

Abstract
Signal transducer and activator of transcription (Stat)3 is a valid anticancer therapeutic target. We have discovered a highly potent chemotype that amplifies the Stat3-inhibitory activity of lead compounds to levels previously unseen. The azetidine-based compounds, including H172 (9f) and H182, irreversibly bind to Stat3 and selectively inhibit Stat3 activity (IC50 0.38–0.98 μM) over Stat1 or Stat5 (IC50 > 15.8 μM) in vitro. Mass spectrometry detected the Stat3 cysteine peptides covalently bound to the azetidine compounds, and the key residues, Cys426 and Cys468, essential for the high potency inhibition, were confirmed by site-directed mutagenesis. In triple-negative breast cancer (TNBC) models, treatment with the azetidine compounds inhibited constitutive and ligand-induced Stat3 signaling, and induced loss of viable cells and tumor cell death, compared to no effect on the induction of Janus kinase (JAK)2, Src, epidermal growth factor receptor (EGFR), and other proteins, or weak effects on cells that do not harbor aberrantly-active Stat3. H120 (8e) and H182 as a single agent inhibited growth of TNBC xenografts, and H278 (hydrochloric acid salt of H182) in combination with radiation completely blocked mouse TNBC growth and improved survival in syngeneic models. We identify potent azetidine-based, selective, irreversible Stat3 inhibitors that inhibit TNBC growth in vivo.
Keywords: Signal transducer and activator of transcription, Small-molecule inhibitors, Covalent modification, Antitumor cell effects, Tumor growth inhibition
1. Introduction
Signal transducer and activator of transcription (Stat) protein family are cytoplasmic transcription factors that respond to cellular stimulation by cytokines and growth factors to promote cell growth and differentiation, inflammation, and immune responses [1]. Classically, upon cytokine and growth factor binding to their cognate receptors, the Stat proteins are activated via the phosphorylation of a critical tyrosine (Tyr, Y) residue by growth factor receptor Tyr kinases, Janus Kinases (JAKs) or Src family kinases. Phosphorylation in turn drives the dimerization of two Stat monomers through a reciprocal phospho-Tyr-Src Homology 2 (SH2) domain interaction. Stat:Stat dimers then translocate to the nucleus and bind to specific DNA-response elements in target gene promoters to regulate gene transcription. By functionally controlling gene expression, the Stat proteins regulate fundamental cellular processes [1].
Unlike classical Stat signaling, which is transient in non-transformed cells, the aberrant-activation of Stat3 occurs in malignant transformation and is implicated in breast and many other human cancers [2]. Aberrantly-active Stat3 promotes tumor progression in part through its direct dysregulation of gene expression and cross-talks with other key proteins, including nuclear factor (NF)-κB, leading to uncontrolled growth and survival of tumor cells, enhanced tumor angiogenesis, and tumor metastasis [2,3]. Stat3 functioning in immune cells in the tumor microenvironment further suppresses tumor immune surveillance [2,3] to promote tumor progression. On-going research continues to uncover unconventional pathways that are regulated by Stat3 to control cell survival and cell death, including the reports that Stat3 is localized in the mitochondria [4] and in the endoplasmic reticulum (ER) to regulate calcium flux [5].
Although Stat3 is a validated target for the discovery of novel anti-cancer drugs [3,6], the discovery and development of potent small molecule inhibitors has proved to be a significant challenge. As a result, to date, no small molecule direct Stat3 inhibitors are in clinical use. Nearly all the current inhibitors of Stat3 are low in potency (micro-molar) [3,7], except the recently reported PROTAC-Stat3 inhibitors, including SD-36, which degrades cellular Stat3, with nanomolar potency and inhibits tumor growth [8,9]. Moreover, most of the current Stat3 inhibitors have unclear mechanisms of inhibition of Stat3 activity, likely indirectly blocking Stat3 phosphorylation via Tyr kinases [3,7]. Most notably, there has been minimal focus on the studies that shed light on how Stat3 inhibitors alter the dynamics of Stat3 processing in tumor cells.
Systematic, hypothesis-driven medicinal chemistry efforts using the previously reported carboxylic acid-based small molecule Stat3 inhibitor leads [10–14] have led to the discovery of a new generation of highly potent and selective Stat3 inhibitors, including H120 (8e), H105 (8f), H172 (9f) [15], and H182. The azetidine compound series irreversibly bind to Stat3 and preferentially and directly inhibit Stat3 DNA-binding activity in vitro, as exemplified by H172 and H182, with IC50 values of 0.38–0.98 μM, and H120 and H105, with IC50 values of 1.75–2.07 μM. Treatment of triple negative breast cancer (TNBC) cells with the compounds inhibited both constitutive and ligand-induced Stat3 activation, and perinuclear aggregation, which led to tumor cell death. Oral or intra-peritoneal delivery of H120 or H182 as a single agent inhibited human TNBC xenografts growth, and H278 (H182 hydrochloric acid salt) combined with radiation therapy completely blocked tumor growth and prolonged survival in mouse TNBC syngeneic models that harbor aberrantly-active Stat3.
2. Materials and Methods
2.1. Chemical synthesis of H182 and H172
Synthesis and detailed characterization of H182 and H172 are described in Supplementary Information, “Methods”.
2.2. Cells and reagents
Mouse fibroblasts transformed by v-Src (NIH3T3/v-Src) or over-expressing the human epidermal growth factor (EGF) receptor (NIH3T3/hEGFR), human breast cancer cell line (MDA-MB-231), the immortalized normal human breast epithelial cell line (MCF-10A) have been previously reported [12,13]. Human breast cancer cells, MCF-7, MDA-MB-468, and HCC1937 were purchased from American Type Culture Collection (ATCC) on February 12, 2020, August 26, 2020, and January 7, 2021, respectively, with authentication and mycoplasma-free certifications. The normal mesothelial cells were obtained from Dr. Michele Carbone and previously reported [16], and the murine breast tumor line (E0771) was purchased from CH3 Biosystems (Amherst, NY) and has been previously reported [17] The E0771 cells were authenticated by short tandem repeat profiling, and species-specific PCR evaluation was performed to rule out any contamination by rat, human, hamster or monkey cells. Mycoplasma tests were conducted on MDA-MB-231 and MCF-10A by IDEXX BioAnalytics (Columbia, MO) in Dec 2019 and both were negative, E0771 cells were screened and certified to be free of pathogens (Mycoplasma, Theiler’s Murine Encephalomyelitis Virus Pneumonia Virus of Mice, Minute Virus of Mice, Mouse Hepatitis Virus, Mouse Parvovirus, Corynebacterium bovis and Sendai Virus), while tests have not been performed on the human mesothelial cells. MCF-10A cells were grown in Dulbecco’s modified Eagle’s medium (DMEM)/F12 with 5% horse serum plus EGF (20 ng/ml), insulin (10 μg/ml), hydrocortisone (0.5 mg/ml), and 100 ng/ml cholera toxin, the mesothelial cells were cultured in DMEM containing 20% fetal bovine serum (FBS) and 1% Penicillin/Streptomycin [16], and the E0771 cells were grown in DMEM with 10% FBS [17]. All other cells were cultured in DMEM plus 10% heat-inactivated FBS. Antibodies against Stat3, pY705-Stat3, pS727-Stat3, pY1173EGFR, EGFR, pY1007/1008JAK2, JAK2, pY416Src, Src, Shc, p-Shc, pS473Akt, Akt, pT202/Y204Erk1/2 (p44/42), Erk1/2, PARP, caspase 3, c-Myc, survivin, β-actin, and α/β-tubulin were purchased from Cell Signaling Technology, Inc. (Danvers, MA), with the exception of VEGF, and GAPDH antibodies, which were purchased from Santa Cruz Biotechnology (Dallas, Texas). Cisplatin and docetaxel were purchased from Sigma-Aldrich (St. Louis, MO).
2.3. Nuclear extract preparation, gel shift assays, and densitometric analysis
These studies were carried out as previously described [11–13] using the 32P-labeled oligonucleotide probes, hSIE (high affinity sis-inducible element from the c-fos gene, m67 variant, 5′-AGCTTCATTTCCCGTAAATCCCTA) that binds Stat1 and Stat3 [11,12] and the mammary gland factor element (MGFe) from the bovine β–casein gene promoter (sense strand, 5′-AGATTTCTAGFAATTCAA) that binds Stat5 and Stat1 [11,12]. Details are presented in Supplementary Information, “Methods”.
