

Summary
Poly(A) tails protect RNAs from degradation and their deadenylation rates determine RNA stability. Although poly(A) tails are generated in the nucleus, deadenylation of tails has mostly been investigated within the cytoplasm. Here, we combined long-read sequencing with metabolic labeling, splicing inhibition and cell fractionation experiments to quantify, separately, the genesis and trimming of nuclear and cytoplasmic tails in vitro and in vivo. We present evidence for genome-wide, nuclear synthesis of tails longer than 200 nt, which are rapidly shortened after transcription. Our data suggests that rapid deadenylation is a nuclear process, and that different classes of transcripts and even transcript isoforms have distinct nuclear tail lengths. For example, many long-noncoding RNAs retain long poly(A) tails. Modeling deadenylation dynamics predicts nuclear deadenylation about 10 times faster than cytoplasmic deadenylation. In summary, our data suggests that nuclear deadenylation might be a key mechanism for regulating mRNA stability, abundance, and subcellular localization.
Subject areas: Molecular physiology, Molecular mechanism of gene regulation, Cell biology
Graphical abstract
Highlights
-
•
Long-read sequencing indicates mRNAs are synthesized with 200–250 nt-long poly(A) tails
-
•
Metabolic labeling shows fast reduction in poly(A) tail length
-
•
Cell fractionation suggests tails shortening happens already in the nucleus
-
•
Modeling of deadenylation dynamics predicts rapid nuclear deadenylation
Molecular physiology; Molecular mechanism of gene regulation; Cell biology.
Introduction
Poly(A) tails are a hallmark of eukaryotic gene expression and protect RNAs from degradation1 while promoting translation of mRNAs,2,3,4,5,6 in particular during early development.7,8,9 Poly(A) tail length is dynamically regulated10 and complete deadenylation typically precedes decapping and exonucleolytic decay.
Poly(A) tails are synthesized post-transcriptionally by Poly(A) Polymerase (PAP) after 3′ end cleavage of the nascent transcript.11 Biochemical studies indicate that PAP is stimulated by the mRNA 3′-end processing factor CPSF12 and the nuclear poly(A) binding protein PABPN1,13 which also acts as a molecular ruler to limit the length of the synthesized tail to ca. 250 nt.14,15 These in vitro studies are in line with radioactive labeling experiments showing similar poly(A) length profiles of newly synthesized RNA.16,17 Poly(A) tails are typically shortened throughout an mRNA lifetime and deadenylation rates determine mRNA stability in a gene-specific manner.16,18,19,20
mRNA 3′-end processing is tightly coupled to other nuclear processing steps such as splicing and RNA export.21 RNA-sequencing studies identified co-transcriptional splicing as the dominant mode for mammalian RNA processing,22,23 whereas a more recent study reports up to 50% of all genes being processed by post-transcriptional splicing, involving complex coordination between individual introns.24,25 Splicing of terminal introns is in this context of particular importance, because it is mechanistically linked to pre-mRNA cleavage and polyadenylation26,27 and in some cases used as a mechanism for regulated nuclear retention of transcripts.28 Splicing of long noncoding RNAs on the other hand is highly complex, generating a multitude of isoforms for individual genes.29 Although being processed in the nucleus through the same pathways as messenger RNAs, lncRNAs are in many cases retained in the nucleus.23,30 Splicing kinetics has been identified as one factor impacting export, but other features such as cis-regulatory sequence elements contribute to active retention of lncRNAs in the nucleus.31,32 Poly(A) tails have been shown to be beneficial but not strictly required for efficient export.33,34
The CCR4-NOT35 and PAN2-PAN336 complexes have been identified as major effectors of cytoplasmic mRNA deadenylation in human and yeast. Initial trimming of poly(A) tails has been attributed to PAN2-PAN3,19,37 whereas CCR4-NOT was shown to completely deadenylate tails.38 Both complexes were found shuttling between cytoplasm and nucleus39 suggesting that poly(A) turnover may have a nuclear component.
A recent study concludes that mRNAs emerge in the cytoplasm with poly(A) tails of ca. 130–150 nt on average,18 which implies drastic nuclear shortening under the assumption that poly(A) tails are globally synthesized at a length of 250 nt.
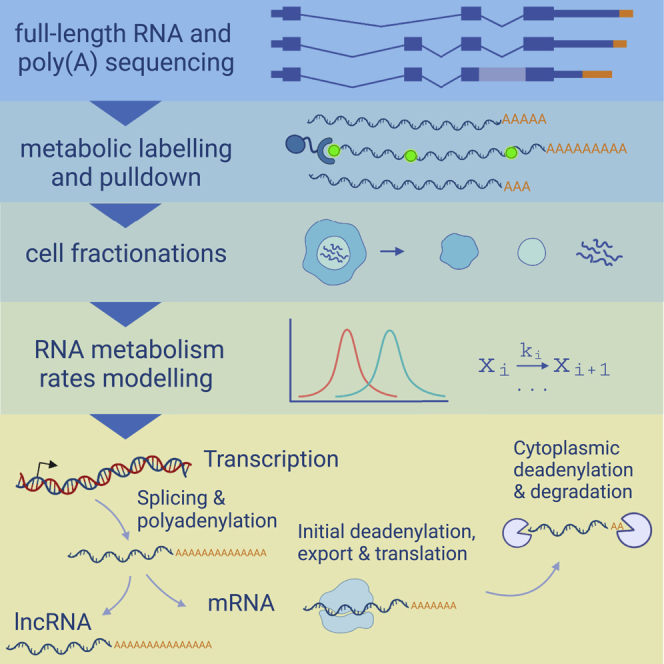
In this study, we investigate nuclear processing of poly(A) tails on a genome-wide scale. We first present evidence for genome-wide synthesis of long poly(A) tails by analyzing full-length mRNA and poly(A) sequencing (FLAM-seq) datasets.40 We validate accumulation of long poly(A) tails by splicing inhibition in HeLa cells. Metabolic labeling of newly synthesized RNA in combination with poly(A) profiling indicates rapid global shortening of poly(A) tails within minutes after synthesis. Subcellular fractionation of HeLa cells and mouse brains reveals nuclear deadenylation as a global mechanism for gene specific diversification of poly(A) tail profiles before export. We finally aggregate nuclear and cytoplasmic poly(A) tail length profiles into a quantitative mathematical model of poly(A) tail and expression dynamics which predicts fast nuclear deadenylation of poly(A) tails, with strong regulatory potential.
Results
Pre-mRNAs are synthesized with long poly(A) tails
To investigate the poly(A) tail length distribution of nascent transcripts, we developed a computational pipeline for extracting unspliced reads from datasets generated by FLAM-seq. We applied stringent criteria to remove ambiguous reads which may not be truly indicative of unspliced transcripts (STAR Methods). We reanalyzed datasets40 of HeLa cells, induced pluripotent stem cells (iPSCs) and cerebral organoids (Figure 1A), and detected between 77 and 1,313 unspliced (‘intronic’) reads per dataset (Figure 1B), which corresponded to 0.03–0.22% of all reads (Figure S1A). Notably, iPSCs and organoids had a significantly higher fraction of unspliced reads indicating potential differences in splicing kinetics.
Figure 1.

Unspliced RNAs have long poly(A) tails
(A) Browser shot example of detected unspliced read in for gene AHSG in Organoid replicate 1 FLAM-seq dataset (number spliced reads = 317; num unspliced = 3).
(B) (top) Bulk polyA tail length density distributions for replicates HeLa S3, iPSC, and Organoid datasets in replicates (bottom) Histogram of intronic poly(A) tail length normalized to total reads per dataset for corresponding datasets and replicates.
(C) Fraction of genes with detected unspliced, intronic reads normalized to total number of genes detected in each dataset (total number per sample in legend). Error bars indicate standard error of the mean for 2 replicates.
(D) (top) bulk poly(A) tail length density distribution of nuclei from HeLa S3 nuclei control and PlaB experiments for replicate datasets. (bottom) histogram of intronic poly(A) tail length normalized to total read counts per dataset for HeLa S3 nuclei control and PlaB splicing inhibition. Number of reads per dataset/replicate for bulk/intronic are displayed inside legend.
(E) Poly(A) tail length distributions for Nanopore direct RNA sequencing.24 Reads grouped by annotated as intronic, poly(A) site, or post poly(A) site reads for sequencing protocols involving enzymatic poly(A) tailing of RNA or no tailing. Reads were categorized as spliced (top) or unspliced (bottom) by our computational pipeline.
Because differences in read length could be linked to different errors in poly(A) quantification, we recorded the raw read length distributions for bulk and intronic reads (Figure S1B). These distributions were very similar across replicate experiments, suggesting consistent poly(A) tail length estimates between spliced and unspliced reads.
Intronic poly(A) tail length distributions were longer than bulk poly(A) distributions in each dataset with a peak at 208 nt for iPSCs and 232 nt for organoids. HeLa datasets also showed longer poly(A) tails for intronic reads (median 151 nt) but with a higher contribution of poly(A) tails shorter than 150 nt, which might be an artifact of transcripts with retained introns, rather than bona fide nascent RNAs.
To investigate whether the detected unspliced, polyadenylated transcripts were representative of transcriptome-wide nascent RNA processing patterns, we first estimated the fraction of genes for which unspliced reads were detected. We found that between 1% and 7% of all genes per dataset had detectable unspliced reads and this number increased to 8% when merging all intronic reads against a background of all detected genes in each dataset (Figure 1C). We found little overlap in detected genes with intronic reads between datasets (Figure S1C), which hinted at an unbiased, random sampling process.
Because our pipeline excluded genes if their introns overlapped with coding or 3′UTR sequences of other isoforms, we quantified the impact of this restriction on our analysis: 49–53% of total detected genes in each dataset had unambiguous intron annotations (Figure S1D), which defined the upper bound of detectable genes with unspliced reads taking into account the average length of FLAM-seq reads.
We downsampled intronic reads and computed the fraction of genes with intronic reads against all detected genes in a dataset. This analysis revealed a nearly linear relationship, showing that higher sequencing depth would further increase the number of genes with intronic reads (Figure S1E).
To test whether detected unspliced reads had differences in intron length we first compared the length distribution of introns which were found in unspliced reads with all annotated introns in the database used for identification of unspliced reads (Figure S1F). We also compared the distribution of intron length derived from Gencode annotation (Gencode 42, 2022) for genes with detected unspliced transcripts against all genes per sample (Figure S1G). In neither case did we find evidence for differences in intron length which ensured that unspliced transcripts were unlikely products of intron-related differences in pre-mRNA processing.
Comparing poly(A) length of intronic reads binned by expression of the associated gene indicates that long tails are represented in each expression domain (Figure S1H), although the fraction of intronic to total reads decreased for more highly expressed genes (Figure S1I).
In summary, we developed a computational pipeline which identifies poly(A) tail length of unspliced transcripts. Our data indicate that unspliced polyadenylated RNAs have poly(A) tails of more than 200 nt in length on average.
Splicing inhibition causes accumulation of unspliced RNA with long tails
To validate our analysis of unspliced RNAs in FLAM-seq datasets, we treated HeLa cells for 3 h with the SF3b inhibitor Pladienolide B (PlaB), which blocks spliceosome assembly on pre-mRNA.41 To enrich unspliced RNAs, we isolated nuclei from control and PlaB-treated cells. We observed a more than 10-fold increase in unspliced reads in RNA extracted from nuclei compared to RNA extracted from whole cells, validating the enrichment of pre-mRNA in nuclear preparations. We found a 3-fold increase in intronic reads on splicing inhibition (from 0.4% to 1.3% of total reads, Figure S2A). We detected 54–304 intronic reads per replicate (Figure 1D) and for 1% of total genes in control and 3.5% in PlaB-treated cells (Figure S2B). We again found little overlap of genes with intronic reads between individual replicates, arguing against biases in detecting unspliced transcripts (Figure S2C).
Poly(A) tail length distributions of intronic reads had a median of 205 nt in control and 242 nt in PlaB-treated HeLa nuclei, with a marked reduction of unspliced RNAs with tails shorter than 150 nt compared to bulk RNA preparations from HeLa cells (Figure 1B). We binned genes by expression and investigated the poly(A) tail length of intronic reads for each bin. We did not observe expression-dependent differences in intronic poly(A) length but noticed a significant increase in poly(A) length between intronic reads from control and PlaB conditions (Figure S2D). Consistent with results from bulk RNA preparations, the relative fractions of unspliced reads decreased with higher gene expression for control samples. This trend was less prominent for PlaB-treated HeLa cells which may indicate differences in the efficacy of PlaB in blocking splicing for different classes of genes (Figure S2E).
