

Abstract
Post-myocardial infarction (MI), cardiac cells work together to regulate wound healing of the infarct. The pathological response to MI yields cardiac remodeling comprised of inflammatory and fibrosis phases, and the interplay in cellular dynamics that underlie these phases have not been elucidated. This study developed a computational model to identify cytokine and cellular dynamics post-MI to predict mechanisms driving post-MI inflammation, resolution of inflammation, and scar formation. Additionally, this study evaluated the interdependence between inflammation and fibrosis. Our model bypassed limitations of in vivo approaches in achieving cellular specificity and performing specific perturbations such as global knockouts of chemical factors. The model predicted that inflammation is a graded response to initial infarct size that is amplified by a positive feedback loop between neutrophils and IL-1β. Resolution of inflammation was driven by degradation of IL-1β, MMP-9, and TGFβ, as well as apoptosis of neutrophils. Inflammation regulated TGFβ secretion directly through immune cell recruitment and indirectly through upregulation of macrophage phagocytosis. Lastly, we found that mature collagen deposition was an ultrasensitive switch in response to inflammation, which was amplified primarily by cardiac fibroblast proliferation. These findings describe the relationship between inflammation and fibrosis and highlight how the two responses work together post-MI. This model revealed that post-MI inflammation and fibrosis are dynamically coupled, which provides rationale for designing novel anti-inflammatory, pro-resolving, or anti-fibrotic therapies that may improve the response to MI.
Keywords: Myocardial infarction, inflammation-fibrosis coupling, intercellular dynamics
Abstract Figure Legend:
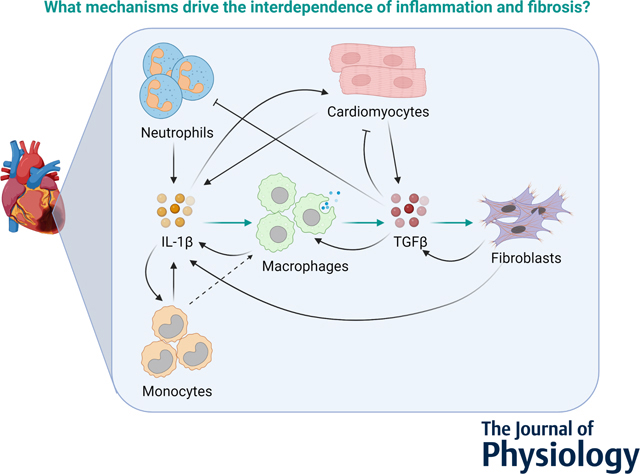
Cardiac cell communication drives wound healing post myocardial infarction. This study developed a computational model of intercellular communication in the heart and applied it to predict mechanisms of inflammation, resolution of inflammation, and fibrosis post-MI. This paper identifies crosstalk between IL-1β and TGFβ as the mechanism that drives inflammation-fibrosis coupling post-MI.
Introduction
In response to myocardial infarction (MI), the cardiac wound healing process is generally divided into three overlapping phases (Forte et al., 2018). The first phase is initiated when dying cardiomyocytes secrete signaling factors that recruit immune cells to the infarct. These infiltrating cells communicate with resident cells through paracrine signaling, working together to remove debris from the infarct. As the inflammatory response resolves, immune cells are removed from the healing infarct and the second phase of wound healing begins. Fibroblast proliferation and activation to myofibroblasts occurs, and extracellular matrix is secreted to form an infarct scar. The first pass scar evolves into a mature scar during the third phase, and myofibroblasts return to a homeostatic state. Understanding how intercellular crosstalk dynamically evolves to lead to a predictable wound healing response may help us to design more effective therapies (Ferrari & Vagnozzi, 2021; Psarras et al., 2019).
Inflammation and fibrosis act sequentially to facilitate healing after injury, and they can be considered two separate phases of infarct healing (Smolgovsky et al., 2021; Suthahar et al., 2017). Past studies have largely focused on inflammation and fibrosis individually as distinct processes, with lesser attention to their interdependence (Schroer et al., 2019; Thackeray et al., 2018). But the presence of abnormally robust inflammation early on enhances fibrosis (Bejerano et al., 2018; Dobaczewski et al., 2010; Salybekov et al., 2018; Yang et al., 2019); and likewise, impaired immune cell behavior yields poor scar formation (Hofmann et al., 2012; Wan et al., 2013). Transforming growth factor β (TGFβ) has varying effects on inflammation; its effect on immune cell infiltration post-MI is cell-specific (Feinberg et al., 2004; Ikeuchi et al., 2004; Rainer et al., 2014; Tan et al., 2010). The effects of TGFβ on inflammation resolution have not been characterized. Overall, experimental studies focused on specific aspects of inflammation or fibrosis and have not provided a whole-system perspective of interdependence. These limitations arise from lack of high cellular specificity in animal models (Dobaczewski et al., 2010), embryonic or early lethality in animal models with genetic deletion of critical factors, inability of animal models to delineate pleiotropic effects with important spatial and temporal dynamics, and the inability of in vitro models to represent complex in vivo environments (Frangogiannis, 2012). While computational systems biology approaches can be used to address these limitations, few in silico studies have modeled immune cell-fibroblast interactions in varying signaling and tissue contexts (Adler et al., 2020; Jin et al., 2011; Y. Wang et al., 2012; Zhou et al., 2018). Further, these studies were either not cardiac or incorporated only a few cell types and cytokines.
Given these challenges, there were three main objectives of this study. First was to elucidate the interplay of cytokines and cell types post-MI. Second was to predict the dynamic mechanisms driving post-MI inflammation, resolution of inflammation, and fibrosis, and third was to evaluate interdependence. To achieve these objectives, we developed a computational model of the cellular crosstalk informed by literature and optimized with ten post-MI datasets from mice. The model was validated using nine additional independent datasets from normal and stressed conditions. We perturbed the model to predict mechanisms that drive inflammation, coordinate inflammation resolution, and stimulate scar formation through an inflammation-fibrosis perspective. The model categorized inflammation as a graded response to initial infarct size and found that inflammation was amplified by a positive feedback loop between neutrophils and IL-1β. Additionally, resolution of inflammation was driven by inherent degradation of IL-1β and neutrophils, MMP-9, and negative feedback from TGFβ. The model predicted that inflammatory cytokines had a significant effect on TGFβ secretion. Lastly, collagen deposition was found to be an ultrasensitive switch in response to initial infarct size, which is amplified primarily by fibroblast proliferation.
Methods
1. Model development
An ordinary differential equation (ODE) model of the healing infarct was constructed using cells, chemical factors, and cell behavior catalogued for mouse models of MI. A literature review of mouse studies and MI was conducted to identify the main components (Anzai et al., 2017; Bujak & Frangogiannis, 2007; DeLeon-Pennell et al., 2017; Forte et al., 2020; Fu et al., 2018; Heidt et al., 2014; Leuschner et al., 2012; Ma et al., 2018; Martin & Blaxall, 2012; Prabhu & Frangogiannis, 2016; Sager et al., 2015; Tian et al., 2015). The components and datasets were chosen by searching the PubMed database for model outputs and the phrase “mice post myocardial infarction”. The cell types in the model included cardiomyocytes, neutrophils, monocytes, macrophages, and fibroblasts. The modeled chemical factors were granulocyte macrophage-colony stimulating factor (GM-CSF), interleukin-1β (IL-1β), matrix metalloproteinase-9 (MMP-9), transforming growth factor-β (TGFβ), tumor necrosis factor-α (TNFα), and collagen. The literature-informed regulation of cell behavior and cell-specific secretion of chemical factors are summarized in Tables 1 and 2.
Table 1:
Cell behavior regulated by secreted factors in the model. The up arrow indicates activation and the down arrow represents inhibition by these regulated factors.
| Regulating Factors | |||||
|---|---|---|---|---|---|
| Cell Behavior | IL-1β | TNFα | GM-CSF | MMP-9 | TGFβ |
| Cardiomyocyte cell death | ↑ | ↑ | ↓ | ||
| Neutrophil infiltration | ↑ | ↑ | ↓ | ||
| Neutrophil removal | ↓ | ↓ | |||
| Monocyte infiltration | ↑ | ↑ | |||
| Monocyte removal | ↓ | ↓ | |||
| Macrophage differentiation | ↑ | ↑ | |||
| Macrophage proliferation | ↑ | ↑ | ↑ | ||
| Macrophage removal | ↓ | ↓ | |||
| Macrophage phagocytosis | ↓ | ↑ | ↑ | ||
| IL-1β secretion by macrophages | ↓ | ||||
| TNFα secretion by macrophages | ↓ | ||||
| TGFβ secretion by macrophages | ↑ | ||||
| Fibroblast proliferation | ↑ | ||||
| TNFα secretion by fibroblasts | ↓ | ||||
| MMP-9 secretion by fibroblasts | ↑ | ||||
| TGFβ secretion by fibroblasts | ↑ | ||||
| Pro-collagen secretion by fibroblasts | ↑ | ||||
| Collagen maturation | ↓ | ||||
Table 2:
Cell sources of secreted factors in the model.
