

Abstract
As a critical component of the tumor microenvironment (TME), cancer‐associated fibroblasts (CAFs) play important roles in cancer initiation and progression. Well‐known signaling pathways, including the transforming growth factor‐β (TGF‐β), Hedgehog (Hh), Notch, Wnt, Hippo, nuclear factor kappa‐B (NF‐κB), Janus kinase (JAK)/signal transducer and activator of transcription (STAT), mitogen‐activated protein kinase (MAPK), and phosphoinositide 3‐kinase (PI3K)/AKT pathways, as well as transcription factors, including hypoxia‐inducible factor (HIF), heat shock transcription factor 1 (HSF1), P53, Snail, and Twist, constitute complex regulatory networks in the TME to modulate the formation, activation, heterogeneity, metabolic characteristics and malignant phenotype of CAFs. Activated CAFs remodel the TME and influence the malignant biological processes of cancer cells by altering the transcriptional and secretory characteristics, and this modulation partially depends on the regulation of signaling cascades. The results of preclinical and clinical trials indicated that therapies targeting signaling pathways in CAFs demonstrated promising efficacy but were also accompanied by some failures (e.g., NCT01130142 and NCT01064622). Hence, a comprehensive understanding of the signaling cascades in CAFs might help us better understand the roles of CAFs and the TME in cancer progression and may facilitate the development of more efficient and safer stroma‐targeted cancer therapies. Here, we review recent advances in studies of signaling pathways in CAFs and briefly discuss some future perspectives on CAF research.
Keywords: Signaling pathways, Cancer‐associated fibroblasts, Cell‐cell interaction, Tumor microenvironment, Therapeutic targets
Abbreviations
- TGF‐β
transforming growth factor‐β
- CAFs
cancer‐associated fibroblasts
- SMAD
Sma‐and Mad‐related protein
- BMP
bone morphogenetic protein
- TME
tumor microenvironment
- ROS
reactive oxygen species
- DIAPH1
diaphanous homolog 1
- LXRα
liver X receptor α
- α‐SMA
α‐smooth muscle actin
- FAP
fibroblast activation protein
- ECM
extracellular matrix
- VCAN
versican
- LAP
latency‐associated polypeptide
- PI3K
phosphoinositide 3‐kinase
- HIF
hypoxia‐inducible factor
- NF‐κB
nuclear factor kappa‐B
- AFP
alpha fetoprotein
- APC
adenomatous polyposis coli
- apCAFs
antigen‐presenting CAFs
- BCKA
branched chain α‐ketoacid
- BMP
bone morphogenetic protein
- BOC
brother of CDON
- CAFs
cancer‐associated fibroblasts
- CCBE1
collagen and calcium binding EGF domains 1
- CCL
C‐C motif chemokine ligand
- CCM3
cerebral cavernous malformations 3
- CDON
cell adhesion associated, oncogene regulated
- CRC
colorectal cancer
- CSCs
cancer stem cells
- CSL
CBF1/Suppressor of Hairless/LAG1
- CTLA4
cytotoxic T‐lymphocyte‐associated protein 4
- CXCL
C‐X‐C chemokine ligand
- CXCR
C‐X‐C chemokine receptor
- DC
dendritic cell
- DDR
DNA damage response
- DIAPH1
diaphanous homolog 1
- DKK3
Dickkopf‐3
- Dvl
Deshevelled
- ECM
extracellular matrix
- EGFR
epidermal growth factor receptor
- EMT
epithelial‐to‐mesenchymal transition
- ERK
extracellular signal regulated kinase
- FAK
focal adhesion kinase
- FAP
fibroblast activation protein
- FGF
Fibroblast growth factor
- FSP1
fibroblast‐specific protein 1
- Fzd
Frizzled
- GAS1
growth arrest specific 1
- GOF
gain‐of‐function
- GSK3β
glycogen synthase kinase 3β
- HCC
hepatocellular carcinoma
- HER2
human epidermal growth factor receptor 2
- HGF
hepatocyte growth factor
- Hh
hedgehog
- HIF
hypoxia‐inducible factor
- HK2
hexokinase 2
- HSF1
heat shock transcription factor 1
- HSPs
heat shock proteins
- iCAFs
inflammatory CAFs
- IDH3α
isocitrate dehydrogenase 3α
- IGF‐1
insulin‐like growth factor 1
- IKK
IκB kinase complex
- IL
interleukin
- INHBA
inhibin subunit beta A
- JAK
Janus kinase
- LDHA
lactate dehydrogenase A
- LIF
leukemia inhibitory factor
- Ln‐γ2
laminin gamma 2
- LRP
LDL receptor related protein
- LRRC15
leucine‐rich repeat containing 15
- LTBR
Lymphotoxin beta receptor
- LXRα
liver X receptors α
- MHC‐II
major histocompatibility complex class II
- MAOA
monoamine oxidase A
- MAPK
mitogen‐activated protein kinase
- MMAE
monomethyl auristatin E
- MMP
matrix metallopeptidase
- MSCs
mesenchymal stem cells
- mTORC
mTOR complex
- myCAFs
myofibroblasts
- ncRNAs
non‐coding RNAs
- NFs
normal fibroblasts
- NF‐κB
nuclear factor kappa‐B
- NICD
Notch intracellular domain
- OS
overall survival
- OSCC
oral squamous cell carcinoma
- PD‐1
programmed cell death‐1
- PD‐L1
programmed death ligand‐1
- PDGFR
platelet‐derived growth factor receptor
- PDK1
phosphoinositide‐dependent protein kinase 1
- PI3K
phosphoinositide 3‐kinase
- PIP2
phosphatidylinositol 4,5‐bisphosphate
- PIP3
phosphatidylinositol 3,4,5‐trisphosphate
- PKM2
pyruvate kinase M2
- PSCs
pancreatic stellate cells
- PTCH
Patched
- rCAFs
restraining CAFs
- RhoA
Ras homolog family member A
- ROSs
reactive oxygen species
- RTKs
receptor tyrosine kinases
- Shh
Sonic hedgehog
- SMAD
Sma‐and Mad‐related protein
- SMO
Smoothened
- SOCS
suppressor of cytokine signaling
- STAT
signal transducers and activators of transcription
- TAK1
TGF‐β activated kinase 1
- TAZ
transcriptional coactivator with PDZ‐binding motif
- TEAD
TEA domain transcription factor
- TFs
transcription factors
- TGF‐β
transforming growth factor‐beta
- THBS2
thrombospondin‐2
- TKIs
tyrosine kinase inhibitors
- TME
tumor microenvironment
- TNBC
triple‐negative breast cancer
- TSC
tuberous sclerosis protein
- TSP‐4
Thrombospondin 4
- USP27X
ubiquitin specific peptidase 27 X‐linked
- VCAN
versican
- VEGF
vascular endothelial growth factor
- YAP
Yes‐associated protein
- ZEB
Zinc finger E‐box binding homeobox
- ZNF37A
Zinc finger protein 37A
- α‐KG
α‐ketoglutarate
- α‐SMA
α‐smooth muscle actin
1. BACKGROUND
Malignant tumors are composed of cancer cells with uncontrolled proliferation and a multiple‐component tumor microenvironment (TME), the complexity of which is similar to that of normal healthy tissues [1]. Precisely because of the increased and accurate understanding of the complexity and heterogeneity of tumors, researchers have gradually realized that cancer progression depends not only on the malignant biological characteristics of cancer cells but also on the TME, which is an indispensable cancer promoter [2]. As an important component of the TME, cancer‐associated fibroblasts (CAFs) are formed by the activation or transformation of precursor cells in tumor tissues [3]. According to their phenotypic features, CAFs are generally classified into two categories: CAFs with a myofibroblastic phenotype (myCAFs), which express α‐smooth muscle actin (α‐SMA) and fibroblast activation protein (FAP) at high levels, and inflammatory CAFs (iCAFs), which show secretory characteristics and functions regulating inflammation [4, 5]. Activated CAFs perform crucial regulatory functions in extracellular matrix (ECM) remodeling, cancer cell proliferation, metabolic reprogramming, invasion, stemness and other malignant behaviors, as well as in tumor angiogenesis, metastasis, immunosuppression and therapeutic resistance [6, 7]. Additionally, researchers believe that multiple groups of heterogeneous CAF subpopulations play diverse roles in promoting or suppressing cancer progression in distinct cancers [8] or in different stages of a specific cancer [9]. Signaling pathways are one of the cruical participants in the aforementioned processes, including CAF formation and activation, the acquisition and maintenance of cancer‐promoting or cancer‐suppressing functions of CAFs, and the mechanism by which CAFs influence cancer cells and the TME.
The dysregulation and mutation of key node molecules in signaling pathways are driving forces of cancer initiation and progression [10, 11]. Cancer cells can undergo independent and uncontrolled proliferation by deregulating growth‐promoting signaling, which is mediated by signaling cascades, for which CAFs are pivotal sources of upstream signals [12, 13]. The role of CAFs in regulating signaling pathways in cancer cells is based on alterations in their transcriptional and secretory characteristics mediated by the internal activation or suppression of signaling activation states, which is partially regulated by cancer cells [14, 15]. Therefore, signaling cascades are communication bridges in the interactions between cancer cells and CAFs, and the regulatory network composed of multiple pathways combines the robustness of cancer cells and the TME to contribute to cancer progression.
In this review, we provide an overview of the definition, origins and heterogeneity of CAFs; summarize the well‐known signaling pathways and transcription factors (TFs) that are crucial in cancers, including the transforming growth factor‐β (TGF‐β), Hedgehog (Hh), Notch, Wnt, Hippo, nuclear factor kappa‐B (NF‐κB), Janus kinase (JAK)/signal transducer and activator of transcription (STAT), mitogen‐activated protein kinase (MAPK), and phosphoinositide 3‐kinase (PI3K)/AKT pathways, hypoxia‐inducible factor (HIF), heat shock transcription factor 1 (HSF1), P53, epithelial‐to‐mesenchymal transition (EMT)‐related TFs Snail, Twist and Zinc finger E‐box binding homeobox (ZEB), and their vital roles in CAF formation and functional acquisition, as well as the contributions of these signaling cascades in CAFs to cancer progression. Moreover, we discuss the potential therapeutic targets of the signaling pathways in CAFs and describe anticipated future research directions.
2. AN OVERVIEW OF CAFS
2.1. Definition of CAFs
Spindle‐shaped cells with the ability to synthesize collagen in connective tissues are named fibroblasts, and they can be activated to play important roles in the process of wound healing, tissue fibrosis and inflammation, such as promoting tissue repair and regeneration. The initiation and progression of cancer are known to inflict irreversible injuries to the body, during which fibroblasts are activated to influence tumor inflammation, fibrosis and numerous biological behaviors during cancer progression [7, 12, 16]. Activated fibroblasts associated with cancer are defined as CAFs [17]. α‐SMA, FAP, fibroblast‐specific protein 1 (FSP1), platelet‐derived growth factor receptor α/β (PDGFRα/β) and vimentin are considered representative CAF markers (Table 1). Nevertheless, none of these markers is sufficient for the specific recognition of CAFs because these molecules are also expressed in other cell types [18]. Compared with normal fibroblasts (NFs), activated CAFs exhibit greater proliferation and migration abilities and higher metabolic levels [19]. These differences allow CAFs to be better adapted to the TME, which supports their corresponding roles in cancer progression. According to most studies, CAFs mainly play a tumor‐promoting role. However, some studies have shown that CAFs also exhibit tumor‐suppressive functions in specific circumstances [6, 20]. For instance, in a pancreatic cancer mouse model, the depletion of α‐SMA+ CAFs accelerated cancer progression, which was characterized by an increase in the numbers of invasive and undifferentiated cancer cells with enhanced hypoxia and stemness, resulting in increased mortality [21]. This evidence indicates that CAFs play a variety of important functions in cancer progression and that they have a dual nature, as indicated by their roles in tumor promotion and inhibition. Elucidating and targeting the functional mechanism of CAFs, such as the initiation of essential signaling pathways, will promote the development of cancer treatments.
TABLE 1.
CAF markers
| Marker | Description | Expression level | Biological effects | Expression pattern (in addition to CAFs) | Refs |
|---|---|---|---|---|---|
| Surface marker | |||||
| Caveolin‐1 | Scaffolding protein | Upregulated or downregulated | Structure component | Adipocytes, endothelial cells, and NFs | [340, 341] |
| FAP | Serine protease | Upregulated | ECM remodeling and fibrogenesis | CD45+ immune cells, macrophages, NFs and quiescent stellate cells | [342, 343, 344, 345] |
| GPR77 | G protein‐coupled receptor | Upregulated | Complement activation and pro‐inflammatory signaling | Polymorphonuclear neutrophils | [251] |
| PDGFRα/β | Growth factor receptor | Upregulated | Receptor tyrosine kinase activity | BM‐MSCs, cancer cells, NFs, pericytes and vascular smooth muscle cells | [346, 347] |
| Podoplanin | Transmembrane glycoprotein | Upregulated | Cell motility, adhesion and vascular remodeling | Cancer cells and endothelial cells | [348, 349] |
| Intracellular marker | |||||
| α‐SMA | Actin isoform | Upregulated | Cell contractility, motility, structural integrity, desmoplasia and ECM remodeling | Cardiomyocytes, NFs, pericytes, quiescent stellate cells and smooth muscle cells | [350, 351] |
| Desmin | Type III intermediate filament protein | Downregulated | Cell contractility, motility and structural integrity | Muscle cells and pericytes | [352] |
| FSP1 | Calcium‐binding protein | Upregulated | Cell motility, collagen induction, ECM remodeling and tissue fibrosis | Cancer cells, epithelial cells undergoing EMT, endothelial cells, NFs and macrophages | [353, 354] |
| Vimentin | Type III intermediate filament protein | Upregulated | Cell motility and structural integrity | Cancer cells, epithelial cells undergoing EMT, endothelial cells, and neural cells | [355, 356] |
| Extracellular marker | |||||
| Type I collagen | ECM protein | Upregulated | ECM remodeling | Osteoblasts, tendon and vascular smooth muscle cells | [357, 358] |
| Tenascin‐C | ECM glycoprotein | Upregulated | Cell adhesion | Cancer cells | [359] |
Abbreviations: CAFs, cancer‐associated fibroblasts; NFs, normal fibroblasts; FAP, fibroblast activation protein; ECM, extracellular matrix; PDGFRα/β: platelet‐derived growth factor receptor α/β; BM‐MSCs, bone marrow‐derived mesenchymal stem cells; α‐SMA, α‐smooth muscle actin; FSP1, fibroblast‐specific protein 1; EMT, epithelial‐to‐mesenchymal transition
2.2. Origins of CAFs
CAFs constitute a population of highly heterogeneous cells that may be closely related to their wide range of origins (Figure 1). NFs residing in tissues are the main source of CAFs. They are transformed into CAFs when stimulated by cancer cells or other components in the TME [22, 23]. Various growth factors, cytokines, chemokines and key molecules involved in cell signal transduction, such as TGF‐β, fibroblast growth factor (FGF), PDGF, Sonic hedgehog (Shh) and interleukin‐6 (IL‐6), induce the transition of NFs into CAFs by activating the related signaling pathways [24]. Stellate cells are special tissue‐resident fibroblasts similar to NFs that may be activated and transformed into CAFs under certain conditions. Based on accumulating evidence, quiescent pancreatic stellate cells (PSCs) and hepatic stellate cells acquire a CAF phenotype and exert their corresponding functions in response to the actions of TGF‐β, IL‐1, PDGF, and other stimuli [25, 26, 27].
FIGURE 1.
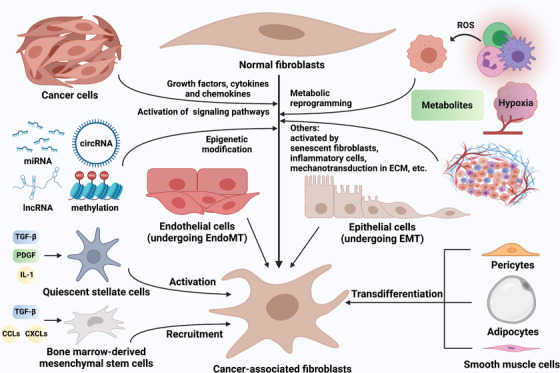
The origins of CAFs. CAFs are formed from a wide range of cell precursors through specific mechanisms. Upon the stimulation of growth factors, cytokines and chemokines such as TGF‐β, FGF, PDGF, and IL‐6, as well as epigenetic modification mediated by non‐coding RNAs and DNA methylation, multiple signaling pathways in tissue‐resident NFs are activated, leading to CAF formation [24, 51, 139, 158]. Metabolic reprogramming caused by cancer cell‐derived ROS and other metabolites, senescent fibroblasts, inflammatory cells and mechanotransduction in ECM also mediated the transformation of NFs into CAFs [231, 335]. In addition, endothelial cells can be transformed into CAFs via the EndoMT, while epithelial cells can be transformed into CAFs via the EMT [33, 36]. BM‐MSCs and quiescent stellate cells are recruited and activated to become CAFs by growth factors, cytokines and chemokines such as TGF‐β, PDGF, IL‐1, CXCL12, CXCL16, CCL2, and CCL5 [285, 336]. Evidence suggests that adipocytes, pericytes and smooth muscle cells can also be transformed into CAFs [37, 41, 42]. Abbreviations: CAFs, cancer‐associated fibroblasts; TGF‐β, transforming growth factor‐β; FGF, fibroblast growth factor; PDGF, platelet‐derived growth factor; IL, interleukin; NFs, normal fibroblasts; ROS, reactive oxygen species; ECM, extracellular matrix; EndoMT, endothelial‐to‐mesenchymal transition; EMT, epithelial‐to‐mesenchymal transition; BM‐MSCs, Bone marrow‐derived mesenchymal stem cells; CXCL, C‐X‐C chemokine ligand; CCL, C‐C motif chemokine ligand
Other irreplaceable sources of CAFs are bone marrow‐derived mesenchymal stem cells (BM‐MSCs). Quante et al. [28] have reported that in gastric cancer, at least 20% of CAFs were derived from BM‐MSCs and were recruited in tumors in a TGF‐β‐ and stromal‐derived factor 1 (SDF1)‐dependent manner. In breast cancer, MSCs obtained the CAF phenotype by activating the myeloid zinc finger 1/TGF‐β1 pathway [29]. Homeobox A9 in ovarian cancer cells stimulated BM‐MSCs to differentiate into CAFs by activating TGF‐β2 transcription [30]. Clearly, the TGF‐β signaling pathway plays a vital role in the transformation of MSCs into CAFs [31], and a TGF‐β inhibitor has been shown to inhibit the tumor‐promoting effect of BM‐MSCs and CAF marker expression [32]. The original sources of CAFs also include a series of mature and differentiated cells. Epithelial cells transdifferentiate into CAFs through the EMT [33, 34], while endothelial cells transdifferentiate via the endothelial‐to‐mesenchymal transition [35, 36]. Other evidence suggests that adipocytes [37, 38, 39], pericytes [40, 41] and smooth muscle cells [42] can also be transformed into CAFs. However, due to the common pedigree of certain cell types and few specific markers, the exact origins of CAFs have not been fully clarified [7]. Although the application of genetic lineage traces [43], fluorescence tags [44] and other technologies has made the study of the transformation of cell phenotypes and types more convenient and the results more credible, the exact biological sources of CAFs still needs to be vigorously explored.
2.3. Heterogeneity and subpopulations of CAFs
CAF heterogeneity is characterized by significant differences in phenotype and function (Figure 2) related to the tumor type, tumor stage, and other properties [45, 46]. The numerous potential origins of CAFs may partially explain their heterogeneity. In addition, the plasticity of CAFs and the activation of different signaling pathways lead to the transformation of CAF phenotypes and functions, which may be another important reason for CAF heterogeneity [47]. Although CAF plasticity is not fully investigated, some studies have confirmed that CAFs can undergo phenotypic and functional alterations [48]. For example, while performing a whole transcriptome analysis, Elwakeel et al. [49] conducted a dynamic analysis of CAF subpopulations from early to late stages of breast cancer. They found that the CAF transcriptome and phenotype changed during cancer initiation and progression. The heterogeneity of CAFs indicates that the CAF population is composed of various cellular subsets with distinct phenotypes and functions. Distinguishing and defining these subpopulations will help us to further understand CAFs [50].
FIGURE 2.
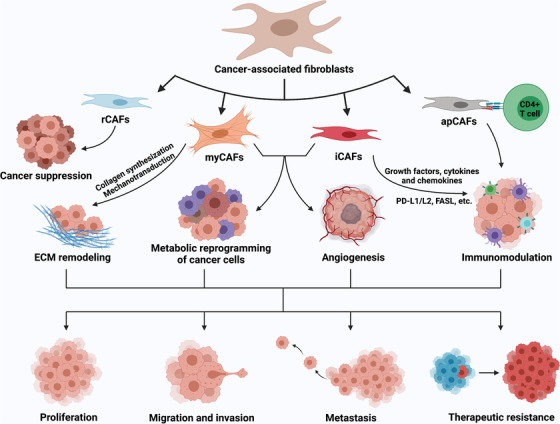
The heterogeneity and functions of CAFs. Due to the wide range of cellular precursors and the differences in activation mechanisms, CAFs show a high degree of heterogeneity and are generally classified into rCAFs, myCAFs, iCAFs and apCAFs [4, 51]. Among these CAFs, rCAFs play a role in cancer suppression; myCAFs mediate ECM remodeling by synthesizing collagen and regulating mechanotransduction; iCAFs perform immunomodulation by changing secretory characteristics; and apCAFs activate CD4+ T cells in an antigen‐specific manner [6, 51]. myCAFs and iCAFs contribute to tumor metabolic reprogramming and angiogenesis through various mechanisms. The joint actions of myCAFs, iCAFs and apCAFs ultimately promote the proliferation, migration, invasion, metastasis and therapeutic resistance of cancer cells, thus facilitating cancer progression. Abbreviations: CAFs, cancer‐associated fibroblasts; rCAFs, restraining CAFs; myCAFs, myofibroblasts; iCAFs, inflammatory CAFs; apCAFs, antigen‐presenting CAFs; ECM, extracellular matrix
Pancreatic cancer is known for its abundant stromal components; therefore, researchers prefer to explore the heterogeneity of CAFs in pancreatic cancer. In 2017, Öhlund et al. [4] defined two CAF subpopulations, myCAFs and iCAFs, in pancreatic cancer. myCAFs, which resided closer to cancer cells, had an α‐SMA+ phenotype and synthesized matrix components, while iCAFs, which resided farther from the tumor core, had a secretory phenotype and secreted a series of tumor‐promoting cytokines and chemokines [4]. In 2019, this same team revealed that myCAFs and iCAFs were interchangeable under certain circumstances [48]. Through single‐cell RNA sequencing, this team confirmed the existence of myCAFs and iCAFs [51]. In addition, a class of CAF subsets expressing major histocompatibility complex class II (MHC‐II) and CD74 and activating CD4+ T cells in an antigen‐specific manner were identified and named “antigen‐presenting CAFs” (apCAFs) [51]. Huang et al. [52] further confirmed that apCAFs directly ligated and induced the differentiation of naive CD4+ T cells into regulatory T cells (Tregs) in an antigen‐specific manner. The discovery of apCAFs suggests that the immunomodulatory effect of CAFs may be mediated by specific subgroups.
CAF subpopulations have been studied most frequently in breast cancer. In estrogen receptor‐positive (ER+) breast cancer, CD146+ CAFs helped to maintain ER expression and hormone‐dependent cell proliferation, while CD146‐ CAFs exerted the opposite effects [53]. As a result of this distinction, differences were observed in the sensitivity of ER+ breast cancer to tamoxifen therapy. Similar to a study of pancreatic cancer [51], a study of breast cancer also identified myCAFs, iCAFs, and apCAFs that expressed MHC‐II [54]. Costa et al. [55] grouped CAFs into four subgroups, in which CAF‐subset 1 (CAF‐S1)‐overexpressing FAP promoted the immunosuppressive environment through multistep mechanisms. The team further typed CAF‐S1 using single‐cell RNA sequencing to better understand the effect of CAF‐S1 on immunotherapy response [56]. In these CAF groups, myofibroblasts in clusters 0 and 3, characterized by ECM proteins and TGF‐β signaling, respectively, showed primary resistance to immunotherapy. Mechanistically, myCAFs in cluster 0 induced increased expression of programmed cell death‐1 (PD‐1) and cytotoxic T‐lymphocyte‐associated protein 4 (CTLA4) in Tregs and increased the proportion of TGF‐β‐myCAFs in cluster 3 [56]. This positive feedback loop eventually led to the formation of an immunosuppressive environment.
According to the phenotypic, functional and spatial heterogeneity of CAFs, the precise targeting of subpopulations that exert a major effect on accelerating cancer progression may be a potentially effective and promising approach for targeted cancer therapy. Moreover, different CAF subsets express distinct markers and exhibit different signaling pathway activation states, which contribute to the development of specific drugs and the effective use of precision therapy.
3. SIGNIFICANCE OF TFS IN CAFS
3.1. HIF
HIF is a heterodimeric TF that leads to increased glycolysis and decreased mitochondrial function, reducing oxygen consumption and ultimately enabling the cell to adapt to a hypoxic microenvironment [57, 58]. Many studies have shown that HIF plays an important role in CAF metabolic reprogramming and mediates the protumorigenic effect of CAFs (Figure 3A).
FIGURE 3.