2.4. Luciferase reporter assay
These studies were performed as previously reported [10–12,18] using the v-Src-transformed NIH3T3/v-Src fibroblasts [10–13,18]. Details are presented in Supplementary Information, “Methods”.
2.5. Site directed mutagenesis to create Stat3 mutants
The prokaryotic pET28-hStat3(127–711)−6 × His plasmid construct that contains the DNA fragment coding for human Stat3 protein residues 127–711 (referring to NM_139276.3) with a C-terminal 6 × His tag, which is flanked by the restriction sites NdeI and XhoI, was kindly provided by Dr. Yuan Chen at the University of California, San Diego [19]. This construct was used to generate the mutants, Stat3C328A, Stat3C426A, Stat3C468A, and Stat3C542S, as described in detail in Supplementary Information “Methods”.
2.6. Protein expression and purification
The expression of un-phosphorylated and tyrosine phosphorylated His-tagged recombinant wtStat3 and mutant Stat3 proteins in BL21 (DE3) bacterial cells is described in detail in Supplementary Information “Methods”.
2.7. SDS-PAGE/Western blotting analysis
These studies were performed as previously described [11–13]. Details are presented in Supplementary Information, “Methods”.
2.8. Cell viability assays
CyQuant assay was performed to evaluate compounds as previously reported [11–13]. Details are presented in Supplementary Information, “Methods”.
2.9. Soft-agar colony formation and clonogenic survival assays
These studies were performed as previously reported [11–13]. Details are presented in Supplementary Information, “Methods”.
2.10. Confocal microscopy studies
These studies were performed as previously reported [18]. Details are presented in Supplementary Information, “Methods”.
2.11. Fluorescence polarization assay (FP)
These studies were conducted as previously reported [11,12] using the labeled phospho-peptide, 5-carboxyfluorescein GpYLPQTV-NH2 (where pY represents phospho-Tyr) as probe and purified recombinant Stat3, with some modifications. Details are presented in Supplementary Information, “Methods”.
2.12. Isothermal titration calorimetry (ITC)
The ITC experiment was carried out as previously described [15,20]. Details are provided in Supplementary Information, “Methods”.
2.13. Wound healing assay for migration
Studies were performed as previously reported [11–13]. Details are presented in Supplementary Information, “Methods”.
2.14. Annexin V binding/apoptosis and flow cytometric analysis
For apoptosis analysis, annexin V/propidium iodide (PI) staining was performed. Details are presented in Supplementary Information, “Methods”.
2.15. Assessment of physicochemical properties
Solubility, human (HLM) and mouse liver microsomal (MLM), and plasma stability studies were performed by Eurofins Cerep Panlabs and Eurofin Discovery Services (www.eurofins.com/PharmaDiscovery), while protein binding studies were performed by Sanford Burnham Prebys Chemical Biology & Drug Discovery Core using standard protocols.
2.16. Mass-spectrometry study for the detection of covalently-bound peptide adducts
Mass Spectrometry studies to detect the peptides bound to Stat3 inhibitors are described in detail in Supplementary Information “Methods”.
2.17. Mice and in vivo tumor studies
Mice were housed in specific pathogen-free conditions in the animal facility. All animal experiments were conducted in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals on a protocol approved by the Institutional Animal Care and Use Committee (IACUC). Subcutaneous xenograft studies were performed as previously reported [12]. Five-week-old female athymic nude mice were purchased from Envigo (Indianapolis, IN) or Charles River (Wilmington, MA) and maintained in the institutional animal facilities approved by the American Association for Accreditation of Laboratory Animal Care. Athymic nude mice were injected in the right flank area with human breast cancer, MDA-MB-231 cells (5 × 106) in 100 μL of PBS, and when tumors have reached 80–120 mm3 (measured by calipers) animals were grouped so that the mean tumor sizes of each group were nearly identical and tumor-bearing mice were treated with different regimen in separate studies as follows: tumor-bearing mice were administered H182 via oral gavage every day at 5 mg/kg for 19 days and 20 mg/kg for additional 14 days, H182 via intraperitoneal (IP) injection every other day at 10 mg/kg for 33 days, and H120 via oral gavage every day at 10–30 mg/kg for 28 days. Tumor sizes were monitored every 3–4 days, measured with calipers, and converted to tumor volume, V, according to the formula V = 0.52 × a2 × b, where a, smallest superficial diameter, and b, largest superficial diameter. For each treatment group, the tumor volumes for each set of measurements were statistically analyzed relative to the control (DMSO-treated; 70% DMSO with sterile PBS) group.
For syngeneic models, 6–10-week-old female C57BL/6 mice were purchased from Jackson Labs (Bar Harbor, ME) and used for the experiments. To generate tumors, 1 × 106 E0771 murine breast cancer cells, which are syngeneic to C57BL/6, were injected in the mammary gland [17]. For analysis of the experimental murine data, mice with insufficient or missing data were censored for the analysis. Radiation was delivered to the tumor only using the X-RAD SmART (PXi, Precision X-Ray, North Branford, CT) once tumors reached 800–1000 mm3. Mice were anesthetized with isoflurane and then placed inside the shielded cabinet. A computed tomography (CT) scan was then performed to determine the location of the tumor. Once the location was determined on the CT, the treatment beams were set up on the mice using the accompanying planning software package (SmART Advanced Treatment Planning System, Precision X-Ray). Collimated opposed tangential beams with an energy of 225 kV were utilized to treat the tumor and minimize the dose to the surrounding normal tissue. The Stat3 inhibitor, H278 (HCl salt of H182) was prepared and delivered intraperitoneally at a concentration of 10 mg/kg. H278 was diluted in a 70% DMSO with sterile PBS and prepared from a 100 mM DMSO stock. Mice were given H278 at 10 mg/kg intraperitoneally every other day alone and in combination with radiation starting 3 days prior to RT and continuing for the duration of treatment. For tumor growth, all measurements at each time point for a given treatment group were averaged and a standard error was calculated. Comparisons between groups were then done using ANOVA with post-hoc analysis. Survival curves were generated using the IACUC mandated endpoint of when a tumor reached >3500 mm3 (or > 20 mm in any one dimension). Mice that reached experimental endpoint (harvested for analysis), but not survival endpoint were censored from that time point forward. Comparisons between groups were then performed using a Log-Rank test. Standard star notations (**p < 0.01) were used to designated level of significance for all comparisons.
3. Results
3.1. Discovery of azetidine lead compound H182 from medicinal chemistry/SAR-directed optimization of carboxylic acid-based Stat3 inhibitors
The insight from having worked extensively on the carboxylic acid-based leads, BP-1–102 [12], SH4–54 and SH5–07 [13] led us to design a new series of structurally unique inhibitors, H120, H105, H172, and H182, which contain a key azetidine ring (Fig. 1A). Compounds, H120, H105, and H172 with the nomenclature 8e, 8f, and 9f, and details of how the novel azetidine-based inhibitors were derived are disclosed in our previous articles [14,15]. Additional information on the synthesis and characterization of H182 and H172 is provided in the Supplementary Information “Methods”.
Fig. 1. Compounds, H105, H120, H172, and H182, and their effects against in vitro Stats DNA-binding activity, and H182 interactions with Stat3.

(A) Structures of H105, H120, H182, and H172; (B, C) EMSA analysis of Stats DNA-binding activities in nuclear extracts of equal total protein containing activated (B) Stat3, or (C) Stat1, Stat3, and Stat5 pre-incubated with increasing concentration of the indicated compounds for 30 min at room temperature prior to incubation with the radiolabeled hSIE probe that binds Stat1 and Stat3 or the MGFe probe that binds Stat1 and Stat5 and then subjecting to native gel electrophoresis; and (D) Isothermal titration calorimetry and the binding isotherm from the integrated thermogram fit using the one-site model in the PEAQ-ITC software generated from the titration of H182 into Stat3, and the signature plots of the thermodynamics parameters, ΔG, ΔH, and −TΔS. Positions of Stats:DNA complexes in gel are labeled; control lanes (0) represent nuclear extracts pre-treated with 10% DMSO. Bands corresponding to Stats:DNA complexes were quantified using ImageJ, calculated as percent of control, and plotted against concentration to derive IC 50 values, which are shown. Data are representative of 2–4 independent determinations.