| Cell Types | |||||
|---|---|---|---|---|---|
| Secreted Factors | Cardiomyocytes | Neutrophils | Monocytes | Macrophages | Fibroblasts |
| IL-1β | ✓ | ✓ | ✓ | ✓ | ✓ |
| TNFα | ✓ | ✓ | ✓ | ✓ | ✓ |
| GM-CSF | ✓ | ✓ | ✓ | ||
| MMP-9 | ✓ | ✓ | ✓ | ✓ | ✓ |
| TGFβ | ✓ | ✓ | ✓ | ||
| Collagen | ✓ | ||||
For each cell type, the model equation consisted of a source term – representing differentiation to the modeled cell type or proliferation, and a removal term – representing apoptosis or emigration (Eq. 1).
| (1) |
Where ci is chemical factor that induces cell proliferation, Kc is the saturation constant for the chemical factor, n is the number of chemical factors regulating proliferation, kcrowd is the crowding effect, λC is the proliferation rate, and αC is the removal rate. The sources for neutrophils and monocytes were considered to be blood neutrophils and monocytes respectively, simulated by a high initial concentration of cells in the blood. A Hill function-like term was used for each cell behavior to model regulation by a chemical factor. The overall cell population in the modeled infarct was controlled by a crowding effect (Eq. 2), as modeled by Jin et al (Jin et al., 2011).
| (2) |
The crowding effect accounts for living (CM) and necrotic cardiomyocytes (CMdebris), recruited (N) and apoptotic neutrophils (Ndebris), monocytes (Mo), macrophages (Mϕ), fibroblasts (F), mature collagen (coll), and a constant term for other cells in the infarct that were not modeled. For each chemical factor, the model equation consisted of source terms that represented secretion by different cell types and a degradation term to account for natural degradation of the chemical factor (Eq. 3).
| (3) |
Where C is the secreted chemical factor, kj,C is the secretion rate of C by cell type j, and kdeg,C is the degradation rate of chemical factor C. For active TGFβ, the model equation consisted of a term for activation from latent TGFβ and degradation of TGFβ (Eq. 4).
| (4) |
Where kTGFβ,act is the activation rate of latent TGFβ to TGFβ and kTGFβ,deg is the degradation rate of active TGFβ. Lastly, collagen dynamics were modeled by two equations – one for pro-collagen (ProColl) secretion (Eq. 5) and the other for collagen maturation (MatColl) and degradation by MMP-9 (Eq. 6).
| (5) |
| (6) |
Where kproColl,F is the secretion rate of pro-collagen by fibroblasts, kcollMat is the maturation rate of pro-collagen to mature collagen, and kdeg,Coll is the degradation rate of mature collagen by MMP-9. In Eq. 5, the terms and show activation of ProColl by TGFβ and MMP-9, while the term models inhibition by mature collagen.
A total of 62 parameters were defined for this set of 43 equations. The parameter values were derived from literature and fit to experimental data (see Tables 3 and 4). The rigor and reproducibility quality of the methods for each individual article reviewed were considered while deriving the raw parameter values from literature. The articles selected for inclusion were also selected based on whether absolute value outputs of the assays (e.g. protein concentrations in ELISAs) were available. For instances when absolute values were not available, assays that provided relative values (e.g. relative expression in Western blots) were selected to guide parameter estimation. The variations in methods across the articles selected did not impact the model dynamics as the literature-derived parameters were optimized to fit experimental output time courses. Datasets with four or more timepoints were selected for calibration and digitized using webplotdigitizer (Rohatgi, 2021). The digitized data were either relative to sham or in absolute values, and were normalized using the maximum value of each dataset. Parameters were optimized using least squares minimization (lsqnonlin with the trust region reflective algorithm) and weighted with standard deviations of the respective calibration datasets. Absolute numbers and concentrations for model outputs were obtained from studies and parameters were manually scaled to ensure that the simulated and experimental outputs were on the same order. The model was implemented in MATLAB R2019b and code is made freely available at https://github.com/saucermanlab/Chowkwale_et_al_IntercellularModel.
Table 3:
Parameter values for cell behavior derived from literature and optimized using parameter estimation.
| Parameter | Description | Value | Unit | Measurement techniques | Citation |
|---|---|---|---|---|---|
| Cardiomyocyte parameters | |||||
| αCM | Apoptosis rate | 0.025 | 1/hour | -- | Estimated value |
| Neutrophil parameters | |||||
| IN | Infiltration rate | 0.095 | 1/hour | -- | Estimated value |
| αN | Removal rate | 0.525 | 1/hour | -- | Estimated value |
| ϕN | Phagocytosis rate | 2.2e-3 | 1/hour | -- | Estimated value |
| Monocyte parameters | |||||
| IMo | Infiltration rate | 0.095 | 1/hour | -- | Estimated value |
| αMo | Removal rate | 0.525 | 1/hour | -- | Estimated value |
| λMo,blood | Proliferation rate in blood | 1e-5 | 1/hour | -- | Estimated value |
| Macrophage parameters | |||||
| dM | Differentiation rate from monocytes | 0.0592 | dimensionless | Flow cytometry | (Nahrendorf et al., 2007) |
| λM | Proliferation rate | 2.3e-4 | 1/hour | Bromodeoxyuridine (BrdU) incorporation assay | (Heidt et al., 2014) |
| αM | Removal rate | 0.0175 | 1/hour | -- | Estimated value |
| ϕM | Phagocytosis rate | 0.015 | 1/hour | -- | Estimated value |
| Fibroblast parameters | |||||
| λF | Proliferation rate | 0.065 | 1/hour | Bromodeoxyuridine (BrdU) incorporation assay | (Virag & Murry, 2003) |
| αF | Removal rate | 1.5e-3 | 1/hour | -- | Estimated value |
Table 4:
Parameter values for secreted factors derived from literature and optimized using parameter estimation.
| Parameter | Description | Value | Unit | Measurement techniques | Citation |
|---|---|---|---|---|---|
| IL-1β parameters | |||||
| kIL-1β,CM | Secretion rate by cardiomyocytes | 7.55e-5 | pg/ml/cell/hour | ELISA | (Horton et al., 2006) |
| kIL-1β,N | Secretion rate by neutrophils | 0.06 | pg/ml/cell/hour | MILLIPLEX MAP mouse cytokine/chemokine panel | (McLoed et al., 2016) |
| kIL-1β,Mo | Secretion rate by monocytes | 9.58e-3 | pg/ml/cell/hour | Immunoblotting | (Gaidt et al., 2016) |
| kIL-1β,Mϕ | Secretion rate by macrophages | 0.015 | pg/ml/cell/hour | ELISA | (Meng & Lowell, 1997) |
| kIL-1β,F | Secretion rate by fibroblasts | 2.25e-3 | pg/ml/cell/hour | -- | Estimated value |
| kIL-1β,deg | Degradation rate | 0.4086 | 1/hour | Ribonuclease protection assay and curve fitting | (Dewald et al., 2004) |
| TNFα parameters | |||||
| kTNFα,CM | Secretion rate by cardiomyocytes | 5e-7 | pg/ml/cell/hour | ELISA | (Horton et al., 2006) |
| kTNFα,N | Secretion rate by neutrophils | 1.2e-3 | pg/ml/cell/hour | Immunofluorescence | (Finsterbusch et al., 2014) |
| kTNFα,Mo | Secretion rate by monocytes | 1.58e-4 | pg/ml/cell/hour | ELISA | (Matic & Simon, 1991) |
| kTNFα,Mϕ | Secretion rate by macrophages | 3.15e-4 | pg/ml/cell/hour | ELISA | (Minshawi et al., 2019) |
| kTNFα,F | Secretion rate by fibroblasts | 9.5e-5 | pg/ml/cell/hour | ELISA | (Del Re et al., 2010) |
| kTNFα,deg | Degradation rate | 0.4786 | 1/hour | Ribonuclease protection assay and curve fitting | (Deten & Zimmer, 2002) |
| GM-CSF parameters | |||||
| kGMCSF,N | Secretion rate by neutrophils | 9.6e-3 | pg/ml/cell/hour | Luminex assay | (Satoh et al., 2006) |
| kGMCSF,Mo | Secretion rate by monocytes | 1.95e-3 | pg/ml/cell/hour | Luminex assay | (Satoh et al., 2006) |
| kGMCSF,F | Secretion rate by fibroblasts | 5.24e-4 | pg/ml/cell/hour | Bio-plex assay | (Rossini et al., 2008) |
| kGMCSF,deg | Degradation rate | 3.15 | 1/hour | qRT-PCR and curve fitting | (Vandervelde et al., 2007) |
| MMP-9 parameters | |||||
| kMMP9,N | Secretion rate by neutrophils | 7.35e-4 | pg/ml/cell/hour | Gelatin zymography | (Chakrabarti et al., 2006) |
| kMMP9,Mo | Secretion rate by monocytes | 6.5e-5 | pg/ml/cell/hour | ELISA | (Nold et al., 2003) |
| kMMP9,Mϕ | Secretion rate by macrophages | 3e-5 | pg/ml/cell/hour | ELISA | (Bellosta et al., 2001) |
| kMMP9,F | Secretion rate by fibroblasts | 6.5e-5 | pg/ml/cell/hour | Immunoblotting | (Somanna et al., 2016) |
| kMMP9,deg | Degradation rate | 0.292 | 1/hour | Gelatin zymography | (F. Wang et al., 2007) |
| Latent TGFβ parameters | |||||
| klTGFB,CM | Secretion rate by cardiomyocytes | 5e-7 | pg/ml/cell/hour | ELISA | (Schindler et al., 1998) |
| klTGFB,Mϕ | Secretion rate by macrophages | 3.15e-4 | pg/ml/cell/hour | ELISA | (Schindler et al., 1998) |
| klTGFB,F | Secretion rate by fibroblasts | 9.5e-5 | pg/ml/cell/hour | Mink lung epithelial cell-bioassay | (Fisher & Absher, 1995) |
| klTGFB,deg | Degradation rate | 0.4786 | 1/hour | Ribonuclease protection assay and curve fitting | (Ikeuchi et al., 2004) |
| TGFβ parameters | |||||
| kTGFβ,act | Activation rate | 5e-7 | 1/hour | Estimated value from TGFβ activation function | (Jin et al., 2011) |
| kTGFβ,deg | Degradation rate | 0.4786 | 1/hour | Ribonuclease protection assay and curve fitting | (Ikeuchi et al., 2004) |
| Collagen and debris parameters | |||||
| kdebris | Debris conversion rate | 1 | 1/hour | -- | Estimated value |
| kproColl,F | Procollagen secretion rate by fibroblasts | 3.15e-4 | pg/ml/cell/hour | Hydroxyproline assay | (Mummidi et al., 2016) |
| kcollMat | Collagen maturation rate | 9.5e-5 | 1/hour | Immunoblotting | (Del Re et al., 2010) |
| kdeg,Coll | Degradation rate of collagen by MMP-9 | 0.4786 | 1/hour | -- | (Rikard et al., 2019) |
2. Model validation
Validation datasets were chosen from separate studies than the calibration datasets but in the same manner (see Model Development). Literature for the perturbation-validation relationships were identified by searching the PubMed database for each modeled cell or chemical factor, output, and the phrase “mice post myocardial infarction” in the PubMed database. Literature with baseline post-MI and perturbation outcomes were selected. For quality and reproducibility, only studies with mouse models of MI and those that were independent from studies used in model development were considered. Validation was performed by running a baseline simulation and a perturbed simulation to the respective timepoints, and comparing the measured output qualitatively between the two simulations. The qualitative, simulated difference – either increase, decrease, or no change from baseline - was then compared to the experimental results. Changes below a threshold were categorized as no change (Zeigler et al., 2016). To determine the validation threshold value, the validation code was run across different thresholds ranging from 0 to 1. Model predictions were robust for thresholds up to 0.05 (Figure 3E), which was then chosen for the validation study. A total of 86 perturbations were selected from 23 published studies for validation (validation table provided in the GitHub repository).
