y, Temkin S, et al. Ovarian Cancer Relies on Glucose Transporter 1 to Fuel Glycolysis and Growth: Anti‐Tumor Activity of BAY‐876. Cancers (Basel). 2018;11(1):33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246. Wu Q, Ba‐Alawi W, Deblois G, Cruickshank J, Duan S, Lima‐Fernandes E, et al. GLUT1 inhibition blocks growth of RB1‐positive triple negative breast cancer. Nat Commun. 2020;11(1):4205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247. Ojelabi OA, Lloyd KP, Simon AH, De Zutter JK, Carruthers A. WZB117 (2‐Fluoro‐6‐(m‐hydroxybenzoyloxy) Phenyl m‐Hydroxybenzoate) Inhibits GLUT1‐mediated Sugar Transport by Binding Reversibly at the Exofacial Sugar Binding Site. J Biol Chem. 2016;291(52):26762–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248. Erber J, Steiner JD, Isensee J, Lobbes LA, Toschka A, Beleggia F, et al. Dual inhibition of GLUT1 and the ATR/Chk1 kinase axis displays synergistic cytotoxicity in KRAS‐mutant cancer cells. Cancer Res. 2019;79(19):4855–68. [DOI] [PubMed] [Google Scholar]
- 249. Yun J, Mullarky E, Lu C, Bosch KN, Kavalier A, Rivera K, et al. Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science. 2015;350(6266):1391–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250. Mueckler M, Thorens B. The SLC2 (GLUT) family of membrane transporters. Mol Aspects Med. 2013;34(2‐3):121–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251. Wise DR, Thompson CB. Glutamine addiction: a new therapeutic target in cancer. Trends Biochem Sci. 2010;35(8):427–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252. Toda K, Nishikawa G, Iwamoto M, Itatani Y, Takahashi R, Sakai Y, et al. Clinical role of ASCT2 (SLC1A5) in KRAS‐mutated colorectal cancer. Int J Mol Sci. 2017;18(8):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253. Vaseva AV, Blake DR, Gilbert TSK, Ng S, Hostetter G, Azam SH, et al. KRAS Suppression‐Induced Degradation of MYC Is Antagonized by a MEK5‐ERK5 Compensatory Mechanism. Cancer Cell. 2018;34(5):807–22.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254. Stine ZE, Walton ZE, Altman BJ, Hsieh AL, Dang CV. MYC, metabolism, and cancer. Cancer Discov. 2015;5(10):1024–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255. Dejure FR, Eilers M. MYC and tumor metabolism: chicken and egg. EMBO J. 2017;36(23):3409–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256. Moreadith RW, Lehninger AL. The pathways of glutamate and glutamine oxidation by tumor cell mitochondria. Role of mitochondrial NAD(P)+‐dependent malic enzyme. J Biol Chem. 1984;259(10):6215–21. [PubMed] [Google Scholar]
- 257. Zhang J, Pavlova NN, Thompson CB. Cancer cell metabolism: the essential role of the nonessential amino acid, glutamine. EMBO J. 2017;36(10):1302–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258. Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, et al. Glutamine supports pancreatic cancer growth through a KRAS‐regulated metabolic pathway. Nature. 2013;496(7443):101–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259. Nicklin P, Bergman P, Zhang B, Triantafellow E, Wang H, Nyfeler B, et al. Bidirectional Transport of Amino Acids Regulates mTOR and Autophagy. Cell. 2009;136(3):521–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260. Najumudeen AK, Ceteci F, Fey SK, Hamm G, Steven RT, Hall H, et al. The amino acid transporter SLC7A5 is required for efficient growth of KRAS‐mutant colorectal cancer. Nat Genet. 2021;53(1):16–26. [DOI] [PubMed] [Google Scholar]
- 261. Welbourne TC. Ammonia production and glutamine incorporation into glutathione in the functioning rat kidney. Can J Biochem. 1979;57(3):233–7. [DOI] [PubMed] [Google Scholar]
- 262. DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, et al. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci. 2007;104(49):19345–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263. Rojo de la Vega M, Chapman E, Zhang DD. NRF2 and the Hallmarks of Cancer. Cancer Cell. 2018;34(1):21–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264. Romero R, Sayin VI, Davidson SM, Bauer MR, Singh SX, Leboeuf SE, et al. Keap1 loss promotes Kras‐driven lung cancer and results in dependence on glutaminolysis. Nat Med. 2017;23(11):1362–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265. Galan‐Cobo A, Sitthideatphaiboon P, Qu X, Poteete A, Pisegna MA, Tong P, et al. LKB1 and KEAP1/NRF2 pathways cooperatively promote metabolic reprogramming with enhanced glutamine dependence inKRAS‐mutant lung adenocarcinoma. Cancer Res. 2019;79(13):3251–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266. Saqcena M, Mukhopadhyay S, Hosny C, Alhamed A, Chatterjee A, Foster DA. Blocking anaplerotic entry of glutamine into the TCA cycle sensitizes K‐Ras mutant cancer cells to cytotoxic drugs. Oncogene. 2015;34(20):2672–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267. Gross MI, Demo SD, Dennison JB, Chen L, Chernov‐Rogan T, Goyal B, et al. Antitumor activity of the glutaminase inhibitor CB‐839 in triple‐negative breast cancer. Mol Cancer Ther. 2014;13(4):890–901. [DOI] [PubMed] [Google Scholar]
