

Abstract
Background and Aims:
Liver regeneration (LR) following partial hepatectomy (PH) occurs via activation of various signaling pathways. Disruption of single pathway can be compensated by activation of another pathway to continue LR. The Wnt-β-catenin pathway is activated early during LR and conditional hepatocyte loss of β-catenin delays LR. Here, we study mechanism of LR in the absence of hepatocyte-β-catenin.
Approach & Results:
Eight-week-old hepatocyte-specific Ctnnb1 knockout mice (β-cateninΔHC) were subjected to PH. These animals exhibited decreased hepatocyte proliferation at 40–120h and decreased cumulative 14-day (14d) BrdU labeling of <40%, but all mice survived suggesting compensation. Insulin-mediated mTORC1 activation was uniquely identified in the β-cateninΔHC mice at 72–96h after PH. Deletion of hepatocyte Raptor, a critical mTORC1 partner, in the β-cateninΔHC mice led to progressive hepatic injury and mortality by 30d. PH on early-stage non-morbid RaptorΔHC-β-cateninΔHC mice led to lethality by 12h. RaptorΔHC mice showed progressive hepatic injury, spontaneous LR with β-catenin activation, but died by 40d. PH on early stage non-morbid RaptorΔHC mice was lethal by 48h. Temporal inhibition of insulin receptor and mTORC1 in β-cateninΔHC or controls after PH was achieved by administration of linsitinib at 48h or rapamycin at 60h post-PH, and completely prevented LR leading to lethality by 12–14d.
Conclusions:
Insulin-mTORC1 activation compensates for β-catenin loss to enable LR after PH. mTORC1 signaling in hepatocytes itself is critical to both homeostasis and LR and is only partially compensated by β-catenin activation. Dual inhibition of β-catenin and mTOR may have notable untoward hepatotoxic side effects.
Keywords: Partial hepatectomy, Wnt signaling, pathway redundancy, liver failure, hepatocyte proliferation
Graphical Abstract
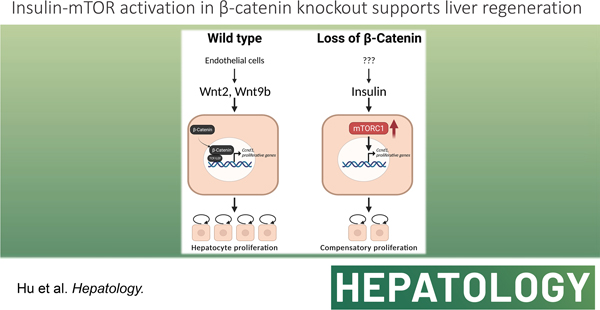
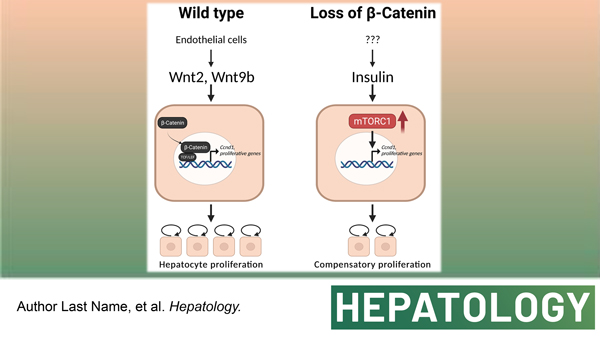
The liver is the largest gland of the body performing around 500 functions related to metabolism, synthesis, storage, and detoxification. Because of the diversity and criticality of the functions as well as its strategic location, liver is bestowed with a capability to regenerate unlike any other organ. The process of liver regeneration (LR) has most commonly been studied by surgical removal of two-thirds of the liver by partial hepatectomy (PH). After PH, the process of LR is initiated by a concerted effort of various mitogenic growth factors like HGF and EGF, and several non-mitogenic cytokines like IL-6, TNFα and others (1). Studies in many transgenic and knockout mouse models have demonstrated molecular and cellular redundancy during LR. In fact, single genetic deletion of growth factors, cytokines, or their receptors or co-receptors, often leads to only a partial delay in LR, suggesting existence of fail-safe mechanisms ensuring LR (1, 2).
The Wnt-β-catenin signaling is a major contributor of LR following PH in rats, mice and zebrafish (3–5), as well as after acetaminophen overdose in mice and patients, and after carbon tetrachloride-induced liver injury (6, 7). Mice with liver-specific knockout of β-catenin, liver-specific double-knockout of LRP5 and LRP6 (co-receptors of Wnt ligands), or hepatic endothelial cell-specific knockout of Wntless, all exhibit delayed LR (5, 8–12). However, LR is eventually restored albeit by unknown mechanisms. The aim of the current study was to validate the delay in LR in the absence of hepatocyte-β-catenin and to identify the compensatory mechanisms enabling LR and allowing survival.
Experimental Procedures
Animals
All animal husbandry and experimental procedures, including animal housing and diet, were performed under the guidelines and approval of the National Institutes of Health and the Institutional Animal Care and Use Committee at the University of Pittsburgh. Mice were fed regular chow in standard caging and kept under a 12-hour light–dark cycle with no enrichment. Ctnnb1flox/flox mice were reported before (5). Raptorflox/flox mice were reported before and were purchased from the Jackson Laboratory (Strain #:013188) (13).
More detailed methods in the online supplement
Results
Acute hepatocyte-specific deletion of β-catenin severely impairs liver regeneration
Previously, others and we have shown that mice with Albumin-Cre driven chronic genetic loss of β-catenin from hepatocytes and cholangiocytes (β-cateninΔLiver), exhibited lower liver weight to body weight ratio (LW/BW), but lacked any other overt phenotype (5, 14). Instead of Albumin-Cre, which might lead to adaption to chronic β-catenin loss from the hepatic epithelial or ‘hepithelial’ cells, we used AAV8-TBG-Cre to acutely and selectively delete β-catenin from hepatocytes (β-cateninΔHC) (Fig. 1A). At baseline, a very small subset of β-cateninΔHC mice had smaller liver size along with stiff and fibrotic livers, which were excluded from the LR studies (Fig. S1A). In majority of the cases, acute hepatocyte-specific deletion did not impact body weight, liver size or LW/BW (Fig. 1B, S1B). Next, β-cateninΔHC and control mice were subjected to PH (Fig. 1A). Control mice showed an average LW/BW recovery to 3.8% at 14 days (14d) after PH, whereas β-cateninΔHC mice recovered to around 2.6% at 96 hours (96h), but no further gain was evident even at 14d of LR (Fig. 1B).
Fig. 1. Acute hepatocyte-specific deletion of β-catenin severely impairs liver regeneration.
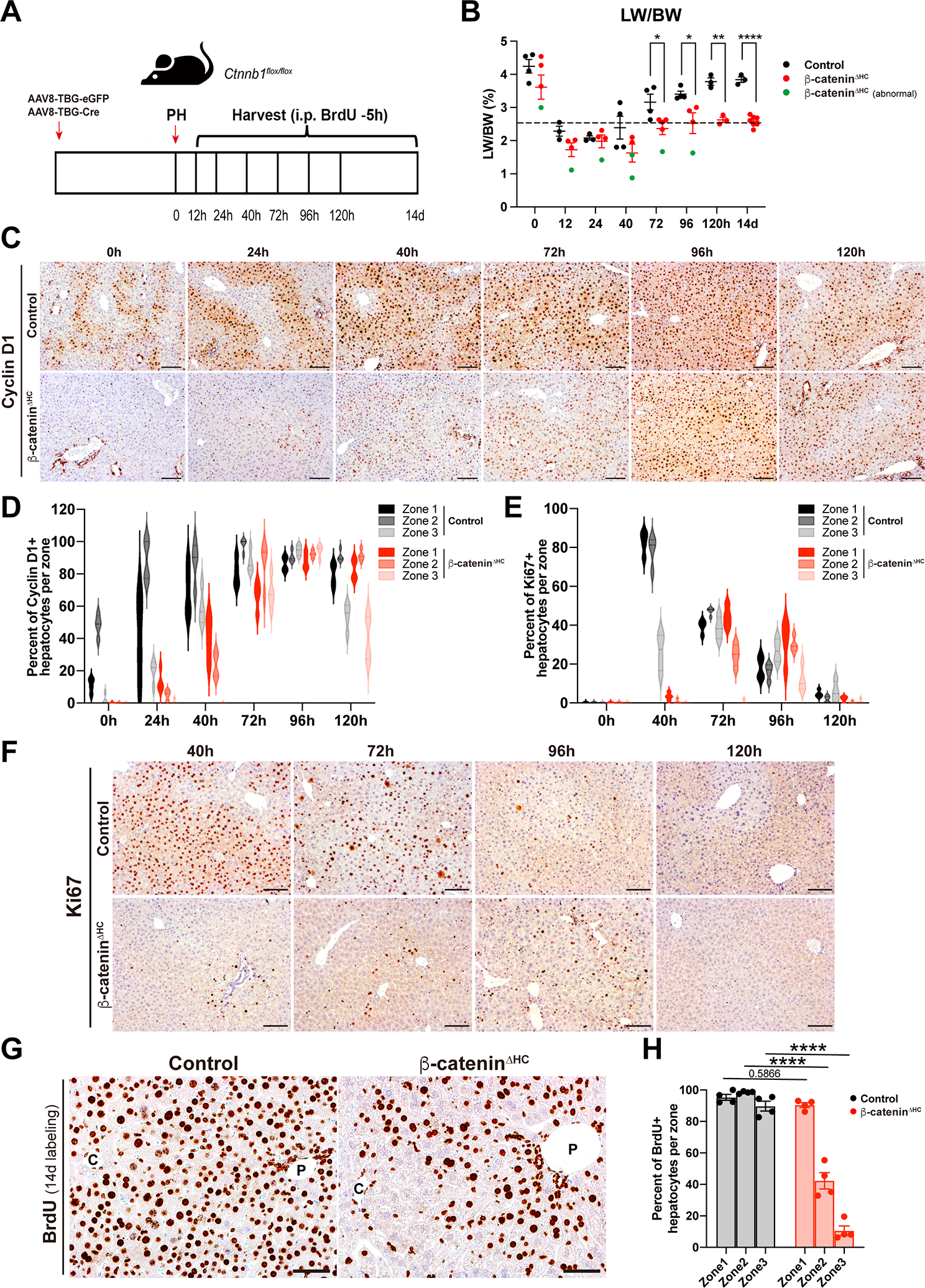
(A) Schema of generation of control and β-cateninΔHC mice model, and partial hepatectomy (PH).
(B) Liver weight to body weight ratio (LW/BW) at different time points at baseline and post-PH in control and β-cateninΔHC mice. Livers of a subset of β-cateninΔHC mice (green dots) were abnormally small, grossly fibrotic and showed poor regeneration. (Multiple unpaired t test, *P < 0.05, **P < 0.01, ****P < 0.0001)
(C) Representative immunohistochemistry (IHC) of Cyclin D1, a proliferative β-catenin target regulating G1-to-S phase transition, in control and β-cateninΔHC mice at different time points. (Scale bars: 100 μm)
(D) Violin plots depicting quantification of Cyclin D1-positive hepatocytes per zone in control and β-cateninΔHC mice at different time points after PH.
(E) Violin plots showing quantification of Ki67-positive hepatocytes per zone in control and β-cateninΔHC mice at different time points after PH.
(F) Representative IHC of Ki67 in control and β-cateninΔHC mice. (Scale bars: 100 μm. Two-way ANOVA, ***P < 0.001, ****P < 0.0001)
(G) Representative IHC of BrdU in control and β-cateninΔHC mice. (Scale bars: 50 μm. Two-way ANOVA, ***P < 0.001, ****P < 0.0001). C: central vein; P: portal vein.
