

Abstract
This review describes, from a chemical point of view, the top “blockbuster” small molecule orphan drugs according to their forecasted sales in 2026. Orphan drugs are intended for the treatment, prevention, or diagnosis of a rare disease or condition. These molecules are mostly addressed to the treatment of rare forms of cancer. The respiratory and central nervous systems represent other common therapeutic subcategories. This work will show how the orphan drugs market has significantly grown and will account for a consistent part of prescriptions by 2026.
Keywords: FDA, EMA, orphan drugs, synthesis, new therapies
1. Introduction
Orphan drugs, by definition, are intended for the treatment, prevention, or diagnosis of a rare disease (RD) or condition [1]. For the Food and Drug Administration (FDA), “supporting the development and evaluation of new treatments for rare diseases is a key priority” [2].
According to the European Medicine Agency (EMA), Between 5000 and 8000 distinct rare diseases exist [3]. In the early 1980s, rare diseases accounted for 5000 types [4].
The Greek-derived term “orphan disease” is used to designate diseases that affect only small numbers of individuals [5]. To date, there is no unique and clear definition of an orphan disease. In the USA, it is defined as one that affects fewer than 200,000 individuals; in Japan, the number of patients having such a disease is 50,000, while Australia sets its limit at 200 [6].
The European Union and the USA have implemented legislation to stimulate the development of drugs for patients suffering from rare diseases. The European Parliament with the Council of 16 December 1999 on Orphan Medicinal Products [7] and the American Orphan Drug Act (1983) [8], followed by the Orphan Drug Regulation (1993) [9], are clear examples of such legislation.
Companies and other drug developers can request an orphan drug designation, and, in the case of the USA, the FDA will grant such a designation if the drug meets specific criteria [10]. Among the benefits of an orphan drug designation are the eligibility for a federal research grant, a grant of 7-year market exclusivity, and up to a 50% tax credit (until 2018) for clinical trials that meet the requirements [2]. From 2018 onwards, the tax credit was reduced from 50% to 25% by the Trump Administration [11]. Having a significant medical benefit is one of the key criteria for the application of the regulation.
2. Top Selling Orphan Drugs (Forecast 2026)
The development and evaluation of new treatments for rare diseases are one of the priorities of pharmaceutical agencies (FDA/EMA). They can grant an orphan drug designation to a drug or biological product to prevent, diagnose, or treat a rare disease or condition. This designation guarantees certain advantages both in terms of tax breaks and for periods of market exclusivity after approval. The majority of orphan diseases possess genetic origins and are life-threatening and/or chronically debilitating [12]. Most new drug modalities first make their mark in rare diseases, as these conditions are often genetically defined.
In addition to their genetic roots, orphan diseases can be caused by helminth, protozoan, and bacterial infections or even by environmental poisoning [13].
From an ethical point of view, the orphan drug concept is intended to help patients suffering from rare conditions. From an economic point of view, pharmaceutical companies can benefit, given the above-mentioned incentives and relaxed regulations.
Novel drug discovery approaches, from gene editing to AI-powered screening and drug design, will play an important role in the development of new orphan drugs [14].
Orphan drug sales growth significantly outpaces that of the wider pharmaceutical market, and Big Pharma’s fortunes are ever more closely linked to orphan drugs.
In support of this evidence, it has to be mentioned that sales have increased about 10% per year between 2005 and 2011 [13], and it is estimated that each of the top 10 “blockbuster” orphans in 2026 will be worth between $3 billion and $13 billion [14].
In 2026, among the top-selling orphan drugs (small molecules) are ibrutinib (brand name Imbruvica®), elexacaftor/tezacaftor/ivacaftor (brand name Trikafta®), olaparib (brand name Lynparza®), ruxolitinib (brand name Jakafi®), venetoclax (brand name Venclexta®), acalabrutinib (brand name Calquence®), and tafamidis (brand name Vyndaqel®) [14] (Figure 1).
Figure 1.

Top selling orphan drugs sales (bn $, forecasted sales for 2026) [14].
The forecast for €mbruvica® is outstanding, not only from the sales point of view but also because the European market will surpass the traditionally dominant USA market.
The goal of this review is to shed more light on the chemistry and sales of orphan blockbusters. Synthetic preparation and therapeutic use of each of these drugs will be discussed.
2.1. Ibrutinib (Brand Name Imbruvica®)
Bruton tyrosine kinase (BTK) signaling plays a fundamental role in B-cell development and in immunoglobulin synthesis. Ibrutinib (10) is an orally bioavailable BTK inhibitor and irreversibly binds to BTK at the Cysteine-481 residue [14]. It exhibits a low nanomolar potency (0.5 nM) against BTK [15]. Ibrutinib has been approved by the FDA for the treatment of several tumors, such as mantle cell lymphoma, chronic lymphocytic leukemia (CLL), Waldenstrom’s macroglobulinemia, marginal zone lymphoma, and chronic graft-versus-host disease in allogeneic stem cell transplantation [16,17].
Its acrylamide moiety reacts with C481 to form a covalent bond in the BTK kinase domain [18]. Ibrutinib was discovered at Celera Genomics. In 2006, Pharmacyclics (today part of AbbVie) initiated the preclinical and clinical development of ibrutinib. In December 2011, Pharmacyclics and the Janssen division of Johnson & Johnson entered into an agreement to jointly develop and market ibrutinib [19].
The synthesis of ibrutinib will b” discussed herein. Two main routes [19], small-laboratory scale and industrial, will be detailed.
The small-scale route [20,21,22,23,24,25] starts from 4-phenoxybenzoyl chloride (1), which is reacted with malononitrile 2 to afford alkenol 3. This is methylated upon the addition of trimethylsilyldiazomethane to yield derivative 4. The addition of hydrazine to 4 yielded the amino-pyrazole 5, which is, in turn, converted to pyrazolo-pyrimidine 6 via adding formamide at 180 °C. The subsequent Mitsunobu reaction between 6 and (S)-tert-butyl 3-hydroxypiperidine-1-carboxylate 7 afforded derivate 8 with a corresponding inversion of configuration at the alcoholic carbon center. The BOC-removal from 8 gave the free piperidine derivative 9. This was acylated with acryloyl chloride to provide ibrutinib (10, Scheme 1). This pathway allowed chemists to explore different heterocycle via Mitsunobu and different substituents at the free N-piperidine moiety.
Scheme 1.

Medicinal chemistry route to ibrutinib (10) [20,21,22,23,24,25].
An alternative pathway (Scheme 2) with fewer steps involves the preparation of synthon 6 in only two steps. The initial 1H-pyrazolo[3,4-d]pyrimidin-4-amine 11 is iodinated using N-iodosuccinimide (NIS) in DMF to afford 12. This was subjected to microwave-assisted Suzuki-Miyaura coupling involving boronic acid 13 and [1,1′-Bis(diphenylphosphino)ferrocene]dichloropalladium(II), Pd(dppf)Cl2] as a catalyst to afford 6. This is then subjected to the Mitsunobu procedure, as seen in Scheme 1.
Scheme 2.

