

Graphical abstract

Keywords: SARS-CoV-2; RdRp inhibitor; Antiviral; 1,4-benzopyrane; Molecular modeling
Abstract
The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has caused a worldwide pandemic. The identification of effective antiviral drugs remains an urgent medical need. In this context, here we report 17 new 1,4-benzopyrone derivatives, which have been designed, synthesized, and characterized for their ability to block the RNA-dependent RNA polymerase (RdRp) enzyme, a promising target for antiviral drug discovery. This compound series represents a good starting point for developing non-nucleoside inhibitors of RdRp. Compounds 4, 5, and 8 were the most promising drug-like candidates with good potency in inhibiting RdRp, improved in vitro pharmacokinetics compared to the initial hits, and no cytotoxicity effects on normal cell (HEK-293). Compound 8 (ARN25592) stands out as the most promising inhibitor. Our results indicate that this new chemical class of 1,4-benzopyrone derivatives deserves further exploration towards novel and potent antiviral drugs for the treatment of SARS-CoV-2 and potentially other viruses.
1. Introduction
The current health emergency due to the pandemic outbreak acute respiratory syndrome coronavirus 2 (SARS-CoV-2) not only affected the world population in terms of human life lost, but also in terms of economic impact on the national health budgets.1, 2 The coronavirus disease 2019 (COVID-19) has left a mark worldwide with>6.2 millions of deaths among over 550 million of cases.3 Most of the patients have mild symptoms, including fever and dry cough, and recover without developing further symptoms. However, up to 5–10 % of cases are characterized by sever symptomatology such as acute respiratory distress syndrome, hypoxia, multiorgan dysfunction syndrome and other serious effects, which could lead toward serious lung lesions.4 The current emergency has prompted the scientific community to search a variety of strategies against the diffusion of this coronavirus.1 For instance, the worldwide vaccination programs began in December 2020 allowing widespread immunization. In addition, the drug repurposing approach helped in the redisposition of some monoclonal antibody against COVID-19.1, 4 Despite all these efforts, the great variability of individual response and, above all, the emergence of uncontrollable drug-resistant mutant strains are still making urgent the search for an effective antiviral treatment.1, 5.
Several studies investigated the pathogenesis of SARS-CoV-2 and identified promising targets to develop an effective drug.6 SARS-CoV-2 is a single-stranded RNA betacoronavirus and the recent publication of its genome sequence revealed that the SARS-CoV-2 genome is closely related to the earlier SARS-CoV (>80 % sequence identity). To a lesser extent, its genome sequence is related to MERS-CoV viruses.7, 8 This information has triggered the identification of druggable targets based on what was already known for SARS-CoV and MERS-CoV.2, 4 These targets include the spike (S), membrane (M), envelope (E), and nucleocapsid (N) viral proteins, which promote the “entry” of the virus in the host.9 Additional targets are the main protease (Mpro) and the RNA-dependent RNA polymerase (RdRp) for the replication of the virus.7 While the recent FDA-approved drug Paxlovid (Pfizer) blocks the Mpro target, 10 additional new drugs targeting SARS-CoV-2 proteins may further boost the development of a multitarget antiviral therapy, as for HIV/AIDS.11
In this context, RdRp represents an ideal target for developing a safe and long-term efficient anti-SARS-CoV-2 agent due to its crucial role in viral life cycle, the lack of homologous proteins in human host cells, and its high conservation across the CoVs family.7, 8 The RdRp enzyme catalyzes the addition of the incoming nucleoside triphosphates (NTPs) through the formation of a phosphodiester bond with the growing strand, in a primer-dependent manner. This enzymatic reaction ensures the synthesis of the large viral RNAs during the virus replication, promoting the progress of the infection in the host.
Several cryogenic electron microscopy (cryo-EM) studies revealed the three-dimensional structure of RdRp at atomic resolution.7, 8 These results are essential for the structure-based design of specific molecular entities targeting RdRp. Notably, RdRp is still an underexplored antiviral target in comparison with other CoVs targets. In this context, RdRp inhibitors are classified into two main categories according to their mode of action and chemical structure: nucleotide or nucleoside inhibitors (NIs) and non-nucleoside inhibitors (NNIs).7 The first group acts as a chain terminator or mutagenic factor by competing with NTP for the incorporation into the strand.12 The NIs include compounds that derive from a large repurposing campaign like Remdesivir, Favipiravir, Galidesivir, Molnupiravir, Ribavirin, Sofosbuvir and Tenofovir.13 Despite the large number of NIs that have been shown to have anti-coronavirus effects in vitro and in vivo, only Remdesivir and Molnupiravir successfully passed clinical trials and were approved.14, 15, 16, 17 The monophosphoramidate nucleoside prodrug Remdesivir has been the first drug approved by both FDA and EMA for the treatment of COVID-19. Also, a broad-spectrum and orally available nucleoside analogue prodrug Molnupiravir, which is known to inhibit the replication of human coronaviruses, has been more recently approved for the treatment of COVID-19. In summary, until now there are only studies concerning the use of repurposed NIs against RdRp, which shared common issues generated by their nucleoside nature such as rapid plasma degradation, high polarity connected to low intestinal permeability (intravenous administration in some cases), and the insurgence of drug resistance.7 Furthermore, their clinical effects are controversial and their efficacy is related only to the early stage of infection, so better antiviral drugs are urgently needed.
In this scenario, NNIs represent a valuable alternative to fight SARS-CoV-2. Indeed, this class of compounds acts through the inhibition of the replication by binding to allosteric sites, or by blocking the association of the NTP.7 Thus, they have several advantages compared to NIs, including the possibility to overcome drug resistance because of the different mechanism of action compared to NIs.7 Indeed, there are few examples on the identification of small molecules as SARS-CoV-2 RdRp NNIs.18, 19, 20, 21, 22 Among these existing NNIs, there are some retrieved from repurposed molecules like suramin, which inhibits SARS-CoV-2 RdRp with an IC50 = 0.26 μM.20 A recent study proved an interesting inhibitory activity toward SARS-CoV-2 RdRp of HeE1-2Tyr, a known potent inhibitor of RdRp Dengue Virus, when tested in both polymerase- and cell-based antiviral assays (IC50RdRp = 27.6 ± 2.1 µM; EC50 Vero cell = 0.65 µM).18 Moreover, two independent studies identified three quinoline-base and 3-thioacetamides indole derivatives with encouraging potency (EC50 in 1–5 µM range) in inhibiting RNA synthesis by SARS-CoV-2 RdRp using a cell based assay.21, 22.
Additional natural products having a promising biological activity against several antiviral targets are the so-called flavonoids, which have been shown to act also against SARS-CoV-2.23, 24, 25, 26, 27 Indeed several studies highlighted the potential capability of quercetin to interfere with SARS-CoV-2 through different mechanisms of action, namely: i) inhibition of the expression of ACE2 receptor, important for cell recognition, ii) inhibition of crucial enzymes of SARS-CoV-2 (3CLPro and RdRp), and iii) an antioxidant, anti-inflammatory ability.28, 29, 30, 31 Despite there are different clinical studies on quercetin and its compositions against COVID-19, the specific effect of quercetin is still not clear.29 In this context, we have recently identified quercetin and luteolin, characterized by 1,4-benzopyrone core, which have shown an appreciable potency in inhibiting SARS-CoV-2 RdRp. In more detail, these flavonoids have shown a one digit micromolar activity against RdRp. These results prompted us to explore these 2 scaffolds. As a result, here we report the design and synthesis of a first set of novel 1,4-benzopyrone compounds, which demonstrate promising inhibitory activity and drug-like properties such as kinetic solubility, plasma and metabolic stability. Importantly, the structure–activity relationship (SAR) of these compounds revealed chemical features that seem crucial for the potency of such a new class of non-nucleoside SARS-CoV-2 RdRp inhibitors.