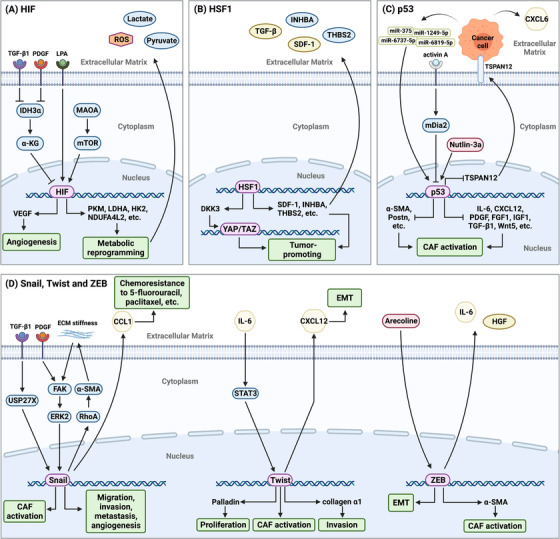
HIF, HSF1, P53, and the EMT‐related TFs Snail, Twist and ZEB in CAFs. (A) HIF in CAFs is activated by the TGF‐β‐ or PDGF‐mediated IDH3α/α‐KG axis [60], MAOA/mTOR axis [61] and LPA [337]. Activated HIF increases the expression of key metabolic enzymes, such as PKM2, LDHA, and HK2 [59], at the transcriptional level to mediate the metabolic reprogramming of CAFs, thus providing the metabolites required for fast‐growing cancer cells and secreting ROS to promote cancer cell migration and invasion [61]. In addition, VEGF is a downstream target of HIF, suggesting that HIF is a key TF regulating tumor angiogenesis. (B) HSF1 activation in CAFs exerts paracrine effects through TGF‐β, SDF1, INHBA and THBS2 and activates related signaling pathways in cancer cells, thus promoting cancer progression [68, 338]. In addition, DKK3, an HSF1 effector, enhances canonical Wnt signaling, resulting in a decrease in YAP/TAZ degradation to subsequently increase ECM remodeling and promote cancer cell growth and invasion [70]. (C) P53 in CAFs are activated by the activin A/mDia2 axis, MDM2 inhibitor Nutlin‐3a, cancer cell‐derived miRNAs, and so on [77, 339]. Activated P53 suppresses the production of a series of growth factors, cytokines and chemokines, resulting in a decrease in the expression of CAF markers and hindering CAF formation. TSPAN12 is highly expressed after P53 activation, and it activates β‐catenin signaling in cancer cells and leads to the secretion of CXCL6, thus promoting tumor invasion [74]. (D) Snail in CAFs is activated by the TGF‐β1/USP27X axis [87], PDGF [86] or ECM stiffness‐mediated FAK/ERK2 axis [85]. Activated Snail feedback regulates ECM stiffness through the RhoA/α‐SMA axis, thus promoting cancer progression [84]. Additionally, Snail+ CAFs can promote the chemoresistance of cancer cells to 5‐fluorouracil and paclitaxel by secreting CCL1 [88]. Twist and ZEB in CAFs are activated under specific conditions and contribute to the regulation of malignant tumor biological behaviors, such as proliferation, invasion, and EMT. Abbreviations: HIF, hypoxia‐inducible factor; HSF1, heat shock transcription factor 1; EMT, epithelial‐to‐mesenchymal transition; TFs, transcription factors; ZEB, Zinc finger E‐box binding homeobox; CAFs, cancer‐associated fibroblasts; TGF‐β, transforming growth factor‐β; PDGF, platelet‐derived growth factor; IDH3α, isocitrate dehydrogenase 3α; α‐KG, α‐ketoglutarate; MAOA, monoamine oxidase A; mTOR, mammalian target of rapamycin; LPA, lysophosphatidic acid; PKM2, pyruvate kinase M2; LDHA, lactate dehydrogenase A; HK2, hexokinase 2; ROS, reactive oxygen species; VEGF, vascular endothelial growth factor; SDF1, stromal‐derived factor 1; INHBA, inhibin subunit beta A; THBS2, thrombospondin‐2; DKK3, Dickkopf‐3; YAP, Yes‐associated protein; TAZ, transcriptional coactivator with PDZ‐binding motif; ECM, extracellular matrix; TSPAN12, Tetraspanin 12; CXCL, C‐X‐C chemokine ligand; USP27X, ubiquitin specific peptidase 27 X‐linked; ERK, extracellular signal regulated kinase; RhoA, Ras homolog family member A; α‐SMA, α‐smooth muscle actin; CCL, C‐C motif chemokine ligand; IL, interleukin; FGF, fibroblast growth factor; IGF, insulin growth factor; STAT, signal transducer and activator of transcription; HGF, hepatocyte growth factor
CAFs show enhanced glycolysis compared with NFs, and cancer cells can meet their metabolic needs by ingesting and processing metabolites secreted by CAFs. Becker et al. [59] found that in breast cancer, hypoxia induced epigenetic reprogramming of HIF‐1α and related glycolytic enzymes, maintaining the metabolic reprogramming phenotype of CAFs. HIF‐1α promoted glycolysis by enhancing glucose uptake, increasing the expression of glycolytic enzymes, and upregulating NDUFA4 mitochondrial complex‐associated like 2 to inhibit oxidative phosphorylation [60]. In addition to providing metabolic fuel for cancer cells, activated HIF‐1α in CAFs might utilize reactive oxygen species (ROSs) to promote the migration and invasion of prostate cancer cells [61]. A recent study revealed that stromal HIF‐1α also affected cancer progression in a hypoxia‐independent manner [62]. The deletion of HIF‐1α in myeloid cells reduced the expression of fibroblast‐activating factors in tumor‐associated macrophages, decreasing the abundance of CAFs and suppressing tumor formation [62].
Interestingly, some recent studies have revealed the regulatory effect of stromal HIF on cancer immunity. In cutaneous squamous cell carcinoma, IL‐17‐induced HIF‐1α transcriptional activation in CAFs driven collagen deposition resistance to anti‐programmed death ligand‐1 (PD‐L1) therapy [63]. In pancreatic cancer, the deletion of CAFs‐HIF‐2α, but not HIF‐1α, moderately reduced tumor fibrosis and significantly reduced intratumoral recruitment of immunosuppressive M2 macrophages and Tregs, thereby improving the tumor response to immunotherapy [64].
3.2. HSF1
HSFs constitute an evolutionarily conserved family of DNA‐binding proteins that regulate gene expression at the transcriptional level to maintain protein stability and reduce cell stress [65, 66]. Various cancers show upregulated levels of activated HSF1, which enables cancer cells to counter imbalances in protein and stress levels in the TME caused by rapid proliferation [67]. However, studies have found that stromal HSF1 plays a broader role in tumor biology, in addition to responding to the aforementioned series of stresses (Figure 3B). Scherz‐Shouval et al. [68] revealed that HSF1 activation in CAFs drove a transcriptional program that promoted malignant phenotypes in cancer cells and a transcriptional process within CAFs that supported malignant cells in mediating cancer progression. In this process, the two central signaling pathways regulated by HSF1, TGF‐β and SDF1, were crucial. In addition, Scherz‐Shouval et al. [68] revealed that the high HSF1 activity in CAFs was associated with a poor prognosis for patients with early‐stage breast or lung cancer. A recent study of oral squamous cell carcinoma (OSCC) confirmed the prognostic role of HSF1 overexpression in CAFs and revealed a series of tumor‐promoting functions mediated by HSF1, such as the induction of the EMT and promotion of cell proliferation, migration and invasion [69]. Dickkopf‐3 (DKK3) is an HSF1 effector that maintains the protumorigenic activity of CAFs through interactions with components of typical signaling pathways. In one mechanistic example, HSF1‐dependent DKK3 upregulation enhanced canonical Wnt signaling, resulting in a decrease in Yes‐associated protein (YAP)/transcriptional coactivator with PDZ‐binding motif (TAZ) degradation and subsequently promoting ECM remodeling and cancer cell growth and invasion [70]. However, the specific mechanisms underlying HSF1 upregulation in CAFs require further study.
3.3. P53
P53 is a TF that is activated by a series of stresses and promotes cell cycle arrest, apoptosis and senescence, thus exerting an antiproliferative effect [71, 72]. P53 is a powerful tumor suppressor, but p53 is also the most frequently mutated gene in cancers. Unsurprisingly, P53 is crucial for the formation and functional maintenance of CAFs (Figure 3C).
Specifically, functional deletion of P53 promotes CAF activation. In prostate cancer, the P53 mutant P53‐N236S significantly increased collagen contraction in fibroblasts by activating STAT3 and led to the overexpression of CAF‐related markers such as α‐SMA [73]. Otomo et al. [74] observed a similar relationship between P53 and α‐SMA; that is, when P53 was knocked down in lung fibroblasts, the expression of α‐SMA increased accordingly. In colorectal cancer (CRC), ROSs produced from P53‐deficient cancer cells induced vascular endothelial growth factor (VEGF) secretion by fibroblasts to regulate angiogenesis and ultimately promote tumor growth [75]. In contrast, a study of pancreatic cancer showed that the activation of P53 induced a series of transcriptional changes that reprogram activated PSCs, driving them into a static state, thereby reducing stromal fibrosis [76]. Exosomal microRNAs (miRNAs) derived from cancer cells can contribute to CAF activation by modulating P53 expression; for example, exosomal miR‐375 from Merkel cell carcinoma cells [77] and exosomal miR‐1249‐5p, miR‐6737‐5p and miR‐6819‐5p from CRC cells exerted this effect [78]. These findings indicate that P53 in CAFs can be regulated by cancer cells, which has been confirmed by many studies [79]. Generally, P53 in CAFs alters transcriptional processes to modulate a series of effects, such as promoting cell migration and invasion [80]. However, a recent study in pancreatic cancer revealed that P53 induced α‐SMA expression through ROS production without functioning as a TF [81]. Therefore, the powerful functions and extensive action mechanisms of P53 in CAFs need to be further explored.
3.4. Snail, Twist and ZEB
The TFs Snail, Twist and ZEB promote the EMT by directly repressing E‐cadherin expression; therefore, these TFs are also known as EMT‐related TFs and are markers for EMT detection [82, 83]. In addition to modulating the EMT, Snail and Twist have been shown to be indispensable in the formation and activation of CAFs, thus promoting tumor development through a wide range of mechanisms (Figure 3D).
Induction by TGF‐β is pivotal for CAFs to activate and obtain the myofibroblast phenotype, and CAFs in which Snail1 has been deleted lack responsiveness to TGF‐β [84]. Through the Snail1/Ras homolog family member A (RhoA)/α‐SMA axis, CAFs expressing Snail1 modulated ECM remodeling, increased ECM hardness and promoted anisotropic fiber orientation to create a TME that supported the directional migration and invasion of cancer cells [84]. In addition, ECM stiffness increased the activity of Rho‐associated coiled‐coil containing protein kinase and indirectly stabilized the Snail1 protein by increasing intracellular tension, integrin aggregation and extracellular signal regulated kinase (ERK) 2 signal transduction [85]. CAFs may be activated continuously by the stably expressed Snail1 protein, maintaining cancer fibrosis and promoting tumor metastasis. Through their ECM remodeling ability, CAFs expressing Snail1 also facilitated tumor angiogenesis in conjunction with PDGFR signaling [86]. In summary, it is obvious that CAFs expressing Snail1 play important roles in the migration and invasion of cancer cells. Interestingly, because of ubiquitin specific peptidase 27 X‐linked (USP27X) deubiquitination, Snail1 was more stable in CAFs and cancer cells than in normal epithelial cells [87]. The abrogation of USP27X repressed Snail1‐dependent CAF activation, reduced cancer metastasis and promoted cellular sensitivity to cisplatin. Li et al. [88] also revealed the role of fibroblasts overexpressing Snail in inducing chemoresistance. In CRC, Snail overexpression induced 3T3 fibroblasts to differentiate into CAFs and reduced tumor sensitivity to 5‐fluorouracil or paclitaxel, which may have been caused by CAF‐derived C‐C motif chemokine ligand (CCL) 1‐induced activation of the TGF‐β/NF‐κB pathway [88].
Twist also activates CAFs and remodels the ECM. NFs stably transfected with Twist1 acquired the characteristics of activated CAFs and increased ECM stiffness. Palladin and collagen α1 were two main mediators of the effect of Twist1 on CAFs: the effect of palladin was closely related to the biomechanical properties and polarity of CAFs, whereas collagen α1 contributed to enhanced migration and invasion [89]. Another study reported that the presence of IL‐6 induced the expression of Twist1 in NFs and drived their transdifferentiation into CAFs by activating STAT3, while C‐X‐C chemokine ligand (CXCL) 12 might be a downstream target of Twist1 [90]. In contrast, in esophageal cancer cells, CXCL12/C‐X‐C chemokine receptor (CXCR) 4 signaling promoted the EMT process through the ERK/AKT‐Twist1‐matrix metallopeptidase (MMP) 1/E‐cadherin axis [91]. Some crosstalk between cancer cells and CAFs in which Twist1 functions may exist.
In contrast to Snail and Twist, ZEB may or may not play a unique role in CAF activation. Chang et al. [92] showed that in the presence of arecoline, the binding between ZEB1 and the α‐SMA promoter increased, which induced myofibroblast transdifferentiation of buccal mucosal fibroblasts. Lobe et al. [93] reported that in vitro, conditioned media from ZEB1‐overexpressing cholangiocarcinoma cells induced myofibroblast proliferation; in vivo, ZEB1‐overexpressing tumor cells formed larger tumors with more abundant stroma. However, the importance and universality of ZEB in CAF activation and maintenance remain to be further explored.
4. SIGNIFICANCE OF SIGNALING PATHWAYS IN CAFS
4.1. Growth factor‐related signaling pathways
4.1.1. TGF‐β
The mature TGF‐β protein is secreted as a latent complex with two copies of latency‐associated polypeptide, which prevents TGF‐β from binding to its receptors [94]. The secretion and deposition of TGF‐β from the latent complex is triggered by an activation mechanism that locally releases activated TGF‐β [95]. After activation, TGF‐β forms a tetramer by interacting with two types of transmembrane kinases (TβRI and TβRII), which phosphorylate serine, threonine and tyrosine residues [96]. In the canonical TGF‐β/Sma‐and Mad‐related protein (SMAD) signaling pathway, TβRI kinase induces the phosphorylation of SMAD2 and 3, while the bone morphogenetic protein (BMP) receptor mediates the phosphorylation of a distinct set of receptor SMADs (R‐SMADs), namely, SMAD1, 5 and 8 [97]. The common SMAD (Co‐SMAD), namely, SMAD4, binds to phosphorylated R‐SMADs to form heteromeric complexes, which are translocated into the nucleus and interact with TFs and co‐regulators to control the expression of target genes [98, 99]. Another group of SMADs, inhibitory SMADs (I‐SMADs), include SMAD6 and 7. I‐SMADs antagonize signal transduction through a variety of mechanisms [100, 101]. In addition to canonical SMAD signaling, TGF‐β activates non‐SMAD signaling pathways, including the PI3K/AKT, ERK and p38/MAPK pathways [102].
TGF‐β plays a vital role in CAF activation and formation and contributes to the maintenance of CAF morphological characteristics and functional phenotypes [103, 104]. In recent years, work has been devoted to elucidating the mechanism by which TGF‐β promotes CAF formation, and a series of interesting results have been presented (Figure 4). During cancer progression, tissue‐resident NFs are gradually transformed into CAFs, and the activation of autocrine signaling pathways mediated by TGF‐β and SDF1 is initiated, which promotes CAF formation via self‐stimulation and cross‐communication [104]. In studies of upstream signaling, the increase in ROS levels induced by long‐term fractionated radiation [105], the loss of glutamine [106], hypoxic conditions [107], exosomes [108], role‐specific miRNAs [109] or integrin [110], or the dysregulation of some molecules, such as diaphanous homolog 1 (DIAPH1) [111], Zinc finger protein 37A (ZNF37A) [112], or liver X receptors α (LXRα) [113], were all shown to lead to the activation of TGF‐β signaling in CAFs from different types of cancers. In addition, non‐coding RNAs (ncRNAs) modulate the signaling crosstalk between CAFs and cancer cells, making a significant contribution to cancer progression (Table 2).
FIGURE 4.
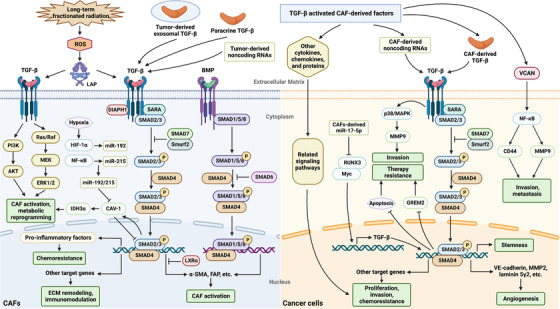
The TGF‐β signaling pathway is involved in the crosstalk between CAFs and cancer cells. TGF‐β signals are transduced in CAFs through canonical and non‐canonical pathways; the former is a SMAD‐dependent pathway mediated by TGF‐β receptors or BMP receptors, while the latter does not require the participation of SMADs. Numerous factors are involved in the activation of TGF‐β signaling in CAFs, including paracrine signaling by the TGF‐β protein in the TME, hypoxic conditions, tumor‐derived exosomes or non‐coding RNAs, increased ROS levels induced by long‐term fractionated radiation, and the dysregulation of molecules such as DIAPH1 and LXRα [104, 105, 106, 107]. Activated TGF‐β signaling in CAFs exerts corresponding biological effects by directly or indirectly modulating the expression of target molecules. For example, TGF‐β signaling upregulates the expression of CAF markers such as α‐SMA and FAP, promoting the activation of CAFs; alters the secretion of proinflammatory factors, driving the acquisition of cell chemoresistance; and modulates a series of other target proteins, mediating ECM remodeling and immunomodulation. Ultimately, TGF‐β signaling in CAFs regulates cancer progression. Furthermore, activated CAFs are among the most important sources of the TGF‐β protein in the TME, and TGF‐β derived from CAFs exerts a pivotal function in initiating TGF‐β signal transduction in cancer cells, which contributes to cancer cell proliferation, stemness maintenance, migration, invasion, tumor angiogenesis, metastasis and the acquisition of chemoresistance. In addition, TGF‐β‐activated CAF‐derived factors, including various cytokines, chemokines, and specific proteins, such as VCAN, regulate cancer progression through various mechanisms. Abbreviations: TGF‐β, transforming growth factor‐β; CAFs, cancer‐associated fibroblasts; SMAD, Sma‐and Mad‐related protein; BMP, bone morphogenetic protein; TME, tumor microenvironment; ROS, reactive oxygen species; DIAPH1, diaphanous homolog 1; LXRα, liver X receptor α; α‐SMA, α‐smooth muscle actin; FAP, fibroblast activation protein; ECM, extracellular matrix; VCAN, versican; LAP: latency‐associated polypeptide; PI3K, phosphoinositide 3‐kinase; HIF, hypoxia‐inducible factor; NF‐κB, nuclear factor kappa‐B
TABLE 2.
ncRNAs involved in signaling crosstalk between CAFs and cancer cells
| ncRNA | Expression level | Cancer type | Biological effects | Signaling cascades | Ref |
|---|---|---|---|---|---|
| The TGF‐β signaling pathway | |||||
| miR‐17‐5p | Upregulated | Colorectal cancer | Metastasis | miR‐17‐5p/RUNX3/Myc/TGF‐β1 | [109] |
| miR‐141 | Downregulated | Breast cancer | Proliferation | TGF‐β1/DNMT3B/miR‐141/TCF12/CXCL12/c‐Myc/Cyclin D1 | [360] |
| miR‐182‐5p | Upregulated | Breast cancer | Metastasis | TGF‐β/miR‐182‐5p/FOXF2 | [361] |
| miR‐192/215 | Upregulated | Head and neck cancer | Metabolism reprogramming | miR‐192/215/Caveolin‐1/TGF‐β/SMAD | [107] |
| miR‐423‐5p | Upregulated | Prostate cancer | Chemoresistance | miR‐423‐5p/TGF‐β/Gremlin 2 | [362] |
| lncRNA CASC9 | Upregulated | Cervical cancer | Metastasis and proliferation | TGF‐β/CASC9/miR‐215/Twist2 | [363] |
| The Hedgehog signaling pathway | |||||
| miR‐10a‐5p | Upregulated | Cervical cancer | Angiogenesis | miR‐10a‐5p/TBX5/Hedgehog | [364] |
| The Notch signaling pathway | |||||
| miR‐200 | Downregulated | Lung cancer | Metastasis | N/A | [204] |
| miR‐221 | Upregulated | Breast cancer | Metastasis and hormone therapy resistance | N/A | [211] |
| The Wnt signaling pathway | |||||
| miR‐34a‐5p | Downregulated | Oral cancer | EMT, proliferation and metastasis | miR‐34a‐5p/AXL/β‐catenin/Snail | [365] |
| miR‐92a‐3p | Upregulated | Colorectal cancer | Stemness, EMT, metastasis and chemoresistance | miR‐92a‐3p/Wnt/β‐catenin/FBXW7 & MOAP1 | [366] |
| miR‐146a | Upregulated | Breast cancer | Invasion and metastasis | miR‐146a/TXNIP/Wnt | [217] |
| miR‐148a | Downregulated | Endometrial cancer | Proliferation and migration | N/A | [367] |
| miR‐148b‐3p | Downregulated | Bladder cancer | Proliferation, metastasis and chemoresistance | miR‐148b‐3p/Wnt/β‐catenin/PTEN | [368] |
| lncRNA CCAL | Upregulated | Colorectal cancer | Chemoresistance | CCAL/HuR/β‐catenin | [369] |
| lncRNA H19 | Upregulated | Colorectal cancer | Stemness and chemoresistance | H19/miR‐141/β‐catenin | [370] |
| The Hippo signaling pathway | |||||
| miR‐92 | Upregulated | Breast cancer | Proliferation, migration and immune suppression | miR‐92/LATS2/YAP1 | [371] |
| The NF‐κB signaling pathway | |||||
| miR‐200b, c | Downregulated | Breast cancer | Proliferation, EMT and invasion | miR‐200b, c/IKKβ/NF‐κB/PAI‐1 | [246] |
| miR‐370‐3p | Upregulated | Breast cancer | Stemness, migration, invasion and EMT | miR‐370‐3p/CYLD/NF‐κB | [372] |
| miR‐630 | Upregulated | Ovarian cancer | Invasion and metastasis | miR‐630/KLF6/NF‐κB | [373] |
| miR‐1247‐3p | Upregulated | Hepatocellular carcinoma | Metastasis | miR‐1247‐3p/B4GALT3/integrin β1/NF‐κB | [249] |
| miR‐6780b | Upregulated | Ovarian cancer | Invasion and metastasis | NF‐κB/miR‐6780b/Dicer | [374] |
| lncRNA TIRY | Upregulated | Oral cancer | Proliferation, invasion and metastasis | TIRY/miR‐14/Wnt/β‐catenin | [375] |
| lncRNA TLR8‐AS1 | Upregulated | Ovarian cancer | Metastasis and chemoresistance | TLR8‐AS1/TLR8/NF‐κB | [376] |
| The JAK/STAT signaling pathway | |||||
| miR‐21 | Upregulated | Hepatocellular carcinoma | CAFs formation | miR‐21/PTEN/PDK1/AKT | [304] |
| miR‐155 | Upregulated | Melanoma | Angiogenesis | miR‐155/SOCS1/JAK2/STAT3 | [377] |
| miR‐210 | Upregulated | Lung cancer | Angiogenesis | N/A | [276] |
| lncRNA NEAT1 | Upregulated | Endometrial cancer | Proliferation and metastasis | NEAT1/miR‐26a/b‐5p/STAT3/YKL‐40 | [378] |
| circ_0088300 | Upregulate | Gastric cancer | Proliferation, migration and invasion | circ_0088300/miR‐1305/JAK/STAT | [379] |
| The PI3K/AKT signaling pathway | |||||
| miR‐21 | Upregulated | Hepatocellular carcinoma | CAFs formation | N/A | [304] |
| miR‐210 | Upregulated | Lung cancer | EMT, migration and invasion | miR‐210/UPF1/PTEN/PI3K/AKT | [380] |
| miR‐590‐3p | Upregulated | Colorectal cancer | Radioresistance | miR‐590‐3p/CLCA4/PI3K/AKT | [381] |
| lncRNA MALAT1 | Upregulated | Gastric cancer | CAFs formation | MALAT1/ELAVL1/PTEN/AKT/mTOR | [382] |
| The MAPK signaling pathway | |||||
| miR‐146a‐5p | Downregulated | Prostate cancer | EMT and metastasis | N/A | [383] |
| miR‐211 | Upregulated | Melanoma | Proliferation and migration | miR‐211/IGF2R/MAPK | [282] |
| miR‐320a | Downregulated | Hepatocellular carcinoma | EMT, proliferation and metastasis | miR‐320a/PBX3/MAPK | [384] |
| miR‐369 | Upregulated | Lung cancer | Migration and invasion | miR‐369/NF1/MAPK | [385] |
Abbreviations: CAFs, cancer‐associated fibroblasts; ncRNAs, non‐coding RNAs; TGF‐β, transforming growth factor‐β; DNMT3B, DNA methyltransferase 3β; TCF12, transcription factor 12; CXCL, C‐X‐C chemokine ligand; FOXF2, Forkhead box F2; SMAD, Sma‐and Mad‐related protein; CASC9, cancer susceptibility 9; TBX5, T‐box transcription factor 5; EMT, epithelial‐to‐mesenchymal transition; FBXW7, F‐box and WD repeat domain containing 7; MOAP1, modulator of apoptosis 1; TXNIP, Thioredoxin interacting protein; PTEN, phosphatase and tensin homolog; LATS2, large tumor suppressor kinase 2; YAP1, Yes‐associated protein 1; NF‐κB, nuclear factor kappa‐B; IKKβ, IκB kinase complex β; PAI‐1, plasminogen activator inhibitor type 1; B4GALT3, beta‐1,4‐galactosyltransferase 3; TLR8, Toll like receptor 8; JAK, Janus kinase; STAT, signal transducer and activator of transcription; PDK1, pyruvate dehydrogenase kinase 1; SOCS1, suppressor of cytokine signaling 1; PI3K, phosphoinositide 3‐kinase; CLCA4, chloride channel accessory 4; ELAVL1, ELAV like RNA binding protein 1; MAPK, mitogen‐activated protein kinase; IGF2R, Insulin like growth factor 2 receptor; NF1, Neurofibromin 1; N/A, not applicable
TGF‐β not only plays an important role in CAF formation but also affects a series of malignant biological behaviors of cancer cells, including proliferation, metabolism, invasion, metastasis, and stemness, by mediating the interplay between CAFs and cancer cells (Figure 4). In stroma‐rich pancreatic cancer, the catabolism induced by the metabolic reprogramming of CAFs and pancreatic cancer cells significantly depended on branched chain α‐ketoacid (BCKA) [114]. The internalization of ECM components caused by the TGF‐β/SMAD5 axis targeting of branched chain amino acid transaminase 1 in CAFs provided the amino acid precursors in CAFs that enable the secretion of BCKA, which exacerbated the cellular demand for BCKA production [114]. In lung cancer, the co‐culture of cancer cells and CAFs induced metabolic reprogramming, in which the glycolytic ability of CAFs was increased and the mitochondrial function of cancer cells was enhanced, and these changes were closely related to changes in TGF‐β signal transduction and ROS levels [115]. In ovarian cancer, TGF‐β upregulated versican expression in CAFs, which activated NF‐κB and accelerated the motility and invasion of cancer cells [116]. Yang et al. [117] showed that in hepatocellular carcinoma (HCC), CAFs promoted the formation of vascular mimicry, which was significantly weakened when TGF‐β or SDF1 signals were abolished. In gastric cancer, more studies have reported the effect of TGF‐β on cancer stemness. According to a recent study, Helicobacter pylori‐activated fibroblasts drove gastric epithelial cells into the differentiation process related to cancer stem cells (CSCs) in a manner partially dependent on TGF‐β signaling, thus promoting tumorigenesis [118]. Co‐culture of gastric cancer cells with CAFs significantly increased the number of spheroid colonies and the expression of CSC markers, and the use of TGF‐β inhibitors reduced these effects, further revealing the role of TGF‐β signaling in the formation and maintenance of stemness mediated by CAFs [119]. Most of the previous evidence showed the tumor‐promoting effect of excessive activation of TGF‐β signaling in CAFs. However, impaired TGF‐β signaling in CAFs has recently been shown to be a possibly important cause of tumor invasion and metastasis, especially in breast cancer and colon cancer. Cancer cells expressing E‐cadherin, MCF‐7 (a breast cancer cell line) and DLD‐1 (a colon cancer cell line) invaded upon integrin α5β1 adhesion to fibronectin fibrils on CAFs, and TGF‐β inhibitors promoted this process by stimulating CAF outgrowth [120]. In breast cancer, TGF‐β signaling in fibroblasts was downregulated by adenosine, which accelerated tumor progression and metastasis via ECM remodeling [121]. In CRC, CAFs secreted collagen and calcium‐binding EGF domain 1 (CCBE1) to promote lymphangiogenesis and VEGFC maturation in cancer cells thus contributing to tumor metastasis, while TGF‐β inhibited CCBE1 transcription [122].