(H) Quantification of BrdU-positive hepatocytes per zone in control and β-cateninΔHC mice after 14d of continuous BrdU administration in drinking water over the course of LR. (Two-way ANOVA, ****P < 0.0001)
Since β-catenin regulates LR in major part by regulating expression of Ccnd1, we first analyzed Cyclin D1 levels and distribution in β-cateninΔHC and control mice by immunohistochemistry (IHC) (5, 15). In control mice, Cyclin D1 was present in zone 2 at baseline, as shown previously (16). During LR, Cyclin D1-positive hepatocytes were seen in the periportal and midzonal region at 24h and also in pericentral regions from 40–96h, while beginning to return to baseline-like localization at 120h after PH (Fig. 1C–D, Fig. S1C). The β-cateninΔHC mice exhibited a diminished induction of Cyclin D1 which gradually increased from 40h to 96h and began to decrease at 120h (Fig. 1C). A more careful characterization revealed the increase to be overall less profound in the β-cateninΔHC mice as compared to controls and to be in the periportal and midzonal regions at 40h and panzonal from 72–96h with continued increase in periportal and midzonal expression even at 120h (Fig. 1D, Fig. S1C). Quantitative polymerase chain reaction (qPCR) analysis also confirmed a rapid Ccnd1 up-regulation after PH in control mice from 12h to 96h, while livers from β-cateninΔHC mice only exhibited scant up-regulation between 40h-120h, peaking at 96h (Fig. S1D).
Hepatocyte proliferation was next assessed by IHC for Ki67 to identify cells in S-phase with careful attention to zonality. In controls, increased Ki67-positive hepatocytes were observed at 40h in periportal and midzonal regions, with overall fewer but panzonal presence at 72–96h after PH (Fig 1E–F, Fig. S1E–F). In β-cateninΔHC, there is a clear lack of any Ki67-positive hepatocytes at 40h with overall lower but maximal positivity seen at 72–96h especially in periportal and midzone regions (Fig. 1E–1F, Fig. S1E–F). Temporal hepatocyte proliferation was assessed by IHC for BrdU, which was injected intraperitoneally 5h prior to euthanasia. Control mice exhibited increased BrdU-positive hepatocytes at 40h followed by a decline thereafter (Fig. S2A–B). In β-cateninΔHC mice, occasional hepatocytes around the portal vein were BrdU-positive at 40h and occasional hepatocytes were BrdU in periportal or midzonal regions at 72–96h (Fig. S2A–B). To address cumulative hepatocyte proliferation over time in control and β-cateninΔHC mice after PH, and to also investigate the localization of proliferating hepatocytes, we performed both IHC for BrdU, and immunofluorescence (IF) for BrdU, HNF4α (hepatocytes), and panCK (cholangiocytes) after 14d of continuous BrdU administration in drinking water over the course of LR. Control mice exhibited around 90–100% of hepatocytes to be BrdU-positive along with a pan-lobular distribution by both IHC and by colocalization of BrdU and HNF4α-positive by IF (Fig. 1G, Fig. S2C–D). Analysis for their zonal localization revealed >90% of hepatocytes to be BrdU-positive in all 3 zones at 14d after PH in the controls (Fig. 1H). In β-cateninΔHC mice, there was preponderance of BrdU-positive or BrdU-HNF4α-dual positive hepatocytes in periportal (~90%) and midzonal (~40%) regions with only <5% hepatocytes in zone 3 being positive (Fig. 1G–H, Fig. S2C–D). These results indicated LR to still occur in the β-cateninΔHC mice even though it was suboptimal, and thus enabled survival of these mice after PH.
Since we observed discordance in LR in β-cateninΔHC versus previously published β-cateninΔLiver mice which showed a notable rebound in LR at 72h after PH (5, 11), we next performed a direct comparison of regeneration kinetics between the two models. Unlike β-cateninΔHC mice, we observed a significant increase of hepatocyte proliferation at 72h post-PH in β-cateninΔLiver mice (Fig. S2E–F), similar to the previously published studies.
Overall, these data suggest LR in β-cateninΔHC mice to be occurring slowly but surely, thus underscoring a fundamental role of β-catenin in murine LR.
Activation of the mTORC1 signaling at 72 hours co-occurs with compensatory increase in LR in β-cateninΔHC mice
Despite impaired LR after PH in β-cateninΔHC mice, increased Cyclin D1 and continued low grade hepatocyte proliferation was evident from 72h onwards with all mice surviving past 14d after PH with around 40% hepatocytes having undergone replication. This suggested activation of a compensatory signaling pathway may be driving LR in the absence of hepatocyte β-catenin. To uncover such mechanisms, livers from controls and β-cateninΔHC mice that underwent PH were first assessed for status of other similar developmental pathways including YAP, NOTCH, and Hedgehog signaling pathways (17–20). We employed qPCR to determine expression of known downstream target genes of the respective pathways as surrogates of the state of their signaling. No differences were observed in the NOTCH and Hedgehog pathways (Fig. S3A–B). We saw an unexpected and significant impairment in YAP activation as observed by failed induction of Cyr61 and Ctgf in the β-cateninΔHC mice during LR as compared to the controls at similar times (Fig. S3C). While intriguing, this observation could not be the contributing mechanism of hepatocyte proliferation in β-cateninΔHC mice during LR.
Next, we assessed the status of some well-known effectors associated with cell proliferation in lysates from control and β-cateninΔHC livers at various time-points after PH. Examining cytokine-induced signaling first, we found no notable differences in phosphorylation status of TNFα-induced NF-κB activity between the controls and β-cateninΔHC (Fig. S3D) (21, 22). Likewise, there were no consistent differences in P-STAT3-Y705 levels between the two groups at all time-points (Fig. S3D). Stress response pathways p38 MAPK and AMPK have also been implicated as mediators of cell proliferation during LR after PH (23, 24). However, we found comparable phosphorylation of both proteins in control and β-cateninΔHC mice during LR (Fig. S3D). Overall, lack of noteworthy and consistent differences in any of these effector molecules between the control and β-cateninΔHC mice after PH precluded any of these as possible mechanisms of ongoing LR in the absence of hepatocyte-β-catenin.
Next, we evaluated the status of the mTOR signaling, which is another pertinent effector downstream of various growth factors that mediates cell proliferation and has been shown to play an important role in liver growth following PH (13, 25, 26). In control mice, the mTORC1 pathway was activated at 12h post-PH shown by increased levels of P-mTOR-S2448 and its downstream effectors P-p70S6K-T389, P-S6-S235/236, P-S6-S240/244, and P-4E-BP1-S65 followed by a decrease in activity from 24h onwards returning to baseline levels at 72h (Fig. 2A–B, S3E), as also reported elsewhere in previous studies (25, 26). Intriguingly, in β-cateninΔHC mice, in addition to the early activation, a second more profound and wider peak of mTORC1 activation was observed at 72–96h as seen by enhanced levels of P-mTOR-S2448 and its downstream effectors P-p70S6K-T389, P-S6-S235/236 and P-S6-S240/244, while P-4E-BP1-S65 was increased at 72h, by WB analyses and their quantification (Fig. 2A–B, S3E).
Fig. 2. Activation of the mTORC1 signaling at 72 hours co-occurs with compensatory increase in LR in β-cateninΔHC mice.
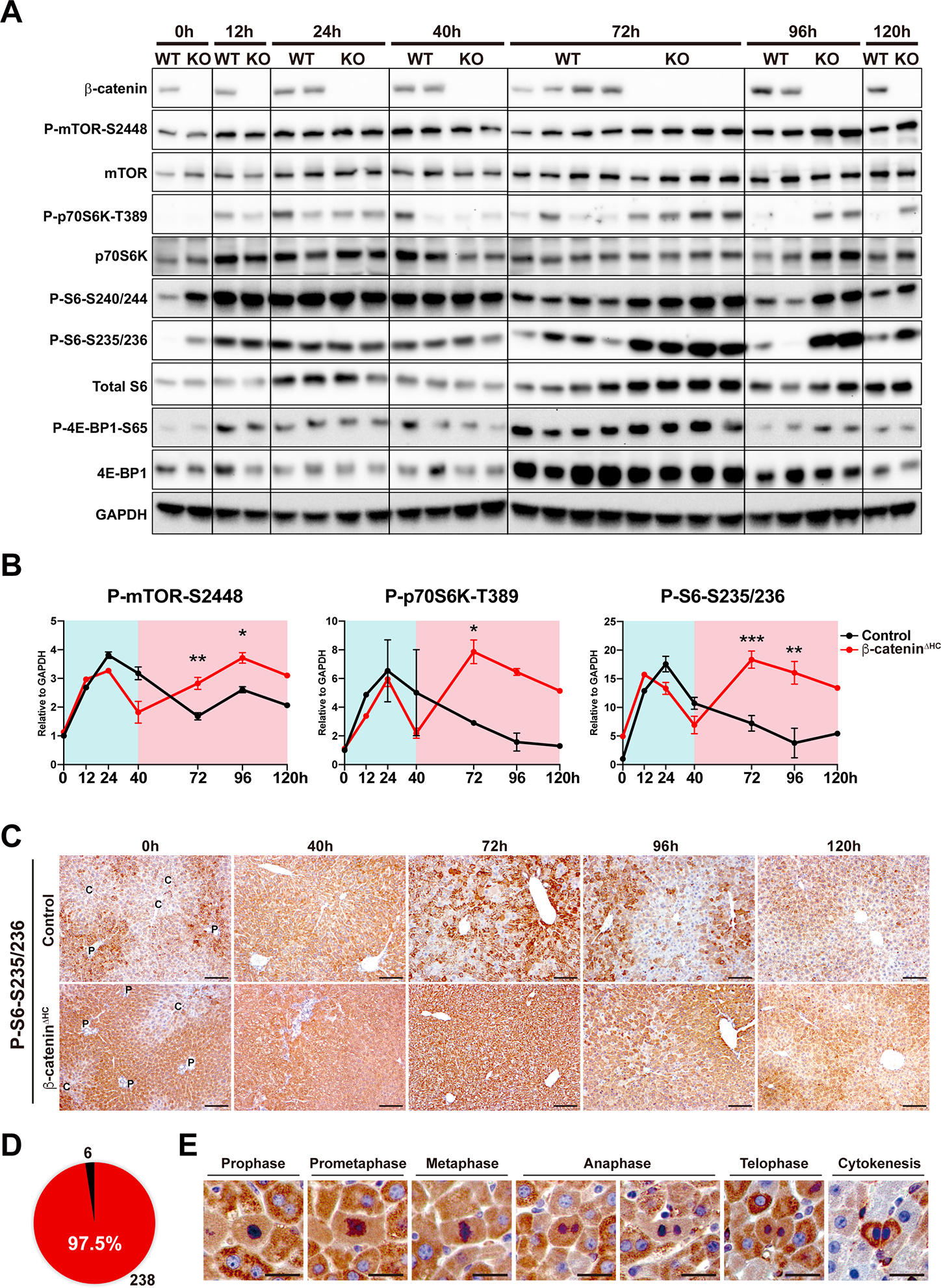
(A) Western blot (WB) analysis of the mTORC1 pathway during LR using whole liver lysates from control and β-cateninΔHC mice.
(B) Quantification of P-mTOR-S2448, P-p70S6K-T389, and P-S6-S235/236 in (A). (Two-way ANOVA, *P < 0.05, **P < 0.01, ***P < 0.001)
(C) Representative IHC of P-S6-S235/236 in control and β-cateninΔHC mice. C: central vein; P: portal vein. (Scale bars: 100 μm)
(D) Number of P-S6-S235/236-positive (red) and -negative (black) proliferative hepatocytes at 72h post-PH in β-cateninΔHC mice.
(E) Representative IHC of P-S6-S235/236-positive proliferative hepatocytes at 72h in β-cateninΔHC mice. (Scale bars: 20 μm)
To address localization, we performed IHC for P-S6-S235/236 in control and β-cateninΔHC livers from different time-points after PH. In controls, IHC for P-S6-S235/236 showed a pan-lobular increase at 40h over baseline followed by a gradual decrease from 72–120h (Fig. 2C). β-cateninΔHC mice exhibited a similar pan-lobular increase in P-S6-S235/236 staining at 40h and showed an even more intense staining from 72–96h (Fig. 2C). Careful quantification showed 97.5% (238 out of 244) of mitotic hepatocytes in β-cateninΔHC mice to be positive for P-S6-S235/236, in all stages of cell cycle phases (Fig. 2D–E). Similar results were also observed by IHC staining of P-S6-S240/244 (Fig. S3F).