Figure 3:

Intercellular model predicted qualitative outcomes of perturbations post myocardial infarction. (A-D) Predicted qualitative response of outputs is shown in the left columns in response to their respective perturbations. Qualitative experimental outcomes are in the right columns. Red indicates an increase from baseline output, blue indicates a decrease, and white indicates no change from baseline. Overall, the model validates 61 of 84 comparisons (72.6%). (E) Robustness of experimental validation comparing model predictions with experimental literature to varying validation thresholds.
3. Simulating perturbations
For perturbations involving chemical factor inhibition or removal of cells, the degradation rates or removal rates were set to 50 and 10 times their original parameter values, respectively. For simulating a decrease in chemical factor concentration or cell counts, the degradation rates or removal rates were set to 25 or 5 times their original parameter values, respectively. A sensitivity analysis was performed by perturbing each main component and analyzing how it affected the system. The perturbed components were neutrophils, monocytes, macrophages, fibroblasts, TNFα, IL-1β, GM-CSF, TGFβ, and MMP-9. The peak values of measured outputs in perturbations were normalized by peak values from the baseline simulation. The perturbed input conditions and measured outputs were then clustered using their average correlation. A simulated infarct size of 1.0 approximated a 30% in vivo infarct size by TTC staining (Takagawa et al., 2007), while reducing it to 0.5 approximated just above 10%. For perturbing various initiating infarct size, the initial and maximum values of fibroblasts, cardiomyocytes, latent TGFβ, collagen, blood neutrophils, and blood monocytes were scaled according to the initial new infarct size.
4. Reduced fibroblast model
Tor elucidate fibroblast proliferation dynamics, a reduced, ordinary differential equation model was created with only fibroblasts (see Equation 7). In this model, fibroblast proliferation is driven by a constant TGFβ concentration, proliferation rate, and crowding effect dependent solely on fibroblast density. The maximum fibroblast density was dependent on initial infarct size. Fibroblast removal was driven by a removal rate parameter. Parameter values were the same as the original model.
| (7) |
Where F represents fibroblasts, λ is fibroblast proliferation rate, d is fibroblast removal rate, IS is infarct size, KTGFβ is the saturation constant, and Fmax is the maximum fibroblast density. The parameters were optimized such that the fibroblast dynamics matched that of the original model at an infarct size of 1. The resulting parameters are shown in Table 5.
Table 5:
Parameter values for the reduced fibroblast model.
| Parameter | Description | Value | Units |
|---|---|---|---|
| λ | Fibroblast proliferation rate | 0.15 | 1/hour |
| d | Fibroblast removal rate | 0.0035 | 1/hour |
| KTGFβ | TGFβ saturation constant | 3 | dimensionless |
| IS | Infarct size | 1 | dimensionless |
| Fmax | Maximum fibroblast density | 7000 | cells |
5. Analyses of reduced fibroblast model
To analyze the stability of the reduced fibroblast model, steady state analysis was performed on Equation 7. The equilibrium without any input to the system is at cells. With a constant input of TGFβ = 10 pg/ml, we obtained a steady state of cells. It is important to note that the steady state value depended on the value of TGFβ, as shown in Table 6 below.
Table 6:
Steady state values for the reduced fibroblast model with various TGFβ inputs.
| Input TGFβ value | Steady state, |
|---|---|
| 0 | 0 |
| 0.1 | 1936 |
| 1 | 6346 |
| 10 | 6787 |
Bifurcation analyses were performed on this reduced system to assess the location and stability of its fixed points with respect to TGFβ concentration. TGFβ was varied from 0 to 20 pg/ml, which is the maximum concentration in model output. Fixed points were calculated by finding the roots of the equation, and the stability of each point was measured by calculating the value of the derivative at that point. Fixed points of the reduced model were solved analytically as and . Evaluating the derivative of Eq. 7 at gives us the following term
| (8) |
Based on the intersection of these fixed points, a transcritical bifurcation will occur at
| (9) |
Applying the parameters in Table 5 to Equation 9, we identified a transcritical bifurcation at TGFβ = 0.0717 pg/ml in the reduced fibroblast model.
Results
1. A model of cardiac intercellular dynamics post-myocardial infarction
An ordinary differential equation (ODE) model of the intercellular dynamics in the healing infarct was constructed (Figure 1). The model represents a section of the infarct with uniform distribution of cells and chemical factors. The model integrates the main cell types from a healing infarct (Forte et al., 2020) and the secreted factors that are known to regulate healing dynamics. In a literature-informed manner, these cells and their corresponding cell behavior were modeled (Figure 1A). This included recruitment to the infarct, differentiation from precursor cell types, proliferation, removal from the infarct, and chemical secretion. The cell-specific secretion of chemical factors (Figure 1B) and regulation of cell behavior (Figure 1C) was simulated based on behavior in mouse models of MI. The detailed procedure for model construction is provided in the Methods section.
Figure 1:

A model of cardiac intercellular dynamics post myocardial infarction. (A) Cellular components of the intercellular model. (B) Network representation of chemical factors secreted by cells, and how the secretion is regulated. (C) Network representation of cell populations regulated by chemical factors, cellular sources, and other model components.
2. Intercellular model predicts dynamics seen in mice post myocardial infarction
The intercellular model predicted the dynamic response to myocardial infarction (Figure 2). The predicted dynamics showed how the cell populations changed over time in response to the changing signaling contexts. The model parameters were first calibrated against population dynamics from post-MI studies in mice and then validated against separate independent studies. The normalized predictions and calibration and validation datasets are shown in Figure 2A. For example, in response to an MI, the macrophage population increases rapidly and peaks around day 3 post-MI but is completely eliminated by day 21 post-MI. The normalized dynamics of all model outputs post-MI are shown in Figure 2B. The cells and cell behavior section shows how neutrophils peak early, followed by monocytes and macrophages, and the gradual increase in fibroblasts. The dynamics of secreted factors illustrate how the inflammatory factors IL-1β, TNFα, and GM-CSF, peak early in the time course, while TGFβ peaks during the inflammation resolution and fibroblast proliferation phases. Collagen secretion is transient while its maturation follows fibroblast population trends and gradually increases.
Figure 2:

Intercellular model predicted dynamics measured in mice post myocardial infarction (MI). (A) Predicted dynamics of selected outputs were extended for 30 days after MI. The independent datasets used to calibrate (red cross, x) and validate (black circle, o) the simulated trends are shown for comparison. (B) The predicted dynamics of all the model outputs for 30 days post-MI are shown. All data are normalized to their respective maximum values.
