

Abstract
Introduction
Primary bilateral macronodular adrenal hyperplasia (PBMAH) is one of the uncommon causes of endogenous Cushing’s syndrome (CS).Pheochromocytoma (PCC) is another adrenal tumor which is derived from neural crest arising in the adrenal medulla. Here we are reporting a case with recurrent overt CS due to PBMAH, 2 years after unilateral adrenalectomy, concomitant with recently developed PCC.
Case Presentation
A 43-year-old woman was admitted to our clinic with a 30 kg weight gain, proximal muscle weakness, menstrual irregularity, easy bruising and excessive hair growth on face and body.The lab results were compatible with a diagnosis of solely ACTH-independent CS. Screening showed bilateral macronodular lesions and she underwent right adrenalectomy. Postoperatively, she had lost weight and her well-being had improved; 2 years later, she developed CS and paroxysmal hypertension. The left adrenal gland was laparoscopically removed. Histopatologically, the lesion was reported as a typical PCC and macronodular-micronodular hyperplasia of the adrenal tissue surrounding that lesion.
Conclusions
Pheochromocytoma with synchronous ACTH-independent CS originating from the same adrenal gland is very rare. To the best of our knowledge,our case is the first one describing the coexistence of overt ACTH-independent CS due to PBMAH and metachronous PCC.The importance of detailed re-evaluation of patients with recurrent ACTH-independent CS is highlighted here.
Keywords: Cushing’s syndrome, Primary bilateral macronodular hyperplasia, Pheochromocytoma
INTRODUCTION
Primary bilateral macronodular adrenal hyperplasia (PBMAH), previously called “adrenocorticotropic hormone (ACTH)-independent macronodular adrenal hyperplasia”, is one of the uncommon causes of endogenous Cushing’s syndrome (CS), accounting for approximately less than 1% of all endogenous causes. As most patients harboring PBMAH present with only bilateral adrenal incidentalomas due to the frequent use of imaging technology, the patients who develop CS generally have subclinical features of the disease with an insidious onset (1). Pheochromocytoma (PCC) is another adrenal tumor which is derived from neural crest arising in the adrenal medulla (2). It is characterized by excess secretion of catecholamines which generally causes various symptoms such as sustained or paroxysmal hypertension, palpitation due to tachycardia, diaphoresis and anxiety (3). The coexistence of PCC and ACTH-independent CS originating from same adrenal gland is very unusual and several etiologies have been described in the literature. Here we are reporting a case with recurrent overt CS due to PBMAH, 2 years after unilateral adrenalectomy, concomitant with recently developed PCC in the same adrenal gland.
CASE PRESENTATION
A 43-year-old Caucasian woman was admitted to our outpatient clinic in November of 2016 with a 30 kg weight gain in 3 years, proximal muscle weakness in the upper and lower limbs, menstrual irregularity, easy bruising and excessive hair growth on face and body for the past 1 year. She had a history of asthma, essential hypertension, type 2 diabetes mellitus and hyperlipidemia for about 23, 10, 4 and 3 years, respectively. She was under treatment with an inhaler budesonide/formoterol 160/4.5 µg/day, oral metformin 1000 mg/day, perindopril 10 mg/day, lercanidipine 10 mg/day and atorvastatin 20 mg/day. She was an active smoker and had a 12 pack-year history of smoking, but no alcohol.
On physical examination, blood pressure (BP) was 130/80 mm Hg, the pulse 90, the respiratory rate 17 and the temperature was 36.9°C. The patient had normal mental status without any neurological deficit except proximal muscle weakness in the upper (4/5) and lower (3/5) extremities. She displayed obvious Cushing’s stigmata with moon facies, plethora, buffalo hump, abdominal obesity (body mass index [BMI] 46.3 kg/m2), hirsutism and a few ecchymoses on her lower extremities. Chest and cardiovascular examinations revealed no abnormalities.