3.2. Azetidine functionalized compounds showed higher potency and selectivity against Stat3:Stat3 DNA-binding activity in vitro over that of Stat1 or Stat5 activity
The activities of H105, H120, H172, and H182 against Stat3, Stat1, or Stat5 DNA-binding activity in vitro were determined by the standard electrophoretic mobility shift assay (EMSA) [11–13]. In this assay, nuclear extracts of equal total protein containing activated Stat3 prepared from NIH3T3/v-Src fibroblasts, or containing activated Stat1, Stat3 and Stat5 prepared from epidermal growth factor (EGF)-stimulated NIH3-T3/hEGFR fibroblasts were pre-incubated with increasing concentration of the compounds at room temperature for 30 min, prior to incubation with the radiolabeled high-affinity sis-inducible element (hSIE) probe that binds Stat3 and Stat1 or the mammary gland factor element (MGFe) probe that binds Stat1 and Stat5 and subjecting to gel shift analysis [10, 12]. Results showed a dose-dependent inhibition of Stat3 DNA-binding activity (Fig. 1B), with IC50 values of 0.66 ± 0.10, 0.98 ± 0.05, 1.75 ± 0.19, and 2.07 ± 0.12 μM, respectively, for H182, H172, H120, and H105. The activities of H172 and H182 are much improved over those (IC50 values of 4–7 μM) of the previous lead inhibitors, BP-1–102, SH5–07 and SH4–54 [12,13]. These data also showed the preferential inhibition of Stat3:Stat3 DNA-binding activity, ahead of the inhibition of Stat1:Stat3 (IC50 of 3.4–8.3 μM), with far lower potencies against Stat1:Stat1 (IC50 > 15.8 μM) and Stat5:Stat5 activities (IC50 values > 19.1 μM) (Fig. 1C).
To demonstrate that H182 and the azetidine compounds directly bind to Stat3, we performed isothermal titration calorimetry (ITC) studies [20] using purified recombinant Stat3 (rStat3), which contains the core fragment of human Stat3 (amino acids 127 to 711) (Fig. 3A), with a C-terminal 6 × His tag [19]. The binding isotherm from the integrated thermogram fit, using the one-site model in the PEAQ-ITC software, generated from the titration of H182 into rStat3 derived the thermodynamics parameters, ΔH of −17.4 kJ/mol, −TΔS of −15.2 kJ/mol, a ΔG of −32.6 kJ/mol (Fig. 1D), suggesting spontaneity of the binding. The ITC study derived a KD of 1.97 μM (Fig. 1D). Together these data indicate that the azetidine-based inhibitors directly bind to Stat3.
Fig. 3. Site-directed mutagenesis identifies cysteine residues in Stat3 that covalently interact with the azetidine inhibitors.

(A) Stat3 domain structure and the cysteine residues identified by mass spectrometry to interact with H098 and H182; NTD, N-terminal domain, CCD, coiled-coil domain, DBD, DNA-binding domain, LD, Linker domain, SH2, Src homology-2 domain, and TAD, transcriptional activation domain, P, phosphate; (B) Stat3 DNA-binding activities in lysates of equal total protein containing pure (i) wild-type (wt) pYStat3, or the mutant pYStat3C328A, pYStat3C426A, pYStat3C468A, or pYStat3C542S pre-incubated with 0–3 μM H182, or (ii) pYStat3C426A or pYStat3C468A pre-incubated with 0–50 μM H182 for 30 min at room temperature prior to incubation with the radiolabeled hSIE probe that binds Stat3, and then subjecting to native gel electrophoresis (upper); protein loading shown by SDS-PAGE (lower); and (C) i) Crystal representation of Stat3 monomer bound to DNA (magenta), highlighting the Cys468 residue (green), which is in the binding domain, within 4.2 Å of direct contact with DNA (PDB code 1BG1), and ii) Modeling representation of Stat3 with Cys468 (green) covalently bound to H182 low energy conformation (blue, with nitrogen in dark blue, oxygen in red, sulfur in yellow, and fluorine in green). (Modeling and visualization used MOE software). Positions of Stat3:DNA complexes or Coomassie-stained protein in gel are labeled; control lanes (0) represent extracts pre-treated with 10% DMSO. Bands corresponding to Stat3:DNA complexes were quantified using ImageJ, calculated as percent of control, and plotted against concentration from which IC50 values were derived. Data are representative of 2–3 independent determinations.
3.3. Covalent binding mechanism of action and irreversible inhibition of Stat3 activity from SAR analysis, supported by biochemical, mass spectrometry, and mutagenesis studies
Substituting the para-fluorine of the inhibitor perfluorophenyl-sulfonamide system strongly compromises binding to Stat3 [15]. For example, replacing the para-fluorine with Cl or H, as in H186 (11a) or H176 (11b), respectively led to loss of Stat3-inhibitory potency, while H142 (5p) [15], with the para-fluorine has strong Stat3-inhibitory activity, IC50 of 0.46 μM (Table 1). Results together suggest that the para-fluorine is essential to maintain high potency, possibly being displaced through a nucleophilic aromatic substitution (SNAr) mechanism of action [15]. Inhibition through a covalent binding mechanism is time-dependent [21,22]. We hypothesized that the potency of inhibition by the azetidines would be influenced by the duration of Stat3 interaction with the azetidines. To investigate the time-dependency of the inhibition, we used H182 to conduct a time-course EMSA analysis in which nuclear extracts of equal total protein were each incubated with 2 μM H182 for 10, 30, and 60 min, prior to incubation with the labeled hSIE probe and subjecting to gel shift analysis. Results showed a progressively stronger inhibition of Stat3 DNA-binding activity with increasing time of incubation from 10 to 60 min, with a complete inhibition at 60 min (Fig. 2Ai). The plot of the percent inhibition with time is shown in Fig. 2Aii. Dose-response studies of H182 against Stat3 DNA-binding activity for the 10 min and 60 min incubation times derived the IC50 values of 1.47 and 0.38 μM, respectively (Fig. 2B), while the IC50 value is 0.66 μM for 30 min incubation (Figs. 1B and 2B). We surmise that the longer incubation time allows for robust interactions between the compounds and Stat3, facilitating the covalent interaction of Stat3:compound with time to completion. The course of the inhibition by the azetidines was further probed. In this case, the Stat3 nuclear extracts were pre-incubated with the hSIE probe for 30 min (to promote the DNA-binding event) prior to the incubation with H182 for 30 min and performing EMSA analysis. H182 was unable to inhibit the pre-formed Stat3:DNA complex up to 100 μM (Fig. 2C), indicating that the Stat3 pre-complexation with the high-affinity oligonucleotide interferes with the ability of H182 to effectively engage with Stat3.
Table 1.
Replacement of p-fluorine with chlorine or hydrogen causes loss of STAT3 activity.

|
Fig. 2. Time-dependency of the inhibition of Stat3 DNA-binding activity by the azetidine compound, H182.

(A–C) EMSA analysis of Stat3 DNA-binding activities in nuclear extracts of equal total protein containing activated Stat3 pre-incubated with (A) 2 μM H182 for 3, 10, 30, and 60 min, or (B) 0.3–2 μM H182 for 10, 30, or 60 min at room temperature prior to incubation with the radiolabeled hSIE probe that binds Stat3; or (C) the hSIE probe for 30 min at room temperature prior to incubation with 0–10 μM H182 for 30 min then subjecting to native gel electrophoresis; Positions of Stat3:DNA complexes in gel are labeled; control lanes (0) represent nuclear extracts pre-treated with 10% DMSO. Bands corresponding to Stat3:DNA complexes were quantified using ImageJ, calculated as percent of control (% of con) and shown or plotted against concentration from which IC50 values were derived, which are shown, or the band intensity is plotted against the time of incubation and shown. Data are representative of 2–4 independent determinations.