Comparing the bulk poly(A) length distributions revealed global shortening of poly(A) tail length of about 11 nt on PlaB treatment (Figure S2F). At the same time, we noticed an accumulation of poly(A) tails longer than 200 nt on PlaB treatment which may be explained by accumulation of unspliced reads which were too short to cover terminal introns (‘false negative’ unspliced reads). Differences in poly(A) length and fold-changes in expression between control and PlaB were not correlated (Figure S2G), but we found several genes for which changes in gene expression coincided with poly(A) tail extension (MYC, IER3, KLF5, RBMX) or with tail shortening (MAT2A, STX10). Transcripts with increasing poly(A) tail length on PlaB treatment were typically less stable (Figure S2H) and had longer 3′UTRs (Figure S2I).
We further compared our analysis to a recently published Nanopore direct RNA sequencing datasets of nascent, chromatin-associated RNA.24 In this study, non-polyadenylated RNA is analyzed by in vitro A-tailing of RNA using an E.Coli polyA polymerase before library preparation and subsequent Nanopore long-read direct RNA sequencing.42 Nanopore reads were (separately for spliced and unspliced reads) sorted into three groups, (1) reads mapping upstream of the poly(A) site (2) reads ending at the poly(A) site and (3) reads that extended downstream of the poly(A) site (Figure 1E). The experimental approach without poly(A) tailing enriched polyadenylated transcripts with long poly(A) tails both for spliced and unspliced reads which ended at annotated polyadenylation sites. As expected, unspliced reads had long poly(A) tails of ca. 200 nt which also validated the poly(A) profiles of intronic reads identified in our FLAM-seq datasets. Poly(A) tailing enriched for reads ending in introns which had poly(A) tails with a median length of 20–35 nt, which were likely the product of the in vitro tailing by E. coli poly(A) polymerase used to generate these data. In these samples, poly(A) tails are short for all reads except those ending in a poly(A) site, which indicated absence of polyadenylation for nascent transcripts which are not cleaved. Both spliced and unspliced reads, which ended at a poly(A) site, had bimodal poly(A) length distributions which could be interpreted as (pre-)mRNAs having either long or no poly(A) tails (Figure 1E).
In summary, we validated that our pipeline captured unspliced pre-mRNAs by inhibiting splicing using PlaB and observed global shortening of poly(A) tails but elongated tails of unspliced transcripts. We further analyzed previously published data and showed that poly(A) profiles of unspliced reads were bimodal in non-poly(A) selected chromatin fractions indicating that transcripts were either not polyadenylated or had ca. 200 nt-long tails at the poly(A) site.
Metabolic labeling reveals poly(A) tail length profiles of newly transcribed RNA
Because poly(A) tail length profiles of unspliced reads were significantly longer than steady-state distributions we investigated the deadenylation kinetics on a genome-wide scale using metabolic labeling of RNA in combination with FLAM-seq (Figure 2A). We first performed 4-thiouridine (4sU) labeling of HEK cells followed by biotinylation of 4sU moieties and separation of labeled pulldown and supernatant fractions using streptavidin-coated beads.43,44
Figure 2.

Metabolic labeling indicates rapid shortening of poly(A) tails after synthesis
(A) Experimental outline for 4sU pulldown and SLAM-Seq metabolic labeling experiments.
(B) Poly(A) tail length distributions for supernatant (SN) and pulldown (PD) fractions after metabolic labeling with 4sU for indicated time points. Dashed distribution indicates poly(A) tail length for intronic reads from HEK metabolic labeling experiments.
(C) Poly(A) tail length distributions of all genes, immediate-early genes (IEGs), long noncoding RNAs (lncRNA) and ribosomal protein encoding genes (ribosomal) for different labeling timepoints in 4-SU pulldown experiments.
We labeled HEK cells for up to 90 min using 4sU at high concentrations (1 mM) to obtain sufficient labeled RNA for performing FLAM-seq. RNA concentrations obtained after pulldown were proportional to labeling time (Figure S3A). Comparing pulldown and supernatant concentrations, 1%–10% of the total RNA pool was captured in pulldown fractions, depending on labeling period, which was in agreement with previous studies.43 We noticed a small fraction of RNA binding to streptavidin beads in control samples without 4sU labeling (0 min), which was also reported before.43 Dot Blots of total RNA before pulldown showed efficient biotinylation and signal intensities proportional to labeling time (Figure S3B).
In the FLAM-seq data, we observed longer poly(A) tails in pulldown fractions compared to supernatant (Figure 2B), with a bimodal distribution peaking at 50 nt and 150 nt. The supernatant distribution had only a single mode at 50 nt. Differences in poly(A) tail length profiles between individual labeling time points were minor, indicating little global poly(A) length dynamics throughout the investigated time course.
We noticed an increased poly(A) tail length in the library prepared from a 0 min control experiment, which could indicate preferential binding of longer tails to streptavidin beads in the absence of labeled RNA. Supernatant samples for individual time points were reproducible and indicated absence of systematic biases in poly(A) tail length profiling between time points (Figure S3C). Comparing transcripts of mitochondrial genes, we observed median lengths of 50 nt in pulldown and supernatant fractions which ensured accurate quantification.45 As mitochondrial poly(A) tail length profiles were similar between pulldown and supernatant fractions independent of labeling period, we concluded that progressive deadenylation is not occurring for mitochondrial transcripts, which had been suggested before.46 Pulldown samples contained the majority of unspliced reads (Figure S3E), which was expected given that splicing is typically completed within minutes.47
Poly(A) tail length of unspliced reads peaked at 200 nt, which validated accurate quantification of long poly(A) tails in pulldown experiments (Figure S3F). We computed differences in median poly(A) tail length per gene between pulldown and supernatant fractions (Figure S3G) finding that poly(A) tails were 24–32 nt longer in pulldown fractions. Comparing poly(A) tail length profiles for different classes of genes, such as genes encoding ribosomal proteins, immediate-early genes (IEGs), or lncRNAs, we found stark differences in their poly(A) tail profiles over the labeling time course (Figure 2C). lncRNAs and IEGs had long poly(A) tails of around 200 nt after 10 min labeling, whereas tails for ribosomal proteins were on average 120 nt. Differences in poly(A) profiles for individual time points were less pronounced for ribosomal transcripts than for IEGs.
As an orthologous approach for profiling poly(A) tail lengths kinetics we applied SLAM-Seq48 in combination with long read sequencing and poly(A) quantification. We were able to successfully discern labeled and unlabeled reads and quantified poly(A) tail length of newly synthesized transcripts after 90 and 180 min of labeling (Figure S3H). Comparing labeled and unlabeled reads we observed that newly synthesized RNAs had longer poly(A) tails than pre-existing transcripts, which was consistent with the pulldown experiments. We noticed several technical challenges which affected the comparison of poly(A) profiles between time points and refer to the STAR Methods for an in-depth assessment (on the long-read implementation of SLAM-seq).
In summary, metabolic labeling experiments indicated that, even for short labeling periods, poly(A) tails of newly synthesized RNAs were significantly shorter than 200 nt for most transcripts. Thus, assuming that mRNAs are synthesized with long poly(A) tails, our data suggest rapid poly(A) deadenylation within minutes after the completion of transcription.
Short poly(A) tails in the nucleus
Our metabolic labeling experiments indicated rapid shortening of poly(A) tails within minutes after synthesis. This raised the question whether initial deadenylation occurs already in the nucleus or after mRNA has been exported, which has been reported to operate on comparable timescales.49,50
We addressed this question by investigating poly(A) tail length profiles in subcellular compartments in vivo and in vitro. We first performed biochemical fractionation of HeLa cells into cytoplasmic, nucleoplasmic and chromatin fractions (Figure 3A). To account for possible experimental variability, we produced 6 biological times 2 technical replicates. We evaluated the purity and absence of contamination for each sample by western blot (Figure S4A) and by quantifying the fraction of mitochondrial transcripts (Figure S4B) and intronic reads (Figure S4C). We did not find evidence for cytoplasmic contamination, although the fraction of mitochondrial and intronic reads differed between biological replicates. Comparing median poly(A) tail length (Figure S4D) and gene expression counts (Figure S4E) further showed high reproducibility between technical replicates.
Figure 3.

Biochemical fractionation reveals nuclear deadenylation
(A) Experimental outline for biochemical fractionation experiments from HeLa S3 cell lines in cytoplasmic, nucleoplasm and chromatin fractions. FLAM-seq libraries were generated from RNA extracted from fractions.
(B) Poly(A) tail length density distributions of cytoplasmic (Cyto), nucleoplasmic (Nuc) and chromatin (Chr) fractions from HeLa S3 cells. Ribbons indicate SD of 12 replicate samples.
(C) Poly(A) tail length profiles of intronic reads in HeLa S3 fractions. Ribbons indicate SD of 12 replicate samples.
(D) Experimental outline for fractionation of mouse brains into nuclei and cytoplasmic fractions using a Dounce homogenizer. FLAM-seq libraries were generated from RNA extracted from fractions.
(E) Poly(A) tail length profiles of mouse brain cytoplasm and nuclei fractions. Bulk distributions include all reads, intronic distributions include unspliced reads.
(F) Poly(A) tail length distributions for lncRNAs in HeLa S3 (top) and mouse brain (bottom) fractionation experiments.
(G) Comparison of median poly(A) tail length per gene profiles between cellular fractions.
(H) Comparison of gene enrichment between nucleus and cytoplasm (inferred from HeLa ENCODE data) and median poly(A) tail length per gene in each fraction.
Comparing length profiles for subcellular fractions indicated variability in poly(A) tail length profiles between biological replicates, whereas technical replicates were in good agreement (Figure S4G). To define experimental parameters which could explain the observed differences between biological replicates we fitted a linear model using variables such as the fraction of intronic reads, RNA concentration and others (STAR Methods) to predict the median poly(A) length for each sample. Median poly(A) length was best explained by a scaling factor for each biological replicate, which described the variation from the average poly(A) tail profile overall replicates. Scaling poly(A) tail length profiles accordingly reduces the observed between biological replicates (Figure S4H). Global biases in poly(A) tail length quantification using FLAM-seq could be caused by differences in PCR cycles, input RNA amounts or efficiency of poly(A) selection. We further assumed that the large number of independent replicates for HeLa fractionation experiments accurately accounts for the variability known to be inherent to biochemical fractionation techniques.51 Bulk poly(A) tail length distributions of HeLa chromatin, nucleoplasm and cytoplasm showed progressively shorter poly(A) tails with a median length of 134 nt, 117 nt and 80 nt respectively (Figure 3B). Poly(A) tail length profiles of intronic reads in chromatin and nucleoplasmic fractions had a median length of 205 nt (Figure 3C), which was in line with poly(A) tail profiles of unspliced reads in whole cell extracts (Figures 1B–1E). Intronic reads in cytoplasmic fractions were rare (0.02% of total reads vs. 0.6% for nuclear fractions) and had shorter tails of 130 nt. Comparing intronic to bulk profiles hence suggested a rapid shortening process already in the nuclear fractions. We did not find substantial differences in poly(A) tail profiles of genes with detected unspliced transcripts versus those without (Figure S5A), illustrating that nuclear shortening is a global mechanism.
To investigate molecular properties differentiating genes in their deadenylation dynamics, we compared gene expression by poly(A) tail length bins between fractions (Figure S5B). As shown before,9,40,52,53 highly expressed genes tended to have shorter poly(A) tails in cytoplasmic fractions, a trend which was also seen in nucleoplasm and chromatin but shifted toward longer poly(A) tails. To ensure that nuclear deadenylation is starting from a uniform initial length distribution at the points of synthesis, we compared poly(A) tail length of intronic reads for genes binned by median bulk poly(A) tail length in chromatin and nucleoplasm and observed comparable intronic poly(A) length of ca. 200 nt for each bin (Figure S5C). We found that short-lived RNAs had longer tails in cytoplasm and a similar trend was seen in nucleoplasm and chromatin (Figure S5D). 3′UTR length increased proportionally to poly(A) tail length both in cytoplasmic and nuclear fractions (Figure S5E). We noticed slightly shorter 3′UTRs for nuclear compared to cytoplasmic transcripts, except for those with very short poly(A) tails of less than 50 nt.
