

Abstract
The structural plasticity of the axon initial segment (AIS) contributes to the homeostatic control of activity and optimizes the function of neural circuits; however, the underlying mechanisms are not fully understood. In this study, we prepared a slice culture containing nucleus magnocellularis from chickens of both sexes that reproduces most features of AIS plasticity in vivo, regarding its effects on characteristics of AIS and cell-type specificity, and revealed that microtubule reorganization via activation of CDK5 underlies plasticity. Treating the culture with a high-K+ medium shortened the AIS and reduced sodium current and membrane excitability, specifically in neurons tuned to high-frequency sound, creating a tonotopic difference in AIS length in the nucleus. Pharmacological analyses revealed that this AIS shortening was driven by multiple Ca2+ pathways and subsequent signaling molecules that converge on CDK5 via the activation of ERK1/2. AIS shortening was suppressed by overexpression of dominant-negative CDK5, whereas it was facilitated by the overexpression of p35, an activator of CDK5. Notably, p35(T138A), a phosphorylation-inactive mutant of p35, did not shorten the AIS. Moreover, microtubule stabilizers occluded AIS shortening during the p35 overexpression, indicating that CDK5/p35 mediated AIS shortening by promoting disassembly of microtubules at distal AIS. This study highlights the importance of microtubule reorganization and regulation of CDK5 activity in structural AIS plasticity and the tuning of AIS characteristics in neurons.
SIGNIFICANCE STATEMENT The structural plasticity of AIS has a strong impact on the output of neurons and plays a fundamental role in the physiology and pathology of the brain. However, the mechanisms linking neuronal activity to structural changes in AIS are not well understood. In this study, we prepared an organotypic culture of avian auditory brainstem, reproducing most AIS plasticity features in vivo, and we revealed that activity-dependent AIS shortening occurs through the disassembly of microtubules at distal AIS via activation of CDK5/p35 signals. This study emphasizes the importance of microtubule reorganization and regulation of CDK5 activity in structural AIS plasticity and tonotopic differentiation of AIS structures in the brainstem auditory circuit.
Keywords: axon initial segment, Ca2+, CDK5, microtubules, p35, plasticity
Introduction
The axon initial segment (AIS) is a highly excitable axonal domain located near the soma and is involved in generating action potentials (Kole and Stuart, 2012). This excitable nature of AIS is attributed to its structural characteristics and the accumulation of voltage-gated Na+ (Nav) channels, which occurs through their interaction with a scaffold protein, ankyrinG, and tethering to the submembranous actin-spectrin meshwork at AIS (Leterrier, 2018). It is now evident that the distribution of AIS, such as length and distance from the soma, shows substantial variation among neurons (Kuba, 2012) and is regulated by neural activity in a homeostatic manner (Grubb et al., 2011), thereby adjusting the membrane excitability of individual neurons.
Structural variation and homeostatic regulation of AIS have been well studied in the nucleus magnocellularis (NM) of the chicken, which is a homolog of the mammalian anteroventral cochlear nucleus. NM neurons are arranged according to their characteristic frequency (CF) and differ in the AIS length along this tonotopic axis, with the length being shorter for higher CF, which optimizes the signal processing of the neurons in each tonotopic region (Kuba and Ohmori, 2009). In addition, the AIS of NM neurons becomes longer after cochlear damage, which maintains the integrity of auditory circuits without afferent inputs (Kuba et al., 2010). We recently revealed that activity-dependent regulation of AIS length also occurs during development; the AIS is shortened by afferent input, specifically in higher-CF neurons, creating a tonotopic difference in AIS length (Akter et al., 2020). As AIS shortening did not affect the periodicity of the submembranous meshwork or the signal intensity of AIS proteins, shortening would occur through reorganization of the distal AIS structure. Ca2+ entry through L-type voltage-gated Ca2+ (Cav) channels and the subsequent activation of calcineurin mediate AIS relocation in hippocampal neurons (Grubb and Burrone, 2010; Evans et al., 2013). However, the mechanisms linking neuronal activity to reorganization of the distal AIS structure remain elusive.
Cytoskeletons in AIS include the submembranous actin-spectrin meshwork and the cytosolic microtubule lattice (Leterrier, 2018). In addition, the localization of ankyrinG at the AIS is stabilized by its interaction with microtubules via end-binding proteins (EB1 and EB3; Fréal et al., 2016) and by its tethering to the extracellular matrix via neurofascin186 (Hedstrom et al., 2007), plasma membrane via the palmitoylation (He et al., 2012), and submembranous meshwork via β4-spectrin (Dzhashiashvili et al., 2007; Yang et al., 2007). Notably, microtubules have a polarized structure and grow and shrink predominantly from one of their ends (plus end) by regulating polymerization and depolymerization (Goodson and Jonasson, 2018). Furthermore, microtubules have a uniform orientation in the axon, with their plus ends facing the distal side. These features are preferable in explaining the reorganization of distal AIS structures, leading to the hypothesis that the disassembly of microtubules plays a role in AIS shortening.
Cyclin-dependent kinase 5 (CDK5) is a member of the CDK family of serine/threonine kinases. CDK5 is ubiquitously expressed in neurons, its activity is regulated by activator proteins p35 and p39, and it is involved in the microtubule reorganization (Shah and Lahiri, 2017). Moreover, activation and/or inhibition of CDK5 has been reported to affect AIS structure (Trunova et al., 2011; Evans et al., 2015; Klinman et al., 2017), suggesting its possible contribution to AIS shortening in NM neurons.
In this study, we explored the molecular mechanisms of AIS shortening using pharmacological screening and overexpression of genes in slice cultures of NM (Adachi et al., 2019). The cultured NM neurons reproduced most features of homeostatic AIS plasticity in vivo; an elevation of activity shortened the AIS length and lowered membrane excitability, specifically in high-CF regions, creating a tonotopic difference in AIS length. We revealed that AIS shortening occurred via destabilization of microtubules at the distal AIS because of activation of CDK5/p35, which was triggered by an elevation of [Ca2+]i and subsequent activation of multiple signaling molecules.
Materials and Methods
Animals
Chickens (Gallus domesticus) of either sex at embryonic day (E) 11 were used in the experiments. The care of experimental animals was in accordance with the regulations for animal experiments at Nagoya University, and the institutional committee approved the experiments.
Organotypic slice culture
The detailed procedure has been previously described (Adachi et al., 2019). Briefly, chick embryos were anesthetized by cooling eggs in ice-cold water, and the brainstem was dissected in high-glucose artificial CSF containing the following (in mm): 75 NaCl, 2.5 KCl, 26 NaHCO3, 1.25 NaH2PO4, 1 CaCl2, 3 MgCl2, and 100 glucose, pH 7.3, bubbled with 95% O2 and 5% CO2. Four to five coronal slices (200 µm) were obtained using a vibratome (VT1200, Leica; Fig. 1A–C). Slices containing high- or low-CF NM regions were collected, transferred onto a Millicell membrane insert (Millipore) in a culture dish (35 mm), and cultured for 10 d in vitro (DIV) in Neurobasal medium (Life Technologies) containing 2% B-27 serum-free supplement (Life Technologies), 1 mm glutamate solution (Life Technologies), and 1% penicillin-streptomycin solution (Wako). During the first 4 d, 5% fetal bovine serum (Biowest) was added, and half of the medium was changed twice a week. NM neurons were depolarized for 3 d from 7 DIV by adding KCl to the medium unless otherwise stated.
Figure 1.