Importantly, TGF‐β signaling in CAFs is also involved in the acquisition of resistance to cancer therapy, including changes in sensitivity to chemotherapeutics, molecular targeting, and immunotherapeutic drugs. These effects have most widely been reported on lung cancer. In non‐small cell lung cancer, treatment with nintedanib significantly repressed adenocarcinoma fibrosis and the its tumor‐promoting effect, but no obvious clinical benefit of nintedanib was observed in squamous cell carcinoma [123]. Further studies revealed that this difference in responsiveness was mediated by differential SMAD3 promoter methylation in CAFs [123]. Li et al. [124] observed that in lung cancer and esophageal cancer, the upregulation of laminin gamma 2 (Ln‐γ2) indicated weakened efficacy of the anti‐PD‐1 antibody, which blocked T cell infiltration by changing the expression of T cell receptors. TGF‐β derived from CAFs activated Ln‐γ2 through a transcriptional mechanism, and the combination of the TGF‐β receptor inhibitor galunisertib and chemotherapeutic drugs enhanced the antitumor activity of anti‐PD‐1 therapy [124]. In addition to lung cancer, a pancancer analysis of ECM gene disorders led to the identification of a group of specific ECM genes, named cancer‐associated ECM (C‐ECM) genes, that are upregulated in cancers, and disruption of the transcriptional program of these genes is associated with TGF‐β signal transduction in CAFs and immunosuppression in other immunoactive cancers, suggesting that C‐ECM genes might predict PD‐1‐blocking failure [125]. A recent study found that TGF‐β suppression induced the remodeling of CAF dynamics and the formation of a new fibroblast population, accompanied by enhanced immune regulation and responsiveness to interferon, which was related to more efficacious PD‐1 immunotherapy [126]. In head and neck cancer, a study found that TGF‐β signaling was activated in cetuximab‐treated CAFs, limiting the efficacy of cetuximab in vitro and in vivo [127]. After blocking the TGF‐β pathway with the SMAD3 inhibitor SIS3, the efficacy of cetuximab was rescued [127]. Bortezomib is an essential drug in multiple myeloma treatment. The presence of CAFs protected plasma cells from bortezomib‐induced apoptosis, which was mediated by TGF‐β [128].
TGF‐β pathway inhibitors have consistently been shown to play a role in limiting cancer progression in most cases, and many attempts to treat cancers have been directed toward TGF‐β pathway activity. In pancreatic cancer, after treatment with the experimental anticancer drug minnelide, TGF‐β signaling in CAFs was dysregulated, which led to a significant reversal of the CAF activation state to a static non‐proliferative state and ultimately resulted in tumor regression [129]. Pei et al. [130] proposed a sequential targeting strategy for the treatment of pancreatic cancer, which first targeted stromal TGF‐β signaling, reversed CAF activation, and weakened the dense matrix barrier to facilitate subsequent drug delivery. Human relaxin‐2 inhibited TGF‐β‐induced PSC differentiation by targeting pSMAD2, thus delaying tumor growth and metastasis [131]. Furthermore, treatment with relaxin‐2 enhanced the effect of gemcitabine after subcutaneous co‐injection (with Panc1 cells and PSCs) on tumor models [131]. In CRC, IL‐1β/TGF‐β1 activated CAF secretion of proinflammatory factors, which changed the chemosensitivity of cancer cells [132]. When neutralizing IL‐1β antibodies were administered with TβRI inhibitors, the non‐canonical TGF‐β pathway mediated by TGF‐β activated kinase 1 (TAK1) was activated, which continued to maintain the tumor‐promoting effect of CAFs. When TAK1 and TβRI inhibitors were applied in combination, the activation of CAFs induced by IL‐1β/TGF‐β1 was blocked, and the chemosensitivity of cancer cells was rescued [132]. Leucine‐rich repeat containing 15 (LRRC15) expression on the stromal fibroblasts of many solid tumors is induced by TGF‐β and has been regarded as a promising target for anti‐stromal therapy [133]. ABBV‐085, a drug targeting LRRC15, is an antibody‐drug conjugate containing monomethyl auristatin E (MMAE), and this conjugate kills cancer cells through MMAE action and increases immune infiltration in the TME [133]. By performing a single‐cell transcriptome analysis, Dominguez et al. [134] clarified that LRRC15+ CAF subsets activated by TGF‐β were associated with adverse reactions to anti‐PD‐L1 therapy, which further implied that LRRC15+ CAFs affected cancer immunity.
In summary, TGF‐β signaling is an essential trigger of CAF activation and formation. TGF‐β signaling in CAFs plays a key role in regulating the malignant biological behavior of cancer cells, thus affecting cancer progression. Furthermore, these effects are bidirectional, which seems to confirm the known dual role of TGF‐β in cancers. Obviously, treatments correctly and efficiently targeting the TGF‐β pathway in CAFs are promising cancer therapies, and increasing efforts are underway to achieve this goal.
4.1.2. FGF
FGFs produce signals by binding to FGF receptors (FGFRs), which exert crucial functions in many diseases [135]. FGFRs are receptor tyrosine kinases (RTKs) that are composed of an extracellular ligand‐binding domain and an intracellular tyrosine kinase domain [136]. In the presence of FGFs, FGFRs activate downstream signaling cascades, such as the PI3K/AKT and Ras/MAPK signaling pathways, to exert their biological effects [137]. Convincing evidence reveals that abnormal FGF signaling contributes to the pathogenesis of many malignancies, and CAFs have been found to be involved in these processes [138]. In gastric cancer, HtrA serine peptidase 1 increased FGF2 expression in cancer cells by activating NF‐κB signaling, which induced the transformation of NFs to CAFs, and α‐SMA was upregulated. However, Akatsu et al. [139] showed that, on the one hand, FGF signaling suppressed the endothelial‐to‐myofibroblast transition induced by TGF‐β, thus attenuating contractile myCAF formation; on the other hand, FGF signaling cooperated with TGF‐β to facilitate CAF formation with migratory and proliferative properties. Bordignon et al. [140] also reported that the function of CAFs was regulated by both FGF and TGF‐β signaling. The activation of FGF signaling mediated by CAFs promoted the proliferation, migration and invasion of cancer cells [141]. For example, in breast cancer, the activation of FGF2/FGFR1 signaling mediated by CAFs under the action of estrogen induced the expression of connective tissue growth factor, leading to the migration and invasion of cancer cells [142]. In addition, FGF signaling has also been reported to be involved in CAF‐mediated cancer therapy resistance. In breast cancer, FGF5 secreted by CAFs activated FGFR2 in cancer cells, eventually resulting in drug resistance to targeted human epidermal growth factor receptor 2 (HER2) therapy through the activation of HER2 by c‐Src [143]. Hormone therapy is an important method of breast cancer treatment; however, the signaling induced by FGF7/FGFR2 has been found to be the basis of CAF‐dependent resistance to tamoxifen [144]. Interestingly, although FGF signaling can result in cancer treatment resistance, it has been shown to enhance the effect of oncolytic virus therapy [145]. CAFs produced FGF2 to initiate signaling cascades in cancer cells, and these cascades reduced the expression of retinoic acid‐inducible gene I and hindered the ability of cancer cells to detect and respond to viruses, enhancing the efficiency of oncolytic viruses [145].
4.1.3. HGF
Met signaling plays crucial roles in tumor survival, growth, angiogenesis and metastasis [146]. As the main cells secreting hepatocyte growth factor (HGF), a vital factor activating Met, CAFs make an important contribution to the activation of Met signaling in cancer cells [147, 148]. As shown by Zhang et al. [149], HGF expression in CAFs was 10‐fold higher than that in NFs, and HGF upregulated CD44 expression through the HGF/Met signaling pathway to promote the adhesion of CRC cells to endothelial cells, thus facilitating tumor metastasis. Another study showed that CAF‐derived HGF potentiated the proliferation, migration and invasion of gastric cancer cells by activating the HGF/Met/STAT3/Twist1 pathway [150]. In addition, the CAF‐mediated HGF signaling pathway plays a major role in cancer treatment resistance. In HCC, HGF secreted by CAFs activated the Met and MEK/ERK1/2 pathways and elevated CD73 expression in cancer cells, inducing resistance to sorafenib and cisplatin [151]. In lung cancer, by secreting HGF and insulin‐like growth factor‐1 (IGF‐1) to activate their corresponding receptors Met and IGF‐1 receptor, CAFs increased the expression and phosphorylation of Annexin A2, which was a key molecule mediating CAF‐induced EMT and gefitinib resistance [152]. Upon long‐term treatment with tyrosine kinase inhibitors (TKIs), cancer cells addicted to epidermal growth factor receptor (EGFR) or Met activation showed a metabolic transition to increased glycolysis and lactate production [153]. These increased lactate levels triggered CAFs to secrete HGF, leading to Met‐dependent signal transduction in cells that maintain their resistance to TKIs. Interestingly, in head and neck squamous cell carcinoma, HGF secreted by CAFs increased extracellular lactate levels in tumors by enhancing glycolysis [154].
4.1.4. PDGF
The PDGF/PDGFR signaling pathway has a variety of functions, including modulating angiogenesis, promoting cancer cell autocrine growth, and regulating tumor stromal fibroblasts [155, 156]. The PDGF/PDGFR signaling pathway is an essential regulator of CAF formation and recruitment in the TME and is crucial in CAF‐mediated ECM remodeling and tumor migration, invasion and metastasis [157, 158]. In breast cancer and CRC, blocking PDGFR significantly suppressed the recruitment of MSCs to tumor tissues and their transformation to CAFs, thus slowing cancer progression [159, 160]. In breast cancer, paracrine PDGF‐CC signaling increased the expression of HGF, IGF binging protein 3 and Stanniocalcin 1 in CAFs, resulting in a malignant cell phenotype that lacked ERα and other luminal markers [161]. Targeting PDGF‐CC could restore cellular sensitivity to hormone therapy in ERα‐negative breast cancers. Cholangiocarcinoma cells have also been documented to stimulate fibroblast migration by secreting PDGF‐DD to activate Rho GTPase and JNK signaling [162]. In cholangiocarcinoma, CAFs stimulated by PDGF‐DD induced lymphatic endothelial cell recruitment and three‐dimensional assembly, increased lymphatic endothelial cell permeability and ultimately accelerated the transendothelial migration of cancer cells [163]. Erdogan et al. [164] revealed that CAFs facilitated the directional migration of prostate and pancreatic cancer cells by rearranging the fibronectin matrix through contraction and traction forces mediated by non‐muscle myosin II and PDGFRα. Lung adenocarcinoma cells undergoing the EMT showed significantly increased levels of secreted PDGF‐BB, which enhanced CAF‐mediated ECM remodeling and promoted tumor invasion [165].
4.1.5. EGF
The EGF/EGFR signaling pathway has been widely studied in cancers. Recent studies have shown that CAFs are involved in regulating cancer progression mediated by the EGF/EGFR pathway [166]. In high‐grade serous ovarian cancer, CAFs recruited ascitic cancer cells with high integrin α5 expression to form metastatic units and accelerate tumor metastasis by secreting EGF to maintain integrin α5 expression in ascitic cancer cells [167]. In head and neck cancer, pharmacological inhibition of EGFR reduced CAF‐induced anchorage‐independent growth and tumor spheroid formation, indicating that EGFR was important for the maintenance of the CSC phenotype [168]. In addition, Magan et al. [169] showed that upon co‐culture with CAFs, head and neck cancer cells showed increased proliferation, and increased EGFR expression and an enhanced therapeutic response to cetuximab were observed. However, another study of CRC revealed that after cetuximab treatment, CAFs increased EGF secretion and rendered neighboring cancer cells resistant to cetuximab through the continuous activation of MAPK signaling [170]. Clearly, the relationship between CAFs and cancer treatment responses is controversial, and further research is needed.
4.2. The Hedgehog signaling pathway
The Hh signaling pathway is evolutionarily conserved and is related to embryonic development, normal tissue repair, the EMT, stem cell maintenance, and other processes [171, 172]. The Hh signaling pathway is mainly composed of three secretory ligands (Shh, Indian hedgehog, and Desert hedgehog), the negative regulatory receptor Patched (PTCH), the positive regulatory receptor Smoothened (SMO), and the TF GLI [173]. In the presence of Hh ligands, PTCH relieves the inhibition of SMO, and activated SMO initiates a signaling cascade that results in the activation and nuclear localization of GLI [174]. Activation and nuclear localization of GLI drive the transcription and expression of Hh target genes, such as cyclin D, c‐myc and BCL2 [175]. In addition to the canonical pathway that is mediated by the PTCH/SMO/GLI axis, non‐canonical pathways affect GLI activity and play a role in the Hh signaling pathway through non‐SMO‐dependent pathways, such as the TGF‐β signaling pathway and MAPK signaling pathway [176].
CAFs not only are potential sources of Hh ligands in the TME but also respond to Hh signaling through GLI1 activation. GLI1 resided only in the nucleus of CAFs but not in NFs, revealing the activation of Hh signaling in CAFs [177]. Interestingly, compared with that in iCAFs, Hh signaling was differentially enhanced in myCAFs. Blocking Hh signaling reduced the number of myCAFs and increased the number of iCAFs, which was related to a decrease in cytotoxic T cells and Treg expansion, revealing the ability of Hh signaling to regulate immune cell infiltration [178]. As an essential signaling pathway in stem cells, the Hh signaling pathway in CAFs makes a significant contribution to the regulation of CSCs, especially in breast cancer. In mouse models of triple‐negative breast cancer (TNBC), Hh ligands produced by cancer cells reprogramed CAFs to provide a TME supportive of CSC acquisition of chemoresistant phenotypes through the expression of FGF5 and the production of fibrillar collagen [179]. Interestingly, CSCs in breast cancer could secrete Shh as a paracrine messenger to activate Hh signaling in CAFs, which produced factors that subsequently accelerate CSC expansion and self‐renewal [180]. In addition to influencing CSCs, Hh signaling in CAFs affects the development of malignant biological behaviors. In gastric cancer, Galectin‐1 upregulated GLI1 expression by binding to a carbohydrate structure in integrin β1, activating Hh signaling and inducing the EMT, migration and invasion of cancer cells [181]. Besides, the activation of Hh signaling increased Forkhead box F1 expression in CAFs, modulating the contractility of these fibroblasts and the production of HGF and FGF2 to stimulate lung cancer cell migration [182]. Studying cholangiocarcinoma, Razumilava et al. [183] reported the effect of non‐canonical Hh signaling on tumor progression and metastasis. GLI needs to be located in cilia before it can be activated, but cilia are not formed by malignant cholangiocarcinoma cell lines. Genetic inhibition of Hh signaling in BDE (ΔLoop2) cells or pharmacological inhibition with vismodegib, a small‐molecule antagonist of SMO, suppressed tumorigenesis and metastasis, indicating a new mechanism for mediating Hh signal transduction in cholangiocarcinoma [183].
As research on Hh signaling inhibitors is relatively mature, many researchers have focused on exploring the role of Hh signaling inhibitors in cancers to identify effective treatments. Compared with that in NFs, SMO expression is upregulated in CAFs. CAFs expressing SMO transduce Shh signals to activate GLI1, while siRNA‐induced knockdown of SMO blocks the induction of GLI1 [184]. Therefore, SMO overexpression may be one of the mechanisms activating Hh signaling in CAFs. Shh could facilitate PSC proliferation and potentiate GLI1 expression, which was abrogated by the SMO inhibitor AZD8542 [185]. In pancreatic cancer, AZD8542 inhibited tumor growth only in the presence of PSCs, suggesting a matrix‐dependent paracrine signaling mechanism, which was subsequently confirmed in prostate and colon cancer models [185]. A large amount of tumor stroma is produced in pancreatic cancer, and it hinders the delivery of chemotherapeutic drugs, but the use of the Shh inhibitor cyclopamine help eliminates stroma‐producing CAFs, thus contributing to the effective delivery of drugs [186]. A recent study revealed that a PTCH‐1‐interacting peptide inhibited the production of ECM and TGF‐β by CAFs and induced cancer cells to express human leukocyte antigen‐ABC and lymphocytes to express interferon‐γ, thereby inhibiting tumor fibrosis and enhancing immune cell infiltration [187]. In breast cancer, the use of the SMO inhibitor vismodegib improved the efficacy of Abraxane and Doxil [188]. Mechanistically, vismodegib therapy normalized the TME and improved vascular function by inhibiting CAF activation to reduce the levels of collagen and hyaluronan. However, blocking Hh signaling may not necessarily suppress cancer growth [189]. In pancreatic cancer, the loss of two Hh coreceptors, growth arrest specific 1 (GAS1) and brother of CDON (BOC), in fibroblasts reduced the reactivity of Hh but accelerated tumor growth in vivo because of an increase in tumor‐associated vascularity [190]. In contrast, the loss of all three coreceptors, GAS1, BOC and cell adhesion associated, oncogene regulated (CDON), resulted in the inhibition of tumorigenesis and angiogenesis. In a Kras‐mutated (G12D) mouse model, ablation of the SMO gene in CAFs led to pancreatic acinar‐ductal metaplasia, which promoted the initiation of pancreatic cancer [191]. In recent years, studies have gradually revealed that, in contrast to the previously described tumor‐promoting effect, the effect of Hh signaling on CAFs also has a tumor‐suppressing function, which may explain the failure of Hh signaling inhibitors [192]. Rhim et al. [193] examined a pancreatic cancer mouse model in which Shh had been knocked out and observed a reduced number of α‐SMA+ myofibroblasts, resulting in increased angiogenesis, cell proliferation and invasion and decreased tumor differentiation. In CRC, the activation of stromal‐specific Hh signaling inhibited advanced cancer progression by regulating BMP signaling and suppressing the colonic stem cell signature [194]. Therefore, more in‐depth studies are needed to determine methods to correctly adjust Hh signaling in CAFs and attenuate cancer progression.
4.3. The Notch signaling pathway
Notch signaling influences numerous cancer biological processes [195], and different cancers and cancer subtypes express different Notch receptors and ligands, which play different roles [196]. Therefore, Notch is considered both a cancer promoter and suppressor. The microenvironment determines whether Notch signaling promotes or suppresses cancer progression [197]. The Notch pathway includes five typical Notch ligands [Jagged 1 (JAG1), JAG2, Delta‐like 1 (DLL1), DLL3 and DLL4] and four Notch receptors (Notch1‐4) [198]. Upon ligand binding, a series of cleavages of Notch receptors are induced to form the Notch intracellular domain (NICD) [199]. Then, NICD is translocated to the nucleus and interacts with CBF1/suppressor of hairless/LAG1 (CSL). NICD binding promotes the recruitment of the coactivation complex to CSL, which eventually leads to the transcriptional activation of CSL response elements [200]. In addition to this classical Notch signaling pathway, non‐canonical Notch signaling is initiated by non‐canonical ligands, in the absence of ligands, or does not require CSL [200, 201].
CSL suppresses transcription when Notch signaling is not activated, and therefore deletion of CSL or the downregulation of its components results in CAF activation since the expression of multiple CAF determinant genes are directly modulated by CSL [202]. The dysregulation of Notch signaling in CAFs affects the proliferation, migration, invasion and angiogenesis of cancer cells and ultimately affects tumor growth and metastasis [203, 204]. The downregulation of CSL in CAFs seems to be closely related to autophagy. Upon autophagy induction, the CSL protein level was usually decreased in the tumor stroma, but the mRNA level was not affected, and studies have shown that endogenous CSL was associated with autophagy and the signaling adaptor p62/Sequestosome 1 [205]. Interestingly, CSL silencing in CAFs induced autophagy by upregulating Unc‐51 like kinase 3 expression [206]. The relationship between autophagy and CSL in CAFs is complex, and it suggests a new possible direction for stromal therapy that remains to be further explored. In addition to autophagy, the DNA damage/repair process is significantly related to CSL in CAFs. CSL expression was negatively regulated by stress/DNA damage induced by ultraviolet radiation A, ROSs, and other factors [207]. P53 is the key effector in the DNA damage response (DDR), which attenuated CSL gene transcription by suppressing CSL promoter activity. In addition, surprisingly, Bottoni et al. [208] showed that, independent of its role in regulating transcription, CSL was part of a multiprotein telomere protective complex required for telomere association and that CSL downregulation in CAFs triggered DNA damage, telomere loss and chromosome fusion. A recent study reported heterogeneous amplification and overexpression of the Notch1 gene in CAFs of skin squamous cell carcinoma. Notch1 overexpression led to the continuous expression of CAF effector genes, which might be caused by the blockade of the DDR and the inhibition of ATM‐forkhead box O3a binding and the downstream signaling cascade [209].
As one of the important regulatory pathways in stem cells, the Notch signaling that is dysregulated by CAFs affects the biological functions of CSCs. In HCC, CAF‐induced Notch3 expression led to lysine‐specific demethylase 1 deacetylation and activation, thus maintaining the self‐renewal and tumorigenicity of CSCs [210]. In breast cancer, exosomal miR‐221 secreted by CAFs decreased ER expression and upregulated Notch3 expression in recipient cancer cells, which induced the production of CD133‐overexpressing CSCs and ultimately promoted hormone therapy resistance [211]. Additionally, DLL1+ breast cancer cells recruited CAFs and promoted Wnt ligand secretion by Notch2/3‐expressing CAFs, resulting in increased Wnt/β‐catenin‐dependent DLL1+ CSC functions to promote metastasis and radioresistance [212].
In summary, unlike other signaling pathways, the Notch pathway activation in CAFs appears to preferentially alter autophagy and the DNA damage/repair process and thus contributes to cancer progression, which suggests novel applications and prospects for targeting the Notch signaling.
4.4. The Wnt signaling pathway
The Wnt signaling pathway regulates organ development and regeneration and stem cell differentiation, which is pivotal for malignancy initiation and progression [213]. The main components of the canonical Wnt pathway include secreted Wnt ligands, the transmembrane receptor Frizzled (Fzd), coreceptor LDL receptor related protein (LRP) 5/6, Dishevelled (Dvl), adenomatous polyposis coli (APC), glycogen synthase kinase 3β (GSK3β), Axin, β‐catenin, and the TFs TCF/LEF [214]. In the absence of Wnt ligand stimulation, Axin, APC and GSK3β form a destructive complex that binds to β‐catenin and phosphorylates it, and β‐catenin is then degraded upon ubiquitination [215]. After Wnts bind to Fzd and its coreceptor LRP5/6, the intracellular protein Dvl is activated, thus inhibiting the degradation activity of the destructive complex and stabilizing the β‐catenin protein in the cytoplasm. Upon accumulation in the cytoplasm, stable β‐catenin enters the nucleus and binds to LEF/TCF, which initiates the transcription of target genes such as c‐myc and cyclin D1 [213]. Furthermore, β‐catenin binds to a variety of TFs in addition to TCF/LEF, such as Forkhead box TFs, SRY‐box TFs, and SMAD, thereby regulating numerous downstream biological processes [216].
Wnt signaling is activated in NFs by various factors and may lead to NF transformation into CAFs [217]. For example, Wnt4 expression was significantly increased in the serum of patients with CRC, and it activated CAFs and induced the EMT by activating β‐catenin [218]. Interestingly, distinct Wnt activities in CAFs also induced the formation of different CAF subtypes: the iCAF subtype was induced by low levels of Wnt, while the myCAF subtype was induced by high levels of Wnt [219]. Transdifferentiated adipocytes are sources of CAFs, and Wnt pathway activation facilitated this process. In breast cancer, activation of the Wnt/β‐catenin pathway caused by Wnt3a secreted by cancer cells resulted in increased secretion of fibronectin and type I collagen and increased expression of the CAF marker FSP1 in adipocytes [37]. In addition, Wnt ligands might promote CAF activation through a non‐canonical Wnt signaling pathway. Avgustinova et al. [220] revealed that Wnt7a, a key factor secreted exclusively by invasive breast cancer cells, induced CAF transformation. However, the activation of canonical Wnt signaling was not detected in this process, while TGF‐β signaling was observed to be enhanced and led to ECM remodeling, which facilitated cancer cell migration and invasion.
The ultimate goal of Wnt‐activated CAFs is tumor promotion; therefore, Wnt signaling in CAFs is innately involved in regulating malignant biological behaviors. CAF‐derived Wnt2 plays a critical role in CRC progression. Aizawa et al. [221] performed immunohistochemical staining of 171 samples from patients with CRC and found that Wnt2 expression in CAFs was significantly associated with the lymph node metastasis (TNM) stage, venous invasion and recurrence. Wnt2 inhibition in CAFs significantly reduced the invasion and migration of CRC cells. The ability of Wnt to enhance the invasiveness of CRC cells might be mediated by its regulation of CAF motility and ECM remodeling [222]. Another study reported that proteins related to angiogenic function, including Angiopoietin‐2, IL‐6, granulocyte colony stimulating factor and placental growth factor, were upregulated by Wnt2 and that Wnt2 knockout in CAFs significantly attenuated angiogenesis [223]. In CRC xenografts, Wnt2 overexpression led to an increase in vessel density and tumor volume. In addition, Wnt2 secreted by CAFs repressed the dendritic cell (DC)‐mediated antitumor T cell response through the suppressor of cytokine signaling 3 (SOCS3)/p‐JAK2/p‐STAT3 axis, while targeting Wnt2 restored antitumor immunity and enhanced anti‐PD‐1 efficacy by increasing the number of active DCs [224]. As shown in Table 2, exosomal ncRNAs derived from CAFs also play an essential role in Wnt signal transduction.
Abnormalities in Wnt signaling are an important driving factor for CRC progression. CAF‐secreted factors, including HGF, enhanced Wnt signaling in CRC cells and induced them to develop CSC phenotypes, thereby restoring the clonogenic ability or tumorigenicity of more differentiated tumor cells with relatively low Wnt signaling activity [225]. As previously stated, Wnt signaling activation in CAFs also exerts a decisive effect on CRC progression, which further reveals the importance of targeting the Wnt pathway in CRC.
4.5. The Hippo signaling pathway
The Hippo signaling pathway, which consists of a group of conserved kinases, suppresses cell growth and controls the size and volume of organs in various species [226]. Increasing evidence has confirmed that Hippo signaling plays critical roles in carcinogenesis, tissue regeneration and immune modulation [226, 227]. Briefly, after receiving a growth inhibition signal, the Hippo pathway modulates the downstream effector YAP/TAZ through a series of kinase phosphorylation events [228]. TEA domain (TEAD) family TFs are the best representatives of the TFs regulated by YAP/TAZ: TEAD TFs transcribe target genes upon activation by YAP/TAZ and promote cell proliferation and survival signaling [229]. Most of the biological functions mediated by YAP/TAZ depend on the interaction of YAP/TAZ and TEADs, including a protumorigenic phenotype acquired upon dysregulation of the Hippo pathway [227].
YAP/TEAD protein complexes affect downstream cytoskeletal proteins by regulating Src transcription, thus transforming NFs into CAFs, which accelerates the proliferation and invasion of epithelial cells and ultimately promotes tumor growth and metastasis [230]. YAP regulated the expression of various cytoskeletal regulators, such as Anillin and Diaphanous related formin 3, thereby contributing to ECM remodeling and stiffening. Interestingly, YAP activation itself was modulated by ECM stiffening, and actomyosin contractility and Src function were required for this effect [231]. Clearly, a feedforward self‐enhancement loop helped maintain the CAF phenotype. Cerebral cavernous malformations 3 (CCM3) located at CAF adhesion sites competed with focal adhesion kinase (FAK) to bind to paxillin and fine‐tuned mechanotransduction and YAP/TAZ activation mediated by the FAK/Src/paxillin axis [232]. The inactivation of CCM3 led to the intensification of tissue remodeling and force transmission to the matrix, resulting in the mutual activation of YAP/TAZ in adjacent cancer cells and ultimately inducing tumor metastasis [232]. Bertero et al. [233] found that ECM stiffening activated glycolysis and glutamine metabolism, thus coordinating the flow of non‐essential amino acids in the tumor niche. The YAP/TAZ‐dependent mechanical transduction pathway influenced the metabolic crosstalk between CAFs and cancer cells and regulated the metabolic reprogramming of cancer cells to support cancer progression [233]. In CAFs, YAP was located in the cytoplasm, while in activated CAFs, it was located in the nucleus and induced the expression of genes needed for tumor promotion. The YAP cytoplasmic‐nuclear shuttle was affected by cell shape changes, YAP nuclear export, and the functions of actin, Src family kinases and XPO1 [234]. In addition, in the absence of ECM stiffness stimuli, Spin90 deficiency led to the recruitment of mDia2 and APC complexes to microtubules, resulting in increased microtubule acetylation, which facilitated the nuclear localization of YAP and ultimately mediated CAF activation [235]. Activated CAFs act on cancer cells by secreting exosomes to modulate YAP activity, which reveals the importance of the YAP pathway in CAFs. For instance, CAF‐derived exosomal Annexin A6 stabilized integrin β1 in cancer cells, leading to ECM network formation and chemoresistance through the integrin β1/FAK/YAP axis [236]. Some typical signaling pathways have been shown to synergize with the YAP/TAZ pathway, and in CAFs, the most common of which is the Wnt/β‐catenin signaling pathway. Liu et al. [237] identified the interaction between YAP and β‐catenin in CAFs by performing coimmunoprecipitation and proximity ligation assays. Wnt/β‐catenin activation in CAFs facilitated the nuclear translocation of YAP, and this nuclear translocation was suppressed when Wnt/β‐catenin signaling was blocked [237]. Ferrari et al. [70] further revealed that DKK3 modulated the synergism between Wnt/β‐catenin signaling and YAP/TAZ signaling.