Alternative synthesis for synthon 6.
The industrial route [26,27] is differentiated from the academic by avoiding the use of non-green Mitsunobu chemistry and expensive (S)-tert-butyl 3-hydroxypiperidine-1-carboxylate.
Moreover, it avoids the use of hydrazine hydrate, a suspected carcinogen that decomposes at higher temperatures.
The synthesis (Scheme 3) starts from the dinitrile 4, which is transformed into pyrazole 15 via adding CBZ-protected€)-hydrazine 14. Then, 15 was transformed into pyrazolo-pyrimidine 17 by mixing it with formamidine 16 at 120 °C. The subsequent removal of the CBZz-protecting group afforded 9 in good yield and purity after recrystallization in methanol (80% yield and 92.5% purity). Finally, 9 was used for subsequent acylation.
Scheme 3.

Industrial route [26,27] to intermediate 9 used in the preparation of ibrutinib.
The forecasted sales for ibrutinib for 2026 are stunning. It is not, therefore, surprising that alternative synthetic pathways have appeared. Among them, and important to be mentioned, is the convergent Mitsunobu/displacement route [28,29,30]. In this route, a Mitsunobu reaction is performed at an earlier stage than the installation of the diphenyl ether group.
2.2. Elexacaftor/Tezacaftor/Ivacaftor (Brand Name Trikafta®)
Launched by Vertex Pharmaceuticals [14], elexacaftor (31)-tezacaftor(48)-ivacaftor(62) is a newly approved triple-combination cystic fibrosis transmembrane conductance regulator (CFTR) [31]. Cystic fibrosis (CF) is the most common, life-limiting autosomal recessive disease in Caucasians and is caused by defects in the production of the CFTR ion channel [32]. The CFTR is a tunnel-shaped chloride channel responsible for controlling the transport of anions and water in and out of epithelial cells [33].
Small-molecule modulators that directly interact with CFTR can aid in protein folding (“correctors”) and/or increasing channel function (“potentiators”) [34]. A recent study in 2022 has shown that when used alone, elexacaftor partially corrected interdomain assembly defects in phenylalanine deleted (Δ508) CFTR, but when combined with a type I corrector (e.g., tezacaftor), it did so fully [35].
2.2.1. Elexacaftor
Elexacaftor has been described as both a corrector and potentiator of CFTR [36]. A first synthetic pathway (Scheme 4) was elaborated by Vertex [31,34,36]. It developed from the reduction of 3,3,3-trifluoro-2,2-dimethylpropanoic acid 18 to alcohol 19, which is used in the Mitsunobu condensation of pyrazolone 21, which is prepared from met€ (E)-3-methoxyacrylate 20 upon the addition of hydrazine hydrate and subsequent BOC protection. Mitsunobu product 22 was subjected to BOC deprotection with HCl to afford hydrochloric salt 23.
Scheme 4.

Intermediate 23 was used under alkaline conditions in the nucleophilic addition to t-Bu ester 25 (prepared from 2,6-dichloropyridine-3-carboxylic acid 24) to yield 26. Deprotection of t-Bu ester 26 by using HCl at reflux generated free carboxylic acid 27. This was activated by 1,1′-carbonyldiimidazole (CDI) and coupled with sulfonamide 28 to afford acyl sulfonamide 29, which was used in the last step, the nucleophilic addition of pyrrolidine salt 30 in the presence of potassium carbonate to produce elexacaftor (31). The authors claim that the overall yield for the seven convergent steps was 29%, with the most problematic step being the Mitsunobu condensation (57%).
An alternative pathway [37] (Scheme 5), again patented by Vertex, employed fewer steps. The starting material is a halogenated nicotinamide derivative 32.
Scheme 5.

Alternative, more expeditious preparation [37] of elexacaftor (31).
SNAr reaction of 32 with pyrrolidine 30 in acetonitrile and K2CO3 afforded amide 33, which was made nucleophilic by treatment with lithium tert-amoxide in 2-MeTHF as a solvent and treated with sulfonyl chloride 34 to yield acyl sulfonamide 35. The eventual copper-catalyzed reaction between 35 and 23 afforded elexacaftor 31. Alternatively, synthon 33 was first converted into pyrazole derivate 36 in a Buchwald–Hartwig amination employing third generation palladacycle catalyst (tBuXPhos Pd G3). Derivate 36 was lastly converted into elexacaftor 31 via the same conditions (lithium tert-amoxide in 2-MeTHF as a solvent, treated with sulfonyl chloride 34) used to synthesize intermediate 35.
2.2.2. Tezacaftor
Tezacaftor, is a CFTR corrector [32]. Its function is to correct the positioning of the CFTR protein on the cell surface to permit proper channel formation and an improved flow of water and salts across the cell membrane [38]. Tezacaftor is approximately 99% bound to plasma proteins, mainly albumin [39].
Its synthesis (Scheme 6) [33,40,41] starts from 2-bromo-5-fluoro-4-nitroaniline 37. Sonogashira coupling [Bis(triphenylphosphine)palladium(II) dichloride] between 37 and alkyne 38 gave disubstituted alkyne 39, which was subjected to cyclization under Pd(Cl)2 conditions to afford indole 40. The indolic NH was alkylated with tosylate 41 in the presence of cesium carbonate to yield 42 and the undesired 43 (the transesterification product). Compound 42 was treated with LiAlH4 to yield the corresponding primary alcohol 44. This underwent nitro reduction to yield indolamine 45. Then, the nucleophilic attack of 45 to acyl chloride 46 in the presence of triethylamine afforded 47. In the last step, the removal of the acetal protecting group afforded tezacaftor 48 in a moderate yield (47%) after chromatography.
Scheme 6.

An alternative pathway for tezacaftor (Scheme 7) involves the initial opening of (S)-epoxide 49 by 2-bromo-5-fluoro-4-nitroaniline (37) catalyzed by zinc perchlorate and 4 Å molecular sieves to afford intermediate 50. Catalytic hydrogenation (platinum catalyst) of the nitro group followed by treatment with p-toluenesulfonic acid generated the anilinium salt 51. The corresponding free base of 51 was subjected to Sonogashira coupling in copper-free conditions with alkyne 52 to yield disubstituted alkyne 53. Indole formation catalyzed by Pd(Cl)2 in acetonitrile afforded intermediate 54. The aromatic amine group of 54 was coupled with acyl chloride 46 in the presence of triethylamine to yield cyclopropyl amide 55. Benzyl groups of 55 were removed under a hydrogen atmosphere (H2, Pd/C)) to generate tezacaftor (48). Yield for the last two steps ranged from 68% to 84% after crystallization from 2-PrOH/heptane [42]. This pathway affords the final product in better yields than those previously shown in Scheme 6.
Scheme 7.

2.2.3. Ivacaftor
Ivacaftor (62) is a potentiator (it enhances the chloride transport) of the cystic fibrosis transmembrane conductance regulator (CFTR), and it is the first drug to be licensed for use that treats an underlying cause of cystic fibrosis [43]. Similar to tezacaftor, ivacaftor is also metabolized extensively in humans. In vitro and in vivo data indicate that ivacaftor is metabolized primarily by CYP3A4 and CYP3A5 [39,44].
Its synthesis (Scheme 8) was first reported by Vertex [45]. The initial 2,4-di-tert-butylphenol (56) was protected with methyl chloroformate at its phenolic function to yield methyl carbonate 57. The nitration afforded mainly derivate 58 (the undesired 6-nitro regioisomer is obtained in a ratio of 1:8). Hydrolysis of 58 provided phenol 59, which underwent reduction of the nitro group under Pd/ammonium formate conditions to yield precursor 60. This was eventually transformed into ivacaftor (62) via condensation with quinoline 61 upon HATU/TEA activation.
Scheme 8.