We grouped 3′UTR isoforms by shorter proximal and longer distal isoforms and observed isoform-specific deadenylation (Figure S5F). In summary, the molecular features differentiating poly(A) tail length in the cytoplasm are also associated with poly(A) tail length in nuclear fractions.
We validated nuclear deadenylation in vivo by performing fractionation of a mouse brain into cytoplasm and nuclei (Figure 3D). We showed absence of cytoplasmic contamination in mouse brain nuclei by western blot and analysis of mitochondrial and intronic read fractions in each fraction (Figures S4A–S4C). When comparing poly(A) tail length distributions of all reads, we found that nuclear tails were longer than cytoplasmic tails (119 nt vs. 146 nt) but markedly shorter than unspliced intronic reads in the nuclear fraction (Figure 3E). Unspliced reads in cytoplasmic fractions had shorter poly(A) tails and could reflect transcripts with retained introns as observed for the HeLa cytoplasm. Inspecting poly(A) tail length of different gene biotypes in HeLa and mouse brains, we found that lncRNAs had overall much longer poly(A) tails in the nuclear fractions, both in HeLa and mouse brain samples (Figure 3F). We found 50–60% of all nuclear lncRNAs with a median poly(A) tail length longer than 200 nt which indicated absence of nuclear deadenylation for those transcripts.
We next compared median poly(A) tail length per gene between fractions (Figure 3G). Chromatin and nuclear poly(A) tails were on average longer than cytoplasmic tails and we observed a larger dynamic range in nuclear compartments. Chromatin tails were longer than nucleoplasmic tails with a minor offset between fractions. A similar trend was observed when comparing mouse brain cytoplasm and nuclei (Figure S5H). Different classes of genes showed distinct poly(A) tail length profiles, as for instance observed for ribosomal protein genes which had short poly(A) tails in the nucleus, whereas tails were markedly longer for immediate-early genes (IEGs) and lncRNA poly(A) tails (Figure S5I).
We finally investigated poly(A) tail length in the context of transcript distributions between nucleus and cytosol. We obtained cytoplasmic-to-nuclear transcript ratios from ENCODE RNA-seq data for HeLa cell fractions38 which we found reflective of the ratios inferred from FLAM-seq fractionation data (Figure S5J). 30% of genes were enriched more than 1.5-fold in the nucleus, which was in line with previous studies investigating subcellular transcript distributions.28 We compared cytoplasmic-to-nuclear enrichments with median poly(A) tail length per gene for each HeLa fraction and found that nuclear tail length is more predictive of transcript enrichment than cytoplasmic tail length (Figure 3H). Transcripts enriched in the cytoplasm tended to have short tails in all fractions, as illustrated for ribosomal protein genes. On the other hand, lncRNAs highly enriched in the nucleus had long poly(A) tails, whereas IEGs had intermediate tail length.
In summary, cellular fractionation experiments revealed nuclear deadenylation of poly(A) tails as a gene-specific process in vitro and in vivo.
Modeling of nuclear and cytoplasmic poly(A) tail dynamics predicts fast initial deadenylation
We conceived a simple mathematical model describing mRNA abundance and tail length in the nucleus and the cytosol of a cell to quantitatively address the hypothesis that poly(A) tails are rapidly deadenylated in the nucleus. We described the steady state expression levels of mRNAs in the two compartments as a function of gains and losses, defined by transcription, export, nuclear/cytoplasmic deadenylation and decay. Each process was defined by a rate, which was initially estimated by building a normal distribution around an informed guess (from published data, Figure S6A Table S1 and STAR Methods). Rates were then inferred by minimizing the square root differences of simulated mRNA abundances and cytoplasmic and nuclear poly(A) tail length profiles measured by FLAM-seq (Figure 4A). We observed that the simplest model, which does not allow for nuclear decay and defines a constant export rate, resulted in a buildup of completely deadenylated mRNAs and a bulk distribution of long tails in the nucleus, which did not fit our data (Figure 4B). Instead, allowing for nuclear decay, or alternatively, setting an export rate dependent on poly(A) tail length, resulted in a better fit for both nuclear and cytoplasmic mRNA distributions (Figure 4B). We varied some of the model assumptions, including potential contamination of nuclear fractions in fractionation experiments, allowing for nuclear decay or tail-dependent export (Figures S6B and S6C), and independently estimated the model parameters from individual replicates (Figure S7A). We observed that our best estimates for the nuclear deadenylation rates were consistently higher than cytoplasmic deadenylation rates (approximately one order of magnitude - ca. 4 nt/min versus 0.5 nt/min, Figure 4C and Table S1). Nuclear deadenylation may represent an additional, robust layer for global mRNA regulation: we predicted that varying the deadenylation rate in the nucleus results in strong changes in cytosolic mRNA levels (Figure S7B). In summary, our quantitative modeling approach predicted nuclear deadenylation kinetics which are an order of magnitude faster than in the cytoplasm, with additional features of nuclear processing such as nuclear decay and tail-length dependent export rates (Figure 4D).
Figure 4.

A mathematical model predicts fast nuclear deadenylation
(A) Model description (see STAR Methods).
(B) Simulations of mRNA levels in the nucleus (green) and cytosol (red) with the basic model assuming constant export (top left), the model assuming constant export and nuclear decay (ndce model; top right), the model assuming tail-dependent export (nndle model; bottom left), compared to the merged FLAM-seq data from 12 fractionation replicates (bottom right). The average of the estimated parameter distributions was used as input. Cytoplasmic contamination in the nuclear fraction of 0% was assumed.
(C) Distributions of cytoplasmic (red) and nuclear (green) deadenylation rates, estimated with models assuming nuclear decay (ndce) or tail-dependent export (nndle), with three contamination values and from 12 fractionation experiments replicates merged (see STAR Methods).
(D) Summary sketch: mRNAs with long poly(A) tail are synthesized and rapidly deadenylated in the nucleus. mRNAs with shortened tails are exported to the cytoplasm where they are translated, until progressive deadenylation leaves mRNAs without poly(A) tails, which are decapped and degraded.
Discussion
Active mRNA degradation is a key mechanism of post-transcriptional gene regulation. Complete deadenylation of poly(A) tails is typically required for decapping and degradation of transcripts. The rates of deadenylation are gene-specific and therefore central determinants of RNA stability.18,19,20 Biochemical and labeling studies indicate the synthesis of poly(A) tails of ca. 250 nt by nuclear poly(A) polymerase,10 which are then exported and deadenylated in the cytoplasm,17,54 although a genome-wide study on deadenylation rates concluded that mRNAs may emerge in the cytoplasm with poly(A) tails of ca. 130–150 nt on average.18
We found that poly(A) tails of nascent transcripts, which we identified from unspliced reads in FLAM-seq data, support the hypothesis of genome-wide synthesis of long poly(A) tails with a tail length of 200–250 nucleotides, consistent across samples and conditions. We validated the detection of unspliced reads by performing splicing inhibition and biochemical fractionation experiments in vivo and in vitro, showing that unspliced transcripts are enriched in the nucleus and increased on splicing inhibition. We further analyzed published Nanopore direct RNA sequencing datasets of highly pure nascent, chromatin-associated transcripts24 and confirmed genome-wide synthesis of long poly(A) tails. A previous study applying Nanopore long-read sequencing on RNA from a human cell line also showed that reads containing intronic sequences had longer poly(A) tails, but these were attributed to possibly aberrant intron-retaining transcripts.42 Our computational pipeline relied on identification of unspliced and polyadenylated transcripts which are likely products of RNAs undergoing post-transcriptional (terminal) intron splicing. The extent of post-transcriptional versus co-transcriptional splicing is a matter of ongoing research. Recent studies indicate the occurrence of post-transcriptional terminal intron splicing for up to 50% of all mammalian genes.24,25
To rule out that our analysis was limited to specific subsets of transcripts, we carefully quantified molecular properties such as intron length or expression levels: these analyses did not detect potential biases in the detection of unspliced transcripts, although we cannot exclude additional confounding factors that we may have not taken into account.
We further rule out the possibility that a larger fraction of unspliced, nuclear reads originated from retained introns: the analysis of newly synthesized, non-poly(A) selected, chromatin-associated mRNAs from Nano-COP experiments showed that also nascent spliced mRNAs have long poly(A) tails which supports our notion of global synthesis of long tail synthesis.
A second explanation of long nuclear tails could be related to nuclear hyperadenylation which has been proposed as a mechanism of nuclear mRNA degradation.55 We yet find it highly unlikely that hyperadenylation generally acts on all messenger RNAs, both spliced and unspliced, as observed in our study.
Splicing inhibition affected bulk poly(A) length 2-fold: first, a mild global shortening was observed which could be explained by continuous deadenylation whereas mature RNA processing of newly synthesized transcripts is inhibited; second, an increase in transcripts with long tails may indicate compensatory mechanisms (e.g. for MYC, IER3, KLF5, genes with long 3′UTRs), or reflect unspliced RNAs not annotated by our pipeline for technical reasons (‘false negatives’). Stabilization of certain transcripts has been observed on transcription inhibition56 and it is conceivable that similar mechanisms may take place on splicing inhibition. PlaB has further been shown to induce cell-cycle arrest and apoptosis, which may explain the observed MYC downregulation.57 We further found poly(A) tail length of unspliced transcripts slightly increased upon splicing inhibition. Certain nuclear RNA decay pathways involve hyperadenylation for clearance of aberrant transcripts by the nuclear exosome,58,59,60 and increased poly(A) length on splicing inhibition may also indicate intermediates of nuclear decay.
Analysis of Nanopore direct RNA sequencing data24 extended our finding of global synthesis of long poly(A) tails beyond post-transcriptional splicing events, because non-polyadenylated nascent transcripts were tailed in vitro for Nanopore sequencing. 3′-end cleaved transcripts appeared either without or with long poly(A) tails. No evidence for the synthesis of intermediate poly(A) tail length significantly shorter than 250 nt was found in nascent RNA fractions. Poly(A) synthesis occurred rapidly, in the order of seconds,61 which may explain emergence of bimodal length profiles at the poly(A) site in these datasets.
We did not find evidence for unspliced transcripts in nuclear fractions with poly(A) tails shorter than 100–150 nucleotides. This was also true on splicing inhibition, which we expected to increase the dwell-time of unspliced mRNAs in the nucleus. We therefore hypothesize that completion of splicing could be required for shortening of tails.
Resolving the kinetics of the early phase of deadenylation using metabolic labeling and pulldown experiments revealed an increased poly(A) tail length compared to the steady-state poly(A) distribution. Surprisingly, the differences in poly(A) tail length for pulldown samples between labeling timepoints were small for most genes. For certain genes, such as IEGs or lncRNAs, a more pronounced shortening throughout the labeling period was observed. This result required for a rapid initial deadenylation step within minutes after synthesis, given the assumption of poly(A) tails being longer than 200 nt at the time of synthesis. We further applied SLAM-seq in combination with FLAM-seq which qualitatively resulted in a similar finding but suffered from technical challenges (see STAR Methods).
We observed significant shortening of tails in nuclear fractions of HeLa cells and mouse brain samples, which aligns with the hypothesis of rapid deadenylation. Our findings contradict earlier hypotheses describing export of non-deadenylated transcripts,17,54 but support more recent models observing emergence of cytoplasmic tails with an average length of 130–150 nt.18
Nanopore direct RNA sequencing24 shows that chromatin-associated nascent RNA retains long poly(A) tails. Our chromatin fractionation experiments on the contrary show significant shortening of tails which can be explained either by a relatively long dwell time on chromatin on which deadenylation occurs or inefficient separation of nuclear and chromatin fractions. A similar argument relates to the 10-min metabolic labeling performed in this study. We hence hypothesize that deadenylation may require a lag phase and/or release from chromatin, which could for instance involve a change in nuclear compartments.
We found evidence for gene-specific nuclear deadenylation because certain classes of genes show differences in tail length even after short labeling time points, when RNA is likely still localized in the nuclear compartment. Yet, differentiating the contributions of RNA export, nuclear deadenylation rates and nuclear decay on nuclear steady-state poly(A) distributions is in general very challenging through the lack of appropriate kinetic data.