Furthermore, the model was validated against perturbations in mouse models of myocardial infarction. The complete annotation of the validation relationships is in the Github repository. Of 84 relationships from 23 unique journal articles, the model accurately predicted qualitative changes in 61 (72.6%) of the new articles. Figure 3 shows model validation for four types of relationships – input-output, knockout, inhibition, and overexpression. The input-output relationships show the effects of a change in cell populations; abnormal neutrophil or macrophage counts led to abnormal collagen deposition. The model predicts these collagen changes accurately. Most chemical factor knockouts lead to a decrease in experimental outputs. This illustrates how critical these factors are in the healing infarct. The model predicts these outcomes accurately, confirming that the critical nature of these factors has been incorporated in the model.
While predictions agreed with experimental outputs for 72.6% of the evaluations, there were some notable exceptions. In particular, some cell population perturbations or TGFβ inhibition did not match, which indicates complexity in the response to MI that will guide further study. For example, experimentally, neutrophil deletion or overabundance has similar results, indicating a biphasic relationship rather than monotonic response curves.
3. Post-MI inflammation reflected a graded response to the initial infarct size
With the number of positive feedback motifs in this system, we asked whether inflammation exhibits all-or-none behavior (like turning an electric toggle switch that is on or off) or graded behavior (like a continuous dimmer switch). All-or-none behavior has been shown in a number of biological systems, including action potentials (Hodgkin & Huxley, 1952) and inflammatory cytokines in the NF-κB pathway (Yde et al., 2011). Many other systems exhibit graded behavior, such as the change in postsynaptic membrane potential in response to neurotransmitter concentrations (Rien et al., 2011). To test whether post-MI inflammation is all-or-none, we perturbed the initial infarct size. With increasing initial infarct size, we observed a roughly linear change in IL-1β peak values, indicating a graded response (Figures 4A and 4B). As neutrophils were the major source of IL-1β during the early timepoints post-MI (Figure 4C), we examined whether neutrophils also exhibited a graded response to infarction. Similar to IL-1β, infiltrating neutrophils are a graded response to MI (Figures 4D and 4E). To assess the functional role of the positive feedback loop between neutrophils and IL-1β, we individually perturbed the removal rates of neutrophils and IL-1β (Figure 4F). The positive feedback increased neutrophil counts as well as IL-1β peak value, thus amplifying inflammation.
Figure 4:

Inflammation was a graded response to infarction. (A) Predicted IL-1β time courses for select initial infarct sizes. (B) Peak values plotted against initial infarct size indicate a graded increase in the inflammatory response. (C) Cell sources for overall IL-1β in the given time periods post-MI. (D) Neutrophil time courses for select initial infarct sizes. (E) Neutrophil peak values plotted against initial infarct size. (F) Perturbations of neutrophil removal rate or IL-1β degradation rate and their effects on the neutrophil-IL-1β positive feedback loop.
4. Inflammation resolution was regulated by both inflammatory and fibrotic model components
While the previous section identified drivers of inflammation, we next examined the regulators of inflammation resolution. We perturbed parameters that directly or indirectly played a role in IL-1β degradation or neutrophil removal. These perturbations were chosen because TGFβ inhibits neutrophil infiltration (Ikeuchi et al., 2004; Rainer et al., 2014) and MMP-9 inhibits neutrophil removal (Iyer et al., 2016). We varied the IL-1β degradation rate and observed a decrease in peak values of the respective IL-1β time courses (Figure 5A, left). For each perturbation, the decrease in resolution time was quantified using the T50 of IL-1β (Figure 5A, right). Varying neutrophil removal rates decreased IL-1β duration but did not have a drastic effect on the peak of IL-1β (Figure 5B). Perturbing MMP-9 degradation rates increased the T90 (Figure 5C) due to excess IL-1β secreted at late timepoints. Inhibiting TGFβ caused a similar increase in IL-1β secreted at late timepoints and increase in T90 (Figure 5D). This TGFβ negative feedback was due to delayed monocyte and macrophage removal from the system (right panel in Figure 5D compared with Figure 4C). This shows that inflammation resolution was driven by the removal of inflammatory components as well as fibrotic components that actively resolved inflammation.
Figure 5:

Inflammation resolution was regulated by removal of inflammatory and enhancement of fibrotic processes. (A) IL-1β time courses for varied IL-1β degradation rates (left) show the changes in peak values. The T50s (right) for the perturbations quantify change in inflammation resolution. (B) IL-1β curves for different neutrophil removal rates show the change in peak values and T50s show changes in inflammation resolution. (C) IL-1β time courses for varied MMP-9 degradation rates (left) and inflammation resolution quantified as T90s (right) (D) Effect of TGFβ inhibition on IL-1β duration (left), quantified by the difference in T90 (middle). Cell sources for overall IL-1β after TGFβ inhibition in the given time periods post-MI are shown on the right.
5. Multiple inflammatory mechanisms drive inflammation-fibrosis coupling post-MI
While studies have demonstrated that inflammation precedes fibrosis, the extent to which they are causally linked has not been clear. To test whether inflammation and fibrosis are directly coupled, we first looked at the predicted post-MI cell contributions of TGFβ (Figure 6A). Macrophages were a major source of TGFβ, followed by fibroblasts and then cardiomyocytes. When we individually inhibited the inflammatory cytokines, we found that IL-1β and GM-CSF had notable effects on TGFβ and its cell sources (Figure 6B). Moreover, the effects of these cytokines on cell-specific TGFβ were quantified, which showed similar regulation of TGFβ secreted by fibroblasts and macrophages. Inflammatory cytokines can directly exert their effects on TGFβ secretion through two different mechanisms – affecting cell populations or through phagocytosis regulation. Our simulation revealed that IL-1β inhibition and GM-CSF inhibition decreased macrophage and fibroblast counts, as well as ingested cell debris (Figure 6C). The decrease in macrophage counts and ingested debris caused a decrease in TGFβ. TNFα did not change macrophage numbers as drastically, and also increased ingested debris. This opposing effect led to an insignificant change in secreted TGFβ. Lastly, we perturbed phagocytosis mechanisms to see the effects on TGFβ secretion (Figure 6D). Together, these simulations identified IL-1β and macrophage phagocytosis of cardiomyocytes as drivers of TGFβ secretion by macrophages.
Figure 6:

Multiple inflammatory mechanisms drive inflammation-fibrosis coupling post-myocardial infarction. (A) Cell sources of TGFβ post MI, followed by fibroblasts. (B) Roles of inflammatory cytokines IL-1β, GM-CSF, and TNFa in TGFβ secretion post MI. (C) Effect of inflammatory cytokines on cell counts, debris, and phagocytosis. (D) Effects of various phagocytosis mechanisms on overall and macrophage TGFβ secretion. The relative area under the curve represents a cumulative sum of the secreted factors in simulated time course.
6. Collagen is an ultrasensitive switch in response to initial infarct size
In the previous sections, we found that inflammation showed a graded response to initial infarct size, and a coupled relationship between inflammation and fibrosis was established. Following this, we wanted to characterize the dynamics of collagen deposition. Firstly, we perturbed initial infarct size and observed that collagen deposition was an ultrasensitive switch, with a Hill coefficient of 9.56 (Figure 7A). Individually inhibiting inflammatory cytokines IL-1β and GM-CSF prevented excess collagen deposition (Figure 7B). Moreover, collagen deposition was desensitized to changes in initial infarct size in the absence of inflammatory cytokines (Figure 7B).
Figure 7:

Mature collagen deposition was an ultrasensitive switch in response to initial infarct size. (A) Peak values of collagen deposition plotted against infarct size indicated an ultrasensitive switch, with a Hill coefficient of 9.56. (B) Roles of inflammatory cytokines IL-1β, GM-CSF, and TNFa in collagen deposition post MI for different initial infarct sizes. (C) Peak values of model components upstream of collagen indicated ultrasensitivity, amplified by fibroblasts. (D) Bifurcation analysis of a reduced model with TGFβ concentration, as the bifurcation parameter revealed a transcritical bifurcation. Filled circles: stable fixed points; open circles: unstable fixed points. Inset: bifurcation analysis with low concentrations of TGFβ. (E and F) Bifurcation analysis of the reduced model with either (E) fibroblasts or (F) crowding term removed from the proliferation term.
To look at what drives the ultrasensitivity of collagen, we plotted model components upstream of collagen deposition – collagen secretion, the number of fibroblasts, and TGFβ. We found that TGFβ, fibroblasts, and collagen secretion were all ultrasensitive to initial infarct size, with the number of fibroblasts being most sensitive (Figure 7C). To identify the dynamics that underlie the ultrasensitivity in a controlled setting, we created a reduced one-variable ordinary differential equation model of fibroblast numbers (see Methods). The reduced model had one differential equation for fibroblasts and was dependent on constant values for proliferation and removal rates, initial infarct size, and TGFβ. Performing a bifurcation analysis using TGFβ concentration as the varied parameter revealed a transcritical bifurcation (Figure 7D). We solved the transcritical bifurcation analytically (refer to Methods section) and found that it occurs at a TGFβ concentration of 0.0717 pg/ml (shown in inset).