Preparation of ivacaftor (62) [45].
Protection of the phenol as the electron-withdrawing carbonate likely also minimizes the ortho/para-directing effect of the oxygen substituent, allowing nitration to occur primarily ortho/para to the tert-butyl groups [32].
An alternative pathway (Scheme 9) [46] encompasses similar steps but in a different order. In particular, the hydrolysis of the carbonate was carried out as the final step. Nitro compound 58 was reduced to aniline 63. This was condensed with quinoline 61 upon activation of the carboxylic acid with propanephosphonic acid anhydride 64 to yield precursor 65. This was eventually transformed after alkaline hydrolysis into ivacaftor 62. No yields were provided for these reported steps. The protection of the phenol as the electron-withdrawing carbonate is likely to minimize its ortho/para-directing effect, as seen in the previous synthesis.
Scheme 9.

Alternative preparation of ivacaftor (62) [46].
2.3. Olaparib
Olaparib (72, brand name Lynparza®) was approved by the FDA for the first-line maintenance treatment of breast cancer susceptibility gene (BRCA)-mutated (BRCAm) advanced ovarian cancer [47]. Olaparib belongs to the N-acyl piperazines class of compounds. The action of olaparib relies on the poly(ADP-ribose) polymerase PARP inhibition [48]. Poly(ADP-ribose) polymerases (PARPs) are a family of enzymes that catalyze the addition of poly(ADP-ribose) subunits onto themselves and other acceptor proteins and are involved in DNA repair [49]. The IC50 value towards PARP of olaparib is 6 nM [50]
Several synthetic strategies have appeared for the preparation of olaparib. Among these, the strategy disclosed by Scinopharm Taiwan [51] will be detailed. The authors of [51] state that the process was improved in terms of yield.
Nitrile derivate 69 (Scheme 10) is a key intermediate to form the phthalazine-one, from which two different pathways are built to the final product, olaparib.
Scheme 10.

Preparation of intermediate 69 [51].
Compound 69 is obtained by first reacting benzofuran 66 with dimethyl phosphonate to yield derivative 67. This was used in the subsequent Horner–Wadsworth–Emmons (HWE) reaction with aldehyde 68 to yield alkene 69.
In the first strategy (Scheme 11) [52], nitrile 69 is converted into the corresponding carboxylic acid by alkaline hydrolysis, and the phthalazine-one is formed upon the addition of hydrazine (compound 70). This is subjected to condensation mediated by (2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU) followed by removal of the BOC group upon addition of HCl to provide the piperazine-containing amide 71. This was eventually treated with cyclopropanecarbonyl chloride and DIPEA as a base to afford olaparib (72).
Scheme 11.

Preparation of olaparib (72) from synthon 69 [52].
In the second strategy (Scheme 12) [53], the carboxylic acid 70, previously obtained from 69, is treated directly with cyclopropyl(piperazin-1-yl)methanone 73 and HBTU to afford olaparib (72).
Scheme 12.

Preparation of olaparib (72) from synthon 69 [53].
2.4. Ruxolitinib (Brand Name Jakafi®)
Ruxolitinib (73) is an orally administered, first-in-class Janus Kinase (JAK) 1 and 2 inhibitor that was recently approved for the treatment of patients with polycythemia vera (PV) [54], which is a rare slow-growing blood cancer characterized by increased erythrocyte production with a poorly understood etiology; IC50 values towards JAK1 and JAK2 are of 2.7, 4.5 nM, respectively [55].
In 2019, the FDA approved ruxolitinib for steroid-refractory acute graft-versus-host disease (SR-aGVHD) [56].
Ruxolitinib is a small molecule composed of a substituted pyrazole linked to a deazapurine core. The retrosynthetic strategy for the preparation of ruxolitinib suggests that the most straightforward and intuitive strategy is linking the deazapurine core with the N-substituted pyrazole (Scheme 13) through a Suzuki reaction [57].
Scheme 13.

Retrosynthetic strategy for the preparation of ruxolitinib (73).
Haydl and co-workers, as an example, used a rhodium-catalyzed regioselective synthesis of pyrazole, which is the crucial synthon for the preparation of ruxolitinib (Scheme 14) [58]. Pyrazole 75 was added to terminal allene 74 to yield enantioenriched allylic pyrazole 76 (90% ee). The reaction was catalyzed by rhodium with ligand JoSPOPhos. The additional presence of pyridinium p-toluenesulfonate (PPTS, 0.2 equiv) played a significant role in obtaining the desired N1 product selectively. The subsequent hydroboration (9-borabicyclo[3.3.1]nonane or 9-BBN) followed by oxidation (H2O2) provided the primary alcohol 77. Swern oxidation of 77 afforded aldehyde 78, in turn, converted into nitrile 79 by the addition of ammonium hydroxide and iodine and purification by recrystallization from heptane. The installation of boron in the place of the bromide group was mediated by bis(pinacolato)diboron (B2pin2) and [1,1′-Bis(diphenylphosphino)ferrocene]palladium(II) dichloride ([PdCl2(dppf)] to yield boronic ester 80. Eventually, Suzuki coupling [catalyzed by bis(triphenylphosphine)palladium chloride] of 80 with chloro-azapurine 81 provided ruxolitinib (73). The yield of the final product (over two steps) was 81%.
Scheme 14.

Preparation of ruxolitinib (73) via rhodium-catalyzed regioselective synthesis of pyrazole. Structures of [{Rh(cod)Cl}2], JoSPOPhos SL-J688-2, and 9-BBN are reported in blue boxes.
Despite the elegant strategy, the reactions performed to synthesize the pyrazole moiety required high amounts of expensive and complex chiral catalysts [59]. Therefore, a more recent patent (see Scheme 15) [60] overcame these issues and made the process more scalable. The selected starting material was cyclopentanecarbaldehyde 82, which was converted to €-alkene 83 in a Knoevenagel reaction involving malonic acid and piperidine/pyridine as a base. This was treated with hydrazine to afford cyclopentyl-pyrazolidinone 84, which was resolved with di-p-toluoyl-L-tartaric acid (L-DTTA) to yield pure enantiomer 85. On the other hand, the 6-methyl-7-deazapurine derivative 86 was treated with Vilsmeier reagent to yield aldehyde 87. This was used with 85 in the presence of alkaline media to yield carboxylic acid precursor 88 of ruxolitinib. This was first treated with oxalyl chloride and ammonia to form the primary amide and later dehydrated to a nitrile upon using POCl3 to yield ruxolitinib. This route allowed the obtainment of 73 in a good yield, avoiding the use of complex and expensive catalysts.
Scheme 15.