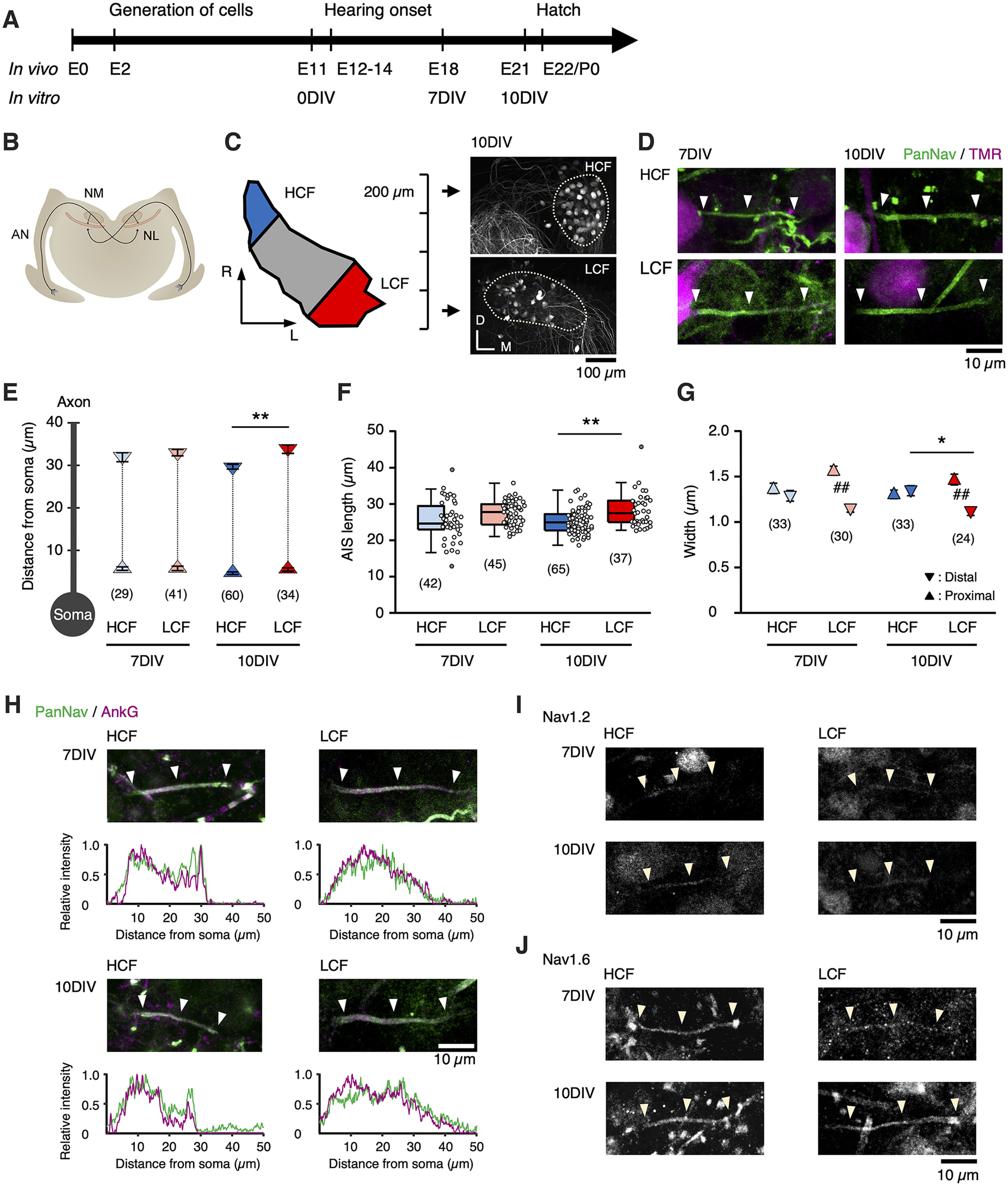
Structural differentiation of AIS in slice culture. A, Development of an avian auditory system in vivo and in vitro. B, Brainstem auditory circuit of chickens. AN, Auditory nerve; NL, nucleus laminaris. C, Left, NM is tonotopically organized along the rostromedial-caudolateral axis. In most rostral and caudal slices, NM was defined as high-CF (HCF) and low-CF (LCF) regions, respectively. Right, NM neurons were retrogradely labeled with dextran TMR (dotted line). R, Rostral; L, lateral; D, dorsal; M, medial. D, AIS immunostained with panNav antibody (green, arrowheads) after visualizing NM neurons (TMR, magenta) for HCF and LCF at 7 DIV and 10 DIV. E, Position of the proximal and distal ends of the AIS. F, G, Length (F) and widths at the proximal and distal ends of AIS (G). Values from individual cells are plotted (open circles) in this and the subsequent figures. H, Double immunostaining of panNav and ankyrinG. Intensity profiles of panNav and ankyrinG signals along the axon are shown. I, J, Immunostaining of Nav1.2 (I) and Nav1.6 (J). Numbers in parentheses indicate the number of cells; *p < 0.05, ** p < 0.01 between tonotopic regions by Kruskal–Wallis test; ##p < 0.01 between proximal and distal ends by Student's t test.
Pharmacology
All pharmacological experiments were performed at 7 DIV unless otherwise stated. The following reagents were used: DNQX, 8-Br-cAMP, nimodipine, 2-APB, KT5720, Rp-cAMPS, W13, doxycycline hyclate, taxol (paclitaxel), and nocodazole (Sigma-Aldrich); d-AP5 and ryanodine (Alomone Labs); TTX and cyclosporin A (Wako); 3-((2-methyl-4-thiazolyl)ethynyl)pyridine (MTEP) hydrochloride and LY367385 (Tocris Bioscience); ω-conotoxin GVIA and ω-agatoxin IVA (Peptide Institute); TTA-P2 (Merck); (S)-(–)-Bay-K-8644, AZD 6244, and PD98059 (Cayman Chemical); forskolin, PMA, GF109203X, TATCN21, roscovitine, okadaic acid, U0126, and jasplakinolide (Calbiochem); taccalonolide AJ and tubacin (MedChemExpress); and 4-aminopyridine (Nacalai). All reagents were added to the culture medium at least 1 h before KCl treatment at a concentration <10 times the half-maximal effective concentration.
Immunohistochemistry
NM neurons were labeled by injecting dextran (MW 3000) conjugated with tetramethylrhodamine (TMR; 10–40% in 0.1 m phosphate buffer adjusted to pH 2.0 with HCl, Life Technologies) into the midline tract region of cultured slices 2 h before fixation. The slices were fixed with a periodate-lysine-paraformaldehyde fixative (0.4% paraformaldehyde, 2.7% lysine HCl, 0.21% NaIO4, and 2.85 mm Na2HPO4) for 12 min at room temperature. Nonspecific binding of the antibodies was reduced by incubating the slices for 4 h with PBS containing 1% donkey serum, 0.05% carrageenan, and 0.3% Triton X-100. The primary antibodies used were as follows: mouse monoclonal panNav antibody (5 µg/ml; Sigma-Aldrich), guinea pig anti-chick polyclonal Nav1.6 antibody (0.49 µg/ml; Kuba et al., 2006), Nav1.2 antibody (0.68 µg/ml; Kuba et al., 2014), rabbit polyclonal ankyrinG antibody (5 µg/ml; Bouzidi et al., 2002), rabbit polyclonal TRITC (TMR) antibody (2.5 µg/ml; Life Technologies), rabbit polyclonal RFP antibody (5 µg/ml; Rockland), and rabbit polyclonal GFP antibody (×500; MBL). After overnight incubation with the primary antibodies at room temperature, the slices were incubated with secondary antibodies conjugated with Alexa Fluor (Life Technologies) for 2 h, mounted on a glass slide, coverslipped, and observed under a confocal laser-scanning microscope (FV1000, Olympus) with a 40×, 0.9 NA objective (Olympus). Serial sections were Z-stacked at a step of 0.8 µm. The distance from the soma and length of the AIS were measured as previously described (Akter et al., 2020). For the quantification of signal intensity, images were captured using the same microscope settings. Intensity profiles were created by measuring the signals along the axons within 50 µm of the soma. The background signal was subtracted.
Electrophysiology
A patch-clamp recording was also performed. The cultures were perfused with an ACSF containing the following (in mm): 125 NaCl, 2.5 KCl, 26 NaHCO3, 1.25 NaH2PO4, 2 CaCl2, 1 MgCl2, and 17 glucose, pH 7.3, when spikes were recorded under whole-cell or cell-attached mode. When sodium current was recorded, [Na+]o was decreased to 27.3 mm by isotonic replacement of Na+ with TEA+ (tetraethylammonium) for whole-cell mode (ENa = +25.8 mV) but not for outside-out mode (ENa = +69.8 mV), whereas [Ca2+]o was decreased to 0.5 mm, and 4-aminopyridine (0.2 mm), Cs+ (5 mm), CdCl2 (0.2 mm), NiCl2 (0.5 mm), SR95531 (10 μm), and DNQX (20 μm) were added for both modes. The pipettes were filled with a K+-based solution containing the following in (mm): 113 K-gluconate, 4.5 MgCl2, 0.1 EGTA, 14 Tris2-phosphocreatine, 4 Na2-ATP, 0.3 Tris-GTP, and 9 HEPES-KOH, pH 7.2, for spike recording, and a Cs+-based solution containing the following (in mm): 150 CsCl, 10 NaCl, 0.2 EGTA, and 10 HEPES-CsOH, pH 7.2, for sodium current recording. The electrode capacitance and series resistance (3–7 MΩ) were electronically compensated by up to 70%. The recording temperatures were 37–38°C for the current clamp and 20°C for the voltage clamp. The liquid junction potential (3.1–8.0 mV) was corrected after the experiments. The data were sampled at 100 kHz and low-pass filtered at 10 kHz.
Spikes were recorded without holding current by applying current pulses at intervals of 1–2 s in increments of 40 pA. The threshold current was defined as the minimum current required for spike generation and the threshold voltage as the voltage corresponding to an inflection point in the time derivative just above the threshold current. The steady-state inactivation curve of the sodium current was fitted to the Boltzmann equation, I/Imax = 1/{1 + exp[– (Vm – V1/2)/S]}, where I is the tail current amplitude, Imax is the maximum tail current amplitude, Vm is the membrane potential, V1/2 is the half-inactivation voltage, and S is the slope factor (Kuba and Ohmori, 2009; Akter et al., 2020).
Plasmid construction
The following plasmids were used: phSyn1-TetOn3G-WPRE, pTRE3G-tdTomato-WPRE, pTRE3G-tdTomato-P2A-dnCDK5-WPRE, pTRE3G-tdTomato-P2A-p35-WPRE, pTRE3G-tdTomato-P2A-p35(T138A)-WPRE, pTRE3G-tdTomato-P2A-CDK5-WPRE, and pTRE3G-mGreenLantern-P2A-CDK5-WPRE. All plasmids were constructed by inserting the following sequences into the plasmid backbone of pCAG-floxedSTOP-tdTomato-WPRE (Egawa and Yawo, 2019) using In-Fusion cloning (Takara Bio): hSyn1 promoter (pLenti Syn hChR2-EYFP-Nav1.2II-III; a gift from Matthew S. Grubb; Grubb and Burrone, 2010), TetOn3G and TRE3GS promoter (pTetOne Vector, catalog #634301, Clontech), mGreenLantern (LifeAct-mGreenLantern, plasmid catalog #164459, Addgene; a gift from Gregory Petsko; Campbell et al., 2020), CDK5 and dominant-negative CDK5 (Cdk5-HA and Cdk5-DN-HA, plasmid #1872 and #1873, Addgene; a gift from Sander van den Heuvel; van den Heuvel and Harlow, 1993), and p35 (pCMV-P35, plasmid #1347, Addgene; a gift from Li-Huei Tsai). Thr138 of p35 was replaced with Ala using PCR-based site-directed mutagenesis for p35(T138A). All constructs were verified by Sanger sequencing.