Collectively, our data identified a second unique wave of mTORC1 activation in the β-cateninΔHC mice after PH, suggesting it to be a likely compensatory mechanism driving LR.
Acute hepatocyte-specific deletion of Raptor and β-catenin causes failure of LR after PH
To unequivocally address the role of mTORC1 signaling in the absence of hepatocyte β-catenin, we took advantage of genetically engineered mouse models. Since Raptor is the defining subunit of mTORC1 essential for its activation, we crossed Raptorflox/flox mice with Ctnnb1flox/flox mice, and generated acute hepatocyte-specific Raptor and β-catenin double deletion mice (RaptorΔHC-β-cateninΔHC) using AAV8-TBG-Cre. Raptorflox/flox mice getting AAV8-TBG-Cre (RaptorΔHC) were used as single knockout controls. Two weeks after virus injection, we performed PH to these mice to investigate their role in LR.
Strikingly, none of the RaptorΔHC-β-cateninΔHC mice survived beyond 6–12h post-PH, and none of the RaptorΔHC mice survived beyond 48h post-PH (Fig. 3A). At the time of death, RaptorΔHC-β-cateninΔHC and RaptorΔHC mice displayed significantly lower LW/BW than controls (Fig. 3B). We examined serum biochemistry from the two groups of animals at the time of their demise after PH. At 6h, the RaptorΔHC-β-cateninΔHC showed lower ALT, comparable AST, higher ALP and lower serum albumin compared to controls at the same time point (Fig. 3C). This suggested ongoing hepatobiliary injury along with hepatic insufficiency and liver failure. Histologically, while the control livers at 6h post-PH were already showing increase in Cyclin D1 staining in midzonal hepatocytes, there were only a few periportal Cyclin D1-positive hepatocytes (Fig. 3D). Despite high serum ALT and AST in controls at 6h which are likely remnants of surgical resection, there was no ongoing cell death within hepatic parenchyma in the controls at 6h as seen by negative TUNEL staining (Fig. 3D). However, clusters of TUNEL-positive hepatocytes were specifically observed in pericentral regions in the RaptorΔHC-β-cateninΔHC livers at 6h post-PH (Fig. 3D). Histology was also reminiscent of ongoing cell death and immune cell infiltration in the pericentral region (Fig. 3E). A notable ductular reaction was only visible in the 6h post-PH RaptorΔHC-β-cateninΔHC livers (Fig. 3E). The hepatocytes were also notably smaller in the RaptorΔHC-β-cateninΔHC livers at this stage (Fig. 3F). All of these features were indicative of ongoing failure of LR and associated acute liver failure.
Fig. 3. Acute hepatocyte-specific deletion of Raptor and β-catenin causes failure of LR after PH.
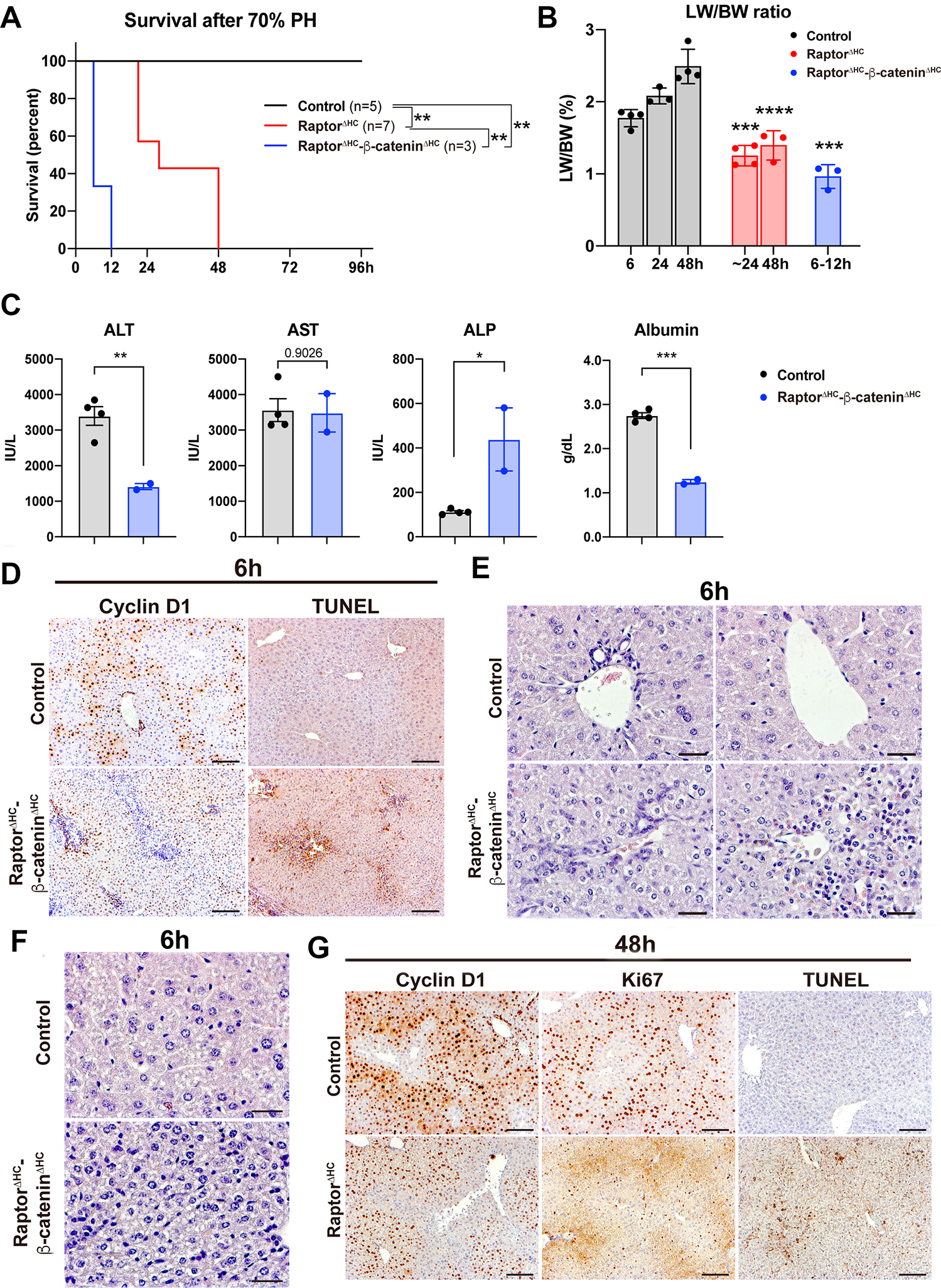
(A) Kaplan-Meier survival curve of control, RaptorΔHC, and RaptorΔHC-β-cateninΔHC mice post-PH. (**P < 0.01)
(B) LW/BW at indicated time points post-PH. (Unpaired t test, ***P < 0.001, ****P < 0.0001)
(C) Serum levels of ALT, AST, ALP, and albumin. (Unpaired t test, *P < 0.05, **P < 0.01, ***P < 0.001)
(D) Representative IHC of Cyclin D1 and TUNEL at 6h post-PH in control and RaptorΔHC-β-cateninΔHC mice. (Scale bars: 100 μm)
(E) Representative H&E staining showing ductular reaction and immune cell infiltration in the 6h post-PH RaptorΔHC-β-cateninΔHC livers. (Scale bars: 25 μm)
(F) Representative H&E staining showing decreased hepatocyte size in the 6h post-PH RaptorΔHC-β-cateninΔHC livers. (Scale bars: 25 μm)
(G) Representative IHC of Cyclin D1, Ki67, and TUNEL at 48h post-PH in control and RaptorΔHC mice. (Scale bars: 100 μm)
Serum biochemistry from RaptorΔHC mice was assessed at around 24h before 100% mortality was observed in these animals. While serum ALT, AST and ALP levels were reducing in controls from the 6h control values, the RaptorΔHC mice showed persistent increases in these markers of hepatobiliary injury which were significantly higher than the time-matched controls (Fig. S4A). IHC analysis showed a notable decrease in numbers of Cyclin D1-positive hepatocytes, Ki67-positive hepatocytes and higher numbers of TUNEL-positive cells in the RaptorΔHC mice as compared to controls at 48h post-PH (Fig. 3G). There was a small decrease in hepatocyte size in the RaptorΔHC mice at this time along with evidence of microsteatosis indicating blunted LR (Fig. S4F).
In short, loss of Raptor from hepatocyte-β-catenin-deficient mice led to early lethality after PH precluding us from addressing any compensatory role of mTORC1 in LR in the β-cateninΔHC mice. Intriguingly, RaptorΔHC mice also succumbed to PH albeit at 48h after surgery suggesting the relevance of early mTORC1 activation after PH in controls. Also, since RaptorΔHC-β-cateninΔHC mice exhibited a much more severe liver injury and accelerated liver failure after PH compared to RaptorΔHC mice, this suggests that β-catenin may be cooperating with the mTORC1 signaling for optimum liver recovery after PH.
Acute hepatocyte-specific deletion of Raptor causes progressive injury, liver regeneration partially by β-catenin activation, and is eventually fatal
Although we observed cooperation of mTORC1 and β-catenin in initiating LR, PH led to death in 100% of RaptorΔHC mice around 48h. Therefore, we decided to examine these mice at baseline. Two weeks after virus injection, livers from the RaptorΔHC mice were assessed for mTORC1 signaling. RaptorΔHC livers showed loss of Raptor, P-mTOR-S2448, P-p70S6K-T389, P-S6-S235/236 and P-S6-S240/244 (Fig. 4A). RaptorΔHC mice at this stage showed slight but significant elevations in serum levels of ALT, AST, and ALP (Fig. 4B). Simultaneously, we observed reduction in serum albumin, glucose, triglycerides, and cholesterol (Fig. 4B, S4B). Histologically, livers from RaptorΔHC mice exhibited weak eosinophilic staining of cytosol by H&E, enhanced cell death by significantly increased TUNEL-positive hepatocytes, elevated CD45-positive immune cells and F4/80-positive macrophage infiltration (Fig. 4C–D). No fibrosis was observed by Sirius red staining or by mRNA expression of Col1a1 and Acta2 (Fig. S4C–E). Because of increased inflammation and pale eosinophilic cytoplasm, we next examined the state of hepatocyte differentiation. Markers of hepatocyte dedifferentiation including Hnf4α-P2 isoform and Afp expression were decreased whereas Hnf4α-P1 isoform was unchanged (Fig. 4E). Further, expression of hepatocyte metabolic genes including those encoding proteins involved in urea cycle (Arg1) and gluconeogenesis (G6pc, Pck1) were decreased as were genes encoding for liver-derived secretory proteins (Alb, Trf, Ttr) (Fig. 4F). Transmission electron microscopy (TEM) revealed mitochondrial swelling, endoplasmic reticulum (ER) whorl formation indicating protein synthesis inhibition and ER stress, increased lysosomes, and increased autophagy (Fig. 4G). Due to the observed indolent injury in RaptorΔHC mice, we next examined state of hepatocyte proliferation. A concurrent attempt at compensatory liver regeneration was observed by the presence of increased Ki67-positive hepatocytes in S-phase of cell cycle in the RaptorΔHC mice (Fig. 4C).
Fig. 4. Acute hepatocyte-specific deletion of Raptor causes hepatopathy.