We next employed nano-LC/MS/MS proteomics approach to study whether the azetidine compounds covalently interact with Stat3 protein’s cysteine residues in vitro and selected H098 (5a) [15] and H182 for these studies. We utilized the same rStat3 protein that was used in the ITC assay, which contains amino acids 127 to 711 and a total of 10 cysteine residues in its amino acid sequence. We needed to detect all these 10 cysteine residues with MS/MS spectra if possible, which could be challenging. Therefore, we used a comprehensive method by way of multiple enzymatic digestions and multiple MS/MS fragmentation techniques in order to maximize Stat3 sequence coverage. Overall, with these exhaustive efforts, we were able to achieve more than 75% sequence coverage with more than 2800 peptide spectrum matches (PSMs) at 5% false discovery rate (FDR), and 8 out of 10 cysteines were detected from these PSMs (data not shown). We hypothesized that if the compounds were able to interact with the sulfhydryl (-SH) group of the Cys residues covalently, each Cys residue will increase its mono-mass by 618.1448 and 627.1563 Da for H098 [15] and H182, respectively (Schemes 1 and 2). We used these mass increases as variable modifications for Cys to perform MASCOT database search and discovered that, indeed, several Cys residues were modified by the two compounds as summarized in Table 2. These experiments were repeated three or two times as indicated with “experimental reproducibility” in the table. The results indicate that compound H098 is able to react with Cys residues C328 and C426, and compound H182 can react with C468 and C542. Therefore, we conclude that compounds H098 and H182 have the ability to covalently interact with Stat3 protein in vitro.
Schemes 1 and 2.

Expected reaction of H098 (5a) or H182 with Stat3 Cys residue.
Table 2.
Cysteine residues in Stat3 protein that were modified by azetidine compounds and detected by MS.
| Compound name | Cys residue | m/z | Mr(expt) | Mr(calc) | ppm | Score | Expect | Peptide | Experimental reproducibility |
|---|---|---|---|---|---|---|---|---|---|
| H098 | C328 | 920.3475 | 1838.6804 | 1838.6800 | 0.23 | 39 | 0.0002 | F.VVERQPCMPM.H + 2 Oxidation (M); STAT3-H098 (C) | 3 out of 3 |
| C426 | 791.2959 | 1580.5772 | 1580.5687 | 5.38 | 22 | 0.0093 | N.GGRANCDASL.I + STAT3-H098 (C) | 3 out of 3 | |
| H182 | C468 | 1121.0018 | 2239.9891 | 2239.9880 | 0.51 | 37 | 0.00036 | H.SLPVVVISNICQMPN.A + STAT3- H182 (C) | 1 out of 2 |
| C542 | 711.2601 | 1420.5057 | 1420.4992 | 4.60 | 18 | 0.046 | Y.SGCQITW.A + STAT3-H182 (C) | 2 out of 2 |
Given the MS data, we conducted site-directed mutagenesis analysis for the effects of Cys mutations (Cys-to-Ala or Cys-to-Ser mutations) on the inhibitory activities or potencies of the compounds. We used the same rStat3 construct that contains the core fragment of human Stat3 protein from amino acids 127 to 711 [19], as shown in Fig. 3A and stated in the “Methods” section to generate the Cys mutants. Purified, phosphorylated wild-type (wt)Stat3, and phosphorylated mutants, Stat3C328A, Stat3C426A, Stat3C468A, and Stat3C542S protein samples of equal amounts were pre-incubated with or without 3 μM H182 for 30 min at room temperature prior to incubation with the radiolabeled hSIE probe that binds Stat3 and subjecting to EMSA analysis. Results showed strong inhibition of the DNA-binding activities of wtpYStat3, pYS-tatC328A, and pYStat3C542S by 3 μM H182, while the DNA-binding activities of pYStat3C426A and pYStat3C468A are unaffected (Fig. 3Bi, compare 0 and 3 μM). These results suggested that C426 and C468 might be key for H182 to bind to and inhibit Stat3 activity. Focusing on the C426 and C468, we next conducted dose-response studies for effects of H182 on Stat3C426A and Stat3C468A. EMSA analysis showed that 10 μM H182 induced near complete inhibition of pYStat3C426A activity, but had no effect on pYStat3C468A activity, which was instead inhibited by 30 μM H182, with IC50 values of 6 μM (pYStat3C426A) and 40 μM (pYStat3C468A), with respect to the inhibition by H182 (Fig. 3Bii). Equal proteins used in the DNA-binding assay/EMSA analysis are shown by SDS-PAGE Coomassie staining (Fig. 3B, lower). Thus, we conclude that though both C426 and C468 are contributing to the binding of Stat3 with the azetidine compounds, the results suggest the C468 represents the predominant residue driving the interactions with the H182. Ribbon diagram of Stat3 monomer bound to DNA (PDB code 1BG1) highlighting Cys468, which is within 4.2 Å of direct contact with DNA as generated by Molecular Operating Environment (MOE) software (MOE 2020.09, Chemical Computing Group, Montreal, Quebec, Canada (2021)) [23] are shown in Fig. 3C.
The Stat3:azetidine compound interaction was investigated further by focusing on how the compounds might interfere with Stat3 dimerization (Stat3:Stat3), which occurs via pTyr peptide:SH2 domain interaction. We employed the Stat3 fluorescence polarization (FP) assay [11, 12,24], which is designed to model the Stat3:Stat3 dimerization [25,26] and uses 5-carboxy fluorescein-labeled high affinity pTyr peptide, GpYLPQTV-NH2 to bind to the SH2 domain of pure recombinant Stat3 (5-fl-GpYLPQTV-NH2:Stat3 complex). The GpYPQTV-NH2 peptide is derived from the interleukin-6 receptor (IL-6R)/gp130 [24,27] and binds with high affinity (KD = 150 nM) to the SH2 domain [27]. As expected, in the FP assay, the presence of 0–10 μM of the unlabeled GpYLPQTV-NH2 peptide (at 30 min incubation with Stat3) led to a classical dose-dependent reduction of the FP signal down to the baseline of 45–50 mP, whether the FP signal measurements were taken at 10, 30, or 60 min after adding probe (Supplementary Fig. S1, GpYLPQTV-NH2), indicative of the disruption of pTyr:SH2 domain interaction [11,26]. By contrast, the presence of 0–10 μM H182 (at 10–60-min incubation with Stat3) led to fluctuations of the FP signal that are of a different pattern from the dose-dependent reduction of the FP signal observed for the unlabeled GpYLPQTV-NH2 peptide (Supplementary Fig. S1, H182), despite the high inhibitory potency (Fig. 1B). Altogether, the Stat3 DNA-binding/EMSA analysis, ITC, and FP results indicate that the azetidine compounds directly bind to Stat3 in a unique and distinct way from SH2 domain-binding molecules.
3.4. Azetidine compounds inhibit constitutive and ligand-induced Stat3 activation and block Stat3 nuclear accumulation with no change in Stat3 protein levels in breast cancer cells
The breast cancer lines, MDA-MB-468 and MDA-MB-231, which harbor constitutively-active Stat3 [3,13], were used in both dose-response (treated with 0–5 μM H182 or H172 for 1 or 2 h) and time-course studies (treated with 1 or 3 μM H182 and H172, or 5 μM H120 and H105 for 0–24 h) to evaluate the effects of the inhibitors on Stat3 activation. Nuclear extracts and whole-cell lysates were prepared, and samples of equal total protein were subjected to Stat3 DNA-binding assay/EMSA analysis using the hSIE probe, or to SDS/PAGE and Western blotting analysis for pY705Stat3, Stat3, or GAPDH [11,12]. H182 and H172 treatment led to dose-dependent inhibition of constitutive Stat3 DNA-binding activity (Fig. 4A, Supplementary Fig. S2B) and Tyr phosphorylation (Fig. 4B, Supplementary Fig. S2A, pYStat3). Depending on the concentration (1, 3, or 5 μM), the inhibitory effects of the azetidine compounds were observed relatively early, at 30–60 min, on both Stat3 DNA-binding activity (Fig. 4C) and Tyr phosphorylation (Fig. 4D, pY705Stat3). Treatment of cells with the higher, 5 μM concentration leads to an even earlier inhibition of Stat3 activity, as early as 8–15 min (Supplementary Fig. S2A, H120). For the treatment with 1 μM H182, or with the relatively weaker H105, we note that there was a rebound of the pY705Stat3 levels at later time points (3–24 h) (Fig. 4Di, Supplementary Fig. S2A, H105), which was not the case when the cells were treated with the higher concentration 3 μM H182 that caused a sustained inhibition of Stat3 activation for up to 48 h (Fig. 4Dii).