To rationalize our results, we have also used molecular docking calculations performed using the available cryo-EM structures of SARS-CoV-2 RdRp. Possible binding modes of these compounds are proposed at two distinct allosteric binding pockets, as previously described (i.e. BRNA and BNTP).32 Overall, these results support the further optimization of such inhibitors toward novel antiviral compounds.
2. Material and methods
2.1. RdRp enzymatic assay
Compounds 1–17 were tested against SARS-CoV-2 RdRp with an in vitro enzymatic inhibition assay in collaboration with BPS Bioscience. The RdRp reactions were conducted in duplicate at 37 °C for 60 min in a 10 μl mixture containing assay buffer (20 mM Tris pH8.0 and 0.01 % Triton X100), RNA duplex, ATP substrate and enzyme, and the test compound. The enzyme was produced by BPS Bioscience, and was formulated as 45 mM Tris–HCl pH 8.0, 124 mM NaCl, 2.4 mM KCl, 4 mM MgCl2, 1 mM TCEP, 10 % glycerol. Typical purity was 95–97 %, and typical concentration was 1 mg/ml. These 10 μl reactions were carried out in wells of 384-well Optiplate (PerkinElmer). A 10 mM stock solution of test compound in DMSO was prepared. Dilutions of this stock solution were prepared in assay buffer (5 % DMSO concentration) and 2 μl of the dilution was added to a 6 μl of RdRp (final concentration 0.08 mg/mL) containing RNAse inhibitor for preincubation (30 min at room temperature with slow shaking). Reaction was started by addition of 2 μl of the substrate mix containing RNA duplex (40 nM) and ATP substrate (3 μM). Final concentration of DMSO was 1 % in all reactions (reference compound–0 % DMSO). After enzymatic reactions, 10 μl of anti-Dig Acceptor beads (PerkinElmer, diluted 1:500 with 1 × detection buffer) were added to the reaction mix. After brief shaking, plate was incubated for 30 min. Finally, 10 μl of AlphaScreen Streptavidin-conjugated donor beads (Perkin, diluted 1:125 with 1 × detection buffer) were added. In 30 min, the samples were measured in AlphaScreen microplate reader (EnSpire Alpha 2390 Multilabel Reader, PerkinElmer). In the absence of the compound, the intensity (Ce) in each data set was defined as 100 % of activity. In the absence of the enzyme, the intensity (C0) in each data set was defined as 0 % of activity. The percent activity in the presence of each compound was calculated according to the following equation: % activity = (C–C0)/(Ce–C0), where C is the intensity in the presence of the compound. As a positive control, the reference compound 6-chloropurine-ribose TP was tested at three different concentrations (0.02 μM, 0.2 μM, and 2 μM).
2.2. Human cell Culture
Human cancer cell lines HEK-293 (epithelial, ATCC CRL-1573) were obtained from ATCC. Cells were grown in Dulbecco's Modified Eagle Medium (DMEM), supplemented with 2 mM l-glutamine, 10 % heat-inactivated FBS and 1 % Penicillin/streptomycin. HEK-293 cell lines were growth in a humidified atmosphere of 5 % CO2, at 37 °C. To assess the antiproliferative activity of the compounds, cells were seeded at a density of 10,000 cells/well in 96-well plates, and cell viability was measured using the MTT assay.
2.3. MTT cell viability assay
Cell viability was measured using the MTT assay. Cells were seeded in 96 well plates. Twenty-four hours after seeding, the cells were treated with compounds or vehicle (DMSO, final concentration 0.5 %) as control and incubated for 24 or 48 h. Then, MTT solution ( 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) was added to a final concentration of 0.5 mg/ml and cells were further incubated for 2 h at 37 °C. After solubilization of the formazan crystals by the addition of ethanol, absorbance was measured at 570 nm (reference 650 nm) in a plate reader (Infinite M200, Tecan spark). Inhibition curves consisted of 8 serial dilutions in triplicate in each case, and results were analyzed as sigmoidal dose–response curves using GraphPad Prism software (version 5.03). Values are reported as the mean ± SD of three independent experiments.
2.4. Aqueous kinetic solubility
The aqueous kinetic solubility was determined from a 10 mM DMSO stock solution of test compound in Phosphate Buffered Saline (PBS) at pH 7.4. The study was performed by incubation of an aliquot of 10 mM DMSO stock solution in PBS (pH 7.4) at a target concentration of 250 µM resulting in a final concentration of 2.5 % DMSO. The incubation was carried out under shaking at 25 °C for 24 h followed by centrifugation at 21.100g for 30 min. The supernatant was analyzed by UPLC/MS for the quantification of dissolved compound by UV at a specific wavelength (215 nm). The analyses were performed on a Waters ACQUITY UPLC/MS SQD system consisting of a SQD (Single Quadrupole Detector) Mass Spectrometer equipped with Electrospray Ionization interface. The analyses were run on an ACQUITY UPLC BEH C18 column (50x2.1mmID, particle size 1.7 µm) with a VanGuard BEH C18 pre-column (5x2.1mmID, particle size 1.7 µm), using 10 mM NH4OAc in H2O at pH 5 adjusted with AcOH (A) and 10 mM NH4OAc in MeCN-H2O (95:5) at pH 5 (B) as mobile phase.
2.5. In vitro microsomal stability
10 mM DMSO stock solution of test compound was pre-incubated at 37˚C for 15 min with mouse liver microsomes added 0.1 M Tris-HCl buffer (pH 7.4). The final concentration was 4.6 µM. After pre-incubation, the co-factors (NADPH, G6P, G6PDH and MgCl2 pre-dissolved in 0.1 M Tris-HCl) were added to the incubation mixture and the incubation was continued at 37˚C for 1 h. At each time point (0, 5, 15, 30, 60 min), 30 µl of incubation mixture was diluted with 200 µl cold CH3CN spiked with 200 nM of internal standard, followed by centrifugation at 3500g for 15 min. The supernatant was further diluted with H2O (1:1) for analysis. The concentration of test compound was quantified by LC/MS-MS on a Waters ACQUITY UPLC/MS TQD system consisting of a TQD (Triple Quadrupole Detector) Mass Spectrometer equipped with an Electrospray Ionization interface. The analyses were run on an ACQUITY UPLC BEH C18 (50x2.1mmID, particle size 1.7 µm) with a VanGuard BEH C18 pre-column (5x2.1mmID, particle size 1.7 µm) at 40 °C, using 0.1 % HCOOH in H2O (A) and 0.1 % HCOOH in CH3CN (B) as mobile phase. Electrospray ionization (ESI) was applied in positive mode. The percentage of test compound remaining at each time point relative to t = 0 was calculated. The half-lives (t½) were determined by an one-phase decay equation using a non-linear regression of compound concentration versus time.