In ovo electroporation
The present procedure was slightly modified from the previous report (Egawa and Yawo, 2019). Briefly, a plasmid cocktail (0.4–0.5 µg/µl of each) was injected into the neural tube of chick embryos at E2 (Hamburger–Hamilton Stage 10–12; Hamburger and Hamilton, 1951) and introduced into the right side of the hindbrain (rhombomere 3–8). Electrical pulses were applied using a pair of electrodes (CUY613P1, Nepa Gene) placed 2 mm apart in parallel. The settings of the electroporator (NEPA21, Nepa Gene) were as follows: poring pulse, 15 V, 30 ms width, 50 ms interval, three pulses, 10% decay; transfer pulse, 5 V, 50 ms width, 50 ms interval, five pulses, 40% decay.
Quantitative RT-PCR
Detailed procedures have been described previously (Kuba et al., 2015). Cultures were incubated with normal or high-K+ medium for 1 (8 DIV) or 3 d (8–10 DIV), and NM tissues in the high-CF region were excised using a fine needle under observation with a dissecting microscope immediately after incubation. Tissues from four animals were pooled for each set of experiments. Total RNA was extracted using a NucleoSpin RNA Plus XS kit (Takara Bio). The quality and concentration of the extracted RNA were measured with an Agilent 2100 Bioanalyzer and NanoDrop2000C (Thermo Fisher Scientific), and cDNA was synthesized using the PrimeScript IV cDNA synthesis kit (Takara Bio). The mRNA levels of CDK5, p35, and GAPDH were quantified using the THUNDERBIRD SYBER qPCR Mix (Toyobo) with an Mx3005P QPCR system (Agilent). The primers used are listed in Table 1. Cycle threshold data of individual molecules were analyzed using GAPDH as an internal standard, and the ratio was calculated between normal and high-K+ media.
Table 1.
Primer sets used in this study
| Primer | Forward | Reverse |
|---|---|---|
| CDK5 | 5′-TGAAGGAGCTGAAGCACAAA-3′ | 5′-CACGATCTCAGGGTCCAGAT-3′ |
| p35 | 5′-GCCAAGAAGAAGAGCTCCAA-3′ | 5′-GGAGAGCGACTTCTTCAGGTT-3′ |
| GAPDH | 5′-CATCCAAGGAGTGAGCCAAG-3′ | 5′-TGGAGGAAGAAATTGGAGGA-3′ |
Statistics
The normality of data and equality of variance were evaluated using the Shapiro–Wilk test and F test, respectively. Statistical significance was determined using a two-tailed Student's t test for comparisons between the two groups. ANOVA or the Kruskal–Wallis test was used for analysis of variance, and the post hoc Tukey's test or Steel–Dwass was test for multiple pairwise comparisons. Values are presented as the mean ± SE. The level of significance was set at p < 0.05.
Results
Cultured NM neurons differed in AIS length among tonotopic regions
We examined the geometry of AIS in high-CF and low-CF regions in cultured NM for 7–10 DIV, corresponding to E 18–21, after visualizing the axon with a retrograde tracer and labeling the AIS with a panNav antibody (Fig. 1A–C). PanNav signals could be seen on retrogradely labeled axons of cultured NM neurons (Fig. 1D). These signals were colocalized with ankyrinG signals (Fig. 1H), confirming that they represented the AIS structure. The length of the AIS was slightly shorter for the high-CF region at 7 DIV, and the difference became significant by 10 DIV (Fig. 1F); it was 25 µm and 27 µm at 7 DIV, whereas it was 25 µm and 28 µm at 10 DIV for the high-CF and low-CF regions, respectively. The AIS length did not change further after 10 DIV during our observation until 14 DIV, indicating that 7 DIV would almost complete the maturation and differentiation of the AIS. Notably, the position of the proximal end of the AIS did not differ between the tonotopic regions regardless of days in culture, whereas that of the distal end was distributed more proximally for the high-CF region, specifically at 10 DIV (Fig. 1E). AIS thickness did not differ tonotopically at the proximal end. In contrast, it was slightly thinner in the low-CF region at the distal end (Fig. 1G). The primary subtype of Nav channels at the AIS was Nav1.6, regardless of age and tonotopic region (Fig. 1I, J). These results agree with in vivo observations (Akter et al., 2020).
Cultured NM neurons reproduced most features of AIS plasticity in vivo
NM neurons in culture show spontaneous synaptic activity in both tonotopic regions (Adachi et al., 2019). We then tested the contribution of these activities to the tonotopic differentiation of AIS length by adding DNQX (20 μm) and TTX (0.1 μm) to the medium for 3 d from 7 DIV. The blockade of spontaneous activity increased the AIS length, specifically in high-CF neurons, abolishing the tonotopic difference in AIS length (Fig. 2A–D); it was 28 µm and 30 µm for high-CF and low-CF neurons, respectively. In addition, when the spontaneous activity was elevated for 3 d from 7 DIV by increasing [K+] in the medium by two times (10.6 mm), the AIS length was shortened in the high-CF neurons (20 µm) but not in the low-CF neurons (31 µm; Fig. 2F–I). Elevating activities by blocking potassium channels with 4-aminopyridine (0.5 mm) caused a similar AIS shortening in high-CF neurons (20.4 ± 0.9 µm, n = 39). These findings confirmed that the activity-dependent reorganization of AIS occurs primarily in high-CF neurons, underlying the tonotopic differentiation of AIS length (Akter et al., 2020). Importantly, these manipulations did not affect the position of the proximal end (Fig. 2C,H), indicating that AIS shortening occurs via cytoskeletal reorganization at the distal end. The subtype of Nav channels did not change during the manipulations either (Fig. 2E,J), which also agreed with the findings in vivo.
Figure 2.
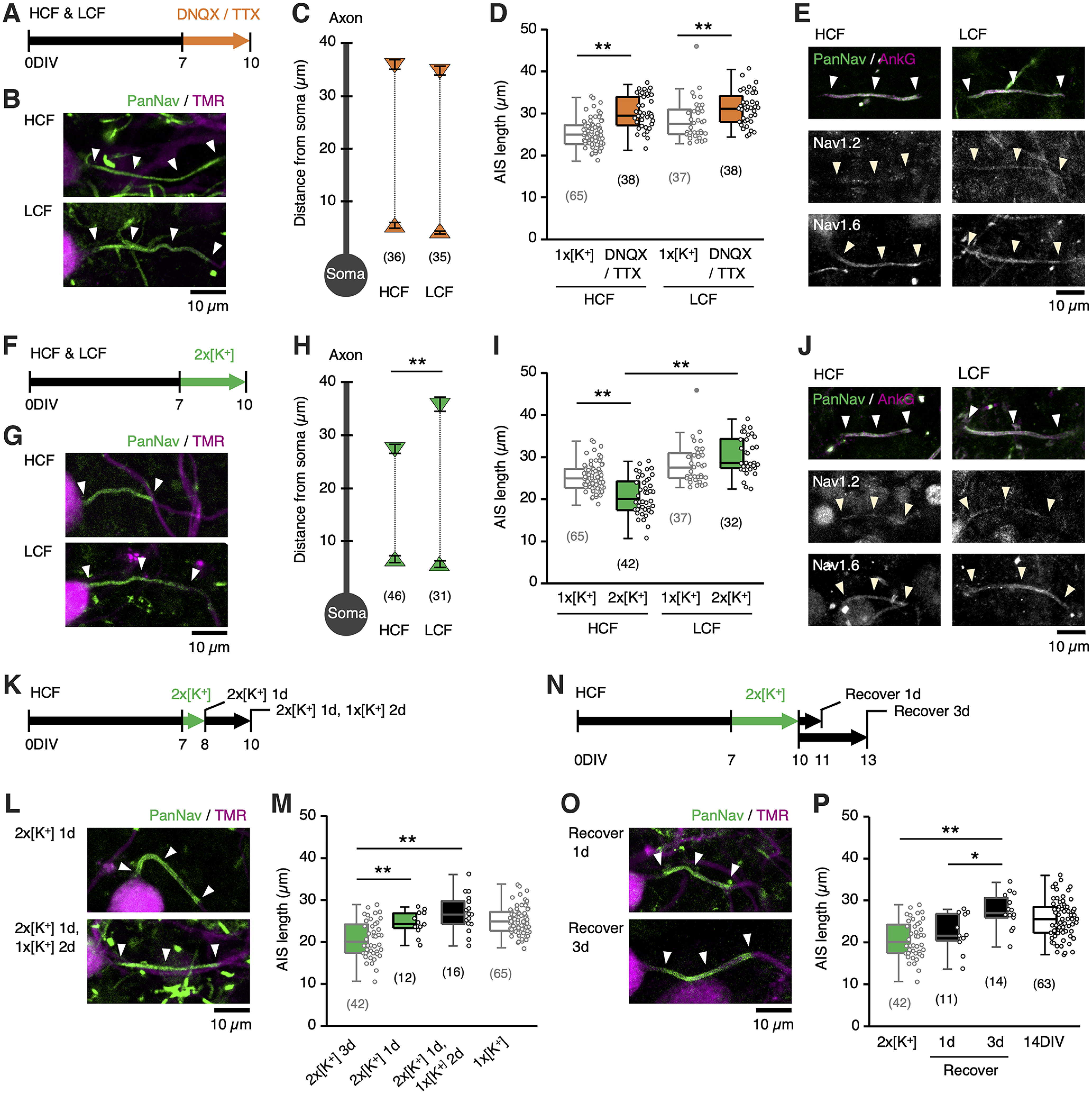
Activity-dependent AIS shortening in high-CF region. A–E, Synaptic and spike activity was blocked by DNQX/TTX for 7–10 DIV. Time course of experiment (A), AIS (green) of NM neurons (magenta) (B), position of proximal and distal ends (C), length (D) of AIS, and immunostaining of panNav and ankyrinG, Nav1.2, and Nav1.6 (E) for the HCF and LCF at 10 DIV. F–J, Membrane was depolarized by increasing [K+]o in the culture medium by two times (10.6 mm, 2 × [K+] medium) for 7–10 DIV. Time course of experiments (F), AIS of NM neurons (G), position of proximal and distal ends (H), length (I) of AIS, and immunostaining of panNav and ankyrinG, Nav1.2, and Nav1.6 (J) for HCF and LCF at 10 DIV. AIS length for the normal (1 × [K+]) medium (light gray) is from Figure 1F (10 DIV). K–M, HCF slices were cultured in 2 × [K+] medium for 1 d (7–8 DIV). Time course of experiments (K), AIS of NM neurons after 2 × [K+] treatment without (top) or with (bottom) subsequent incubation in 1 × [K+] medium for 2 d (L), and AIS length (M). N–P, HCF slices were incubated in 1 × [K+] medium for 1 or 3 d after 2 × [K+] treatment for 7–10 DIV. Time course of the experiment (N), AIS of NM neurons at 1 d (top) and 3 d (bottom) after 2 × [K+] treatment (O), and AIS length (P). The AIS length for the 2 × [K+] medium (green) is from Figure 2I. Numbers in parentheses indicate the number of cells. Arrowheads indicate the AIS; **p < 0.01 compared with control by Kruskal–Wallis test (D, I), between tonotopic regions by Student's t test (H); *p < 0.05, **p < 0.01 by one-way ANOVA and post hoc test (M, P).