Preparation of ruxolitinib (73) suitable for industrial production. Di-p-toluoyl-L-tartaric acid (L-DTTA) is shown in the blue box [60].
2.5. Venetoclax (Brand Name Venclexta®)
Venetoclax (103) is an oral selective inhibitor of the prosurvival protein BCL-2 and, therefore, restores the apoptotic ability of malignant cells. It was developed by AbbVie. Venetoclax is approved in the USA for use as monotherapy in patients with chronic lymphocytic leukemia (CLL) [61]. Venetoclax addresses the same blood cancer subtype, chronic lymphocytic leukemia (CLL), as ibrutinib. Food increases venetoclax bioavailability by 3- to 5-fold, depending on the fat content, but no specific recommendation in terms of fat content in the meal is needed for the intake of venetoclax [62].
Recently, an acceptable yield and high-quality pathway with a desired solid polymorph property were published. This pathway also avoids the formation of undesired impurities [63,64].
The synthesis (Scheme 16) starts from 3,3-dimethylcyclohexan-1-one 89, which was treated with Vilsmeier reagent to afford chlorinated aldehyde 90. This was used in the next step without purification in the Suzuki-Miyaura coupling with boronic acid 91 to yield intermediate 92. The subsequent reductive amination with tert-butyl piperazine-1-carboxylate and sodium triacetoxyborohydride afforded amine 93. The BOC-protecting group was cleaved by 37% HCl in isopropanol to yield salt 94, which was rendered a free base (95) upon treatment with K3PO4.
Scheme 16.

Preparation of venetoclax (103) suitable for industrial production [61,63]. Amphos is shown in the blue box.
On the other side, 4-bromo-2-fluoro-1-iodobenzene 96 was metalated with isopropylmagnesium chloride, followed by the addition of Boc2O to yield ester 97. This underwent SNAr with 98 in the presence of tBuONa as a base to yield diaryl ether 99. This was subjected to Buchwald–Hartwig amination under Amphos and Pd2(dba)3 as a ligand and catalyst, respectively, to yield disubstituted piperazine 100. This was hydrolyzed under alkaline conditions (tBuOK/H2O) to form 101. The last stage involved the condensation between carboxylic acid and sulfonamide 102 under activation by EDC/DMAP and N,N-dimethylethylenediamine (DMEDA) to yield venetoclax. The last stage step was 84%. The presence of DMEDA led to the reformation of the desired product (from impurity) along with the corresponding DMEDA amide impurity (Figure 2) that was easily purified. This pathway, in summary, has led to a highly efficient and cost-effective synthesis.
Figure 2.
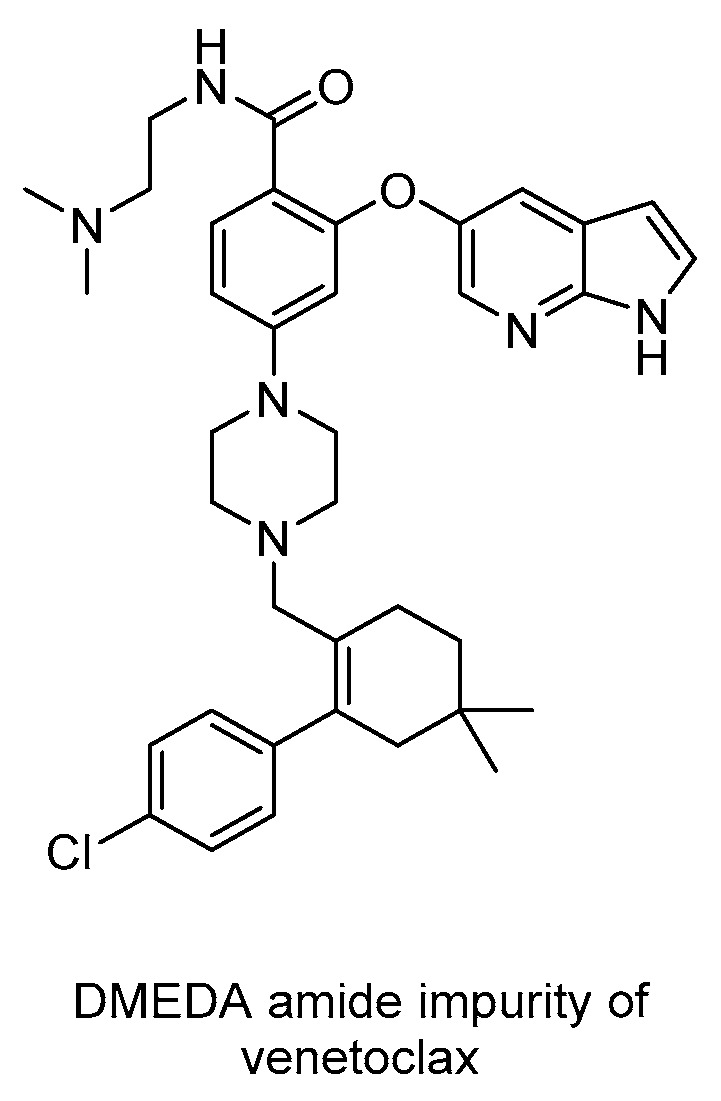
Structure of DMEDA amide impurity encountered in the synthesis of venetoclax (103).
2.6. Acalabrutinib (Brand Name Calquence®)
Developed by Acerta Pharma, acalabrutinib (114, Calquence®) is a Bruton’s tyrosine kinase inhibitor for the treatment of various hematological and solid malignancies [65].
In November 2019, the FDA approved acalabrutinib (owned by AstraZeneca) for adults with chronic lymphocytic leukemia (CLL) or small lymphocytic lymphoma (SLL) [66].
It is a selective, irreversible inhibitor (binding of acalabrutinib to the C481) of BTK that has improved pharmacologic features, including favorable plasma exposure, rapid oral absorption, a short half-life, and the absence of irreversible targeting to alternative kinases, such as an epidermal growth factor receptor (EGFR), tyrosine-protein kinase TEC, and IL-2-inducible tyrosine kinase (ITK) [67].
The half maximal inhibitory concentration (IC50) for BTK is 5.1 nM for acalabrutinib. The inhibition is weaker than that exhibited by ibrutinib [68].
Chemically, it contains an electron-deficient alkyne (responsible for irreversible inhibition) and the central imidazo-pyrazine.
Its synthesis (Scheme 17) [69,70] starts from (3-chloropyrazin-2-yl)methanamine 104 that is condensed with CBZ-L-proline 105 in the presence of HATU to afford the amide 106. This was intra-molecularly cyclized under POCl3/1,3-dimethyl-2-imidazolidinone (DMI) conditions in acetonitrile to afford chloro-imidazopyrazine 107. This was subjected to bromination (N-bromosuccinimide, NBS) on the imidazole scaffold to provide bromo derivative 108. Treatment of 108 with ammonia in isopropanol afforded amine 109. This was used in the subsequent microwave-assisted Suzuki-Miyaura coupling with boronic acid 110 using Pd(dppf)Cl2 as a catalyst to yield intermediate 111. The deprotection of CBZ-protection mediated by HBr in acetic acid followed by neutralization in NaOH afforded amine 112. Finally, 112 was condensed with 2-butynoic acid 113 in the presence of HATU to yield acalabrutinib (114).
Scheme 17.

The reported yield for this synthetic pathway was low and showed certain issues. For example, the process needed chromatography for the purification of the final stage and employed the corrosive hydrobromic acid in the deprotection stage. To overcome such a burden, more efficient pathways (cost-efficient and scalable) have been proposed.
For example, it has been reported that the product can be obtained via the unprotected pyrrolidine moiety (109b, Scheme 18), which can be easily purified by industrially viable purification methods, avoiding tedious chromatographic purification [69].
Scheme 18.