An exception were lncRNAs, with more than 50% of nuclear lncRNAs not showing poly(A) tail shortening. Many lncRNAs function in the nucleus, for instance NEAT1 in paraspeckle formation,62 and further undergo distinct processing events compared to mRNAs, which may suggest coupling between nuclear deadenylation and other processing steps.29,63,64 In particular, we found a negative correlation between cytoplasmic-to-nuclear enrichment and poly(A) tail length in nuclear fractions, which lets us hypothesize nuclear deadenylation as a mechanism controlling RNA export which could be utilized to retain nuclear lncRNAs and/or to determine export rates via gene-specific deadenylation rates.
Quantitative modeling of deadenylation dynamics and mRNA steady state distributions predicts that nuclear deadenylation is by an order of magnitude faster than cytoplasmic deadenylation, which is expected given the significant shortening observed for chromatin and nucleoplasmic fractions within relatively short dwell times in the nucleus until RNA is exported. Of interest, allowing poly(A) tail-length dependent export rates or nuclear mRNA decay dramatically improves the goodness-of-fit. Nuclear decay is mediated by the nuclear exosome which has a broad range with up to a 1,000 mRNA targets reported in yeast,65 arguing for widespread nuclear decay which is consistent with our model. Similarly, poly(A) tails have been shown to increase export rates in in vitro experiments,33 although little is known about the impact of poly(A) tail length itself on export rates.
Several deadenylase enzymes have been identified with CCR4-NOT and PAN2-PAN3 being most relevant for canonical deadenylation. A biphasic model has been proposed for yeast and mammalian deadenylation19,37 in which PAN2-PAN3 first trims long tails, and CCR4-NOT subsequently deadenylates tails until degradation. Whether this process is coordinated across nucleus and cytoplasm is not known, although both PAN2-PAN3 and CCR4-NOT are able to shuttle between cytoplasm and nucleus39 and have a divergent set of target genes in yeast.66 A recent study investigating poly(A) tail dynamics of serum response genes after transcription induction identified the CNOT1 subunit of the CCR4-NOT complex as a nuclear deadenylase complex in mouse fibroblasts.67 Whether the same enzyme complexes operate in different cellular compartments with different processivity is yet unknown.
In summary, we present evidence for rapid deadenylation of mRNAs occurring in the nucleus of mammalian cells (Figure 4C). We propose that mRNAs are initially synthesized with a tail of 200–250 nt, which is then shortened in a splicing-dependent manner, with possible implications in the control of mRNA export. Similar to cytoplasmic deadenylation, nuclear poly(A) tail shortening is a gene-specific process which may serve as a nuclear control instance specifying the fate of mRNAs before they enter the cytoplasm.
Limitations of the study
Our genome-wide estimate of 200–250 nt long poly(A) tails at synthesis derives from a) using intronic reads from FLAM-seq datasets as a proxy for nascent RNA and b) reanalysis of Nanopore direct RNA sequencing data performed on purified nascent RNA. Importantly, these estimates are compatible with previous in vitro and radioactive labeling data.
Furthermore, we base our conclusion that mRNA can be deadenylated already in the nucleus on a) the aforementioned assumption that tails are synthesized at 200–250 nt for all mRNAs, b) published observations that mRNAs at the time of appearance in the cytosol have an average length of 130–150 nt, c) the observation that RNA metabolically labeled for 10–90 min has also a mode of ca. 150 nt; d) steady state nuclear tail length profile has an average of ca. 120 nt.
An alternative interpretation of all these data is that tail length at synthesis is mRNA/gene-specific and the observed 200–250 nt long tails result from an aberrant hyperadenylated population of mRNAs. Furthermore, the “short” nuclear tails might result from heavy contamination of nuclear RNA with cytosolic RNA, which would be compatible with the observation of nuclearly enriched lncRNAs retaining long tails, and similarly could be argued for the labeled/unlabeled RNA analysis. As discussed throughout the manuscript, we carefully controlled for these possibilities and found no evidence for them, but we cannot formally exclude that these or other confounders might still be in place. In addition, our quantitative modeling was performed without regard for gene-specific effects (we estimated weighted average rates across all genes), therefore different complements of mRNAs with different kinetic rates in nucleus and cytoplasm could confound the estimated rates for those compartments.
Another limitation is the lack of mechanistic insight explaining how this rapid initial deadenylation takes place (i.e., which enzymes are involved). We think that answering this question may also address the previous points: if we or others were to find the nucleases responsible for the observed effects and perturb their activity, we would be able to observe whether such perturbation causes a genome-wide nuclear accumulation of 200–250 nt-long tails at the steady state, which would further confirm whether mRNAs are generally produced with a long tail that is rapidly shortened after synthesis in the nucleus.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| GAPDH Monoclonal Anti GAPDH clone 71.1 | Sigma Aldrich | Cat#G8795-200UL |
| TDP43 Polyclonal | Proteintech | Cat#10782-2-AP |
| BCAP31 (BAP31) Polyclonal | Proteintech | Cat#11200-1-AP |
| Chemicals, peptides, and recombinant proteins | ||
| Pladeinolide B | Biomol | Cat#Cay16538-100 |
| 4-Thiouridine | Chemgenes | Cat#N-RP-2304 |
| Pierce High Sensitivity Strep-HRP | Thermo Fisher | Cat#21130 |
| EZ-Link Biotin HPDP | Thermo Fisher | Cat#21341 |
| Dynabeads MyOne Streptavidin C1 | Thermo Fisher | Cat#65001 |
| Critical commercial assays | ||
| Nuclei EZ Prep kit | Sigma Aldrich | Cat#NUC101-1KT |
| USB poly(A) length assay kit | Thermo Fisher | Cat#764551KT |
| TruSeq mRNA preparation kit | Illumina | Cat#RS-122-2102 |
| SMARTScribe Reverse Transcriptase kit | Takara | Cat#639537 |
| Advantage 2 DNA polymerase mix | Takara | Cat#639201 |
| Deposited data | ||
| HeLa, iPSC, Organoid FLAM-seq data | Legnini et al.40 | GEO: GSE126465 |
| All FLAM-seq data generated in this work | This paper | GEO: GSE188539 |
| Nascent Nanopore RNA data | Drexler et al.24 | GEO: GSE123191 |
| Experimental models: Cell lines | ||
| HEK Flp-In 293 T-Rex cells | Invitrogen | Cat#R78007 |
| HeLa S3 cells | Rajewsky Lab | N/A |
| Experimental models: Organisms/strains | ||
| Mouse | MDC animal facility | C57BL/6N |
| Oligonucleotides | ||
| RT Primer 1: GGTAATACGACTCACTATAGCGA GANNNNNNNNNNCCCCCCCCCTTT |
Legnini et al.40 | N/A |
| RT primer 2: TGAGTCGGCAGAGAACTGGCGAA NNNNNNNNNNCCCCCCCCCTTT |
Legnini et al.40 | N/A |
| Template switching oligo (Iso-TSO): iCiGiCAAGCA GTGGTATCAACGCAGAGTACATrGrGrG |
Legnini et al.40 | N/A |
| PCR Primer 1 (RV, used in case RT Primer 1 was used): GGTAATACGACTCACTATAGCGAG |
Legnini et al.40 | N/A |
| PCR Primer 2 (RV, used in case RT Primer 2 was used): TGAGTCGGCAGAGAACTGGCGAA |
Legnini et al.40 | N/A |
| PCR Primer IIA (FW): AAGCAGTGGTATCAACGCAGAGT | Legnini et al.40 | N/A |
| Software and algorithms | ||
| FLAMAnalysis | Rajewsky Lab Github | https://github.com/rajewsky-lab/FLAMAnalysis |
| Additional code used in this work | This paper | Mendeley data: https://doi.org/10.17632/drwmy4y8f5.1 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Nikolaus Rajewsky (rajewsky@mdc-berlin.de).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Cell lines
HeLa S3 cells were previously kindly provided by the lab of Giuseppe Macino (Sapienza University of Rome). HEK Flp-In 293 T-REx cells were previously purchased. Both lines were cultured in DMEM (high glucose, glutamax) supplemented with 10% FBS in a 37°C, 5% CO2 incubator and passaged every 2–3 days with 0.05% trypsin. Cell lines were not authenticated.
Mouse samples
Mouse tissues were dissected from one male at day of birth (P0). The strain was generated and maintained on the pure C57BL/6N background. All animals used for breeding were kept in a pathogen-free facility in a 12 h light–dark cycle with ad libitum food and water. Animal care and mouse work were conducted according to the guidelines of the Institutional Animal Care and Use Committee of the Max Delbrück Center for Molecular Medicine, the Landesamt für Gesundheit und Soziales of the federal state of Berlin, Germany.
Method details
FLAM-seq library preparation and PacBio sequencing
FLAM-seq library preparation was performed according to.40 In brief, polyadenylated RNA was isolated from total RNA using the Illumina Truseq mRNA preparation kit. A GI tail was appended to selected mRNA using tailing reagents from the USB poly(A) length assay kit. Tailed RNA was purified using 1.8x RNAClean beads and reverse transcribed using the SMARTScribe Reverse Transcriptase kit and isoTSO and RT primer 1 or 2. cDNA was purified using 0.6x Ampure XP beads and PCR amplified using PCR primer 1 or 2 and PCR Primer IIA using the Advantage 2 DNA polymerase mix. FLAM-seq libraries were purified 2x using 0.6x Ampure XP beads.
PacBio adapters ligation, sequencing on a Sequel instrument and data preprocessing to generate fastq files was performed according to standard procedures by the BIMSB Genomics platform.
A detailed protocol for the FLAM-seq method can be found at https://doi.org/10.21203/rs.2.10045/v1.
Splicing inhibition using PlaB
Two 10 cm dishes of HeLa S3 cells per replicate cultured in DMEM medium were supplemented with 10 μL 100 nm Pladienolide B (PlaB, dissolved in DMSO) or DMSO (control). Cells were incubated for 3 h at 37°C and 5% CO2.
Cells were once washed with 5 mL cold PBS. 2 mL cold PBS, supplemented with 1:200 Ribolock and 1:100 Proteinase Inhibitor, was added to each plate and cells were scraped from dishes and collected in 2 mL tubes. Cells were spun for 5 minat 300 g and supernatant was removed.
For isolation of nuclei, the cell pellet was carefully resuspended in 500 μL 1x lysis buffer (140 mM NaCl, 1–5 mM MgCl2, 10 mM Tris HCl pH 7.5, 0.5% NP40, suppl. with Ribolock 1:20 and Proteinase inihibitor cocktail 1:100) and incubated for 5 min on ice. A cushion of 500 μL 1x lysis buffer/50% sucrose solution was pipetted under the cell lysate. Lysates were centrifuged through the cushion for 10 minat 16.000 g. The supernatant was removed and the nuclei pellet was again resuspended in 500 μL 1x lysis buffer and centrifuged through a sucrose cushion.
RNA from isolated nuclei was extracted using Trizol and Phenol-Chloroform extraction. A DNAse cleanup step was added to remove gDNA, using Turbo DNA-free kit.
FLAM-seq libraries were prepared as described above.
RNA metabolic labeling and pulldown
HEK Flp-In T-rex cells cultured in four 15 cm dishes in DMEM medium. 4-Thiouridine (4sU, dissolved in DMSO) was added at a final concentration of 1 mM. Incubation was performed for 10, 15, 20, 45 and 90 minutes. For the 0 min control, DMSO was added.
Cells were washed once with 10 mL cold PBS. 5 mL Trizol was added per dish and RNA was extracted using direct-zol RNA miniprep kit with DNAse gDNA removal.
Two biotinylation reactions (labeling buffer, 1 mg/mL biotin-EZ-link) with each 100 μg total RNA input were prepared for each sample and incubated for 2 h at RT in the dark.
Biotinylated RNA was purified using Phenol-Chloroform extraction. RNA was denatured for 3 minat 70°C and placed on ice.
Pulldown experiments were performed using MyOne Streptavidin C1 beads. 120 μL bead suspension were washed three times with 150 μL MPG buffer on a magnetic rack. Biotinylated RNA in 40 μL H2O was added to 120 μL MPG buffer on Streptavidin beads and incubated for 15 minat RT with rotation. Supernatants were separated from beads with bound biotinylated RNA by incubating on a magnetic rack for 1 min and collecting the supernatant as unbound fraction. Beads were washed three times with 150 μL pre-warmed MPG buffer (37°C). 150 μL 100 mM DTT was added and incubated for 5 min for elution of biotinylated RNAs as bound fractions. RNA from bound and unbound fractions were purified by Phenol-Chloroform extraction. FLAM-seq libraries were prepared from bound and unbound fractions as described above.