We then characterized the progression of AIS shortening during high-K+ treatment in high-CF neurons (Fig. 2K–M). One-day treatment with a high-K+ medium did not change the AIS length (24 µm). Furthermore, an additional 2 d incubation in a normal medium did not shorten the AIS either (26 µm), suggesting that AIS shortening would require elevation of activities for substantial periods (>1 d). We also examined recovery from the shortening by incubating the cultures in a normal medium after 3 d of treatments with a high-K+ medium (Fig. 2N–P). The AIS returned to its original length within 3 d of incubation in the normal medium; the length was 22 µm and 27 µm for 1 and 3 d incubation, respectively, confirming the reversibility of the shortening.
We evaluated the effects of AIS shortening on the biophysical features of the high-CF neurons. We recorded the whole-cell sodium current under voltage clamp at –20 mV with a prepulse between −85 and −20 mV. The current amplitude was reduced by 40% in the high-K+-treated group compared with the control, whereas the voltage dependence of inactivation did not differ between the two groups (Fig. 3A–D). These results suggest that the same subtype of Nav channels mediates the current, supporting the immunohistochemical findings (Figs. 1I,J, 2J). Importantly, we could not detect sodium current in outside-out patches from the soma in both control (13 cells) and high-K+-treated (15 cells) groups (Fig. 3E), indicating that the reduction of whole-cell sodium current in the high-K+-treated group would primarily reflect a decrease of axonal Nav channels because of the AIS shortening. We also recorded action potentials in response to somatic current injection under current clamp, which revealed an elevation of threshold current, decreased amplitude, and maximum dV/dt in the high-K+-treated group. Despite a decreasing tendency, threshold voltage did not differ significantly between the groups (Fig. 3F–K). The absence of a change in threshold voltage might be related to the rich expression of Kv1 channels in the neurons (Kuba et al., 2015), which would facilitate the underestimation of threshold voltage at the AIS when monitored at the soma (Kole and Stuart, 2008). Notably, the control neurons showed a barrage of spontaneous synaptic and spike activities without stimuli, but these activities did not occur in the high-K+-treated neurons (Fig. 3L). As these activities were still more prominent in the control neurons under the blockade of Kv1 current with dendrotoxin (40 nm; Fig. 3M), the results indicated that AIS shortening by high-K+ treatment reduced sodium current and membrane excitability of neurons, thereby contributing to homeostatic control of activity in the circuit. Thus, cultured NM neurons reproduced most features of AIS plasticity in vivo and should be a good model for examining the molecular mechanisms of plasticity.
Figure 3.

AIS shortening reduced sodium current and membrane excitability. A, Time course of the experiments. HCF slices were cultured in 2 × [K+] medium for 7–10 DIV, and whole-cell recordings were made at 10 DIV. B, Whole-cell sodium currents were recorded at –20 mV with a prepulse (30 ms) from –85 mV to –20 mV. Control (left) and 2 × [K+] (right). C, Voltage dependence of inactivation. Values were fitted to the Boltzmann equation, and V1/2 was specified. D, Amplitude of sodium current. E, Outside-out sodium currents were not detected at –30 mV with a prepulse (30 ms) from –85 mV to –20 mV. Control (left) and 2 × [K+] (right). Membrane capacitance was 13.3 ± 1.1 pF (n = 10) and 13.8 ± 1.4 pF (n = 7) for whole-cell membrane from control and 2 × [K+], respectively (p = 0.73), whereas 2.4 ± 0.5 pF (n = 13) and 2.1 ± 0.3 pF (n = 15) for outside-out patches (p = 0.56). F, Spike responses to somatic current injection just above the threshold current for the control and 2 × [K+]. The injected current and membrane potential were specified for each trace. Arrowheads indicate thresholds. G, dV/dt and membrane potential relationship of the action potential in F. Arrowheads indicate the threshold voltage. H–K, Threshold current (H), threshold voltage (I), amplitude (J), and maximum dV/dt (K) of spikes. Resting membrane potential was –63.8 ± 0.7 mV (n = 17) and –67.1 ± 0.7 mV (n = 18; p < 0.01) for control and 2 × [K+], respectively. L, M, Spontaneous activities recorded under a current clamp without (L) and with (M) DTX (40 nm). Spontaneous spike bursts appeared in the control but not in the 2 × [K+]-treated neurons (10 s). The numbers in parentheses indicate the number of cells; *p < 0.05, **p < 0.01 by Student's t test.
Ca2+ entry via multiple pathways mediated AIS shortening
High-K+ treatment increases spontaneous synaptic input, which elevates [Ca2+]i in cultured NM neurons via activation of glutamate receptors and/or Cav channels (Adachi et al., 2019; Fig. 4B). We explored the triggers of AIS shortening during high-K+ treatment using specific inhibitors of these receptors and channels in high-CF neurons (Fig. 4A–D). Inhibition of ionotropic glutamate receptors with DNQX (20 μm) and AP-5 (50 μm) suppressed AIS shortening (25 µm). These receptors cause Ca2+ influx not only by permeating Ca2+ but also by activating Cav channels via depolarization. L-type and T-type channels are localized preferentially at postsynaptic membranes, whereas P/Q-type and N-type Cav channels are known to locate at presynaptic as well as postsynaptic membranes (Catterall, 2000), which were consistent with the effects of specific blockers on spontaneous spikes in the culture (Fig. 4G,H) and the previous observations in the neurons (Koyano et al., 1996; Lu and Rubel, 2005). Inhibitors of these Cav channels occluded the effects of high-K+ treatment when they were applied separately, and the effects were maximized when applied as a cocktail (Fig. 4D). The lengths were 27, 28, 28, 28, and 31 µm for inhibitors of L- (nimodipine, 10 μm), P/Q- (ω-agatoxin IVA, 0.2 μm), N- (ω-conotoxin GVIA, 2 μm), T-type (TTA-P2, 2 μm) channels, and a cocktail, respectively. In contrast, an activator of L-type Cav channels (Bay-K, 1 μm) caused AIS shortening (23 µm) in the normal medium (i.e., without high-K+ treatment; Fig. 4E), confirming the importance of elevation of [Ca2+]i in AIS shortening.
Figure 4.