4.6. The NF‐κB signaling pathway
The NF‐κB family of TFs is involved in a variety of biological processes, including inflammation, immune response, and regulation of cellular functions [238]. These biological processes associate closely with cancer initiation and progression; therefore, NF‐κB signaling is not only an essential regulatory pathway in inflammation and immunity but also a crucial factor in malignancy. The NF‐κB family consists of five members, RelA (p65), RelB, cRel, NF‐κB1 (p50) and NF‐κB2 (p52), which exert their physiological functions by forming dimers [239]. In the canonical pathway, NF‐κB activation is initiated by the binding of ligands (such as TNF‐α, IL‐1, and lipopolysaccharide) to their respective receptors (such as the TNF, IL‐1 and Toll‐like receptors) [240]. The non‐canonical NF‐κB pathway is activated by a small number of cytokines from the TNF family, such as B cell activating factor of the tumor necrosis factor family and CD40 ligand, to induce the synthesis of NF‐κB‐inducing kinases [241, 242]. The typical targets of canonical NF‐κB signaling include genes encoding cytokines, chemokines, growth factors, and CXCLs, which exert crucial functions in controlling innate immunity and inflammation, while non‐canonical pathways contribute to the development of secondary lymphoid organs and the maturation of B lymphocytes [243, 244].
CAFs in skin, breast and pancreatic cancers present a proinflammatory gene signature that depends on the activation of NF‐κB signaling [245]. CAFs with a proinflammatory gene signature have the ability to promote macrophage recruitment, angiogenesis and tumor growth, which are attenuated when NF‐κB signaling is suppressed. NF‐κB signaling is vital for the acquisition and maintenance of tumor‐promoting functions in CAFs, as evidenced by dysregulated NF‐κB signaling inducing iCAF formation and CAF secretion of factors such as cytokines, chemokines, and growth factors that regulate tumor growth (Table 3) [246, 247]. For instance, lymphotoxin in ovarian cancer cells induced chemokine expression in CAFs mediated by the Lymphotoxin beta receptor (LTBR)/NF‐κB axis, through which CXCL11 enhanced the proliferation and migration of cancer cells [248]. In HCC, the increase in serum exosomal miR‐1247‐3p levels was related to lung metastasis. Fang et al. [249] found that tumor‐derived miR‐1247‐3p targeted B4GALT3, activating integrin β1/NF‐κB signaling in CAFs. Subsequently, CAFs facilitated cancer progression by secreting proinflammatory cytokines, including IL‐6 and IL‐8. Similarly, integrin β like 1‐enriched exosomes derived from CRC cells stimulated TNF‐α induced protein 3‐mediated NF‐κB signaling to sensitize CAFs, which then produced large amounts of proinflammatory cytokines to enhance metastatic tumor growth [250].
TABLE 3.
Cytokines and chemokines involved in signaling crosstalk between CAFs and cancer cells
| Cytokine or chemokine | Cancer type | Biological effects | Signaling cascades | Ref |
|---|---|---|---|---|
| The Notch signaling pathway | ||||
| IL‐8 | Ovarian cancer | Stemness and chemoresistance | N/A | [386] |
| The Wnt signaling pathway | ||||
| CXCL12 | Ovarian cancer | EMT and chemoresistance | CXCL12/CXCR4/Wnt/β‐catenin | [387] |
| IL‐1β | Bladder cancer | Proliferation and invasion | N/A | [388] |
| Ovarian cancer | Immune suppression | PS1/Wnt/β‐catenin/IL‐1β | [389] | |
| The NF‐κB signaling pathway | ||||
| CCL5 | Lung cancer | Proliferation | HIF‐1α/NF‐κB/CCL5 | [390] |
| CXCL11 | Ovarian cancer | Proliferation and migration | LTBR/NF‐κB/CXCL11 | [248] |
| IL‐1β | Oral cancer | Proliferation, migration and invasion | IL‐1β/NF‐κB/CCL22/CCR4 | [391] |
| IL‐8 | Breast cancer | Invasion and EMT | IL‐8/NF‐κB/S100A8 | [392] |
| Nasopharyngeal carcinoma | Radioresistance | N/A | [253] | |
| Gastric cancer | Chemoresistance | N/A | [252] | |
| The JAK/STAT signaling pathway | ||||
| IL‐6 | Esophageal squamous cell carcinoma | Chemoresistance | N/A | [270] |
| Gallbladder cancer | Angiogenesis | IL‐6/JAK/STAT3/NOX4 | [264] | |
| Breast cancer | Proliferation | IL‐6/STAT3/AUF1/ATR | [393] | |
| Colorectal cancer | Proliferation | IL‐6/STAT3/Periostin | [394] | |
| Hepatocellular carcinoma | EMT | IL‐6/IL6R/STAT3/TG2 | [395] | |
| Hepatocellular carcinoma | Immune suppression | IL6/STAT3/PD‐L1 | [267] | |
| IL‐17a | Gastric cancer | Migration and invasion | N/A | [271] |
| The PI3K/AKT signaling pathway | ||||
| CCL26 | Pancreatic cancer | Migration | N/A | [396] |
| CXCL5 | Colorectal cancer | Immune suppression | CXCL5/CXCR2/PI3K/AKT/PD‐L1 | [314] |
| The MAPK signaling pathway | ||||
| CCL20 | Pancreatic cancer | Immune suppression | N/A | [292] |
| CXCL1 | Esophageal squamous cell carcinoma | Radioresistance | N/A | [397] |
| CXCL6 | Hepatocellular carcinoma | Stemness | CXCL6/ERK1/2/CLCF1 | [14] |
| IL32 | Breast cancer | EMT and invasion | IL32/integrin β3/p38 MAPK | [289] |
| IL‐33 | Gastric cancer | EMT, migration and invasion | IL‐33/ST2L/ERK1/2/SP1/ZEB2 | [398] |
Abbreviations: CAFs, cancer‐associated fibroblasts; IL, interleukin; CXCL, C‐X‐C chemokine ligand; EMT, epithelial‐to‐mesenchymal transition; CXCR, C‐X‐C chemokine receptor; PS1, Presenilin 1; NF‐κB, nuclear factor kappa‐B; CCL, C‐C motif chemokine ligand; HIF, hypoxia‐inducible factor; LTBR, Lymphotoxin beta receptor; CCR, C‐C motif chemokine receptor; JAK, Janus kinase; STAT, signal transducers and activators of transcription; NOX4, NADPH oxidase 4; TG2, Transglutaminase 2; PD‐L1, programmed death ligand‐1; PI3K, phosphoinositide 3‐kinase; MAPK, mitogen‐activated protein kinase; ERK, extracellular signal regulated kinase; CLCF1, Cardiotrophin like cytokine factor 1; ZEB, Zinc finger E‐box binding homeobox; N/A, not applicable
Importantly, NF‐κB signaling in CAFs is of unprecedented importance in cancer treatment resistance. Various factors leading to NF‐κB pathway activation in CAFs exhibit the potential to promote drug resistance. Su et al. [251] identified and defined a new subpopulation of CAFs, CD10+GPR77+ CAFs, which was significantly associated with chemoresistance and poorer survival of patients with breast or lung cancer. CD10+GPR77+ CAFs were driven by the continuous activation of NF‐κB through p65 phosphorylation and acetylation, which were maintained by complement signaling through the C5a receptor GPR77. CD10+GPR77+ CAFs induced CSC enrichment by secreting IL‐6 and IL‐8, providing a survival niche for CSCs and ultimately promoting cancer initiation and chemoresistance [251]. Similarly, IL‐8 was expressed at high levels in patients with gastric cancer presenting chemoresistance. CAF‐derived IL‐8 potentiated the chemoresistance of gastric cancer by activating NF‐κB signaling [252]. Interestingly, in nasopharyngeal carcinoma, CAFs reduced radiation‐induced DNA damage through the IL‐8/NF‐κB axis, thus inducing the acquisition of radiation resistance [253]. In addition, the NF‐κB pathway affects the metabolic characteristics of CAFs, leading to an increase in aerobic glycolysis and autophagy, thus providing support for cancer cell growth and invasion and contributing to drug resistance [254]. Lactate, a product of glycolysis, induced CAFs to secrete HGF in an NF‐κB‐dependent manner, and HGF activated Met‐dependent signal transduction in cancer cells and maintained their resistance to TKIs [153]. Lactate in cancer cells also mediated brain derived neurotrophic factor production by CAFs in an NF‐κB‐dependent manner, activating TrkB/Nrf2 signaling in cancer cells to reduce their sensitivity to anlotinib [255]. These findings confirm the relationship between NF‐κB signaling, metabolism and drug resistance.
In general, the NF‐κB pathway in CAFs modulates cancer progression and promotes treatment resistance by regulating inflammatory factors in the TME and a series of chemokines and cytokines. Approaches targeting NF‐κB signaling itself or downstream chemokines and cytokines may contribute to the treatment of cancers with inflammatory characteristics.
4.7. The JAK/STAT signaling pathway
The JAK/STAT signaling pathway is mainly stimulated and activated by cytokines [256], all of which have corresponding receptors on the membrane. These receptors themselves have no kinase activity but contain an intracellular binding site for JAKs [257]. In general, cytokines and growth factors bind to tyrosine kinase‐related receptors and subsequently activate receptor‐bound JAKs, resulting in autophosphorylation of JAKs and receptor tyrosine kinase residues [258]. The phosphorylation site on the receptor binds to the SH2 site of STATs, and receptor‐bound STATs are phosphorylated by JAK to form dimers, which translocate into the nucleus and act as activated TFs to regulate the expression of target genes [259, 260]. Different combinations of JAKs [JAK1, JAK2, JAK3 and tyrosine kinase 2 (TYK2)] and STATs (STAT1, 2, 3, 4, 5A, 5B and 6) are activated by different ligands and receptors with a high degree of specificity, thereby regulating basic biological processes such as cell proliferation, differentiation, apoptosis and immunomodulation [261, 262].
In the TME, the IL‐6/JAK/STAT3 signaling pathway drives cancer cell proliferation, invasion and metastasis and considerably suppresses the antitumor immune response [263], and CAFs play crucial roles in these processes. As the main sources of secreted IL‐6, CAFs play a role in regulating cancer progression by secreting IL‐6 to activate STAT3 in cancer cells (Table 3). For example, IL‐6 secreted by CAFs accelerated the growth and angiogenesis of gallbladder cancer by activating the JAK/STAT3 signaling pathway in cancer cells and upregulating the expression of NOX4, a key gene involved in vasculogenic mimicry [264]. Importantly, CAFs affect cancer immunity through this pathway [265]. By activating STAT3, IL‐6 in CAFs induced the differentiation and formation of immunosuppressive T cells called CD73+ γδ Tregs, the infiltration of which weakened the tumor‐killing function of CD8+ T cells and was significantly related to a poor prognosis for patients [266]. Interestingly, CD73+ γδ Tregs could stimulate CAFs to secrete IL‐6 through the adenosine/adenosine A2b receptor/p38MAPK signaling pathway, thus forming an IL‐6/adenosine positive feedback loop [266]. In HCC, CAFs modulated the survival, activation and function of neutrophils in tumor tissues through the IL‐6/STAT3/PD‐L1 axis [267]. CAFs are also involved in the differentiation of myeloid‐derived suppressor cells (MDSCs) [268]. CAFs recruited monocytes through the SDF1a/CXCR4 pathway and induced them to differentiate into MDSCs, which impaired T cell proliferation and changed the T cell phenotype and function in an IL‐6/STAT3‐dependent manner [269]. In esophageal squamous cell carcinoma, IL‐6 and exosomal miR‐21 secreted from CAFs synergistically promoted MDSC production by activating STAT3, resulting in the resistance of tumor cells to cisplatin [270]. In addition to secreting IL‐6, CAFs alter STAT3 activity in cancer cells through a variety of other mechanisms to exert their biological effects [132]. For instance, CAFs secreted IL‐17a to promote the migration and invasion of gastric cancer cells by activating the JAK2/STAT3 signaling pathway [271]. IL‐11 secreted by CAFs enhanced the migration and invasion of gastric cancer cells by activating the JAK/STAT3 and MAPK/ERK pathways [272].
On the other hand, activation of the JAK/STAT pathway in NFs induces the activation and formation of CAFs, thus modulating cancer progression [273, 274]. PSCs secreted the autocrine leukemia inhibitory factor (LIF) and activated the JAK/STAT signaling pathway upon stimulation with IL‐1, thus accelerating iCAF formation [48]. Albrengues et al. [275] showed that under the action of LIF, NFs were reprogrammed into CAFs with a preinvasive phenotype, thus promoting ECM remodeling and cancer cell invasion. Mechanistically, LIF activated an epigenetic switch that led to the structural activation of JAK1/STAT3 signaling and resulted in the continuous preinvasion activity of CAFs [275]. In addition, exosomes derived from cancer cells can induce the transformation of NFs into CAFs and regulate the function of CAFs by activating the JAK/STAT pathway in CAFs. For example, exosomal miR‐210 derived from lung cancer cells increased the expression of angiogenic factors such as MMP9, FGF2 and VEGFA in CAFs by activating the JAK2/STAT3 signaling pathway, which ultimately promoted angiogenesis [276].
4.8. The MAPK signaling pathway
MAPKs are normally expressed in cells where they convert extracellular signals such as those from growth factors and stress stimuli into intracellular responses, contributing to embryonic development, tissue homeostasis and inflammation [277, 278]. Four main branches of the MAPK signaling pathway have been identified: the ERK, JNK, p38/MAPK and ERK5 branches [279]. Among these pathways, the JNK and p38/MAPK pathways have similar functions related to inflammation, cell apoptosis and proliferation, while ERK mainly regulates cell growth and differentiation [280]. The major driver of MAPK signaling pathway activation is the phosphorylation cascade involving MAPK kinase kinase (MAPKKK), MAPK kinase (MAPKK), and MAPK, while ERK activation is altered by the canonical Ras/Raf/MEK/ERK axis [281].
ERK activation in fibroblasts results in their transformation into CAFs and contributes to tumor growth [282, 283]. For example, when melanosomal miR‐211 was transferred to NFs, it directly targeted IGF2 receptor and activates MAPK signaling, which drove NFs to acquire CAF phenotypic characteristics, including increased proliferation, migration and proinflammatory gene expression, and ultimately promoting the growth of melanoma [282]. In fact, CAFs preferentially exert their tumor‐promoting functions by regulating the MAPK signaling pathway in cancer cells. Ligorio et al. [284] combined single‐cell RNA and protein analyses to study the role of CAFs in regulating heterogeneity in pancreatic cancer and found that a significant single‐cell population shifted toward the invasion‐related EMT and proliferative phenotypes with the activation of MAPK and STAT3 signaling. In neuroblastoma, CAFs promoted the proliferation, survival and chemoresistance of neuroblastoma cells in vitro through a mechanism depending on the co‐activation of JAK2/STAT3 and MEK/ERK signaling in cancer cells [285]. Exosomal ncRNAs also mediate the effect of CAFs on MAPK signaling in cancer cells (Table 2).
The JNK/p38 MAPK signaling pathway plays a pivotal role in the formation and activation of CAFs. In lung cancer, p38‐dependent fibroblast‐specific hyaluronan synthesis regulated the activation of fibroblasts, thus accelerating cancer cell proliferation [286]. P38 activation maintained the expression of tumor‐promoting factors by CAFs, and this process depended on AUF1 binding and stabilized the mRNAs encoding related factors to exert posttranscriptional regulatory effects [287]. However, Hong et al. [288] revealed that in breast cancer, p38 signaling suppressed MSC migration to the primary tumor and metastatic site, inhibited the transformation of MSCs into CAFs, and ultimately repressed tumor metastasis. Therefore, p38 signaling may exert distinct effects on the formation and activation of CAFs under different conditions, and further research is needed to clarify the mechanism. Similar to the ERK pathway, the p38 MAPK pathway mediates the interaction between CAFs and cancer cells to influence cancer progression [289]. In ovarian cancer, CAFs stimulated glycogen mobilization in cancer cells, thus promoting glycolysis, and this process depended on p38α MAPK activation in CAFs [290]. P38α MAPK in CAFs affected tumor growth and metastasis by altering glycogen metabolism because glycogen was one of the energy sources in cancer cells, promoting metastatic foci growth. Another study showed that under hypoxic conditions, lactate in CAFs mediated the metabolic coupling between CAFs and breast cancer cells to enhance the mitochondrial activity of cancer cells by activating the TGF‐β1/p38 MAPK/MMP2/9 axis, thus promoting the invasion of cancer cells [291]. In addition, JNK signaling activation in CAFs suppressed CCL20 secretion, resulting in a reduction in CD8+ T cell infiltration and subsequently affecting antitumor immunity [292]. In summary, the JNK/p38 MAPK signaling pathway modulates CAF activation and the crosstalk between activated CAFs and cancer cells through various mechanisms. Strategies targeting this pathway are important to normalize the TME and treat malignant diseases.
The MAPK signaling pathway usually cooperates with the AKT signaling pathway [293, 294]. For example, in gastric cancer, tumor‐educated neutrophils activated the AKT and p38 pathways in MSCs by secreting IL‐17, IL‐23 and TNF‐α, which induced MSC transformation into CAFs to facilitate tumor growth and metastasis [295]. In lung cancer, vascular cell adhesion molecule‐1 secreted by CAFs activated the AKT and MAPK signaling pathways in cancer cells by binding to integrin α4β1, promoting the growth and invasion of cancer cells [296]. In pancreatic cancer, the combination of MEK inhibitors and STAT3 inhibitors alleviated stromal inflammation and enriched CAF phenotypes with mesenchymal stem cell‐like properties to overcome immunotherapy resistance [297]. The combined use of MEK inhibitors (e.g., trametinib), STAT3 inhibitors (e.g., ruxolitinib) and PD‐1 inhibitor (e.g., nivolumab) to treat a patient with chemotherapy‐refractory metastatic pancreatic cancer has yielded clinical benefits. Therefore, the effect of crosstalk with other signaling pathways should also be considered in efforts to target MAPK signaling.
4.9. The PI3K/AKT signaling pathway
The PI3K/AKT signaling pathway is the most commonly activated pathway in cancers and promotes the growth, survival, and, particularly, the metabolism of cancer cells [298]. The PI3K/AKT pathway is usually triggered by the activation of membrane receptors such as RTKs or G protein‐coupled receptors [299]. Activated PI3K on the plasma membrane stimulates the phosphorylation of phosphatidylinositol 4,5‐bisphosphate (PIP2) to produce phosphatidylinositol 3,4,5‐trisphosphate (PIP3) [300]. As a second messenger, PIP3 recruits AKT to the plasma membrane, where it is fully activated upon phosphorylation by the action of phosphoinositide‐dependent protein kinase 1 (PDK1) and mTOR complex 2 (mTORC2) [301, 302]. Activated AKT phosphorylates tuberous sclerosis protein 1 (TSC1) and TSC2 to dissociate the TSC1/TSC2 complex, negatively regulating mTOR activity. Eventually, AKT leads to the activation of mTORC1, which increases protein and lipid synthesis and decreases autophagy, thus supporting cell growth and proliferation [300]. In addition to the classical TSC1/TSC2/mTOR axis, AKT regulates a series of other molecules, such as GSK3, Forkhead box TFs and IκB kinase complex (IKK), to participate in a wide range of functions [303].
The activation of the AKT signaling pathway stimulates CAF formation and infiltration in the TME. Numerous upstream factors, such as exosomal miR‐21 derived from HCC cells [304] and soluble carcinoembryonic antigen released from CRC cells [305], sensitized the AKT pathway to activate CAFs and contribute to cancer progression. Activation of AKT signaling in CAFs affects cancer progression in many ways. Yamamura et al. [306] reported that Girdin, an AKT substrate, was activated by AKT in CAFs. When Girdin activity was inhibited, CAF infiltration and tumor growth were significantly decreased. In OSCC, integrin β2 was expressed at high levels in CAFs and enhanced the glycolytic activity of CAFs by mechanically regulating the PI3K/AKT/mTOR pathway [307]. Subsequently, lactate secreted by CAFs was metabolized by cancer cells to produce nicotinamide adenine dinucleotide, which supported cell proliferation. In lung cancer, the activation of the AKT/mTORC1 signaling pathway increased MDM2 translation in CAFs, thereby accelerating cell invasion [308]. In breast cancer, C3a/C3aR signaling promoted metastatic cytokine secretion and ECM generation by CAFs by activating the PI3K/AKT pathway, ultimately facilitating tumor metastasis [309]. Zhang et al. [310] utilized CUDC‐907 to target the PI3K/AKT pathway in CAFs, successfully suppressed cancer progression and observed a decrease in the expression of CAF markers. These outcomes confirm the importance of the PI3K/AKT pathway in CAFs for cancer progression and emphasize the potential of therapies targeting this pathway.
More commonly, CAFs modulate cancer progression by altering the activity of the PI3K/AKT pathway in cancer cells. In CRC cells, blocking the PI3K/AKT pathway reversed the accelerated progression caused by co‐culture with CAFs [311]. Thrombospondin 4 (TSP‐4) secreted by CAFs bound to integrin α2 on gallbladder cancer cells to induce AKT‐dependent phosphorylation of HSF1, thus maintaining the malignant phenotype of these cancer cells, including their proliferation, EMT and stemness [312]. Interestingly, activated HSF1 signaling increased TGF‐β1 expression to enhance CAF activation and recruitment and elevated TSP‐4 expression in CAFs, forming a positive feedback loop. Chemoresistance has also been shown to be related to the activation of the PI3K/AKT pathway in cancer cells mediated by CAFs. Li et al. [313] revealed that CAFs regulated microtubule‐directed chemoresistance in breast cancer by secreting collagen to activate the integrin β1/PI3K/AKT signaling pathway. In addition, CAF‐derived CXCL5 promoted PD‐L1 expression in cancer cells by activating the PI3K/AKT signaling pathway, thus forming an immunosuppressive microenvironment [314]. In summary, CAF‐mediated activation of the PI3K/AKT pathway in cancer cells regulates cancer progression through multiple mechanisms, and therapies targeting the PI3K/AKT pathway show promise for application.
5. SIGNALING PATHWAYS IN CAFS AS POTENTIAL THERAPEUTIC TARGETS IN THE CLINIC
As mentioned above, TGF‐β is crucial for the activation, formation, and phenotypic maintenance of CAFs. Numerous preclinical models have shown the therapeutic benefit of targeting TGF‐β, which has prompted a series of clinical trials (Table 4). In general, the new approach involves incorporating TGF‐β inhibitors into tried‐and‐true treatment regimens in an effort to improve efficacy. A phase Ib/II clinical trial reported that the combination of galunisertib, a TGF‐β receptor I kinase inhibitor, and gemcitabine prolonged the overall survival (OS) of patients with unresectable pancreatic cancer (NCT01373164) [315]. In patients with advanced HCC, galunisertib combined with sorafenib showed an acceptable safety profile and prolonged OS (NCT01246986) [316]. For patients with advanced HCC who were not eligible to receive sorafenib, galunisertib monotherapy had a controllable safety profile, and longer survival was related to lower baseline alpha fetoprotein (AFP) and response in AFP or TGF‐β1 levels (NCT01246986) [317]. For patients with locally advanced rectal cancer, the addition of galunisertib to neoadjuvant chemotherapy improved the complete response rate to 32% with good tolerability (NCT02688712) [318]. Additionally, studies examining TGF‐β inhibitors in combination with immunotherapy are ongoing. The phase I study initiated by Melisi et al. (NCT02734160) [319] evaluated the safety and activity of galunisertib combined with the anti‐PD‐L1 antibody durvalumab in metastatic pancreatic cancer, but further randomized trials have not been launched. The safety of SHR‐1701, a bifunctional fusion protein targeting PD‐L1 and TGF‐β, has been certified in a phase I study for recurrent or metastatic cervical cancer and has shown encouraging antitumor activity (NCT03774979) [320]. M7824 is also a protein that simultaneously targets PD‐L1 and TGF‐β. The results of NCT02517398 showed that M7824 had a manageable safety profile and showed encouraging efficacy in patients with heavily pretreated advanced solid tumors [321]. A series of phase II clinical trials for SHR‐1701 or M7824 are ongoing (e.g., NCT04624217 and NCT05300269).
TABLE 4.
Summary of completed clinical trials targeting the TGF‐β pathway, Hedgehog pathway, and CXCL12/CXCR4 axis in CAFs
| Target | Agent | Combination therapy | Cancer type | Phase | Outcome | Clinical Trial Identifier |
|---|---|---|---|---|---|---|
| TGF‐β pathway | Galunisertib | Gemcitabine | Unresectable pancreatic cancer | Ib/II | Galunisertib‐gemcitabine combination prolonged OS, with minimal added toxicity | NCT01373164 |
| Galunisertib | Sorafenib | Advanced hepatocellular carcinoma | II | Galunisertib‐sorafenib combination showed acceptable safety and prolonged OS | NCT01246986 | |
| Galunisertib | Neoadjuvant chemotherapy | Locally advanced rectal cancer | II | Galunisertib‐ neoadjuvant chemotherapy combination improved the complete response rate to 32%, with acceptable toxicity | NCT02688712 | |
| Galunisertib | Durvalumab | Metastatic pancreatic cancer | Ib | Galunisertib‐durvalumab combination was tolerable | NCT02734160 | |
| SHR‐1701 | N/A | Metastatic cervical cancer | I | SHR‐1701 exhibited encouraging antitumor activity and controllable safety | NCT03774979 | |
| M7824 | N/A | Advanced solid tumors | I | M7824 exhibits encouraging antitumor activity and controllable safety | NCT02517398 | |
| Hedgehog pathway | IPI‐926 | Gemcitabine | Metastatic pancreatic cancer | II | The use of IPI‐926 reduced the OS | NCT01130142 |
| Vismodegib | Gemcitabine | Metastatic pancreatic cancer | Ib/II | Vismodegib‐gemcitabine combination did not prolong OS | NCT01064622 | |
| Vismodegib | Gemcitabine plus nab‐paclitaxel | Metastatic pancreatic cancer | II | Vismodegib‐chemotherapy combination did not prolong OS | NCT01088815 | |
| Sonidegib | Docetaxel | Triple‐negative advanced breast cancer | Ib | Sonidegib‐docetaxel combination showed antitumor activity in 3 (of 10) patients | NCT02027376 | |
| CXCL12/CXCR4 axis | BL‐8040 | Pembrolizumab and chemotherapy | Metastatic pancreatic cancer | IIa | BL‐8040‐pembrolizumab combination may expand the benefit of chemotherapy | NCT02826486 |
| Plerixafor | Bortezomib | Relapsed/refractory multiple myeloma | I/II | Plerixafor‐bortezomib combination improved the objective response rate, with acceptable toxicity | NCT00903968 | |
| Balixafotide | Eribulin | HER2‐negative metastatic breast cancer | I | combination exhibited Balixafotide‐eribulin encouraging antitumor activity and controllable safety | NCT01837095 |
Abbreviations: TGF‐β, transforming growth factor‐β; OS, overall survival; CXCL12, C‐X‐C chemokine ligand 12; CXCR4, C‐X‐C chemokine receptor 4; CAFs, cancer‐associated fibroblasts; HER2, human epidermal growth factor receptor 2; N/A, not applicable
Stroma remodeling and deposition caused by the activation of the Hh pathway in CAFs is one of the crucial factors leading to chemotherapy resistance. Olive et al. [322] reported that the application of IPI‐926, an SMO inhibitor, considerably enhanced the therapeutic effect of chemotherapy on a mouse model of pancreatic cancer by increasing the intratumoral concentration of gemcitabine. Unfortunately, the phase II clinical trial of IPI‐926 combined with gemcitabine reported the opposite results; namely, the use of IPI‐926 reduced the OS of patients with metastatic pancreatic cancer (NCT01130142). This disappointing result also led to the early termination of the phase I study of IPI‐926 plus FOLFIRINOX for advanced pancreatic adenocarcinoma (NCT01383538) [323]. The combination of vismodegib, another Hh pathway inhibitor, with gemcitabine or gemcitabine plus nab‐paclitaxel to treat metastatic pancreatic cancer also produced negative results (NCT01064622 and NCT01088815) [324, 325]. In TNBC, Hh‐dependent CAF activation and ECM remodeling promote the formation of a CSC‐supportive niche, leading to docetaxel resistance [179]. Based on the good effect on the preclinical model, a phase Ib study of the SMO inhibitor sonidegib combined with docetaxel was launched and showed antitumor activity in 3 (of 10) patients with advanced TNBC (NCT02027376) [326]. Another clinical trial applying the SMO inhibitor vismodegib combined with neoadjuvant chemotherapy to treat patients with TNBC is ongoing (NCT02694224). In the previous section, we have described the immunomodulatory effect of activated Hh signaling in CAFs, and clinical trials of Hh pathway inhibitors combined with immunotherapy are also ongoing (NCT04007744 and NCT04827953).