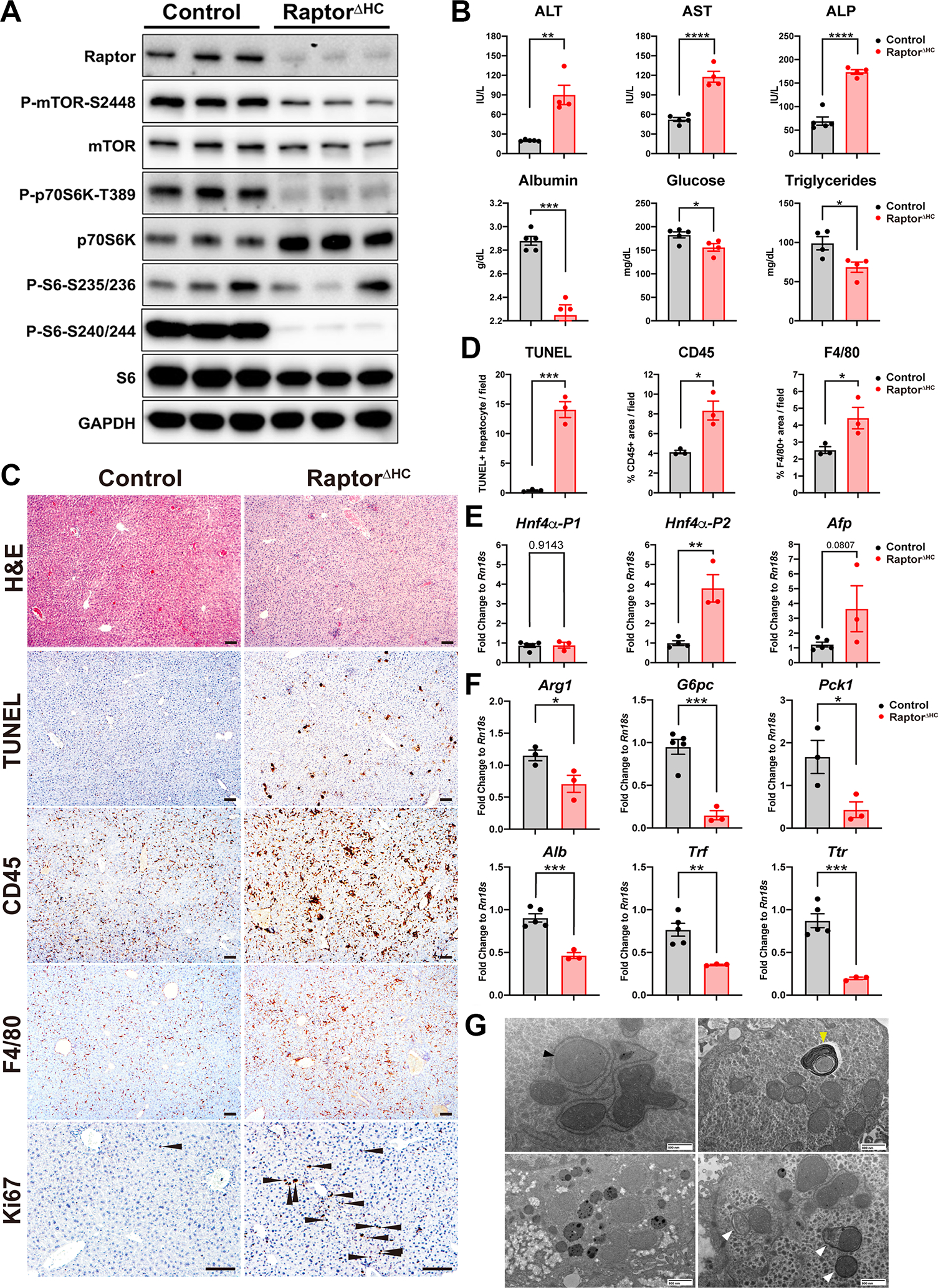
(A) WB of the mTORC1 pathway using whole liver lysates from control and RaptorΔHC mice.
(B) Serum levels of ALT, AST, ALP, albumin, glucose, and triglycerides in control and RaptorΔHC mice at two weeks after virus injection. (Unpaired t test, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001)
(C) Representative H&E and IHC showing increased cell death (TUNEL), immune cell (CD45) and macrophage (F4/80) infiltration, and cell proliferation (Ki67, arrowhead: Ki67-positive hepatocytes) in RaptorΔHC mice. (Scale bars: 100 μm)
(D) Quantification of (C). (Unpaired t test, *P < 0.05, ***P < 0.001)
(E) qPCR of HNF4α-P1, P2, and Afp showing dedifferentiation of RaptorΔHC livers. (Unpaired t test, **P < 0.01)
(F) qPCR of genes in urea cycle (Arg1), gluconeogenesis (G6pc, Pck1), and genes encoding liver-derived secretory proteins (Alb, Trf, Ttr). (Unpaired t test, *P < 0.05, **P < 0.01, ***P < 0.001)
(G) Representative transmission electron microscopy (TEM) images of RaptorΔHC livers showing mitochondrial swelling (black arrowhead), endoplasmic reticulum whorl formation (yellow arrowhead), increased lysosomes, and increased autophagy (white arrowheads). (Scale bars were indicated in each panel)
Next, we asked if liver repair by enhanced cell cycle entry of hepatocytes in RaptorΔHC mice could be due to activation of the Wnt-β-catenin signaling. WB analysis was performed on whole liver lysates from the livers of controls and RaptorΔHC mice. We observed an accumulation of β-catenin and active non-phosphorylated form of β-catenin together with its proliferative targets Cyclin D1 and Myc (Fig. 5A). A concomitant decrease in the known metabolic targets of β-catenin like GS and CYP2E1 was also observed in the RaptorΔHC livers (Fig. 5A). Accumulation of β-catenin was also verified by IHC staining (Fig. 5B). While control livers showed midzonal Cyclin D1 staining, RaptorΔHC livers showed enhanced periportal and midzonal Cyclin D1 positive hepatocytes with only 1–2 layers of hepatocytes around the central vein remaining consistently negative for this marker of G1-to-S transition (Fig. 5B). Pericentral zonation of GS and CYP2E1 was maintained but overall weaker in RaptorΔHC mice and in agreement with WB (Fig. 5B). In line with the changes at the protein level, we observed increased mRNA expression of β-catenin proliferative target genes Ccnd1 and Myc, a universal β-catenin target Axin2, and decreased mRNA of hepatocyte-specific differentiation/metabolic targets Glul and Cyp2e1 (Fig. 5C). β-catenin is well known to be regulated post-translationally by phosphorylation-mediated degradation. Interestingly, in addition to decreased degradation as indicated by increased non-phosphorylated β-catenin (Fig. 5A), we also observed a modest but significant increase in the mRNA expression of Ctnnb1, the gene encoding for β-catenin (Fig. 5C).
Fig. 5. Acute hepatocyte-specific deletion of Raptor causes liver regeneration partially by β-catenin activation and is eventually fatal.
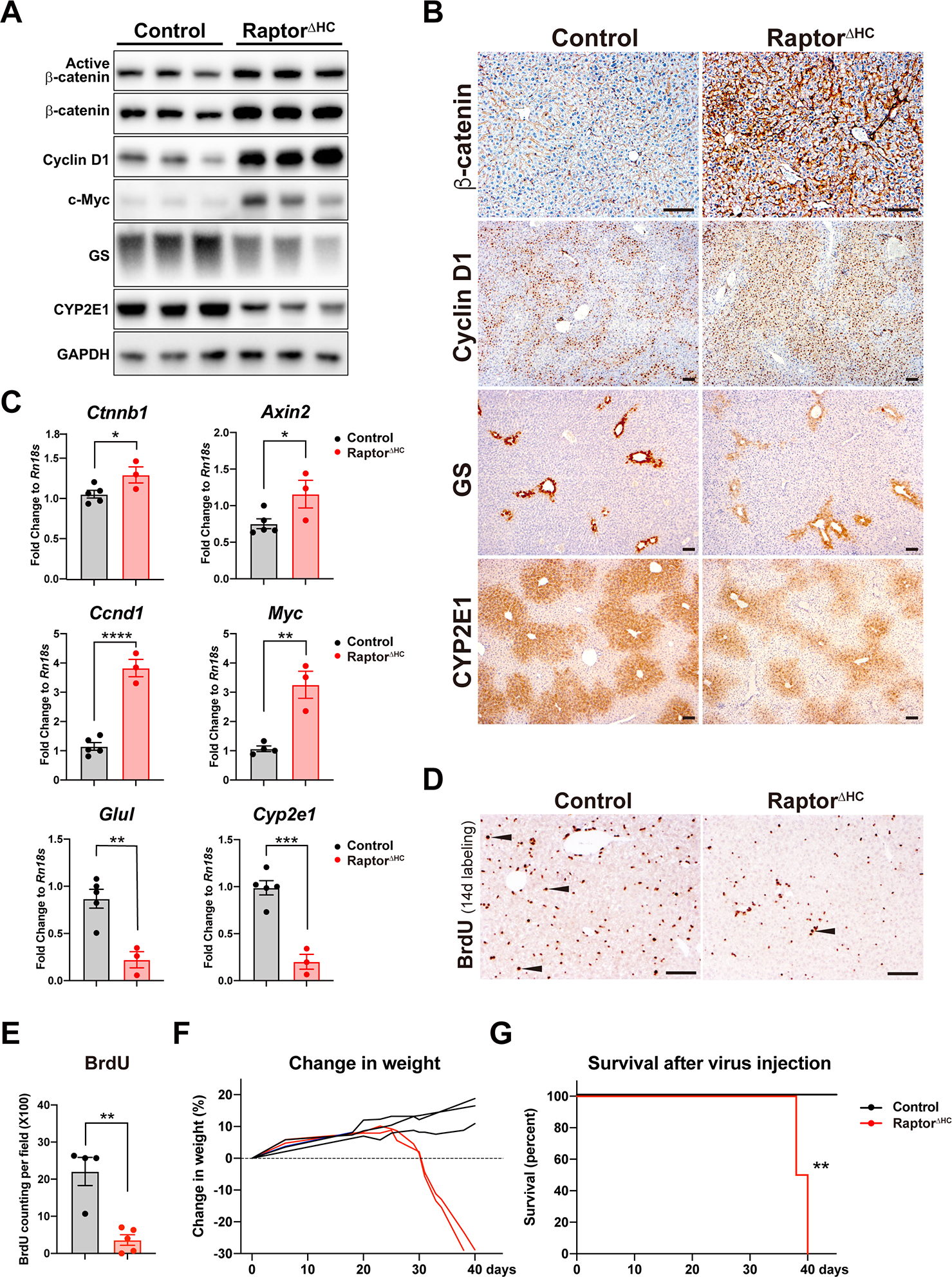
(A) WB of the Wnt-β-catenin pathway using whole liver lysates from control and RaptorΔHC mice.
(B) Representative IHC of β-catenin and its downstream targets Cyclin D1, GS, and CYP2E1 in control and RaptorΔHC mice. (Scale bars: 100 μm)
(C) qPCR of the Wnt-β-catenin pathway genes in control and RaptorΔHC mice. (Unpaired t test, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001)
(D) Representative IHC of BrdU and (E) quantification of BrdU-positive hepatocytes in control and RaptorΔHC mice after 14d of continuous BrdU administration in drinking water after virus injection. Arrowhead: BrdU-positive hepatocytes. (Scale bars: 100 um. Unpaired t test, **P < 0.01)
(F) RaptorΔHC mice exhibited progressive weight loss from 20d to 40d after virus injection.
(G) Kaplan-Meier survival curve of control and RaptorΔHC mice after virus injection. (**P < 0.01)
Because of the evidence of activation of pro-proliferative β-catenin signaling in the RaptorΔHC mice, we next wanted to directly assess the ongoing cell proliferation. BrdU was administered in drinking water for 14d to controls and RaptorΔHC mice. Intriguingly, fewer hepatocytes labeled with BrdU in the RaptorΔHC mice despite an increase in Cyclin D1- and Ki67-positive hepatocytes suggesting a failure of completion of cell cycle underscoring an important role of mTORC1 signaling in successful cell division (Fig. 5D–E).
Considering that despite compensatory β-catenin activation in the RaptorΔHC mice did not lead to successful increase in hepatocyte proliferation (Fig. 5D–E), we next decided to follow these acute hepatocyte Raptor-deleted mice for extended periods of time. Indeed, after 20d of AAV8-TBG-Cre injection, the RaptorΔHC mice stopped gaining weight but after 30d lost body weight progressively (Fig. 5F). In fact, they became increasing morbid and died by 40d thus showing significantly shorter lifespan (Fig. 5G).
Thus overall, an acute loss of Raptor and in turn mTORC1 signaling in hepatocytes leads to a global impairment of metabolic, synthetic, secretory functions, and limits overall liver repair.