To identify which aspects of the model contribute to this bifurcation, we separately removed fibroblasts and the crowding term from the source term of the equation (Figures 7E and 7F). In case of removing the fibroblast variable, the system had one stable fixed point whose value increased non-linearly to very low fibroblast numbers. On removing the crowding effect, the fibroblasts proliferated uncontrollably as it only had one unstable fixed point at zero fibroblasts. This prediction was consistent with previous experiments that deleted the Hippo pathway in mice, which resulted in similar spontaneous fibroblast proliferation and excessive MI fibrosis (Xiao et al., 2019). Thus, the reduced model identified the inherent nature of proliferation as the cause of ultrasensitivity in the collagen deposition.
Discussion
Wound healing post-MI is a complex process involving a tightly controlled inflammatory response, activation of fibroblasts, and crosstalk between resident and recruited cells. Excessive wound healing can lead to future infarctions, arrhythmias, and eventually heart failure (Talman & Ruskoaho, 2016). Optimal therapeutic wound healing will be driven by a more complete understanding of how cells interact with each other to govern cardiac function in health and dysfunction in disease (Ferrari & Vagnozzi, 2021). Towards this goal, we developed a computational model of intercellular dynamics, which was validated using time course and perturbation data from post-MI mouse studies. The model predicted that inflammation is a graded response to initial infarct size, while fibrosis is an ultrasensitive switch in response to initial infarct size. The model predicts that inflammation is amplified by neutrophil-IL-1β positive feedback, and resolution is driven by IL-1β degradation, neutrophil apoptosis, MMP-9, and TGFβ negative feedback. TGFβ secretion and mature collagen deposition are dependent on inflammatory cytokines and macrophage phagocytosis. This confirms an interdependence between inflammation and fibrosis post-MI, leading to the concept of inflammation-fibrosis coupling. Additionally, mature collagen deposition was amplified by fibroblast proliferation and limited by cell crowding.
Inflammation and fibrosis are considered important pathophysiological phases of post-MI wound healing. However, the interplay of their nonlinear dynamics has not been analyzed. In abstract tissue, models predicted that inflammatory signaling may be excitable, with all-or-none responses beyond a threshold (Yde et al., 2011). In contrast, our model analyses predict that post-MI inflammation is a graded response to initial infarct size. Moreover, the inflammatory response was proportional to the initial infarct size, which avoids overresponse in the system. Identifying the cell circuits that regulate post-MI inflammation provides therapeutic targets that overcome the general challenges in anti-inflammatory therapies (Huang & Frangogiannis, 2018). In addition to immune cells, fibroblasts have also been shown to exhibit nonlinear dynamics such as bistability (Yeo et al., 2018). This led to an interest in systems properties of fibroblast behavior post myocardial infarction in the current study. Fibrosis, represented by collagen secretion, was found to be ultrasensitive to initial infarct size. Fibroblast proliferation increased overall collagen deposition in the infarct, and was found to be ultrasensitive to smaller TGFβ concentrations. Components amplifying collagen sensitivity - fibroblast proliferation, TGFβ, and initial infarct size – can provide targets for anti-fibrotic therapies post MI while ensuring that there is sufficient wound healing, eventually preventing rupture.
The intense inflammatory response post-MI, including the roles of immune cells and cytokines, has been characterized and extensively reviewed (Ong et al., 2018; Smolgovsky et al., 2021). However, the interplay of cytokines and immune cells has not been elucidated in a systemic manner. Moreover, the model predictions may have a number of implications regarding therapeutics that target inflammation or fibrosis post-MI. The model predicted that the positive feedback loop between neutrophils and IL-1β amplifies inflammation; this can be a target of anti-inflammatory therapies (Huang & Frangogiannis, 2018). Anakinra, an IL-1β receptor antagonist, has been used clinically as treatment post myocardial infarction (Abbate et al., 2008), as well as other pathologies such as autoinflammatory diseases, heart disease, diabetes (Cavalli & Dinarello, 2018). Similar to the model prediction, the use of anakinra to block IL-1β has shown reduction in neutrophils and white blood cells in patients (Del Buono et al., 2022). Inflammation resolution is characterized by apoptosis of neutrophils and their subsequent clearance from the infarct. This indicates a role for pro-resolving macrophages (Ong et al., 2018). There have been several studies looking at pro-resolving therapies (Al-Darraji et al., 2018; Bejerano et al., 2018; Cheng et al., 2020; Salybekov et al., 2018), however these prematurely initiate the proliferative phase of wound healing instead of directly targeting inflammation resolution (Leoni & Soehnlein, 2018). These indirect targets have effects outside of the infarct that can be harmful to patients with higher cardiovascular risk (Kain et al., 2014). Hence, direct mechanisms that drive inflammation resolution identified by the model, such as IL-1β, MMP-9, and TGFβ degradation and neutrophil apoptosis, provide novel targets for pro-resolving therapies. While the effect of inflammation on fibrosis has been described (Bejerano et al., 2018; Dobaczewski et al., 2010; Hofmann et al., 2012; Salybekov et al., 2018; Wan et al., 2013; Yang et al., 2019), the mechanisms through which inflammation regulates fibrotic model components had not been characterized. Our model shows that inflammatory cytokines mediate TGFβ secretion through a direct effect on macrophage cell counts, and indirectly by regulating phagocytosis. Additionally, mature collagen deposition was found to be ultrasensitive to TGFβ, and desensitized to initial infarct size in the absence of inflammatory cytokines. To the best of our knowledge, a coupling relationship between inflammation and fibrosis had not been established prior to this study.
Our model predicts dynamics for acute, transient inflammation post myocardial infarction. In the presence of chronic inflammation due to conditions such as cardiometabolic defects, aging, or co-medications, there is a dysregulation of inflammation resolution (Halade & Lee, 2022; Kolpakov et al., 2020). This model could be further extended to predict intercellular dynamics in the presence of chronic post-MI inflammation. Moreover, the model does not include tissue-resident macrophages (Dick et al., 2019; Jia et al., 2022; Nahrendorf et al., 2007). Tissue-resident macrophages are lost post-MI, but they recover to pre-infarct levels by around 4 weeks after infarction (Dick et al., 2019). Sufficient data was not available to model the dynamics of tissue-resident macrophages. However, the model accurately simulates the behavior of monocyte-derived macrophages post myocardial infarction.
Our use of the term inflammation-fibrosis coupling is inspired by cardiac excitation-contraction coupling (Bers, 2002), while acknowledging that this phenomenon is on a different spatiotemporal scale. The transient dynamics of inflammation are reminiscent of an action potential. But in contrast to all-or-none action potentials, post-MI inflammation appears graded. Analogous to how the action potential is driven by positive feedback between Na+ current and voltage, here we find that post-MI inflammation is amplified by a neutrophil-IL-1β positive feedback loop. Action potential repolarization (driven by K+ currents) is dynamically similar to inflammation resolution (by IL-1β degradation and neutrophil apoptosis). Membrane depolarization triggers release of Ca2+, which activates the myofilaments that cause contraction. Similarly, TGFβ is essential for fibrotic activity and plays a direct role in fibroblast proliferation, which then leads to collagen deposition. Intriguingly, we predicted that collagen is ultrasensitive to TGFβ, much the way force production is ultrasensitive to Ca2+. The intensity of the coupling effect can be modulated by altering amplitude of inflammation or altering sensitivity of fibroblasts to TGFβ. In conclusion, we discerned a set of mechanisms driving inflammation, fibrosis, and inflammation-fibrosis by building and applying a comprehensive model of intercellular dynamics post myocardial infarction.
Supplementary Material
Key points.
Inflammation and matrix remodeling are two processes involved in wound healing after a heart attack.
Cardiac cells work together to facilitate these processes; this is done by secreting cytokines that then regulate the cells themselves or other cells surrounding them.
This study developed a computational model of the dynamics of cardiac cells and cytokines to predict mechanisms through which inflammation and matrix remodeling is regulated.
We show the roles of various cytokines and signaling motifs in driving inflammation, resolution of inflammation, and fibrosis.
The novel concept of inflammation-fibrosis coupling, based on the model prediction that inflammation and fibrosis are dynamically coupled, provides rationale for future studies and designing therapeutics to improve the response after a heart attack.
Funding
American Heart Association Predoctoral Fellowship (#830266), National Institutes of Health HL137755, HL162925, GM115458, and HL137319, and the Department of Veterans Affairs 5I01BX000505.
First author profile
Mukti Chowkwale (she/her) is currently a PhD candidate in the Cardiac Systems Pharmacology Group in the Biomedical Engineering department at the University of Virginia. She has a background in computer engineering (Bachelor’s degree, University of Pune, India) and biomedical engineering (Master’s degree, Binghamton University, NY). She enjoys developing computational models of biological processes, and applying them to uncover novel biological dynamics. She wants to apply her knowledge to inflammation and fibrosis in cardiac diseases and women’s health. Her current research focuses on systems biology approaches to delineate inter- and intra-cellular dynamics in the heart.
Footnotes
Additional Information
Competing interests
None
Data availability statement
The model code and the scripts used to generate the figures are available at https://github.com/saucermanlab/Chowkwale_et_al_IntercellularModel.