Routine laboratory studies revealed normal complete blood count (CBC) except a mild leukocytosis, serum electrolyte levels, liver and kidney function tests, as described in Table 1. Basal morning serum cortisol and adrenocorticotropic hormone (ACTH) levels were 18.6 µg/dL and <5 pg/mL, respectively. Her diurnal cortisol rhythm was impaired (serum cortisol at 23:00: 20.8 µg/dL) and after a 48-hour, 2-mg dexamethasone suppression test (DST), serum cortisol level was 24.3 µg/dL. The results were compatible with a diagnosis of ACTH-independent CS. A contrast-enhanced computed tomography (CT) scan showed bilateral macronodular lesions of which 2 nodules on the right side were 3.9 and 2.8 cm in diameter and two on the left side with diameters of 3.0 and 2.6 cm (Fig. 1). A diagnosis of CS due to primary bilateral macronodular adrenal hyperplasia (PBMAH) was made. No genetic testing could be performed. She was evaluated for aberrant stimulators for 3 consecutive days and all tests for aberrant hormone receptors such as beta-adrenergic receptors, glucose-dependent insulinotropic peptide, vasopressin, serotonin, glucagon and angiotensin II receptors were negative (1). Screening for pheochromocytoma (PCC) and primary aldosteronism was performed and both of them were ruled out (Table 1). Her bone mineral density for L1-4 vertebrae had decreased to -2.1SD of Z-score. As the gold standard treatment for CS due to PBMAH, the removal of the right adrenal gland, which was greater than the left adrenal gland in terms of maximum diameter was decided and she underwent a right adrenalectomy by the transabdominal route. Intraoperatively, there occurred no complication such as hypertensive crisis. The histopathological examination of the gland was reported as macronodular adrenal hyperplasia with positive immunostaining for ACTH and vimentin. Postoperatively, plasma cortisol and ACTH levels were 16.7 µg/dL and 6 pg/mL, respectively. In the absence of nausea and vomiting with adequate cortisol level, the patient was discharged without glucocorticoid replacement therapy. At the 3-month follow-up, her muscle weakness, moon facies, plethora, buffalo hump, hirsutism and ecchymoses diminished. She also noted that she had lost weight and her well-being had improved, but no menstruation, yet. 24-hour urinary free cortisol and morning plasma ACTH levels were 368 µg/24hr (58-403) and 9.4 pg/mL, respectively. Morning serum cortisol level after 2-day 2-mg DST was 4.4 µg/dL. She was asked to follow up in 3 months without any medical treatment, but never completed this follow-up.
Table 1.
Comparison of TAC and MDA level in pregnant EMF exposed rats, Control and Sham Groups
| Pre-operative (1st operation) | Remission of CS | Pre-operative (2nd operation) | Remission of CS* and PCC | Normal Range | |
|---|---|---|---|---|---|
| Time | 12/2016 | 03/2017 | 10/2019 | 01/2020 | |
| fP-glucose (mg/dL) | 218 | 80 | 133 | 101 | 74-106 |
| S-sodium (mEq/L) | 140.7 | 139 | 144 | 141 | 136-146 |
| S-potassium (mEq/L) | 4.3 | 4.7 | 3.9 | 4.4 | 3.5-5.1 |
| S-creatinine (mg/dL) | 0.67 | 0.52 | 0.55 | 0.64 | 0.51-0.95 |
| S-urea (mg/dL) | 43 | 26 | 35 | 30 | 17-43 |
| S-AST (U/L) | 21 | 13 | 21 | 11 | <35 |
| S-ALT (U/L) | 16 | 11 | 29 | 13 | <35 |
| Hemoglobin | 14.6 | 12.8 | 13.4 | 12.5 | 12.5-16.3 |
| WBC count x 103 | 15.1 | 7.1 | 12.5 | 10.7 | 3.6-10.2 |
| Platelet count x 103 | 272 | 387 | 396 | 575 | 152-348 |
| Morning S-cortisol (µg/dL) | 18.6 | 12.8 | 5-18 | ||
| Midnight S-cortisol (µg/dL) | 20.8 | 14.4 | <7.5 | ||
| Morning P-ACTH (pg/mL) | <5 | 9.4 | <5 | 0-45 | |
| 2-day 2mg DST (µg/dL) | 24.3 | 4.4 | 13.8 | <1.8 | |
| U-cortisol (µg/24hr) | 466 | 368 | 830 | 58-403 | |
| U- metanephrine (µg/24hr) | 412 | 2332 | 138 | <350 | |
| U-normetanephrine (µg/24hr) | 632 | 1012 | 368 | <600 | |
| P-aldosterone (ng/dL) | 4.1 | 5.5 | 3.7-43.2 | ||
| P-renin activity (ng/ml/h) | 5.26 | 3.46 | 0.06-4.69 |
CS-Cushing’s syndrome, DST-dexamethasone suppression test, f-fasting, P-plasma, PCC-Pheochromocytoma, S-serum, U-urine, WBC-white blood cell. * Under treatment with oral prednisolone 10mg/day.