Fig. 4. Effects of azetidine-based compounds on the induction of Stat3 phosphorylation, DNA-binding and transcriptional activities in breast cancer cells.

(A, C) Nuclear extracts of equal total protein prepared from the indicated breast cancer lines untreated (DMSO, 0) or treated with H172 or H182 at 1–5 μM for 1 h (A), or 1 μM for 0.5–6 h (C) and subjected to EMSA analysis of Stat3 DNA-binding activity using the radiolabeled hSIE probe that binds Stat3; (B, D, E, G) SDS-PAGE and immunoblotting analysis of whole-cell lysates of equal total protein prepared from the indicated human breast cancer lines untreated (DMSO, 0, -) or (B) treated with 0.5–3 μM H172 or H182 for 2 h, (D) treated with 1 or 3 μM H182 for 0.5–48 h, (E) treated with 1 or 2 μM H182 for 0.5–48 h; or (G) serum-starved for 24 h and pre-treated with 1–3 μM H182 for 1 h prior to stimulation with 100 ng/ml EGF for 12 min, and probing for pY705Stat3, Stat3, tubulin, or GAPDH; (F) Immunofluorescence staining with laser-scanning confocal microscopy analysis of MDA-MB-231 and MDA-MB-468 lines untreated (0) or treated with 1–2 μM H182 for 12 h, stained for DAPI (blue) and Stat3 (red). Images were captured for DAPI, Stat3, and merged; and (H) Luciferase reporter activity in the cytosolic extracts of equal total protein prepared from the v-Src-transformed mouse fibroblasts, NIH3T3/v-Src transiently transfected with the Stat3-dependent luciferase reporter, pLucTKS3 or Stat3-independent luciferase reporter, pLucSRE, untreated (DMSO, 0) or treated with 1–3 μM H182 for 1 h. Luciferase reporter activity was assayed with a luminometer, normalized to protein concentration, and plotted as percent of control. Positions of Stat3:DNA complex or proteins in gel are labeled; control lane (0, -) represents nuclear extracts, whole-cell lysates, or cytosolic extracts prepared from 0.5% DMSO-treated cells. Values, mean ± S.D., n = 3, *p < 0.05. Bands corresponding to Stat3:DNA complexes or proteins in gel were quantified using ImageJ, calculated as percent of control, and shown. Data are representative of 2–4 independent determinations.
During confluence of cultured cells, the cell-to-cell adhesion and cadherin engagement trigger increased Tyr705Stat3 phosphorylation in a variety of cell lines [28,29]. To investigate the impact of cell-to-cell adhesion and cadherin engagement, parallel studies were conducted in which cells in culture were untreated or treated with 1 or 2 μM H182 for 0.5–48 h, and whole-cell lysates prepared and immunoprobed for pTyr705Stat3. Results showed that with longer duration of culture the pTyr705Stat3 levels increased (Fig. 4E), and that comparing the untreated versus the treated side-by-side, the treatment with H182 led to inhibition of pTyr705Stat3 (Fig. 4E). The inhibition of pTyr705Stat3 at 48-h treatment with H182 was minimal at the concentration of 2 μM and undetectable at 1 μM (Fig. 4E). After 48-h incubation, the cell confluency of the DMSO-treated samples reached to around 60% for MDA-MB-468 cells and 80% for MDA-MB-231 cells. These results indicate that cell-to-cell adhesion and cadherin engagement are part of the mechanism for the re-bounce in pTyr705Stat3 levels following prolonged treatment. We next determined the effects of the azetidine inhibitors on the intracellular localization of Stat3 by performing immunofluorescence staining with laser-scanning confocal microscopy analysis on both MDA-MB-231 and MDA-MB-468 lines treated with H182 for 12 h. In the untreated cells, Stat3 (red punctate) staining was predominantly localized in the nucleus of the tumor cells, with weak presence in the cytoplasm (Fig. 4F, 0, Stat3, red merged with blue DAPI nuclear staining). Treatment with 1 or 2 μM H182 led to decreased nuclear Stat3 staining and Stat3 aggregation at the perinuclear region (Fig. 4F, H182, Stat3, red merged with blue DAPI nuclear staining). These results indicate that the azetidine compounds disrupt Stat3 nuclear accumulation and promote the aggregation of Stat3 at the perinuclear region, consistent with our previous studies [18]. Treatment with 1–3 μM H182 further inhibited EGF-induced pY705Stat3 in MDA-MB-468 cells in a dose-dependent manner (Fig. 4G). Total Stat3 protein levels in all of these studies were unchanged, and GAPDH, tubulin or β-actin levels show equal protein loading. The percent changes induced by H182 treatment on Stat3 DNA-binding activity and phosphorylation in the treated cells are shown.
We next evaluated the effects of the azetidine compound, H182 on Stat3 transcriptional activity. We used mouse fibroblasts transformed by the viral Src oncoprotein (NIH3T3/vSrc) that harbor constitutive Stat3 activation, which were transiently transfected with the Stat3-dependent luciferase reporter, pLucTKS3 [25,30]. Transfected cells were treated with H182 and the luciferase activity was assayed in the cytosolic extract preparations of equal total protein [12,13,30]. Results showed dose-dependent inhibition of the Stat3-driven luciferase reporter expression by the treatment of cells with 1–3 μM H182 for 1 h (Fig. 4H, pLuckTKS3), with significant (p < 0.05) decreases at 2 and 3 μM treatments. By contrast, the luciferase reporter assay of the cytosolic extracts from NIH3T3/vSrc fibroblasts transiently transfected with the Stat3-independent pLucSRE luciferase reporter [25,30] showed no changes in the expression of the reporter following similar treatments with H182 (Fig. 4H, pLucSRE).
3.5. Azetidine compounds have little or no effects on pS727Stat3, Stat1, JAK2, EGFR, Shc, Erk1/2, Src, or Akt induction in breast cancer cells
We sought to assess any non-specific effects of the azetidine compounds. Breast cancer MDA-MB-231 and MDA-MB-468 lines were treated with H182 for 2 h at concentrations of 0–3 μM that inhibit constitutive and ligand-induced Stat3 activation, stimulated or not with EGF for 12 min, and whole-cell lysates were prepared. Samples of equal total protein were subjected to SDS-PAGE and Western blotting analysis. Treatment with 0.5–3 μM H182 for 2 h had no inhibitory effect on the constitutive or EGF-induced pS727Stat3, pYStat1, pY416Src, pY1068EGFR, pShc, pErk1/2MAPK, pS473Akt, and pJAK2 in TNBC cells (Fig. 5A–C), while pY705Stat3 was inhibited (Fig. 5C pYStat3) in the TNBC cells. The total protein levels were unchanged, and the tubulin, β-actin, and GAPDH levels show equal protein loading.
Fig. 5. Lack of non-specific effects on other signaling pathways in breast cancer cells.

SDS-PAGE and immunoblotting analysis of whole-cell lysates of equal total protein prepared from the indicated human breast cancer lines untreated (DMSO, 0, -) or treated with (A, C) 0.5–3 μM H182 for 2 h, or (B) 1–3 μM H182 for 1 h prior to stimulation with 100 ng/ml EGF for 12 min, and probing for pY705Stat3, pS727Stat3, Stat3, pYStat1, Stat1, pY1068EGFR, EGFR, pJAK2, JAK2, pY416Src, Src, pShc, Shc, pErk1/2, Erk1/2, pS473Akt, Akt, or GAPDH, tubulin, and β-Actin. Positions of proteins in gel are labeled; control lane (0, -) represents whole-cell lysates prepared from 0.5% DMSO-treated cells. Data are representative of 3–4 independent determinations.