Dot blots were prepared by spotting 5 μg RNA from from each sample before and after biotinylation on a Amersham Hyperbond N+ membrane. The membrane was dried and crosslinked for with 2 × 1,200 μJ at 254 nm. The membrane was incubated with methylene blue for 10 min in order to record images of RNA transfer per spot.
The membrane was blocked 20 min in Blocking Solution and probed for 10 min with 1:10.000 dilution of Strep-HRP antibody. Membranes were washed with each 2x for 5 min with 10%, 1% and 0.1% Blocking Solution. Signals of biotinylated RNA were recorded by chemiluminescence detection upon addition of ECL Select reagent.
SLAM-seq
HeLa S3 cells were cultured in DMEM medium. Cells were seeded on 6-well plates until reaching 70% confluency. For metabolic labeling the medium was supplemented with 500 μM 4sU and or DMSO control and incubated for 0 min, 90 min, 180 min. Cells were harvested, and RNA was extracted using Trizol and Phenol-Chloroform extraction.
Polyadenylated RNA was extracted from 10 μg total RNA per sample using TruSeq RNA purification beads (Illumina) and eluted in 15 μL H2O.
First, GI tailing was performed using the USB poly(A) length kit. 4 μL 5x tail buffer mix was added to 2 μL 10x tail enzyme mix and incubated for 1 hat 37°C. 1.5 μL Stop solution was added to quench the reaction. GI-tailed RNA was cleaned up using a 1.8x ratio of RNAClean XP beads. Then, for alkylation reactions introducing T-C conversions, 15 μL GI-tailed RNA was incubated with 5 μL 100 mM iodoacetamide, 25 μL DMSO and 5 μL 500 mm NaPO4 pH 8.0 buffer for 15 minat 50°C. The reaction was quenched by addition of 1 μL 1 M DTT. RNA was purified using a 1.8x ratio of RNAClean XP beads and then FLAM-seq libraries were prepared as described above.
HeLa S3 cell fractionation
HeLa S3 cells were seeded in two 10 cm dishes per replicate in DMEM medium. Cells were washed once with cold PBS. 2 mL cold PBS, supplemented with 1:200 Ribolock and 1:100 Protease Inhibitor, was added and cells were scraped from dishes and collected in 2 mL tubes. 50 μL cell suspension was collected as input fraction.
The cell pellet was carefully resuspended in 500 μL 1x lysis buffer (140 mM NaCl, 1–5 mM MgCl2, 10 mM Tris HCl pH 7.5, 0.5% NP40) and incubated for 5 min on ice. A cushion of 500 μL 1x lysis buffer/50% sucrose solution was pipetted under the cell lysate. Lysates were centrifuged into the cushion for 10 minat 16,000 g. The supernatant was collected as cytoplasmic fraction. The nuclei pellet was again resuspended in 500 μL 1x lysis buffer and centrifuged through a sucrose cushion, the procedure was repeated once. Pellets were carefully resuspended in 100 μL Nuclear Buffer I (NaCl 75 mM, EDTA 0.5 mM, Tris HCl pH 7.9 20 mM, DTT 0.85 mM, Glycerol 50%, suppl. with Ribolock 1:20 and Proteinase inihibitor cocktail 1:100). 1 mL Nuclear Buffer II (NaCl 300 mM, MgCl2 7.5 mM, EDTA 0.2 mM, Urea 1 M, NP40 1%, DTT 1 mM, HEPES pH 7.9 20 mM, suppl. with Ribolock 1:20 and Proteinase inihibitor cocktail 1:100) was added, tubes were inverted 5 times and incubated for 15 min on ice. Suspensions were centrifuged for 10 minat 16.000 g. Supernatants were collected as nucleoplasmic fraction. Chromatin pellets were resuspended in 500 μL H2O. 50 μL of each fraction were collected for Western Blot analysis. 5 vol. Trizol were added to resuspended fractions and RNA was extracted by Phenol-Chloroform extraction. FLAM-seq libraries were prepared as described above.
For Western Blot analysis, 15 μL suspension from input, cytoplasmic and chromatin fraction, as well as 30 μL nucleoplasmic fraction were loaded on 12% SDS-PAGE gels and run according to standard protocols.68 Blotting was performed using BioRad Trans-Blot Turbo Transfer System and standard settings for 2 mini gels. Membranes were blocked using 5% skim milk in TBS-T buffer for 1 h. Membranes were then probed with BCAP31, GAPDH and TBP-43 antibodies in 5% skim milk/TBS-T overnight. Membranes were washed 3x in TBS-T and incubated with 1:10.000 anti-mouse- or anti-rabbit-HRP antibodies in 5% skim milk/TBS-T buffer. Membranes were washed 3x with TBS-T buffer. Membranes were probed with ECL Select Solution and imaged.
For certain experiments membranes were stripped and re-probed with antibodies.
Mouse brain cell fractionation
The method for separation of nuclei and cytoplasmic fractions from the mouse brain was adapted from Sigma-Aldrich single nuclei isolation protocol (Nuclei EZ prep nuclei isolation kit). The mouse brain tissue was thawed in a Petri dish and 1 mL EZ lysis buffer was added. A razor blade was used to cut the brain into two hemispheres, which each were used as replicates. The tissue was transferred in cold Dounce homogenizer and chopped with 20–25 strokes with pester A and 15 strokes with pester B. Homogenate was transferred into a 15 mL Falcon tube with wide-bore pipette. An aliquot was saved for RNA quality control. After addition of 2 mL of EZ lysis buffer, the falcon was vortexed briefly and the sample was incubated 5 min on ice. An aliquot of the whole cell lysate was saved for Western Blot. The tube was centrifuged at 500 g for 5 minat 4°C and supernatant was collected as cytoplasmic fraction. An aliquot was saved for Western Blot analysis. The nuclei pellet was resuspended in 4 mL EZ lysis buffer, incubated 5 min on ice and centrifuged again at 500 g for 5 minat 4°C. The supernatant was carefully aspired and the pellet was resuspended in 200 μL PBS-BSA 0.01%. An aliquot of the nuclei fractions was saved for Western Blot analysis. 10 μL nuclei sample was stained with DAPI to assess the quality of the nuclei suspension. All steps were performed on ice to minimize nuclei damage and maintain RNA quality. Western blot analysis of mouse brain fractions was performed as described above. RNA was extracted from fractions by Phenol-Chloroform extraction. FLAM-seq libraries were prepared as described above.
Quantification and statistical analysis
A detail description of the analyses is provided in the following sections. Further information on statistical tests, number of replicates and distribution parameters are provided in the figure legends.
Processing of FLAM-seq datasets
FLAM-seq datasets were processed using the FLAMAnalysis pipeline (https://github.com/rajewsky-lab/FLAMAnalysis) to identify gene identities as well as poly(A) tail length and tail sequence for each read. For analysis of 3′UTR isoforms, datasets were processed as previously described40 in order to group reads by 3′UTR isoforms. For human datasets, reads were mapped against human genome reference hg38 and human Gencode annotation version 28, both complemented with sequences from Thermo Fisher ERCC spike ins. For mouse datasets, reads were mapped against mouse GRCm38 and mouse Gencode annotation GRCm38.101.
Identification and analysis of unspliced reads
For identification of unspliced reads from FLAM-seq datasets, reads were required to overlap with a curated set of intron coordinates as well as 3′UTRs in order to exclude events of partial splicing or intron retention.
Intron sets were curated as follows. We first downloaded all introns, exons and 3′UTR annotations from UCSCtable browser (hg38, Gencode v24 for human, mm10, Gencove VM23 for mouse), then filtered for only protein coding genes, for which we presumed a better annotation, and filtered out those overlapping between each other with bedtools intersect.69 Along the same line, we filtered out all introns overlapping with annotated exons, also with bedtools intersect, not allowing any overlap.
Intronic reads were filtered using bedtools intersect -a bam_alignment -b intron.bed, filtering out overlaps shorter than 50 nt and retaining only unspliced reads, and restricted to those reads overlapping annotated 3′UTRs, in order to filter out potential artifacts such as internally primed reads, through bedtools intersect -a intronic_reads.bed -b 3utrs.bed.
For visualization of intronic poly(A) tail length distributions, intronic read counts were normalized to absolute reads counts for each dataset.
For the downsampling analysis, intronic reads and detected genes in HeLa S3, iPS and Organoid datasets were merged. Intronic reads were downsampled and for each downsampled read set the number of detected genes as a fraction of total genes in the merged dataset was calculated.
To investigate, for how many detected genes in FLAM-seq datasets identification of unspliced reads is possible, coordinates of introns in the curated intron set were compared to gencode annotation of genes using bedtools intersect -s -wa curated_introns.bed -wb gencode_genes.bed.
Intersected coordinates were then filtered for genes where the intron end is <3 kbp away from the annotated gene end, to get a realistic estimate of detectable genes since>95% of all reads are shorter than 3 kb.
In search of potential biases in our analysis, we compared the length distribution of introns to which unspliced reads were mapped to all introns (Figure S1F). We also compared the distribution of intron length derived from Gencode annotation for genes with detected unspliced transcripts against all genes per sample (Figure S1G).
For investigation of gene expression dependent changes in poly(A) tail length, genes from merged datasets for HeLa S3, iPS and Organoid datasets were binned by gene counts. Intronic reads with respective poly(A) tail lengths were assigned to genes in each expression bin (Figures S1H and S1I).
Analysis of nano-cop nascent RNA nanopore sequencing
We analyze previously published Nanopore direct RNA sequencing datasets24 to investigate poly(A) tail length of nascent, chromatin associated RNA. Fastq records and processed data were downloaded from GEO: GSE123191. Fastq reads were mapped using Minimap2 (version 2.16-r922)70 for direct RNA sequencing minimap2 -ax splice -uf -k14 -t 8 index fastq > sam.
Aligned reads were annotated using featureCounts and human gencode version 28.gtf annotation
featureCounts -L -g gene_name -s 0 -t gene -O --fracOverlap 0.3 -R CORE -a gtf -o out sam
Reads were grouped by provided read end annotation defining how read ends overlap with gene features (i.e. intronic, poly(A) site). Reads were annotated by provided poly(A) tail length estimates. For extraction of unspliced reads, we applied our computation pipeline as described above.
Browser visualization of FLAM-seq alignments
Visualization of FLAM-seq alignments including poly(A) tails on genome browsers was achieved by appending poly(A) tail sequences to the alignments, and loading these new alignments on IGV (or Gviz, or UCSC genome browser), taking advantage of the fact that these browsers mark mismatches with a different color for each nucleotide (thus coloring mismatched Ts, from the complementary DNA of the poly(A) tail, with a different color than the bulk of the aligned read). Briefly, the poly(A) tail sequences are retrieved from the output of the FLAMAnalysis pipeline (the cleaned_tail_lengths.txt file) and appended to the corresponding read of the bam file, also produced by the FLAMAnalysis pipeline. The CIGAR string is modified accordingly, appending a stretch of mismatches with the same length as the appended poly(A) tail.
Analysis of intron length
For comparison of intron length between genes with unspliced reads and all genes in each dataset, intron lengths of all annotated transcript isoforms for each gene were extracted from Homo Sapiens Gencode GTF annotation v28. Intron distributions for genes with detected unspliced reads were compared to the background of all detected genes in each dataset.
For comparing length of non-exon overlapping intron sequences (used for identification of unspliced reads, s. above), length distribution of introns overlapping with unspliced reads were compared to all introns in the curated intron set.
Analysis of half-life/3′UTR length for PlaB splicing inhibition
For analysis of gene half-lives between genes with altered median poly(A) tail length upon splicing inhibition, genes were first grouped into bins based on differences in median poly(A) tail length between PlaB treated and control samples. Genes with differences >50 nt were grouped in “shorter poly(A) PlaB” or “longer poly(A) PlaB” bins while remaining genes were classified as “unchanged poly(A) PlaB”. Control datasets comprised random genes of identical sample site as “shorter”/”longer” bins.
RNA half-life estimated per gene for HeLa cell lines were taken from Tani et al.71 and plotted for each bin.
3′UTR isoforms for reads from FLAM-seq datasets were annotated as previously described40 and 3′UTR length was plotted for each bin.