Figure 1.

Contrast-enhanced CT scan images at initial diagnosis . Four nodular lesions at bilateral adrenal glands: (A)3.9cm, (B)2.8cm, (C)2.6cm and (D)3.0cm in diameter.
In October 2019, she was referred to our hospital again with the similar symptoms to her previous admission. She stated that after the operation, the symptoms of CS had disappeared. During this 2.5-year period, menstruation started, skin texture had improved and weight gain had stopped, but no significant weight loss achieved. Blood glucose and blood pressure levels were under control with her previous oral antidiabetic and antihypertensive drugs. However, about 6 months prior to the latest admission, menstruation ceased abruptly. She began bruising more easily and gaining weight with central redistribution of body fat with decreased muscle strength. She also noted that she started developing hypertensive crises (BP: 200/100 mm Hg) approximately once a month and monitoring high blood glucose levels in the previous 9 months.
Physical examination revealed the classical features of CS which were present in her previous admission including central obesity (BMI 48.4 kg/m2), plethora, round facies, buffalo hump, hirsutism and ecchymoses on both upper and lower limbs. Vital signs (BP, pulse rate, respiratory rate and temperature) and routine laboratory test results (CBC, electrolytes, liver and renal function tests) were within normal range, but only a mild leukocytosis (Table 1). The basal plasma ACTH level was less than 5 pg/mL and the midnight serum cortisol was impaired. 2-day 2-mg DST demonstrated inadequate suppression of cortisol and 24-h urinary free cortisol was elevated (Table 1). Contrast-enhanced adrenal magnetic resonance imaging (MRI) demonstrated 2 nodules in the left adrenal gland, measuring 25x21mm and 28x28mm, of which the greater lesion was with areas of necrosis (Fig. 2). Additionally, her 24-h urine metanephrine and normetanephrine levels were significantly elevated (2332 µg/24hr [<350] and 1012 µg/24hr [<600], respectively). The clinical diagnosis was consequently PCC and the recurrence of CS due to PBMAH. Thereafter, gallium-68 DOTA-Tyr3-octreotate (68Ga-DOTATATE) positron emission tomography/computed tomography (PET/CT) was performed. It revealed 2.8x3 cm left adrenal mass with increased tracer accumulation (Fig. 2).
Figure 2.

68Ga-DOTATATE PET/CT and contrast-enhanced adrenal MRI images. (A) 68Ga-DOTATATE PET/CT axial image, demonstrating the elevated tracer uptake in one of the nodular lesions, in which the uptake is mostly accumulated in the necrotic area, (B) T2-weighted SPAIR axial MRI image, showing the necrotic area in the left adrenal mass, (C) 68Ga-DOTATATE PET/CT axial image, displaying the other nodular mass with the normal physiologic uptake, (D) T2-weighted SPAIR axial MRI image, showing the other nodular mass on the left adrenal gland with homogenous intensity.
After adequate α and β-adrenoreceptor blockade treatment (doxazosin 2x4mg/day and followed by propranolol 2x40 mg/day after orthostatic hypotension was maintained with doxazosin), the left adrenal gland was laparoscopically removed under glucocorticoid replacement therapy without any intraoperative complications. In cross section, the left adrenal contained two different lesions each located in the cortex and medulla (Fig. 3). Histopathological examination reported the lesion as a typical PCC measuring 2.8 cm in diameter and macronodular-micronodular hyperplasia of the adrenal tissue surrounding that lesion (Fig. 4). In the PCC the tumor consisted of abundant fine, granular red-purple cytoplasm cells, which generally showed nested (zelballen), trabecular or solid arrangement. Immunostaining revealed positive staining for chromogranin, synaptophysin with a Ki-67 index of 4% and negative for ACTH in the lesion. Additionally, ACTH staining was weakly positive in the surrounding micronodular hyperplastic focus. Postoperatively, the hydrocortisone therapy (2x10 mg/day) was maintained and fludrocortisone acetate 0.1 mg/day was initiated. At the 3-month follow-up, the proximal muscle strength, blood pressure and blood glucose control had improved. She also started losing weight (about 3 kg in 3 months) and menstruating again. 24-h urine metanephrine and normetanephrine levels were within reference range, suggestive of the remission of the PCC. She is scheduled for a follow-up every 3 months.