Activated CAFs are key regulators of the stromal CXCL12/CXCR4 axis, which is closely related to immune cell infiltration in the TME. A preclinical study of pancreatic cancer revealed that FAP+ CAFs produced CXCL12, which prevented cancer cells from being detected and eliminated by T cells. The combination of the CXCR4 inhibitor plerixafor and anti‐PD‐L1 treatment might improve the efficacy of immunotherapy [327]. Garg et al. [328] performed pancreatic injections of a combination of pancreatic cancer cells and PSCs in a mouse model, documenting that PSCs secreted CXCL12 in an NF‐κB‐dependent manner, which decreased the infiltration of cytotoxic T cells and increased tumor growth. This effect was confirmed in human samples and was abolished by plerixafor [328, 329]. Based on the support from preclinical results, clinical trials focusing on the CXCL12/CXCR4 axis monotherapy or in combination with immunotherapy are ongoing (e.g., NCT04177810, NCT02907099, NCT03168139, NCT02826486, NCT 02179970, and NCT00903968). NCT02826486, a phase IIa study evaluating the efficacy and safety of the CXCR4 antagonist BL‐8040 combined with pembrolizumab and chemotherapy in patients with metastatic pancreatic cancer, reported that BL‐8040 increased the tumor infiltration of CD8+ T cells and reduced the numbers of MDSCs and circulating Tregs, which might expand the benefit of chemotherapy [330]. NCT02179970, a phase I study designed to evaluate the safety of the continued use of plerixafor by patients with advanced pancreatic or CRC has been completed. The continuous administration of plerixafor increased the intratumoral accumulation of CD8+ T cells and natural killer cells to induce an integrated immune response [331]. The results from NCT00903968 also showed the safety and effectiveness of using plerixafor in combination with bortezomib in patients with relapsed/refractory multiple myeloma [332]. The safety of balixafortide, another CXCR4 antagonist, in combination with eribulin in the treatment of HER2‐negative metastatic breast cancer has also been confirmed, and its potential to improve outcomes has been documented (NCT01837095) [333].
Since the treatments that target CAF markers, such as FAP, do not directly act on the key signaling pathways we introduced and have already been extensively summarized [334], they will not be discussed further in this review. In addition, clinical trials of IL and STAT inhibitors are also widely ongoing. However, in contrast to the TGF‐β pathway, Hh pathway, and CXCL12/CXCR4 axis, researchers have not clearly determined to what extent the therapeutic effects of IL and STAT inhibitors are related to CAFs. Notably, due to its particularity, evaluations of the efficacy of treatments targeting the stroma are mainly based on preclinical models. Accurate assessment of how medications affect the TME in clinical studies is still challenging.
6. CONCLUSIONS
The dysregulation of specific signaling pathways in cancer cells is one of the key contributors to cancer initiation and progression, and this dysregulation may be induced by various circumstances, including the actions of CAFs. Through paracrine signaling, cell‐cell interactions and exosome release, CAFs trigger the necessary upstream signals to activate or repress specific pathways in cancer cells, thus regulating a variety of malignant biological behaviors. Importantly, the function of CAFs in regulating cancer initiation and progression is modulated in many respects by the dysregulation of signaling pathways, most of which are triggered by cancer cells, thus forming a feedback loop between the tumor center and the stroma that regulates cancer progression. Although numerous studies described in the present review have conducted substantial work on the mechanisms by which CAFs are involved in cancer signal transduction, research on the complex regulatory networks in the TME formed by multiple pathways is still lacking. This lack of information is evident when applying treatments targeting tumor‐stromal signaling pathways. The failure of targeted therapy may result from the activation of potential compensatory signaling when a specific pathway is blocked in CAFs. The treatment effectiveness may be considerably increased when the interplay of multiple pathways is recognized to apply combined or sequential targeted therapy. Despite the challenges, strategies designed to target stromal signaling pathways have shown considerable promise, regardless of whether the therapy is focused on single or multiple targets or administered in conjunction with other therapies, such as chemotherapy and immunotherapy. The development of efficient treatments will be aided by obtaining a thorough understanding of the signaling pathways in CAFs and the regulatory role of the TME in cancer progression.
DECLARATIONS
AUTHOR CONTRIBUTIONS
ZLF, QCM and JX collected the related studies and drafted the manuscript. WW, BZ, JL, CL, JH and YJZ participated in the design of the review. SS and XJY initiated the study and revised the manuscript. All authors have read and agreed on the published version of the manuscript.
CONFLICT OF INTEREST
The authors declare that they have no competing interests.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable.
CONSENT FOR PUBLICATION
Not applicable.
ACKNOWLEDGMENTS
We thank American Journal Experts (AJE) for their assistance with language editing, and all figures were created with BioRender.com.
Fang Z, Meng Q, Xu J, Wang W, Zhang B, Liu J, et al. Signaling pathways in cancer‐associated fibroblasts: recent advances and future perspectives. Cancer Commun. 2023;43:3–41. 10.1002/cac2.12392
Zengli Fang, Qingcai Meng, and Jin Xu These authors contributed equally to this work and share first authorship.
Contributor Information
Xianjun Yu, Email: yuxianjun@fudanpci.org.
Si Shi, Email: shisi@fudanpci.org.
DATA AVAILABILITY STATEMENT
Not applicable.
REFERENCES
- 1. Junttila MR, de Sauvage FJ. Influence of tumour micro‐environment heterogeneity on therapeutic response. Nature. 2013;501:346–54. [DOI] [PubMed] [Google Scholar]
- 2. Hinshaw DC, Shevde LA. The Tumor Microenvironment Innately Modulates Cancer Progression. Cancer Res. 2019;79:4557–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sahai E, Astsaturov I, Cukierman E, DeNardo DG, Egeblad M, Evans RM, et al. A framework for advancing our understanding of cancer‐associated fibroblasts. Nat Rev Cancer. 2020;20:174–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Öhlund D, Handly‐Santana A, Biffi G, Elyada E, Almeida AS, Ponz‐Sarvise M, et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med. 2017;214:579–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ishii G, Ochiai A, Neri S. Phenotypic and functional heterogeneity of cancer‐associated fibroblast within the tumor microenvironment. Adv Drug Deliv Rev. 2016;99:186–96. [DOI] [PubMed] [Google Scholar]
- 6. Chen X, Song E. Turning foes to friends: targeting cancer‐associated fibroblasts. Nat Rev Drug Discov. 2019;18:99–115. [DOI] [PubMed] [Google Scholar]
- 7. Kalluri R. The biology and function of fibroblasts in cancer. Nat Rev Cancer. 2016;16:582–98. [DOI] [PubMed] [Google Scholar]
- 8. Bu L, Baba H, Yoshida N, Miyake K, Yasuda T, Uchihara T, et al. Biological heterogeneity and versatility of cancer‐associated fibroblasts in the tumor microenvironment. Oncogene. 2019;38:4887–901. [DOI] [PubMed] [Google Scholar]
- 9. Boyd LNC, Andini KD, Peters GJ, Kazemier G, Giovannetti E. Heterogeneity and plasticity of cancer‐associated fibroblasts in the pancreatic tumor microenvironment. Semin Cancer Biol. 2021. [DOI] [PubMed] [Google Scholar]
- 10. Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med. 2004;10:789–99. [DOI] [PubMed] [Google Scholar]
- 11. Martin GS. Cell signaling and cancer. Cancer Cell. 2003;4:167–74. [DOI] [PubMed] [Google Scholar]
- 12. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. [DOI] [PubMed] [Google Scholar]
- 13. Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. 2012;21:309–22. [DOI] [PubMed] [Google Scholar]
- 14. Song M, He J, Pan QZ, Yang J, Zhao J, Zhang YJ, et al. Cancer‐Associated Fibroblast‐Mediated Cellular Crosstalk Supports Hepatocellular Carcinoma Progression. Hepatology. 2021;73:1717–35. [DOI] [PubMed] [Google Scholar]
- 15. Hawinkels LJ, Paauwe M, Verspaget HW, Wiercinska E, van der Zon JM, van der Ploeg K, et al. Interaction with colon cancer cells hyperactivates TGF‐β signaling in cancer‐associated fibroblasts. Oncogene. 2014;33:97–107. [DOI] [PubMed] [Google Scholar]
- 16. Dvorak HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med. 1986;315:1650–9. [DOI] [PubMed] [Google Scholar]
- 17. Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6:392–401. [DOI] [PubMed] [Google Scholar]
- 18. Nurmik M, Ullmann P, Rodriguez F, Haan S, Letellier E. In search of definitions: Cancer‐associated fibroblasts and their markers. Int J Cancer. 2020;146:895–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kobayashi H, Enomoto A, Woods SL, Burt AD, Takahashi M, Worthley DL. Cancer‐associated fibroblasts in gastrointestinal cancer. Nat Rev Gastroenterol Hepatol. 2019;16:282–95. [DOI] [PubMed] [Google Scholar]
- 20. Pallangyo CK, Ziegler PK, Greten FR. IKKβ acts as a tumor suppressor in cancer‐associated fibroblasts during intestinal tumorigenesis. J Exp Med. 2015;212:2253–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Özdemir BC, Pentcheva‐Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR, et al. Depletion of carcinoma‐associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell. 2014;25:719–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Arina A, Idel C, Hyjek EM, Alegre ML, Wang Y, Bindokas VP, et al. Tumor‐associated fibroblasts predominantly come from local and not circulating precursors. Proc Natl Acad Sci U S A. 2016;113:7551–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhang W, Wang H, Sun M, Deng X, Wu X, Ma Y, et al. CXCL5/CXCR2 axis in tumor microenvironment as potential diagnostic biomarker and therapeutic target. Cancer Commun (Lond). 2020;40:69–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fang Z, Xu J, Zhang B, Wang W, Liu J, Liang C, et al. The promising role of noncoding RNAs in cancer‐associated fibroblasts: an overview of current status and future perspectives. J Hematol Oncol. 2020;13:154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tan HX, Gong WZ, Zhou K, Xiao ZG, Hou FT, Huang T, et al. CXCR4/TGF‐β1 mediated hepatic stellate cells differentiation into carcinoma‐associated fibroblasts and promoted liver metastasis of colon cancer. Cancer Biol Ther. 2020;21:258–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Omary MB, Lugea A, Lowe AW, Pandol SJ. The pancreatic stellate cell: a star on the rise in pancreatic diseases. J Clin Invest. 2007;117:50–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yin C, Evason KJ, Asahina K, Stainier DY. Hepatic stellate cells in liver development, regeneration, and cancer. J Clin Invest. 2013;123:1902–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Quante M, Tu SP, Tomita H, Gonda T, Wang SS, Takashi S, et al. Bone marrow‐derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell. 2011;19:257–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Weber CE, Kothari AN, Wai PY, Li NY, Driver J, Zapf MA, et al. Osteopontin mediates an MZF1‐TGF‐β1‐dependent transformation of mesenchymal stem cells into cancer‐associated fibroblasts in breast cancer. Oncogene. 2015;34:4821–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ko SY, Barengo N, Ladanyi A, Lee JS, Marini F, Lengyel E, et al. HOXA9 promotes ovarian cancer growth by stimulating cancer‐associated fibroblasts. J Clin Invest. 2012;122:3603–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Barcellos‐de‐Souza P, Comito G, Pons‐Segura C, Taddei ML, Gori V, Becherucci V, et al. Mesenchymal Stem Cells are Recruited and Activated into Carcinoma‐Associated Fibroblasts by Prostate Cancer Microenvironment‐Derived TGF‐β1. Stem Cells. 2016; 34:2536–47. [DOI] [PubMed] [Google Scholar]
- 32. Shangguan L, Ti X, Krause U, Hai B, Zhao Y, Yang Z, et al. Inhibition of TGF‐β/Smad signaling by BAMBI blocks differentiation of human mesenchymal stem cells to carcinoma‐associated fibroblasts and abolishes their protumor effects. Stem Cells. 2012;30:2810–9. [DOI] [PubMed] [Google Scholar]
- 33. Rhim AD, Mirek ET, Aiello NM, Maitra A, Bailey JM, McAllister F, et al. EMT and dissemination precede pancreatic tumor formation. Cell. 2012;148:349–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fischer KR, Durrans A, Lee S, Sheng J, Li F, Wong ST, et al. Epithelial‐to‐mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature. 2015;527:472–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yeon JH, Jeong HE, Seo H, Cho S, Kim K, Na D, et al. Cancer‐derived exosomes trigger endothelial to mesenchymal transition followed by the induction of cancer‐associated fibroblasts. Acta Biomater. 2018;76:146–53. [DOI] [PubMed] [Google Scholar]
- 36. Zeisberg EM, Potenta S, Xie L, Zeisberg M, Kalluri R. Discovery of endothelial to mesenchymal transition as a source for carcinoma‐associated fibroblasts. Cancer Res. 2007;67:10123–8. [DOI] [PubMed] [Google Scholar]
- 37. Bochet L, Lehuédé C, Dauvillier S, Wang YY, Dirat B, Laurent V, et al. Adipocyte‐derived fibroblasts promote tumor progression and contribute to the desmoplastic reaction in breast cancer. Cancer Res. 2013;73:5657–68. [DOI] [PubMed] [Google Scholar]
- 38. Jotzu C, Alt E, Welte G, Li J, Hennessy BT, Devarajan E, et al. Adipose tissue derived stem cells differentiate into carcinoma‐associated fibroblast‐like cells under the influence of tumor derived factors. Cell Oncol (Dordr). 2011;34:55–67. [DOI] [PubMed] [Google Scholar]
- 39. Tang H, Chu Y, Huang Z, Cai J, Wang Z. The metastatic phenotype shift toward myofibroblast of adipose‐derived mesenchymal stem cells promotes ovarian cancer progression. Carcinogenesis. 2020;41:182–93. [DOI] [PubMed] [Google Scholar]
- 40. Ning X, Zhang H, Wang C, Song X. Exosomes Released by Gastric Cancer Cells Induce Transition of Pericytes Into Cancer‐Associated Fibroblasts. Med Sci Monit. 2018;24:2350–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hosaka K, Yang Y, Seki T, Fischer C, Dubey O, Fredlund E, et al. Pericyte‐fibroblast transition promotes tumor growth and metastasis. Proc Natl Acad Sci U S A. 2016;113:E5618–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rinkevich Y, Mori T, Sahoo D, Xu PX, Bermingham JR, Jr., Weissman IL. Identification and prospective isolation of a mesothelial precursor lineage giving rise to smooth muscle cells and fibroblasts for mammalian internal organs, and their vasculature. Nat Cell Biol. 2012;14:1251–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Alcolea MP, Jones PH. Tracking cells in their native habitat: lineage tracing in epithelial neoplasia. Nat Rev Cancer. 2013;13:161–71. [DOI] [PubMed] [Google Scholar]
- 44. LeBleu VS, Taduri G, O'Connell J, Teng Y, Cooke VG, Woda C, et al. Origin and function of myofibroblasts in kidney fibrosis. Nat Med. 2013;19:1047–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Liu T, Han C, Wang S, Fang P, Ma Z, Xu L, et al. Cancer‐associated fibroblasts: an emerging target of anti‐cancer immunotherapy. J Hematol Oncol. 2019;12:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Öhlund D, Elyada E, Tuveson D. Fibroblast heterogeneity in the cancer wound. J Exp Med. 2014;211:1503–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ping Q, Yan R, Cheng X, Wang W, Zhong Y, Hou Z, et al. Cancer‐associated fibroblasts: overview, progress, challenges, and directions. Cancer Gene Ther. 2021. [DOI] [PubMed] [Google Scholar]
- 48. Biffi G, Oni TE, Spielman B, Hao Y, Elyada E, Park Y, et al. IL1‐Induced JAK/STAT Signaling Is Antagonized by TGFβ to Shape CAF Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancer Discov. 2019;9:282–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Elwakeel E, Brüggemann M, Fink AF, Schulz MH, Schmid T, Savai R, et al. Phenotypic Plasticity of Fibroblasts during Mammary Carcinoma Development. Int J Mol Sci. 2019;20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pereira BA, Vennin C, Papanicolaou M, Chambers CR, Herrmann D, Morton JP, et al. CAF Subpopulations: A New Reservoir of Stromal Targets in Pancreatic Cancer. Trends Cancer. 2019;5:724–41. [DOI] [PubMed] [Google Scholar]
- 51. Elyada E, Bolisetty M, Laise P, Flynn WF, Courtois ET, Burkhart RA, et al. Cross‐Species Single‐Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen‐Presenting Cancer‐Associated Fibroblasts. Cancer Discov. 2019;9:1102–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Huang H, Wang Z, Zhang Y, Pradhan RN, Ganguly D, Chandra R, et al. Mesothelial cell‐derived antigen‐presenting cancer‐associated fibroblasts induce expansion of regulatory T cells in pancreatic cancer. Cancer Cell. 2022;40:656–73.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Brechbuhl HM, Finlay‐Schultz J, Yamamoto TM, Gillen AE, Cittelly DM, Tan AC, et al. Fibroblast Subtypes Regulate Responsiveness of Luminal Breast Cancer to Estrogen. Clin Cancer Res. 2017;23:1710–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sebastian A, Hum NR, Martin KA, Gilmore SF, Peran I, Byers SW, et al. Single‐Cell Transcriptomic Analysis of Tumor‐Derived Fibroblasts and Normal Tissue‐Resident Fibroblasts Reveals Fibroblast Heterogeneity in Breast Cancer. Cancers (Basel). 2020;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Costa A, Kieffer Y, Scholer‐Dahirel A, Pelon F, Bourachot B, Cardon M, et al. Fibroblast Heterogeneity and Immunosuppressive Environment in Human Breast Cancer. Cancer Cell. 2018;33:463–79.e10. [DOI] [PubMed] [Google Scholar]
- 56. Kieffer Y, Hocine HR, Gentric G, Pelon F, Bernard C, Bourachot B, et al. Single‐Cell Analysis Reveals Fibroblast Clusters Linked to Immunotherapy Resistance in Cancer. Cancer Discov. 2020;10:1330–51. [DOI] [PubMed] [Google Scholar]
- 57. Ratcliffe PJ. Oxygen sensing and hypoxia signalling pathways in animals: the implications of physiology for cancer. J Physiol. 2013;591:2027–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hypoxia Denko NC., HIF1 and glucose metabolism in the solid tumour. Nat Rev Cancer. 2008;8:705–13. [DOI] [PubMed] [Google Scholar]
- 59. Becker LM, O'Connell JT, Vo AP, Cain MP, Tampe D, Bizarro L, et al. Epigenetic Reprogramming of Cancer‐Associated Fibroblasts Deregulates Glucose Metabolism and Facilitates Progression of Breast Cancer. Cell Rep. 2020;31:107701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Zhang D, Wang Y, Shi Z, Liu J, Sun P, Hou X, et al. Metabolic reprogramming of cancer‐associated fibroblasts by IDH3α downregulation. Cell Rep. 2015;10:1335–48. [DOI] [PubMed] [Google Scholar]
- 61. Du Y, Long Q, Zhang L, Shi Y, Liu X, Li X, et al. Curcumin inhibits cancer‐associated fibroblast‐driven prostate cancer invasion through MAOA/mTOR/HIF‐1α signaling. Int J Oncol. 2015;47:2064–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Rohwer N, Jumpertz S, Erdem M, Egners A, Warzecha KT, Fragoulis A, et al. Non‐canonical HIF‐1 stabilization contributes to intestinal tumorigenesis. Oncogene. 2019;38:5670–85. [DOI] [PubMed] [Google Scholar]
- 63. Chen X, Zhao J, Herjan T, Hong L, Liao Y, Liu C, et al. IL‐17‐induced HIF1α drives resistance to anti‐PD‐L1 via fibroblast‐mediated immune exclusion. J Exp Med. 2022;219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Garcia Garcia CJ, Huang Y, Fuentes NR, Turner MC, Monberg ME, Lin D, et al. Stromal HIF2 Regulates Immune Suppression in the Pancreatic Cancer Microenvironment. Gastroenterology. 2022;162:2018–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Akerfelt M, Morimoto RI, Sistonen L. Heat shock factors: integrators of cell stress, development and lifespan. Nat Rev Mol Cell Biol. 2010;11:545–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Anckar J, Sistonen L. Regulation of HSF1 function in the heat stress response: implications in aging and disease. Annu Rev Biochem. 2011;80:1089–115. [DOI] [PubMed] [Google Scholar]
- 67. Dai C, Sampson SB. HSF1: Guardian of Proteostasis in Cancer. Trends Cell Biol. 2016;26:17–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Scherz‐Shouval R, Santagata S, Mendillo ML, Sholl LM, Ben‐Aharon I, Beck AH, et al. The reprogramming of tumor stroma by HSF1 is a potent enabler of malignancy. Cell. 2014;158:564–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wang Q, Zhang YC, Zhu LF, Pan L, Yu M, Shen WL, et al. Heat shock factor 1 in cancer‐associated fibroblasts is a potential prognostic factor and drives progression of oral squamous cell carcinoma. Cancer Sci. 2019;110:1790–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ferrari N, Ranftl R, Chicherova I, Slaven ND, Moeendarbary E, Farrugia AJ, et al. Dickkopf‐3 links HSF1 and YAP/TAZ signalling to control aggressive behaviours in cancer‐associated fibroblasts. Nat Commun. 2019;10:130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Levine AJ, Oren M. The first 30 years of p53: growing ever more complex. Nat Rev Cancer. 2009;9:749–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kastenhuber ER, Lowe SW. Putting p53 in Context. Cell. 2017;170:1062–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Liu Q, Yu B, Tian Y, Dan J, Luo Y, Wu X. P53 Mutant p53(N236S) Regulates Cancer‐Associated Fibroblasts Properties Through Stat3 Pathway. Onco Targets Ther. 2020;13:1355–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Otomo R, Otsubo C, Matsushima‐Hibiya Y, Miyazaki M, Tashiro F, Ichikawa H, et al. TSPAN12 is a critical factor for cancer‐fibroblast cell contact‐mediated cancer invasion. Proc Natl Acad Sci U S A. 2014;111:18691–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Hayashi Y, Tsujii M, Kodama T, Akasaka T, Kondo J, Hikita H, et al. p53 functional deficiency in human colon cancer cells promotes fibroblast‐mediated angiogenesis and tumor growth. Carcinogenesis. 2016;37:972–84. [DOI] [PubMed] [Google Scholar]
- 76. Saison‐Ridinger M, DelGiorno KE, Zhang T, Kraus A, French R, Jaquish D, et al. Reprogramming pancreatic stellate cells via p53 activation: A putative target for pancreatic cancer therapy. PLoS One. 2017;12:e0189051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Fan K, Spassova I, Gravemeyer J, Ritter C, Horny K, Lange A, et al. Merkel cell carcinoma‐derived exosome‐shuttle miR‐375 induces fibroblast polarization by inhibition of RBPJ and p53. Oncogene. 2021;40:980–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Yoshii S, Hayashi Y, Iijima H, Inoue T, Kimura K, Sakatani A, et al. Exosomal microRNAs derived from colon cancer cells promote tumor progression by suppressing fibroblast TP53 expression. Cancer Sci. 2019;110:2396–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Bar J, Feniger‐Barish R, Lukashchuk N, Shaham H, Moskovits N, Goldfinger N, et al. Cancer cells suppress p53 in adjacent fibroblasts. Oncogene. 2009;28:933–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Arandkar S, Furth N, Elisha Y, Nataraj NB, van der Kuip H, Yarden Y, et al. Altered p53 functionality in cancer‐associated fibroblasts contributes to their cancer‐supporting features. Proc Natl Acad Sci U S A. 2018;115:6410–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Huang X, He C, Hua X, Kan A, Mao Y, Sun S, et al. Oxidative stress induces monocyte‐to‐myofibroblast transdifferentiation through p38 in pancreatic ductal adenocarcinoma. Clin Transl Med. 2020;10:e41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Peinado H, Olmeda D, Snail Cano A., Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer. 2007;7:415–28. [DOI] [PubMed] [Google Scholar]
- 83. Qin Q, Xu Y, He T, Qin C, Xu J. Normal and disease‐related biological functions of Twist1 and underlying molecular mechanisms. Cell Res. 2012;22:90–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Stanisavljevic J, Loubat‐Casanovas J, Herrera M, Luque T, Peña R, Lluch A, et al. Snail1‐expressing fibroblasts in the tumor microenvironment display mechanical properties that support metastasis. Cancer Res. 2015;75:284–95. [DOI] [PubMed] [Google Scholar]
- 85. Zhang K, Grither WR, Van Hove S, Biswas H, Ponik SM, Eliceiri KW, et al. Mechanical signals regulate and activate SNAIL1 protein to control the fibrogenic response of cancer‐associated fibroblasts. J Cell Sci. 2016;129:1989–2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Herrera A, Herrera M, Guerra‐Perez N, Galindo‐Pumariño C, Larriba MJ, García‐Barberán V, et al. Endothelial cell activation on 3D‐matrices derived from PDGF‐BB‐stimulated fibroblasts is mediated by Snail1. Oncogenesis. 2018;7:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Lambies G, Miceli M, Martínez‐Guillamon C, Olivera‐Salguero R, Peña R, Frías CP, et al. TGFβ‐Activated USP27X Deubiquitinase Regulates Cell Migration and Chemoresistance via Stabilization of Snail1. Cancer Res. 2019;79:33–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Li Z, Chan K, Qi Y, Lu L, Ning F, Wu M, et al. Participation of CCL1 in Snail‐Positive Fibroblasts in Colorectal Cancer Contribute to 5‐Fluorouracil/Paclitaxel Chemoresistance. Cancer Res Treat. 2018;50:894–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. García‐Palmero I, Torres S, Bartolomé RA, Peláez‐García A, Larriba MJ, Lopez‐Lucendo M, et al. Twist1‐induced activation of human fibroblasts promotes matrix stiffness by upregulating palladin and collagen α1(VI). Oncogene. 2016;35:5224–36. [DOI] [PubMed] [Google Scholar]