References
- Abbate A, Salloum FN, Vecile E, Das A, Hoke NN, Straino S, Biondi-Zoccai GGL, Houser J-E, Qureshi IZ, Ownby ED, Gustini E, Biasucci LM, Severino A, Capogrossi MC, Vetrovec GW, Crea F, Baldi A, Kukreja RC, & Dobrina A. (2008). Anakinra, a Recombinant Human Interleukin-1 Receptor Antagonist, Inhibits Apoptosis in Experimental Acute Myocardial Infarction. Circulation, 117(20), 2670–2683. 10.1161/CIRCULATIONAHA.107.740233 [DOI] [PubMed] [Google Scholar]
- Adler M, Mayo A, Zhou X, Franklin RA, Meizlish ML, Medzhitov R, Kallenberger SM, & Alon U. (2020). Principles of Cell Circuits for Tissue Repair and Fibrosis. IScience, 23(2). 10.1016/j.isci.2020.100841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Darraji A, Haydar D, Chelvarajan L, Tripathi H, Levitan B, Gao E, Venditto VJ, Gensel JC, Feola DJ, & Abdel-Latif A. (2018). Azithromycin therapy reduces cardiac inflammation and mitigates adverse cardiac remodeling after myocardial infarction: Potential therapeutic targets in ischemic heart disease. PloS One, 13(7), e0200474. 10.1371/journal.pone.0200474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anzai A, Choi JL, He S, Fenn AM, Nairz M, Rattik S, McAlpine CS, Mindur JE, Chan CT, Iwamoto Y, Tricot B, Wojtkiewicz GR, Weissleder R, Libby P, Nahrendorf M, Stone JR, Becher B, & Swirski FK. (2017). The infarcted myocardium solicits GM-CSF for the detrimental oversupply of inflammatory leukocytes. The Journal of Experimental Medicine, 214(11), 3293–3310. 10.1084/jem.20170689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bejerano T, Etzion S, Elyagon S, Etzion Y, & Cohen S. (2018). Nanoparticle Delivery of miRNA-21 Mimic to Cardiac Macrophages Improves Myocardial Remodeling after Myocardial Infarction. Nano Letters, 18(9), 5885–5891. 10.1021/acs.nanolett.8b02578 [DOI] [PubMed] [Google Scholar]
- Bellosta S, Canavesi M, Favari E, Cominacini L, Gaviraghi G, Fumagalli R, Paoletti R, & Bernini F. (2001). Lacidipine [correction of Lalsoacidipine] modulates the secretion of matrix metalloproteinase-9 by human macrophages. The Journal of Pharmacology and Experimental Therapeutics, 296(3), 736–743. [PubMed] [Google Scholar]
- Bers DM. (2002). Cardiac excitation–contraction coupling. Nature, 415(6868), 198–205. 10.1038/415198a [DOI] [PubMed] [Google Scholar]
- Bujak M, & Frangogiannis NG. (2007). The role of TGF-beta signaling in myocardial infarction and cardiac remodeling. Cardiovascular Research, 74(2), 184–195. 10.1016/j.cardiores.2006.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalli G, & Dinarello CA. (2018). Anakinra Therapy for Non-cancer Inflammatory Diseases. Frontiers in Pharmacology, 9, 1157. 10.3389/fphar.2018.01157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti S, Zee JM, & Patel KD. (2006). Regulation of matrix metalloproteinase-9 (MMP-9) in TNF-stimulated neutrophils: Novel pathways for tertiary granule release. Journal of Leukocyte Biology, 79(1), 214–222. 10.1189/jlb.0605353 [DOI] [PubMed] [Google Scholar]
- Cheng Y, Luo D, Zhao Y, & Rong J. (2020). N-Propargyl caffeate amide (PACA) prevents cardiac fibrosis in experimental myocardial infarction by promoting pro-resolving macrophage polarization. Aging, 12(6), 5384–5398. 10.18632/aging.102959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Buono MG, Damonte JI, Trankle CR, Kadariya D, Carbone S, Thomas G, Turlington J, Markley R, Canada JM, Biondi‐Zoccai GG, Kontos MC, Van Tassell BW, & Abbate A. (2022). Effect of interleukin-1 blockade with anakinra on leukocyte count in patients with ST-segment elevation acute myocardial infarction. Scientific Reports, 12(1), 1254. 10.1038/s41598-022-05374-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Re DP, Matsuda T, Zhai P, Gao S, Clark GJ, Van Der Weyden L, & Sadoshima J. (2010). Proapoptotic Rassf1A/Mst1 signaling in cardiac fibroblasts is protective against pressure overload in mice. The Journal of Clinical Investigation, 120(10), 3555–3567. 10.1172/JCI43569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLeon-Pennell KY, Meschiari CA, Jung M, & Lindsey ML. (2017). Matrix Metalloproteinases in Myocardial Infarction and Heart Failure. Progress in Molecular Biology and Translational Science, 147, 75–100. 10.1016/bs.pmbts.2017.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deten A, & Zimmer H-G. (2002). Heart function and cytokine expression is similar in mice and rats after myocardial infarction but differences occur in TNFalpha expression. Pflugers Archiv: European Journal of Physiology, 445(2), 289–296. 10.1007/s00424-002-0930-x [DOI] [PubMed] [Google Scholar]
- Dewald O, Ren G, Duerr GD, Zoerlein M, Klemm C, Gersch C, Tincey S, Michael LH, Entman ML, & Frangogiannis NG. (2004). Of mice and dogs: Species-specific differences in the inflammatory response following myocardial infarction. The American Journal of Pathology, 164(2), 665–677. 10.1016/S0002-9440(10)63154-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick SA, Macklin JA, Nejat S, Momen A, Clemente-Casares X, Althagafi MG, Chen J, Kantores C, Hosseinzadeh S, Aronoff L, Wong A, Zaman R, Barbu I, Besla R, Lavine KJ, Razani B, Ginhoux F, Husain M, Cybulsky MI, … Epelman S. (2019). Self-renewing resident cardiac macrophages limit adverse remodeling following myocardial infarction. Nature Immunology, 20(1), 29–39. 10.1038/s41590-018-0272-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobaczewski M, Xia Y, Bujak M, Gonzalez-Quesada C, & Frangogiannis NG. (2010). CCR5 signaling suppresses inflammation and reduces adverse remodeling of the infarcted heart, mediating recruitment of regulatory T cells. The American Journal of Pathology, 176(5), 2177–2187. 10.2353/ajpath.2010.090759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinberg MW, Shimizu K, Lebedeva M, Haspel R, Takayama K, Chen Z, Frederick JP, Wang X-F, Simon DI, Libby P, Mitchell RN, & Jain MK. (2004). Essential role for Smad3 in regulating MCP-1 expression and vascular inflammation. Circulation Research, 94(5), 601–608. 10.1161/01.RES.0000119170.70818.4F [DOI] [PubMed] [Google Scholar]
- Ferrari I, & Vagnozzi RJ. (2021). Mechanisms and strategies for a therapeutic cardiac immune response. Journal of Molecular and Cellular Cardiology, 158, 82–88. 10.1016/j.yjmcc.2021.05.013 [DOI] [PubMed] [Google Scholar]
- Finsterbusch M, Voisin M-B, Beyrau M, Williams TJ, & Nourshargh S. (2014). Neutrophils recruited by chemoattractants in vivo induce microvascular plasma protein leakage through secretion of TNF. The Journal of Experimental Medicine, 211(7), 1307–1314. 10.1084/jem.20132413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher SA, & Absher M. (1995). Norepinephrine and ANG II stimulate secretion of TGF-beta by neonatal rat cardiac fibroblasts in vitro. The American Journal of Physiology, 268(4 Pt 1), C910–917. 10.1152/ajpcell.1995.268.4.C910 [DOI] [PubMed] [Google Scholar]
- Forte E, Furtado MB, & Rosenthal N. (2018). The interstitium in cardiac repair: Role of the immune-stromal cell interplay. Nature Reviews. Cardiology, 15(10), 601–616. 10.1038/s41569-018-0077-x [DOI] [PubMed] [Google Scholar]
- Forte E, Skelly DA, Chen M, Daigle S, Morelli KA, Hon O, Philip VM, Costa MW, Rosenthal NA, & Furtado MB. (2020). Dynamic Interstitial Cell Response during Myocardial Infarction Predicts Resilience to Rupture in Genetically Diverse Mice. Cell Reports, 30(9), 3149–3163.e6. 10.1016/j.celrep.2020.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frangogiannis NG. (2012). Regulation of the inflammatory response in cardiac repair. Circulation Research, 110(1), 159–173. 10.1161/CIRCRESAHA.111.243162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu X, Khalil H, Kanisicak O, Boyer JG, Vagnozzi RJ, Maliken BD, Sargent MA, Prasad V, Valiente-Alandi I, Blaxall BC, & Molkentin JD. (2018). Specialized fibroblast differentiated states underlie scar formation in the infarcted mouse heart. The Journal of Clinical Investigation, 128(5), 2127–2143. 10.1172/JCI98215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaidt MM, Ebert TS, Chauhan D, Schmidt T, Schmid-Burgk JL, Rapino F, Robertson AAB, Cooper MA, Graf T, & Hornung V. (2016). Human Monocytes Engage an Alternative Inflammasome Pathway. Immunity, 44(4), 833–846. 10.1016/j.immuni.2016.01.012 [DOI] [PubMed] [Google Scholar]