2.6. In vitro plasma stability
10 mM DMSO stock solution of test compound was diluted 50-fold with DMSO-H2O (1:1) and incubated at 37˚C for 2 h with mouse plasma added 5 % DMSO (pre-heated at 37˚C for 10 min). The final concentration was 2 µM. At each time point (0, 5, 15, 30, 60, 120 min), 50 µl of incubation mixture was diluted with 200 µl cold CH3CN spiked with 200 nM of internal standard, followed by centrifugation at 3500g for 20 min. The supernatant was further diluted with H2O (1:1) for analysis. The concentration of test compound was quantified by LC/MS-MS on a Waters ACQUITY UPLC/MS TQD system consisting of a TQD (Triple Quadrupole Detector) Mass Spectrometer equipped with an Electrospray Ionization interface. The analyses were run on an ACQUITY UPLC BEH C18 (50x2.1mmID, particle size 1.7 µm) with a VanGuard BEH C18 precolumn (5x2.1mmID, particle size 1.7 µm) at 40 °C, using 0.1 % HCOOH in H2O (A) and 0.1 % HCOOH in CH3CN (B) as mobile phase. Electrospray ionization (ESI) was applied in positive mode. The response factors, calculated on the basis of the internal standard peak area, were plotted over time. When possible, response vs time profiles were fitted with Prism (GraphPad Software, Inc., USA) to estimate compounds half-life in plasma.
2.7. Computational methods
In order to perform the molecular docking, the SARS-CoV-2 RdRp was retrieved from PDB database (PDB ID 7D4F)20 and prepared for docking using Schrödinger's Protein Preparation Wizard tool.33 The preparation consisted in: i) adding the hydrogen atoms, ii) eliminating water molecules that are not involved in ligand-binding interaction, and iii) assigning atomic charges. Subsequently, the energy minimized 3D molecular structures of all 22 small molecules (i.e. 20 synthesized compounds in addition to luteolin and quercetin) were generated and prepared for docking using LigPrep tool. 34 Eventually, the SARS-CoV-2 RdRp structure (PDB ID 7D4F)20 was used for docking the 22 small molecules. The grid was centered on the suramin’s center of mass, either bound to the BRNA or the BNTP pocket, and the docking was performed using Glide XP methodology.35, 36.
2.8. Chemical characterization of compound 8 by NMR and HRMS
Compound 8 was obtained through the synthetic procedure described in detail in the supplementary material and characterized by NMR and HRMS. 1H NMR (400 MHz, DMSO–d6) δ 7.61 (t, J = 8.4 Hz, 1H), 6.99 (dd, J = 8.5, 0.9 Hz, 1H), 6.78 (d, J = 7.9 Hz, 1H), 6.74 (d, J = 2.2 Hz, 1H), 6.69 (d, J = 8.1 Hz, 1H), 6.63 (dd, J = 8.1, 2.1 Hz, 1H), 6.33 (s, 1H), 4.01 (q, J = 7.2 Hz, 1H), 1.53 (d, J = 7.1 Hz, 3H). 13C NMR (101 MHz, DMSO–d6) δ 183.2 (Cq), 174.3 (Cq), 159.9 (Cq), 156.2 (Cq), 145.3 (Cq), 144.5 (Cq), 135.9 (CH), 131.8 (Cq), 118.3 (CH), 115.7 (CH), 114.8 (CH), 110.8 (CH), 109.8 (Cq), 107.2 (CH), 107.0 (CH), 42.6 (CH2), 18.4 (CH2, recovered from HSQC). HRMS (AP-ESI) m/z calculated for C17H15O5 [M + H]+ 299.0919, found 299.0924.
3. Results and discussion
3.1. Exploring the structure of 1,4-benzopyrone scaffold
Luteolin and quercetin feature a 1,4-benzopyrone core constituted by the fused ring A and C, substituted in 2 position by a catechol moiety and by two hydroxyl groups in 5 and 7 position (Figure 1 ). Quercetin differs from luteolin for just one additional hydroxyl group in 3 position. Inspired by the inhibitory activity of such a natural scaffold,32 we initially build our SAR study exploring a larger and different chemical space compared to the flavonoids. We synthesized a new set of 17 1,4-benzopyrone derivatives, which maintain a catechol in 2 position and a β-hydroxy-ketone motif on the core. These two structural motifs likely form key interactions with the target. However, we also added other chemical functionalities to improve the inhibitory activity toward the target. As depicted in Figure 1, we mainly explored the positions C2 and C3: 6 analogues were generated by the insertion of several flexible substituents on the carbon 3 (compound 1–6, Scaffold A) of the benzopyrone, while other 11 derivatives embedded different spacers, with a diverse degree of polarity, flexibility and branching (7–17, Scaffold B) in between the benzopyrone core and the catechol ring. Notably, our new compounds were all measured for their inhibitor activity against SARS-CoV-2 RdRp using an enzymatic biochemical assay.
Figure 1.

SARS-CoV-2 RdRp inhibitors (luteolin and quercetin) and newly designed compounds (Scaffold A and B).
To begin, we synthesized six analogues to evaluate the effect of different alkyl and alkoxy chains on position 3 of ring C (compounds 1–6, Table 1 ). Such chains differ in length and polarity. At first, the role of the hydroxyl group of quercetin was investigated by replacing it with a simple methyl group. In this way, we generated compound 1, which has no H-bond acceptor (HBA) and H-bond donor (HBD) abilities, while retaining the steric hindrance. Notably, compound 1 returned an IC50 of 4.3 ± 1.1 µM, which is comparable to quercetin, IC50 = 6.9 ± 1.0 µM. Prompted by these initial data, we decided to elongate the alkyl chain through the insertion of either a propyl and butyl group, generating compounds 2 and 3 (Table 1). The transition to longer alkyl chains slightly decreased the activity, displaying an IC50 values of 8.8 ± 0.7 µM and 6.0 ± 1.1 µM, respectively. At this stage, we decided to reinsert the HBA oxygen atom in position 3, generating compound 4 with a butoxy group. This retained the activity, with an IC50 = 5.3 ± 1.0 µM. Finally, we wanted to investigate the role of a H-bond donor (HBD) group introducing a hydroxyl group at the terminal position of the chain, as in 5 and 6. The substitution of the terminal methyl group of compound 4 with a hydroxyl group generated derivate 6, which maintained the activity, with an IC50 = 5.6 ± 0.1 µM. Shortening the length of the chain, as in 5, also maintained a similar activity, with an IC50 = 6.1 ± 1.0 µM.
Table 1.
Structures and activity against SARS-CoV-2 RdRp of 1,4-benzopyrone derivatives 1–6.a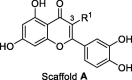 .
.
| Entry | Compound | R1 | IC50 (µM) |
|---|---|---|---|
| 1 | 1 | -Me | 4.3 ± 1.1 |
| 2 | 2 | -nPr | 8.8 ± 0.7 |
| 3 | 3 | -nBu | 6.0 ± 1.1 |
| 4 | 4 | -OBu | 5.3 ± 1.0 |
| 5 | 5 | 6.1 ± 1.0 | |
| 6 | 6 | 5.6 ± 0.1 |
a) See related dose–response curves in Supplementary material.