Preparation of intermediate 112. This method avoids the deprotection stage, thus avoiding use of corrosive HBr [69].
In this way, the deprotection of the CBZ group is not necessary, thereby providing an improvement to the preparation seen in Scheme 18. The thus-obtained precursor 112 can be converted into the product acalabrutinib (114) in the same way seen in Scheme 17, but the authors pointed out that acalabrutinib may be isolated from the reaction mixture by conventional methods, such as, but not limited to, filtration and/or centrifugation.
2.7. Tafamidis (Brand Name Vyndaqel®)
Tafamidis (117) is a small inhibitor discovered and developed by Scripps Research Centre in association with Pfizer in the treatment of transthyretin familial amyloid polyneuropathies (TTR-FAP). Recently, the FDA approved tafamidis for the treatment of heart disease (cardiomyopathy) that is caused by transthyretin-mediated amyloidosis (ATTR-CM) in adults. ATTR is caused by an abnormal deposit of specific proteins known as amyloids in the body’s organs and tissues [71].
Its main chemical feature is certainly a relatively low MW (MW = 308) and the easy synthetic pathway (Scheme 19) [72] to obtain it.
Scheme 19.

Preparation of tafamidis (117) [72].
4-amino-3-hydroxybenzoic acid 115 was mixed with 3,5-dichlorobenzoyl chloride 116 in the presence of pyridine to yield the acylated form of 115. The subsequent ring closure is mediated by p-toluenesulfonic acid monohydrate (TsOH-H2O), followed by esterification with diazomethane and final hydrolysis afforded tafamidis (117). Before hydrolysis, chromatography was used to purify the ester.
Another interesting approach (Scheme 20) involved the use of a nitro intermediate 118. This was treated with 116 in the presence of potassium carbonate, and the corresponding ester was treated with zinc/methanesulfonic acid (MsOH). The use of a simple zinc-MsOH as a catalyst renders the protocol suitable for large-scale synthesis, providing a valuable synthetic tool for industrial application reductive cyclization reaction. The stoichiometry of the reaction was also very important: the main product benzoxazole-based 117 was obtained when using Zn (5 moles) and MsOH (15 times) at 100–110 °C. Interestingly, when using acetic acid (60 °C) instead of MsOH, the N-acylated corresponding intermediate 120 was obtained (Figure 3) [71].
Scheme 20.