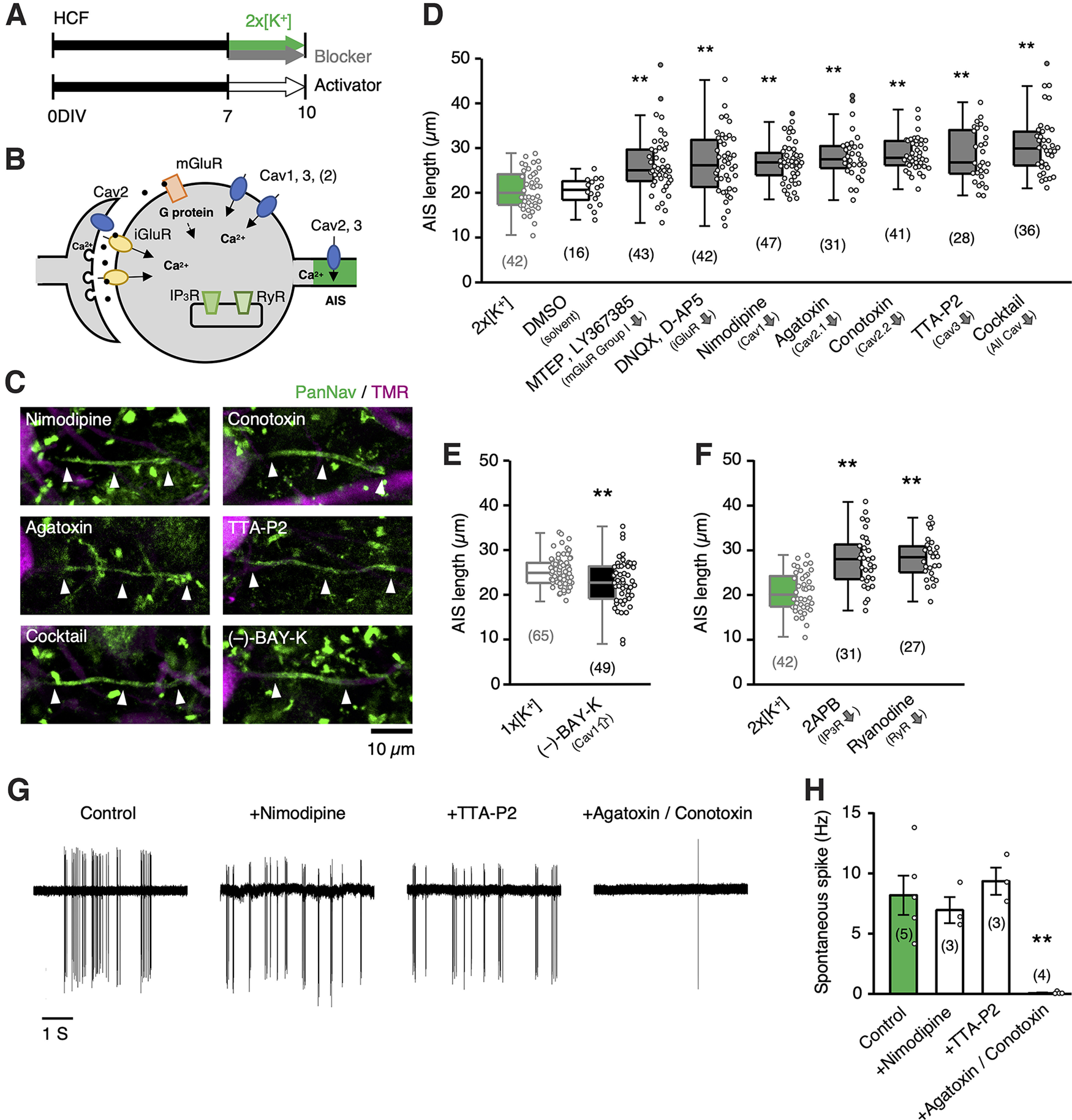
AIS shortening occurred via activation of glutamate receptors and Cav channels. A, Time course of the experiments. HCF slices were incubated with blockers of glutamate receptors, Cav channels in a 2 × [K+] medium, or with an activator in a normal (1 × [K+]) medium for 7–10 DIV. B, Schematic drawing of Ca2+ sources in NM neurons. C, AIS of NM neurons. D–F, Length of AIS. The numbers in parentheses indicate the number of cells. The AIS lengths for 1 × [K+] (light gray) and 2 × [K+] (green) media are from Figures 1F (10 DIV) and 2I, respectively. G, Effects of Cav channel blockers on spontaneous spikes recorded under a cell-attached clamp in ACSF containing 10 mm KCl. H, Spontaneous spikes are occluded by a cocktail of P/Q- and N-type Cav channel blockers; **p < 0.01 compared with 2 × [K+] (D, F) by Kruskal–Wallis test, control (H) by one-way ANOVA and post hoc test, or by Student's t test (E).
The inhibition of group I metabotropic glutamate receptors (mGluRs) with MTEP (50 nm) and LY367385 (20 μm) also reduced the effects of high-K+ treatment (26 µm; Fig. 4D). In addition, inhibitors of both inositol trisphosphate (IP3) receptors (2-APB, 50 μm) and ryanodine receptors (ryanodine, 50 μm) showed similar effects, with a length of 28 µm and 28 µm, respectively (Fig. 4F). These results are supported by the expression of group I mGluRs in neurons (Zirpel et al., 2000) and may emphasize the importance of [Ca2+]i elevation in AIS shortening.
Intracellular signals of AIS shortening
We explored intracellular signals of AIS shortening downstream of Ca2+ by testing the effects of kinase inhibitors during high-K+ treatment in the culture (Fig. 5A,B). To our surprise, AIS shortening was sensitive to multiple kinase inhibitors (Fig. 5C,D), with a length of 26 µm with KT5720 (0.5 μm), 28 µm with Rp-cAMPS (100 μm) for protein kinase A (PKA), 29 µm with GF109203X (50 nm) for protein kinase C (PKC), and 30 µm with TATCN21 (5 μm) for calmodulin-dependent kinase II (CaMKII), suggesting substantial cross talk among the kinases. However, AIS shortening was less sensitive to phosphatase inhibitors; the length was 22 µm with okadaic acid (20 nm) for PP1/PP2A, and 26 µm with cyclosporin A (50 nm) for PP2B (Fig. 5F). PKA and CaMKII were activated via calmodulin (CaM) in a Ca2+-dependent manner (Xia and Storm, 2005). Consistently, inhibition of CaM with W13 (50 μm) blocked the effects of high-K+ treatment (26 µm; Fig. 5D). Moreover, activation of either PKA or PKC alone mimicked the effects of high-K+ treatment (Fig. 5C,G); the AIS length was 21 µm for forskolin (10 μm, adenylate cyclase activator) and 21 µm for PMA (0.5 μm, PKC activator). These results confirmed the involvement of PKA/PKC/CaMKII in AIS shortening.
Figure 5.
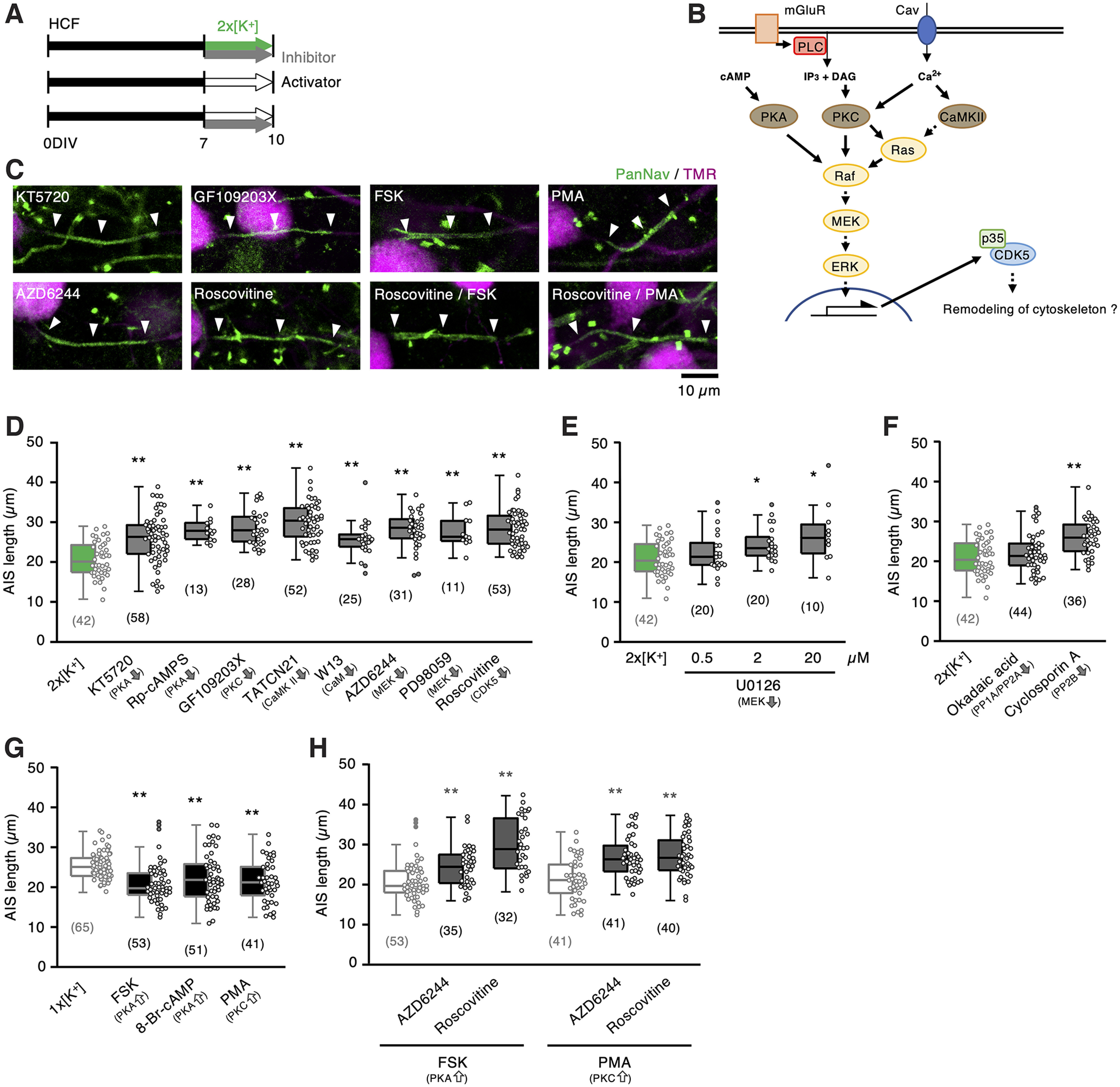
AIS shortening occurred via activation of MEK and CDK5 pathways. A, Time course of the experiments. HCF slices were incubated with kinase inhibitors in a 2 × [K+] medium or with activators in a normal (1 × [K+]) medium for 7–10 DIV. B, MEK signaling pathway. C, AIS of NM neurons. D–F, Length of AIS. Effects of kinase inhibitors (D), concentration dependence of U0126 (E), and phosphatase inhibitors (F), respectively in the 2 × [K+] medium. Control (2 × [K+], green) is from Figure 2I. G, Activators of PKA and PKC shortened AIS in the 1 × [K+] medium. Control (1 × [K+], light gray) is from Figure 1F. H, MEK or CDK5 inhibitors occluded AIS shortening by forskolin (FSK) or PMA. AIS lengths for FSK and PMA alone (light gray) are from Figure 5G. Numbers in parentheses indicate the number of cells; *p < 0.05, **p < 0.01 compared with 2 × [K+] (D–F) by one-way ANOVA and post hoc test, 1 × [K+] (G), and FSK or PMA alone (H) by Kruskal–Wallis test.
