

Summary
Accurate prediction of protein–ligand binding affinity is crucial in structure-based drug design but remains some challenges even with recent advances in deep learning: (1) Existing methods neglect the edge information in protein and ligand structure data; (2) current attention mechanisms struggle to capture true binding interactions in the small dataset. Herein, we proposed SEGSA_DTA, a SuperEdge Graph convolution-based and Supervised Attention-based Drug–Target Affinity prediction method, where the super edge graph convolution can comprehensively utilize node and edge information and the multi-supervised attention module can efficiently learn the attention distribution consistent with real protein-ligand interactions. Results on the multiple datasets show that SEGSA_DTA outperforms current state-of-the-art methods. We also applied SEGSA_DTA in repurposing FDA-approved drugs to identify potential coronavirus disease 2019 (COVID-19) treatments. Besides, by using SHapley Additive exPlanations (SHAP), we found that SEGSA_DTA is interpretable and further provides a new quantitative analytical solution for structure-based lead optimization.
Subject areas: Classification of proteins, Molecular interaction, Biocomputational method
Graphical abstract
Highlights
-
•
The SuperEdge GNN is used to fuse node and edge features of protein structure
-
•
More information of protein-ligand interactions is used to assist model training
-
•
SHapley Additive exPlanations (SHAP) provides more reasonable explanation for the model
Classification of proteins; Molecular interaction; Biocomputational method
Introduction
Protein–ligand binding affinity calculation is crucial to the discovery and optimization of lead compounds based on the protein structure. With the recent advancements in data storage and computing power, various computational methods have been developed to calculate the binding affinity of a given protein–ligand complex,1 including molecular docking, molecular dynamics simulation and molecular mechanics/quantum mechanics. It is widely known that molecular dynamics simulation methods and molecular mechanics/quantum mechanics methods can calculate the protein–ligand binding affinity with high accuracy. Nevertheless, the extensive computational resources required by these methods greatly limit the ligand search space in the process of drug design.2 Molecular docking uses a scoring function to evaluate the binding conformation obtained by each docking to predict binding affinity and uses significantly fewer computational resources. Molecular docking can thereby quickly calculate the binding affinity between multiple ligands and proteins and is commonly used for high-throughput virtual screening of drug molecules based on the protein structure. Nonetheless, the computational efficiency of molecular docking comes at the expense of accuracy.
In recent years, deep learning has achieved great success in many fields such as computer vision, speech recognition and natural language processing. Deep learning is also widely used in numerous aspects of drug design,3 including de novo molecule design and chemical synthesis. Currently, most deep learning-based protein–ligand binding affinity prediction models, such as DeepDTA,4 DeepAffinity5 and DeepDTAF,6 only use protein and ligand sequence information to predict protein–ligand binding affinity. It is known that protein structure information is essential for protein-ligand binding, yet these sequence-based methods lack sufficient structure information. Alternatively, several studies have utilized co-crystal structures of protein–ligand complexes to predict binding affinity.7 Nevertheless, obtaining co-crystal structure in protein–ligand complexes is a time-consuming practice, and thus the protein structure-based model is still needed. Moreover, with the recent development of graph neural networks, the advantages of using graphs to represent proteins and ligands are gradually being highlighted in deep learning-based models.8 The graphical representation retains the structure information of proteins and ligands,9,10 and graph convolutional neural network (GCN)11 is commonly used to learn the structure pattern. However, current graph neural network-based methods for protein–ligand binding affinity prediction focus more on the nodes, that is, they only use graph neural networks to embed node features and neglect the influence of edge features. How to effectively exploit the edge information of proteins and ligands in graph neural networks, especially in graph convolutional networks, still needs further investigation.
Graph-based methods are useful for learning feature representations, but a protein or ligand may contain a large number of features (residues or individual atoms), each of which might affect binding affinity differently regardless of whether they are directly involved with binding, and thus complicate graph-based predictions. The attention mechanism12 is one of the most powerful approaches to solve this problem. By introducing an attention mechanism, the prediction model can focus on features that are most relevant to the prediction target and at the same time improve the accuracy and interpretability of the model.13,14,15 However, because of the small size of the structure dataset and the lack of detailed knowledge concerning protein–ligand interactions, most of the existing methods are not yet able to effectively learn the attention distribution and accurately capture the true interaction information between proteins and ligands, limiting the predictive performance.16 Several studies in the fields of visual question answering17,18 and natural language processing19,20 have demonstrated that training attention mechanism in a supervised manner can result in more effective attention distribution and improve model performance significantly, but its effectiveness in building a better protein–ligand binding affinity prediction model remains unclear.
In this study, to predict protein–ligand binding affinity using structures of proteins and ligands, we developed a deep neural network SEGSA_DTA, a SuperEdge Graph convolution-based and Supervised Attention-based Drug–Target Affinity prediction method. Super edge graph convolution was adopted to learn feature representations of both nodes and edges from graph structures of proteins and ligands. Multiple supervised attention blocks were built with prior knowledge of protein–ligand non-covalent interaction and residue contribution for ligand binding, making it possible to learn the attention distribution effectively. A Comprehensive evaluation on the multiple datasets indicates that SEGSA_DTA offers superior performance compared to existing state-of-the-art methods. Also, we applied SEGSA_DTA to repurpose FDA-approved drugs to discover potential anti-coronavirus disease 2019 (COVID-19) treatments, some of which have been reported to possess inhibitory activity against the main protease of SARS-CoV-2 (SARS-CoV-2 Mpro). Moreover, in the case study, we found that SEGSA_DTA reveals the selective binding mechanism of ligands to proteins and can provide a new quantitative analytical solution for structure-based lead optimization.
Results
Simultaneously modeling binding affinity, non-covalent interaction and residue contribution
SEGSA_DTA is a multi-task learning network with three tasks, predicting protein–ligand binding affinity, non-covalent interaction and residue contribution simultaneously. Protein–ligand non-covalent interaction and residue contribution values are key factors affecting protein–ligand binding, and they have a strong correlation with binding affinity, hence SEGSA_DTA regards binding affinity prediction as the main task and protein–ligand non-covalent interaction prediction and residue contribution prediction as two auxiliary tasks.
Regarding the model structure (Figure 1A), SEGSA_DTA contains three modules: (1) The feature extraction module (Figure 1B) is first used to extract pocket and ligand features from each protein–ligand pair. In this module, the super edge graph convolution network is designed, in which in addition to the graph convolution network of nodes, an extra graph convolution network is used to extract features from neighbor edges, forming the so-called super edge and fusing it into node information to produce a more informative representation. (2) Then, the pocket and ligand features are processed by the interaction module to produce the pocket and ligand vectors, predicting the non-covalent interaction and the residue contribution (Figure 1C). The interaction module is structurally a multi-supervised attention module, including a supervised bidirectional attention block (pale blue background color) and a supervised unidirectional attention block (pale yellow background color), whose attention distributions are supervised by the prior knowledge of qualitative protein–ligand non-covalent interaction and quantitative residue contribution, respectively. (3) Finally, the pocket and ligand vectors are concatenated and fed into the prediction module, a fully connected deep neural network with two hidden layers, to predict the binding affinity (STAR Methods).
Figure 1.

Architecture of SEGSA_DTA
(A) Architecture overview of SEGSA_DTA. The SEGSA_DTA is shown to predict the binding affinity of protein–ligand pairs. In this model, a feature extraction module is first used to extract pocket and ligand features from each protein–ligand pair. Each row of the pocket or ligand features represents a residue or an atom. Then, the pocket and ligand features are processed by the interaction module to produce the pocket and ligand vectors, with the predicted non-covalent interactions and the predicted residue contributions. Finally, the pocket and ligand vectors are concatenated and fed into the prediction module, a fully connected deep neural network with two hidden layers, to predict the binding affinity.
(B) Feature extraction module. The input protein graph and ligand graph are subjected to super edge graph convolution network to extract protein and ligand features, respectively. The pocket features are selected from the protein features according to protein pocket residues.
(C) Interaction module. The interaction module is structurally a multi-supervised attention module, including a supervised bidirectional attention block (pale blue background color) and a supervised unidirectional attention block (pale yellow background color).