After evaluating the inhibitory activity of this first subset of derivatives, we assessed additional new molecules with different spacers between the benzopyrone core and ring B. This strategy aimed at moving further away from the flavonoid class, through unexplored modifications of such a well-known scaffold. Therefore, additional modifications have been inserted to test the flexibility and role of the catechol function, more distant from the bicyclic core. In principle, these structural features may allow new interactions with unexplored portions of the binding site.
To access easily to scaffold of type B (Figure1), we removed the hydroxyl group in position 7. Interestingly, compound 7 with a simple methylene bridge in 2 position, exhibited a potency of IC50 = 8.5 ± 0.7 µM, comparable to quercetin and only two fold worse of luteolin (IC50 = 4.6 ± 0.3 µM). Thus, this compound was a good starting point for our second subclass of derivatives. Therefore, we decided to explore several hydrocarbon lipophilic spacers as for compounds 8–12 (Table 2 ). Firstly, a ramification on the methylene bridge was inserted to understand the influence of lipophilic carbon chains with different lengths and degree of steric hindrance on this position. Replacing hydrogen with methyl (compound 8) has shown an IC50 = 4.1 ± 1.4 µM (Table 2, entry 2; Figure 2 A), quite similar to luteolin. On the other hand, the chain extension to four aliphatic carbons (compound 9) annihilated the activity (IC50 > 100 µM), while the presence of a more lipophilic terminal trifluoromethyl group (compound 10) displayed no inhibition. These data demonstrate that a long ramification, with at least four carbon linear chain, and an increased lipophilicity were not tolerated on that position. Notably, the ramification of the methylene spacer introduces a stereocenter, which may open new avenues for future ramifications and investigations. In this regard, we clarify that here we report the racemic synthesis of such compounds. However, an asymmetric route or chiral separation of enantiomers could be performed with extra efforts to evaluate specifically the stereochemical impact on the biological activity.
Table 2.
Structures and activity against SARS-CoV-2 RdRp of 1,4-benzopyrone derivatives 7–17.a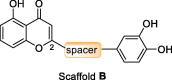 .
.
| Entry | Compound | spacer | IC50 (µM) |
|---|---|---|---|
| 1 | 7 | –CH2- | 8.5 ± 0.7 |
| 2 | 8b |  |
4.1 ± 1.4 |
| 3 | 9 | 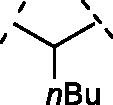 |
> 100 |
| 4 | 10 | 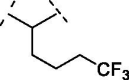 |
no inhibition |
| 5 | 11 | 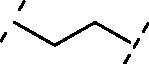 |
5.8 ± 0.7 |
| 6 | 12 |  |
7.0 ± 1.2 |
| 7 | 13 | 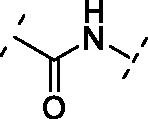 |
7.1 ± 0.8 |
| 8 | 14 | 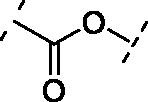 |
2.0 ± 1.0 |
| 9 | 15 | 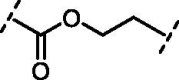 |
44.1 ± 1.1 |
| 10 | 16 | 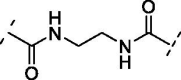 |
15.9 ± 1.1 |
| 11 | 17 | 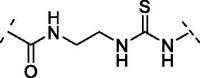 |
3.4 ± 1.2 |
a) See related dose–response curves of all compounds with the exception of 8 in Supplementary material.
b) See related dose–response curve in Figure 2A.
Figure 2.

Dose-response curves of compound 8 related to the enzymatic inhibition of RdRp (A, left), and to cytotoxicity assay of 8 on HEK-293 for 24 and 48 h (B, right).
Increasing the length of spacer in linear fashion with an ethylene spacer as in 11 maintained the activity with an IC50 = 5.8 ± 0.7 µM, like compound 8 and luteolin. Then, the modulation of the flexibility through a vinyl spacer in compound 12 returned an IC50 of 7.0 ± 1.2 µM. At this point, we wanted to evaluate how the insertion of a polar motif affects the activity. Interestingly, the amide spacer in 13 retained an IC50 of 7.1 ± 0.8 µM, while the ester counterpart 14 increased about 4-fold the activity, showing an IC50 of 2.0 ± 1.0 µM compared to 12 and 13. This suggests that the right balance between flexibility, polarity and hydrogen-bonding motifs in the bridge between benzopyrone and catechol units can be used to modulate the activity of these analogues. Surprisingly, moving away the B-ring of two carbon units from the ester as in 15 dropped 22-fold the potency (IC50 = 44.1 ± 1.1 µM) compared to the rigid ester 14. On the other hand, the presence of two amide functional groups interconnected through an ethylene chain in analogue 16 decreased twice the potency (IC50 = 15.9 ± 1.1 µM) compared to the simple amide 13. The replacement of the second amide function with a thiourea directly bonded to the catechol restored the activity, improved the IC50 from over 15 µM of compound 16 to 3.4 ± 1.2 µM of 17.
3.2. Chemistry
The 17 new derivatives were synthesized exploiting 5 different syntheses of 5–6 steps as outlined in Scheme 1, Scheme 2, Scheme 3, Scheme 4, Scheme 5, Scheme 6 . As described in Scheme 1, the synthesis of compounds 1–3 started from the commercial available 3,5-dimethoxyphenol 18 and proper acyl chlorides 19a-c, with the specific chain of the final target compound already embedded into it. In the first step, the Friedel-Craft acylation of 18 with 19a-c in the presence of alluminium trichloride as Lewis acid in dichloromethane afforded intermediates 20a-c in 57–59 % yields, which underwent to aldol condensation with vetraldehyde 21 in basic conditions (KOH in MeOH), giving chalcone intermediates 22a-c in 35–50 % yields. Then, oxidative iodine-catalyzed cyclization generated chromone intermediates 23a-c in good 50–70 % yields. Lastly, desired final products 1–3 were obtained in 30–76 % yields by demethylation of 23a-c with molten pyridinium hydrochloride at 190 °C.
Scheme 1.

Synthesis of compounds 1–3. Reagents and conditions: (a) AlCl3, DCM, r.t., 1–6 h, (b) vetraldehyde 21, KOH, MeOH, r.t., 72 h, (c) I2 cat., DMSO, 120 °C, 2 h, (d) pyridinium chloride, 190 °C, 5 h.
Scheme 2.

Synthesis of compounds 4–6. Reagents and conditions: (a) BnBr, K2CO3, DMF, r.t., overnight, (b) HCl conc., EtOH, reflux, 2 h, (c) alkyl halide RX 27a-c, K2CO3, DMF, r.t., 3 h, (d) Et3SiH, Pd\C (20 % w\w), DCM, MeOH, r.t., overnight.
Scheme 3.

General synthetic scheme for compounds 7–10: a) LDA or LiHMDS, R-I, THF, −78 °C to rt; b) NaH, THF, reflux; c) LDA, THF, −78 °C to rt; d) HCl(aq) 37 %, MeOH, rt; e) H2SO4, CH3COOH, 100 °C; f) BBr3, DCM, 0 °C to rt; g) LiOH; THF/H2O, 50 °C; h) SOCl2, DCM, reflux; i) 30, LDA, THF, −78 °C to rt.
Scheme 4.