These kinases can activate the extracellular signal-regulated kinase (ERK1/2) pathway in neurons (Miningou and Blackwell, 2020), and ERK1/2 is known as an upstream molecule of CDK5 (Shah and Lahiri, 2017). Thus, we hypothesized that ERK1/2 integrates PKA/PKC/CaMKII signals and mediates AIS shortening via CDK5 activation. Inhibition of either mitogen-activated protein kinase kinase (MEK1/2) or CDK5 suppressed AIS shortening during the high-K+ treatment (Fig. 5C,D), with a length of 28 µm with AZD6244 (10 μm) for MEK1/2 and 28 µm with roscovitine (2 μm) for CDK5. Other MEK1/2 inhibitors showed similar dose-dependent effects (Fig. 5D,E); the length was 23 µm (0.5 μm), 25 µm (2 μm), and 27 µm (20 μm) with U0126, and 28 µm with PD98059 (100 μm). More importantly, these inhibitors occluded AIS shortening during the activation of either PKA or PKC (Fig. 5C,H); the lengths were 24 µm and 30 µm for AZD6244 and roscovitine, respectively, in the presence of forskolin, and 27 µm and 27 µm in the presence of PMA. These results support the hypothesis that ERK1/2 and CDK5 contribute to the shortening of AIS downstream of PKA/PKC/CaMKII in NM neurons.
Activation of CDK5 was required for AIS shortening
To confirm the involvement of CDK5 in AIS shortening, we introduced dominant-negative CDK5 (dnCDK5) in right-sided NM neurons by in ovo electroporation at E2 and overexpressed the genes under Tet-On control in high-CF regions in the culture, whereas doxycycline (DOX, 2 μm) was added for 4 d in the high-K+ medium (Fig. 6A,B). The overexpression of dnCDK5 occluded the AIS shortening during the high-K+ treatment with little changes in the maximum intensity of panNav signals at the AIS (Fig. 6C,D,L); the length was 28 µm in dnCDK5-positive neurons, whereas it was 24 µm for neurons in the opposite (nonelectroporated) side and 22 µm for those expressing tdTomato alone.
Figure 6.

AIS shortening occurred in a manner dependent on CDK5 activity. A, Time course of the experiments. Plasmids were introduced into NM neurons at E2, HCF slices were prepared at E 11, and DOX was added to the culture medium at 6–10 DIV. B, tdTomato (red) was expressed in NM neurons (ipsi) in slice culture stained with panNav antibody (white). Dotted line indicates the midline. C–K, AIS of NM neurons with (ipsi) or without (contra) overexpression of dnCDK5 in 2 × [K+] medium (C), and of CDK5 (E), p35 (G), p35(T138A) (I), or CDK5 and p35 (K) in normal (1 × [K+]) medium. Plasmids used are shown in C–K. Note the absence of Nav signals at AIS in CDK5 and p35 double-positive neurons (K, left). Intensity profiles of Nav signal are the average of 10 cells (D, F, H, J). L, M, Length of AIS in 2 × [K+] (L) and 1 × [K+] (M) media. Numbers in parentheses indicate the number of cells. N, Ratio of mRNA level of CDK5 and p35 between 2 × [K+] and 1 × [K+] media. Numbers in parentheses represent the number of experiments in N. O, AIS length of NM neurons from LCF with (ipsi) or without (contra) overexpression of p35 in normal (1 × [K+]) medium. Plasmid in G was used; *p < 0.05, **p < 0.01 compared with mock by Student's t test (L, N) and one-way ANOVA and post hoc test (M). Ipsi, Ipsilateral; contra, contralateral.
Overexpression of CDK5 did not affect the AIS length (28 µm), whereas overexpression of p35 caused AIS shortening (23 µm) in normal medium (Fig. 6E–H,M). Notably, a mutation in the phosphorylation site of p35(T138A) occluded AIS shortening (30 µm; Fig. 6I,J,M). In addition, overexpression of p35 and CDK5 eliminated AIS (34 of 34 cells; Fig. 6K,M), suggesting the importance of CDK5/p35 activity in regulating AIS length in the high-CF neurons. In contrast, in low-CF neurons, overexpression of p35 did not affect AIS length (Fig. 6O); the lengths were 28 µm and 30 µm for control and overexpression of p35, respectively, indicating that molecules downstream of CDK5 would differ between high- and low-CF neurons and are responsible for the tonotopic difference in the AIS length in NM. As high-K+ treatment did not affect the mRNA levels of CDK5 and p35 in NM (Fig. 6N), posttranscriptional regulation of these molecules might be involved in the process.
Microtubule reorganization contributed to AIS shortening
Microtubules contribute to the assembly of AIS structures by anchoring ankyrinG via EB1 and EB3 (Leterrier et al., 2011; Fréal et al., 2016). Thus, we tested the possibility that CDK5 mediates AIS shortening via the disassembly of microtubules. We incubated the cultures with microtubule-stabilizing agents, taxol (50 nm), and taccalonolide AJ (50 nm), and found that these microtubule stabilizers suppressed the AIS shortening during the high-K+ treatment in the high-CF neurons (Fig. 7A–D); the AIS length was 27 µm with taxol and 28 µm with taccalonolide AJ. AIS shortening was also occluded (25 µm) by tubacin (0.1 μm), an inhibitor of HDAC6, an enzyme that destabilizes microtubules via deacetylation of tubulin. In addition, taxol occluded AIS shortening by overexpression of p35 (29 µm; Fig. 7F,G,I, left) or by the activators of PKA (28 µm) or PKC (29 µm; Fig. 7D, right). Notably, taxol occluded the elimination of AIS after the overexpression of p35 together with CDK5 (22 µm; Fig. 7F,H,I, right), consistent with the idea that the elimination was attributed to the facilitation of AIS shortening rather than the toxicity of strong CDK5/p35 signals. Moreover, the inhibition of PP1/PP2A by okadaic acid, which promotes phosphorylation of p35 at T138 (Kamei et al., 2007), shortened AIS (22 µm), and this AIS shortening was suppressed by taxol (29 µm; Fig. 7B,E), suggesting that phosphorylation of p35 at T138 underlies AIS shortening via interaction with microtubules. We also treated the cultures with a microtubule destabilizing agent, nocodazole (10 μm), which completely eliminated AIS, confirming the importance of microtubule integrity in maintaining AIS structure. In contrast, jasplakinolide, which promotes actin polymerization and stabilization, did not occlude (22 µm) AIS shortening in the high-K+ medium (Fig. 7C,D, left).
Figure 7.

CDK5 mediated AIS shortening via reorganization of microtubules. A, Time course of the experiments. HCF slices were incubated with stabilizers of microtubules (MT) or actin during treatment with 2 × [K+] medium, FSK or PMA, or okadaic acid for 7–10 DIV. B, Pharmacological manipulation of microtubule dynamics. C, Effects of MT and actin filament stabilizers on AIS of NM neurons in 2 × [K+] medium. D, E, MT stabilizers occluded AIS shortening by 2 × [K+] medium (D, left), by FSK, PMA (D, right), or by okadaic acid (E). AIS lengths for 2 × [K+] (green), FSK or PMA alone (light gray), and normal (1 × [K+]) medium (light gray) were from Figures 2I, 5D, and 1F (10 DIV), respectively; **p < 0.01 compared with 2 × [K+] (D, left), 1 × [K+] (E) by one-way ANOVA and post hoc test, FSK or PMA alone (D, right) by Kruskal–Wallis test. F–I, Taxol occluded AIS shortening by overexpression of p35 (G) or p35 together with CDK5 (H). Time course of experiments (F) and AIS length (I). p35 and p35 together with CDK5 are from Figure 6M; **p < 0.01 by Student's t test. Numbers in parentheses indicate the number of cells.
Discussion
In the slice culture of NM, AIS shortening occurred in high-CF neurons but not in low-CF neurons during high-K+ treatment, creating a tonotopic difference in AIS length, as observed in vivo (Kuba and Ohmori, 2009). As spontaneous synaptic activity and Ca2+ influx did not differ between the neurons during high-K+ treatment (Adachi et al., 2019), the results implied that the ability of AIS plasticity is determined intrinsically in a cell-specific manner and is equipped explicitly in high-CF neurons (Akter et al., 2020). The cell-type specificity of AIS plasticity has also been reported in other brain regions, such as the olfactory bulb, sensory cortex, and hippocampus (Grubb and Burrone, 2010; Gutzmann et al., 2014; Chand et al., 2015). AIS shortening progressed over time in the order of days via reorganization of the distal AIS structure, which reduced the membrane excitability of neurons, suggesting its involvement in homeostatic control of neural activity. Thus, structural AIS plasticity in cultured NM neurons reproduces most features of AIS plasticity in vivo (Kuba et al., 2010). With accessibility to pharmacological and genetic manipulations, slice culture of NM would be a powerful tool for exploring the molecular mechanisms of AIS plasticity.
Ca2+ pathways of AIS plasticity
Relocation of AIS occurs via Ca2+ entry through L-type Cav channels in dissociated neurons from the hippocampus and olfactory bulb (Grubb and Burrone, 2010; Evans et al., 2013; Chand et al., 2015). Because these Cav channels are preferentially localized in somatodendritic domains (Catterall, 2000), these findings suggest the contribution of global [Ca2+]i within neurons to AIS plasticity. However, AIS shortening in NM neurons occurs via multiple Ca2+ pathways, such as Ca2+ entry through several types of Cav channels and glutamate receptors, and Ca2+ release from Ca2+ stores, confirming the importance of global [Ca2+]i elevation within neurons for AIS shortening. The variation in Ca2+ pathways among studies might be related to differences in the characteristics of neurons (i.e., composition of channels and receptors), phenotype of AIS plasticity (relocation vs shortening), and/or preparation (dissociated vs organ culture). The present observations also suggested a possibility that local [Ca2+]i at the AIS is involved in the AIS shortening as P/Q-, N-, and T-type Cav channels are known to localize at the AIS (Bender and Trussell, 2009; Yu et al., 2010), whereas cisterna organelle, a Ca2+ store at the AIS, is coupled with T-type Cav channels and elevates [Ca2+]i at the AIS (Lipkin et al., 2021).