SEGSA_DTA demonstrates state-of-the-art scoring power
Scoring power, an important assessment of binding affinity prediction, aims to evaluate the linear correlation between the predicted binding affinity and the true one. To test the scoring power of SEGSA_DTA, we compared three state-of-the-art deep learning-based methods on the core set v.2016, including DeepDTAF,6 Pafnucy7 and GraphDTA.9 These three methods are implemented using the official open source repository and ensure that the performance of the obtained models is consistent with the results of the original papers.
As shown in Table 1, SEGSA_DTA demonstrates the best performance overall, which achieves the best results on Pearson correlation coefficient (Pearson), standard deviation in regression (SD), RMSE and MAE, and achieves competitive performance with DeepDTAF on the concordance index (CI), differing by only 0.5%.
Table 1.
Scoring power of SEGSA_DTA and other methods on the core set v.2016
| Method | RMSE | MAE | Pearson | SD | CI |
|---|---|---|---|---|---|
| GraphDTA | 5.649 | 5.226 | 0.123 | 2.161 | 0.539 |
| Pafnucy | 1.423 | 1.136 | 0.774 | 1.378 | 0.788 |
| DeepDTAF | 1.355 | 1.073 | 0.789 | 1.337 | 0.799 |
| SEGSA_DTA | 1.319 | 1.063 | 0.799 | 1.308 | 0.794 |
Bold indicates the best prediction performance.
It is worth highlighting that SEGSA_DTA outperforms all other compared methods on two key indicators of scoring power, Pearson and SD.22 According to the Pearson, SEGSA_DTA achieves a strong correlation value of 0.799, whereas the Pearson of GraphDTA (Table S1), Pafnucy, and DeepDTAF were 0.123, 0.774, and 0.789, respectively (Figure 2A). In terms of SD (the smaller the SD, the better the model performance), SEGSA_DTA achieves the lowest SD score, which is 85.3% (GraphDTA), 7.0% (Pafnucy) and 2.9% (DeepDTAF) lower than the other methods respectively.
Figure 2.

Performance of SEGSA and effectiveness of the model structure
(A) Distributions of predicted binding affinities on the core set v.2016.
(B) Virtual screening power of SEGSA_DTA and other methods on the DUD-Ehand.
(C) Performance of SEGSA_DTA with different edge features on the validation set of 5-fold cross-validation. Data are represented as mean ±95% confidence interval. For the four metrics, the pvalues of other models compared with SEGSA_DTA are calculated. The pvalues for all cases are less than 0.0001, except for the of ligEdge_DTA (pvalue 0.692 > 0.05).
(D) Performance of SEGSA_DTA with different supervised attention on the validation set of five-fold cross-validation. Data are represented as mean ±95% confidence interval. For the four metrics, the pvalues of other models compared with SEGSA_DTA are less than 0.0001.
To further illustrate the generalization ability of our model. Two new external independent test datasets Mpro_37 (SARS-CoV-2 Mpro structure and its 37 reported ligands)23 and PIM1_89 (PIM-1 kinase structure and its reported 89 ligands)24,25,26 are created. On these two datasets, the experimental results show that compared with some reported methods (Pafnuity, DeepDTAF, GraphDTA), SEGSA_ DTA has obtained the best Pearson correlation coefficient, with an average increase of 21.13% and 8.4% respectively (Figures S1 and S2). The above results illustrate the superior scoring power of SEGSA_DTA.
SEGSA_DTA demonstrates impressive virtual screening power
Virtual Screening is one of the main applications of protein–ligand binding affinity prediction. It refers that using the score function or model to evaluate the binding between small molecules in the small molecule library and specific proteins, so as to discover small molecules that potentially bind to the protein as lead compounds for subsequent drug development. For the virtual screening power of the binding affinity prediction task, the goal is to assess the ability of the model to identify small molecules that actually bind to a given target protein in a random small molecule library. The predicted affinity of the small molecule that actually bind (active ligand) should be greater than that of the small molecule that cannot bind (decoy ligand). And the widely used metric to assess virtual screening power is AUC,27 which is explained from the perspective of sorting here.
To evaluate the virtual screening power of SEGSA_DTA, we compared SEGSA_DTA against two popular docking programs, i.e. Glide,28 LeDock29 and the above three deep learning-based methods, i.e. DeepDTAF,6 Pafnucy7 and GraphDTA9 on an independent external validation set, DUD-Ehand. DUD-Ehand is a subset of DUD-E30 containing Kinase and Gpcr with a total of 16 target proteins and corresponding 6,872 active ligands and 243,390 decoy ligands (STAR Methods).
As shown in Figure 2B, our method SEGSA_DTA outperforms LeDock and all other deep learning-based methods (Table S2). Notably, the AUC of SEGSA_DTA and Glide are 0.705 and 0.776, respectively. It is no surprise that Glide shows better performance than ours as the GlideScore of Glide is an empirical scoring function with many terms, including force field contributions and terms rewarding or penalizing interactions known to influence ligand binding, like solvation,28 and thus provides a more complete and comprehensive description of ligand binding. However, from the perspective of computational time cost, for the DUD-Ehand containing 16 target proteins and 250,262 ligands, Glide requires approximately fifteen days, whereas SEGSA_DTA takes only approximately 3 h with the GPU environment. SEGSA_DTA balances the speed and accuracy to provide a model that is much faster and performs in the same order of magnitude as Glide.
Furthermore, we analyzed the performance of SEGSA_DTA and Glide on each target protein of DUD-Ehand (Figure 2B). Our model has an AUC bigger than 0.5 for all 16 proteins, which means that our model is better than random selection. Our model achieved an AUC >0.7 on 11 proteins, whereas Glide achieved an AUC >0.7 on 13 proteins. In addition, compared with Glide, our model achieved a better AUC for five proteins, including SRC, AKT1, CSF1R, CXCR4 and DRD3. The above results indicated the outstanding virtual screening power of SEGSA_DTA.
SEGSA_DTA shows drug repurposing ability for COVID-19
The COVID-19 pandemic, since its occurrence in December 2019, has caused over 157 million cases worldwide with more than 3.3 million deaths as of May 2021. There is an urgent need to find effective anti-COVID-19 drugs. Drug repurposing is a drug discovery strategy to identify new uses of existing drugs. Compared with traditional drug development, it can significantly shorten the drug development cycle, reduce costs and avoid risks.31 Using the above trained SEGSA_DTA model, we calculated the binding affinity of 1,697 FDA-approved drugs from the DrugBank database32 to SARS-CoV-2 Mpro. Full calculation results are in the Data S1. Table 2 lists drugs with top-100 binding affinity to SARS-CoV-2 Mpro that are HIV protease inhibitors, histamine H2 receptor antagonists or ACE inhibitors. Through literature searches,33,34,35,36 we found that some drugs, such as nelfinavir, cimetidine, and spirapril, have been reported to be potential inhibitors of SARS-CoV-2 Mpro. The predicted ranking of Glide and other methods on these three types of drugs was also explored. For these drugs, the predicted ranking of SEGASA _DTA is the closest to that of Glide, especially for HIV protease inhibitors. This further indicates that SEGASA_DTA model provide much more robust generalization performance than previous reported models.
Table 2.