Metabolic labeling experiments
4sU metabolic labeling and pulldown experiments were analyzed by first quantifying poly(A) tail length for each read using the FLAMAnalysis pipeline. Intronic reads were extracted as described above. Poly(A) tail lengths between pulldown and supernatants were compared by calculating the differences in median poly(A) tail length of genes in pulldown datasets with the merged supernatant dataset for genes with more than 3 counts.
For analysis of SLAM-Seq datasets, reads were first processed applying the FLAM-seq pipeline to retain only reads with valid poly(A) tails. In a next step, filtered alignment files (∗_cleaned.bam) were annotated with an MD tag using samtools calmd72 with hg38 reference genome used for FLAMAnalysis pipeline. To exclude low quality reads with aberrant mutation patterns reads with >30 overall mutations and read quality <85 were excluded. We clipped the first 20 bases of each read as we observed an enrichment of mutations towards the read start which reflect mapping artefacts.
As proposed by Jürges et al.73 we estimated the ‘labeling rate’ plabel of T-C conversions from the fraction of observed T-C mutations overall bases sequences in a given dataset. We estimated the background (T-C) ‘error’ rate perror as the average mutation rate for all non-T-C mutations. For each read, we estimated the likelihood of an observed number of T-C mutations under a ‘labeling’ model versus an ‘error’ model. We modeled the probability of observing k T-C mutations in a read containing n Ts under each model as a binomial with B(n;k;plabel) or B(n;k;perror). We set a cutoff for flagging each read as ‘labeled’ if the log likelihood >1.15, which minimizes the number of false positives in datasets without 4sU labeling. We then produced a matrix with labeled and unlabeled reads for each gene.
Median poly(A) tail length per gene was compared between labelled reads and all reads per gene in each dataset by subtracting the median for genes with detected labeled reads and more than 3 counts in total.
Analysis of biochemical fractionation data
FLAM-seq datasets from biochemical fractionation experiments were processed using the FLAMAnalysis pipeline. 3′UTR isoforms for reads mapped to a given gene were annotated as described previously.40 Intronic reads were identified for each dataset as described above. lncRNA genes were extracted based on bioMart74 gene biotype annotation.
For comparisons of all poly(A) tail length profiles between fractions, poly(A) tail length densities were calculated for each replicate and fraction for poly(A) tail length bins of 10 nt. Mean and standard deviations were calculated for each poly(A) bin and plotted. Average density distributions for intronic reads calculated similarly and smoothed using R ‘smooth’ function.
To investigate sources of variation for poly(A) tail length profiles between different biological replicates we first calculated a scaling factor for each replicate as average scaled median poly(A) length for each fraction of a replicate. We applied a linear model to predict poly(A) tail length in a fraction of the variables ‘scaling factor’, ‘RNA concentration’, ‘fraction intronic reads’ and ‘cellular compartment’ and evaluate model performance using different predictors to identify factors best explaining variability in poly(A) tail length distributions. Scaled poly(A) tail length distributions were calculated by dividing poly(A) tail length for each read by the scaling factor the fraction.
For analysis of poly(A) tail length of genes with or without associated intronic reads, bulk reads were split by genes in each bin for each replicate and average densities were calculated as above.
Analysis of gene counts by poly(A) tail length bin was performed by binning median poly(A) tail length per gene and aggregating gene counts for genes in each bin. For analysis of intronic poly(A) tail length by poly(A) bin, genes were grouped by median poly(A) length bin and poly(A) tail length of intronic reads for genes in each bin were aggregated.
For analysis of gene half-life by poly(A) tail bin, half-life data of HeLa cells were downloaded from Tani et al.71 and plotted for each gene in poly(A) tail length bins.
For comparison of median poly(A) tail length per gene between fractions, genes were filtered to have more than 5 counts in each fraction. Cytoplasmic-to-nuclear ratios for each gene were taken from processed ENCODE data38 and plotted again median poly(A) tail length in each fraction for genes with >25 counts.
All above analyses were performed on a merged dataset of all replicates for each fraction.
Modelling of tail length distributions
To understand whether our hypothesis of a rapid nuclear deadenylation process is consistent with the observed tail length distributions at synthesis and at steady state, we conceived a simple model describing the amount of mRNA in a cell with a given tail length as a function of transcription, export, deadenylation and decay. For simplicity, we model the total amount of mRNA in the nucleus and in the cytosol as a function of global rates, thus not estimating gene-specific parameters as for example in,18 where much deeper sequencing data were produced for both steady state and metabolic labeling experiments.
In this model, we assume that a single cell contains 200,000 mRNA molecules at steady state, sorted between nucleoplasm, chromatin and cytosol according to certain ratios estimated by experimental quantification of total RNA masses obtained from 12 fractionation experiments (assuming the total amount of RNA in the fractions reflects the amount of mRNA, see Table S1). We use these ratios to compute the amount of mRNA molecules in the nucleus and in the cytosol, at the steady state, for each tail length between 1 and 501 nt, directly from FLAM-seq data. To account for variable contamination of the nuclear fraction with cytoplasmic mRNA, we scaled these distributions according to a contamination of 0, 25 and 50%. We then simulate the amount of nuclear and cytosolic mRNA for each tail length, as resulting from the steady state solutions:
| (Equation 1) |
| (Equation 2) |
of the ODEs:
| (Equation 3) |
| (Equation 4) |
The amount of nuclear mRNA xN with tail length i is a function of a transcription rate k0 (the only zeroth-order process in the chain), a deadenylation rate k1 and the amount of mRNA with tail length i +1, an export rate kE, and in one case, to which we will come, a decay rate kd. Conversely, the amount of cytoplasmic mRNA xC with tail length i is a function of the export rate and the amount of nuclear mRNA with tail length i, a cytoplasmic deadenylation rate k2 and the amount of cytoplasmic mRNA with tail length i +1, and a decay rate kd. Some of these rates are tail-length dependent. k0, the transcription rate, is the product of a synthesis rate a and a synthesis tail length probability function (slp). The decay rate is computed as a logistic function of parameters ad, md and vd, as previously done in Eisen et al.,18 since it describes the fact that tails are shortened to a certain length before decay by decapping and exonucleolytic activities can proceed. Nuclear and cytoplasmic deadenylation rates are not tail length-dependent. For the export rate kE, we allow two possibilities. We first set it as a constant (i.e. not depending on tail length) and we observed that the model would produce a large buildup of completely deadenylated molecules in the nucleus, which does not reflect common knowledge and observations. To account for this, we either allowed decay of short-tailed mRNAs in the nucleus (“nuclear decay – constant export” or ndce), or we added a second term to the export rate, modelled as a logistic of parameters aE, mE and vE, which would reflect a second scenario where mRNAs with a shortened tail are exported more efficiently (“no nuclear decay – logistic export” or nndle). Both these models were able to reproduce the experimentally determined poly(A) tail profiles, cutting down the residuals with the data of one order of magnitude with respect to the basic model with no nuclear decay and constant export.
With these two models, we proceeded to estimate the parameters by optimization. We used the sum of the squared differences between the simulated and measured nuclear and cytoplasmic mRNA amounts as a cost function to minimize. For minimizing it, we used the limited memory, box-constrained version of the Broyden–Fletcher–Goldfarb–Shanno method (L-BFGS-B) available within the optimx R package. We first make a guess of the initial parameters as explained below, then sample 1,000 times from a normal distribution built around each of these initial guesses with a standard deviation of 50% (and limited by generous lower and upper limits: if the sampling exceeds the limits, there are substituted with the initial guess), then run the optimization function to estimate a distribution for each parameter. This process is repeated taking into account three different contamination rates of FLAM-seq nuclear data, and for the two proposed models allowing nuclear decay and keeping export constant, or not allowing nuclear decay and adding a logistic export term. An example for the two models, along with that of the basic model which does not allow nuclear decay nor tail-dependent export, is shown in Figure 4B. We tested the models’ sensitivity to the parameters and observed that it behaved as expected (for example, increasing the synthesis rate increases the amount of mRNA in both nucleus and cytosol, increasing export rate increases at amount of mRNA in the cytosol at the expense of the nucleus etc.). Interestingly, given the tail dependency of decay, we observed that the cytoplasmic steady state mRNA level is strongly dependent on both nuclear and cytoplasmic deadenylation rates. In Figure S4E, we show as an example that doubling or halving the nuclear deadenylation rate results in changes of cytoplasmic mRNA level.
The parameters to initialize the optimization were estimated as follows. The slp is estimated with a loess fit of all intronic reads from nucleoplasm and chromatin of 12 fractionation experiments, and peaks at ca. 215 nt (Figure S6A). A global synthesis rate is computed as the sum of all synthesis rates of all genes from Eisen et al.,18 rescaled according to gene expression in our dataset (i.e. we kept only genes which were expressed in our data, and increased the total transcription rate by the ratio between the expression of these genes and all genes in our data). The global transcription rate was further scaled to estimate how many mRNA molecules are produced every minute in a cell containing 200,000 molecules at the steady state (Figure S6A). A similar estimate (640 mol/min as compared to 712 mol/min) was obtained from a completely independent project carried on in our lab (Haiyue Liu, manuscript in preparation), where single-cell RNA-sequencing of cells labeled with 4-thiouridine was used to estimate transcription rates in single cells. The deadenylation rates k1 and k2 were set equal, according to the distribution of deadenylation rates from Eisen et al.,18 weighted for gene expression in our data (Figure S6A). The decay rate parameters were also set identical to Eisen et al.,18 with the ad expressing the rate itself also weighted for gene expression in our data (Figure S6A). For the export rate, we set it around 0.02 min−1, estimating a nuclear half-life of ca. 30 min and consistent with metabolic labeling-based measurements in Drosophila by.50 For the logistic export rate, we adjusted the aE, mE and vE parameters from initial optimizations to 1.2, 5 and 10. These values are somehow arbitrary and are introduced, as previously stated, to eliminate the buildup of deadenylated nuclear molecules which happens if no nuclear turnover or no length-dependent export is allowed. As previously described, we sample 1,000 times from the distributions built around the initial guesses, and use these samples to initialize optimization with the merged FLAM-seq data from 12 fractionation experiments, with the two ndce and nndle models and with three values of contamination. We then retain the iterations with successful convergence and compute the parameters distributions shown in Figures S6B and S6C. The merged estimates from the two models and the three contaminations were used for the distribution shown in Figure 4C. With both models and only 0.25 contamination, we separately repeated the optimization for the 12 individual fractionation experiments and show the means in Figure S6D.
On the long-read implementation of SLAM-seq
SLAM-seq is a fast and robust protocol for investigating RNA dynamics over time by labeling newly synthesized RNA, which can be identified in high-throughput sequencing experiments through introduction of T-C conversions in labeled RNA.48 We combine the SLAM-Seq approach with our FLAM-seq method to simultaneously query poly(A) tail length of labeled reads.
Newly synthesized RNA incorporates 4sU which is added to the cell medium. 4sU moieties within RNA are alkylated by iodoacetamide (IAA) causing T-C mismatches during cDNA synthesis at the positions of 4sU incorporation. We perform 4sU labeling in HeLa S3 cell lines and subsequent poly(A) selection and GI tailing before IAA alkylation. Alkylation reactions are performed at basic pH and 50°C which may cause hydrolysis. While this has been carefully investigated for quantification of gene expression,48 it is unclear whether poly(A) length quantification could be affected by harsh reaction conditions. To minimize the effects of possible hydrolysis on poly(A) tails, we adapted the FLAM-seq protocol such that we append a GI tail to RNA poly(A) tails before alkylation. RNA fragmentation in the tail would cause loss of the GI tail and exclude those RNAs from being reverse transcribed using the FLAM-seq poly(C) RT primer.
We performed labeling of HeLa S3 cell lines for 0 (control), 90 and 180 minutes. We developed a computational pipeline to extract mutations with respect to a reference genome and applied a likelihood model similar to the GRAND-SLAM approach73 to categorize reads as newly synthesized (“labeled”) or pre-existing (“unlabeled”) based on observed T-C mutations over a background model of expected T-C mutations estimated from all mutations in a dataset.
We observed an increasing number of average T-C mutations per read proportional to the 4sU labeling time. The average number of total mutations per read increases similarly, yet we observed more non-T-C mutations for longer labeling (Figure S3I). Calculating the distribution of mutation events overall possible nucleotide conversions showed the expected enrichment of T-C conversions for 90 min and 180 min labeling while non-T-C mutations occurred at similar frequencies (Figure S3J). Overall, we categorized 15% of all reads as labeled after 90 min 4sU and ca. 22% after 180 min labeling. The fraction of labeled reads appeared slightly higher than the fraction of labeled total RNA in pulldown experiments (10% for 90 min labeling) and may be influenced by differences in labeling between ribosomal RNA, which makes up the majority of signal in pulldowns, and mRNA in SLAM-Seq.