Dual loss of Raptor and β-catenin from hepatocytes leads to accelerated liver injury, failure, and lethality
Considering the compensatory activation of β-catenin in RaptorΔHC mice at baseline, and the striking phenotype of deleting β-catenin in addition to Raptor deficiency in RaptorΔHC-β-cateninΔHC mice post-PH, we decided to monitor these mice for longer time without any intervention. RaptorΔHC-β-cateninΔHC mice started losing weight from 10d after virus injection. Males lost 20% body weight and females lost 25% body weight by 30d, at which time they died thus exhibiting significantly shorter survival (Fig. 6A–B). Control and β-cateninΔHC mice had no weight loss and showed normal survival during the course of the study (Fig. 6A–B).
Fig. 6. Dual loss of Raptor and β-catenin from hepatocytes leads to accelerated liver injury, failure, and lethality.
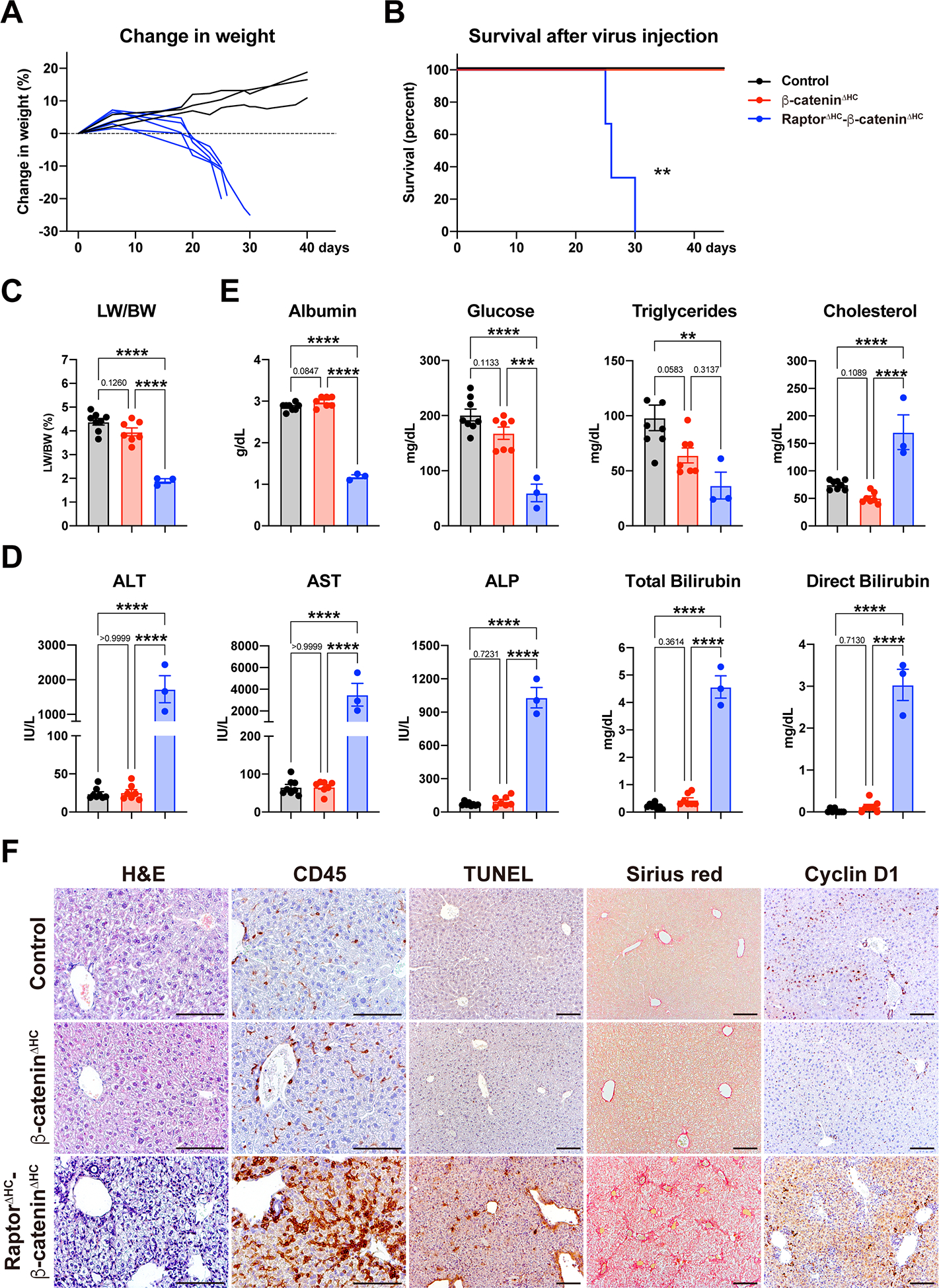
(A) RaptorΔHC-β-cateninΔHC mice showed rapid weight loss from 10d to 30d after virus injection.
(B) Kaplan-Meier survival curve of control, β-cateninΔHC, and RaptorΔHC-β-cateninΔHC mice after virus injection. (**P < 0.01)
(C) LW/BW of control, β-cateninΔHC, and RaptorΔHC-β-cateninΔHC mice 30d after virus injection. (One-way ANOVA, ****P < 0.0001)
(D) Serum levels of ALT, AST, ALP, Total Bilirubin, and Direct Bilirubin showing severe hepatocellular and cholestatic injury in RaptorΔHC-β-cateninΔHC mice. (One-way ANOVA, ****P < 0.0001)
(E) Serum metabolic profiles showing hepatic insufficiency in RaptorΔHC-β-cateninΔHC mice. (One-way ANOVA, **P < 0.01, ***P < 0.001, ****P < 0.0001)
(F) Representative H&E and IHC of CD45, TUNEL, Sirius red, and Cyclin D1. (Scale bars: 100 μm)
At the time of required euthanasia, livers of the RaptorΔHC-β-cateninΔHC mice were pale and small, and the LW/BW was only 1.89% as compared to controls and β-cateninΔHC mice at around 4–4.25% (Fig. 6C). RaptorΔHC-β-cateninΔHC mice showed significantly elevated ALT, AST, ALP, total and direct bilirubin, indicating severe hepatocellular and cholestatic injury (Fig. 6D). β-cateninΔHC and control mice showed no abnormal serum biochemistry at this time. RaptorΔHC-β-cateninΔHC mice also showed a significant decrease in serum levels of albumin, which was one-third of the controls or β-cateninΔHC mice suggesting severe defect in synthetic ability (Fig. 6E). These mice also displayed hypoglycemia and hypotriglyceridemia, all of which are signs of hepatic insufficiency (Fig. 6E). Serum cholesterol levels were significantly upregulated in the RaptorΔHC-β-cateninΔHC mice and likely due to cholestatic injury in these mice, which may lead to decreased bile acid synthesis through feedback inhibition of de novo bile acid synthesis from cholesterol leading to higher serum cholesterol (Fig. 6E). No such defects were evident in either control or β-cateninΔHC mice.
Next, we evaluated liver histology in the three groups. Both the control mice and the β-cateninΔHC mice showed normal histology overall (Fig. 6F). RaptorΔHC-β-cateninΔHC mice exhibited decreased size of liver lobules, weak eosinophilic staining of cytosol, decreased size of hepatocyte nucleus, and massive immune cell infiltration under H&E (Fig. 6F). CD45 staining showed a pan-lobular infiltration of immune cells with more cells in the pericentral region which were also F4/80 positive (Fig. 6F, S4G). Sirius red staining revealed massive portal and sinusoidal fibrosis with massive induction of αSMA-positive stellate cells (Fig. 6F, S4G). TUNEL staining revealed significantly increased cell death (Fig. 6F). β-catenin deletion in RaptorΔHC mice prevented cell proliferation as no nuclear Ki67- or Cyclin D1-positive hepatocytes were observed in the RaptorΔHC-β-cateninΔHC livers (Fig. 6F, S4G).
HNF4α is an important regulator of hepatocyte functions. Hnf4α can be transcribed in two isoforms. P1 isoform is expressed in the adult liver while P2 isoform is expressed in the fetal liver and in liver cancer, and can serve as a dominant-negative effector of the P1 form (27–29). We observed down-regulation of Hnf4α-P1 and an upregulation of Hnf4α-P2, along with a significant decrease in HNF4α positive target gene Ugt2b1 and increase in its negative target gene Akr1b7 in the RaptorΔHC-β-cateninΔHC mice, suggesting loss of an overall differentiation of the liver (Fig. S4H).
Collectively, these results show that Raptor cooperates with β-catenin in maintaining hepatic homeostasis. Loss of both proteins in mature hepatocytes leads to rapid deterioration in the hepatic function and acute liver failure. While this is a novel finding, this model is unable to allow us to investigate specifically the role and regulation of mTORC1 signaling that appears to allow LR in the absence of hepatocyte β-catenin.
Insulin/IGF1 drives mTORC1 activation and promotes compensatory adaptation in the β-cateninΔHC after PH
To further determine the mechanism and significance of delayed mTORC1 activation during LR in the β-cateninΔHC mice, we next tested various growth factors that are known to drive mTORC1 activation (30). To address the upstream effector of sustained mTORC1 activation in the β-cateninΔHC mice, we first assessed status of well-known growth factor drivers of LR including hepatocyte growth factor (HGF) and epidermal growth factor (EGF) (1, 2). HGF and EGF induce phosphorylation of receptor tyrosine kinases MET and EGFR respectively to activate Ras/Raf/MEK/ERK and/or PI3K/AKT to activate the mTORC1 signaling. We tested lysates from regenerating livers from controls and β-cateninΔHC mice for P-MET-Y1349 as a readout for MET activation but did not find any unique increase in the β-cateninΔHC livers at any time-point (Fig. S5A). We also examined P-EGFR-Y1068 as an indicator of EGFR activation and did not observe any distinct increase in β-cateninΔHC livers as well (Fig. S5B).
Insulin/Insulin-like growth factor 1 (IGF1) signaling pathway is known to regulate cell survival, growth, and proliferation and a well-known contributor to LR after PH (31, 32). In fact, the liver is the key organ in insulin-mediated regulation of metabolism and growth, and is the main secretory site of IGF1 and insulin-like growth factor binding proteins (IGFBPs) (31, 33). We asked whether compensatory mTORC1 activation evident in LR at 72h after PH in the β-cateninΔHC mice could be insulin/IGF1 dependent. Indeed, a strong insulin receptor/IGF1 receptor (INSR/IGF1R) activation was observed as seen by enhanced levels of P-INSR β-Y1150/1151 and P-IGF1R β-Y1135/1136 (Fig. 7A–B). Insulin signaling is known to phosphorylate GSK3β at serine-9 (34), which was also observed in the β-cateninΔHC livers at 72h after PH (Fig. 7A). Next, we examined gene expression of key ligands and receptors individually within the insulin growth factor signaling pathway. Igf1 expression was comparably elevated in both β-cateninΔHC and control mice at 24h post-PH (Fig. S5C). No significant differences in the expression of IGF2 (Igf2), IGF receptors Igf1r and Igf2r, and several IGFBP genes including Igfbp1, Igfbp3, Igfbp4, Igfbp5, and Igfbp6 were observed between control and β-cateninΔHC mice (Fig. S5C). IGFBP2 is known to act in part through IGF1 pathway to control hepatocyte proliferation (35). Interestingly, Igfbp2 exhibited a transient drop from 12h to 96h in β-cateninΔHC mice, while it stayed stable during LR in control mice (Fig. S5C). This could be a mechanism of impairment of hepatocyte proliferation in the β-cateninΔHC mice but could not be the basis of enhanced proliferation through higher P-INSR/IGF1R evident in the mouse livers especially at 72h post-PH. No difference was evident in the expression of insulin receptor (Insr) either across LR process between the control and β-cateninΔHC mice (Fig. S5C). In the absence of a clear upstream stimuli, we next examined insulin levels in the serum of control and β-cateninΔHC mice at 72h. Interestingly, we found significantly higher serum levels of insulin in the β-cateninΔHC as compared to control mice, which also coincided with high P-INSR β-Y1150/1151 and P-GSK3β-S9 levels (Fig. 7C). As a possible mechanism behind higher insulin levels, we next examined the expression of genes involved in gluconeogenesis positing that high glucose output in β-cateninΔHC mice to counteract the well-known hypoglycemia that occurs post PH, may be the trigger behind higher serum insulin levels (36). In fact, gluconeogenesis genes including phosphoenolpyruvate carboxykinase 1 (Pck1) and glucose 6-phosphatase (G6pc), which are normally periportally expressed, are negatively regulated by the Wnt-β-catenin signaling (37). We observed greater increase in the extent of expression of both genes especially at 24h-40h after PH in the β-cateninΔHC livers supporting our argument (Fig. S5D).