Alternative preparation of Zn-mediated tafamidis (117) [71].
Figure 3.
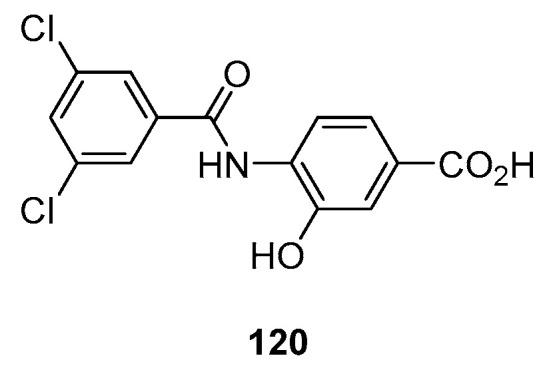
Structure of intermediate obtained by using Zn/acetic acid.
3. Concluding Remarks
In this review, the top-selling orphan drugs (forecast for the year 2026) are discussed. The biological activity and, in particular, the synthesis of each derivative are detailed.
When possible, more than a single synthetic pathway was discussed: more efficient and scalable processes were discussed together with small scale-driven oriented ones. The majority (five out of seven) of the presented drugs belong to the oncology therapeutic category. The remaining two target diseases in the respiratory (elexacaftor/tezacaftor/ivacaftor) and central nervous system (tafamidis) disease categories.
The pyrazolo-pyrimidine nucleus, present in ibrutinib, and imidazo-pyrazine, present in acalabrutinib, represent well-studied scaffolds investigated by academia and pharmaceutical companies alike. The easy accessibility to these scaffolds and their well-explored structure–activity relationship (SAR) represent advantages from a synthetic point of view. These two drugs also share the presence of a Michael acceptor (acrylamide and alkyne, respectively) that makes them irreversible inhibitors, characterized by a Michael acceptor moiety able to form a covalent bond with the conserved Cys481 residue in the ATP binding site. Novel covalent warheads and a fine-tuning of their chemical neighborhood could allow for targeting specific amino acid residues and improve toxicity issues due to off-target binding. BTK inhibitors, such as ibrutinib and acalabrutinib, possess higher efficacy in patients with a high risk of disease and offer better tolerability in elderly and fragile patients [73].
The pyrrolo-pyrimidine scaffold of ruxolitinib also shares a similar central core, related to ibrutinib and acalbrutinib, with the difference of being a deazapurine.
Bruton tyrosine kinase and Janus Kinase (Jak) inhibitors (ruxolitinib) have seen a very prolific yield in terms of drug approval. These two targets offer a very promising opportunity in terms of reactivity and selectivity.
The triple-combination elexacaftor-tezacaftor-ivacaftor offers a nice example of how a synergistic effect brings an advantage in terms of drug efficacy and safety. Correctors of protein folding and/or potentiators act together, increasing CFTR channel function. Olaparib for BRCAm advanced ovarian cancer possesses a N-acylpiperazine moiety and a relatively easy synthetic preparation.
Venetoclax shares the piperazine moiety revealing that this aza-heterocycle is a very important synthon for many drugs.
In certain leukemias and certain solid tumors, apoptosis is a prominent (if not exclusive) mechanism associated with the induction of tumor remission. In addition, the expression of apoptotic modulators within a tumor appears to correlate with the sensitivity to traditional cancer therapies [74].
Therefore, molecules such as olaparib and venetoclax are of great importance in cancer therapy.
Benzoxazole tafamidis possess a particularly small size and a very easily accessible chemistry.
New tools and drug discovery approaches, from gene editing to artificial intelligence (AI)-powered screening and drug design, will provide the possibility for getting more and more orphan drug candidates in the future. With the implementation of AI in the manufacturing of pharmaceutical products, personalized therapies with precise doses, release parameters, and other required aspects can be manufactured according to individual patient needs [75]. As an example, the British company Benevolent Bio used a technological platform to screen out 5 compounds from 100 potential compounds that can treat ALS and confirmed that 4 of them were effective in curing motor neurodegeneration [76].
The benefits of orphan drug designation (for example, 7-year market exclusivity and up to a 50% tax credit for clinical trials that qualify) are encouraging pharma companies to invest in treating rare diseases. In addition, many rare diseases are still underexplored and could open the discovery of many other compounds in the next future.
Author Contributions
D.B.T., C.S. contributed to the conception of this review. All the authors analyzed the literature D.B.T., L.S. and C.S. wrote the manuscript. D.B.T. and L.S. completed the figure drawing. D.B.T., L.S., C.S., O.R., L.B. and F.M. revised the manuscript. All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Funding Statement
Project “DELPHI Star Labs” in the frame of the financial support of “Dipartimenti di Eccellenza 2018–2022” support the post-doc position of D.B.T.
Footnotes
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
References
- 1.Milne C.-P., Cabanilla L.A. Comprehensive Medicinal Chemistry II. Elsevier; Amsterdam, The Netherlands: 2007. Orphan Drugs and Generics; pp. 655–680. [Google Scholar]
- 2.Designating an Orphan Product: Drugs and Biological Products. [(accessed on 26 October 2022)]; Available online: https://www.fda.gov/industry/medical-products-rare-diseases-and-conditions/designating-orphan-product-drugs-and-biological-products.
- 3.EMA Orphan Designation: Overview. [(accessed on 26 October 2022)]. Available online: https://www.ema.europa.eu/en/human-regulatory/overview/orphan-designation-overview.
- 4.The Story behind the Orphan Drug Act. [(accessed on 26 October 2022)]; Available online: https://www.fda.gov/industry/fdas-rare-disease-day/story-behind-orphan-drug-act.
- 5.Aronson J.K. Rare Diseases and Orphan Drugs. Br. J. Clin. Pharmacol. 2006;61:243–245. doi: 10.1111/j.1365-2125.2006.02617.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lavandeira A. Orphan Drugs: Legal Aspects, Current Situation. Haemophilia. 2002;8:194–198. doi: 10.1046/j.1365-2516.2002.00643.x. [DOI] [PubMed] [Google Scholar]
- 7.European Commission . Regulation (EC) No 141/2000 of the European Parliament and of the Council of 16 December 1999 on Orphan Medicinal Products. Volume 018 European Commission; Brussels, Belgium: 1999. [Google Scholar]
- 8.Asbury C.H. The Orphan Drug Act: The First 7 Years. JAMA. 1991;265:893–897. doi: 10.1001/jama.1991.03460070075046. [DOI] [PubMed] [Google Scholar]
- 9.Mariz S., Reese J.H., Westermark K., Greene L., Goto T., Hoshino T., Llinares-Garcia J., Sepodes B. Worldwide Collaboration for Orphan Drug Designation. Nat. Rev. Drug Discov. 2016;15:440–441. doi: 10.1038/nrd.2016.80. [DOI] [PubMed] [Google Scholar]
- 10.Rare Diseases at FDA. [(accessed on 26 October 2022)]; Available online: https://www.fda.gov/patients/rare-diseases-fda.
- 11.U.S. Congress. H.R. 1—An Act to Provide for Reconciliation Pursuant to Titles II and V of the Concurrent Resolution on the Budget for Fiscal Year 2018. [(accessed on 22 November 2022)]; Available online: https://www.congress.gov/115/plaws/publ97/PLAW-115publ97.pdf.
- 12.Zhang M., Zhu C., Jacomy A., Lu L.J., Jegga A.G. The Orphan Disease Networks. Am. J. Hum. Genet. 2011;88:755–766. doi: 10.1016/j.ajhg.2011.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kontoghiorghe C.N. World Health Dilemmas: Orphan and Rare Diseases, Orphan Drugs and Orphan Patients. WJM. 2014;4:163. doi: 10.5662/wjm.v4.i3.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Orphan Drugs: From Niche to Mainstream. [(accessed on 9 November 2022)]. Available online: https://www.pharmexec.com/view/orphan-drugs-from-niche-to-mainstream.