Less than 1% of reads were flagged as “labeled” in 0 min control experiments, indicating high specificity of our computational approach (Figure S3K).
Comparing poly(A) tail length distributions between labeled and total reads from SLAM-Seq experiments showed a modest increase in poly(A) tail length of newly synthesized RNAs as observed in HEK pulldown experiments (Figure S3H).
Unspliced reads had longer poly(A) tails, yet with a significant number of reads having tails shorter than 100 nt as observed for bulk HeLa S3 datasets (Figure S3L).
Poly(A) tail length distributions of mitochondrial transcripts also peaked at 50 nt both for labeled and total reads categories (Figure S3M). Comparing the median poly(A) length for each gene between labeled and all reads showed a median increase in poly(A) length for labeled reads of 10–15 nt for 90 min and 180 min labeling respectively (Figure S3K). We did note an overall shift in global poly(A) tail length profiles between individual timepoints, which is unexpected since the SLAM-Seq protocol should preserve the steady-state. 0 min and 90 min labeling samples had longer tails (median 102–106 nt) than 180 min labeling (median 66 nt), which we attributed to variability introduced by the experimental setup which required extensive handling of ultra-low RNA quantities.
We conclude that combining the SLAM-Seq method with full-length RNA sequencing enables parallel investigation of RNA dynamics and poly(A) tail length but will require further optimization of the experimental protocol to avoid batch effects between labeling timepoints.
Acknowledgments
We thank C. A. Cerda Jara and A. Boltengagen from the Rajewsky lab for preparation of mouse brain subcellular fractions and preliminary analysis, C. Quedenau from the BIMSB Genomics platform for PacBio Sequencing, S. Ayoub and M. Schott from the Rajewsky lab for help with library preparations, A. Rybak-Wolf and R. P. Alacaraz from the BIMSB Organoids platform for help with fractionations, N. Karaiskos and M. Jens from the Rajewsky lab, D. Schwabe from the Falcke lab and F. Silverii (GFZ) for helpful discussions on modeling & data analysis, E. Conti and I. Schäfer (MPI Biochemistry) and E. Wahle (Halle University) for helpful discussions and data interpretation. Illustrations in Figures 2A, 3A, 3D, and 4A and the graphical abstract were created with Biorender. I.L. was supported by an EMBO long term fellowship (ALTF1235-2016). J.A. was supported by the MDC-NYU PhD exchange program. M.P. was supported by an Erasmus Mundus fellowship.
Author contributions
J.A.: Study design, Experiments, Computational analysis, Manuscript.
I.L.: Study design, Experiments, Computational analysis.
M.P.: Experiments, Computational analysis.
N.R.: Study design and Supervision.
Declaration of interests
The authors declare no competing interests. A patent application for FLAM-seq was filed before this work (WO2020069791A1).
Published: January 20, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.105878.
Supplemental information
Data and code availability
-
•
All raw and processed FLAM-seq data have been deposited at Gene Expression Omnibus (GEO) and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. This paper also analyzes existing, publicly available data. The accession numbers for the datasets are available in the key resources table.
-
•
FLAM-seq datasets were processed using the FLAMAnalysis pipeline available at https://github.com/rajewsky-lab/FLAMAnalysis. Additional code used for analyzing the data is described in the STAR Methods and is available at Mendeley data along with the processed datasets used for the analyses. Accession numbers are listed in the key resources table.
-
•
Additional raw data and/or specific information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Garneau N.L., Wilusz J., Wilusz C.J. The highways and byways of mRNA decay. Nat. Rev. Mol. Cell Biol. 2007;8:113–126. doi: 10.1038/nrm2104. [DOI] [PubMed] [Google Scholar]
- 2.Munroe D., Jacobsen A. Tales of poly(A): a review. Gene. 1990;91:151–158. doi: 10.1016/0378-1119(90)90082-3. [DOI] [PubMed] [Google Scholar]
- 3.Preiss T., Muckenthaler M., Hentze M.W. Poly(A)-tail-promoted translation in yeast: implications for translational control. RNA. 1998;4:1321–1331. doi: 10.1017/s1355838298980669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beilharz T.H., Preiss T. Widespread use of poly(A) tail length control to accentuate expression of the yeast transcriptome. RNA. 2007;13:982–997. doi: 10.1261/rna.569407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weill L., Belloc E., Bava F.A., Méndez R. Translational control by changes in poly(A) tail length: recycling mRNAs. Nat. Struct. Mol. Biol. 2012;19:577–585. doi: 10.1038/nsmb.2311. [DOI] [PubMed] [Google Scholar]
- 6.Chorghade S., Seimetz J., Emmons R., Yang J., Bresson S.M., Lisio M.D., Parise G., Conrad N.K., Kalsotra A. Poly(A) tail length regulates PABPC1 expression to tune translation in the heart. Elife. 2017;6:e24139. doi: 10.7554/eLife.24139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Palatnik C.M., Wilkins C., Jacobson A. Translational control during early Dictyostelium development: possible involvement of poly(A) sequences. Cell. 1984;36:1017–1025. doi: 10.1016/0092-8674(84)90051-5. [DOI] [PubMed] [Google Scholar]
- 8.Sheets M.D., Fox C.A., Hunt T., Vande Woude G., Wickens M. The 3’-untranslated regions of c-mos and cyclin mRNAs stimulate translation by regulating cytoplasmic polyadenylation. Genes Dev. 1994;8:926–938. doi: 10.1101/gad.8.8.926. [DOI] [PubMed] [Google Scholar]
- 9.Subtelny A.O., Eichhorn S.W., Chen G.R., Sive H., Bartel D.P. Poly(A)-tail profiling reveals an embryonic switch in translational control. Nature. 2014;508:66–71. doi: 10.1038/nature13007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eckmann C.R., Rammelt C., Wahle E. Control of poly(A) tail length. Wiley Interdiscip. Rev. RNA. 2011;2:348–361. doi: 10.1002/wrna.56. [DOI] [PubMed] [Google Scholar]
- 11.Wahle E. Purification and characterization of a mammalian polyadenylate polymerase involved in the 3’ end processing of messenger RNA precursors. J. Biol. Chem. 1991;266:3131–3139. [PubMed] [Google Scholar]
- 12.Christofori G., Keller W. Poly(A) polymerase purified from HeLa cell nuclear extract is required for both cleavage and polyadenylation of pre-mRNA in vitro. Mol. Cell Biol. 1989;9:193–203. doi: 10.1128/mcb.9.1.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wahle E. A novel poly(A)-binding protein acts as a specificity factor in the second phase of messenger RNA polyadenylation. Cell. 1991;66:759–768. doi: 10.1016/0092-8674(91)90119-j. [DOI] [PubMed] [Google Scholar]
- 14.Keller R.W., Kühn U., Aragón M., Bornikova L., Wahle E., Bear D.G. The nuclear poly(A) binding protein, PABP2, forms an oligomeric particle covering the length of the poly(A) tail. J. Mol. Biol. 2000;297:569–583. doi: 10.1006/jmbi.2000.3572. [DOI] [PubMed] [Google Scholar]
- 15.Kühn U., Gündel M., Knoth A., Kerwitz Y., Rüdel S., Wahle E. Poly(A) tail length is controlled by the nuclear Poly(A)-binding protein regulating the interaction between Poly(A) polymerase and the cleavage and polyadenylation specificity factor. J. Biol. Chem. 2009;284:22803–22814. doi: 10.1074/jbc.M109.018226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sheiness D., Puckett L., Darnell J.E. Possible relationship of poly(A) shortening to mRNA turnover. Proc. Natl. Acad. Sci. USA. 1975;72:1077–1081. doi: 10.1073/pnas.72.3.1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brawerman G., Diez J. Metabolism of the polyadenylate sequence of nuclear RNA and messenger RNA in mammalian cells. Cell. 1975;5:271–280. doi: 10.1016/0092-8674(75)90102-6. [DOI] [PubMed] [Google Scholar]
- 18.Eisen T.J., Eichhorn S.W., Subtelny A.O., Lin K.S., McGeary S.E., Gupta S., Bartel D.P. The dynamics of cytoplasmic mRNA metabolism. Mol. Cell. 2020;77:786–799.e10. doi: 10.1016/j.molcel.2019.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brown C.E., Sachs A.B. Poly(A) tail length control in Saccharomyces cerevisiae occurs by message-specific deadenylation. Mol. Cell Biol. 1998;18:6548–6559. doi: 10.1128/mcb.18.11.6548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cao D., Parker R. Computational modeling of eukaryotic mRNA turnover. RNA. 2001;7:1192–1212. doi: 10.1017/s1355838201010330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Misra A., Green M.R. From polyadenylation to splicing: dual role for mRNA 3’ end formation factors. RNA Biol. 2016;13:259–264. doi: 10.1080/15476286.2015.1112490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ameur A., Zaghlool A., Halvardson J., Wetterbom A., Gyllensten U., Cavelier L., Feuk L. Total RNA sequencing reveals nascent transcription and widespread co-transcriptional splicing in the human brain. Nat. Struct. Mol. Biol. 2011;18:1435–1440. doi: 10.1038/nsmb.2143. [DOI] [PubMed] [Google Scholar]
- 23.Tilgner H., Knowles D.G., Johnson R., Davis C.A., Chakrabortty S., Djebali S., Curado J., Snyder M., Gingeras T.R., Guigó R. Deep sequencing of subcellular RNA fractions shows splicing to be predominantly co-transcriptional in the human genome but inefficient for lncRNAs. Genome Res. 2012;22:1616–1625. doi: 10.1101/gr.134445.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Drexler H.L., Choquet K., Churchman L.S. Splicing kinetics and coordination revealed by direct nascent RNA sequencing through nanopores. Mol. Cell. 2020;77:985–998.e8. doi: 10.1016/j.molcel.2019.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Coté A., Coté C., Bayatpour S., Drexler H.L., Alexander K.A., Chen F., Wassie A.T., Boyden E.S., Berger S., Churchman L.S., Raj A. The spatial distributions of pre-mRNAs suggest post-transcriptional splicing of specific introns within endogenous genes. bioRxiv. 2020 doi: 10.1101/2020.04.06.028092. Preprint at. [DOI] [Google Scholar]
- 26.Niwa M., Berget S.M. Mutation of the AAUAAA polyadenylation signal depresses in vitro splicing of proximal but not distal introns. Genes Dev. 1991;5:2086–2095. doi: 10.1101/gad.5.11.2086. [DOI] [PubMed] [Google Scholar]
- 27.Vagner S., Vagner C., Mattaj I.W. The carboxyl terminus of vertebrate poly(A) polymerase interacts with U2AF 65 to couple 3’-end processing and splicing. Genes Dev. 2000;14:403–413. [PMC free article] [PubMed] [Google Scholar]
- 28.Bahar Halpern K., Caspi I., Lemze D., Levy M., Landen S., Elinav E., Ulitsky I., Itzkovitz S. Nuclear retention of mRNA in mammalian tissues. Cell Rep. 2015;13:2653–2662. doi: 10.1016/j.celrep.2015.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Deveson I.W., Brunck M.E., Blackburn J., Tseng E., Hon T., Clark T.A., Clark M.B., Crawford J., Dinger M.E., Nielsen L.K., et al. Universal alternative splicing of noncoding exons. Cell Syst. 2018;6:245–255.e5. doi: 10.1016/j.cels.2017.12.005. [DOI] [PubMed] [Google Scholar]