- 268. Shen Y‐A, Hong J, Asaka R, Asaka S, Hsu F‐C, Suryo Rahmanto Y, et al. Inhibition of the MYC‐Regulated Glutaminase Metabolic Axis Is an Effective Synthetic Lethal Approach for Treating Chemoresistant Ovarian Cancers. Cancer Res. 2020;80(20):4514–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269. Jacque N, Ronchetti AM, Larrue C, Meunier G, Birsen R, Willems L, et al. Targeting glutaminolysis has antileukemic activity in acute myeloid leukemia and synergizes with BCL‐2 inhibition. Blood. 2015;126(11):1346–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270. Momcilovic M, Bailey ST, Lee JT, Fishbein MC, Braas D, Go J, et al. The GSK3 Signaling Axis Regulates Adaptive Glutamine Metabolism in Lung Squamous Cell Carcinoma. Cancer Cell. 2018;33(5):905–21.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271. Emberley ED, Bennett M, Chen J, Gross M, Huang T, Makkouk A, et al. Abstract 3509: The glutaminase inhibitor CB‐839 synergizes with CDK4/6 and PARP inhibitors in pre‐clinical tumor models. Cancer Res. 2018;78(13 Supplement):3509. [Google Scholar]
- 272. Schulte ML, Fu A, Zhao P, Li J, Geng L, Smith ST, et al. Pharmacological blockade of ASCT2‐dependent glutamine transport leads to antitumor efficacy in preclinical models. Nat Med. 2018;24(2):194–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273. Bröer A, Fairweather S, Bröer S. Disruption of Amino Acid Homeostasis by Novel ASCT2 Inhibitors Involves Multiple Targets. Front Pharmacol. 2018;9(785). 10.3389/fphar.2018.00785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274. Biancur DE, Paulo JA, Małachowska B, Quiles Del Rey M, Sousa CM, Wang X, et al. Compensatory metabolic networks in pancreatic cancers upon perturbation of glutamine metabolism. Nat Commun. 2017;8(1):15965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275. Davidson SM, Papagiannakopoulos T, Olenchock BA, Heyman JE, Keibler MA, Luengo A, et al. Environment impacts the metabolic dependencies of ras‐driven non‐small cell lung cancer. Cell Metab. 2016;23(3):517–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276. Best SA, Ding S, Kersbergen A, Dong X, Song JY, Xie Y, et al. Distinct initiating events underpin the immune and metabolic heterogeneity of KRAS‐mutant lung adenocarcinoma. Nat Commun. 2019;10(1):1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277. Finicle BT, Jayashankar V, Edinger AL. Nutrient scavenging in cancer. Nat Rev Cancer. 2018;18(10):619–33. [DOI] [PubMed] [Google Scholar]
- 278. Kamphorst JJ, Nofal M, Commisso C, Hackett SR, Lu W, Grabocka E, et al. Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer Res. 2015;75(3):544–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279. Levy JMM, Towers CG, Thorburn A. Targeting autophagy in cancer. Nat Rev Cancer. 2017;17(9):528–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280. Guo JY, Chen H‐Y, Mathew R, Fan J, Strohecker AM, Karsli‐Uzunbas G, et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011;25(5):460–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281. Commisso C, Davidson SM, Soydaner‐Azeloglu RG, Parker SJ, Kamphorst JJ, Hackett S, et al. Macropinocytosis of protein is an amino acid supply route in Ras‐transformed cells. Nature. 2013;497(7451):633–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282. Bryant KL, Stalnecker CA, Zeitouni D, Klomp JE, Peng S, Tikunov AP, et al. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat Med. 2019;25(4):628–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283. Kinsey CG, Camolotto SA, Boespflug AM, Guillen KP, Foth M, Truong A, et al. Protective autophagy elicited by RAF→MEK→ERK inhibition suggests a treatment strategy for RAS‐driven cancers. Nat Med. 2019;25(4):620–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284. Lee CS, Lee LC, Yuan TL, Chakka S, Fellmann C, Lowe SW, et al. MAP kinase and autophagy pathways cooperate to maintain RAS mutant cancer cell survival. Proc Natl Acad Sci U S A. 2019;116(10):4508–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285. Jayashankar V, Edinger AL. Macropinocytosis confers resistance to therapies targeting cancer anabolism. Nat Commun. 2020;11(1):1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286. O'Hara MH, Karasic TB, Vasilevskaya I, Redlinger M, Loaiza‐Bonilla A, Teitelbaum UR, et al. Phase II trial of the autophagy inhibitor hydroxychloroquine with FOLFOX and bevacizumab in front line treatment of metastatic colorectal cancer. J Clin Oncol. 2017;35(15_suppl):3545–5. [Google Scholar]
- 287. Karasic TB, O'Hara MH, Loaiza‐Bonilla A, Reiss KA, Teitelbaum UR, Borazanci E, et al. Effect of Gemcitabine and nab‐Paclitaxel With or Without Hydroxychloroquine on Patients With Advanced Pancreatic Cancer. JAMA Oncol. 2019;5(7):993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288. Rosenfeld MR, Grossman SA, Brem S, Mikkelsen T, Wang D, Piao S, et al. Pharmacokinetic analysis and pharmacodynamic evidence of autophagy inhibition in patients with newly diagnosed glioblastoma treated on a phase I trial of hydroxychloroquine in combination with adjuvant temozolomide and radiation (ABTC 0603). J Clin Oncol. 2010;28(15_suppl):3086–6. [Google Scholar]
- 289. McAfee Q, Zhang Z, Samanta A, Levi SM, Ma XH, Piao S, et al. Autophagy inhibitor Lys05 has single‐agent antitumor activity and reproduces the phenotype of a genetic autophagy deficiency. Proc Natl Acad Sci U S A. 2012;109(21):8253–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290. Rebecca VW, Nicastri MC, Fennelly C, Chude CI, Barber‐Rotenberg JS, Ronghe A, et al. PPT1 promotes tumor growth and is the molecular target of chloroquine derivatives in cancer. Cancer Discov. 2019;9(2):220–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.