- 15.Ponader S., Chen S.-S., Buggy J.J., Balakrishnan K., Gandhi V., Wierda W.G., Keating M.J., O’Brien S., Chiorazzi N., Burger J.A. The Bruton Tyrosine Kinase Inhibitor PCI-32765 Thwarts Chronic Lymphocytic Leukemia Cell Survival and Tissue Homing in Vitro and in Vivo. Blood. 2012;119:1182–1189. doi: 10.1182/blood-2011-10-386417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Paydas S. Management of Adverse Effects/Toxicity of Ibrutinib. Crit. Rev. Oncol./Hematol. 2019;136:56–63. doi: 10.1016/j.critrevonc.2019.02.001. [DOI] [PubMed] [Google Scholar]
- 17.Nocco S., Andriano T.M., Bose A., Chilov M., Godwin K., Dranitsaris G., Wu S., Lacouture M.E., Roeker L.E., Mato A.R., et al. Ibrutinib-Associated Dermatologic Toxicities: A Systematic Review and Meta-Analysis. Crit. Rev. Oncol./Hematol. 2022;174:103696. doi: 10.1016/j.critrevonc.2022.103696. [DOI] [PubMed] [Google Scholar]
- 18.Ghosh A.K., Samanta I., Mondal A., Liu W.R. Covalent Inhibition in Drug Discovery. ChemMedChem. 2019;14:889–906. doi: 10.1002/cmdc.201900107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hughes D.L. Patent Review of Manufacturing Routes to Recently Approved Oncology Drugs: Ibrutinib, Cobimetinib, and Alectinib. Org. Process Res. Dev. 2016;20:1855–1869. doi: 10.1021/acs.oprd.6b00304. [DOI] [Google Scholar]
- 20.Honigberg L., Verner E., Pan Z. Inhibitors of Bruton’s Tyrosine Kinase. US2010022561(A1) U.S. Patent. 2010 January 28;
- 21.Pan Z., Li S.J., Schereens H., Honigberg L., Verner E. Bruton’s Tyrosine Kinase Activity Probe and Method of Using. European Patent. No. EP2089391A4. Office Patent. 2010 March 17;
- 22.Chen W., Loury D.J., Mody T.D. Pyrazolo-Pyrimidine Inhibitors of Bruton’s Tyrosine Kinase. US7718662B1. U.S. Patent. 2010 May 18;
- 23.Pan Z., Scheerens H., Li S.-J., Schultz B.E., Sprengeler P.A., Burrill L.C., Mendonca R.V., Sweeney M.D., Scott K.C.K., Grothaus P.G., et al. Discovery of Selective Irreversible Inhibitors for Bruton’s Tyrosine Kinase. ChemMedChem. 2007;2:58–61. doi: 10.1002/cmdc.200600221. [DOI] [PubMed] [Google Scholar]
- 24.Liu H., Pan Z. Innovative Drug Synthesis. John Wiley & Sons, Ltd.; Hoboken, NJ, USA: 2015. Ibrutinib (Imbruvica): The First-in-Class Btk Inhibitor for Mantle Cell Lymphoma, Chronic Lymphocytic Leukemia, and Waldenstrom’s Macroglobulinemia; pp. 157–166. [Google Scholar]
- 25.Owens T.D. Bioactive Carboxylic Compound Classes. John Wiley & Sons, Ltd.; Hoboken, NJ, USA: 2016. Ibrutinib, a Carboxylic Acid Amide Inhibitor of Bruton’s Tyrosine Kinase; pp. 197–208. [Google Scholar]
- 26.Pye P., Haim C.B., Conza M., Houpis I.N. Processes and Intermediates for Preparing a Medicament. US20140275126A1. U.S. Patent. 2014 September 18;
- 27.Pye P., Haim C.B., Conza M., Houpis I.N. Processes and Intermediates for Preparing a Medicament. US9156847B2. U.S. Patent. 2015 October 13;
- 28.Fang T., Chen Y., Jiang M., Zhang W., Sun K., Chen X. Synthesis Method of Ibrutinib. CN107674079B. CN Patent. 2019 December 13;
- 29.Jaychandra S., Sebastian S., Rao J., Naidu H., Adla M., Mannava S.R., Sabbella S.R., Dandala R. Process for the Preparation of Ibrutinib. WO2016132383A1. 2016 August 25;
- 30.Sharma K., Thanki B.P., Khanna M.S., Prasad M. A Process for the Preparation of Ibrutinib. WO2016079693A1. WO Patent. 2016 May 25;
- 31.Ridley K., Condren M. Elexacaftor-Tezacaftor-Ivacaftor: The First Triple-Combination Cystic Fibrosis Transmembrane Conductance Regulator Modulating Therapy. J. Pediatr. Pharmacol. Ther. 2020;25:192–197. doi: 10.5863/1551-6776-25.3.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sala M.A., Jain M. Tezacaftor for the Treatment of Cystic Fibrosis. Expert Rev. Respir. Med. 2018;12:725–732. doi: 10.1080/17476348.2018.1507741. [DOI] [PubMed] [Google Scholar]
- 33.Hughes D.L. Patent Review of Synthetic Routes and Crystalline Forms of the CFTR-Modulator Drugs Ivacaftor, Lumacaftor, Tezacaftor, and Elexacaftor. Org. Process Res. Dev. 2019;23:2302–2322. doi: 10.1021/acs.oprd.9b00326. [DOI] [Google Scholar]
- 34.Shaughnessy C.A., Zeitlin P.L., Bratcher P.E. Elexacaftor Is a CFTR Potentiator and Acts Synergistically with Ivacaftor during Acute and Chronic Treatment. Sci. Rep. 2021;11:19810. doi: 10.1038/s41598-021-99184-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fiedorczuk K., Chen J. Molecular Structures Reveal Synergistic Rescue of Δ508 CFTR by Trikafta Modulators. Science. 2022;378:284–290. doi: 10.1126/science.ade2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Laselva O., Bartlett C., Gunawardena T.N.A., Ouyang H., Eckford P.D.W., Moraes T.J., Bear C.E., Gonska T. Rescue of Multiple Class II CFTR Mutations by Elexacaftor+tezacaftor+ivacaftor Mediated in Part by the Dual Activities of Elexacaftor as Both Corrector and Potentiator. Eur. Respir. J. 2021;57:2002774. doi: 10.1183/13993003.02774-2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Abela A.R., Alcacio T., Anderson C., Angell P.T., Baek M., Clemens J.J., Cleveland T., Ferris L.A., Grootenhuis P.D.J., Gross R.S., et al. Modulator of Cystic Fibrosis Transmembrane Conductance Regulator, Pharmaceutical Compositions, Methods of Treatment, and Process for Making the Modulator. WO2018107100A1. WO Patent. 2018 June 14;
- 38.Tezacaftor (VX-661) for Cystic Fibrosis. [(accessed on 9 November 2022)]. Available online: https://cysticfibrosisnewstoday.com/tezacaftor-vx-661-for-cystic-fibrosis.
- 39.Highlights of Prescribing Information. [(accessed on 30 December 2022)]; Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/210491s007lbl.pdf.
- 40.Looker A., Littler B.J., Choudhury A., Harrison C., Veluri R., Ryan M.P., Jiang L., Luss-Lusis E. Modulators of Atp-Binding Cassette Transporters. US20130116238A1. U.S. Patent. 2013 May 9;
- 41.Modulators of ATP-Binding Cassette Transporters Patent Application. [(accessed on 3 November 2022)]. Available online: https://uspto.report/patent/app/20050148648.
- 42.Tanoury G.J., Harrison C., Littler B.J., Rose P.J., Huges R.M., Jung Y.C., Siesel D.A., Lee E.C., Belmont D.T. Process of Producing Cycloalkylcarboxamido-Indole Compounds. WO2011US33396. WO Patent. 2011 April 21;
- 43.Deeks E.D. Ivacaftor: A Review of Its Use in Patients with Cystic Fibrosis. Drugs. 2013;73:1595–1604. doi: 10.1007/s40265-013-0115-2. [DOI] [PubMed] [Google Scholar]
- 44.Fohner A.E., McDonagh E.M., Clancy J.P., Whirl Carrillo M., Altman R.B., Klein T.E. PharmGKB Summary: Ivacaftor Pathway, Pharmacokinetics/Pharmacodynamics. Pharm. Genom. 2017;27:39–42. doi: 10.1097/FPC.0000000000000246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lai C.K., Song T.V., Li J.J. Ivacaftor (Kalydeco): A CFTR Potentiator for the Treatment of Cystic Fibrosis. In: Li J.J., Johnson D.S., editors. Innovative Drug Synthesis. John Wiley & Sons, Inc.; Hoboken, NJ, USA: 2015. pp. 303–316. [Google Scholar]
- 46.Arekar S.G., Johnston S.C., Krawiec M., Medek A., Mudunuri P., Sullivan M.J. Solid Forms of N-[2,4-Bis(1,1-Dimethylethyl)-5-Hydroxyphenyl]-1,4-Dihydro-4-Oxoquinoline-3-Carboxamide. WO2011116397A1. WO Patent. 2011 September 22;