Parts of drugs with top-100 binding affinity to SARS-CoV-2 Mpro
| DrugType | Drug BankID |
Drug Name | ours | Glide | DeepDTAF | Pafnucy | GraphDTA |
|---|---|---|---|---|---|---|---|
| HIV protease inhibitor | DB01232 | Saquinavir | 6 | 36 | 147 | 1069 | 888 |
| DB00220 | Nelfinavir | 12 | 48 | 181 | 777 | 1219 | |
| DB00224 | Indinavir | 17 | 38 | 657 | 353 | 152 | |
| DB01601 | Lopinavir | 18 | 64 | 183 | 121 | 314 | |
| Histamine H2 receptor antagonist | DB00501 | Cimetidine | 8 | 1654 | 1160 | 1576 | 926 |
| DB00585 | Nizatidine | 13 | 1471 | 1059 | 1521 | 243 | |
| DB00863 | Ranitidine | 35 | 1420 | 1136 | 1525 | 147 | |
| DB01069 | Promethazine | 89 | 504 | 1373 | 717 | 291 | |
| ACE inhibitor | DB01348 | Spirapril | 11 | 151 | 694 | 863 | 1302 |
| DB00492 | Fosinopril | 84 | 985 | 834 | 1076 | 1566 | |
| DB00584 | Enalapril | 87 | 287 | 1113 | 776 | 1152 | |
| DB00881 | Quinapril | 81 | 365 | 809 | 922 | 1284 | |
| DB00519 | Trandolapril | 90 | 67 | 907 | 1216 | 1386 | |
| DB13166 | Zofenopril | 91 | 597 | 644 | 904 | 1579 |
Edge awareness effectively enhance binding affinity prediction performance
To investigate whether the edge information of the protein and ligand can benefit the binding affinity prediction, we removed both the edge features and the graph convolution on the edges (noEdge_DTA), trained it and SEGSA_DTA on the training set with the same 5-fold cross-validation setting, and reported the average performance on the validation set.
As shown in Figure 2C, and are metrics for the binding affinity prediction, whereas and are the metrics for non-covalent interaction prediction and residue contribution prediction, respectively. Compared to noEdge_DTA, SEGSA_DTA showed superior performances in all three tasks (Table S3). The performance of noEdge_DTA model on DUD-Ehand dataset was also examined. Compared to SEGSA_DTA with an average AUC of 0.705, the noEdge_DTA model only achieved an average AUC of 0.592, which is a drop of 11.3% (Figure 2B). All these strongly suggests that edge features contain crucial information about protein–ligand binding and that the introduction of super-edge graph convolution can effectively exploit this information to improve performance in all three tasks, particularly for the binding affinity prediction.
We further examined the effect of the edge information of the protein or ligand on the model performance. Compared to pro-Edge_DTA with only protein edge features, ligEdge_DTA with only ligand edge features outperformed pro-Edge_DTA on all tasks, indicating that ligand edge features are more important to the model performance relative to protein edge features. Besides, the comparison of noEdge_DTA and pro-Edge_DTA shows that their performance on each task metric is very close, indicating that it is difficult to effectively improve model performance by introducing protein edge features alone. Moreover, comparing with pro-Edge_DTA and ligEdge_DTA, SEGSA_DTA, which introduces both protein and ligand edge features, has the best overall performance, which to some extent indicates that protein edge features and ligand edge features need to exchange information with each other to model the protein–ligand binding process more comprehensively.
Multi-supervised attention module significantly improves the model performance on all three tasks
To verify the effect of the supervised attention in SEGSA_DTA, we compared performances of SEGSA_DTA (our proposed model with supervised training for the two attentions), interSA_DTA (only the supervised training of the bidirectional attention), contriSA_DTA (only the supervised training of the unidirectional attention) and noSA_DTA (no supervised training for the two attentions). The four models were trained with the same five-fold cross-validation settings on the training set. Figure 2D shows the average performance on the validation set. SEGSA_DTA is superior to other models in all indicators, suggesting that the supervised attentions are of significant benefit to the model (Table S4). Without knowledge of non-covalent interaction (contriSA_DTA) or residue contribution (interSA_DTA), main task performance decreased and corresponding auxiliary task performance dropped significantly. This indicates that it is difficult for the attention mechanism to capture the real knowledge of protein–ligand interactions without supervision in our study.
Furthermore, the main task performance of noSA_DTA is comparable to that of interSA_DTA but better than that of contriSA_DTA, demonstrating that only introducing a certain type of knowledge (supervision signal) is insufficient for improving the main task performance. The reason may be that there is a potential synergy promotion relationship between the two types of knowledge.
Selective binding mechanism of ligands to targets can be revealed by SEGSA_DTA
Here, we further explored the reason for the superior performance of SEGSA_DTA. The binding selectivity of ligands to proteins is an important part of lead compound optimization. Improving the binding selectivity of ligands may improve ligand binding efficacy and reduce the side effects of ligands. The difference in the physicochemical properties of amino acid residues in protein pockets is one of several important reasons that affect the binding selectivity.37 To date, several studies have employed the attention mechanism in ligand-protein interaction prediction models.5,14,16 With the learned attention weight, these models can reveal the importance of residues to the interaction between the protein and ligand, but it is impossible to characterize whether the residues promote or inhibit the interaction between the protein and ligand.
SHAP is an additive attribution method inspired by cooperative game theory.38 SHAP interprets the model output as the sum of the attributed values (SHAP values) of all input features (for more details, see STAR Methods). Compared with the attention weight, the biggest advantage of SHAP values is that they not only can reflect the magnitude of effect of each feature but also show the direction of the effect, i.e., positive or negative. In the case study, we used SHAP values to analyze the interaction between the key residue in the pockets of proteins belong to three different protein families (COXs, 5-HTs and TREKs) and their ligands to verify whether SEGSA_DTA could explain the binding selectivity of the ligands (Table S5). And the verification strategy is to determine whether the SHAP value of the residue of protein with weak ligand binding will increase, when it mutates into the residue at same position of the protein with strong ligand binding.
COXs. SC-558 is a selective COX-2 inhibitor. The structural analysis indicated that SC-558 has a weak inhibitory activity on COX-1 because of the V523I substitution39 (Figure 3A), which is consistent with the analysis results of SHAP values in our model. When the residue Val at position 523 of COX-2 was replaced with the residue Ile, the SHAP value of the interaction between the residue and SC-558 decreased from −0.008 to −0.208 (Figure 3D). This indicated that a V523I substitution is bad for SC-558 binding to COX-2.
Figure 3.

Selective binding mechanism of ligands
(A–C) Protein–ligand complex structures reveal the key residues for binding selectivity of ligands to COX-2 (PDB:5COX), 5-HT1 (PDB:4IAR) and TREK-1 (PDB:4WTK), respectively. The yellow part in the figure represents the substituted residue, which corresponds to the residue number and the protein PDB number in red.
(D–F) The SHAP values of pocket residues were calculated using our model.
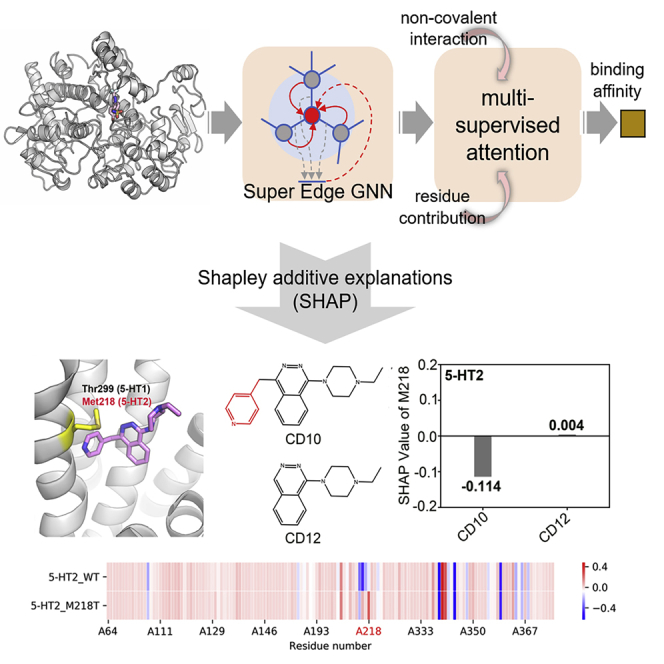
5-HTs. 5-HT receptors are G-protein-coupled receptors and mainly include two subtypes: 5-HT1 and 5-HT2. The biggest difference between 5-HT1 and 5-HT2 in the ligand binding pocket is the large residue Met at position 218 in 5-HT2 (Figure 3B). On the basis of this large residue, CD10 was designed to selectively binding to 5-HT140 The SHAP value showed that Met218 has a negative effect on CD10 binding to 5-HT2 and that M218T substitution has a positive effect (Figure 3E).