3.6. Compounds inhibited anchorage-dependent and independent growth, induced apoptosis, and suppressed Stat3 target gene expression in TNBC cells harboring constitutively-active Stat3
Aberrantly-active Stat3 promotes tumor cell growth and proliferation, survival, migration, invasion, and metastasis [2,3]. We investigated the antitumor effects of the azetidine analogs by treating human breast cancer MDA-MB-231 and MDA-MB-468 lines harboring aberrantly-active Stat3 with increasing concentrations of H105, H120, H172, or H182 for 72 h and performing CyQuant assay for viable cell numbers [13]. All four compounds dose-dependently reduced viable cell numbers (Fig. 6Ai and ii, Supplementary Fig. S3B). H182 had the highest potency against both lines, with an IC50 of 1 μM (Fig. 6Ai and ii), H172 was 2-fold more potent against MDA-MB-468 (IC50 of 1.5 μM) than against MDA-MB-231 (IC50 of 3.3 μM) (Fig. 6Ai and ii), while H120 and H105 had IC50 range of 2.6–3.6 μM (Supplementary Fig. S3B). The combined treatment with 1 μM H182 and increasing doses of docetaxel [31] or cisplatin [32] led to a shift of the chemotherapeutic agents’ dose-response curves to the left (Fig. 6B), indicating enhanced responses to the chemotherapeutic drugs when used in combination with H182. The IC50 values improved from 1.5 to 0.3 μM for docetaxel alone and docetaxel with H182, respectively, and from 7.0 to 3.2 μM for cisplatin alone and cisplatin with H182, respectively (Fig. 6B). These results are consistent with the role of aberrantly-active Stat3 in suppressing drug sensitivity and promoting drug resistance [33], and together suggest that the azetidine compounds may be used in combination with chemotherapy to enhance therapeutic response. By contrast, treatments with the azetidine compounds showed relatively weaker effects on the viable cell numbers of cells that do not harbor aberrantly-active Stat3. The potencies (IC50s) of H172 and H182 are 3.8–4.0 μM on normal human breast epithelial MCF-10A cells, 3.6–4.3 μM on breast cancer HCC1937 cells (Fig. 6Aiii, iv), and 4.9–9.2 μM on normal human mesothelial cells (Supplementary Fig. S3A), while the IC50s of H120 and H105 on MCF-7 or MCF-10A cells are 7.7 - >10 μM (Supplementary Fig. S3C). Furthermore, treatment of MDA-MB-231 cells with 0.3–2 μM H182 potently and dose-dependently inhibited both anchorage-independent growth in soft-agar (Fig. 6C), and anchorage-dependent colony formation (Fig. 6D). In a scratch assay, one-time treatment of MDA-MB-231 cells with H182 inhibited the cell migration into the denuded area within 22 h of treatment (Fig. 6E). Immunoblotting analysis of whole-cell lysates from MDA-MB-231 cells treated with H182, single concentration (1 μM) for 0–24 h or increasing concentration for 2 h showed moderate levels of cleaved poly ADP-ribose polymerase (PARP) and caspase 3 (Fig. 6Fi and ii), while annexin V/propidium iodide (PI) staining with flow cytometric analysis showed roughly 12% of double-positive staining for cells treated with 3 μM H182 for 3 h (Fig. 6Fiii; Supplementary Fig. S3D, H182, 3 μM) indicative of late apoptosis. Moreover, consistent with constitutively-active Stat3’s role in dysregulating gene expression [1–3], immunoblotting analysis showed that the expression of classical Stat3 downstream genes, including c-Myc, survivin and vascular endothelial growth factor (VEGF), was rapidly suppressed in MDA-MB-231 cells in response to H182 treatment at doses that inhibit Stat3 activity (Supplementary Fig. S4). Together these studies show that the azetidine compounds inhibit aberrantly-active Stat3 signaling and functions in TNBC cells and induce antitumor cell effects in vitro.
Fig. 6. Azetidine-based compounds suppress anchorage-dependent and independent growth, colony survival, and migration, and induce apoptosis of breast cancer cells harboring aberrantly-active Stat3.
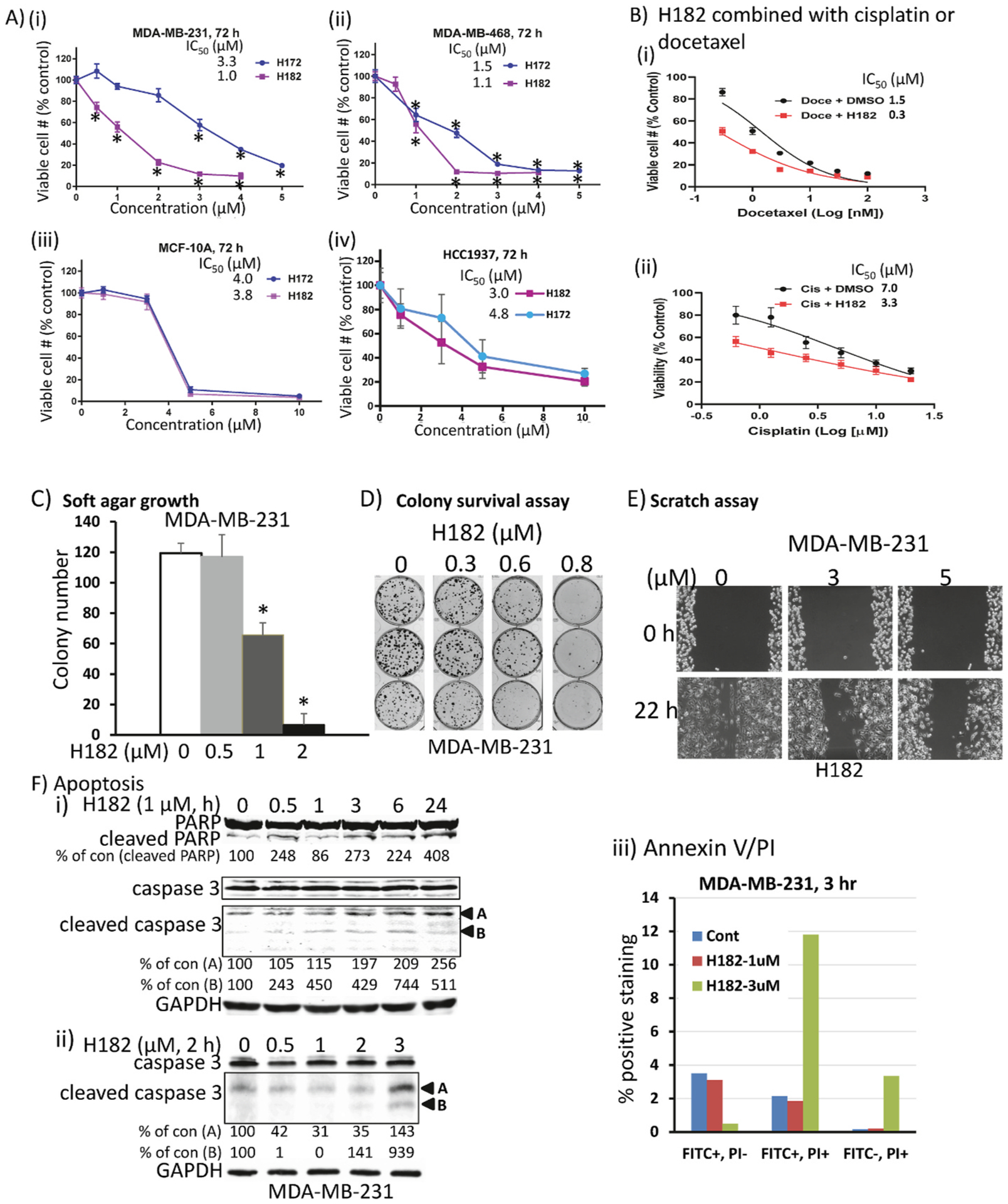
(A) Human breast camcer, MDA-MB-231 and MDA-MB-468 cells (i and ii) that harbor constitutively-active Stat3 or normal human breast epithelial, MCF-10A or breast cancer HCC1937 cells (iii and iv) that do not in 96-well culture were treated once with increasing concentration of H172 or H182 for 72 h, or (B) MDA-MB-231 cells in 96-well culture, untreated (DMSO) or pre-treated with 1 μM H182 for 6 h prior to treating with 0–100 nM docetaxel (i) or 0–20 μM cisplatin (ii) for a total of 72 h, and viable cell numbers were assayed using CyQuant cell proliferation kit and plotted as % viable cell numbers against concentration, from which IC50 values were derived; (C–E) MDA-MB-231 cells seeded (C) in soft-agar were treated every 3–4 days with 0–2 μM H182 and allowed to grow until large colonies were visible, which were stained with crystal violet, counted and plotted, (D) as single-cell culture and treated once with 0–0.8 μM H182 and allowed to culture until large colonies were visible, which were stained with crystal violet and imaged, or (E) in culture and wounded, and treated once with 0–5 μM H182, and allowed to migrate to the denuded area over a 22-h period and imaged; and (F) MDA-MB-231 cells in culture treated once with H182 at (i) 1 μM for 0–24 h or (ii) 0–3 μM for 2 h, and whole-cell lysates prepared and subjected to SDS-PAGE and immunoblotting analysis for PARP, cleaved PARP, caspase 3, cleaved caspase 3, and GAPDH, or (iii) 0–3 μM for 3 h, and samples prepared and processed for Annexin V binding and flow cytometry. Positions of proteins in gel are labeled; control (0) represents samples from 0.5% DMSO-treated cells. Values, mean ± S.D., n = 3–6, *p < 0.01. Data are representative of 3 independent determinations.