- 47.Arora S., Balasubramaniam S., Zhang H., Berman T., Narayan P., Suzman D., Bloomquist E., Tang S., Gong Y., Sridhara R., et al. FDA Approval Summary: Olaparib Monotherapy or in Combination with Bevacizumab for the Maintenance Treatment of Patients with Advanced Ovarian Cancer. Oncologist. 2021;26:e164–e172. doi: 10.1002/onco.13551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rohit Deepak Nalawade; Bhagwan Dilip Devkar Olaparib an Anticancer Drug: A Review. World J. Adv. Res. Rev. 2021;11:329–336. doi: 10.30574/wjarr.2021.11.2.0406. [DOI] [Google Scholar]
- 49.Harrision D., Gravells P., Thompson R., Bryant H.E. Poly(ADP-Ribose) Glycohydrolase (PARG) vs. Poly(ADP-Ribose) Polymerase (PARP)— unction in Genome Maintenance and Relevance of Inhibitors for Anti-Cancer Therapy. Front. Mol. Biosci. 2020;7:191. doi: 10.3389/fmolb.2020.00191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Menear K.A., Adcock C., Boulter R., Cockcroft X.L., Copsey L., Cranston A., Dillon K.J., Drzewiecki J., Garman S., Gomez S., et al. 4-[3-(4-cyclopropanecarbonylpiperazine-1-carbonyl)-4-fluorobenzyl]-2Hphthalazin-1-one: A novel bioavailable inhibitor of poly (ADP-ribose) polymerase-1. J. Med. Chem. 2008;51:6581–6591. doi: 10.1021/jm8001263. [DOI] [PubMed] [Google Scholar]
- 51.Hsiao T.Y., Chang Y.H. Processes for Preparing Olaparib. WO2018038680A1. WO Patent. 2018 March 1;
- 52.Martin N.M.B., Smith G.C., Jackson S.P., Loh V.J.M., Cockcroft X.-L.F., Matthews I.T.W., Menear K.A., Kerrigan F., Ashworth A. Phthalazinone Derivatives. US7449464B2. U.S. Patent. 2008 November 11;
- 53.Menear K.A., Ottridge A.P., Londesbrough D.J., Hallett M.R., Mulholland K.R., Pittam J.D., Laffan D.D.P., Ashworth I.W., Jones M.F., Cherryman J.H. Phthalazinone Derivative. US8247416B2. U.S. Patent. 2012 August 21;
- 54.McKeage K. Ruxolitinib: A Review in Polycythaemia Vera. Drugs. 2015;75:1773–1781. doi: 10.1007/s40265-015-0470-2. [DOI] [PubMed] [Google Scholar]
- 55.Purandare A.V., Lorenzi M.V., Lombardo L.J. Annual Reports in Medicinal Chemistry. Volume 45. Elsevier; Amsterdam, The Netherlands: 2010. Janus Kinase 2 (JAK2) Inhibitors for the Treatment of Myeloproliferative Neoplasm (MPN) pp. 210–227. [Google Scholar]
- 56.Przepiorka D., Luo L., Subramaniam S., Qiu J., Gudi R., Cunningham L.C., Nie L., Leong R., Ma L., Sheth C., et al. FDA Approval Summary: Ruxolitinib for Treatment of Steroid-Refractory Acute Graft-Versus-Host Disease. Oncologist. 2020;25:e328–e334. doi: 10.1634/theoncologist.2019-0627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Coricello A., Mesiti F., Lupia A., Maruca A., Alcaro S. Inside Perspective of the Synthetic and Computational Toolbox of JAK Inhibitors: Recent Updates. Molecules. 2020;25:3321. doi: 10.3390/molecules25153321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Haydl A.M., Xu K., Breit B. Regio- and Enantioselective Synthesis of N-Substituted Pyrazoles by Rhodium-Catalyzed Asymmetric Addition to Allenes. Angew. Chem. Int. Ed. 2015;54:7149–7153. doi: 10.1002/anie.201501758. [DOI] [PubMed] [Google Scholar]
- 59.Lin Q., Meloni D., Pan Y., Xia M., Rodgers J., Shepard S., Li M., Galya L., Metcalf B., Yue T.-Y., et al. Enantioselective Synthesis of Janus Kinase Inhibitor INCB018424 via an Organocatalytic Aza-Michael Reaction. Org. Lett. 2009;11:1999–2002. doi: 10.1021/ol900350k. [DOI] [PubMed] [Google Scholar]
- 60.Zhang X., Zhang A., Zhou Z., YANG L., Yao H., Zhu X., Wang H. Synthesis Process of Ruxolitinib. US10562904B2. U.S. Patent. 2020 February 18;
- 61.Deeks E.D. Venetoclax: First Global Approval. Drugs. 2016;76:979–987. doi: 10.1007/s40265-016-0596-x. [DOI] [PubMed] [Google Scholar]
- 62.Salem A.H., Agarwal S.K., Dunbar M., Nuthalapati S., Chien D., Freise K.J., Wong S.L. Effect of low and high-fat meals on the pharmacokinetics of venetoclax, a selective first-in-class BCL-2 inhibitor. J. Clin. Pharmacol. 2016;56:1355–1361. doi: 10.1002/jcph.741. [DOI] [PubMed] [Google Scholar]
- 63.Ku Y.-Y., Chan V.S., Christesen A., Grieme T., Mulhern M., Pu Y.-M., Wendt M.D. Development of a Convergent Large-Scale Synthesis for Venetoclax, a First-in-Class BCL-2 Selective Inhibitor. J. Org. Chem. 2019;84:4814–4829. doi: 10.1021/acs.joc.8b02750. [DOI] [PubMed] [Google Scholar]
- 64.Chan V.S., Christesen A.C., Grieme T.A., Ku Y.-Y., Mulhern M.M., Pu Y.-M.M. Processes For The Preparation Of An Apoptosis-Inducing Agent. US20140275540A1. U.S. Patent. 2014 September 18;
- 65.Markham A., Dhillon S. Acalabrutinib: First Global Approval. Drugs. 2018;78:139–145. doi: 10.1007/s40265-017-0852-8. [DOI] [PubMed] [Google Scholar]
- 66.FDA Approves Acalabrutinib for CLL and SLL. [(accessed on 28 October 2022)]; Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/project-orbis-fda-approves-acalabrutinib-cll-and-sll.
- 67.Byrd J.C., Harrington B., O’Brien S., Jones J.A., Schuh A., Devereux S., Chaves J., Wierda W.G., Awan F.T., Brown J.R., et al. Acalabrutinib (ACP-196) in Relapsed Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2016;374:323–332. doi: 10.1056/NEJMoa1509981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Patel V., Balakrishnan K., Bibikova E., Ayres M., Keating M.J., Wierda W.G., Gandhi V. Comparison of Acalabrutinib, A Selective Bruton Tyrosine Kinase Inhibitor, with Ibrutinib in Chronic Lymphocytic Leukemia Cells. Clin. Cancer Res. 2017;23:3734–3743. doi: 10.1158/1078-0432.CCR-16-1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Budideti S.R., Konduri S.K.M., Sanapureddy J.M.R., Remella R.K., Thoota S.K., Kothamunireddygari S., Muddasani P.R., Nannapaneni V.C. Novel Process for the Preparation of Acalabrutinib and Its Intermediates. WO2021111465A1. WO Patent. 2021 June 10;
- 70.Barf T.A., Jans C., de Man A., Oubrie A.A., Raaijmakers H.C.A., Rewinkel J., Sterrenburg J.-G., Wijkmans J.C.H.M. 4-Imidazopyridazin-1-Yl-Benzamides and 4-Imidazotriazin-1-Yl-Benzamides as Btk-Inhibitors. WO2013010868. WO Patent. 2013 January 24;
- 71.Karumanchi K., Natarajan S.K., Gadde S., Vanchanagiri K., Moturu K.V.R. A New Synthesis of Tafamidis via Zinc-MsOH Mediated Reductive Cyclisation Strategy. J. Chem. Sci. 2021;133:48. doi: 10.1007/s12039-021-01910-9. [DOI] [Google Scholar]
- 72.Simões C.J.V., Almeida Z.L., Costa D., Jesus C.S.H., Cardoso A.L., Almeida M.R., Saraiva M.J., Pinho e Melo T.M.V.D., Brito R.M.M. A Novel Bis-Furan Scaffold for Transthyretin Stabilization and Amyloid Inhibition. Eur. J. Med. Chem. 2016;121:823–840. doi: 10.1016/j.ejmech.2016.02.074. [DOI] [PubMed] [Google Scholar]
- 73.Burge J.A. BTK Inhibitors: Present and future. Cancer J. 2019;25:386–393. doi: 10.1097/PPO.0000000000000412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sellers W.R., Fisher D.E. Apoptosis and cancer drug targeting. J. Clin. Investig. 1999;104:1655–1661. doi: 10.1172/JCI9053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jiang J., Ma X., Ouyang D., Williams III R.O. Emerging Artificial Intelligence (AI) Technologies Used in the Development of Solid Dosage Forms. Pharmaceutics. 2022;14:2257. doi: 10.3390/pharmaceutics14112257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Liu Q. Artificial intelligence and drug discovery. Acad. J. Second Mil. Med. Univ. 2018;39:869–872. doi: 10.16781/j.0258-879x.2018.08.0869. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.