Synthesis of building blocks 38a-b. Reagents an conditions: (a) NaH, EtOAc/THF, reflux; (b) HCl (37%), MeOH, overnight; (c) diethyloxalate, NaOEt, EtOH; (d) LiOH, THF/H2O.
Scheme 5.

Synthesis of derivatives 11–12. Reagents and conditions: a) 3,4‑dimethoxy benzaldehyde, NaOEt, EtOH, 50 °C; b) NH4CO2H, Pd(OH)2/C; MeOH, 80 °C; (c) BBr3 (1 M in DCM), DCM, from 0 °C to rt.
Scheme 6.

Synthesis of derivatives 13–17. (a) 41a, HATU, DIPEA, DMF/DCM; (b) 41b or 41c, DCC, DMAP, DCM; c) BBr3 (1 M in DCM), DCM; (d) N-Boc ethylendiamine 41d, DIPEA, PyBOP, DMF/DCM; (e) HCl (4 M in 1,4-dioxane); (f) 3,4‑dimethoxy benzoic acid, DIPEA, PyBOP, DMF/DCM; (g) 45, DIPEA, EtOH, reflux.
Quercetin derivatives 4–6 were synthesized using commercially available rutin 24 according to the synthetic route shown in Scheme 2. Rutin 24 features the four hydroxyl groups in 3′, 4′, 5 and 7 position of our initial hits and the oxygen in 3 position of quercetin masked as glycoside with rutinose. The use of an already functionalized starting material limited this synthetic route. However, this strategy represented a valid late-stage diversification approach to rapidly explore position 3. In the first step, phenolic groups on positions 7, 3′ and 4′ were protected by benzylation to generate intermediate 25. Then, rutinose was removed by acid hydrolysis to obtain intermediate 26 in excellent 85 % yield over 2 steps. Following acid hydrolysis, the C-3 hydroxyl group was regioselectively alkylated with the proper alkyl halide 27a-c in the presence of potassium carbonate as a base to obtain 28a-c. Finally, debenzylation was subsequently performed group with triethylsilane and palladium on carbon, obtaining desired compounds 4–6 in 11–62 % yields over two steps.
The synthesis of compounds 7–10 involved three key steps that are the Claisen-type condensation between the α-substituted carbonyl compound and the 1-(2-hydroxy-6-methoxyphenyl)ethan-1-one 30, followed by dehydrative cyclization and deprotection of the methoxide groups (Scheme 3). Compound 7 was isolated with 92 % of yield after deprotection of intermediate 32a, which was formed starting from condensation of 29a and 30 with NaH as base and subsequent cyclization with HCl(aq) 37 %. On the contrary, the first Claisen-type condensation to obtain the final compounds 8–10 required different chemical conditions. This was probably due to a different reactivity of the Claisen acceptor related to the presence of a more hindered group next to electrophile carbonyl. In order to have the right condensation partner 29b for the construction of compound 8, the α-alkylation of ester 29a using LiHMDS and iodomethane was needed as a first step. Ester 29b did not undergo to the desired conversion into intermediate 31b using the condensation conditions previously used. Thus, LDA was used as a base, instead of NaH, for the following Claisen-type step to give intermediate 31b. This has been cyclized by means of dehydrative mixture H2SO4/CH3COOH yielding compound 32b in 24 % of yield after two steps. After deprotection with BBr3, the desired compound 8 was obtained in 78 % of yield. Instead, the condensation partners for the synthesis of compounds 9 and 10 were made using a different synthetic pathway, with acyl chloride 35, 36. This was because the corresponding esters turned out to be unreactive on Claisen reaction conditions. Therefore, the α-alkylation of 29a with 1‑iodobutane and 1,1,1-trifluoro-4‑iodobutane was used to deliver the α-substituted esters, which have been hydrolyzed generating the corresponding carboxylic acid 33 and 34 with 52 % and 48 % of yield, respectively, after two steps. In order to make them more reactive, they were converted in the acyl chlorides 35 and 36 and slowly added into a basic mixture of 30, in which the corresponding enolate was preforming. Later, cyclization of intermediates 31c and 31d in the same strong acid dehydrative mixture provided the protected compounds 32c and 32d in 29 % and 24 % yields after two steps, which were treated with BBr3 in order to isolate final compounds 9 and 10 with 92 % and 82 % of yield, respectively.
We envisioned the intermediate 38a-b as two useful building blocks to access to compounds 11–17 starting from the same starting material 30 (Scheme 4). The cross-Claisen condensation of 2-hydroxy-6-methoxyacetophenone 30 with ethyl acetate in basic condition afforded the β-diketone 37a, which underwent to dehydrative cyclization in strong acid environment to yield 2-methylchromone 38a with 84 % yield after 2 steps (Scheme 4). The same sequence of cross-Claisen-dehydrative cyclizations using 30 and ethyl oxalate in presence of NaOEt in EtOH allowed the direct construction of the benzopyrone bicyclic core of 37b with an ester function in 2 position, with 79 % yield. The ester 37b was then hydrolyzed using classic conditions (LiOH in THF/H2O) to give carboxylic acid 38b.
As depicted in Scheme 5, we then exploited the pronucleophilic nature of the methyl group in 38a to access to the divergent intermediate (E)-39 in a stereoselective fashion (10:1 ratio E/Z isomers), performing a vinylogous aldol condensation with 3,4-dimethoxybenzaldehyde in basic condition. The next reduction of the double C—C bond of 39 with ammonium formate and palladium hydroxide on carbon, gave the methylated precursor 40 in 63 % yield, which after final deprotection with boron tribromide led to the final compound 11, with a completely satured hydrocarbon spacer. Alternately, the direct deprotection of (E)-39 with boron tribromide led to the formation of the final compound 12, with a vinyl spacer.
On the other hand, building block 38b was subjected to a divergent synthetic strategy to access to final derivatives 13–17 (Scheme 6). The key amide coupling or Steglich esterification between 38b and a suitable amine or alcohol was exploited to obtain an amide or ester intermediate 42a-d. Amide coupling between 38b and 3,4-dimethoxyaniline 41a using HATU, DIPEA in a DMF/DCM mixture generated amide 42a with 90 % yield, while the Steglich reaction with 3,4-dimethoxyphenol 41b and the elongated alcohol 41c with DCC and DMAP afforded the ester intermediate 42b-c with 25 % and 44 % yield, respectively. Importantly, alcohol 41c has been synthesized by reduction of methyl 2-(3,4-dimethoxyphenyl)acetate 29a with LiAlH4 (See Supplementary material). However, final deprotection with boron tribromide of precursors 42a-c led to the formation of the desired deprotected compounds 13–15 with 40–84 % range yield. Noteworthy, with the aim to evaluate the progressive elongation of one carbon unit of the ester derivatives, we were able to synthesize the benzylic ester counterpart of compound type 42. We noticed its degradation in the last step, restoring the carboxylic acid 38b and corresponding methyl ester after quenching, without isolation of the desired compound. Probably, the methylene group between the ester function and the catechol favors the in situ formation of the quinone methide during the deprotection, promoting the hydrolysis or the transesterification reaction in presence of methanol. To overcome this issue, we extended the distance between the ester function and the catechol of two more carbon units as in final analog 15.