TREKs. TREKs are two-pore domain potassium channels and include three subtypes: TREK-1, TREK-2 and TRAAK. A rather unique structural feature of these channels is the extracellular cap domain. Based on the cap domain of the channel TREK-1, TKDC was designed to have a strong inhibitory activity against TREK-1 and weak inhibitory activity against TRAAK.41 Biological experiments showed that the weak inhibitory activity of the ligand TKDC on the channel TRAAK was because of the repulsion between the negatively charged sulfonic acid group of TKDC and the negatively charged residue Glu38 on TRAAK (Figure 3C). The SHAP value of our model shows results consistent with these interactions. The interaction between residue Glu38 of the channel TRAAK and the ligand TKDC has a negative SHAP value: −0.024. When the residue E38 of TRAAK was replaced with the residue Thr, the absolute value of this negative SHAP value decreased. In addition, our model found that the negatively charged residue Glu41, which is near residue Glu38 but not a residue in the ligand binding pocket, was also not conducive to the binding of TKDC to TRAAK (Figure 3F). This indicates that our model can also explore the effect of non-pocket residues on ligand binding.
SEGSA_DTA provides a new guidance for structural-based lead optimization
The objective of structural-based lead optimization is to modify ligand structure to make the ligand more compatible with the protein pocket to improve the protein–ligand binding affinity. Thus, quantitative analysis of the interaction between residues of protein pockets and ligands is critical. Accurate calculation of interaction strength between residues in the protein pocket and ligand remains a challenge in computer-aided drug design. Quantum chemistry calculation and alchemical-free energy methods are two common but rigorous computational approaches, which are either very expensive in time and resource or inconvenient to use.42,43 In this case study, we found that the SHAP values of the residues calculated with SEGSA_DTA may provide a guidance for structural-based lead optimization (Table S6).
TRAAK. Ligand 28NH was obtained by removing the sulfonic acid group on TKDC (Figure 4A). Compared with TKDC, the inhibitory activity of 28NH on TRAAK was significantly increased.41 Through the calculation and analysis of the SHAP values, we observed that TRAAK residue Glu38(E38) had a negative SHAP value with TKDC and a positive SHAP value with 28NH (Figure 4B). This shows that our model SEGSA_DTA can explain the structure–activity relationship between ligands and TRAAK channels.
Figure 4.

Guidance for structural-based lead optimization
(A) The structure of TKDC and its modification compound 28NH.
(B) The SHAP value of residue E38 of TRAAK channel for TKDC and 28NH binding.
(C) The structure of CD10 and its modification compound CD12.
(D) The SHAP values of residue M218 of 5-HT2 for CD10 and CD12 binding. Differences between ligand structures are marked in red.
5-HT2. It is reported that the activation activity of ligand CD12 on 5-HT2 is significantly improved when the groups on CD10 that conflict with residue Met218 (M218) are removed40(Figure 4C). The SHAP value calculated by our model also showed the same prediction results. The SHAP value of 5-HT2 residue M218 interaction with CD12 is greater than that with CD10 (Figure 4D).
Discussion
Protein–ligand binding affinity prediction is one of the most critical tasks in the early stages of drug design. The accuracy of its prediction directly determines the success rate of drug design and is the cornerstone of all later stages of drug design. Because protein–ligand binding is a complex physiological process, building accurate and efficient computational models for the binding affinity prediction has always been a major challenge in drug design.
Unlike other existing method, our proposed model SEGSA_DTA takes full advantage of the edge information and prior knowledge of protein–ligand interactions though the super edge graph convolution and multi-supervised attention module. Also, the comparison results of ablation experiments prove that our proposed model structure can improve the performance of protein-ligand binding affinity prediction based on protein and ligand structure data. The prediction accuracy of SEGSA_DTA on the PDBbind v.2016 core set has a slightly improvement, but it is worth mentioning that SEGSA_DTA performs significantly better than the reported model on the other three external test sets (DUDEhand, Mpro_37 and PIM1_89), and shows the drug virtual screening ability comparable to the current popular but expensive software Glide. All these indicate that SEGASA_DTA has a higher prediction accuracy and stronger generality on binding affinity prediction.
SHAP value is used for model interpretation. Through this method, we find SEGSA_DTA can well capture the residues that play a key role in ligand binding. However, as the dimensions of protein characteristics are much larger than those of ligands and our model pays more attention to the information of protein, the SHAP value of ligand is not consistent with expert knowledge (Table S7). In future research, we will try to enhance the model’s information learning on ligands to further improve the prediction accuracy of binding affinity.
Edge information, in conjunction with the node information, fundamentally determines protein–ligand binding,44,45 but current deep learning-based binding affinity prediction methods rarely pay attention to edge information. Our study also demonstrates the edge information plays an essential role for predicting protein–ligand binding affinity. The proposed super edge graph convolution can comprehensively utilize node and edge features in the protein and ligand structure data, and the experimental results demonstrate its effectiveness. However, the super edge graph convolution is relatively simple in terms of the network structure. In the future investigation, it is promising to adopt other strategies to better exploit the edge information, thereby further improving the model performance.
Limitations of the study
In this study, many amino acid features were utilized. Whether they are all actually beneficial for the prediction needs further exploration. In addition, all information learned by the SEGSA_DTA comes from the static crystal structure. How to integrate the dynamic behavior information of protein–ligand complex in the model structure is still not well solved. Finally, more datasets are needed to verify the generalization ability and interpretability of SEGSA_DTA.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Software and algorithms | ||
| SEGSA_DTA | This paper | https://github.com/koyurion/SEGSA_DTA |
| DeepDTAF | (Wang et al., 2021)1 | https://github.com/KailiWang1/DeepDTAF |
| GraphDTA | (Nguyen et al., 2021)9 | https://github.com/thinng/GraphDTA |
| Pafnucy | (Stepniewska-Dziubinska et al., 2018)7 | https://gitlab.com/cheminfIBB/pafnucy |
| LeDock | (Zhang and Zhao, 2016)28 | http://www.lephar.com/software.htm |
| Glide | (Friesner et al., 2006)27 | https://www.schrodinger.com/glide |
| PLIP | (Salentin et al., 2015)45 | https://github.com/pharmai/plip |
| Rosetta | (Fleishman et al., 2011)46 | https://rosettacommons.org/ |
| SHAP | (Lundberg and Lee, 2017)37 | https://github.com/slundberg/shap |
| Other | ||
| Database: AAindex | (Kawashima et al., 2008)43 | https://www.genome.jp/aaindex/ |
| Database: PDBbind | (Wang et al., 2005)49 | Version 2019; http://www.pdbbind.org.cn/ |
| Database: RCSB Protein Data Bank | (Burley et al., 2021)51 | https://www.rcsb.org/ |
| Database: DUD-E | (Mysinger et al., 2012)29 | http://dude.docking.org/ |
| Database: DrugBank | (Wishart et al., 2018)31 | https://go.drugbank.com/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Qichao Luo (luoqichao@ahmu.edu.cn).
Materials availability
This study did not generate new unique reagents.
Method details
Extraction of the non-covalent interaction between proteins and ligands
The binding between proteins and ligands is mainly controlled by the non-covalent interaction, which plays a key role in protein–ligand binding affinity prediction. We used PLIP46 to analyze the non-covalent bond in protein–ligand complexes and extracted seven non-covalent bond interaction types between protein residues and ligand atoms: hydrogen bond, water bridge, hydrophobic interaction, π-stacking, π-cation interaction, saltbridge and halogen bond. On the basis of the extracted interactions, we built a protein–ligand non-covalent interaction matrix as the label of the non-covalent interaction prediction, where 1 indicates the presence of a non-covalent bond interaction and 0 indicates absence.
Quantitatively analyze the contribution of each residue to ligand binding
To quantitatively analyze the protein–ligand interaction, we used Rosetta47 to calculate the protein–ligand binding energy and decomposed it into van der Waals (VDW) energy, hydrogen bond energy () and electrostatic interaction energy (). VDW was calculated as the sum of attractive and repulsive components ( and , respectively). To reveal the spatial distribution of binding interaction between the protein and ligand, VDW, hydrogen bond and electrostatic interaction energies were further mapped on a per residue basis to the channel by Rosetta’s residue_energy_breakdown utility (STAR Methods). We regard amino acid residues with energy allocation as protein pocket residues. Then, the contribution of pocket residue can be obtained by the following formulae:
| (Equation 1) |
| (Equation 2) |
where , , , are the , , , of pocket residue , respectively. The stands for absolute value function, stands for the number of pocket residues.