Figure 3.
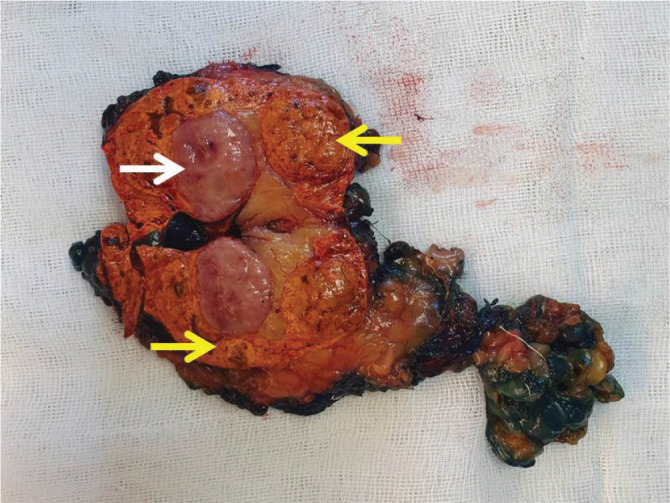
Macroscopic coexistence of adrenocortical nodular lesions and pheochromocytoma. Macroscopically; pheochromocytoma (white arrow) and micro-macronodular hyperplasia of adrenal tissue around this nodule (yellow arrows).
Figure 4.
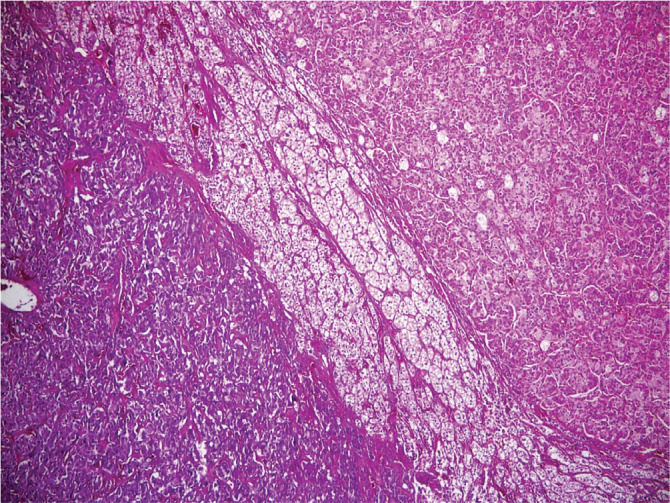
Hematoxylin- Eosin staining shows the association of pheochromocytoma and adrenocortical macronodular hyperplasia. Pheochromocytoma in the lower left and macronodular hyperplasia in the upper right (H&E x 40).
DISCUSSION
Pheochromocytoma with synchronous ACTH-independent CS originating from the same adrenal gland is very rare. There are various etiologies described in the literature which demonstrate the coexistence of adrenal adenoma and PCC (4–6), ACTH-secreting PCC (7–9), corticomedullary mixed tumors (10,11) and the coexistence of adrenocortical hyperplasia and PCC (12–14). To the best of our knowledge, our case is the first one describing the coexistence of overt ACTH-independent CS due to PBMAH and metachronous PCC.
The coexistence of ACTH-independent CS with primary adrenocortical hyperplasia and PCC has been shown in only 3 different case reports in which all of them demonstrated unilateral adrenocortical hyperplasia. The first one was reported in 1960, representing a case who was admitted to the hospital with weight loss, hirsutism, polydipsia, polyuria and emotional lability (14). Despite the absence of clinical symptoms/findings, lab results were compatible with CS and PCC. The patient underwent a left adrenalectomy after the suprarenal mass was revealed by intravenous urogram and histopathological examination revealed a benign PCC coexisting with hyperplastic cortex. Patients with ACTH-independent CS originating from only one adrenal gland generally required glucocorticoid therapy for an extended period of time following removal of that hyperfunctioning adrenal gland; however, similar to our presented patient, in this case report, she did not require any glucocorticoid replacement postoperatively. As a result, it was predicted that she had bilateral adrenal hyperplasia.