- Halade GV, & Lee DH. (2022). Inflammation and resolution signaling in cardiac repair and heart failure. EBioMedicine, 79. 10.1016/j.ebiom.2022.103992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidt T, Courties G, Dutta P, Sager HB, Sebas M, Iwamoto Y, Sun Y, Da Silva N, Panizzi P, van der Laan AM, van der Lahn AM, Swirski FK, Weissleder R, & Nahrendorf M. (2014). Differential contribution of monocytes to heart macrophages in steady-state and after myocardial infarction. Circulation Research, 115(2), 284–295. 10.1161/CIRCRESAHA.115.303567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkin AL, & Huxley AF. (1952). A quantitative description of membrane current and its application to conduction and excitation in nerve. The Journal of Physiology, 117(4), 500–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann U, Beyersdorf N, Weirather J, Podolskaya A, Bauersachs J, Ertl G, Kerkau T, & Frantz S. (2012). Activation of CD4+ T lymphocytes improves wound healing and survival after experimental myocardial infarction in mice. Circulation, 125(13), 1652–1663. 10.1161/CIRCULATIONAHA.111.044164 [DOI] [PubMed] [Google Scholar]
- Horton J, Maass D, White J, & Sanders B. (2006). Effect of aspiration pneumonia-induced sepsis on post-burn cardiac inflammation and function in mice. Surgical Infections, 7(2), 123–135. 10.1089/sur.2006.7.123 [DOI] [PubMed] [Google Scholar]
- Huang S, & Frangogiannis NG. (2018). Anti‐inflammatory therapies in myocardial infarction: Failures, hopes and challenges. British Journal of Pharmacology, 175(9), 1377–1400. 10.1111/bph.14155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeuchi M, Tsutsui H, Shiomi T, Matsusaka H, Matsushima S, Wen J, Kubota T, & Takeshita A. (2004). Inhibition of TGF-beta signaling exacerbates early cardiac dysfunction but prevents late remodeling after infarction. Cardiovascular Research, 64(3), 526–535. 10.1016/j.cardiores.2004.07.017 [DOI] [PubMed] [Google Scholar]
- Iyer RP, de Castro Brás LE, Patterson NL, Bhowmick M, Flynn ER, Asher M, Cannon PL, Deleon-Pennell KY, Fields GB, & Lindsey ML. (2016). Early matrix metalloproteinase-9 inhibition post-myocardial infarction worsens cardiac dysfunction by delaying inflammation resolution. Journal of Molecular and Cellular Cardiology, 100, 109–117. 10.1016/j.yjmcc.2016.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia D, Chen S, Bai P, Luo C, Liu J, Sun A, & Ge J. (2022). Cardiac Resident Macrophage-Derived Legumain Improves Cardiac Repair by Promoting Clearance and Degradation of Apoptotic Cardiomyocytes After Myocardial Infarction. Circulation, 145(20), 1542–1556. 10.1161/CIRCULATIONAHA.121.057549 [DOI] [PubMed] [Google Scholar]
- Jin Y-F, Han H-C, Berger J, Dai Q, & Lindsey ML. (2011). Combining experimental and mathematical modeling to reveal mechanisms of macrophage-dependent left ventricular remodeling. BMC Systems Biology, 5, 60. 10.1186/1752-0509-5-60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kain V, Prabhu SD, & Halade GV. (2014). Inflammation revisited: Inflammation versus resolution of inflammation following myocardial infarction. Basic Research in Cardiology, 109(6), 444. 10.1007/s00395-014-0444-7 [DOI] [PubMed] [Google Scholar]
- Kolpakov MA, Guo X, Rafiq K, Vlasenko L, Hooshdaran B, Seqqat R, Wang T, Fan X, Tilley DG, Kostyak JC, Kunapuli SP, Houser SR, & Sabri A. (2020). Loss of Protease-Activated Receptor 4 Prevents Inflammation Resolution and Predisposes the Heart to Cardiac Rupture After Myocardial Infarction. Circulation, 142(8), 758–775. 10.1161/CIRCULATIONAHA.119.044340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leoni G, & Soehnlein O. (2018). (Re) Solving Repair After Myocardial Infarction. Frontiers in Pharmacology, 9. https://www.frontiersin.org/article/10.3389/fphar.2018.01342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leuschner F, Rauch PJ, Ueno T, Gorbatov R, Marinelli B, Lee WW, Dutta P, Wei Y, Robbins C, Iwamoto Y, Sena B, Chudnovskiy A, Panizzi P, Keliher E, Higgins JM, Libby P, Moskowitz MA, Pittet MJ, Swirski FK, … Nahrendorf M. (2012). Rapid monocyte kinetics in acute myocardial infarction are sustained by extramedullary monocytopoiesis. The Journal of Experimental Medicine, 209(1), 123–137. 10.1084/jem.20111009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Mouton AJ, & Lindsey ML. (2018). Cardiac macrophage biology in the steady-state heart, the aging heart, and following myocardial infarction. Translational Research : The Journal of Laboratory and Clinical Medicine, 191, 15–28. 10.1016/j.trsl.2017.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin ML, & Blaxall BC. (2012). Cardiac Intercellular Communication: Are myocytes and fibroblasts fair-weather friends? Journal of Cardiovascular Translational Research, 5(6), 768–782. 10.1007/s12265-012-9404-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matic M, & Simon SR. (1991). Tumor necrosis factor release from lipopolysaccharide-stimulated human monocytes: Lipopolysaccharide tolerance in vitro. Cytokine, 3(6), 576–583. 10.1016/1043-4666(91)90484-u [DOI] [PubMed] [Google Scholar]
- McLoed AG, Sherrill TP, Cheng D-S, Han W, Saxon JA, Gleaves LA, Wu P, Polosukhin VV, Karin M, Yull FE, Stathopoulos GT, Georgoulias V, Zaynagetdinov R, & Blackwell TS. (2016). Neutrophil-Derived IL-1β Impairs the Efficacy of NF-κB Inhibitors against Lung Cancer. Cell Reports, 16(1), 120–132. 10.1016/j.celrep.2016.05.085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng F, & Lowell CA. (1997). Lipopolysaccharide (LPS)-induced macrophage activation and signal transduction in the absence of Src-family kinases Hck, Fgr, and Lyn. The Journal of Experimental Medicine, 185(9), 1661–1670. 10.1084/jem.185.9.1661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minshawi F, White MRH, Muller W, Humphreys N, Jackson D, Campbell BJ, Adamson A, & Papoutsopoulou S. (2019). Human TNF-Luc reporter mouse: A new model to quantify inflammatory responses. Scientific Reports, 9(1), 193. 10.1038/s41598-018-36969-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mummidi S, Das NA, Carpenter AJ, Kandikattu H, Krenz M, Siebenlist U, Valente AJ, & Chandrasekar B. (2016). Metformin inhibits aldosterone-induced cardiac fibroblast activation, migration and proliferation in vitro, and reverses aldosterone+salt-induced cardiac fibrosis in vivo. Journal of Molecular and Cellular Cardiology, 98, 95–102. 10.1016/j.yjmcc.2016.07.006 [DOI] [PubMed] [Google Scholar]
- Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo J-L, Libby P, Weissleder R, & Pittet MJ. (2007). The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. The Journal of Experimental Medicine, 204(12), 3037–3047. 10.1084/jem.20070885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nold M, Goede A, Eberhardt W, Pfeilschifter J, & Mühl H. (2003). IL-18 initiates release of matrix metalloproteinase-9 from peripheral blood mononuclear cells without affecting tissue inhibitor of matrix metalloproteinases-1: Suppression by TNF alpha blockage and modulation by IL-10. Naunyn-Schmiedeberg’s Archives of Pharmacology, 367(1), 68–75. 10.1007/s00210-002-0648-5 [DOI] [PubMed] [Google Scholar]
- Ong S-B, Hernández-Reséndiz S, Crespo-Avilan GE, Mukhametshina RT, Kwek X-Y, Cabrera-Fuentes HA, & Hausenloy DJ. (2018). Inflammation following acute myocardial infarction: Multiple players, dynamic roles, and novel therapeutic opportunities. Pharmacology & Therapeutics, 186, 73–87. 10.1016/j.pharmthera.2018.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabhu SD, & Frangogiannis NG. (2016). The Biological Basis for Cardiac Repair After Myocardial Infarction: From Inflammation to Fibrosis. Circulation Research, 119(1), 91–112. 10.1161/CIRCRESAHA.116.303577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Psarras S, Beis D, Nikouli S, Tsikitis M, & Capetanaki Y. (2019). Three in a Box: Understanding Cardiomyocyte, Fibroblast, and Innate Immune Cell Interactions to Orchestrate Cardiac Repair Processes. Frontiers in Cardiovascular Medicine, 6, 32. 10.3389/fcvm.2019.00032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainer PP, Hao S, Vanhoutte D, Lee DI, Koitabashi N, Molkentin JD, & Kass DA. (2014). Cardiomyocyte-specific transforming growth factor β suppression blocks neutrophil infiltration, augments multiple cytoprotective cascades, and reduces early mortality after myocardial infarction. Circulation Research, 114(8), 1246–1257. 10.1161/CIRCRESAHA.114.302653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rien D, Kern R, & Kurtz R. (2011). Synaptic transmission of graded membrane potential changes and spikes between identified visual interneurons. The European Journal of Neuroscience, 34(5), 705–716. 10.1111/j.1460-9568.2011.07801.x [DOI] [PubMed] [Google Scholar]