Feature representation
Since proteins and ligands are natural graph structures, in this study, proteins and ligands are represented as graphs for subsequent feature extraction using graph convolution networks.
Protein representation
For proteins, we regarded each residue as a node in the graph. If the distance between the atom of two residues was less than 6Å, then an edge was considered to exist between these two residues. The AAindex database44 contains various physicochemical and biochemical characteristics of amino acids and pairs of amino acids. The AAindex1 contains 566 amino acid features; we retrieved 511 features that had no missing values to form the feature vector of the residue node. Additionally, the AAindex3 contains 47 contact potential features of amino acid pairs; we retrieved 40 features that did not contain asymmetrical features nor missing values to form the edge feature vector (for more details, see Data S2).
Ligand representation
We used RDKit to convert ligands in the SDF format to a molecular graph with atoms as nodes and chemical bonds as edges. Each atom was characterized by eight features, including the type of atom, the degree of the atom, the partial charge, the total charge of the implicit hydrogens on the atom, the type of hybridization of the atom, whether it is aromatic, the number of attached hydrogens and the chirality of the atom. Each chemical bond has three features, including the type of bond (single, double, triple or aromatic), whether the bond is conjugated and whether the bond forms part of a ring. Each atom was represented by a 26-dimensional feature vector and the bond was represented by a 6-dimensional feature vector (Table S8).
Model structure of SEGSA_DTA
Feature extraction module
In the feature extraction module (Figure 1B), the super edge graph convolution network is used to produce informative feature representations of proteins and ligands. The structure data of proteins and ligands are rich in edge information, which is of great significance for protein–ligand binding. However, the existing graph convolution networks only focus on node information, which cannot meet the characteristics of protein and ligand structure data. Therefore, this study proposes a super edge graph convolution network to solve this problem. In addition to the graph convolution network of nodes, the super edge graph convolution network uses an extra graph convolution network to extract features from neighbor edges to form the so-called super edge, which is fused into node information, producing a more informative representation. The specific implementation formulae of super edge graph convolution network are as follow,
| (Equation 3) |
| (Equation 4) |
| (Equation 5) |
where represents the -th layer of super edge graph convolution layer, and , , represent node features, edge features, and super edge features, respectively. denotes neural network weight, denotes ReLU, denotes the adjacency matrix with self-loop, is the degree matrix of , and denotes the concatenation operation.
For the adjacency matrix of ligands, the self-loop is filled with 1, the neighbor atoms with 1 and the non-neighbor with 0. But for the of proteins, neighbor residues fill the inverse of the distance between the atoms of adjacent residues, and the rest is set in line with the of the ligand. This is because the edge between ligand atoms (i.e. chemical bonds) are real, but the edge between protein residues are not, which is determined by an inter-residue distance threshold. That is, if the distance between the atoms of the residues is less than 6A, the two residues are considered to have edges. Therefore, the spatial structure information between residues is enhanced by introducing the information of inter-residue distance into the adjacency matrix to turn the protein unweighted graph into a weighted graph.
It is worth noting that for the normalization strategy, the better performing (the random wandering normalized Laplacian operator) is selected for normalization in our study, comparing with the symmetric normalized Laplacian operator . In contrast to which only considers the degree of the current node itself, also considers the degree of neighboring nodes, which weakens the influence of neighboring nodes with large degrees on the current node. And for protein–ligand binding, the nodes with large degrees are also generally more important, so this may be the reason why the normalization strategy performs better.
It is well known that the pocket residues in the protein play a key role in protein–ligand binding. For protein graph convolution networks, GCN was usually performed on pocket residues only, ignoring the effect that residues near the pocket had on the pocket residues.10 To incorporate such information, we performed the graph convolution on all residues of the protein and selected pocket residues to feed into the interaction module. More specifically, the graph convolution for proteins or ligands comprised a stack of two graph convolution layers. The first graph convolution layer is a super edge graph convolution layer, where each residue or atom aggregates information from its neighbor nodes and neighbor edges to update its own features. In the second graph convolution layer, each node aggregates information from its neighbor nodes only because the edge information has already been passed to the relevant nodes via the super edge graph convolution layer. Finally, we obtained the protein pocket residue features by selecting pocket residues from the protein and the ligand atom features .
Interaction module
The interaction module (Figure 1C) is structurally a multi-supervised attention module, consisting of a supervised bidirectional attention block (pale blue background color) and a supervised unidirectional attention block (pale yellow background color), whose attention distributions are supervised by the protein–ligand non-covalent interaction and residue contribution, respectively.
In the supervised bidirectional attention block, the bidirectional attention mechanism is used to learn the interaction between proteins and ligands in a dual direction. In specific operations, we first transformed the previous learned protein pocket residue feature and ligand atom feature using a neural network layer activated with Leakly ReLU, respectively, and then performed inner product and sigmoid to obtain the protein–ligand interaction matrix as the predicted value for the non-covalent interaction prediction task.
| (Equation 6) |
where stands for sigmoid, stands for Leakly ReLU, stands for matrix transpose, and stands for neural network weight. Each row of the represents a ligand atom and each column represents a protein pocket residue.
On the basis of the interaction matrix, we generated the attention weight () of the protein pocket on each ligand atom by adding up the interaction values of all residues on each atom (the summation of in the row direction, ) and then applying the softmax48 function. And then, using the generated attention weight , the ligand atom features was weighted and summed using to obtain the ligand feature vector , which conducts the attention mechanism of protein to the ligand atom.
| (Equation 7) |
| (Equation 8) |
Similarly, the attention mechanism of the ligand to each protein pocket residue was performed. The attention weight () of the ligand on each protein pocket residue was calculated as the sum of the interaction values of all atoms on each residue (the summation of in the column direction, ) and followed by the application of softmax function. We then use the attention weight to perform a weighted summation of the protein pocket residue features to yield the pocket residue feature vector .
| (Equation 9) |
| (Equation 10) |
Additionally, in the supervised unidirectional attention block, we exploited ligand vector ( and protein pocket residues () to implement the unidirectional attention mechanism, highlighting important residues by the ligand vector. In this case, we regarded the generated attention weight as the binding energy contribution of residues and used it as the prediction value for the residual contribution prediction task. Afterward, we obtained another protein pocket vector (), which was concatenated with to get the final protein pocket vector ().
| (Equation 11) |
| (Equation 12) |
| (Equation 13) |
Prediction module
Through the feature extraction module and interaction module, SEGSA_DTA learned the structural information of proteins and ligands, and exchanged the information between the protein and ligand to model the protein–ligand interaction process, producing the informative and representative protein feature vector and ligand feature vector . In the prediction module (Figure 1A), we concatenated the and generated in the interaction module, and fed them into a fully connected deep neural network (fc-DNN) with two hidden layers activated by ReLU, where the numbers of nodes were 512 and 256, respectively. The output layer had one node for predicting binding affinity.
| (Equation 14) |
Training protocol
Since our proposed SEGSA_DTA model is a multi-task learning-based framework, the loss function of the model comprised three components, namely the loss of binding affinity prediction (), the loss of the protein–ligand non-covalent interaction prediction () and the loss of the residue contribution prediction (), as shown below:
| (Equation 15) |
where and are hyperparameters for the trade-off of different loss components, and are the mean square error loss function and Lnon-covalent is the cross-entropy loss function.
To train our network, we applied the Adam method as the optimizer. And, we searched the optimal hyperparameters for the binding affinity prediction on the training set via 5-fold cross-validation (For optimal hyperparameter settings, see Table S9). During the search of hyperparameters, to reduce training time, we set the maximum training epoch to 300; if the performance metric of the main task on the validation set did not improve in 30 epochs, the training process was terminated early (Figure S3). After hyperparameter tuning, we used the entire training set to retrain the model to make full use of the data, with the maximum number of training epochs set to 500. The model was implemented by PyTorch50 and accelerated using the GPU (GeForce GTX 3090 and 2080Ti).