3.7. H120 and H182 inhibited growth of breast tumors in mice, enhanced response to radiation and prolonged survival
Constitutively-active Stat3 promotes primary tumor development and the progression to therapy resistance [3]. We proceeded to evaluate the in vivo antitumor efficacy against tumor growth in both human xenograft and mouse syngeneic models of TNBC. In four separate studies, we tested the efficacy of H182, H278 (HCl salt of H182), and H120. For these xenografts, the human breast cancer, MDA-MB-231 cells were injected subcutaneously into athymic nude mice. When tumors were palpable (80–120 mm3), mice were grouped so that the mean tumor sizes were identical between groups and treated as follows: A) intraperitoneal (IP) injections of H182 at 10 mg/kg every other day for 33 days; B) oral gavage of H182 every day at 5 mg/kg for 20 days, and then increased to 20 mg/kg for additional 12 days; and C) oral gavage of H120 at 20 mg/kg, daily for 14 days, and then increased to 30 mg/kg, daily for additional 14 days. These doses were chosen based in part on our previous studies with the earlier lead inhibitors [12,34]. In all three independent studies, significant tumor growth inhibition was observed for H182 and H120 (Fig. 7Ai–Ci). No notable changes in body weights (Fig 7Aii-Cii) or obvious signs of toxicity, such as anorexia, lethargy or change in body condition score were observed.
Fig. 7. In vivo antitumor efficacy against the growth of breast cancer xenografts and syngeneic tumors in mice.

(A–C) Plots of tumor growth (i) and body weights (ii) of mice bearing subcutaneous human breast, MDA-MB-231 tumor xenografts. Anti-tumor effects of A) H182 administered via i.p., 10 mg/kg, every 2 or 3 days (*p < 0.05; values, mean ± S.D., n = 5; vehicle, 70/30, DMSO/normal saline v/v), B) H182 administered via oral gavage, 5 mg/kg, every day for 20 days, followed by 20 mg/kg every day for 12 days (**p < 0.01; values, mean ± SEM., n = 5; vehicle, 60/40, DMSO/normal saline v/v), or C) H120 administered via oral gavage, 20 mg/kg, every day for 14 days, followed by 30 mg/kg every day for 14 days (*p < 0.05; values, mean ± S.D., n = 5; vehicle, 5/95, DMSO/sesame oil v/v), compared to vehicle alone (control); and D) mouse bearing orthotopic E0771 mammary tumors were treated with H278, 10 mg/kg, via i.p., every other day, and after one week, a localized kV irradiation (16 Gy) was added or not, and the H278 treatment was continued for the duration of study. Mean tumor burden ± SEM (i) and survival (ii) for the indicated treatments were assessed every 3 days until endpoint (**p < 0.01, mean ± S.D., n = 3 (WT), 3 (vehicle), 5 (vehicle + RT), 7 (H278), and 7 (H278 + RT), vehicle alone (control, 70/30, DMSO/normal saline v/v).
Given that Stat3 activity promotes chemotherapy resistance [35,36], and the results that H182 in combination with cisplatin or docetaxel had a synergistic effect against breast cancer cells in vitro (Fig. 6B), we were interested to determine the antitumor efficacy of combining H182 and radiation therapy. Syngeneic E0771 murine breast cancer cells were injected into the mammary gland of C57BL/6 mice [17], and when tumors were palpable (800–1000 mm3), mice were grouped so that the mean tumor sizes were identical between groups. Treatment was initiated as follows: wt (no treatment), vehicle, vehicle and radiation (16 Gy) delivered focally to the tumor using the X-RAD SmART (PXi, Precision X-Ray, North Branford, CT), H278 (HCl salt of H182, 10 mg/kg, I.P., every other day) alone, and H278 (10 mg/kg, I.P., every other day) together with radiation (16 Gy). Both tumor growth and survival were monitored and showed that while H278 alone did not have significant effect, the combination of H278 and focal radiation completely inhibited tumor growth and improved survival in this mouse model of breast cancer (Fig. 7D).
We then profiled tumor tissues for Stat3 activity and Stat3 target gene expression as pharmacodynamic markers of therapeutic response. Tissue lysates of equal total protein prepared from the extracted tumors were subjected to Stat3 DNA-binding assay/EMSA analysis or SDS-PAGE and immunoblotting analysis. Results showed that H182 treatment suppressed Stat3 DNA-binding activity in tumor tissues (Supplementary Fig. S5A, treated T vs. untreated control, C), in parallel with the suppression of the expression of Stat3 downstream genes, including c-Myc, VEGF, and survivin (Supplementary Figs. S5B and T), compared to untreated control tumors. These results validate the inhibition of Stat3 activity and together indicate that the inhibition of constitutively-active Stat3 functions leads to the suppression of growth of TNBC in mice and sensitizes tumors to radiation.
3.8. Initial evaluation of DMPK parameters of the azetidine compounds
The in vivo activity of a compound is influenced by its overall bioavailability, which is also driven by parameters, such as solubility, permeability, and metabolism [37,38]. In order to assess the physicochemical properties of the azetidine compounds, we conducted the industry standard evaluation of H182 and H172 for solubility, metabolism, CYP inhibition, plasma protein binding, plasma stability, and CEREP SafetyScreen44™ [39] via the contract clinical research organization (CRO), Eurofins-CEREP. The results of these studies showed that H172 has aqueous solubility of 104 and 123 μg/ml, while H182 has solubility of 130 and 128 μg/ml, respectively, for simulated gastric fluid (SGF) and simulated intestinal fluid (SIF) (Table 3). These values are all above the industry cut-off of 60 μg/ml [40]. However, H172 and H182 have low solubility of 6.3 and 3.0 μg/ml, respectively, in PBS (Table 3). For metabolic stability, H182 has half-life >150 and 21 min for mouse liver microsome (MLM) and human liver microsome (HLM) assays, respectively, both of which are above the industry cut-off of HLM t ½ >15 min [41,42]. It has CLint in human hepatocytes of 16 μL/min/106 cells, which falls into moderate stability range [43]. Metabolic stability for H172 was not determined.
Table 3.