Fig. 7. Insulin-insulin receptor drives compensatory mTORC1 activation in β-cateninΔHC mice.
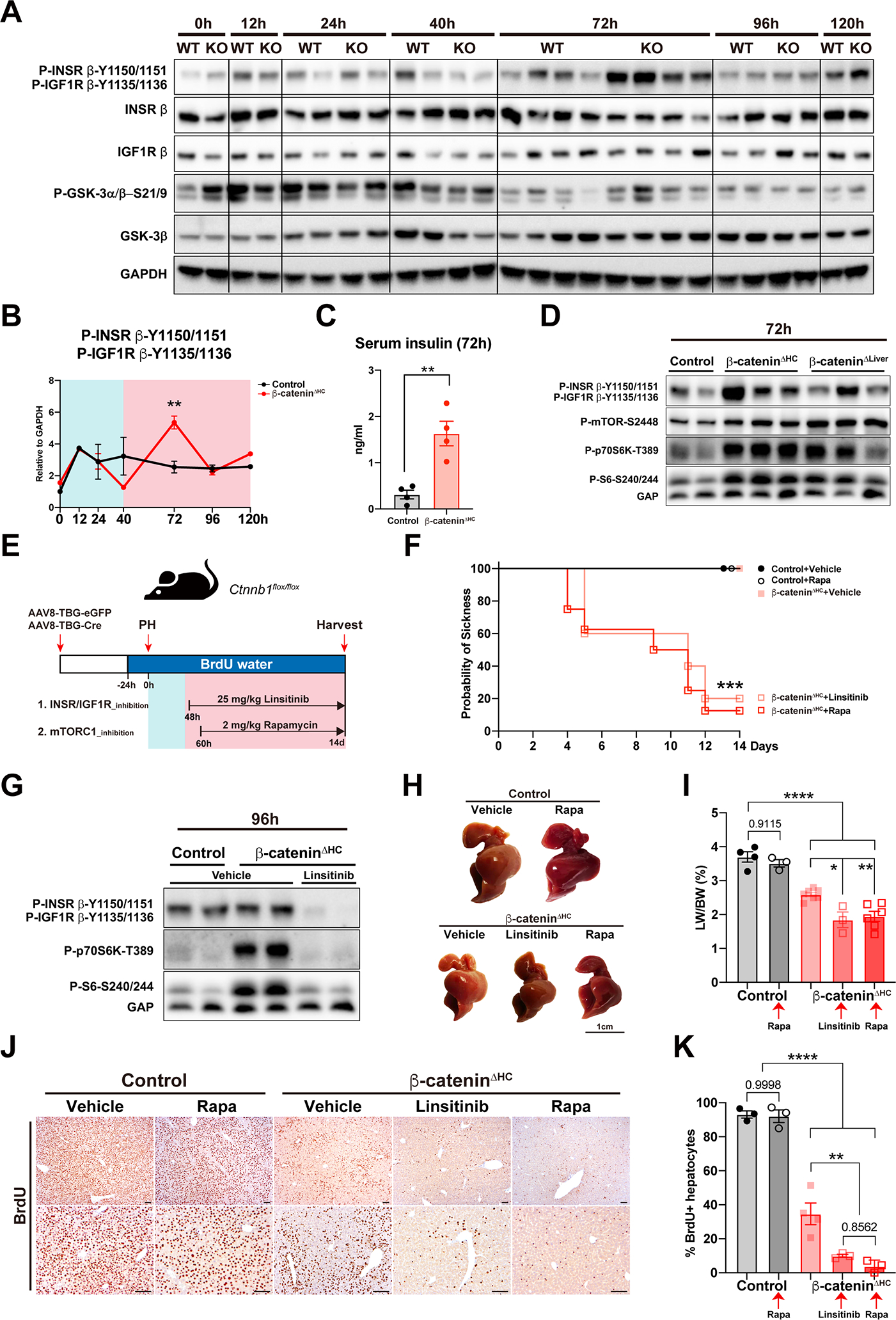
(A) WB showing high levels of P-INSR β-Y1150/1151, P-IGF1R β-Y1135/1136, and P-GSK3α/β-S21/9 at 72h using whole liver lysates from control and β-cateninΔHC mice.
(B) Quantification of P-INSR β-Y1150/1151 and P-IGF1R β-Y1135/1136 in (A). (Two-way ANOVA, **P < 0.01)
(C) ELISA of serum insulin levels at 72h post-PH in control and β-cateninΔHC mice. (Unpaired t test, **P < 0.01)
(D) WB of P-INSR β-Y1150/1151, P-IGF1R β-Y1135/1136, P-mTOR-S2448, P-p70S6K-T389, and P-S6-S240/244 at 72h post-PH using whole liver lysates from control, β-cateninΔHC, and β-cateninΔLiver mice.
(E) Schema showing experimental design of INSR/IGF1R and mTORC1 inhibition.
(F) Kaplan-Meier survival curve of vehicle or drug-treated control and β-cateninΔHC mice. (***P < 0.001)
(G) WB of P-INSR β-Y1150/1151, P-IGF1R β-Y1135/1136, P-p70S6K-T389, and P-S6-S240/244 at 96h post-PH using whole liver lysates from vehicle or drug-treated control and β-cateninΔHC mice.
(H) Gross images of livers from vehicle or drug-treated control and β-cateninΔHC mice at 14d post-PH. (Scale bars: 1 cm)
(I) LW/BW of vehicle or drug-treated control and β-cateninΔHC mice. (One-way ANOVA, *P < 0.05, **P < 0.01, ****P < 0.0001)
(J) Representative IHC and (K) quantification of BrdU in vehicle or drug-treated control and β-cateninΔHC mice. (Scale bars: 100 μm. One-way ANOVA, **P < 0.01, ****P < 0.0001)
To further validate our observations, we next examined status of mTORC1 signaling in the β-cateninΔLiver mice at 72h after PH, when there was a profound rebound of hepatocyte proliferation in these mice. Like β-cateninΔHC mice, β-cateninΔLiver mice showed increased levels of P-INSR β-Y1150/1151, P-mTOR-S2448, P-p70S6K-T389, and P-S6-S240/244 at 72h after PH (Fig. 7D).
Thus insulin-insulin receptor-mTORC1 axis seems to be activated in the absence of β-catenin in hepatocytes after PH and may be playing a role in enabling LR.
Temporal insulin or mTORC1 blockade completely prevents LR in β-cateninΔHC after PH leading to mortality
To conclusively address role of insulin-INSR-mTORC1 as the driver of LR in β-catenin-deficient mice, we administered INSR/IGF1R inhibitor linsitinib or mTORC1-specific inhibitor rapamycin to control and β-cateninΔHC mice at 48h or 60h post-PH, after the conclusion of the first peak of mTORC1 activation, and daily thereafter (Fig. 7E). Either treatment led to significantly enhanced morbidity and mortality in the β-cateninΔHC mice while no effect was observed on the control mice (Fig. 7F). Linsitinib completely blocked P-INSR β-Y1150/1151, P-IGF1R β-Y1135/1136, P-p70S6K-T389, and P-S6-S240/244 in the β-cateninΔHC mice, proving its high specificity and efficacy (Fig. 7G). Both linsitinib and rapamycin treatment prevented recovery of liver mass following PH (Fig. 7H–I) and led to a failure of hepatocyte proliferation in β-cateninΔHC mice as noted by a 14-day BrdU pulse (Fig. 7J–K), while there was a negligible effect seen in the controls.
In conclusion, we have identified insulin signaling driving mTORC1 activation leading to acute compensatory adaptation in the absence of hepatocyte β-catenin to ensure growth of the liver following PH.
Discussion
PH and living donor liver transplantation can be performed clinically because of liver’s capacity to regenerate. Preclinical models of LR have revealed multiple signaling pathways that are activated to drive the regeneration process. Redundancy among pathways during LR is also known such that inhibition of one pathway is usually compensated by activation of another, enabling continued LR. HGF/Met loss has been shown to be compensated by EGF/EGFR activation and EGF/EGFR loss by HGF/Met activation (38, 39). IL-6 or TNFα loss is compensated by unknown factor/s, but these animals also regenerate albeit with a delay (22, 40). Likewise, elimination of β-catenin from the hepithelial cells is also compensated during LR by heretofore unknown factor. Understanding these redundancies is of relevance since hepatocellular cancers (HCCs) commonly display an activation of similar pathways and inhibitors of several of these pathways are either available or on the horizon for clinical use. Judicious use of combinations in HCC will be important to prevent untoward adverse effects caused by inhibition of ongoing LR in the background diseased liver.
We also observed differences between use of Albumin-Cre mice and AAV8-TBG-Cre for floxed-gene deletion in the liver. Gene deletion by the former in the hepithelial cells begins at around embryonic day 14–16 when cholangiocytes still express Albumin and continues 2–4 weeks after birth (41), while the latter allows acute gene deletion from hepatocytes within 1–2 weeks of administration. Other than also impacting expression in cholangiocytes, genetic elimination by Albumin-cre leads to chronic adaptation in the liver cells which may confound true functions of the deleted gene. Use of acute approaches of genetic deletion disallow any adaptation and thus may report more accurate function of a gene. For example, chronic deletion of integrin-linked kinase induced chronic injury, adaptation and hepatocyte proliferation, while adenoviral-cre-mediated acute elimination led to acute widespread hepatic necrosis (42, 43). We observed differences in phenotypes when β-catenin or Raptor were deleted by AAV8-TBG-Cre versus Albumin-Cre. Mice with Albumin-Cre Raptor deletion (RaptorΔLiver) showed delayed LR after PH without impact on survival (44). However, acute Raptor deletion led to a 100% mortality by 48h after PH. β-CateninΔHC mice generated acutely showed 5–6-fold lower Ki67-positive hepatocytes compared to controls at 72h peaking at 96h (still 6-fold lower than the peak of hepatocyte proliferation in controls at 40h), whereas β-cateninΔLiver mice rebound by 72h after PH to the same level as controls at 40h (5, 11). Thus, care should be taken when interpreting results of gene deletion attained acutely versus chronically.
We also observed what appears to be a spatiotemporal wave of hepatocyte proliferation during LR after PH. Using zonal analysis for staining for Cyclin D1 and Ki67, this wave of proliferation appears at 40h from the periportal and midzonal regions and extends to the pericentral zone by 96h in controls. However, in the β-cateninΔHC mice it begins in the same directionality but at 72h and is overall severely blunted. Based on long-term BrdU incorporation, control livers show hepatocyte proliferation in all three zones, while β-cateninΔHC mice showed blunted but definite proliferation mostly in periportal zone and midzone of the hepatic lobules.
To find the mechanisms driving LR in the absence of hepatocyte β-catenin, we screened many candidates and finally identified a novel compensatory regulation of the mTORC1 and β-catenin signaling in murine livers. We found insulin being the driver of the mTORC1 activation in β-cateninΔHC mice, as linsitinib could completely block INSR activation, mTORC1 activation, and LR, leading to liver failure and demise of the animals. Insulin is produced by the pancreas and is constantly available through the portal vein. Plasma insulin levels are increased after PH in rats and dogs (45, 46). Intraportal infusion of insulin augments liver graft regeneration in patients undergoing living donor liver transplantation (47). Although there are other growth factors serving as possible stimulators of the mTORC1, pharmacological blockage of INSR using linsitinib was able to completely shut down the delayed mTORC1 signaling in β-cateninΔHC mice after PH, like mTORC1 inhibition itself, proving the significance of this unique axis in driving LR in the absence of hepatocyte β-catenin. The reason of high insulin level is unknown at this stage and requires further investigation.