In order to avoid overfitting, we have applied many techniques: i) Regularization. We adopted the L2 regularization and set the L2 weight decoy to 0.0001; ii) Dropout. We set different dropouts for the feature extraction module (dropout = 0.1) and prediction module (dropout = 0.3). Among them, the dropout value of the feature extraction module closer to the input layer is slightly lower to prevent discarding a large amount of potentially useful feature information; iii) Early stop. The early stop setting was as described in the hyperparameter search in the previous paragraph; iv) Multi-task leaning. Our model employed the multi-task learning strategy, which can also reduce the risk of overfitting.
Quantification and statistical analysis
Dataset preparation
PDBbind database
PDBbind (version 2019, the general set) database contains complex structures and corresponding binding affinity expressed with Kd, Ki, or IC50.51 Since the IC50 values are influenced by experimental conditions and may contain substantial noise, we instead used the negative log value of the Kd and Ki data (-log Kd,-log Ki) as the target for binding affinity prediction. We obtained a 3D structure of the complexes and ligands from the RCSB PDB database52,53 and screened the data according to the following criteria: (i) a ligand requires the standard PDB ligand id and the corresponding binding affinity must be accurate and not a range value; (ii) the crystal structure resolution of the complex should be no greater than 2.5 Å; (iii) a ligand can be processed using RDKit (https://www.rdkit.org), and its molecular weight must be less than 500. After screening, a total of 5,482 protein-ligand pairs were obtained (For more details, see Table S10).
PDBbind v.2016 core set (core set v.2016)51 is a high-quality benchmark dataset for protein–ligand binding affinity prediction, containing 290 protein–ligand pairs. To evaluate the model performance on core set v.2016, we removed 246 protein–ligand pairs that were duplicated with core set v.2016 in the above 5,482 protein–ligand pairs, and the final training set contained 5,236 protein-ligand pairs.
DUD-E database
DUD-E30 database is a widely adopted benchmark for structure-based virtual screening. The DUD-E database comprises 22,886 active ligands and their affinities against 102 targets divided into seven subsets. Each target has an average of 224 active ligands, and each active ligand has an average of 50 decoy ligands.
However, some recent studies have pointed out that in the context of machine learning and deep learning, DUD-E is difficult to evaluate and prone to produce optimistic estimates of performance.54,55 Even when the model only inputs ligand information and no protein information, the model can also provide good enough predictions.54 This may be caused by two reasons: i) DUD-E may have underlying biases such as analogue bias, which likely lead to poor generalizability When we train and evaluate the model based on DUD-E, the model is likely to learn just these biases and thus get an over-optimistic estimate of prediction performance. ii) The characteristic of DUD-E that the number of proteins is very small and each protein corresponds to a mass of different ligands. For most data samples in DUD-E, the protein features are the same while the ligand features are different, which will make the model focus more on the ligand information and rarely or even ignore the protein information. Even if DUD-E does not suffer from these biases, the model trained on it will also yield optimistic estimates. Therefore, DUD-E is not appropriate as a training set for machine learning.
To eliminate the influence of the underlying biases and the aforementioned characteristic of DUD-E on the model, the final model for evaluating virtual screen power was trained on the PDBbind training set and core set v.2016 where each protein corresponds to only one ligand. We used all 16 manually collected protein targets in the Kinase and Gpcr subsets as the independent external validation set (DUD-Ehand). After removing ligands that could not be processed by our method and other compared methods, the DUD-Ehand dataset contains 16 proteins, 6,872 active ligands and 243,390 decoy ligands, for a total of 250,262 ligands.
Evaluation metrics
Root-mean-square error (RMSE), mean absolute error (MAE) and concordance index (CI) were used to assess the prediction error for binding affinity prediction, whereas the Pearson correlation coefficient and SD in regression (SD)21,22 that are two vital indicators of scoring power were used as the correlation metrics. The area under the receiver operating characteristic curve (AUC) was used to assess the virtual screening power.27 The CI is defined as:
| (Equation 16) |
where is a normalization constant equal to the number of pairwise sample pairs whose true affinities are different. and stand for the true affinity and predicted affinity of i-th sample, respectively. is a step function defined as follows:
| (Equation 17) |
The SD is defined by following equation:
| (Equation 18) |
where denotes the number of samples. The and are the slope and intercept of the regression line of the linear regression equation between the true affinity and the predicted affinity , respectively.
We used the average AUC and the average RMSE to evaluate the non-covalent interaction prediction and the residue contribution prediction, respectively. Given a dataset with N samples, the average AUC was defined as follows:
| (Equation 19) |
where indicates the AUC between the true non-covalent interaction labels and the predictions of the i-th sample. The average RMSE was defined as follows:
| (Equation 20) |
where indicates the RMSE between the true contribution values and the predictions of the i-th sample.
The performance assessment of SEGSA_DTA with different edge features and The performance assessment of SEGSA_DTA with different supervised attention are given as mean ±95% confidence interval, which was estimated by calculation of repeating the random sampling with replacement 100 times. Besides, the significance for the difference in these two experiments was estimated by Student’s t test and the statistical significance is defined as p value<0.05.
Shapley additive explanations
Shapley additive explanations (SHAP) is an additive attribution method inspired by cooperative game theory. SHAP interprets the model output as the sum of the attributed values (SHAP values) of all input features, that is:
| (Equation 21) |
where is the model output of , is the explanation model defined by SHAP, D is the number of input features, indicates whether a feature exists or not (0 or 1), is the SHAP value of feature . SHAP value of is the average marginal contribution of across all combinations.
| (Equation 22) |
where is the set of all input features, is one of all combinations of features for which is excluded.
In this study, we applied the GradientExplainer method of SHAP (see https://github.com/slundberg/shap), whichfurther combines the advantages of Integrated Gradients and SmoothGrad. And it is the only method that supports Pytorch well.
Acknowledgments
This study was supported by the Major Research Plan of the National Natural Science Foundation of China (Grant No.91746204) to YH; the Science and Technology Development in Guangdong Province, China (Grant No.2017B030308008) to YH; Guangdong Engineering Technology Research Center for Big Data Precision Healthcare, China (Grant No.603141789047) to YH; the Research Startup Fund of Anhui Medical University, China (Grant No.900206) to QL; Youth Science Fund of Anhui Province Natural Science (Grant No.2208085QH276) to QL; and the National Science Foundation of USA (Grant No. 2014554) to ML; and the Youth Science Fund of the National Natural Science Foundation of China (Grant No.61802149) to LW.
Author contributions
Y.G.: Conceptualization, Methodology, Software, Validation, Investigation, Visualization, Writing – Original Draft. X. Z.: Investigation, Resources, Data Curation, Writing – Review and Editing. A.X.: Investigation, Resources, Writing – Review and Editing, Visualization. W.C.: Investigation, Writing – Review and Editing. K.L.: Writing – Review and Editing. L.W.: Writing – Review and Editing. S.M.: Software, Writing – Review and Editing. Y.H.: Methodology, Writing – Review and Editing, Supervision, Project administration. M.L.: Methodology, Writing – Review and Editing, Supervision, Project administration. Q.L.: Conceptualization, Methodology, Formal analysis, Investigation, Resources, Writing – Review and Editing, Visualization, Project administration.
Declaration of interests
The authors have declared no competing interests.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: January 20, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.105892.
Contributor Information
Yong Hu, Email: yonghu@jnu.edu.cn.
Mei Liu, Email: mei.liu@ufl.edu.
Qichao Luo, Email: luoqichao@ahmu.edu.cn.
Supplemental information
Data and code availability
-
•
This paper analyzes existing, publicly available data. These accession numbers for the datasets are listed in the key resources table.