- 90. Lee KW, Yeo SY, Sung CO, Kim SH. Twist1 is a key regulator of cancer‐associated fibroblasts. Cancer Res. 2015;75:73–85. [DOI] [PubMed] [Google Scholar]
- 91. Zhu X, Han S, Wu S, Bai Y, Zhang N, Wei L. Dual role of twist1 in cancer‐associated fibroblasts and tumor cells promoted epithelial‐mesenchymal transition of esophageal cancer. Exp Cell Res. 2019;375:41–50. [DOI] [PubMed] [Google Scholar]
- 92. Chang YC, Tsai CH, Lai YL, Yu CC, Chi WY, Li JJ, et al. Arecoline‐induced myofibroblast transdifferentiation from human buccal mucosal fibroblasts is mediated by ZEB1. J Cell Mol Med. 2014;18:698–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Lobe C, Vallette M, Arbelaiz A, Gonzalez‐Sanchez E, Izquierdo L, Pellat A, et al. Zinc Finger E‐Box Binding Homeobox 1 Promotes Cholangiocarcinoma Progression Through Tumor Dedifferentiation and Tumor‐Stroma Paracrine Signaling. Hepatology. 2021;74:3194–212. [DOI] [PubMed] [Google Scholar]
- 94. Derynck R, Turley SJ, Akhurst RJ. TGFβ biology in cancer progression and immunotherapy. Nat Rev Clin Oncol. 2021;18:9–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Derynck R, Budi EH. Specificity, versatility, and control of TGF‐β family signaling. Sci Signal. 2019;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Heldin CH, Moustakas A. Signaling Receptors for TGF‐β Family Members. Cold Spring Harb Perspect Biol. 2016;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Itoh S, Thorikay M, Kowanetz M, Moustakas A, Itoh F, Heldin CH, et al. Elucidation of Smad requirement in transforming growth factor‐beta type I receptor‐induced responses. J Biol Chem. 2003;278:3751–61. [DOI] [PubMed] [Google Scholar]
- 98. Shi Y, Hata A, Lo RS, Massagué J, Pavletich NP. A structural basis for mutational inactivation of the tumour suppressor Smad4. Nature. 1997;388:87–93. [DOI] [PubMed] [Google Scholar]
- 99. Hill CS. Transcriptional Control by the SMADs. Cold Spring Harb Perspect Biol. 2016;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Zhang S, Fei T, Zhang L, Zhang R, Chen F, Ning Y, et al. Smad7 antagonizes transforming growth factor beta signaling in the nucleus by interfering with functional Smad‐DNA complex formation. Mol Cell Biol. 2007;27:4488–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Nakao A, Afrakhte M, Morén A, Nakayama T, Christian JL, Heuchel R, et al. Identification of Smad7, a TGFbeta‐inducible antagonist of TGF‐beta signalling. Nature. 1997;389:631–5. [DOI] [PubMed] [Google Scholar]
- 102. Zhang YE. Non‐Smad Signaling Pathways of the TGF‐β Family. Cold Spring Harb Perspect Biol. 2017;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Desmoulière A, Geinoz A, Gabbiani F, Gabbiani G. Transforming growth factor‐beta 1 induces alpha‐smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J Cell Biol. 1993;122:103–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Kojima Y, Acar A, Eaton EN, Mellody KT, Scheel C, Ben‐Porath I, et al. Autocrine TGF‐beta and stromal cell‐derived factor‐1 (SDF‐1) signaling drives the evolution of tumor‐promoting mammary stromal myofibroblasts. Proc Natl Acad Sci U S A. 2010;107:20009–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Shimura T, Sasatani M, Kawai H, Kamiya K, Kobayashi J, Komatsu K, et al. Radiation‐Induced Myofibroblasts Promote Tumor Growth via Mitochondrial ROS‐Activated TGFβ Signaling. Mol Cancer Res. 2018;16:1676–86. [DOI] [PubMed] [Google Scholar]
- 106. Mestre‐Farrera A, Bruch‐Oms M, Peña R, Rodríguez‐Morató J, Alba‐Castellón L, Comerma L, et al. Glutamine‐Directed Migration of Cancer‐Activated Fibroblasts Facilitates Epithelial Tumor Invasion. Cancer Res. 2021;81:438–51. [DOI] [PubMed] [Google Scholar]
- 107. Zhu G, Cao B, Liang X, Li L, Hao Y, Meng W, et al. Small extracellular vesicles containing miR‐192/215 mediate hypoxia‐induced cancer‐associated fibroblast development in head and neck squamous cell carcinoma. Cancer Lett. 2021;506:11–22. [DOI] [PubMed] [Google Scholar]
- 108. Huang Q, Hsueh CY, Shen YJ, Guo Y, Huang JM, Zhang YF, et al. Small extracellular vesicle‐packaged TGFβ1 promotes the reprogramming of normal fibroblasts into cancer‐associated fibroblasts by regulating fibronectin in head and neck squamous cell carcinoma. Cancer Lett. 2021;517:1–13. [DOI] [PubMed] [Google Scholar]
- 109. Zhang Y, Wang S, Lai Q, Fang Y, Wu C, Liu Y, et al. Cancer‐associated fibroblasts‐derived exosomal miR‐17–5p promotes colorectal cancer aggressive phenotype by initiating a RUNX3/MYC/TGF‐β1 positive feedback loop. Cancer Lett. 2020;491:22–35. [DOI] [PubMed] [Google Scholar]
- 110. Peng C, Zou X, Xia W, Gao H, Li Z, Liu N, et al. Integrin αvβ6 plays a bi‐directional regulation role between colon cancer cells and cancer‐associated fibroblasts. Biosci Rep. 2018;38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Liu D, Fu X, Wang Y, Wang X, Wang H, Wen J, et al. Protein diaphanous homolog 1 (Diaph1) promotes myofibroblastic activation of hepatic stellate cells by regulating Rab5a activity and TGFβ receptor endocytosis. Faseb J. 2020;34:7345–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Liu J, Huang Z, Chen HN, Qin S, Chen Y, Jiang J, et al. ZNF37A promotes tumor metastasis through transcriptional control of THSD4/TGF‐β axis in colorectal cancer. Oncogene. 2021;40:3394–407. [DOI] [PubMed] [Google Scholar]
- 113. Morén A, Bellomo C, Tsubakihara Y, Kardassis D, Mikulits W, Heldin CH, et al. LXRα limits TGFβ‐dependent hepatocellular carcinoma associated fibroblast differentiation. Oncogenesis. 2019;8:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Zhu Z, Achreja A, Meurs N, Animasahun O, Owen S, Mittal A, et al. Tumour‐reprogrammed stromal BCAT1 fuels branched‐chain ketoacid dependency in stromal‐rich PDAC tumours. Nat Metab. 2020;2:775–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Cruz‐Bermúdez A, Laza‐Briviesca R, Vicente‐Blanco RJ, García‐Grande A, Coronado MJ, Laine‐Menéndez S, et al. Cancer‐associated fibroblasts modify lung cancer metabolism involving ROS and TGF‐β signaling. Free Radic Biol Med. 2019;130:163–73. [DOI] [PubMed] [Google Scholar]
- 116. Yeung TL, Leung CS, Wong KK, Samimi G, Thompson MS, Liu J, et al. TGF‐β modulates ovarian cancer invasion by upregulating CAF‐derived versican in the tumor microenvironment. Cancer Res. 2013;73:5016–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Yang J, Lu Y, Lin YY, Zheng ZY, Fang JH, He S, et al. Vascular mimicry formation is promoted by paracrine TGF‐β and SDF1 of cancer‐associated fibroblasts and inhibited by miR‐101 in hepatocellular carcinoma. Cancer Lett. 2016;383:18–27. [DOI] [PubMed] [Google Scholar]
- 118. Krzysiek‐Maczka G, Targosz A, Szczyrk U, Wrobel T, Strzalka M, Brzozowski T, et al. Long‐Term Helicobacter pylori Infection Switches Gastric Epithelium Reprogramming Towards Cancer Stem Cell‐Related Differentiation Program in Hp‐Activated Gastric Fibroblast‐TGFβ Dependent Manner. Microorganisms. 2020;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Hasegawa T, Yashiro M, Nishii T, Matsuoka J, Fuyuhiro Y, Morisaki T, et al. Cancer‐associated fibroblasts might sustain the stemness of scirrhous gastric cancer cells via transforming growth factor‐β signaling. Int J Cancer. 2014;134:1785–95. [DOI] [PubMed] [Google Scholar]
- 120. Miyazaki K, Togo S, Okamoto R, Idiris A, Kumagai H, Miyagi Y. Collective cancer cell invasion in contact with fibroblasts through integrin‐α5β1/fibronectin interaction in collagen matrix. Cancer Sci. 2020;111:4381–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Vasiukov G, Novitskaya T, Zijlstra A, Owens P, Ye F, Zhao Z, et al. Myeloid Cell‐Derived TGFβ Signaling Regulates ECM Deposition in Mammary Carcinoma via Adenosine‐Dependent Mechanisms. Cancer Res. 2020;80:2628–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Song J, Chen W, Cui X, Huang Z, Wen D, Yang Y, et al. CCBE1 promotes tumor lymphangiogenesis and is negatively regulated by TGFβ signaling in colorectal cancer. Theranostics. 2020;10:2327–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Ikemori R, Gabasa M, Duch P, Vizoso M, Bragado P, Arshakyan M, et al. Epigenetic SMAD3 repression in tumor‐associated fibroblasts impairs fibrosis and response to the antifibrotic drug nintedanib in lung squamous cell carcinoma. Cancer Res. 2020;80:276–90. [DOI] [PubMed] [Google Scholar]
- 124. Li L, Wei JR, Dong J, Lin QG, Tang H, Jia YX, et al. Laminin γ2‐mediating T cell exclusion attenuates response to anti‐PD‐1 therapy. Sci Adv. 2021;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Chakravarthy A, Khan L, Bensler NP, Bose P, De Carvalho DD. TGF‐β‐associated extracellular matrix genes link cancer‐associated fibroblasts to immune evasion and immunotherapy failure. Nat Commun. 2018;9:4692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Grauel AL, Nguyen B, Ruddy D, Laszewski T, Schwartz S, Chang J, et al. TGFβ‐blockade uncovers stromal plasticity in tumors by revealing the existence of a subset of interferon‐licensed fibroblasts. Nat Commun. 2020;11:6315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Yegodayev KM, Novoplansky O, Golden A, Prasad M, Levin L, Jagadeeshan S, et al. TGF‐Beta‐Activated Cancer‐Associated Fibroblasts Limit Cetuximab Efficacy in Preclinical Models of Head and Neck Cancer. Cancers (Basel). 2020;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Frassanito MA, De Veirman K, Desantis V, Di Marzo L, Vergara D, Ruggieri S, et al. Halting pro‐survival autophagy by TGFβ inhibition in bone marrow fibroblasts overcomes bortezomib resistance in multiple myeloma patients. Leukemia. 2016;30:640–8. [DOI] [PubMed] [Google Scholar]
- 129. Dauer P, Zhao X, Gupta VK, Sharma N, Kesh K, Gnamlin P, et al. Inactivation of cancer‐associated‐fibroblasts disrupts oncogenic signaling in pancreatic cancer cells and promotes its regression. Cancer Res. 2018;78:1321–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Pei Y, Chen L, Huang Y, Wang J, Feng J, Xu M, et al. Sequential Targeting TGF‐β signaling and kras mutation increases therapeutic efficacy in pancreatic cancer. Small. 2019;15:e1900631. [DOI] [PubMed] [Google Scholar]
- 131. Mardhian DF, Storm G, Bansal R, Prakash J. Nano‐targeted relaxin impairs fibrosis and tumor growth in pancreatic cancer and improves the efficacy of gemcitabine in vivo. J Control Release. 2018;290:1–10. [DOI] [PubMed] [Google Scholar]
- 132. Guillén Díaz‐Maroto N, Sanz‐Pamplona R, Berdiel‐Acer M, Cimas FJ, García E, Gonçalves‐Ribeiro S, et al. Noncanonical TGFβ Pathway Relieves the Blockade of IL1β/TGFβ‐Mediated Crosstalk between Tumor and Stroma: TGFBR1 and TAK1 Inhibition in Colorectal Cancer. Clin Cancer Res. 2019;25:4466–79. [DOI] [PubMed] [Google Scholar]
- 133. Purcell JW, Tanlimco SG, Hickson J, Fox M, Sho M, Durkin L, et al. LRRC15 Is a Novel Mesenchymal Protein and Stromal Target for Antibody‐Drug Conjugates. Cancer Res. 2018;78:4059–72. [DOI] [PubMed] [Google Scholar]
- 134. Dominguez CX, Müller S, Keerthivasan S, Koeppen H, Hung J, Gierke S, et al. Single‐Cell RNA Sequencing Reveals Stromal Evolution into LRRC15(+) Myofibroblasts as a Determinant of Patient Response to Cancer Immunotherapy. Cancer Discov. 2020;10:232–53. [DOI] [PubMed] [Google Scholar]
- 135. Dieci MV, Arnedos M, Andre F, Soria JC. Fibroblast growth factor receptor inhibitors as a cancer treatment: from a biologic rationale to medical perspectives. Cancer Discov. 2013;3:264–79. [DOI] [PubMed] [Google Scholar]
- 136. Katoh M. Fibroblast growth factor receptors as treatment targets in clinical oncology. Nat Rev Clin Oncol. 2019;16:105–22. [DOI] [PubMed] [Google Scholar]
- 137. Katoh M, Nakagama H. FGF receptors: cancer biology and therapeutics. Med Res Rev. 2014;34:280–300. [DOI] [PubMed] [Google Scholar]
- 138. Musumeci M, Coppola V, Addario A, Patrizii M, Maugeri‐Saccà M, Memeo L, et al. Control of tumor and microenvironment cross‐talk by miR‐15a and miR‐16 in prostate cancer. Oncogene. 2011;30:4231–42. [DOI] [PubMed] [Google Scholar]
- 139. Akatsu Y, Takahashi N, Yoshimatsu Y, Kimuro S, Muramatsu T, Katsura A, et al. Fibroblast growth factor signals regulate transforming growth factor‐β‐induced endothelial‐to‐myofibroblast transition of tumor endothelial cells via Elk1. Mol Oncol. 2019;13:1706–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Bordignon P, Bottoni G, Xu X, Popescu AS, Truan Z, Guenova E, et al. Dualism of FGF and TGF‐β Signaling in Heterogeneous Cancer‐Associated Fibroblast Activation with ETV1 as a Critical Determinant. Cell Rep. 2019;28:2358–72.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Chen L, Wang YY, Li D, Wang C, Wang SY, Shao SH, et al. LMO2 upregulation due to AR deactivation in cancer‐associated fibroblasts induces non‐cell‐autonomous growth of prostate cancer after androgen deprivation. Cancer Lett. 2021;503:138–50. [DOI] [PubMed] [Google Scholar]
- 142. Santolla MF, Vivacqua A, Lappano R, Rigiracciolo DC, Cirillo F, Galli GR, et al. GPER Mediates a Feedforward FGF2/FGFR1 Paracrine Activation Coupling CAFs to Cancer Cells toward Breast Tumor Progression. Cells. 2019;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Fernández‐Nogueira P, Mancino M, Fuster G, López‐Plana A, Jauregui P, Almendro V, et al. Tumor‐Associated Fibroblasts Promote HER2‐Targeted Therapy Resistance through FGFR2 Activation. Clin Cancer Res. 2020;26:1432–48. [DOI] [PubMed] [Google Scholar]
- 144. Turczyk L, Kitowska K, Mieszkowska M, Mieczkowski K, Czaplinska D, Piasecka D, et al. FGFR2‐Driven Signaling Counteracts Tamoxifen Effect on ERα‐Positive Breast Cancer Cells. Neoplasia. 2017;19:791–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Ilkow CS, Marguerie M, Batenchuk C, Mayer J, Ben Neriah D, Cousineau S, et al. Reciprocal cellular cross‐talk within the tumor microenvironment promotes oncolytic virus activity. Nat Med. 2015;21:530–6. [DOI] [PubMed] [Google Scholar]
- 146. Gao CF, Vande Woude GF. HGF/SF‐Met signaling in tumor progression. Cell Res. 2005;15:49–51. [DOI] [PubMed] [Google Scholar]
- 147. Feldmann K, Maurer C, Peschke K, Teller S, Schuck K, Steiger K, et al. Mesenchymal Plasticity Regulated by Prrx1 Drives Aggressive Pancreatic Cancer Biology. Gastroenterology. 2021;160:346–61.e24. [DOI] [PubMed] [Google Scholar]
- 148. Affo S, Nair A, Brundu F, Ravichandra A, Bhattacharjee S, Matsuda M, et al. Promotion of cholangiocarcinoma growth by diverse cancer‐associated fibroblast subpopulations. Cancer Cell. 2021;39:866–82.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Zhang R, Qi F, Shao S, Li G, Feng Y. Human colorectal cancer‐derived carcinoma associated fibroblasts promote CD44‐mediated adhesion of colorectal cancer cells to endothelial cells by secretion of HGF. Cancer Cell Int. 2019;19:192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Ding X, Ji J, Jiang J, Cai Q, Wang C, Shi M, et al. HGF‐mediated crosstalk between cancer‐associated fibroblasts and MET‐unamplified gastric cancer cells activates coordinated tumorigenesis and metastasis. Cell Death Dis. 2018;9:867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Peng H, Xue R, Ju Z, Qiu J, Wang J, Yan W, et al. Cancer‐associated fibroblasts enhance the chemoresistance of CD73(+) hepatocellular carcinoma cancer cells via HGF‐Met‐ERK1/2 pathway. Ann Transl Med. 2020;8:856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Yi Y, Zeng S, Wang Z, Wu M, Ma Y, Ye X, et al. Cancer‐associated fibroblasts promote epithelial‐mesenchymal transition and EGFR‐TKI resistance of non‐small cell lung cancers via HGF/IGF‐1/ANXA2 signaling. Biochim Biophys Acta Mol Basis Dis. 2018;1864:793–803. [DOI] [PubMed] [Google Scholar]
- 153. Apicella M, Giannoni E, Fiore S, Ferrari KJ, Fernández‐Pérez D, Isella C, et al. Increased lactate secretion by cancer cells sustains non‐cell‐autonomous adaptive resistance to MET and EGFR Targeted Therapies. Cell Metab. 2018;28:848–65.e6. [DOI] [PubMed] [Google Scholar]
- 154. Kumar D, New J, Vishwakarma V, Joshi R, Enders J, Lin F, et al. Cancer‐associated fibroblasts drive glycolysis in a targetable signaling loop implicated in head and neck squamous cell carcinoma progression. Cancer Res. 2018;78:3769–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Pietras K, Sjöblom T, Rubin K, Heldin CH, Ostman A. PDGF receptors as cancer drug targets. Cancer Cell. 2003;3:439–43. [DOI] [PubMed] [Google Scholar]
- 156. Crawford Y, Kasman I, Yu L, Zhong C, Wu X, Modrusan Z, et al. PDGF‐C mediates the angiogenic and tumorigenic properties of fibroblasts associated with tumors refractory to anti‐VEGF treatment. Cancer Cell. 2009;15:21–34. [DOI] [PubMed] [Google Scholar]
- 157. Yoon H, Tang CM, Banerjee S, Yebra M, Noh S, Burgoyne AM, et al. Cancer‐associated fibroblast secretion of PDGFC promotes gastrointestinal stromal tumor growth and metastasis. Oncogene. 2021;40:1957–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Shinagawa K, Kitadai Y, Tanaka M, Sumida T, Onoyama M, Ohnishi M, et al. Stroma‐directed imatinib therapy impairs the tumor‐promoting effect of bone marrow‐derived mesenchymal stem cells in an orthotopic transplantation model of colon cancer. Int J Cancer. 2013;132:813–23. [DOI] [PubMed] [Google Scholar]
- 159. Camorani S, Hill BS, Fontanella R, Greco A, Gramanzini M, Auletta L, et al. Inhibition of Bone Marrow‐Derived Mesenchymal Stem Cells Homing Towards Triple‐Negative Breast Cancer Microenvironment Using an Anti‐PDGFRβ Aptamer. Theranostics. 2017;7:3595–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Zhang Z, Ren X, Lu X, Wang D, Hu X, Zheng Y, et al. GZD856, a novel potent PDGFRα/β inhibitor, suppresses the growth and migration of lung cancer cells in vitro and in vivo. Cancer Lett. 2016;375:172–8. [DOI] [PubMed] [Google Scholar]
- 161. Roswall P, Bocci M, Bartoschek M, Li H, Kristiansen G, Jansson S, et al. Microenvironmental control of breast cancer subtype elicited through paracrine platelet‐derived growth factor‐CC signaling. Nat Med. 2018;24:463–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Cadamuro M, Nardo G, Indraccolo S, Dall'olmo L, Sambado L, Moserle L, et al. Platelet‐derived growth factor‐D and Rho GTPases regulate recruitment of cancer‐associated fibroblasts in cholangiocarcinoma. Hepatology. 2013;58:1042–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Cadamuro M, Brivio S, Mertens J, Vismara M, Moncsek A, Milani C, et al. Platelet‐derived growth factor‐D enables liver myofibroblasts to promote tumor lymphangiogenesis in cholangiocarcinoma. J Hepatol. 2019;70:700–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Erdogan B, Ao M, White LM, Means AL, Brewer BM, Yang L, et al. Cancer‐associated fibroblasts promote directional cancer cell migration by aligning fibronectin. J Cell Biol. 2017;216:3799–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Neri S, Miyashita T, Hashimoto H, Suda Y, Ishibashi M, Kii H, et al. Fibroblast‐led cancer cell invasion is activated by epithelial‐mesenchymal transition through platelet‐derived growth factor BB secretion of lung adenocarcinoma. Cancer Lett. 2017;395:20–30. [DOI] [PubMed] [Google Scholar]
- 166. Cirillo F, Lappano R, Bruno L, Rizzuti B, Grande F, Guzzi R, et al. AHR and GPER mediate the stimulatory effects induced by 3‐methylcholanthrene in breast cancer cells and cancer‐associated fibroblasts (CAFs). J Exp Clin Cancer Res. 2019;38:335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Gao Q, Yang Z, Xu S, Li X, Yang X, Jin P, et al. Heterotypic CAF‐tumor spheroids promote early peritoneal metastatis of ovarian cancer. J Exp Med. 2019;216:688–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Álvarez‐Teijeiro S, García‐Inclán C, Villaronga M, Casado P, Hermida‐Prado F, Granda‐Díaz R, et al. Factors Secreted by Cancer‐Associated Fibroblasts that Sustain Cancer Stem Properties in Head and Neck Squamous Carcinoma Cells as Potential Therapeutic Targets. Cancers (Basel). 2018;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Magan M, Wiechec E, Roberg K. CAFs affect the proliferation and treatment response of head and neck cancer spheroids during co‐culturing in a unique in vitro model. Cancer Cell Int. 2020;20:599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Garvey CM, Lau R, Sanchez A, Sun RX, Fong EJ, Doche ME, et al. Anti‐EGFR Therapy Induces EGF Secretion by Cancer‐Associated Fibroblasts to Confer Colorectal Cancer Chemoresistance. Cancers (Basel). 2020;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Ingham PW, McMahon AP. Hedgehog signaling in animal development: paradigms and principles. Genes Dev. 2001;15:3059–87. [DOI] [PubMed] [Google Scholar]
- 172. Beachy PA, Hymowitz SG, Lazarus RA, Leahy DJ, Siebold C. Interactions between Hedgehog proteins and their binding partners come into view. Genes Dev. 2010;24:2001–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Varjosalo M, Taipale J. Hedgehog: functions and mechanisms. Genes Dev. 2008;22:2454–72. [DOI] [PubMed] [Google Scholar]
- 174. Zhou Q, Kalderon D. Hedgehog activates fused through phosphorylation to elicit a full spectrum of pathway responses. Dev Cell. 2011;20:802–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Amakye D, Jagani Z, Dorsch M. Unraveling the therapeutic potential of the Hedgehog pathway in cancer. Nat Med. 2013;19:1410–22. [DOI] [PubMed] [Google Scholar]
- 176. Takebe N, Miele L, Harris PJ, Jeong W, Bando H, Kahn M, et al. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: clinical update. Nat Rev Clin Oncol. 2015;12:445–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Guimaraes VSN, Vidal MTA, de Faro Valverde L, de Oliveira MG, de Oliveira Siquara da Rocha L, Coelho PLC, et al. Hedgehog pathway activation in oral squamous cell carcinoma: cancer‐associated fibroblasts exhibit nuclear GLI‐1 localization. J Mol Histol. 2020;51:675–84. [DOI] [PubMed] [Google Scholar]
- 178. Steele NG, Biffi G, Kemp SB, Zhang Y, Drouillard D, Syu L, et al. Inhibition of Hedgehog Signaling Alters Fibroblast Composition in Pancreatic Cancer. Clin Cancer Res. 2021;27:2023–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Cazet AS, Hui MN, Elsworth BL, Wu SZ, Roden D, Chan CL, et al. Targeting stromal remodeling and cancer stem cell plasticity overcomes chemoresistance in triple negative breast cancer. Nat Commun. 2018;9:2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Valenti G, Quinn HM, Heynen G, Lan L, Holland JD, Vogel R, et al. Cancer Stem Cells Regulate Cancer‐Associated Fibroblasts via Activation of Hedgehog Signaling in Mammary Gland Tumors. Cancer Res. 2017;77:2134–47. [DOI] [PubMed] [Google Scholar]
- 181. Chong Y, Tang D, Xiong Q, Jiang X, Xu C, Huang Y, et al. Galectin‐1 from cancer‐associated fibroblasts induces epithelial‐mesenchymal transition through β1 integrin‐mediated upregulation of Gli1 in gastric cancer. J Exp Clin Cancer Res. 2016;35:175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Saito RA, Micke P, Paulsson J, Augsten M, Peña C, Jönsson P, et al. Forkhead box F1 regulates tumor‐promoting properties of cancer‐associated fibroblasts in lung cancer. Cancer Res. 2010;70:2644–54. [DOI] [PubMed] [Google Scholar]
- 183. Razumilava N, Gradilone SA, Smoot RL, Mertens JC, Bronk SF, Sirica AE, et al. Non‐canonical Hedgehog signaling contributes to chemotaxis in cholangiocarcinoma. J Hepatol. 2014;60:599–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Walter K, Omura N, Hong SM, Griffith M, Vincent A, Borges M, et al. Overexpression of smoothened activates the sonic hedgehog signaling pathway in pancreatic cancer‐associated fibroblasts. Clin Cancer Res. 2010;16:1781–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Hwang RF, Moore TT, Hattersley MM, Scarpitti M, Yang B, Devereaux E, et al. Inhibition of the hedgehog pathway targets the tumor‐associated stroma in pancreatic cancer. Mol Cancer Res. 2012;10:1147–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Zhao J, Wang H, Hsiao CH, Chow DS, Koay EJ, Kang Y, et al. Simultaneous inhibition of hedgehog signaling and tumor proliferation remodels stroma and enhances pancreatic cancer therapy. Biomaterials. 2018;159:215–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Oyama Y, Onishi H, Koga S, Murahashi M, Ichimiya S, Nakayama K, et al. Patched 1‐interacting Peptide Represses Fibrosis in Pancreatic Cancer to Augment the Effectiveness of Immunotherapy. J Immunother. 2020;43:121–33. [DOI] [PubMed] [Google Scholar]
- 188. Mpekris F, Papageorgis P, Polydorou C, Voutouri C, Kalli M, Pirentis AP, et al. Sonic‐hedgehog pathway inhibition normalizes desmoplastic tumor microenvironment to improve chemo‐ and nanotherapy. J Control Release. 2017;261:105–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Neesse A, Bauer CA, Öhlund D, Lauth M, Buchholz M, Michl P, et al. Stromal biology and therapy in pancreatic cancer: ready for clinical translation? Gut. 2019;68:159–71. [DOI] [PubMed] [Google Scholar]