Different coupling agent PyBOP was used for N-Boc-diamine 41d, yielding amide 42d. As shown in Scheme 6, suitable manipulations of amide 42d gave final products 16 and 17. The Boc removal in acidic conditions released the free amine 43, which represented a further divergent point of the synthetic plan. Indeed, an additional amide coupling with 3,4‑dimethoxy benzoic acid afforded di-amide 44, otherwise the reaction with isothiocyanate 45 allowed the insertion of thiourea functionality in compound 46. Notably, isothiocyanate 45 was easily prepared in one step using thiocarbonyl diimidazole TCDI and 3,4-dimethoxyaniline 41a with 51 % yield (See Supplementary material). Finally the methyl deprotection of 44 and 46 with classic conditions (BBr3 in DCM) gave the desired products 16 and 17.
3.3. Metabolic stability and chemical solubility.
After the initial evaluation of the new set of benzopyrone derivatives for their inhibitory activity against RdRp in vitro, we selected the compounds with one-digit micromolar activity for further evaluation of their drug-likeness. At first, we assessed their kinetic solubility (Sk) in neutral buffer. As previously reported, the natural compounds (luteolin and quercetin) had a poor kinetic solubility in the range of 16–21 µM (Table 3 , entry 1, 2).32 While the introduction of alkyl groups in 3 position, as in compounds 1–3, decreased the solubility with an increasing chain length, the presence of alkoxy or hydroxylalkoxy groups as in 4–6 increased about 7 to 11-fold the solubility in the range of 156–231 µM, compared to the hits.
Table 3.
Kinetic solubility, plasma stability, and microsomal stability of selected compounds.
| Entry | Compound | Kinetic solubility Sk (µM) |
Mouse plasma stability t1/2 (minutes) |
Mouse microsomal stability t1/2 (minutes) |
|---|---|---|---|---|
| 1 | luteolin | 21 | >120 | >60 |
| 2 | quercetin | 16 | 7 | >60 |
| 3 | 1 | 32 | – | – |
| 4 | 2 | 13 | – | – |
| 5 | 3 | < 1 | – | – |
| 6 | 4 | 156 | >120 | >60 |
| 7 | 5 | 167 | >120 | >60 |
| 8 | 6 | 231 | 90 | 60 |
| 9 | 7 | 63 | >120 | >60 |
| 10 | 8 | 165 | >120 | >60 |
| 11 | 11 | 67 | >120 | >60 |
| 12 | 12 | <1 | – | – |
| 13 | 13 | 65 | 83 ± 13 | 60 |
| 14 | 14 | <1 | – | – |
| 15 | 17 | <1 | – | – |
Looking at analogues with a spacer embedded in their scaffold, compound 7 with a methylene bridge improved 3 to 4-fold the solubility, with Sk = 63 µM, similarly to the carbon elongated analogue 11 (Sk = 67 µM) and the amide 13 (Sk = 65 µM). Interestingly the methyl substituent of 8 improved Sk to 165 µM. On the contrary, the esters 12, 14 and thiourea 17 drastically decreased the solubility to 1 µM.
Then, we evaluated the chemical stability of the active compounds with acceptable or good Sk (>60 µM) using mouse serum and mouse liver microsomes. These molecules had good or excellent plasma stability of 80–90 min or > 120 min, and very good microsomal stability (60 or > 60 min). Overall, these selected new derivatives showed good DMPK proprieties, thus providing us crucial information on the portions that could be modified to further improve their pharmacokinetic profile, without affecting their inhibitory potency.
Additionally we selected 4, 5, 6 and 8 as best compounds in terms of potency, solubility and stability to evaluate their cytotoxicity in normal cell. In particular, we performed a cytotoxicity assay on HEK-293 for 24 and 48 h. These compounds did not appear to be cytotoxic for the normal cells even at high concentrations as 100 µM (Figure 2B, and see Supplementary material ). Nicely these findings strengthen this class of new compounds as promising antiviral agents.
3.4. Molecular modeling
To explore the possible binding mode of our compounds and gain further insights into the structure–activity relationship, we performed molecular docking of 22 molecules (i.e. structures 1–17 including the enantiomers of derivatives 8–10, in addition to luteolin and quercetin) against SARS-CoV-2 RdRp. Based on our previous results obtained from testing luteolin and quercetin,32 we decided to dock the compounds into the two allosteric pockets, i.e. BRNA and BNTP, identified in a recent cryoelectron microscopy (cryo-EM) structure of SARS-CoV-2 RdRp in complex with suramin (PDB ID 7D4F).20.
Comparison of binding poses in BRNA and BNTP pockets. In line with our previous study,32 we docked our compounds in either BRNA and BNTP binding pockets. The resulting docking scores for all 22 molecules are shown in the Supplementary material (Table S1). Overall, the compounds belonging to the two subset of derivatives showed different binding modes. In particular, the docking poses in both BRNA and BNTP pockets adopted by the first set of derivatives (i.e. compounds 1 to 6 in addition to luteolin and quercetin, for a total of 8 molecules) spatially overlap, thus establishing interactions with the same residues (Figure 3 and Figure S1). For the BRNA binding pocket, the main interactions between the small molecules and the protein involve the side chains of Asn496, Asn497, Lys500, Arg569, Gln573, Lys577, Arg583 and Tyr689 residues. For the BNTP pocket the main residues involved in the interactions with the docked structures are Lys438, His439, Ser549, Lys551, Arg555, Ser814, His816, and Arg836. However, the binding pose of compound 2 is unaligned with the other compounds from the same subset. It overlaps with the docking poses of part of second subset of derivatives (Figure S2). Overall, the predicted binding modes for the first subset of compounds are in line with the results obtained in our previous study on luteolin and quercetin.32 Remarkably, many of these interactions were previously identified as important for binding stability of luteolin and quercetin by MD simulations, as reported in ref 32. Additionally, this result is in good agreement with the IC50 values, which are similar to those obtained for luteolin and quercetin (see Table 2).32.
Figure 3.

XP Glide docking poses for all 22 compounds in both BRNA (in light blue, transparent surface) and BNTP (in light green, transparent surface) pockets of RdRp enzyme. (Left) The first subset of derivatives (i.e. compounds 1–6 in addition to luteolin and quercetin) docked into BRNA in dark blue licorice and BNTP in pink licorice. (Right) The second subset of derivatives (i.e. compounds 7–17) docked into BRNA in dark blue licorice and BNTP in pink licorice. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
On the other hand, the second subset of derivatives (i.e. compounds 7 to 17) adopts a wider variety of binding modes (Figure 3 and Figure S2). The introduction of a spacer has conferred different degrees of freedom to the molecules, which depend on the physicochemical properties of the specific spacer. This has resulted in diverse binding poses for both BRNA and BNTP pockets.