-
•
All original code has been deposited at GitHub (https://github.com/koyurion/SEGSA_DTA) and is publicly available as of the date of publication. DOIs are listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Wang D.D., Zhu M., Yan H. Computationally predicting binding affinity in protein-ligand complexes: free energy-based simulations and machine learning-based scoring functions. Brief. Bioinform. 2021;22:bbaa107. doi: 10.1093/bib/bbaa107. [DOI] [PubMed] [Google Scholar]
- 2.Huang K., Luo S., Cong Y., Zhong S., Zhang J.Z.H., Duan L. An accurate free energy estimator: based on MM/PBSA combined with interaction entropy for protein-ligand binding affinity. Nanoscale. 2020;12:10737–10750. doi: 10.1039/c9nr10638c. [DOI] [PubMed] [Google Scholar]
- 3.Elton D.C., Boukouvalas Z., Fuge M.D., Chung P.W. Deep learning for molecular design - a review of the state of the art. Mol. Syst. Des. Eng. 2019;4:828–849. doi: 10.1039/c9me00039a. [DOI] [Google Scholar]
- 4.Öztürk H., Özgür A., Ozkirimli E. DeepDTA: deep drug-target binding affinity prediction. Bioinformatics. 2018;34:i821–i829. doi: 10.1093/bioinformatics/bty593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Karimi M., Wu D., Wang Z., Shen Y. DeepAffinity: interpretable deep learning of compound–protein affinity through unified recurrent and convolutional neural networks. Bioinformatics. 2019;35:3329–3338. doi: 10.1093/bioinformatics/btz111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang K., Zhou R., Li Y., Li M. DeepDTAF: a deep learning method to predict protein-ligand binding affinity. Brief. Bioinform. 2021;22:bbab072. doi: 10.1093/bib/bbab072. [DOI] [PubMed] [Google Scholar]
- 7.Stepniewska-Dziubinska M.M., Zielenkiewicz P., Siedlecki P. Development and evaluation of a deep learning model for protein–ligand binding affinity prediction. Bioinformatics. 2018;34:3666–3674. doi: 10.1093/bioinformatics/bty374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lim S., Lu Y., Cho C.Y., Sung I., Kim J., Kim Y., Park S., Kim S. A review on compound-protein interaction prediction methods: data, format, representation and model. Comput. Struct. Biotechnol. J. 2021;19:1541–1556. doi: 10.1016/j.csbj.2021.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nguyen T., Le H., Quinn T.P., Nguyen T., Le T.D., Venkatesh S. GraphDTA: predicting drug-target binding affinity with graph neural networks. Bioinformatics. 2021;37:1140–1147. doi: 10.1093/bioinformatics/btaa921. [DOI] [PubMed] [Google Scholar]
- 10.Torng W., Altman R.B. Graph convolutional neural networks for predicting drug-target interactions. J. Chem. Inf. Model. 2019;59:4131–4149. doi: 10.1021/acs.jcim.9b00628. [DOI] [PubMed] [Google Scholar]
- 11.Kipf T.N., Welling M. Semi-supervised classification with graph convolutional networks. ArXiv. 2017 doi: 10.48550/arXiv.1609.02907. Preprint at. [DOI] [Google Scholar]
- 12.Vaswani A., Shazeer N., Parmar N., Uszkoreit J., Jones L., Gomez A.N., Kaiser Ł., Polosukhin I. Advances in Neural Information Processing Systems. NIPS; 2017. Attention is all you need; pp. 5998–6008. [Google Scholar]
- 13.Gao K.Y., Fokoue A., Luo H., Iyengar A., Dey S., Zhang P. Proceedings of the Twenty-Seventh International Joint Conference on Artificial Intelligence. 2018. Interpretable drug target prediction using deep neural representation; pp. 3371–3377. [DOI] [Google Scholar]
- 14.Tsubaki M., Tomii K., Sese J. Compound-protein interaction prediction with end-to-end learning of neural networks for graphs and sequences. Bioinformatics. 2019;35:309–318. doi: 10.1093/bioinformatics/bty535. [DOI] [PubMed] [Google Scholar]
- 15.Yang L., Yang G., Chen X., Yang Q., Yao X., Bing Z., Niu Y., Huang L., Yang L. Deep scoring neural network replacing the scoring function components to improve the performance of structure-based molecular docking. ACS Chem. Neurosci. 2021;12:2133–2142. doi: 10.1021/acschemneuro.1c00110. [DOI] [PubMed] [Google Scholar]
- 16.Li S., Wan F., Shu H., Jiang T., Zhao D., Zeng J. MONN: a multi-objective neural network for predicting compound-protein interactions and affinities. Cell Systems. 2020;10:308–322.e11. doi: 10.1016/j.cels.2020.03.002. [DOI] [Google Scholar]
- 17.Liu Y., Zhang X., Zhao Z., Zhang B., Cheng L., Li Z. ALSA: adversarial learning of supervised attentions for visual question answering. IEEE Trans. Cybern. 2022;52:4520–4533. doi: 10.1109/tcyb.2020.3029423. [DOI] [PubMed] [Google Scholar]
- 18.Patro B.N., S K.G., Jain A., Namboodiri V.P. Proceedings of the IEEE/CVF Winter Conference on Applications of Computer Vision. 2021. Self supervision for attention networks; pp. 726–735. [DOI] [Google Scholar]
- 19.Li P., Yu H., Zhang W., Xu G., Sun X. SA-NLI: a supervised attention based framework for natural language inference. Neurocomputing. 2020;407:72–82. doi: 10.1016/j.neucom.2020.03.092. [DOI] [Google Scholar]
- 20.Liu L., Utiyama M., Finch A., Sumita E. Proceedings of COLING 2016, the 26th International Conference on Computational Linguistics: Technical Papers. 2016. Neural machine translation with supervised attention; pp. 3093–3102. [Google Scholar]
- 21.Li Y., Han L., Liu Z., Wang R. Comparative assessment of scoring functions on an updated benchmark: 2. Evaluation methods and general results. J. Chem. Inf. Model. 2014;54:1717–1736. doi: 10.1021/ci500081m. [DOI] [PubMed] [Google Scholar]
- 22.Günther S., Reinke P.Y.A., Fernández-García Y., Lieske J., Lane T.J., Ginn H.M., Koua F.H.M., Ehrt C., Ewert W., Oberthuer D., et al. X-ray screening identifies active site and allosteric inhibitors of SARS-CoV-2 main protease. Science. 2021;372:642–646. doi: 10.1126/science.abf7945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cheney I.W., Yan S., Appleby T., Walker H., Vo T., Yao N., Hamatake R., Hong Z., Wu J.Z. Identification and structure–activity relationships of substituted pyridones as inhibitors of Pim-1 kinase. Bioorg. Med. Chem. Lett. 2007;17:1679–1683. doi: 10.1016/j.bmcl.2006.12.086. [DOI] [PubMed] [Google Scholar]
- 24.Ishchenko A., Zhang L., Le Brazidec J.-Y., Fan J., Chong J.H., Hingway A., Raditsis A., Singh L., Elenbaas B., Hong V.S., et al. Structure-based design of low-nanomolar PIM kinase inhibitors. Bioorg. Med. Chem. Lett. 2015;25:474–480. doi: 10.1016/j.bmcl.2014.12.041. [DOI] [PubMed] [Google Scholar]
- 25.Oyallon B., Brachet-Botineau M., Logé C., Bonnet P., Souab M., Robert T., Ruchaud S., Bach S., Berthelot P., Gouilleux F., et al. Structure-based design of novel quinoxaline-2-carboxylic acids and analogues as Pim-1 inhibitors. Eur. J. Med. Chem. 2018;154:101–109. doi: 10.1016/j.ejmech.2018.04.056. [DOI] [PubMed] [Google Scholar]
- 26.Pereira J.C., Caffarena E.R., dos Santos C.N. Boosting docking-based virtual screening with deep learning. J. Chem. Inf. Model. 2016;56:2495–2506. doi: 10.1021/acs.jcim.6b00355. [DOI] [PubMed] [Google Scholar]
- 27.Friesner R.A., Murphy R.B., Repasky M.P., Frye L.L., Greenwood J.R., Halgren T.A., Sanschagrin P.C., Mainz D.T. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006;49:6177–6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 28.Zhang N., Zhao H. Enriching screening libraries with bioactive fragment space. Bioorg. Med. Chem. Lett. 2016;26:3594–3597. doi: 10.1016/j.bmcl.2016.06.013. [DOI] [PubMed] [Google Scholar]