The latter two case reports interestingly describe two different Turkish women. The patient represented by Finkenstedt et al. had evident ACTH-independent CS and PCC(13). Unilateral macronodular adrenal hyperplasia was displayed by adrenal MRI, but unexpectedly, adrenal cortex was reported as totally normal by histopathology. The pathophysiologic mechanism here was uncertain, but it was hypothesized that adrenocortical tissue was stimulated by IL-6, since pathological examination revealed IL-6 positive immunostaining of PCC. The third and final case was reported in 2005, demonstrating a 52-year-old woman who was complaining solely of heart palpitations and heat intolerance without any feature of CS (12). Laboratory outcomes were suggestive of PCC and possibly subclinical CS (ACTH<5pg/mL and serum cortisol level was suppressed to 3.5 µg/dL following 2-day 2-mg DST with normal 24-h urinary free cortisol and diurnal rhythm of cortisol) and a CT revealed a 4cm solid mass in the right adrenal gland. After a right adrenalectomy, histopathological examination displayed PCC and adrenocortical hyperplasia.
ACTH-secreting PCC, also called ectopic ACTH syndrome due to PCC, is the most common cause of all defined etiologies. In a very recent case report, it was stated that to date, there were less than 100 cases with ACTH-secreting PCC reported (7). In a retrospective study, Falhammar et al. examined 164 cases with CS and only one of them (0.6%) was due to ACTH-secreting PCC. Hypercortisolism is a well-known pathologic mechanism which can induce adrenocortical hyperplasia. A case with bilateral nodular adrenal hyperplasia was reported in 1985 which was diagnosed with PCC and ACTH-dependent CS due to ACTH-secreting PCC (15). Furthermore, adrenocortical hyperplasia induced by excess epinephrine secretion from PCC was also indicated in a case report by Cope et al. (16). However, a rat research, conducted one year after that case report demonstrated that chronic administration of epinephrine did not stimulate developing adrenocortical hyperplasia (17). In our patient, bilateral macronodular hyperplasia was primary and developed much earlier than the catecholamine secretion from PCC.
A corticomedullary mixed tumor is another rare adrenal tumor and less than 20 patients were reported until now, in which 4 of them were diagnosed with mixed corticomedullary carcinoma (10,11). Lastly, the coexistence of PCC and adrenal adenoma causing overt CS is also very uncommon, described in only 2 case reports (5,6). In addition to that, Hasassri et al. retrospectively evaluated the data of 413 subjects with PCC and encountered only 16 patients with PCC and synchronous cortical adenoma (4). Only 4 of them indicated autonomous glucocorticoid secretion on biochemical analysis, though none of them presented any symptom/finding suggestive of CS.
In conclusion, to our knowledge, the patient presented here is the first case, diagnosed with overt CS due to PBMAH and PCC. Furthermore, considering the disease progression timeline and observing the metachronous occurrence of PCC makes our case report even more unique. The importance of detailed re-evaluation of patients with recurrent ACTH-independent CS is highlighted here. There is no doubt if the patient was not diagnosed with PCC preoperatively and gotten prepared properly prior to the second adrenalectomy, there could have been a more severe or even fatal outcome.
Acknowledgment
We would like to thank Internal Medicine residents and the rest of General Surgery team who contributed to the management of the patient.
Conflict of interest
The authors declare that they have no conflict of interest.