We speculate glucose metabolic perturbations after surgical hepatic resection to be highly relevant in this process of sensing liver loss and triggering LR. The development of “physiological hypoglycemia” after PH has been shown to be pertinent for initiating LR and any attempt to interfere with it disrupts the process (48, 49). The development of hypoglycemia is likely due to the loss of liver mass since liver is the fundamental organ regulating systemic glucose homeostasis and may simply be due to loss of hepatic mass culminating into limited glycogen reserves along with decreased gluconeogenic output (reviewed in (36)). The hypoglycemia begins to be curtailed by adaptive responses in the form of increased hepatic gluconeogenesis and by suppression of liver glycolytic activity, which together likely limits development of sustained life-threatening hypoglycemia (50). Eventually, as hepatocyte proliferation occurs and liver mass begins to be restored, these perturbations in glucose metabolism resolve. However, in hepatocytes lacking β-catenin, the response to the physiological hypoglycemia after PH may be exacerbated. Gluconeogenesis genes are normally located in the periportal zone of the hepatic metabolic lobule and absent in pericentral hepatocytes where normal Wnt-β-catenin signaling is observed at baseline (51). The Wnt-β-catenin-TCF signaling has been shown to directly suppress gluconeogenesis in the liver through inhibition of genes like fructose-1,6-bisphosphatase (Fbp1), phosphoenolpyruvate carboxykinase 1 (Pck1), and glucose 6-phosphatase (G6pc) (52–54). In our recent unpublished work, β-catenin-deficient livers exhibit expansion of periportal genes to other metabolic zones, a phenomenon labeled as periportalization of the liver. It is conceivable that β-cateninΔHC mice after PH and in response to hypoglycemia have an exaggerated adaptation due to higher gluconeogenesis (shown by greater increases in expression of some gluconeogenesis genes) that may lead to episodic hyperglycemia, and trigger enhanced insulin secretion leading to the observed hyperinsulinemia in β-cateninΔHC mice seen at 72h after PH. Higher levels of P-GSK3β-S9 at this time also validate the ongoing insulin action on hepatocytes but the overall impact of inactivation of GSK3β in regulating glucose-glycogen balance remains incompletely understood and outside the scope of the current study (55, 56). This observed increase in serum insulin leads to engagement of insulin receptor on hepatocytes causing their activation and downstream mTORC1 activation which collaterally contributes to hepatocyte proliferation. This is an intriguing example of integration of metabolism and proliferation in the hepatocytes.
While we were trying to address the role of mTORC1 in β-cateninΔHC mice, we also were able to study RaptorΔHC mice as a genetic model of mTORC1 deficiency, in some detail. Acute elimination of hepatocyte-Raptor led to notable hepatopathy which was a result of progressive loss of hepatocyte identity and injury leading to gradual metabolic impairment, loss of weight and eventually demise by 40d. A global decrease in Hnf4α expression and function along with expression of several key genes essential for metabolic homeostasis, were seen in the RaptorΔHC livers. Eventually, there was evidence of ER stress, mitophagy and other forms of autophagy, leading to a complex phenotype encompassing cell death, immune response and compensatory LR. There appears to be evidence of Wnt-β-catenin activation leading to enhanced cell proliferation, however due to a more universal requirement of mTOR signaling in control of RNA translation and ribosomal biogenesis, eventually the activation of regenerative pathway becomes inconsequential leading to hepatic failure and mortality in mice. Intriguingly, mTORC1 signaling has been previously shown to inhibit WNT signaling (31). And ectopic activation of WNT signaling can also lead to perturbations in zonal targets of β-catenin such that zone 1 could begin to express zone 3 target genes (57). However, we don’t observe ectopic expression of WNT target genes in zone1 in RaptorΔHC livers despite enhanced ccnd1 expression. It is conceivable that the imbalance in the P1 and P2 isoforms of HNF4α observed in the RaptorΔHC livers may uncouple WNT signaling from the metabolic gene expression along with the global impact of absent mTOR on control of RNA translation and ribosomal biogenesis. Future work is needed to further dissect how loss of mTORC1 uncouples WNT signaling from regulating various metabolic and proliferative events. Nonetheless, the function of β-catenin in RaptorΔHC mice is directly evident when both Raptor and β-catenin are simultaneously deleted from hepatocytes, which resulted in accelerated morbidity and mortality.
Our findings have high translational value. While liver is innately bestowed with a capacity to regenerate and there appear to be notable fail-safe mechanisms in place, the redundancies are finite and should be carefully elucidated to prevent untoward adverse effects on liver function when using combinatorial therapies. For example, simultaneous blockade of EGF and HGF signaling in the liver led to notable morbidity and mortality (58, 59). Chronic use of mTOR inhibitors (which is not equal to genetic elimination of mTOR in hepatocytes) such as in post-transplantation indications can have metabolic side effects including dyslipidemias, metabolic syndrome and diabetes and their non-judicious use may profoundly impair hepatic function based on our findings (60). Eventually, based on our current study, we caution the use of combinatorial therapies that may concomitantly target mTORC1 and β-catenin signaling in hepatocytes. Anti-β-catenin therapies are on the horizon for treatment of subsets of cancers (61). mTOR inhibitors including more recent bi-steric and specific inhibitors of mTORC1 are also being discovered for use as anticancer agents (62). It is enticing to combine these therapies for treatment of cancers such as HCC. However, such combinations may have perilous consequences on liver function, although our studies are based only on genetic elimination of effectors of the mTOR and Wnt pathway rather than therapeutic inhibition, which may be overall less profound. However, adverse effect due to hepatocyte-specific deletion of β-catenin and Raptor alerts us to a possibility of liver injury, while therapeutic inhibition achieved by systemic administration of inhibitors of the two pathways may not be as thorough, it may confound hepatic toxicity profile due to impact on extrahepatic tissues. Also, we have previously shown that mutated- and active-β-catenin HCCs exhibit Glutamine Synthetase-Glutamine-mTORC1 activation, and these tumors respond to mTOR inhibitors, precluding the need if using anti-β-catenin agents (63). Anti-β-catenin therapy by itself is highly effective in controlling tumor burden in β-catenin-driven preclinical HCC models although such therapeutics are not yet available for use in patients (64–66). Taken together, there is no advantage of combining the two therapies, but if combined, caution should be taken. Thus, careful elucidation of the cooperative physiological functions of specific pathways may be of high significance and relevance.
Supplementary Material
Financial support statement:
This work was supported by NIH grants 1R01DK62277, 1R01DK116993, 1R01CA251155, 1R01CA204586, and Endowed Chair for Experimental Pathology to SPM, and by NIH grant 1P30DK120531 to Pittsburgh Liver Research Center (PLRC) for services provided by the ACTIC and CBRPC.
Abbreviations
- ALP
alkaline phosphatase
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- IF
immunofluorescence
- IGF1
insulin -like growth factor 1
- IGFBPs
insulin-like growth factor binding proteins; IHC, immunohistochemistry
- INSR
insulin receptor
- LR
liver regeneration
- LW/BW
liver weight to body weight ratio
- mTOR
mechanistic target of rapamycin
- mTORC1
mTOR complex 1
- PH
partial hepatectomy
- qPCR
quantitative polymerase chain reaction
- Raptor
regulatory-associated protein of mTOR
- TUNEL
terminal deoxynucleotidyl transferase dUTP nick end labeling
- WB
western blot
Footnotes
Conflict of interest statement: No conflicts to declare relevant to the current study.
References
- 1.Michalopoulos GK. Novel insights into liver homeostasis and regeneration. Nat Rev Gastroenterol Hepatol 2021;18:369–370. [DOI] [PubMed] [Google Scholar]
- 2.Michalopoulos GK, Bhushan B. Liver regeneration: biological and pathological mechanisms and implications. Nat Rev Gastroenterol Hepatol 2021;18:40–55. [DOI] [PubMed] [Google Scholar]
- 3.Monga SP, Pediaditakis P, Mule K, Stolz DB, Michalopoulos GK. Changes in WNT/beta-catenin pathway during regulated growth in rat liver regeneration. Hepatology 2001;33:1098–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goessling W, North TE, Lord AM, Ceol C, Lee S, Weidinger G, Bourque C, et al. APC mutant zebrafish uncover a changing temporal requirement for wnt signaling in liver development. Dev Biol 2008;320:161–174. [DOI] [PubMed] [Google Scholar]
- 5.Tan X, Behari J, Cieply B, Michalopoulos GK, Monga SP. Conditional deletion of beta-catenin reveals its role in liver growth and regeneration. Gastroenterology 2006;131:1561–1572. [DOI] [PubMed] [Google Scholar]
- 6.Apte U, Singh S, Zeng G, Cieply B, Virji MA, Wu T, Monga SP. Beta-catenin activation promotes liver regeneration after acetaminophen-induced injury. Am J Pathol 2009;175:1056–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhao L, Jin Y, Donahue K, Tsui M, Fish M, Logan CY, Wang B, et al. Tissue Repair in the Mouse Liver Following Acute Carbon Tetrachloride Depends on Injury-Induced Wnt/beta-Catenin Signaling. Hepatology 2019;69:2623–2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leibing T, Geraud C, Augustin I, Boutros M, Augustin HG, Okun JG, Langhans CD, et al. Angiocrine Wnt signaling controls liver growth and metabolic maturation in mice. Hepatology 2018;68:707–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ma R, Martinez-Ramirez AS, Borders TL, Gao F, Sosa-Pineda B. Metabolic and non-metabolic liver zonation is established non-synchronously and requires sinusoidal Wnts. Elife 2020;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Preziosi M, Okabe H, Poddar M, Singh S, Monga SP. Endothelial Wnts regulate beta-catenin signaling in murine liver zonation and regeneration: A sequel to the Wnt-Wnt situation. Hepatol Commun 2018;2:845–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sekine S, Gutierrez PJ, Lan BY, Feng S, Hebrok M. Liver-specific loss of beta-catenin results in delayed hepatocyte proliferation after partial hepatectomy. Hepatology 2007;45:361–368. [DOI] [PubMed] [Google Scholar]
- 12.Yang J, Mowry LE, Nejak-Bowen KN, Okabe H, Diegel CR, Lang RA, Williams BO, et al. beta-catenin signaling in murine liver zonation and regeneration: a Wnt-Wnt situation! Hepatology 2014;60:964–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sengupta S, Peterson TR, Laplante M, Oh S, Sabatini DM. mTORC1 controls fasting-induced ketogenesis and its modulation by ageing. Nature 2010;468:1100–1104. [DOI] [PubMed] [Google Scholar]
- 14.Sekine S, Lan BY, Bedolli M, Feng S, Hebrok M. Liver-specific loss of beta-catenin blocks glutamine synthesis pathway activity and cytochrome p450 expression in mice. Hepatology 2006;43:817–825. [DOI] [PubMed] [Google Scholar]
- 15.Torre C, Benhamouche S, Mitchell C, Godard C, Veber P, Letourneur F, Cagnard N, et al. The transforming growth factor-alpha and cyclin D1 genes are direct targets of beta-catenin signaling in hepatocyte proliferation. J Hepatol 2011;55:86–95. [DOI] [PubMed] [Google Scholar]
- 16.Alvarado TF, Puliga E, Preziosi M, Poddar M, Singh S, Columbano A, Nejak-Bowen K, et al. Thyroid Hormone Receptor beta Agonist Induces beta-Catenin-Dependent Hepatocyte Proliferation in Mice: Implications in Hepatic Regeneration. Gene Expr 2016;17:19–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grijalva JL, Huizenga M, Mueller K, Rodriguez S, Brazzo J, Camargo F, Sadri-Vakili G, et al. Dynamic alterations in Hippo signaling pathway and YAP activation during liver regeneration. Am J Physiol Gastrointest Liver Physiol 2014;307:G196–204. [DOI] [PubMed] [Google Scholar]