- 29.Mysinger M.M., Carchia M., Irwin J.J., Shoichet B.K. Directory of useful decoys, enhanced (DUD-E): better ligands and decoys for better benchmarking. J. Med. Chem. 2012;55:6582–6594. doi: 10.1021/jm300687e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pushpakom S., Iorio F., Eyers P.A., Escott K.J., Hopper S., Wells A., Doig A., Guilliams T., Latimer J., McNamee C., et al. Drug repurposing: progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019;18:41–58. doi: 10.1038/nrd.2018.168. [DOI] [PubMed] [Google Scholar]
- 31.Wishart D.S., Feunang Y.D., Guo A.C., Lo E.J., Marcu A., Grant J.R., Sajed T., Johnson D., Li C., Sayeeda Z., et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 2018;46:D1074–D1082. doi: 10.1093/nar/gkx1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ohashi H., Watashi K., Saso W., Shionoya K., Iwanami S., Hirokawa T., Shirai T., Kanaya S., Ito Y., Kim K.S., et al. Potential anti-COVID-19 agents, cepharanthine and nelfinavir, and their usage for combination treatment. iScience. 2021;24:102367. doi: 10.1016/j.isci.2021.102367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sargolzaei M. Effect of nelfinavir stereoisomers on coronavirus main protease: molecular docking, molecular dynamics simulation and MM/GBSA study. J. Mol. Graph. Model. 2021;103:107803. doi: 10.1016/j.jmgm.2020.107803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Qu C., Fuhler G.M., Pan Y. Could histamine H1 receptor antagonists be used for treating COVID-19? Int. J. Mol. Sci. 2021;22:5672. doi: 10.3390/ijms22115672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sencanski M., Perovic V., Pajovic S.B., Adzic M., Paessler S., Glisic S. Drug repurposing for candidate SARS-CoV-2 main protease inhibitors by a novel in silico method. Molecules. 2020;25:3830. doi: 10.3390/molecules25173830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huggins D.J., Sherman W., Tidor B. Rational approaches to improving selectivity in drug design. J. Med. Chem. 2012;55:1424–1444. doi: 10.1021/jm2010332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lundberg S.M., Lee S.-I. Advances in Neural Information Processing Systems. NIPS; 2017. A unified approach to interpreting model predictions; pp. 4768–4777. [Google Scholar]
- 38.Kurumbail R.G., Stevens A.M., Gierse J.K., McDonald J.J., Stegeman R.A., Pak J.Y., Gildehaus D., Miyashiro J.M., Penning T.D., Seibert K., et al. Structural basis for selective inhibition of cyclooxygenase-2 by anti-inflammatory agents. Nature. 1996;384:644–648. doi: 10.1038/384644a0. [DOI] [PubMed] [Google Scholar]
- 39.Rodríguez D., Brea J., Loza M.I., Carlsson J. Structure-based discovery of selective serotonin 5-HT1B receptor ligands. Structure. 2014;22:1140–1151. doi: 10.1016/j.str.2014.05.017. [DOI] [PubMed] [Google Scholar]
- 40.Luo Q., Chen L., Cheng X., Ma Y., Li X., Zhang B., Li L., Zhang S., Guo F., Li Y., Yang H. An allosteric ligand-binding site in the extracellular cap of K2P channels. Nat. Commun. 2017;8:378. doi: 10.1038/s41467-017-00499-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ehrlich S., Göller A.H., Grimme S. Towards full quantum-mechanics-based protein–ligand binding affinities. ChemPhysChem. 2017;18:898–905. doi: 10.1002/cphc.201700082. [DOI] [PubMed] [Google Scholar]
- 42.Abel R., Wang L., Mobley D.L., Friesner R.A. A critical review of validation, blind testing, and real-world use of alchemical protein-ligand binding free energy calculations. Curr. Top. Med. Chem. 2017;17:2577–2585. doi: 10.2174/1568026617666170414142131. [DOI] [PubMed] [Google Scholar]
- 43.Kawashima S., Pokarowski P., Pokarowska M., Kolinski A., Katayama T., Kanehisa M. AAindex: amino acid index database, progress report 2008. Nucleic Acids Res. 2008;36:D202–D205. doi: 10.1093/nar/gkm998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xiong Z., Wang D., Liu X., Zhong F., Wan X., Li X., Li Z., Luo X., Chen K., Jiang H., Zheng M. Pushing the boundaries of molecular representation for drug discovery with the graph attention mechanism. J. Med. Chem. 2020;63:8749–8760. doi: 10.1021/acs.jmedchem.9b00959. [DOI] [PubMed] [Google Scholar]
- 45.Salentin S., Schreiber S., Haupt V.J., Adasme M.F., Schroeder M. PLIP: fully automated protein–ligand interaction profiler. Nucleic Acids Res. 2015;43:W443–W447. doi: 10.1093/nar/gkv315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fleishman S.J., Leaver-Fay A., Corn J.E., Strauch E.-M., Khare S.D., Koga N., Ashworth J., Murphy P., Richter F., Lemmon G., et al. RosettaScripts: a scripting language interface to the Rosetta macromolecular modeling suite. PLoS One. 2011;6:e20161. doi: 10.1371/journal.pone.0020161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bridle J.S. In: Neurocomputing NATO ASI Series. Soulié F.F., Hérault J., editors. Springer; 1990. Probabilistic interpretation of feedforward classification network outputs, with relationships to statistical pattern recognition; pp. 227–236. [DOI] [Google Scholar]
- 48.Paszke A., Gross S., Massa F., Lerer A., Bradbury J., Chanan G., Killeen T., Lin Z., Gimelshein N., Antiga L., et al. PyTorch: an imperative style, high-performance deep learning library. Advances in Neural Information Processing Systems, NIPS. 2019;32:8026–8037. [Google Scholar]
- 49.Wang R., Fang X., Lu Y., Yang C.-Y., Wang S. The PDBbind database: methodologies and updates. J. Med. Chem. 2005;48:4111–4119. doi: 10.1021/jm048957q. [DOI] [PubMed] [Google Scholar]
- 50.Burley S.K., Bhikadiya C., Bi C., Bittrich S., Chen L., Crichlow G.V., Christie C.H., Dalenberg K., Di Costanzo L., Duarte J.M., et al. RCSB Protein Data Bank: powerful new tools for exploring 3D structures of biological macromolecules for basic and applied research and education in fundamental biology, biomedicine, biotechnology, bioengineering and energy sciences. Nucleic Acids Res. 2021;49:D437–D451. doi: 10.1093/nar/gkaa1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Feng Z., Chen L., Maddula H., Akcan O., Oughtred R., Berman H.M., Westbrook J. Ligand Depot: a data warehouse for ligands bound to macromolecules. Bioinformatics. 2004;20:2153–2155. doi: 10.1093/bioinformatics/bth214. [DOI] [PubMed] [Google Scholar]
- 52.Chen L., Cruz A., Ramsey S., Dickson C.J., Duca J.S., Hornak V., Koes D.R., Kurtzman T. Hidden bias in the DUD-E dataset leads to misleading performance of deep learning in structure-based virtual screening. PLoS One. 2019;14:e0220113. doi: 10.1371/journal.pone.0220113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sieg J., Flachsenberg F., Rarey M. In need of bias control: evaluating chemical data for machine learning in structure-based virtual screening. J. Chem. Inf. Model. 2019;59:947–961. doi: 10.1021/acs.jcim.8b00712. [DOI] [PubMed] [Google Scholar]
- 54.Xia J., Tilahun E.L., Reid T.-E., Zhang L., Wang X.S. Benchmarking methods and data sets for ligand enrichment assessment in virtual screening. Methods. 2015;71:146–157. doi: 10.1016/j.ymeth.2014.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pahikkala T., Airola A., Pietilä S., Shakyawar S., Szwajda A., Tang J., Aittokallio T. Toward more realistic drug-target interaction predictions. Brief. Bioinform. 2015;16:325–337. doi: 10.1093/bib/bbu010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
This paper analyzes existing, publicly available data. These accession numbers for the datasets are listed in the key resources table.
-
•
All original code has been deposited at GitHub (https://github.com/koyurion/SEGSA_DTA) and is publicly available as of the date of publication. DOIs are listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.