References
- 1.Lacroix A. ACTH-independent macronodular adrenal hyperplasia. Vol. 23. Best Practice and Research: Clinical Endocrinology and Metabolism. Baillière Tindall. 2009:245–259. doi: 10.1016/j.beem.2008.10.011. [DOI] [PubMed] [Google Scholar]
- 2.Farrugia FA, Charalampopoulos A. Pheochromocytoma. Endocr Regul. 2019;53(3):191–212. doi: 10.2478/enr-2019-0020. [DOI] [PubMed] [Google Scholar]
- 3.Manger WM, Gifford RW. Pheochromocytoma. J Clin Hypertens. 2002;4(1):62–72. doi: 10.1111/j.1524-6175.2002.01452.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hasassri ME, Pandian TK, Bobr AA, Bancos I, Young WF, Jr, Richards ML, Farley DR, Thompson GB, McKenzie TJ. Pheochromocytoma with Synchronous Ipsilateral Adrenal Cortical Adenoma. World J Surg. 2017;41(12):3147–3153. doi: 10.1007/s00268-017-4110-8. [DOI] [PubMed] [Google Scholar]
- 5.Higuchi K, Sakamoto S, Kato H, Takahashi Y, Tanaka T, Ichikawa T. Cushing’s tumor meets pheochromocytoma in one adrenal gland. Int J Urol. 2015;22(1):135–136. doi: 10.1111/iju.12620. [DOI] [PubMed] [Google Scholar]
- 6.Ghander C, Tenenbaum F, Tissier F, Silvera S, Lalej D, Dousset B, Groussin L. When adrenal Cushing’s and phaeochromocytoma meet. Lancet. 2012;380(9854):1683. doi: 10.1016/S0140-6736(12)60438-3. [DOI] [PubMed] [Google Scholar]
- 7.Lee MN, Wan WY, Chormanski DC, Kravchenko MI. Adrenal Adenoma Anarchy: A Case of an ACTH-Secreting Pheochromocytoma. Case Rep Endocrinol. 2020;2020:4869467. doi: 10.1155/2020/4869467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gabi JN, Milhem MM, Tovar YE, Karem ES, Gabi AY, Khthir RA. Severe Cushing Syndrome Due to an ACTH-Producing Pheochromocytoma: A Case Presentation and Review of the Literature. J Endocr Soc. 2018;2(7):621–630. doi: 10.1210/js.2018-00086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Falhammar H, Calissendorff J, Höybye C. Frequency of Cushing’s syndrome due to ACTH-secreting adrenal medullary lesions: a retrospective study over 10 years from a single center. Endocrine. 2017;55(1):296–302. doi: 10.1007/s12020-016-1127-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Donatini G, Van Slycke S, Aubert S, Carnaille B. Corticomedullary mixed tumor of the adrenal gland-A clinical and pathological chameleon: Case report and review of literature. Updates Surg. 2013;65(2):161–164. doi: 10.1007/s13304-011-0132-1. [DOI] [PubMed] [Google Scholar]
- 11.Alsabek MB, Alhmaidi R, Ghazzawi B, Hamed G, Alseoudi A. Mixed corticomedullary adrenal carcinoma–case report: Comparison in features, treatment and prognosis with the other two reported cases. Int J Surg Case Rep. 2017;31:254–261. doi: 10.1016/j.ijscr.2017.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Erem C, Hacihasanoglu A, Ersöz HO, Reis AK, Calik A, Ukinç K, Koçak M. Pheochromocytoma combined with pre-clinical Cushing’s syndrome in the same adrenal gland. J Endocrinol Invest. 2005;28(6):561–565. doi: 10.1007/BF03347247. [DOI] [PubMed] [Google Scholar]
- 13.Finkenstedt G, Gasser RW, Höfle G, Lhotta K, Kölle D, Gschwendtner A, Janetschek G. Pheochromocytoma and sub-clinical Cushing’s syndrome during pregnancy: Diagnosis, medical pre-treatment and cure by laparoscopic unilateral adrenalectomy. J Endocrinol Invest. 1999;22(7):551–557. doi: 10.1007/BF03343608. [DOI] [PubMed] [Google Scholar]
- 14.Williams GA, Crockett CL, Butler WWSS, Crispell KR. The coexistence of pheochromocytoma and adrenocortical hyperplasia. J Clin Endocrinol Metab. 1960;20(4):622–631. doi: 10.1210/jcem-20-4-622. [DOI] [PubMed] [Google Scholar]
- 15.Bruining HA, Ong EGL, Gershuny AR, Lamberts SWJ. Cushing’s syndrome and pheochromocytoma caused by an adrenal tumor, also containing met-enkephalin and somatostatin: A case report. World J Surg. 1985;9(4):639–641. doi: 10.1007/BF01656073. [DOI] [PubMed] [Google Scholar]
- 16.Cope O, Labbe JP, Raker JW, Bland EF. Pheochromocytoma and adrenal cortical adenoma. Report of a case with both tumors and discussion of their relation. J Clin Endocrinol Metab. 1952;12(7):875–880. doi: 10.1210/jcem-12-7-875. [DOI] [PubMed] [Google Scholar]
- 17.Coutinho HB, Baker BL, Ingle DJ. Effect of Continuous Injection of Epinephrine on Adrenal Cortex and Anterior Hypophysis. Proc Soc Exp Biol Med. 1953;84(1):137–140. doi: 10.3181/00379727-84-20567. [DOI] [PubMed] [Google Scholar]