- 18.Kohler C, Bell AW, Bowen WC, Monga SP, Fleig W, Michalopoulos GK. Expression of Notch-1 and its ligand Jagged-1 in rat liver during liver regeneration. Hepatology 2004;39:1056–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lu L, Finegold MJ, Johnson RL. Hippo pathway coactivators Yap and Taz are required to coordinate mammalian liver regeneration. Exp Mol Med 2018;50:e423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ochoa B, Syn WK, Delgado I, Karaca GF, Jung Y, Wang J, Zubiaga AM, et al. Hedgehog signaling is critical for normal liver regeneration after partial hepatectomy in mice. Hepatology 2010;51:1712–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Campbell JS, Riehle KJ, Brooling JT, Bauer RL, Mitchell C, Fausto N. Proinflammatory cytokine production in liver regeneration is Myd88-dependent, but independent of Cd14, Tlr2, and Tlr4. J Immunol 2006;176:2522–2528. [DOI] [PubMed] [Google Scholar]
- 22.Sakamoto T, Liu Z, Murase N, Ezure T, Yokomuro S, Poli V, Demetris AJ. Mitosis and apoptosis in the liver of interleukin-6-deficient mice after partial hepatectomy. Hepatology 1999;29:403–411. [DOI] [PubMed] [Google Scholar]
- 23.Campbell JS, Argast GM, Yuen SY, Hayes B, Fausto N. Inactivation of p38 MAPK during liver regeneration. Int J Biochem Cell Biol 2011;43:180–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Merlen G, Gentric G, Celton-Morizur S, Foretz M, Guidotti JE, Fauveau V, Leclerc J, et al. AMPKalpha1 controls hepatocyte proliferation independently of energy balance by regulating Cyclin A2 expression. J Hepatol 2014;60:152–159. [DOI] [PubMed] [Google Scholar]
- 25.Espeillac C, Mitchell C, Celton-Morizur S, Chauvin C, Koka V, Gillet C, Albrecht JH, et al. S6 kinase 1 is required for rapamycin-sensitive liver proliferation after mouse hepatectomy. J Clin Invest 2011;121:2821–2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiang YP, Ballou LM, Lin RZ. Rapamycin-insensitive regulation of 4e-BP1 in regenerating rat liver. J Biol Chem 2001;276:10943–10951. [DOI] [PubMed] [Google Scholar]
- 27.Argemi J, Latasa MU, Atkinson SR, Blokhin IO, Massey V, Gue JP, Cabezas J, et al. Defective HNF4alpha-dependent gene expression as a driver of hepatocellular failure in alcoholic hepatitis. Nat Commun 2019;10:3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nishikawa T, Bell A, Brooks JM, Setoyama K, Melis M, Han B, Fukumitsu K, et al. Resetting the transcription factor network reverses terminal chronic hepatic failure. J Clin Invest 2015;125:1533–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Parviz F, Matullo C, Garrison WD, Savatski L, Adamson JW, Ning G, Kaestner KH, et al. Hepatocyte nuclear factor 4alpha controls the development of a hepatic epithelium and liver morphogenesis. Nat Genet 2003;34:292–296. [DOI] [PubMed] [Google Scholar]
- 30.Gruppuso PA, Sanders JA. Regulation of liver development: implications for liver biology across the lifespan. J Mol Endocrinol 2016;56:R115–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Adamek A, Kasprzak A. Insulin-Like Growth Factor (IGF) System in Liver Diseases. Int J Mol Sci 2018;19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Desbois-Mouthon C, Wendum D, Cadoret A, Rey C, Leneuve P, Blaise A, Housset C, et al. Hepatocyte proliferation during liver regeneration is impaired in mice with liver-specific IGF-1R knockout. FASEB J 2006;20:773–775. [DOI] [PubMed] [Google Scholar]
- 33.Kineman RD, Del Rio-Moreno M, Sarmento-Cabral A. 40 YEARS of IGF1: Understanding the tissue-specific roles of IGF1/IGF1R in regulating metabolism using the Cre/loxP system. J Mol Endocrinol 2018;61:T187–T198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sutherland C, Leighton IA, Cohen P. Inactivation of glycogen synthase kinase-3 beta by phosphorylation: new kinase connections in insulin and growth-factor signalling. Biochem J 1993;296 ( Pt 1):15–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wei Y, Wang YG, Jia Y, Li L, Yoon J, Zhang S, Wang Z, et al. Liver homeostasis is maintained by midlobular zone 2 hepatocytes. Science 2021;371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang J, Rudnick DA. Elucidating the metabolic regulation of liver regeneration. Am J Pathol 2014;184:309–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jin T Current understanding and dispute on the function of the Wnt signaling pathway effector TCF7L2 in hepatic gluconeogenesis. Genes Dis 2016;3:48–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Paranjpe S, Bowen WC, Bell AW, Nejak-Bowen K, Luo JH, Michalopoulos GK. Cell cycle effects resulting from inhibition of hepatocyte growth factor and its receptor c-Met in regenerating rat livers by RNA interference. Hepatology 2007;45:1471–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Paranjpe S, Bowen WC, Tseng GC, Luo JH, Orr A, Michalopoulos GK. RNA interference against hepatic epidermal growth factor receptor has suppressive effects on liver regeneration in rats. Am J Pathol 2010;176:2669–2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shimizu T, Togo S, Kumamoto T, Makino H, Morita T, Tanaka K, Kubota T, et al. Gene expression during liver regeneration after partial hepatectomy in mice lacking type 1 tumor necrosis factor receptor. J Surg Res 2009;152:178–188. [DOI] [PubMed] [Google Scholar]
- 41.Postic C, Magnuson MA. DNA excision in liver by an albumin-Cre transgene occurs progressively with age. Genesis 2000;26:149–150. [DOI] [PubMed] [Google Scholar]
- 42.Gkretsi V, Apte U, Mars WM, Bowen WC, Luo JH, Yang Y, Yu YP, et al. Liver-specific ablation of integrin-linked kinase in mice results in abnormal histology, enhanced cell proliferation, and hepatomegaly. Hepatology 2008;48:1932–1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gkretsi V, Mars WM, Bowen WC, Barua L, Yang Y, Guo L, St-Arnaud R, et al. Loss of integrin linked kinase from mouse hepatocytes in vitro and in vivo results in apoptosis and hepatitis. Hepatology 2007;45:1025–1034. [DOI] [PubMed] [Google Scholar]
- 44.Umemura A, Park EJ, Taniguchi K, Lee JH, Shalapour S, Valasek MA, Aghajan M, et al. Liver damage, inflammation, and enhanced tumorigenesis after persistent mTORC1 inhibition. Cell Metab 2014;20:133–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cohen DM, Jaspan JB, Polonsky KS, Lever EG, Moossa AR. Pancreatic hormone profiles and metabolism posthepatectomy in the dog. Evidence for a hepatotrophic role of insulin, glucagon, and pancreatic polypeptide. Gastroenterology 1984;87:679–687. [PubMed] [Google Scholar]
- 46.Kotsis T, Nastos C, Stamatis K, Chondroudaki I, Pafiti A, Frangou M, Kotsovolou V, et al. Insulin Metabolism and Assessment of Hepatic Insulin Extraction During Liver Regeneration. A Study in a Rat Model. J Invest Surg 2020;33:69–76. [DOI] [PubMed] [Google Scholar]
- 47.Xu MQ, Yan LN, Li B, Wen TF, Zeng Y, Zhao JC, Wang WT, et al. Initial clinical effect of intraportal insulin administration on liver graft regeneration in adult patients underwent living donor right lobe liver transplantation. Transplant Proc 2009;41:1698–1702. [DOI] [PubMed] [Google Scholar]
- 48.Huang J, Schriefer AE, Cliften PF, Dietzen D, Kulkarni S, Sing S, Monga SP, et al. Postponing the Hypoglycemic Response to Partial Hepatectomy Delays Mouse Liver Regeneration. Am J Pathol 2016;186:587–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Weymann A, Hartman E, Gazit V, Wang C, Glauber M, Turmelle Y, Rudnick DA. p21 is required for dextrose-mediated inhibition of mouse liver regeneration. Hepatology 2009;50:207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brinkmann A, Katz N, Sasse D, Jungermann K. Increase of the gluconeogenic and decrease of the glycolytic capacity of rat liver with a change of the metabolic zonation after partial hepatectomy. Hoppe Seylers Z Physiol Chem 1978;359:1561–1571. [DOI] [PubMed] [Google Scholar]
- 51.Soto-Gutierrez A, Gough A, Vernetti LA, Taylor DL, Monga SP. Pre-clinical and clinical investigations of metabolic zonation in liver diseases: The potential of microphysiology systems. Exp Biol Med (Maywood) 2017;242:1605–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ip W, Shao W, Chiang YT, Jin T. The Wnt signaling pathway effector TCF7L2 is upregulated by insulin and represses hepatic gluconeogenesis. Am J Physiol Endocrinol Metab 2012;303:E1166–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ip W, Shao W, Song Z, Chen Z, Wheeler MB, Jin T. Liver-specific expression of dominant-negative transcription factor 7-like 2 causes progressive impairment in glucose homeostasis. Diabetes 2015;64:1923–1932. [DOI] [PubMed] [Google Scholar]
- 54.Norton L, Fourcaudot M, Abdul-Ghani MA, Winnier D, Mehta FF, Jenkinson CP, Defronzo RA. Chromatin occupancy of transcription factor 7-like 2 (TCF7L2) and its role in hepatic glucose metabolism. Diabetologia 2011;54:3132–3142. [DOI] [PubMed] [Google Scholar]
- 55.Patel S, Doble BW, MacAulay K, Sinclair EM, Drucker DJ, Woodgett JR. Tissue-specific role of glycogen synthase kinase 3beta in glucose homeostasis and insulin action. Mol Cell Biol 2008;28:6314–6328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wan M, Leavens KF, Hunter RW, Koren S, von Wilamowitz-Moellendorff A, Lu M, Satapati S, et al. A noncanonical, GSK3-independent pathway controls postprandial hepatic glycogen deposition. Cell Metab 2013;18:99–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sun T, Annunziato S, Bergling S, Sheng C, Orsini V, Forcella P, Pikiolek M, et al. ZNRF3 and RNF43 cooperate to safeguard metabolic liver zonation and hepatocyte proliferation. Cell Stem Cell 2021;28:1822–1837 e1810. [DOI] [PubMed] [Google Scholar]
- 58.Paranjpe S, Bowen WC, Mars WM, Orr A, Haynes MM, DeFrances MC, Liu S, et al. Combined systemic elimination of MET and epidermal growth factor receptor signaling completely abolishes liver regeneration and leads to liver decompensation. Hepatology 2016;64:1711–1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tsagianni A, Mars WM, Bhushan B, Bowen WC, Orr A, Stoops J, Paranjpe S, et al. Combined Systemic Disruption of MET and Epidermal Growth Factor Receptor Signaling Causes Liver Failure in Normal Mice. Am J Pathol 2018;188:2223–2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pallet N, Legendre C. Adverse events associated with mTOR inhibitors. Expert Opin Drug Saf 2013;12:177–186. [DOI] [PubMed] [Google Scholar]
- 61.Krishnamurthy N, Kurzrock R. Targeting the Wnt/beta-catenin pathway in cancer: Update on effectors and inhibitors. Cancer Treat Rev 2018;62:50–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lee BJ, Mallya S, Dinglasan N, Fung A, Nguyen T, Herzog LO, Thao J, et al. Efficacy of a Novel Bi-Steric mTORC1 Inhibitor in Models of B-Cell Acute Lymphoblastic Leukemia. Front Oncol 2021;11:673213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Adebayo Michael AO, Ko S, Tao J, Moghe A, Yang H, Xu M, Russell JO, et al. Inhibiting Glutamine-Dependent mTORC1 Activation Ameliorates Liver Cancers Driven by beta-Catenin Mutations. Cell Metab 2019;29:1135–1150 e1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Delgado E, Okabe H, Preziosi M, Russell JO, Alvarado TF, Oertel M, Nejak-Bowen KN, et al. Complete response of Ctnnb1-mutated tumours to beta-catenin suppression by locked nucleic acid antisense in a mouse hepatocarcinogenesis model. J Hepatol 2015;62:380–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ganesh S, Koser ML, Cyr WA, Chopda GR, Tao J, Shui X, Ying B, et al. Direct Pharmacological Inhibition of beta-Catenin by RNA Interference in Tumors of Diverse Origin. Mol Cancer Ther 2016;15:2143–2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tao J, Zhang R, Singh S, Poddar M, Xu E, Oertel M, Chen X, et al. Targeting beta-catenin in hepatocellular cancers induced by coexpression of mutant beta-catenin and K-Ras in mice. Hepatology 2017;65:1581–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.