- 190. Mathew E, Zhang Y, Holtz AM, Kane KT, Song JY, Allen BL, et al. Dosage‐dependent regulation of pancreatic cancer growth and angiogenesis by hedgehog signaling. Cell Rep. 2014;9:484–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Liu X, Pitarresi JR, Cuitiño MC, Kladney RD, Woelke SA, Sizemore GM, et al. Genetic ablation of Smoothened in pancreatic fibroblasts increases acinar‐ductal metaplasia. Genes Dev. 2016;30:1943–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Yang Z, Peng YC, Gopalan A, Gao D, Chen Y, Joyner AL. Stromal hedgehog signaling maintains smooth muscle and hampers micro‐invasive prostate cancer. Dis Model Mech. 2017;10:39–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA, et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell. 2014;25:735–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Gerling M, Büller NV, Kirn LM, Joost S, Frings O, Englert B, et al. Stromal Hedgehog signalling is downregulated in colon cancer and its restoration restrains tumour growth. Nat Commun. 2016;7:12321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Ranganathan P, Weaver KL, Capobianco AJ. Notch signalling in solid tumours: a little bit of everything but not all the time. Nat Rev Cancer. 2011;11:338–51. [DOI] [PubMed] [Google Scholar]
- 196. Yang L, Shi P, Zhao G, Xu J, Peng W, Zhang J, et al. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct Target Ther. 2020;5:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Hu Y, Wang Z, Liu Y, Pan S, Zhang H, Fang M, et al. Melatonin reduces hypoxic‐ischaemic (HI) induced autophagy and apoptosis: An in vivo and in vitro investigation in experimental models of neonatal HI brain injury. Neurosci Lett. 2017;653:105–12. [DOI] [PubMed] [Google Scholar]
- 198. Bray SJ. Notch signalling: a simple pathway becomes complex. Nat Rev Mol Cell Biol. 2006;7:678–89. [DOI] [PubMed] [Google Scholar]
- 199. Kopan R, Ilagan MX. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell. 2009;137:216–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Majumder S, Crabtree JS, Golde TE, Minter LM, Osborne BA, Miele L. Targeting Notch in oncology: the path forward. Nat Rev Drug Discov. 2020. [DOI] [PubMed] [Google Scholar]
- 201. Ayaz F, Osborne BA. Non‐canonical notch signaling in cancer and immunity. Front Oncol. 2014;4:345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Procopio MG, Laszlo C, Al Labban D, Kim DE, Bordignon P, Jo SH, et al. Combined CSL and p53 downregulation promotes cancer‐associated fibroblast activation. Nat Cell Biol. 2015;17:1193–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Pelon F, Bourachot B, Kieffer Y, Magagna I, Mermet‐Meillon F, Bonnet I, et al. Cancer‐associated fibroblast heterogeneity in axillary lymph nodes drives metastases in breast cancer through complementary mechanisms. Nat Commun. 2020;11:404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Xue B, Chuang CH, Prosser HM, Fuziwara CS, Chan C, Sahasrabudhe N, et al. miR‐200 deficiency promotes lung cancer metastasis by activating Notch signaling in cancer‐associated fibroblasts. Genes Dev. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Goruppi S, Jo SH, Laszlo C, Clocchiatti A, Neel V, Dotto GP. Autophagy Controls CSL/RBPJκ Stability through a p62/SQSTM1‐Dependent Mechanism. Cell Rep. 2018;24:3108–14.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Goruppi S, Procopio MG, Jo S, Clocchiatti A, Neel V, Dotto GP. The ULK3 Kinase Is Critical for Convergent Control of Cancer‐Associated Fibroblast Activation by CSL and GLI. Cell Rep. 2017;20:2468–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Menietti E, Xu X, Ostano P, Joseph JM, Lefort K, Dotto GP. Negative control of CSL gene transcription by stress/DNA damage response and p53. Cell Cycle. 2016;15:1767–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Bottoni G, Katarkar A, Tassone B, Ghosh S, Clocchiatti A, Goruppi S, et al. CSL controls telomere maintenance and genome stability in human dermal fibroblasts. Nat Commun. 2019;10:3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Katarkar A, Bottoni G, Clocchiatti A, Goruppi S, Bordignon P, Lazzaroni F, et al. NOTCH1 gene amplification promotes expansion of Cancer Associated Fibroblast populations in human skin. Nat Commun. 2020;11:5126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Liu C, Liu L, Chen X, Cheng J, Zhang H, Zhang C, et al. LSD1 Stimulates Cancer‐Associated Fibroblasts to Drive Notch3‐Dependent Self‐Renewal of Liver Cancer Stem‐like Cells. Cancer Res. 2018;78:938–49. [DOI] [PubMed] [Google Scholar]
- 211. Sansone P, Berishaj M, Rajasekhar VK, Ceccarelli C, Chang Q, Strillacci A, et al. Evolution of Cancer Stem‐like Cells in Endocrine‐Resistant Metastatic Breast Cancers Is Mediated by Stromal Microvesicles. Cancer Res. 2017;77:1927–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Nandi A, Debnath R, Nayak A, To TKJ, Thacker G, Reilly M, et al. Dll1‐mediated Notch signaling drives tumor cell crosstalk with cancer associated fibroblasts to promote radioresistance in breast cancer. Cancer Res. 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Kahn M. Can we safely target the WNT pathway? Nat Rev Drug Discov. 2014;13:513–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Klaus A, Birchmeier W. Wnt signalling and its impact on development and cancer. Nat Rev Cancer. 2008;8:387–98. [DOI] [PubMed] [Google Scholar]
- 215. Rubinfeld B, Albert I, Porfiri E, Fiol C, Munemitsu S, Polakis P. Binding of GSK3beta to the APC‐beta‐catenin complex and regulation of complex assembly. Science. 1996;272:1023–6. [DOI] [PubMed] [Google Scholar]
- 216. Le NH, Franken P, Fodde R. Tumour‐stroma interactions in colorectal cancer: converging on beta‐catenin activation and cancer stemness. Br J Cancer. 2008;98:1886–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Yang SS, Ma S, Dou H, Liu F, Zhang SY, Jiang C, et al. Breast cancer‐derived exosomes regulate cell invasion and metastasis in breast cancer via miR‐146a to activate cancer associated fibroblasts in tumor microenvironment. Exp Cell Res. 2020;391:111983. [DOI] [PubMed] [Google Scholar]
- 218. Yang D, Li Q, Shang R, Yao L, Wu L, Zhang M, et al. WNT4 secreted by tumor tissues promotes tumor progression in colorectal cancer by activation of the Wnt/β‐catenin signalling pathway. J Exp Clin Cancer Res. 2020;39:251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Mosa MH, Michels BE, Menche C, Nicolas AM, Darvishi T, Greten FR, et al. A Wnt‐Induced Phenotypic Switch in Cancer‐Associated Fibroblasts Inhibits EMT in Colorectal Cancer. Cancer Res. 2020;80:5569–82. [DOI] [PubMed] [Google Scholar]
- 220. Avgustinova A, Iravani M, Robertson D, Fearns A, Gao Q, Klingbeil P, et al. Tumour cell‐derived Wnt7a recruits and activates fibroblasts to promote tumour aggressiveness. Nat Commun. 2016;7:10305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Aizawa T, Karasawa H, Funayama R, Shirota M, Suzuki T, Maeda S, et al. Cancer‐associated fibroblasts secrete Wnt2 to promote cancer progression in colorectal cancer. Cancer Med. 2019;8:6370–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Kramer N, Schmöllerl J, Unger C, Nivarthi H, Rudisch A, Unterleuthner D, et al. Autocrine WNT2 signaling in fibroblasts promotes colorectal cancer progression. Oncogene. 2017;36:5460–72. [DOI] [PubMed] [Google Scholar]
- 223. Unterleuthner D, Neuhold P, Schwarz K, Janker L, Neuditschko B, Nivarthi H, et al. Cancer‐associated fibroblast‐derived WNT2 increases tumor angiogenesis in colon cancer. Angiogenesis. 2020;23:159–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Huang TX, Tan XY, Huang HS, Li YT, Liu BL, Liu KS, et al. Targeting cancer‐associated fibroblast‐secreted WNT2 restores dendritic cell‐mediated antitumour immunity. Gut. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Vermeulen L, De Sousa EMF, van der Heijden M, Cameron K, de Jong JH, Borovski T, et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat Cell Biol. 2010;12:468–76. [DOI] [PubMed] [Google Scholar]
- 226. Yu FX, Zhao B, Guan KL. Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell. 2015;163:811–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Dey A, Varelas X, Guan KL. Targeting the Hippo pathway in cancer, fibrosis, wound healing and regenerative medicine. Nat Rev Drug Discov. 2020;19:480–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Ma S, Meng Z, Chen R, Guan KL. The Hippo Pathway: Biology and Pathophysiology. Annu Rev Biochem. 2019;88:577–604. [DOI] [PubMed] [Google Scholar]
- 229. Hong W, Guan KL. The YAP and TAZ transcription co‐activators: key downstream effectors of the mammalian Hippo pathway. Semin Cell Dev Biol. 2012;23:785–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Shen T, Li Y, Zhu S, Yu J, Zhang B, Chen X, et al. YAP1 plays a key role of the conversion of normal fibroblasts into cancer‐associated fibroblasts that contribute to prostate cancer progression. J Exp Clin Cancer Res. 2020;39:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Calvo F, Ege N, Grande‐Garcia A, Hooper S, Jenkins RP, Chaudhry SI, et al. Mechanotransduction and YAP‐dependent matrix remodelling is required for the generation and maintenance of cancer‐associated fibroblasts. Nat Cell Biol. 2013;15:637–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Wang S, Englund E, Kjellman P, Li Z, Ahnlide JK, Rodriguez‐Cupello C, et al. CCM3 is a gatekeeper in focal adhesions regulating mechanotransduction and YAP/TAZ signalling. Nat Cell Biol. 2021;23:758–70. [DOI] [PubMed] [Google Scholar]
- 233. Bertero T, Oldham WM, Grasset EM, Bourget I, Boulter E, Pisano S, et al. Tumor‐Stroma Mechanics Coordinate Amino Acid Availability to Sustain Tumor Growth and Malignancy. Cell Metab. 2019;29:124–40.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Ege N, Dowbaj AM, Jiang M, Howell M, Hooper S, Foster C, et al. Quantitative Analysis Reveals that Actin and Src‐Family Kinases Regulate Nuclear YAP1 and Its Export. Cell Syst. 2018;6:692–708.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. You E, Huh YH, Kwon A, Kim SH, Chae IH, Lee OJ, et al. SPIN90 Depletion and Microtubule Acetylation Mediate Stromal Fibroblast Activation in Breast Cancer Progression. Cancer Res. 2017;77:4710–22. [DOI] [PubMed] [Google Scholar]
- 236. Uchihara T, Miyake K, Yonemura A, Komohara Y, Itoyama R, Koiwa M, et al. Extracellular Vesicles from Cancer‐Associated Fibroblasts Containing Annexin A6 Induces FAK‐YAP Activation by Stabilizing β1 Integrin, Enhancing Drug Resistance. Cancer Res. 2020;80:3222–35. [DOI] [PubMed] [Google Scholar]
- 237. Liu T, Zhou L, Yang K, Iwasawa K, Kadekaro AL, Takebe T, et al. The β‐catenin/YAP signaling axis is a key regulator of melanoma‐associated fibroblasts. Signal Transduct Target Ther. 2019;4:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Vallabhapurapu S, Karin M. Regulation and function of NF‐kappaB transcription factors in the immune system. Annu Rev Immunol. 2009;27:693–733. [DOI] [PubMed] [Google Scholar]
- 239. Ghosh S, May MJ, Kopp EB. NF‐kappa B and Rel proteins: evolutionarily conserved mediators of immune responses. Annu Rev Immunol. 1998;16:225–60. [DOI] [PubMed] [Google Scholar]
- 240. Perkins ND, Gilmore TD. Good cop, bad cop: the different faces of NF‐kappaB. Cell Death Differ. 2006;13:759–72. [DOI] [PubMed] [Google Scholar]
- 241. Sun SC. The non‐canonical NF‐κB pathway in immunity and inflammation. Nat Rev Immunol. 2017;17:545–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242. Sun SC. Non‐canonical NF‐κB signaling pathway. Cell Res. 2011;21:71–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. Baud V, Karin M. Is NF‐kappaB a good target for cancer therapy? Hopes and pitfalls. Nat Rev Drug Discov. 2009;8:33–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244. Taniguchi K, Karin M. NF‐κB, inflammation, immunity and cancer: coming of age. Nat Rev Immunol. 2018;18:309–24. [DOI] [PubMed] [Google Scholar]
- 245. Erez N, Truitt M, Olson P, ST Arron, Hanahan D. Cancer‐Associated Fibroblasts Are Activated in Incipient Neoplasia to Orchestrate Tumor‐Promoting Inflammation in an NF‐kappaB‐Dependent Manner. Cancer Cell. 2010;17:135–47. [DOI] [PubMed] [Google Scholar]
- 246. Sun Y, Yang D, Xi L, Chen Y, Fu L, Sun K, et al. Primed atypical ductal hyperplasia‐associated fibroblasts promote cell growth and polarity changes of transformed epithelium‐like breast cancer MCF‐7 cells via miR‐200b/c‐IKKβ signaling. Cell Death Dis. 2018;9:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247. Gu J, Li X, Zhao L, Yang Y, Xue C, Gao Y, et al. The role of PKM2 nuclear translocation in the constant activation of the NF‐κB signaling pathway in cancer‐associated fibroblasts. Cell Death Dis. 2021;12:291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248. Lau TS, Chung TK, Cheung TH, Chan LK, Cheung LW, Yim SF, et al. Cancer cell‐derived lymphotoxin mediates reciprocal tumour‐stromal interactions in human ovarian cancer by inducing CXCL11 in fibroblasts. J Pathol. 2014;232:43–56. [DOI] [PubMed] [Google Scholar]
- 249. Fang T, Lv H, Lv G, Li T, Wang C, Han Q, et al. Tumor‐derived exosomal miR‐1247–3p induces cancer‐associated fibroblast activation to foster lung metastasis of liver cancer. Nat Commun. 2018;9:191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250. Ji Q, Zhou L, Sui H, Yang L, Wu X, Song Q, et al. Primary tumors release ITGBL1‐rich extracellular vesicles to promote distal metastatic tumor growth through fibroblast‐niche formation. Nat Commun. 2020;11:1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251. Su S, Chen J, Yao H, Liu J, Yu S, Lao L, et al. CD10(+)GPR77(+) Cancer‐Associated Fibroblasts Promote Cancer Formation and Chemoresistance by Sustaining Cancer Stemness. Cell. 2018;172:841–56.e16. [DOI] [PubMed] [Google Scholar]
- 252. Zhai J, Shen J, Xie G, Wu J, He M, Gao L, et al. Cancer‐associated fibroblasts‐derived IL‐8 mediates resistance to cisplatin in human gastric cancer. Cancer Lett. 2019;454:37–43. [DOI] [PubMed] [Google Scholar]
- 253. Huang W, Zhang L, Yang M, Wu X, Wang X, Huang W, et al. Cancer‐associated fibroblasts promote the survival of irradiated nasopharyngeal carcinoma cells via the NF‐κB pathway. J Exp Clin Cancer Res. 2021;40:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254. Wu X, Zhou Z, Xu S, Liao C, Chen X, Li B, et al. Extracellular vesicle packaged LMP1‐activated fibroblasts promote tumor progression via autophagy and stroma‐tumor metabolism coupling. Cancer Lett. 2020;478:93–106. [DOI] [PubMed] [Google Scholar]
- 255. Jin Z, Lu Y, Wu X, Pan T, Yu Z, Hou J, et al. The cross‐talk between tumor cells and activated fibroblasts mediated by lactate/BDNF/TrkB signaling promotes acquired resistance to anlotinib in human gastric cancer. Redox Biol. 2021;46:102076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256. O'Shea JJ, Gadina M, Schreiber RD. Cytokine signaling in 2002: new surprises in the Jak/Stat pathway. Cell. 2002;109(Suppl): S121–31. [DOI] [PubMed] [Google Scholar]
- 257. Ernst M, Jenkins BJ. Acquiring signalling specificity from the cytokine receptor gp130. Trends Genet. 2004;20:23–32. [DOI] [PubMed] [Google Scholar]
- 258. Quintás‐Cardama A, Verstovsek S. Molecular pathways: Jak/STAT pathway: mutations, inhibitors, and resistance. Clin Cancer Res. 2013;19:1933–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259. Levy DE, Darnell JE, Jr . Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol. 2002;3:651–62. [DOI] [PubMed] [Google Scholar]
- 260. Villarino AV, Kanno Y, O'Shea JJ. Mechanisms and consequences of Jak‐STAT signaling in the immune system. Nat Immunol. 2017;18:374–84. [DOI] [PubMed] [Google Scholar]
- 261. O'Shea JJ, Plenge R. JAK and STAT signaling molecules in immunoregulation and immune‐mediated disease. Immunity. 2012;36:542–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262. Rochman Y, Spolski R, Leonard WJ. New insights into the regulation of T cells by gamma(c) family cytokines. Nat Rev Immunol. 2009;9:480–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263. Johnson DE, O'Keefe RA, Grandis JR. Targeting the IL‐6/JAK/STAT3 signalling axis in cancer. Nat Rev Clin Oncol. 2018;15:234–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264. Pan MS, Wang H, Ansari KH, Li XP, Sun W, Fan YZ. Gallbladder cancer‐associated fibroblasts promote vasculogenic mimicry formation and tumor growth in gallbladder cancer via upregulating the expression of NOX4, a poor prognosis factor, through IL‐6‐JAK‐STAT3 signal pathway. J Exp Clin Cancer Res. 2020;39:234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265. Cheng JT, Deng YN, Yi HM, Wang GY, Fu BS, Chen WJ, et al. Hepatic carcinoma‐associated fibroblasts induce IDO‐producing regulatory dendritic cells through IL‐6‐mediated STAT3 activation. Oncogenesis. 2016;5:e198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266. Hu G, Cheng P, Pan J, Wang S, Ding Q, Jiang Z, et al. An IL6‐Adenosine Positive Feedback Loop between CD73(+) γδTregs and CAFs Promotes Tumor Progression in Human Breast Cancer. Cancer Immunol Res. 2020;8:1273–86. [DOI] [PubMed] [Google Scholar]
- 267. Cheng Y, Li H, Deng Y, Tai Y, Zeng K, Zhang Y, et al. Cancer‐associated fibroblasts induce PDL1+ neutrophils through the IL6‐STAT3 pathway that foster immune suppression in hepatocellular carcinoma. Cell Death Dis. 2018;9:422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268. Mace TA, Ameen Z, Collins A, Wojcik S, Mair M, Young GS, et al. Pancreatic cancer‐associated stellate cells promote differentiation of myeloid‐derived suppressor cells in a STAT3‐dependent manner. Cancer Res. 2013;73:3007–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269. Deng Y, Cheng J, Fu B, Liu W, Chen G, Zhang Q, et al. Hepatic carcinoma‐associated fibroblasts enhance immune suppression by facilitating the generation of myeloid‐derived suppressor cells. Oncogene. 2017;36:1090–101. [DOI] [PubMed] [Google Scholar]
- 270. Zhao Q, Huang L, Qin G, Qiao Y, Ren F, Shen C, et al. Cancer‐associated fibroblasts induce monocytic myeloid‐derived suppressor cell generation via IL‐6/exosomal miR‐21‐activated STAT3 signaling to promote cisplatin resistance in esophageal squamous cell carcinoma. Cancer Lett. 2021;518:35–48. [DOI] [PubMed] [Google Scholar]
- 271. Zhang J, Li S, Zhao Y, Ma P, Cao Y, Liu C, et al. Cancer‐associated fibroblasts promote the migration and invasion of gastric cancer cells via activating IL‐17a/JAK2/STAT3 signaling. Ann Transl Med. 2020;8:877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272. Wang X, Che X, Liu C, Fan Y, Bai M, Hou K, et al. Cancer‐associated fibroblasts‐stimulated interleukin‐11 promotes metastasis of gastric cancer cells mediated by upregulation of MUC1. Exp Cell Res. 2018;368:184–93. [DOI] [PubMed] [Google Scholar]
- 273. Heichler C, Scheibe K, Schmied A, Geppert CI, Schmid B, Wirtz S, et al. STAT3 activation through IL‐6/IL‐11 in cancer‐associated fibroblasts promotes colorectal tumour development and correlates with poor prognosis. Gut. 2020;69:1269–82. [DOI] [PubMed] [Google Scholar]
- 274. Shen J, Zhai J, You Q, Zhang G, He M, Yao X, et al. Cancer‐associated fibroblasts‐derived VCAM1 induced by H. pylori infection facilitates tumor invasion in gastric cancer. Oncogene. 2020;39:2961–74. [DOI] [PubMed] [Google Scholar]
- 275. Albrengues J, Bertero T, Grasset E, Bonan S, Maiel M, Bourget I, et al. Epigenetic switch drives the conversion of fibroblasts into proinvasive cancer‐associated fibroblasts. Nat Commun. 2015;6:10204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276. Fan J, Xu G, Chang Z, Zhu L, Yao J. miR‐210 transferred by lung cancer cell‐derived exosomes may act as proangiogenic factor in cancer‐associated fibroblasts by modulating JAK2/STAT3 pathway. Clin Sci (Lond). 2020;134:807–25. [DOI] [PubMed] [Google Scholar]
- 277. Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M, Berman K, et al. Mitogen‐activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev. 2001;22:153–83. [DOI] [PubMed] [Google Scholar]
- 278. Peluso I, Yarla NS, Ambra R, Pastore G, Perry G. MAPK signalling pathway in cancers: Olive products as cancer preventive and therapeutic agents. Semin Cancer Biol. 2019;56:185–95. [DOI] [PubMed] [Google Scholar]
- 279. Yuan J, Dong X, Yap J, Hu J. The MAPK and AMPK signalings: interplay and implication in targeted cancer therapy. J Hematol Oncol. 2020;13:113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280. Kyriakis JM, Avruch J. Mammalian MAPK signal transduction pathways activated by stress and inflammation: a 10‐year update. Physiol Rev. 2012;92:689–737. [DOI] [PubMed] [Google Scholar]
- 281. Sebolt‐Leopold JS, Herrera R. Targeting the mitogen‐activated protein kinase cascade to treat cancer. Nat Rev Cancer. 2004;4:937–47. [DOI] [PubMed] [Google Scholar]
- 282. Dror S, Sander L, Schwartz H, Sheinboim D, Barzilai A, Dishon Y, et al. Melanoma miRNA trafficking controls tumour primary niche formation. Nat Cell Biol. 2016;18:1006–17. [DOI] [PubMed] [Google Scholar]
- 283. Zhao XP, Wang M, Song Y, Song K, Yan TL, Wang L, et al. Membrane microvesicles as mediators for melanoma‐fibroblasts communication: roles of the VCAM‐1/VLA‐4 axis and the ERK1/2 signal pathway. Cancer Lett. 2015;360:125–33. [DOI] [PubMed] [Google Scholar]
- 284. Ligorio M, Sil S, Malagon‐Lopez J, Nieman LT, Misale S, Di Pilato M, et al. Stromal Microenvironment Shapes the Intratumoral Architecture of Pancreatic Cancer. Cell. 2019;178:160–75.e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285. Borriello L, Nakata R, Sheard MA, Fernandez GE, Sposto R, Malvar J, et al. Cancer‐Associated Fibroblasts Share Characteristics and Protumorigenic Activity with Mesenchymal Stromal Cells. Cancer Res. 2017;77:5142–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286. Brichkina A, Bertero T, Loh HM, Nguyen NT, Emelyanov A, Rigade S, et al. p38MAPK builds a hyaluronan cancer niche to drive lung tumorigenesis. Genes Dev. 2016;30:2623–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287. Alspach E, Flanagan KC, Luo X, Ruhland MK, Huang H, Pazolli E, et al. p38MAPK plays a crucial role in stromal‐mediated tumorigenesis. Cancer Discov. 2014;4:716–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288. Hong B, Li H, Zhang M, Xu J, Lu Y, Zheng Y, et al. p38 MAPK inhibits breast cancer metastasis through regulation of stromal expansion. Int J Cancer. 2015;136:34–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289. Wen S, Hou Y, Fu L, Xi L, Yang D, Zhao M, et al. Cancer‐associated fibroblast (CAF)‐derived IL32 promotes breast cancer cell invasion and metastasis via integrin β3‐p38 MAPK signalling. Cancer Lett. 2019;442:320–32. [DOI] [PubMed] [Google Scholar]
- 290. Curtis M, Kenny HA, Ashcroft B, Mukherjee A, Johnson A, Zhang Y, et al. Fibroblasts Mobilize Tumor Cell Glycogen to Promote Proliferation and Metastasis. Cell Metab. 2019;29:141–55.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291. Sun K, Tang S, Hou Y, Xi L, Chen Y, Yin J, et al. Oxidized ATM‐mediated glycolysis enhancement in breast cancer‐associated fibroblasts contributes to tumor invasion through lactate as metabolic coupling. EBioMedicine. 2019;41:370–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292. Sato T, Shibata W, Hikiba Y, Kaneta Y, Suzuki N, Ihara S, et al. c‐Jun N‐terminal kinase in pancreatic tumor stroma augments tumor development in mice. Cancer Sci. 2017;108:2156–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293. Blanco‐Gómez A, Hontecillas‐Prieto L, Corchado‐Cobos R, García‐Sancha N, Salvador N, Castellanos‐Martín A, et al. Stromal SNAI2 Is Required for ERBB2 Breast Cancer Progression. Cancer Res. 2020;80:5216–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294. Tan MWY, Sng MK, Cheng HS, Low ZS, Leong BJJ, Chua D, et al. Deficiency in fibroblast PPARβ/δ reduces nonmelanoma skin cancers in mice. Cell Death Differ. 2020;27:2668–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295. Zhang J, Ji C, Li W, Mao Z, Shi Y, Shi H, et al. Tumor‐Educated Neutrophils Activate Mesenchymal Stem Cells to Promote Gastric Cancer Growth and Metastasis. Front Cell Dev Biol. 2020;8:788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296. Zhou Z, Zhou Q, Wu X, Xu S, Hu X, Tao X, et al. VCAM‐1 secreted from cancer‐associated fibroblasts enhances the growth and invasion of lung cancer cells through AKT and MAPK signaling. Cancer Lett. 2020;473:62–73. [DOI] [PubMed] [Google Scholar]
- 297. Datta J, Dai X, Bianchi A, De Castro Silva I, Mehra S, Garrido V, et al. Combined MEK and STAT3 inhibition uncovers stromal plasticity by enriching for cancer‐associated fibroblasts with mesenchymal stem cell‐like features to overcome immunotherapy resistance in pancreatic cancer. Gastroenterology. 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298. Courtney KD, Corcoran RB, Engelman JA. The PI3K pathway as drug target in human cancer. J Clin Oncol. 2010;28:1075–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299. Fruman DA, Chiu H, Hopkins BD, Bagrodia S, Cantley LC, Abraham RT. The PI3K Pathway in Human Disease. Cell. 2017;170:605–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300. Janku F, Yap TA, Meric‐Bernstam F. Targeting the PI3K pathway in cancer: are we making headway? Nat Rev Clin Oncol. 2018;15:273–91. [DOI] [PubMed] [Google Scholar]
- 301. Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB, et al. Characterization of a 3‐phosphoinositide‐dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol. 1997;7:261–9. [DOI] [PubMed] [Google Scholar]
- 302. Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor‐mTOR complex. Science. 2005;307:1098–101. [DOI] [PubMed] [Google Scholar]