The AIS length roughly correlated with the level of activity and/or depolarization in the cultured NM neurons; it became shorter during high-K+ treatment and longer in the presence of DNQX and TTX (Fig. 2), which agreed with our previous observations in vivo (Kuba et al., 2010). This may indicate that the AIS length is determined by [Ca2+]i; the length is longer for lower [Ca2+]i. Blockade of the single Ca2+ pathway occluded AIS shortening and even elongated AIS, whereas blockade of multiple Ca2+ pathways showed only a minor additional effect. These results may indicate that the machinery of AIS shortening has a steep relationship with [Ca2+]i; it is highly sensitive to [Ca2+]i and is easily saturated with a slight change in [Ca2+]i. This idea is compatible with our previous observation that [Ca2+]i was elevated to only ∼200 nm by depolarization corresponding to that during the high-K+ treatment (Adachi et al., 2019). Of note, 1 d treatment with the high-K+ medium did not cause AIS shortening even 3 d after the treatment. These results indicate that long-lasting changes in basal Ca2+ levels are required for structural AIS changes, which are preferable for the homeostatic control of neural activity in a circuit.
Multiple signaling pathways of AIS plasticity
One surprising finding of this study was that multiple kinase inhibitors occluded AIS shortening. Although these inhibitors are known to affect molecules other than the target at higher concentrations (Bain et al., 2003), we do not think that the results were attributable to the nonspecific effects of these drugs for the following reasons. First, the effects depended on the drug concentration, and the concentration of inhibitors was kept as low as possible during the experiments (see above, Materials and Methods). Second, multiple inhibitors could reproduce the effects of specific molecules. Third, activators caused the opposite effects. Thus, the present results would indicate multiple pathways for AIS shortening and strong cross talk among these pathways while converging on ERK1/2 and CDK5. Although major signals in physiological schemes still need to be determined, the presence of multiple pathways and their cross talk would be advantageous in reliably inducing AIS reorganization and adjusting activity at an appropriate level in various situations.
Roles of CDK5 in AIS plasticity
Several signaling molecules have been reported to regulate AIS structure and/or protein localization. Among them, formin2, GSK3β, and pMLC are known to affect AIS length, and their inhibition causes the shortening of AIS (Tapia et al., 2013; Berger et al., 2018; Zhang et al., 2021). Importantly, however, the AIS shortening during inhibition of these molecules resulted from the uniform reduction of ankyrinG signals along the AIS and progressed rather rapidly (within hours). In contrast, AIS shortening in NM occurs without changes in the signal intensity of AIS proteins and requires much longer periods (a few days). These results indicate that the reorganization of AIS structure in NM is restricted to the distal part of AIS and includes rate-limiting steps, suggesting the involvement of different mechanisms in the process.
We found that CDK5 activation is critical for inducing AIS shortening in NM. Overexpression of CDK5 alone did not shorten AIS, whereas overexpression of p35 caused substantial AIS shortening in the culture. This is compatible with reports that ERK1/2 can upregulate both CDK5 and p35, but CDK5 is ubiquitously expressed, and its activity is primarily regulated by the level of p35 expression (Shah and Lahiri, 2014). It is important to note that activation of CDK5 promoted AIS shortening to a different extent among tonotopic regions, with the effects being more prominent in high-CF neurons, which would underlie the tonotopic difference in AIS length in NM. In addition, the effects of CDK5 on AIS length were opposite in previous reports; in the Drosophila mushroom body, activation of CDK5 increased the length of AIS-like structures (Trunova et al., 2011), whereas inhibition of CDK5 decreased the AIS length in dissociated hippocampal neurons (Evans et al., 2015; Klinman et al., 2017). What then underlies the variation in CDK5-mediated effects on AIS length among neurons? AIS length decreases because of reorganization of the distal AIS structure in NM (Akter et al., 2020), and CDK5 mediates this process via the disassembly of microtubules (Fig. 7). CDK5/p35 may regulate microtubule remodeling in pleiotropic manner depending on autophosphorylation (Shah and Lahiri, 2017). CDK5 phosphorylates p35 at S8, allowing its translocation from the plasma membrane and interaction with microtubules for microtubule polymerization (Hou et al., 2007; Asada et al., 2012). However, CDK5 also phosphorylates p35 at T138, preventing this interaction and inhibiting microtubule polymerization (He et al., 2008), whereas T138 is dephosphorylated by phosphatases PP1 and PP2A (Kamei et al., 2007), which agreed with the positive and negative effects on the AIS shortening by okadaic acid and p35(T138A), respectively, in high-CF NM neurons (Figs. 6, 7). Therefore, one possible explanation for the discrepancy in CDK5-mediated effects is that the levels of these phosphatases differ among neurons, being lower in NM neurons, suppressing microtubule polymerization and facilitating AIS shortening. Importantly, substrates of CDK5 include those that regulate microtubule organization, either positively or negatively. For example, phosphorylation of doublecortin at S297 negatively regulates its microtubule-binding affinity, promoting the depolymerization of microtubules (Tanaka et al., 2004). In contrast, phosphorylation of ndel1 facilitates dynein-mediated retrograde transport and promotes the polarization of microtubules, leading to elongation of the AIS (Klinman et al., 2017). Thus, another possible explanation is that CDK5 is differentially coupled with its substrates in individual neurons, creating cell-specific effects of CDK5 on AIS length. We found that inhibition of HDAC6 by tubacin occluded AIS shortening (Fig. 7). HDAC6 interacts with EB1 (Zilberman et al., 2009), which mediates both capping of microtubule plus ends and association of ankyrinG with microtubules at the AIS (Leterrier et al., 2011; Fréal et al., 2016). Moreover, HDAC6 interferes with forming AIS via the hyperacetylation of tubulins (Tapia et al., 2010). Therefore, it is important to determine whether HDAC6 is involved in AIS shortening downstream of CDK5 in NM.
Footnotes
This work was supported by Ministry of Education, Culture, Sports, Science, and Technology Grants 19H04747 and 21H02577 (H.K.) and the Takeda Science Foundation (H.K.). We thank the Division for Medical Research Engineering, Nagoya University Graduate School of Medicine, for use of NanoDrop 2000, Agilent 2100 Bioanalyzer, and the Mx3005P qPCR system.
The authors declare no competing financial interests.
References
- Adachi R, Yamada R, Kuba H (2019) Tonotopic differentiation of coupling between Ca2+ and Kv1.1 expression in brainstem auditory circuit. iScience 13:199–213. 10.1016/j.isci.2019.02.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akter N, Fukaya R, Adachi R, Kawabe H, Kuba H (2020) Structural and functional refinement of the axon initial segment in avian cochlear nucleus during development. J Neurosci 40:6709–6721. 10.1523/JNEUROSCI.3068-19.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asada A, Saito T, Hisanaga S (2012) Phosphorylation of p35 and p39 by Cdk5 determines the subcellular location of the holokinase in a phosphorylation-site-specific manner. J Cell Sci 125:3421–3429. [DOI] [PubMed] [Google Scholar]
- Bain J, McLauchlan H, Elliott M, Cohen P (2003) The specificities of protein kinase inhibitors: an update. Biochem J 371:199–204. 10.1042/BJ20021535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender KJ, Trussell LO (2009) Axon initial segment Ca2+ channels influence action potential generation and timing. Neuron 61:259–271. 10.1016/j.neuron.2008.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger SL, Leo-Macias A, Yuen S, Khatri L, Pfennig S, Zhang Y, Agullo-Pascual E, Caillol G, Zhu M, Rothenberg E, Melendez-Vasquez CV, Delmar M, Leterrier C, Salzer JL (2018) Localized myosin II activity regulates assembly and plasticity of the axon initial segment. Neuron 97:555–570.e6. 10.1016/j.neuron.2017.12.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouzidi M, Tricaud N, Giraud P, Kordeli E, Caillol G, Deleuze C, Couraud F, Alcaraz G (2002) Interaction of the Nav1.2a subunit of the voltage-dependent sodium channel with nodal ankyrinG. In vitro mapping of the interacting domains and association in synaptosomes. J Biol Chem 277:28996–29004. 10.1074/jbc.M201760200 [DOI] [PubMed] [Google Scholar]
- Campbell BC, Nabel EM, Murdock MH, Lao-Peregrin C, Tsoulfas P, Blackmore MG, Lee FS, Liston C, Morishita H, Petsko GA (2020) mGreenLantern: a bright monomeric fluorescent protein with rapid expression and cell filling properties for neuronal imaging. Proc Natl Acad Sci U S A 117:30710–30721. 10.1073/pnas.2000942117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall WA (2000) Structure and regulation of voltage-gated Ca2+ channels. Annu Rev Cell Dev Biol 16:521–555. 10.1146/annurev.cellbio.16.1.521 [DOI] [PubMed] [Google Scholar]
- Chand AN, Galliano E, Chesters RA, Grubb MS (2015) A distinct subtype of dopaminergic interneuron displays inverted structural plasticity at the axon initial segment. J Neurosci 35:1573–1590. 10.1523/JNEUROSCI.3515-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzhashiashvili Y, Zhang Y, Galinska J, Lam I, Grumet M, Salzer J (2007) Nodes of Ranvier and axon initial segments are ankyrin G-dependent domains that assemble by distinct mechanisms. J Cell Biol 177:857–870. 10.1083/jcb.200612012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egawa R, Yawo H (2019) Analysis of neuro-neuronal synapses using embryonic chick ciliary ganglion via single-axon tracing, electrophysiology, and optogenetic techniques. Curr Protoc Neurosci 87:e64. 10.1002/cpns.64 [DOI] [PubMed] [Google Scholar]