Among these compounds, 9 and 10 have a stereocenter within the spacer, and show either low or no inhibitory activity, respectively (see Table 2). Notably, the molecular docking results for the BRNA pocket predicted the binding energy values of both enantiomers of 9 and 10 (i.e. (S)/(R)-9 and (S)/(R)-10) within the last six values (see Table S1). This result qualitatively reflects the experimental IC50 values (Table 2). In particular, the enantiomers (R)-9 and (R)-10 bind similarly to the pocket, with the bulky chain of the spacer positioned in a small cleft of the protein (Figure 4 ). Additionally, such cleft is formed by several polar residues, such as Asn568, His572, Gln573, Lys577, Asp684 and Thr686, thus adversely interacting with the lipophilic n-Bu chain of the molecule (R)-9. For both molecules, the B ring establishes interactions with Asn497, Lys500 and Arg569, while the benzopyrone core interacts with Tyr689 residue. On the other hand, the enantiomers (S)-9 and (S)-10 bind differently (Figure 4). Indeed, the (S)-9 molecule is positioned with the B ring in the polar cleft, consequently exposing the aliphatic chain towards the solvent bulk. The (S)-10 molecule binding mode requires the benzopyrone core positioned in the cleft, the bulky chain solvent exposed and the B ring interacting with Lys500. Overall, all four binding modes show relevant destabilizing binding modes, in agreement with their very week IC50 values.
Figure 4.

Docking poses for the (R)-9/10 and (S)-9/10 enantiomers in BNTP pocket. Polar residues in the small cleft are represented in white licorice.
Conversely, all four (S)/(R)-9 and (S)/(R)-10 molecules bind similarly in the BNTP pocket. In all four poses the benzopyrone core interacts with His439, Lys545, Ser549, Arg555 and Arg836. The B ring form interactions with Lys593 and Asp865 and the lipophilic chain of (S)/(R)-9 is positioned close to the lipophilic Ala840 and Leu462 residues, or Phe441, Ala457 and Ala840 residues, respectively. Thus, the binding modes predicted for (S)/(R)-9 and (S)/(R)-10 molecules in the BNTP pocket show more stabilizing interactions with the target compared to the binding modes for the BRNA pocket. All together, these results offer a structural rationale for the low or absent inhibitory effect of 9 and 10 chiral molecules, supporting the BRNA pocket as the potential binding pocket for this class of inhibitors.
By analysing the binding modes obtained for both enantiomers (S)/(R)-8 in the BRNA pocket, it appears that in both cases the short aliphatic methyl group is solvent exposed (Figure S3). However, for (R)-8, the B ring is positioned in the polar cleft of the protein, thus orienting the methyl group within 6 Å from two lipophilic residues, i.e. Ala580 and Ile589. On the other hand, (S)-8 binds with the benzopyrone core in the polar cleft and the methyl group results to be completely solvent exposed, missing the supplementary lipophilic interactions that stabilized the system (see Figure S3). Overall, the analysis of the docking poses for (S)/(R)-8 molecules in the BRNA pocket suggest that the (R)-8 enantiomer binds more stably in the pocket.
4. Conclusion
Here, we have reported the design, synthesis, and an extensive experimental − computational characterization of a novel chemical class of RdRp inhibitors.32 The resulting SAR elucidates the key structural features that enhance the potency and the drug-like profile of our new benzopyrone derivatives. Most of these new analogues exhibit a promising one-digit micromolar potency, favorable solubility, and good in vitro stability values. Moreover, based on docking calculations, we propose possible binding modes of these inhibitors, which are consistent with our SAR.
Indeed, we present two subset of molecules to explore a new chemical space centered on a benzopyrone core. The first subset includes the functionalization of position 3 of the bicycle with different alkyl and alkoxy chains, generating compounds 1–6, all with good potency. The second subset, compounds 7–17, covers a well-diversified chemical space with the introduction of a spacer moiety with a different flexibility degree. This last effort generated derivative 8 as the most interesting inhibitor of this novel chemical series, which has good potency and favorable in vitro pharmacokinetic properties with IC50 comparable or better than previous described non-nucleoside inhibitors (NNI).18, 21, 22 Additionally, in our MMT assay, compounds 4, 5, 6 and 8 displayed no cytotoxicity on normal cells (HEK-293).
In conclusion, a new class SARS-CoV-2 RdRp inhibitors has been designed and characterized as a promising starting point to develop new antiviral agents. Even if most of the new benzopyrone derivatives cover a narrow range of IC50 values (3.4–15.9 µM), this study provides a promising starting point to fine-tune the activity of this new chemical class for activity against RdRp SARS-CoV-2. These compounds show a good drug-like profile and potency against the target. Indeed compound 8 (ARN25592) represents a promising and viable lead, which deserves further investigation and optimization, as supported by the modeling study of its two enantiopure forms. Notably, such micromolar activity versus SARS-CoV-2 RdRp is not expected to be highly specific for this target but it certainly indicates that ARN25592 constitutes a proper initial compound for lead generation campaigns where to improve potency in the nanomolar range, which should then also generate specificity for RdRp. Therefore, taken together, these results constitute the basis for the structure-based design and further development of new molecular entities against SARS-CoV-2 RdRp, paving the way for the further expansion of this novel chemical class of compounds towards new antiviral therapeutics.
Declaration of Competing Interest
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests: [Marco De Vivo reports financial support, administrative support, article publishing charges, and equipment, drugs, or supplies were provided by Italian Institute of Technology. Marco De Vivo reports a relationship with Italian Institute of Technology that includes: employment. Marco De Vivo has patent #IT 102022000000920 pending to Italian Institute of Technology. Nicoletta Brindani has patent #IT 102022000000920 pending to Italian Institute of Technology. Andrea Menichetti has patent #IT 102022000000920 pending to Italian Institute of Technology. None.].
Acknowledgments
We thank S. Venzano and BPS bioscience for the technical support.
Footnotes
Detailed synthetic procedure, significative 1H and 13C spectra, analytical chromatograms, and dose-response curves of inhibitory activity and cytotoxicity of compounds can be find in supplemataty material. Supplementary data to this article can be found online at https://doi.org/10.1016/j.bmc.2023.117179.
Appendix A. Supplementary material
The following are the Supplementary data to this article:
Data availability
Data will be made available on request.
References
- 1.Phillips N. The coronavirus is here to stay - here's what that means. Nature. 2021;590:382–384. doi: 10.1038/d41586-021-00396-2. [DOI] [PubMed] [Google Scholar]
- 2.Wu F., Zhao S., Yu B., et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579:265–269. doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.https://covid19.who.int/.