- 303. Manning BD, Toker A. AKT/PKB Signaling: Navigating the Network. Cell. 2017;169:381–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304. Zhou Y, Ren H, Dai B, Li J, Shang L, Huang J, et al. Hepatocellular carcinoma‐derived exosomal miRNA‐21 contributes to tumor progression by converting hepatocyte stellate cells to cancer‐associated fibroblasts. J Exp Clin Cancer Res. 2018;37:324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305. Abdul‐Wahid A, Cydzik M, Fischer NW, Prodeus A, Shively JE, Martel A, et al. Serum‐derived carcinoembryonic antigen (CEA) activates fibroblasts to induce a local re‐modeling of the extracellular matrix that favors the engraftment of CEA‐expressing tumor cells. Int J Cancer. 2018;143:1963–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306. Yamamura Y, Asai N, Enomoto A, Kato T, Mii S, Kondo Y, et al. Akt‐Girdin signaling in cancer‐associated fibroblasts contributes to tumor progression. Cancer Res. 2015;75:813–23. [DOI] [PubMed] [Google Scholar]
- 307. Zhang X, Dong Y, Zhao M, Ding L, Yang X, Jing Y, et al. ITGB2‐mediated metabolic switch in CAFs promotes OSCC proliferation by oxidation of NADH in mitochondrial oxidative phosphorylation system. Theranostics. 2020;10:12044–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308. Kamer I, Daniel‐Meshulam I, Zadok O, Bab‐Dinitz E, Perry G, Feniger‐Barish R, et al. Stromal‐MDM2 Promotes Lung Cancer Cell Invasion through Tumor‐Host Feedback Signaling. Mol Cancer Res. 2020;18:926–37. [DOI] [PubMed] [Google Scholar]
- 309. Shu C, Zha H, Long H, Wang X, Yang F, Gao J, et al. C3a‐C3aR signaling promotes breast cancer lung metastasis via modulating carcinoma associated fibroblasts. J Exp Clin Cancer Res. 2020;39:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310. Zhang W, Zhang Y, Tu T, Schmull S, Han Y, Wang W, et al. Dual inhibition of HDAC and tyrosine kinase signaling pathways with CUDC‐907 attenuates TGFβ1 induced lung and tumor fibrosis. Cell Death Dis. 2020;11:765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311. Thongchot S, Singsuksawat E, Sumransub N, Pongpaibul A, Trakarnsanga A, Thuwajit P, et al. Periostin regulates autophagy through integrin α5β1 or α6β4 and an AKT‐dependent pathway in colorectal cancer cell migration. J Cell Mol Med. 2020;24:12421–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312. Shi Y, Sun L, Zhang R, Hu Y, Wu Y, Dong X, et al. Thrombospondin 4/integrin α2/HSF1 axis promotes proliferation and cancer stem‐like traits of gallbladder cancer by enhancing reciprocal crosstalk between cancer‐associated fibroblasts and tumor cells. J Exp Clin Cancer Res. 2021;40:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313. Li X, Li Q, Yu X, Li H, Huang G. Reverse of microtubule‐directed chemotherapeutic drugs resistance induced by cancer‐associated fibroblasts in breast cancer. Onco Targets Ther. 2019;12:7963–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314. Li Z, Zhou J, Zhang J, Li S, Wang H, Du J. Cancer‐associated fibroblasts promote PD‐L1 expression in mice cancer cells via secreting CXCL5. Int J Cancer. 2019;145:1946–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315. Melisi D, Garcia‐Carbonero R, Macarulla T, Pezet D, Deplanque G, Fuchs M, et al. Galunisertib plus gemcitabine vs. gemcitabine for first‐line treatment of patients with unresectable pancreatic cancer. Br J Cancer. 2018;119:1208–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316. Kelley RK, Gane E, Assenat E, Siebler J, Galle PR, Merle P, et al. A Phase 2 Study of Galunisertib (TGF‐β1 Receptor Type I Inhibitor) and Sorafenib in Patients With Advanced Hepatocellular Carcinoma. Clin Transl Gastroenterol. 2019;10:e00056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317. Faivre S, Santoro A, Kelley RK, Gane E, Costentin CE, Gueorguieva I, et al. Novel transforming growth factor beta receptor I kinase inhibitor galunisertib (LY2157299) in advanced hepatocellular carcinoma. Liver Int. 2019;39:1468–77. [DOI] [PubMed] [Google Scholar]
- 318. Yamazaki T, Gunderson AJ, Gilchrist M, Whiteford M, Kiely MX, Hayman A, et al. Galunisertib plus neoadjuvant chemoradiotherapy in patients with locally advanced rectal cancer: a single‐arm, phase 2 trial. Lancet Oncol. 2022;23:1189–200. [DOI] [PubMed] [Google Scholar]
- 319. Melisi D, Oh DY, Hollebecque A, Calvo E, Varghese A, Borazanci E, et al. Safety and activity of the TGFβ receptor I kinase inhibitor galunisertib plus the anti‐PD‐L1 antibody durvalumab in metastatic pancreatic cancer. J Immunother Cancer. 2021;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320. Feng J, Tang D, Wang J, Zhou Q, Peng J, Lou H, et al. SHR‐1701, a bifunctional fusion protein targeting PD‐L1 and TGF‐β, for recurrent or metastatic cervical cancer: a clinical expansion cohort of phase 1 study. Clin Cancer Res. 2022. [DOI] [PubMed] [Google Scholar]
- 321. Strauss J, Heery CR, Schlom J, Madan RA, Cao L, Kang Z, et al. Phase I Trial of M7824 (MSB0011359C), a Bifunctional Fusion Protein Targeting PD‐L1 and TGFβ, in Advanced Solid Tumors. Clin Cancer Res. 2018;24:1287–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322. Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324:1457–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323. Ko AH, LoConte N, Tempero MA, Walker EJ, Kate Kelley R, Lewis S, et al. A Phase I Study of FOLFIRINOX Plus IPI‐926, a Hedgehog Pathway Inhibitor, for Advanced Pancreatic Adenocarcinoma. Pancreas. 2016;45:370–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324. Catenacci DV, Junttila MR, Karrison T, Bahary N, Horiba MN, Nattam SR, et al. Randomized Phase Ib/II Study of Gemcitabine Plus Placebo or Vismodegib, a Hedgehog Pathway Inhibitor, in Patients With Metastatic Pancreatic Cancer. J Clin Oncol. 2015;33:4284–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325. De Jesus‐Acosta A, Sugar EA, O'Dwyer PJ, Ramanathan RK, Von Hoff DD, Rasheed Z, et al. Phase 2 study of vismodegib, a hedgehog inhibitor, combined with gemcitabine and nab‐paclitaxel in patients with untreated metastatic pancreatic adenocarcinoma. Br J Cancer. 2020;122:498–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326. Ruiz‐Borrego M, Jimenez B, Antolín S, García‐Saenz JA, Corral J, Jerez Y, et al. A phase Ib study of sonidegib (LDE225), an oral small molecule inhibitor of smoothened or Hedgehog pathway, in combination with docetaxel in triple negative advanced breast cancer patients: GEICAM/2012–12 (EDALINE) study. Invest New Drugs. 2019;37:98–108. [DOI] [PubMed] [Google Scholar]
- 327. Feig C, Jones JO, Kraman M, Wells RJ, Deonarine A, Chan DS, et al. Targeting CXCL12 from FAP‐expressing carcinoma‐associated fibroblasts synergizes with anti‐PD‐L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci U S A. 2013;110:20212–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328. Garg B, Giri B, Modi S, Sethi V, Castro I, Umland O, et al. NFκB in Pancreatic Stellate Cells Reduces Infiltration of Tumors by Cytotoxic T Cells and Killing of Cancer Cells, via Up‐regulation of CXCL12. Gastroenterology. 2018;155:880–91.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329. Ene‐Obong A, Clear AJ, Watt J, Wang J, Fatah R, Riches JC, et al. Activated pancreatic stellate cells sequester CD8+ T cells to reduce their infiltration of the juxtatumoral compartment of pancreatic ductal adenocarcinoma. Gastroenterology. 2013;145:1121–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330. Bockorny B, Semenisty V, Macarulla T, Borazanci E, Wolpin BM, Stemmer SM, et al. BL‐8040, a CXCR4 antagonist, in combination with pembrolizumab and chemotherapy for pancreatic cancer: the COMBAT trial. Nat Med. 2020;26:878–85. [DOI] [PubMed] [Google Scholar]
- 331. Biasci D, Smoragiewicz M, Connell CM, Wang Z, Gao Y, Thaventhiran JED, et al. CXCR4 inhibition in human pancreatic and colorectal cancers induces an integrated immune response. Proc Natl Acad Sci U S A. 2020;117:28960–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332. Ghobrial IM, Liu CJ, Zavidij O, Azab AK, Baz R, Laubach JP, et al. Phase I/II trial of the CXCR4 inhibitor plerixafor in combination with bortezomib as a chemosensitization strategy in relapsed/refractory multiple myeloma. Am J Hematol. 2019;94:1244–53. [DOI] [PubMed] [Google Scholar]
- 333. Pernas S, Martin M, Kaufman PA, Gil‐Martin M, Gomez Pardo P, Lopez‐Tarruella S, et al. Balixafortide plus eribulin in HER2‐negative metastatic breast cancer: a phase 1, single‐arm, dose‐escalation trial. Lancet Oncol. 2018;19:812–24. [DOI] [PubMed] [Google Scholar]
- 334. Chen Y, McAndrews KM, Kalluri R. Clinical and therapeutic relevance of cancer‐associated fibroblasts. Nat Rev Clin Oncol. 2021;18:792–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335. Costa A, Scholer‐Dahirel A, Mechta‐Grigoriou F. The role of reactive oxygen species and metabolism on cancer cells and their microenvironment. Semin Cancer Biol. 2014;25:23–32. [DOI] [PubMed] [Google Scholar]
- 336. Shi Y, Du L, Lin L, Wang Y. Tumour‐associated mesenchymal stem/stromal cells: emerging therapeutic targets. Nat Rev Drug Discov. 2017;16:35–52. [DOI] [PubMed] [Google Scholar]
- 337. Radhakrishnan R, Ha JH, Jayaraman M, Liu J, Moxley KM, Isidoro C, et al. Ovarian cancer cell‐derived lysophosphatidic acid induces glycolytic shift and cancer‐associated fibroblast‐phenotype in normal and peritumoral fibroblasts. Cancer Lett. 2019;442:464–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338. Grunberg N, Pevsner‐Fischer M, Goshen‐Lago T, Diment J, Stein Y, Lavon H, et al. Cancer‐Associated Fibroblasts Promote Aggressive Gastric Cancer Phenotypes via Heat Shock Factor 1‐Mediated Secretion of Extracellular Vesicles. Cancer Res. 2021;81:1639–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339. Cangkrama M, Wietecha M, Mathis N, Okumura R, Ferrarese L, Al‐Nuaimi D, et al. A paracrine activin A‐mDia2 axis promotes squamous carcinogenesis via fibroblast reprogramming. EMBO Mol Med. 2020;12:e11466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340. Goetz JG, Minguet S, Navarro‐Lérida I, Lazcano JJ, Samaniego R, Calvo E, et al. Biomechanical remodeling of the microenvironment by stromal caveolin‐1 favors tumor invasion and metastasis. Cell. 2011;146:148–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341. Cohen AW, Hnasko R, Schubert W, Lisanti MP. Role of caveolae and caveolins in health and disease. Physiol Rev. 2004;84:1341–79. [DOI] [PubMed] [Google Scholar]
- 342. Lynch MD, Watt FM. Fibroblast heterogeneity: implications for human disease. J Clin Invest. 2018;128:26–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343. Cremasco V, Astarita JL, Grauel AL, Keerthivasan S, MacIsaac K, Woodruff MC, et al. FAP Delineates Heterogeneous and Functionally Divergent Stromal Cells in Immune‐Excluded Breast Tumors. Cancer Immunol Res. 2018;6:1472–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344. Arnold JN, Magiera L, Kraman M, Fearon DT. Tumoral immune suppression by macrophages expressing fibroblast activation protein‐α and heme oxygenase‐1. Cancer Immunol Res. 2014;2:121–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345. Huber MA, Kraut N, Park JE, Schubert RD, Rettig WJ, Peter RU, et al. Fibroblast activation protein: differential expression and serine protease activity in reactive stromal fibroblasts of melanocytic skin tumors. J Invest Dermatol. 2003;120:182–8. [DOI] [PubMed] [Google Scholar]
- 346. Weissmueller S, Manchado E, Saborowski M, JPt Morris, Wagenblast E, Davis CA, et al. Mutant p53 drives pancreatic cancer metastasis through cell‐autonomous PDGF receptor β signaling. Cell. 2014;157:382–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347. Pietras K, Pahler J, Bergers G, Hanahan D. Functions of paracrine PDGF signaling in the proangiogenic tumor stroma revealed by pharmacological targeting. PLoS Med. 2008;5:e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348. Krishnan H, Rayes J, Miyashita T, Ishii G, Retzbach EP, Sheehan SA, et al. Podoplanin: An emerging cancer biomarker and therapeutic target. Cancer Sci. 2018;109:1292–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349. Neri S, Ishii G, Hashimoto H, Kuwata T, Nagai K, Date H, et al. Podoplanin‐expressing cancer‐associated fibroblasts lead and enhance the local invasion of cancer cells in lung adenocarcinoma. Int J Cancer. 2015;137:784–96. [DOI] [PubMed] [Google Scholar]
- 350. Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Myofibroblasts and mechano‐regulation of connective tissue remodelling. Nat Rev Mol Cell Biol. 2002;3:349–63. [DOI] [PubMed] [Google Scholar]
- 351. Lazard D, Sastre X, Frid MG, Glukhova MA, Thiery JP, Koteliansky VE. Expression of smooth muscle‐specific proteins in myoepithelium and stromal myofibroblasts of normal and malignant human breast tissue. Proc Natl Acad Sci U S A. 1993;90:999–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352. Eyden B. The myofibroblast: phenotypic characterization as a prerequisite to understanding its functions in translational medicine. J Cell Mol Med. 2008;12:22–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353. Zhang J, Chen L, Liu X, Kammertoens T, Blankenstein T, Qin Z. Fibroblast‐specific protein 1/S100A4‐positive cells prevent carcinoma through collagen production and encapsulation of carcinogens. Cancer Res. 2013;73:2770–81. [DOI] [PubMed] [Google Scholar]
- 354. Österreicher CH, Penz‐Österreicher M, Grivennikov SI, Guma M, Koltsova EK, Datz C, et al. Fibroblast‐specific protein 1 identifies an inflammatory subpopulation of macrophages in the liver. Proc Natl Acad Sci U S A. 2011;108:308–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355. Satelli A, Li S. Vimentin in cancer and its potential as a molecular target for cancer therapy. Cell Mol Life Sci. 2011;68:3033–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356. Vuoriluoto K, Haugen H, Kiviluoto S, Mpindi JP, Nevo J, Gjerdrum C, et al. Vimentin regulates EMT induction by Slug and oncogenic H‐Ras and migration by governing Axl expression in breast cancer. Oncogene. 2011;30:1436–48. [DOI] [PubMed] [Google Scholar]
- 357. Kfoury Y, Scadden DT. Mesenchymal cell contributions to the stem cell niche. Cell Stem Cell. 2015;16:239–53. [DOI] [PubMed] [Google Scholar]
- 358. Chen Y, Kim J, Yang S, Wang H, Wu CJ, Sugimoto H, et al. Type I collagen deletion in αSMA(+) myofibroblasts augments immune suppression and accelerates progression of pancreatic cancer. Cancer Cell. 2021;39:548–65.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359. O'Connell JT, Sugimoto H, Cooke VG, MacDonald BA, Mehta AI, LeBleu VS, et al. VEGF‐A and Tenascin‐C produced by S100A4+ stromal cells are important for metastatic colonization. Proc Natl Acad Sci U S A. 2011;108:16002–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360. Tang X, Tu G, Yang G, Wang X, Kang L, Yang L, et al. Autocrine TGF‐β1/miR‐200s/miR‐221/DNMT3B regulatory loop maintains CAF status to fuel breast cancer cell proliferation. Cancer Lett. 2019;452:79–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361. Lu JT, Tan CC, Wu XR, He R, Zhang X, Wang QS, et al. FOXF2 deficiency accelerates the visceral metastasis of basal‐like breast cancer by unrestrictedly increasing TGF‐β and miR‐182–5p. Cell Death Differ. 2020;27:2973–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362. Shan G, Gu J, Zhou D, Li L, Cheng W, Wang Y, et al. Cancer‐associated fibroblast‐secreted exosomal miR‐423–5p promotes chemotherapy resistance in prostate cancer by targeting GREM2 through the TGF‐β signaling pathway. Exp Mol Med. 2020;52:1809–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363. Zhang J, Wang Q, Quan Z. Long non‐coding RNA CASC9 enhances breast cancer progression by promoting metastasis through the meditation of miR‐215/TWIST2 signaling associated with TGF‐β expression. Biochem Biophys Res Commun. 2019;515:644–50. [DOI] [PubMed] [Google Scholar]
- 364. Zhang X, Wang Y, Wang X, Zou B, Mei J, Peng X, et al. Extracellular vesicles‐encapsulated microRNA‐10a‐5p shed from cancer‐associated fibroblast facilitates cervical squamous cell carcinoma cell angiogenesis and tumorigenicity via Hedgehog signaling pathway. Cancer Gene Ther. 2021;28:529–42. [DOI] [PubMed] [Google Scholar]
- 365. Li YY, Tao YW, Gao S, Li P, Zheng JM, Zhang SE, et al. Cancer‐associated fibroblasts contribute to oral cancer cells proliferation and metastasis via exosome‐mediated paracrine miR‐34a‐5p. EBioMedicine. 2018;36:209–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366. Hu JL, Wang W, Lan XL, Zeng ZC, Liang YS, Yan YR, et al. CAFs secreted exosomes promote metastasis and chemotherapy resistance by enhancing cell stemness and epithelial‐mesenchymal transition in colorectal cancer. Mol Cancer. 2019;18:91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367. Aprelikova O, Palla J, Hibler B, Yu X, Greer YE, Yi M, et al. Silencing of miR‐148a in cancer‐associated fibroblasts results in WNT10B‐mediated stimulation of tumor cell motility. Oncogene. 2013;32:3246–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 368. Shan G, Zhou X, Gu J, Zhou D, Cheng W, Wu H, et al. Downregulated exosomal microRNA‐148b‐3p in cancer associated fibroblasts enhance chemosensitivity of bladder cancer cells by downregulating the Wnt/β‐catenin pathway and upregulating PTEN. Cell Oncol (Dordr). 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369. Deng X, Ruan H, Zhang X, Xu X, Zhu Y, Peng H, et al. Long noncoding RNA CCAL transferred from fibroblasts by exosomes promotes chemoresistance of colorectal cancer cells. Int J Cancer. 2020;146:1700–16. [DOI] [PubMed] [Google Scholar]
- 370. Ren J, Ding L, Zhang D, Shi G, Xu Q, Shen S, et al. Carcinoma‐associated fibroblasts promote the stemness and chemoresistance of colorectal cancer by transferring exosomal lncRNA H19. Theranostics. 2018;8:3932–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371. Dou D, Ren X, Han M, Xu X, Ge X, Gu Y, et al. Cancer‐Associated Fibroblasts‐Derived Exosomes Suppress Immune Cell Function in Breast Cancer via the miR‐92/PD‐L1 Pathway. Front Immunol. 2020;11:2026. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 372. Ren Z, Lv M, Yu Q, Bao J, Lou K, Li X. MicroRNA‐370–3p shuttled by breast cancer cell‐derived extracellular vesicles induces fibroblast activation through the CYLD/Nf‐κB axis to promote breast cancer progression. Faseb j. 2021;35:e21383. [DOI] [PubMed] [Google Scholar]
- 373. Cui Y, Wang D, Xie M. Tumor‐Derived Extracellular Vesicles Promote Activation of Carcinoma‐Associated Fibroblasts and Facilitate Invasion and Metastasis of Ovarian Cancer by Carrying miR‐630. Front Cell Dev Biol. 2021;9:652322. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 374. Yang Z, Jin P, Xu S, Zhang T, Yang X, Li X, et al. Dicer reprograms stromal fibroblasts to a pro‐inflammatory and tumor‐promoting phenotype in ovarian cancer. Cancer Lett. 2018;415:20–9. [DOI] [PubMed] [Google Scholar]
- 375. Jin N, Jin N, Bu W, Li X, Liu L, Wang Z, et al. Long non‐coding RNA TIRY promotes tumor metastasis by enhancing epithelial‐to‐mesenchymal transition in oral cancer. Exp Biol Med (Maywood). 2020;245:585–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376. Xu Q, Lin YB, Li L, Liu J. LncRNA TLR8‐AS1 promotes metastasis and chemoresistance of ovarian cancer through enhancing TLR8 mRNA stability. Biochem Biophys Res Commun. 2020;526:857–64. [DOI] [PubMed] [Google Scholar]
- 377. Zhou X, Yan T, Huang C, Xu Z, Wang L, Jiang E, et al. Melanoma cell‐secreted exosomal miR‐155‐5p induce proangiogenic switch of cancer‐associated fibroblasts via SOCS1/JAK2/STAT3 signaling pathway. J Exp Clin Cancer Res. 2018;37:242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378. Fan JT, Zhou ZY, Luo YL, Luo Q, Chen SB, Zhao JC, et al. Exosomal lncRNA NEAT1 from cancer‐associated fibroblasts facilitates endometrial cancer progression via miR‐26a/b‐5p‐mediated STAT3/YKL‐40 signaling pathway. Neoplasia. 2021;23:692–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379. Shi H, Huang S, Qin M, Xue X, Guo X, Jiang L, et al. Exosomal circ_0088300 Derived From Cancer‐Associated Fibroblasts Acts as a miR‐1305 Sponge and Promotes Gastric Carcinoma Cell Tumorigenesis. Front Cell Dev Biol. 2021;9:676319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380. Yang F, Yan Y, Yang Y, Hong X, Wang M, Yang Z, et al. MiR‐210 in exosomes derived from CAFs promotes non‐small cell lung cancer migration and invasion through PTEN/PI3K/AKT pathway. Cell Signal. 2020;73:109675. [DOI] [PubMed] [Google Scholar]
- 381. Chen X, Liu Y, Zhang Q, Liu B, Cheng Y, Zhang Y, et al. Exosomal miR‐590–3p derived from cancer‐associated fibroblasts confers radioresistance in colorectal cancer. Mol Ther Nucleic Acids. 2021;24:113–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382. Wang Z, Wang X, Zhang T, Su L, Liu B, Zhu Z, et al. LncRNA MALAT1 promotes gastric cancer progression via inhibiting autophagic flux and inducing fibroblast activation. Cell Death Dis. 2021;12:368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383. Zhang Y, Zhao J, Ding M, Su Y, Cui D, Jiang C, et al. Loss of exosomal miR‐146a‐5p from cancer‐associated fibroblasts after androgen deprivation therapy contributes to prostate cancer metastasis. J Exp Clin Cancer Res. 2020;39:282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384. Zhang Z, Li X, Sun W, Yue S, Yang J, Li J, et al. Loss of exosomal miR‐320a from cancer‐associated fibroblasts contributes to HCC proliferation and metastasis. Cancer Lett. 2017;397:33–42. [DOI] [PubMed] [Google Scholar]
- 385. Guo L, Li B, Yang J, Shen J, Ji J, Miao M. Fibroblast‑derived exosomal microRNA‑369 potentiates migration and invasion of lung squamous cell carcinoma cells via NF1‑mediated MAPK signaling pathway. Int J Mol Med. 2020;46:595–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386. Ji Z, Tian W, Gao W, Zang R, Wang H, Yang G. Cancer‐associated fibroblast‐derived interleukin‐8 promotes ovarian cancer cell stemness and malignancy through the notch3‐mediated signaling. Front Cell Dev Biol. 2021;9:684505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387. Zhang F, Cui JY, Gao HF, Yu H, Gao FF, Chen JL, et al. Cancer‐associated fibroblasts induce epithelial‐mesenchymal transition and cisplatin resistance in ovarian cancer via CXCL12/CXCR4 axis. Future Oncol. 2020;16:2619–33. [DOI] [PubMed] [Google Scholar]
- 388. Yang F, Guo Z, He C, Qing L, Wang H, Wu J, et al. Cancer‐associated fibroblasts promote cell proliferation and invasion via paracrine Wnt/IL1β signaling pathway in human bladder cancer. Neoplasma. 2021;68:79–86. [DOI] [PubMed] [Google Scholar]
- 389. Zhang H, Jiang R, Zhou J, Wang J, Xu Y, Zhang H, et al. CTL Attenuation Regulated by PS1 in Cancer‐Associated Fibroblast. Front Immunol. 2020;11:999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390. Zhang Y, Bian Y, Wang Y, Wang Y, Duan X, Han Y, et al. HIF‐1α is necessary for activation and tumour‐promotion effect of cancer‐associated fibroblasts in lung cancer. J Cell Mol Med. 2021;25:5457–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 391. Huang YH, Chang CY, Kuo YZ, Fang WY, Kao HY, Tsai ST, et al. Cancer‐associated fibroblast‐derived interleukin‐1β activates protumor C‐C motif chemokine ligand 22 signaling in head and neck cancer. Cancer Sci. 2019;110:2783–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392. Lim H, Koh M, Jin H, Bae M, Lee SY, Kim KM, et al. Cancer‐associated fibroblasts induce an aggressive phenotypic shift in non‐malignant breast epithelial cells via interleukin‐8 and S100A8. J Cell Physiol. 2021. [DOI] [PubMed] [Google Scholar]
- 393. Al‐Ansari MM, Al‐Saif M, Arafah M, Eldali AM, Tulbah A, Al‐Tweigeri T, et al. Clinical and functional significance of tumor/stromal ATR expression in breast cancer patients. Breast Cancer Res. 2020;22:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394. Ma H, Wang J, Zhao X, Wu T, Huang Z, Chen D, et al. Periostin Promotes Colorectal Tumorigenesis through Integrin‐FAK‐Src Pathway‐Mediated YAP/TAZ Activation. Cell Rep. 2020;30:793–806.e6. [DOI] [PubMed] [Google Scholar]
- 395. Jia C, Wang G, Wang T, Fu B, Zhang Y, Huang L, et al. Cancer‐associated Fibroblasts induce epithelial‐mesenchymal transition via the Transglutaminase 2‐dependent IL‐6/IL6R/STAT3 axis in Hepatocellular Carcinoma. Int J Biol Sci. 2020;16:2542–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 396. Chen X, An Y, Zhang Y, Xu D, Chen T, Yang Y, et al. CCL26 is upregulated by nab‐paclitaxel in pancreatic cancer‐associated fibroblasts and promotes PDAC invasiveness through activation of the PI3K/AKT/mTOR pathway. Acta Biochim Biophys Sin (Shanghai). 2021;53:612–9. [DOI] [PubMed] [Google Scholar]
- 397. Zhang H, Yue J, Jiang Z, Zhou R, Xie R, Xu Y, et al. CAF‐secreted CXCL1 conferred radioresistance by regulating DNA damage response in a ROS‐dependent manner in esophageal squamous cell carcinoma. Cell Death Dis. 2017;8:e2790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 398. Zhou Q, Wu X, Wang X, Yu Z, Pan T, Li Z, et al. The reciprocal interaction between tumor cells and activated fibroblasts mediated by TNF‐α/IL‐33/ST2L signaling promotes gastric cancer metastasis. Oncogene. 2020;39:1414–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.