- Evans MD, Sammons RP, Lebron S, Dumitrescu AS, Watkins TBK, Uebele VN, Renger JJ, Grubb MS (2013) Calcineurin signaling mediates activity-dependent relocation of the axon initial segment. J Neurosci 33:6950–6963. 10.1523/JNEUROSCI.0277-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans MD, Dumitrescu AS, Kruijssen DLH, Taylor SE, Grubb MS (2015) Rapid modulation of axon initial segment length influences repetitive spike firing. Cell Rep 13:1233–1245. 10.1016/j.celrep.2015.09.066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fréal A, Fassier C, Le Bras B, Bullie E, De Gois S, Hazan J, Hoogenraad CC, Couraud F (2016) Cooperative interactions between 480 kDa ankyrin-G and EB proteins assemble the axon initial segment. J Neurosci 36:4421–4433. 10.1523/JNEUROSCI.3219-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodson HV, Jonasson EM (2018) Microtubules and microtubule-associated proteins. Cold Spring Harb Perspect Biol 10:a022608. 10.1101/cshperspect.a022608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubb MS, Burrone J (2010) Activity-dependent relocation of the axon initial segment fine-tunes neuronal excitability. Nature 465:1070–1074. 10.1038/nature09160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubb MS, Shu Y, Kuba H, Rasband MN, Wimmer VC, Bender KJ (2011) Short- and long-term plasticity at the axon initial segment. J Neurosci 31:16049–16055. 10.1523/JNEUROSCI.4064-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutzmann A, Ergül N, Grossmann R, Schultz C, Wahle P, Engelhardt M (2014) A period of structural plasticity at the axon initial segment in developing visual cortex. Front Neuroanat 8:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamburger V, Hamilton HL (1951) A series of normal stages in the development of the chick embryo. J Morphol 88:49–92. [PubMed] [Google Scholar]
- He L, Hou Z, Qi RZ (2008) Calmodulin binding and cdk5 phosphorylation of p35 regulate its effect on microtubules. J Biol Chem 283:13252–13260. 10.1074/jbc.M706937200 [DOI] [PubMed] [Google Scholar]
- He M, Jenkins P, Bennett V (2012) Cysteine 70 of ankyrin-G is s-palmitoylated and is required for function of ankyrin-G in membrane domain assembly. J Biol Chem 287:43995–44005. 10.1074/jbc.M112.417501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedstrom KL, Xu X, Ogawa Y, Frischknecht R, Seidenbecher CI, Shrager P, Rasband MN (2007) Neurofascin assembles a specialized extracellular matrix at the axon initial segment. J Cell Biol 178:875–886. 10.1083/jcb.200705119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Z, Li Q, He L, Lim HY, Fu X, Cheung NS, Qi DX, Qi RZ (2007) Microtubule association of the neuronal p35 activator of Cdk5. J Biol Chem 282:18666–18670. 10.1074/jbc.C700052200 [DOI] [PubMed] [Google Scholar]
- Kamei H, Saito T, Ozawa M, Fujita Y, Asada A, Bibb JA, Saido TC, Sorimachi H, Hisanaga S (2007) Suppression of calpain-dependent cleavage of the CDK5 activator p35 to p25 by site-specific phosphorylation. J Biol Chem 282:1687–1694. 10.1074/jbc.M610541200 [DOI] [PubMed] [Google Scholar]
- Klinman E, Tokito M, Holzbaur ELF (2017) CDK5-dependent activation of dynein in the axon initial segment regulates polarized cargo transport in neurons. Traffic 18:808–824. 10.1111/tra.12529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kole MHP, Stuart GJ (2008) Is action potential threshold lowest in the axon? Nat Neurosci 11:1253–1255. 10.1038/nn.2203 [DOI] [PubMed] [Google Scholar]
- Kole MHP, Stuart GJ (2012) Signal processing in the axon initial segment. Neuron 73:235–247. 10.1016/j.neuron.2012.01.007 [DOI] [PubMed] [Google Scholar]
- Koyano K, Funabiki K, Ohmori H (1996) Voltage-gated ionic currents and their roles in timing coding in auditory neurons of the nucleus magnocellularis of the chick. Neurosci Res 26:29–45. 10.1016/0168-0102(96)01071-1 [DOI] [PubMed] [Google Scholar]
- Kuba H (2012) Structural tuning and plasticity of the axon initial segment in auditory neurons. J Physiol 590:5571–5579. 10.1113/jphysiol.2012.237305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuba H, Ohmori H (2009) Roles of axonal sodium channels in precise auditory time coding at nucleus magnocellularis of the chick. J Physiol 587:87–100. 10.1113/jphysiol.2008.162651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuba H, Ishii TM, Ohmori H (2006) Axonal site of spike initiation enhances auditory coincidence detection. Nature 444:1069–1072. 10.1038/nature05347 [DOI] [PubMed] [Google Scholar]
- Kuba H, Oichi Y, Ohmori H (2010) Presynaptic activity regulates Na+ channel distribution at the axon initial segment. Nature 465:1075–1078. 10.1038/nature09087 [DOI] [PubMed] [Google Scholar]
- Kuba H, Adachi R, Ohmori H (2014) Activity-dependent and activity-independent development of the axon initial segment. J Neurosci 34:3443–3453. 10.1523/JNEUROSCI.4357-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuba H, Yamada R, Ishiguro G, Adachi R (2015) Redistribution of Kv1 and Kv7 enhances neuronal excitability during structural axon initial segment plasticity. Nat Commun 6:8815. 10.1038/ncomms9815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leterrier C (2018) The axon initial segment: an updated viewpoint. J Neurosci 38:2135–2145. 10.1523/JNEUROSCI.1922-17.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leterrier C, Vacher H, Fache M, d'Ortoli SA, Castets F, Autillo-Touati A, Dargent B (2011) End-binding proteins EB3 and EB1 link microtubules to ankyrin G in the axon initial segment. Proc Natl Acad Sci U S A 108:8826–8831. 10.1073/pnas.1018671108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipkin AM, Cunniff MM, Spratt PWE, Lemke SM, Bender KJ (2021) Functional microstructure of Cav-mediated calcium signaling in the axon initial segment. J Neurosci 41:3764–3776. 10.1523/JNEUROSCI.2843-20.2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Rubel EW (2005) Activation of metabotropic glutamate receptors inhibits high-voltage-gated calcium channel currents of chicken nucleus magnocellularis neurons. J Neurophysiol 93:1418–1428. 10.1152/jn.00659.2004 [DOI] [PubMed] [Google Scholar]
- Miningou N, Blackwell KT (2020) The road to ERK activation: do neurons take alternate routes? Cell Signal 68:109541. 10.1016/j.cellsig.2020.109541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah K, Lahiri DK (2014) Cdk5 activity in the brain—multiple paths of regulation. J Cell Sci 127:2391–2400. 10.1242/jcs.147553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah K, Lahiri DK (2017) A tale of the good and bad: remodeling of the microtubule network in the brain by cdk5. Mol Neurobiol 54:2255–2268. 10.1007/s12035-016-9792-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T, Serneo FF, Tseng HC, Kulkarni AB, Tsai LH, Gleeson JG (2004) Cdk5 phosphorylation of doublecortin Ser297 regulates its effect on neuronal migration. Neuron 41:215–227. 10.1016/s0896-6273(03)00852-3 [DOI] [PubMed] [Google Scholar]
- Tapia M, Wandosell F, Garrido JJ (2010) Impaired function of HDAC6 slows down axonal growth and interferes with axon initial segment development. PLOS One 5:e12908. 10.1371/journal.pone.0012908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapia M, Del Puerto A, Puime A, Sánchez-Ponce D, Fronzaroli-Molinieres L, Pallas-Bazarra N, Carlier E, Giraud P, Debanne D, Wandosell F, Garrido JJ (2013) GSK3 and β-catenin determines functional expression of sodium channels at the axon initial segment. Cell Mol Life Sci 70:105–120. 10.1007/s00018-012-1059-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trunova S, Baek B, Giniger E (2011) Cdk5 regulates the size of an axon initial segment-like compartment in mushroom body neurons of the Drosophila central brain. J Neurosci 31:10451–10462. 10.1523/JNEUROSCI.0117-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Heuvel S, Harlow E (1993) Distinct roles for cyclin-dependent kinases in cell cycle control. Science 262:2050–2054. 10.1126/science.8266103 [DOI] [PubMed] [Google Scholar]
- Xia Z, Storm DR (2005) The role of calmodulin as a signal integrator for synaptic plasticity. Nat Rev Neurosci 6:267–276. 10.1038/nrn1647 [DOI] [PubMed] [Google Scholar]
- Yang Y, Ogawa Y, Hedstrom KL, Rasband MN (2007) βIV spectrin is recruited to axon initial segments and nodes of Ranvier by ankyrinG. J Cell Biol 176:509–519. 10.1083/jcb.200610128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Maureira C, Liu X, McCormick D (2010) P/Q and N channels control baseline and spike-triggered calcium levels in neocortical axons and synaptic boutons. J Neurosci 30:11858–11869. 10.1523/JNEUROSCI.2651-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Ciorraga M, Mendez P, Retana D, Boumedine-Guignon N, Achón B, Russier M, Debanne D, Garrido JJ (2021) Formin activity and mDia1 contribute to maintain axon initial segment composition and structure. Mol Neurobiol 58:6153–6169. 10.1007/s12035-021-02531-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilberman Y, Ballestrem C, Carramusa L, Mazitschek R, Khochbin S, Bershadsky A (2009) Regulation of microtubule dynamics by inhibition of the tubulin deacetylase HDAC6. J Cell Sci 122:3531–3541. 10.1242/jcs.046813 [DOI] [PubMed] [Google Scholar]
- Zirpel L, Janowiak MA, Taylor DA, Parks TN (2000) Developmental changes in metabotropic glutamate receptor-mediated calcium homeostasis. J Comp Neurol 421:95–106. [DOI] [PubMed] [Google Scholar]