- 4.Gil C., Ginex T., Maestro I., et al. COVID-19: Drug targets and potential treatments. J Med Chem. 2020;63:12359–12386. doi: 10.1021/acs.jmedchem.0c00606. [DOI] [PubMed] [Google Scholar]
- 5.Dolgin E. Omicron is supercharging the COVID vaccine booster debate. Nature. 2021 doi: 10.1038/d41586-021-03592-2. [DOI] [PubMed] [Google Scholar]
- 6.Lamers M.M., Haagmans B.L. SARS-CoV-2 pathogenesis. Nat Rev Microbiol. 2022;20:270–284. doi: 10.1038/s41579-022-00713-0. [DOI] [PubMed] [Google Scholar]
- 7.Cannalire R., Cerchia C., Beccari A.R., Di Leva F.S., Summa V. Targeting SARS-CoV-2 Proteases and Polymerase for COVID-19 Treatment: State of the Art and Future Opportunities. J Med Chem. 2020 doi: 10.1021/acs.jmedchem.0c01140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gao Y., Yan L., Huang Y., et al. Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science. 2020;368:779–782. doi: 10.1126/science.abb7498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou Y.W., Xie Y., Tang L.S., et al. Therapeutic targets and interventional strategies in COVID-19: mechanisms and clinical studies. Signal Transduct Target Ther. 2021;6:317. doi: 10.1038/s41392-021-00733-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Owen D.R., Allerton C.M.N., Anderson A.S., et al. An oral SARS-CoV-2 M(pro) inhibitor clinical candidate for the treatment of COVID-19. Science. 2021;374:1586–1593. doi: 10.1126/science.abl4784. [DOI] [PubMed] [Google Scholar]
- 11.de Castro S., Camarasa M.J. Polypharmacology in HIV inhibition: can a drug with simultaneous action against two relevant targets be an alternative to combination therapy? Eur J Med Chem. 2018;150:206–227. doi: 10.1016/j.ejmech.2018.03.007. [DOI] [PubMed] [Google Scholar]
- 12.Sofia M.J., Chang W., Furman P.A., et al. Nucleoside, nucleotide, and non-nucleoside inhibitors of hepatitis C virus NS5B RNA-dependent RNA-polymerase. J Med Chem. 2012;55:2481–2531. doi: 10.1021/jm201384j. [DOI] [PubMed] [Google Scholar]
- 13.Vicenti I., Zazzi M., Saladini F. SARS-CoV-2 RNA-dependent RNA polymerase as a therapeutic target for COVID-19. Expert Opin Ther Pat. 2021;31:325–337. doi: 10.1080/13543776.2021.1880568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beigel J.H., Tomashek K.M., Dodd L.E., et al. Remdesivir for the Treatment of Covid-19 - Final Report. N Engl J Med. 2020;383:1813–1826. doi: 10.1056/NEJMoa2007764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang Y.Z., Du G., Du R., Zhao J., Jin Y., Fu S., et al. Remdesivir in adults with severe COVID-19: a randomised, double-blind, placebo-controlled, multicentre trial. Lancet. 2020;395:1569–1578. doi: 10.1016/S0140-6736(20)31022-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schafer A., Martinez D.R., Won J.J., et al. Therapeutic efficacy of an oral nucleoside analog of remdesivir against SARS-CoV-2 pathogenesis in mice. bioRxiv. 2021 doi: 10.1101/2021.09.13.460111. [DOI] [Google Scholar]
- 17.Jayk Bernal A., Gomes da Silva M.M., Musungaie D.B., et al. Molnupiravir for Oral Treatment of Covid-19 in Nonhospitalized Patients. N Engl J Med. 2021 doi: 10.1056/NEJMoa2116044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dejmek M., Konkolova E., Eyer L., et al. Non-Nucleotide RNA-Dependent RNA Polymerase Inhibitor That Blocks SARS-CoV-2 Replication. Viruses. 2021;13 doi: 10.3390/v13081585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jin Y.H., Min J.S., Jeon S., et al. Lycorine, a non-nucleoside RNA dependent RNA polymerase inhibitor, as potential treatment for emerging coronavirus infections. Phytomedicine. 2021;86 doi: 10.1016/j.phymed.2020.153440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yin W., Luan X., Li Z., et al. Structural basis for inhibition of the SARS-CoV-2 RNA polymerase by suramin. Nat Struct Mol Biol. 2021;28:319–325. doi: 10.1038/s41594-021-00570-0. [DOI] [PubMed] [Google Scholar]
- 21.Zhang G.N., Zhao J., Li Q., et al. Discovery and optimization of 2-((1H-indol-3-yl)thio)-N-benzyl-acetamides as novel SARS-CoV-2 RdRp inhibitors. Eur J Med Chem. 2021;223 doi: 10.1016/j.ejmech.2021.113622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao J., Zhang Y., Wang M., et al. Quinoline and Quinazoline Derivatives Inhibit Viral RNA Synthesis by SARS-CoV-2 RdRp. ACS Infect Dis. 2021;7:1535–1544. doi: 10.1021/acsinfecdis.1c00083. [DOI] [PubMed] [Google Scholar]
- 23.Russo M., Moccia S., Spagnuolo C., et al. Roles of flavonoids against coronavirus infection. Chem Biol Interact. 2020;328 doi: 10.1016/j.cbi.2020.109211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lalani S., Poh C.L. Flavonoids as Antiviral Agents for Enterovirus A71 (EV-A71) Viruses. 2020;12 doi: 10.3390/v12020184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kaul, T. N.; Middleton, E., Jr.; Ogra, P. L., Antiviral effect of flavonoids on human viruses. J Med Virol 1985, 15 (1), 71–79. DOI: 10.1002/jmv.1890150110. [DOI] [PubMed]
- 26.Mouffouk C., Mouffouk S., Mouffouk S., et al. Flavonols as potential antiviral drugs targeting SARS-CoV-2 proteases (3CL(pro) and PL(pro)), spike protein, RNA-dependent RNA polymerase (RdRp) and angiotensin-converting enzyme II receptor (ACE2) Eur J Pharmacol. 2021;891 doi: 10.1016/j.ejphar.2020.173759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Muchtaridi M., Fauzi M., Khairul Ikram N.K., et al. Natural Flavonoids as Potential Angiotensin-Converting Enzyme 2 Inhibitors for Anti-SARS-CoV-2. Molecules. 2020;25 doi: 10.3390/molecules25173980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Di Petrillo A., Orru G., Fais A., Fantini M.C. Quercetin and its derivates as antiviral potentials: A comprehensive review. Phytother Res. 2022;36:266–278. doi: 10.1002/ptr.7309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Imran, M.; Thabet, H. K.; Alaqel, S. I.; Alzahrani, A. R.; Abida, A.; Alshammari, M. K.; Kamal, M.; Diwan, A.; Asdaq, S. M. B.; Alshehri, S., The Therapeutic and Prophylactic Potential of Quercetin against COVID-19: An Outlook on the Clinical Studies, Inventive Compositions, and Patent Literature. Antioxidants (Basel) 2022, 11 (5). DOI: 10.3390/antiox11050876. [DOI] [PMC free article] [PubMed]
- 30.Derosa G., Maffioli P., D'Angelo A., Di Pierro F. A role for quercetin in coronavirus disease 2019 (COVID-19) Phytother Res. 2021;35:1230–1236. doi: 10.1002/ptr.6887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Manjunath S.H., Thimmulappa R.K. Antiviral, immunomodulatory, and anticoagulant effects of quercetin and its derivatives: Potential role in prevention and management of COVID-19. J Pharm Anal. 2022;12:29–34. doi: 10.1016/j.jpha.2021.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Munafo F., Donati E., Brindani N., et al. Quercetin and luteolin are single-digit micromolar inhibitors of the SARS-CoV-2 RNA-dependent RNA polymerase. Sci Rep. 2022;12:10571. doi: 10.1038/s41598-022-14664-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schrödinger Release 2021-4: Protein Preparation Wizard; Epik, Schrödinger, LLC, New York, NY, 2021; Impact, Schrödinger, LLC, New York, NY; Prime, Schrödinger, LLC, New York, NY, 2021.
- 34.Schrödinger Release 2021-3: LigPrep, Schrödinger, LLC, New York, NY, 2021.
- 35.Friesner R.A., Banks J.L., Murphy R.B., et al. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 36.Halgren T.A., Murphy R.B., Friesner R.A., et al. Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J Med Chem. 2004;47:1750–1759. doi: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be made available on request.