- 30.Djebali S., Davis C.A., Merkel A., Dobin A., Lassmann T., Mortazavi A., Tanzer A., Lagarde J., Lin W., Schlesinger F., et al. Landscape of transcription in human cells. Nature. 2012;489:101–108. doi: 10.1038/nature11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lubelsky Y., Ulitsky I. Sequences enriched in Alu repeats drive nuclear localization of long RNAs in human cells. Nature. 2018;555:107–111. doi: 10.1038/nature25757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Palazzo A.F., Lee E.S. Sequence determinants for nuclear retention and cytoplasmic export of mRNAs and lncRNAs. Front. Genet. 2018;9:440–516. doi: 10.3389/fgene.2018.00440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dower K., Kuperwasser N., Merrikh H., Rosbash M. A synthetic A tail rescues yeast nuclear accumulation of a ribozyme-terminated transcript. RNA. 2004;10:1888–1899. doi: 10.1261/rna.7166704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wilusz J.E., JnBaptiste C.K., Lu L.Y., Kuhn C.D., Joshua-Tor L., Sharp P.A. A triple helix stabilizes the 3′ ends of long noncoding RNAs that lack poly(A) tails. Genes Dev. 2012;26:2392–2407. doi: 10.1101/gad.204438.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Collart M.A. The Ccr4-Not complex is a key regulator of eukaryotic gene expression. Wiley Interdiscip. Rev. RNA. 2016;7:438–454. doi: 10.1002/wrna.1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wolf J., Passmore L.A. mRNA deadenylation by Pan2–Pan3. Biochem. Soc. Trans. 2014;42:184–187. doi: 10.1042/BST20130211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yamashita A., Chang T.C., Yamashita Y., Zhu W., Zhong Z., Chen C.Y.A., Shyu A.B. Concerted action of poly(A) nucleases and decapping enzyme in mammalian mRNA turnover. Nat. Struct. Mol. Biol. 2005;12:1054–1063. doi: 10.1038/nsmb1016. [DOI] [PubMed] [Google Scholar]
- 38.Yi H., Park J., Ha M., Lim J., Chang H., Kim V.N. PABP cooperates with the CCR4-NOT complex to promote mRNA deadenylation and block precocious decay. Mol. Cell. 2018;70:1081–1088.e5. doi: 10.1016/j.molcel.2018.05.009. [DOI] [PubMed] [Google Scholar]
- 39.Goldstrohm A.C., Wickens M. Multifunctional deadenylase complexes diversify mRNA control. Nat. Rev. Mol. Cell Biol. 2008;9:337–344. doi: 10.1038/nrm2370. [DOI] [PubMed] [Google Scholar]
- 40.Legnini I., Alles J., Karaiskos N., Ayoub S., Rajewsky N. FLAM-seq: full-length mRNA sequencing reveals principles of poly(A) tail length control. Nat. Methods. 2019;16:879–886. doi: 10.1038/s41592-019-0503-y. [DOI] [PubMed] [Google Scholar]
- 41.Martínez-Montiel N., Rosas-Murrieta N.H., Martínez-Montiel M., Gaspariano-Cholula M.P., Martínez-Contreras R.D. Microbial and natural metabolites that inhibit splicing: a powerful alternative for cancer treatment. BioMed Res. Int. 2016;2016:3681094. doi: 10.1155/2016/3681094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Workman R.E., Tang A.D., Tang P.S., Jain M., Tyson J.R., Razaghi R., Zuzarte P.C., Gilpatrick T., Payne A., Quick J., et al. Nanopore native RNA sequencing of a human poly(A) transcriptome. Nat. Methods. 2019;16:1297–1305. doi: 10.1038/s41592-019-0617-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dölken L., Ruzsics Z., Rädle B., Friedel C.C., Zimmer R., Mages J., Hoffmann R., Dickinson P., Forster T., Ghazal P., Koszinowski U.H. High-resolution gene expression profiling for simultaneous kinetic parameter analysis of RNA synthesis and decay. RNA. 2008;14:1959–1972. doi: 10.1261/rna.1136108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rabani M., Levin J.Z., Fan L., Adiconis X., Raychowdhury R., Garber M., Gnirke A., Nusbaum C., Hacohen N., Friedman N., et al. Metabolic labeling of RNA uncovers principles of RNA production and degradation dynamics in mammalian cells. Nat. Biotechnol. 2011;29:436–442. doi: 10.1038/nbt.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Temperley R.J., Wydro M., Lightowlers R.N., Chrzanowska-Lightowlers Z.M. Human mitochondrial mRNAs-like members of all families, similar but different. Biochim. Biophys. Acta. 2010;1797:1081–1085. doi: 10.1016/j.bbabio.2010.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Temperley R.J., Seneca S.H., Tonska K., Bartnik E., Bindoff L.A., Lightowlers R.N., Chrzanowska-Lightowlers Z.M.A. Investigation of a pathogenic mtDNA microdeletion reveals a translation-dependent deadenylation decay pathway in human mitochondria. Hum. Mol. Genet. 2003;12:2341–2348. doi: 10.1093/hmg/ddg238. [DOI] [PubMed] [Google Scholar]
- 47.Rabani M., Raychowdhury R., Jovanovic M., Rooney M., Stumpo D.J., Pauli A., Hacohen N., Schier A.F., Blackshear P.J., Friedman N., et al. High-resolution sequencing and modeling identifies distinct dynamic RNA regulatory strategies. Cell. 2014;159:1698–1710. doi: 10.1016/j.cell.2014.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Herzog V.A., Reichholf B., Neumann T., Rescheneder P., Bhat P., Burkard T.R., Wlotzka W., von Haeseler A., Zuber J., Ameres S.L. Thiol-linked alkylation of RNA to assess expression dynamics. Nat. Methods. 2017;14:1198–1204. doi: 10.1038/nmeth.4435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mor A., Suliman S., Ben-Yishay R., Yunger S., Brody Y., Shav-Tal Y. Dynamics of single mRNP nucleocytoplasmic transport and export through the nuclear pore in living cells. Nat. Cell Biol. 2010;12:543–552. doi: 10.1038/ncb2056. [DOI] [PubMed] [Google Scholar]
- 50.Chen T., van Steensel B. Comprehensive analysis of nucleocytoplasmic dynamics of mRNA in Drosophila cells. PLoS Genet. 2017;13:e1006929. doi: 10.1371/journal.pgen.1006929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jadot M., Boonen M., Thirion J., Wang N., Xing J., Zhao C., Tannous A., Qian M., Zheng H., Everett J.K., et al. Accounting for protein subcellular localization: a compartmental map of the rat liver proteome. Mol. Cell. Proteomics. 2017;16:194–212. doi: 10.1074/mcp.M116.064527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chang H., Lim J., Ha M., Kim V.N. TAIL-seq: genome-wide determination of poly(A) tail length and 3’ end modifications. Mol. Cell. 2014;53:1044–1052. doi: 10.1016/j.molcel.2014.02.007. [DOI] [PubMed] [Google Scholar]
- 53.Lima S.A., Chipman L.B., Nicholson A.L., Chen Y.H., Yee B.A., Yeo G.W., Coller J., Pasquinelli A.E. Short poly(A) tails are a conserved feature of highly expressed genes. Nat. Struct. Mol. Biol. 2017;24:1057–1063. doi: 10.1038/nsmb.3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sheiness D., Darnell J.E. Polyadenylic acid segment becomes shorter with age. Nat. New Biol. 1973;241:265–268. doi: 10.1038/newbio241265a0. [DOI] [PubMed] [Google Scholar]
- 55.Bresson S.M., Hunter O.V., Hunter A.C., Conrad N.K. Canonical poly(A) polymerase activity promotes the decay of a wide variety of mammalian nuclear RNAs. PLoS Genet. 2015;11:e1005610–e1005625. doi: 10.1371/journal.pgen.1005610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shyu A.B., Greenberg M.E., Belasco J.G. The c-fos transcript is targeted for rapid decay by two distinct mRNA degradation pathways. Genes Dev. 1989;3:60–72. doi: 10.1101/gad.3.1.60. [DOI] [PubMed] [Google Scholar]
- 57.Zhang Q., Di C., Yan J., Wang F., Qu T., Wang Y., Chen Y., Zhang X., Liu Y., Yang H., Zhang H. Inhibition of SF3b1 by pladienolide B evokes cycle arrest, apoptosis induction and p73 splicing in human cervical carcinoma cells. Artif. Cell Nanomed. Biotechnol. 2019;47:1273–1280. doi: 10.1080/21691401.2019.1596922. [DOI] [PubMed] [Google Scholar]
- 58.Fasken M.B., Corbett A.H. Mechanisms of nuclear mRNA quality control. RNA Biol. 2009;6:237–241. doi: 10.4161/rna.6.3.8330. [DOI] [PubMed] [Google Scholar]
- 59.Meola N., Domanski M., Karadoulama E., Chen Y., Gentil C., Pultz D., Vitting-Seerup K., Lykke-Andersen S., Andersen J.S., Sandelin A., Jensen T.H. Identification of a nuclear exosome decay pathway for processed transcripts. Mol. Cell. 2016;64:520–533. doi: 10.1016/j.molcel.2016.09.025. [DOI] [PubMed] [Google Scholar]
- 60.Schmid M., Jensen T.H. Controlling nuclear RNA levels. Nat. Rev. Genet. 2018;19:518–529. doi: 10.1038/s41576-018-0013-2. [DOI] [PubMed] [Google Scholar]
- 61.Wahle E. Poly(A) tail length control is caused by termination of processive synthesis. J. Biol. Chem. 1995;270:2800–2808. doi: 10.1074/jbc.270.6.2800. [DOI] [PubMed] [Google Scholar]
- 62.Fox A.H., Nakagawa S., Hirose T., Bond C.S. Paraspeckles: where long noncoding RNA meets phase separation. Trends Biochem. Sci. 2018;43:124–135. doi: 10.1016/j.tibs.2017.12.001. [DOI] [PubMed] [Google Scholar]
- 63.Mukherjee N., Calviello L., Hirsekorn A., de Pretis S., Pelizzola M., Ohler U. Integrative classification of human coding and noncoding genes through RNA metabolism profiles. Nat. Struct. Mol. Biol. 2017;24:86–96. doi: 10.1038/nsmb.3325. [DOI] [PubMed] [Google Scholar]
- 64.Schlackow M., Nojima T., Gomes T., Dhir A., Carmo-Fonseca M., Proudfoot N.J. Distinctive patterns of transcription and RNA processing for human lincRNAs. Mol. Cell. 2017;65:25–38. doi: 10.1016/j.molcel.2016.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Delan-Forino C., Spanos C., Rappsilber J., Tollervey D. Substrate specificity of the TRAMP nuclear surveillance complexes. Nat. Commun. 2020;11:3122. doi: 10.1038/s41467-020-16965-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tudek A., Krawczyk P.S., Mroczek S., Tomecki R., Turtola M., Matylla-Kulińska K., Jensen T.H., Dziembowski A. Global view on the metabolism of RNA poly(A) tails in yeast Saccharomyces cerevisiae. Nat. Commun. 2021;12:4951. doi: 10.1038/s41467-021-25251-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Singhania R., Thorn G.J., Williams K., Gandhi R.D., Daher C., Barthet-Barateig A., Parker H.N., Utami W., Al-Siraj M., Barrett D.A., Wattis J.A. Nuclear poly(A) tail size is regulated by Cnot1 during the serum response. bioRxiv. 2019 doi: 10.1101/773432. Preprint at. [DOI] [Google Scholar]
- 68.Laemmli U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 69.Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018;34:3094–3100. doi: 10.1093/bioinformatics/bty191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tani H., Mizutani R., Salam K.A., Tano K., Ijiri K., Wakamatsu A., Isogai T., Suzuki Y., Akimitsu N. Genome-wide determination of RNA stability reveals hundreds of short-lived noncoding transcripts in mammals. Genome Res. 2012;22:947–956. doi: 10.1101/gr.130559.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jürges C., Dölken L., Erhard F. Dissecting newly transcribed and old RNA using GRAND-SLAM. Bioinformatics. 2018;34:i218–i226. doi: 10.1093/bioinformatics/bty256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Durinck S., Moreau Y., Kasprzyk A., Davis S., De Moor B., Brazma A., Huber W. BioMart and Bioconductor: a powerful link between biological databases and microarray data analysis. Bioinformatics. 2005;21:3439–3440. doi: 10.1093/bioinformatics/bti525. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All raw and processed FLAM-seq data have been deposited at Gene Expression Omnibus (GEO) and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. This paper also analyzes existing, publicly available data. The accession numbers for the datasets are available in the key resources table.
-
•
FLAM-seq datasets were processed using the FLAMAnalysis pipeline available at https://github.com/rajewsky-lab/FLAMAnalysis. Additional code used for analyzing the data is described in the STAR Methods and is available at Mendeley data along with the processed datasets used for the analyses. Accession numbers are listed in the key resources table.
-
•
Additional raw data and/or specific information required to reanalyze the data reported in this paper is available from the lead contact upon request.