- Rikard SM, Athey TL, Nelson AR, Christiansen SLM, Lee J-J, Holmes JW, Peirce SM, & Saucerman JJ. (2019). Multiscale Coupling of an Agent-Based Model of Tissue Fibrosis and a Logic-Based Model of Intracellular Signaling. Frontiers in Physiology, 10. 10.3389/fphys.2019.01481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohatgi A. (2021). WebPlotDigitizer (4.5) [Computer software]. https://automeris.io/WebPlotDigitizer
- Rossini A, Zacheo A, Mocini D, Totta P, Facchiano A, Castoldi R, Sordini P, Pompilio G, Abeni D, Capogrossi MC, & Germani A. (2008). HMGB1-stimulated human primary cardiac fibroblasts exert a paracrine action on human and murine cardiac stem cells. Journal of Molecular and Cellular Cardiology, 44(4), 683–693. 10.1016/j.yjmcc.2008.01.009 [DOI] [PubMed] [Google Scholar]
- Sager HB, Heidt T, Hulsmans M, Dutta P, Courties G, Sebas M, Wojtkiewicz GR, Tricot B, Iwamoto Y, Sun Y, Weissleder R, Libby P, Swirski FK, & Nahrendorf M. (2015). Targeting Interleukin-1β Reduces Leukocyte Production After Acute Myocardial Infarction. Circulation, 132(20), 1880–1890. 10.1161/CIRCULATIONAHA.115.016160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salybekov AA, Kawaguchi AT, Masuda H, Vorateera K, Okada C, & Asahara T. (2018). Regeneration-associated cells improve recovery from myocardial infarction through enhanced vasculogenesis, anti-inflammation, and cardiomyogenesis. PloS One, 13(11), e0203244. 10.1371/journal.pone.0203244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh M, Shimoda Y, Maesawa C, Akatsu T, Ishikawa Y, Minami Y, Hiramori K, & Nakamura M. (2006). Activated toll-like receptor 4 in monocytes is associated with heart failure after acute myocardial infarction. International Journal of Cardiology, 109(2), 226–234. 10.1016/j.ijcard.2005.06.023 [DOI] [PubMed] [Google Scholar]
- Schindler H, Diefenbach A, Röllinghoff M, & Bogdan C. (1998). IFN-gamma inhibits the production of latent transforming growth factor-beta1 by mouse inflammatory macrophages. European Journal of Immunology, 28(4), 1181–1188. [DOI] [PubMed] [Google Scholar]
- Schroer AK, Bersi MR, Clark CR, Zhang Q, Sanders LH, Hatzopoulos AK, Force TL, Majka SM, Lal H, & Merryman WD. (2019). Cadherin-11 blockade reduces inflammation-driven fibrotic remodeling and improves outcomes after myocardial infarction. JCI Insight, 4(18). 10.1172/jci.insight.131545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smolgovsky S, Ibeh U, Tamayo TP, & Alcaide P. (2021). Adding insult to injury—Inflammation at the heart of cardiac fibrosis. Cellular Signalling, 77, 109828. 10.1016/j.cellsig.2020.109828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somanna NK, Valente AJ, Krenz M, Fay WP, Delafontaine P, & Chandrasekar B. (2016). The Nox1/4 Dual Inhibitor GKT137831 or Nox4 Knockdown Inhibits Angiotensin-II-Induced Adult Mouse Cardiac Fibroblast Proliferation and Migration. AT1 Physically Associates With Nox4. Journal of Cellular Physiology, 231(5), 1130–1141. 10.1002/jcp.25210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suthahar N, Meijers WC, Silljé HHW, & de Boer RA. (2017). From Inflammation to Fibrosis—Molecular and Cellular Mechanisms of Myocardial Tissue Remodelling and Perspectives on Differential Treatment Opportunities. Current Heart Failure Reports, 14(4), 235–250. 10.1007/s11897-017-0343-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takagawa J, Zhang Y, Wong ML, Sievers RE, Kapasi NK, Wang Y, Yeghiazarians Y, Lee RJ, Grossman W, & Springer ML. (2007). Myocardial Infarct Size Measurement in the Mouse Chronic Infarction Model: Comparison of Area- and Length-Based Approaches. Journal of Applied Physiology (Bethesda, Md. : 1985), 102(6), 2104–2111. 10.1152/japplphysiol.00033.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talman V, & Ruskoaho H. (2016). Cardiac fibrosis in myocardial infarction—From repair and remodeling to regeneration. Cell and Tissue Research, 365(3), 563–581. 10.1007/s00441-016-2431-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan SM, Zhang Y, Connelly KA, Gilbert RE, & Kelly DJ. (2010). Targeted inhibition of activin receptor-like kinase 5 signaling attenuates cardiac dysfunction following myocardial infarction. American Journal of Physiology-Heart and Circulatory Physiology, 298(5), H1415–H1425. 10.1152/ajpheart.01048.2009 [DOI] [PubMed] [Google Scholar]
- Thackeray JT, Hupe HC, Wang Y, Bankstahl JP, Berding G, Ross TL, Bauersachs J, Wollert KC, & Bengel FM. (2018). Myocardial Inflammation Predicts Remodeling and Neuroinflammation After Myocardial Infarction. Journal of the American College of Cardiology, 71(3), 263–275. 10.1016/j.jacc.2017.11.024 [DOI] [PubMed] [Google Scholar]
- Tian M, Yuan Y-C, Li J-Y, Gionfriddo MR, & Huang R-C. (2015). Tumor necrosis factor-α and its role as a mediator in myocardial infarction: A brief review. Chronic Diseases and Translational Medicine, 1(1), 18. 10.1016/j.cdtm.2015.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandervelde S, van Luyn MJA, Rozenbaum MH, Petersen AH, Tio RA, & Harmsen MC. (2007). Stem cell-related cardiac gene expression early after murine myocardial infarction. Cardiovascular Research, 73(4), 783–793. 10.1016/j.cardiores.2006.11.030 [DOI] [PubMed] [Google Scholar]
- Virag JI, & Murry CE. (2003). Myofibroblast and endothelial cell proliferation during murine myocardial infarct repair. The American Journal of Pathology, 163(6), 2433–2440. 10.1016/S0002-9440(10)63598-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan E, Yeap XY, Dehn S, Terry R, Novak M, Zhang S, Iwata S, Han X, Homma S, Drosatos K, Lomasney J, Engman DM, Miller SD, Vaughan DE, Morrow JP, Kishore R, & Thorp EB. (2013). Enhanced efferocytosis of apoptotic cardiomyocytes through myeloid-epithelial-reproductive tyrosine kinase links acute inflammation resolution to cardiac repair after infarction. Circulation Research, 113(8), 1004–1012. 10.1161/CIRCRESAHA.113.301198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Keimig T, He Q, Ding J, Zhang Z, Pourabdollah-Nejad S, & Yang X-P. (2007). Augmented healing process in female mice with acute myocardial infarction. Gender Medicine, 4(3), 230–247. 10.1016/s1550-8579(07)80043-x [DOI] [PubMed] [Google Scholar]
- Wang Y, Yang T, Ma Y, Halade GV, Zhang J, Lindsey ML, & Jin Y-F. (2012). Mathematical modeling and stability analysis of macrophage activation in left ventricular remodeling post-myocardial infarction. BMC Genomics, 13(S6), S21. 10.1186/1471-2164-13-S6-S21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y, Hill MC, Li L, Deshmukh V, Martin TJ, Wang J, & Martin JF. (2019). Hippo pathway deletion in adult resting cardiac fibroblasts initiates a cell state transition with spontaneous and self-sustaining fibrosis. Genes & Development, 33(21–22), 1491–1505. 10.1101/gad.329763.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Wang B, Li N, Zhou Q, Zhou W, & Zhan Z. (2019). Salvia miltiorrhiza and Carthamus tinctorius Extract Prevents Cardiac Fibrosis and Dysfunction after Myocardial Infarction by Epigenetically Inhibiting Smad3 Expression. Evidence-Based Complementary and Alternative Medicine: ECAM, 2019, 6479136. 10.1155/2019/6479136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yde P, Høgh Jensen M, & Trusina A. (2011). Analyzing inflammatory response as excitable media. Physical Review E, 84(5), 051913. 10.1103/PhysRevE.84.051913 [DOI] [PubMed] [Google Scholar]
- Yeo S-Y, Lee K-W, Shin D, An S, Cho K-H, & Kim S-H. (2018). A positive feedback loop bi-stably activates fibroblasts. Nature Communications, 9(1), 3016. 10.1038/s41467-018-05274-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeigler AC, Richardson WJ, Holmes JW, & Saucerman JJ. (2016). A computational model of cardiac fibroblast signaling predicts context-dependent drivers of myofibroblast differentiation. Journal of Molecular and Cellular Cardiology, 94, 72–81. 10.1016/j.yjmcc.2016.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Franklin RA, Adler M, Jacox JB, Bailis W, Shyer JA, Flavell RA, Mayo A, Alon U, & Medzhitov R. (2018). Circuit design features of a stable two-cell system. Cell, 172(4), 744–757.e17. 10.1016/j.cell.2018.01.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The model code and the scripts used to generate the figures are available at https://github.com/saucermanlab/Chowkwale_et_al_IntercellularModel.