DMPK properties of H182 and H172.
| Solubilitya | Metabolic Stabilitya | CYP Inhibition | Plasma Protein Binding | Plasma Stability | SafetyScreen44b | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| ID | PBS pH 7.4 (μg/mL) | SGF (μg/mL) | SIF (μg/mL) | MLM t1/2 (min) | HLM t1/2 (min) | Human Hepatocytes CLint (μL/min/106 cells) | IC50 or % Inh at 10 μM | (%) | (% Remaining) | Receptors over 80% Inhibition at 10 μM IC50 (μM) |
| H182 | 3.0 | 132 | 128 | >150 | 21 | 14.6 (t1/2 68 min) | CYP3A (Midazolam): IC50 6.9 μM CYP3A (Testosterone): IC50 1.2 μM CYP2C9: 33% CYP2D6: 9% | Human: 99.75 Mouse: 99.90 | Human: 53% at 1 h 10% at 2 h Mouse: 69% at 1 h 23% at 2 h | hERGc >10 GR 0.11 NET 0.40 |
| H172 | 6.3 | 104 | 123 | Nd | Nd | nd | nd | nd | nd | nd |
nd, not determined.
Cut-offs: Solubility >60 μg/mL; Metabolic Stability HLM half life >15 min, Human Hepatocytes CLint 3.5–19.0 μL/min/106 cells.
SafetyScreen44™ according to Eurofins-CEREP Scientific.
hERG IC50 results from Qpatch assay.
For plasma stability, H182 shows a decay from 100% to 53% and 69%, respectively, in human and mouse plasma after 1 h, and to 10% and 23%, respectively, after 2 h (Table 3). H172 was not evaluated. Moreover, H182 plasma protein binding is over 99% for both human and mouse, and it has cytochrome (CYP) inhibition profiles as follows: CYP3A, IC50 6.9 and 1.2 μM at midazolam and testosterone sites, respectively; and the CYP2C9 or CYP2D6 inhibition by 10 μM H182 is 33% or 9%, respectively. Further, the CEREP SafetyScreen44™ assay showed only the hERG (human Ether-à-go-go-Related Gene), glucocorticoid receptor (GR), and norepinephrine transporter (NET) had over 80% inhibition at 10 μM H182. We thus conducted dose-response studies for IC50, which gave hERG IC50 > 10 μM (Qpatch assay), GR IC50 0.11 μM, and NET IC50 0.40 μM (Table 3). These results together suggest the azetidine compounds overall have good properties, though we can optimize the drug metabolism and pharmacokinetic (DMPK) properties further to make the antitumor efficacy even stronger.
4. Discussion
Described herein are four members of the new class of azetidine-based small molecules [15], H105, H120, H172, and H182, which directly inhibit Stat3 activity, with a range of potencies in the nanomolar to low micromolar. While many Stat3 inhibitors have been reported [10, 13,14,18,44–48], the in vitro potencies (IC50s) of 0.38–0.98 μM make H172 and H182 two of the most potent, direct Stat3 inhibitors. The conformational constraints engendered by the azetidine ring led to stronger interactions with Stat3, which result in enhanced potency. The observation that replacing the perfluorobenzene-sulfonamide para-f-fluorine atom with any other substituent compromises the Stat3-inhibitory potency [15], suggests that the azetidine-based compounds engage in covalent interactions with Stat3. This hypothesis is supported by the time-dependency of the potency of covalent inhibitors [49], with the potency of Stat3 inhibition by H182 improving from 1.47 μM to 0.66 μM–0.38 μM for 10 min, 30 min, and 60 min incubation with Stat3, respectively. Nano-LC/MS/MS proteomics approach shows that the previously reported azetidine compound H098 reacted with C328 and C426, and the compound H182 reacted with C468 and C542, indicating that the azetidine compounds are capable to covalently interact with Stat3 protein in vitro. These findings were confirmed by site-directed mutagenesis that showed the Cys426 and Cys468 residues to be essential for the high potency, though based on the site-directed mutagenesis, Cys468 alone appears to be sufficient to impact the binding to the compounds (Fig. 3Cii). The higher potency with incubation time suggests the irreversible Stat3:H182 adduct formation leads to the exhaustion of the target (Stat3) [49]. The recent successes with the allosteric irreversible inhibitors of mutant KRas that are in clinical trials highlight the value of covalent inhibitors as an approach to effectively inhibit otherwise challenging and intractable targets [50,51].
The cellular activities of the new azetidine analogs are similarly improved over the previous Stat3 inhibitors [3]. H182 shows cellular activities at 1 μM. By interacting with Stat3, the azetidine compounds inhibits constitutively-active ligand-induced Stat3 phosphorylation, DNA-binding activity, and transcriptional activity in human TNBC cells. The compounds further inhibited Stat3 nuclear accumulation and promoted Stat3 perinuclear aggregation in the tumor cells. By contrast, the lack of inhibitory responses against pYStat1, pS727Stat3, pYStat5, JAK2, Src, Shc, pEGFR, Erk1/2 and Akt by the azetidine compounds at concentrations that inhibit Stat3 activity is indicative of the minimal off-target effects. This is consistent with the overall selectivity profile of H182, as determined also from the CEREP SafetyScreen44™ in vitro panel, which showed that apart from GR and NET receptors (IC50s 0.11 and 0.40 μM, respectively), data on hERG inhibition and on the affinities for a number of other receptors, including GPCR, ion channels, transporters, neurotransmitters, NHR, and kinases (data not shown) indicated good off-target properties. This correlates with the in vivo studies that showed little evidence of toxicity in mice.
In concordance with the in vitro activities, H182 and H120 formulations for oral or intraperitoneal administrations are efficacious against human TNBC xenografts as a standalone, and H278 (HCl salt of H182) in combination with radiation therapy completely blocked tumor growth and prolonged survival in mouse TNBC in syngeneic models. The Stat3-dependency of the antitumor effects is supported by the suppression of the Stat3 target genes, c-Myc, VEGF, and survivin in vitro and in vivo in response to treatment with H182. Physicochemical property limitations are some of the factors that have precluded the clinical development of small molecule Stat3 inhibitors [3]. Present studies suggest H182 and H172 present favorable solubility in SIF and GIF environments, while metabolic stability may be modest. H182 shows favorable human liver microsome (HLM) and mouse liver microsome (MLM), half-life (t1/2) of 21 and 150 min, greater than the cut-off 15 min [41,42]. However, the human hepatocyte assay with CLint 16 μL/min/106 cells, indicates moderate to low stability (CLint 19 μL/min/106 cells). In addition, plasma stability was 10% and 23% after 2 h in human and mouse, respectively. Currently, these two parameters (hepatocyte and plasma stability) are being addressed to further improve the PK profile. H182 and H172 therefore represent suitable candidates for further PK optimization towards the goal of clinical development for treating TNBC and other tumors that are dependent on aberrantly-active Stat3 for the tumor phenotype. The azetidine-based small molecules are a new class of inhibitors that potently and irreversibly bind to Stat3, leading to tumor cell death and tumor growth inhibition in vivo. Industry standard CEREP SafetyScreen44 that showed a lack of inhibitory activities against a wide array of targets suggests a relatively low toxicity of the azetidine-based compounds.
Statistical analysis.
Statistical analysis was performed on mean values using Prism GraphPad Software, Inc. (La Jolla, CA). The significance of differences between groups was determined by the paired t-test at *p < 0.05, or **p < 0.01.
Supplementary Material
Acknowledgement
We thank all colleagues and members of our laboratory for the stimulating discussions. We thank the Cedars-Sinai Biobank and Pathology Core facility for the assistance in performing confocal microscopy study and the Flow Cytometry Core for the assistance in cell apoptotic analysis, and the IIGB proteomics core facility at the University of California, Riverside for the mass spectrometry study, which is supported by NIH S10 OD010669 grant. This work was supported by the National Institutes of Health/National Cancer Institute Grant CA208851 (JT), LEIDOS Biomedical Research/NCI Contract 19X122Q (JT), and Cedars-Sinai Start-up funds (JT).
Abbreviations
- Stat
signal transducer and activator of transcription
- PBST
phosphate-buffered saline tween-20
- PBS
phosphate-buffered saline
- EMSA
electrophoretic mobility shift assay
- EGFR
epidermal growth factor receptor
- JAK
Janus kinase
- SH2
Src homology 2
- MAPK
mitogen-activated protein kinase
- Erk
extracellular signal-regulated kinase
- FBS
fetal bovine serum
- PARP
poly ADP-ribose polymerase
Footnotes
Declaration of competing interest
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests:
James Turkson reports a relationship with Novella, LLC that includes: equity or stocks. Co-authors have nothing to declare. Co-owners-James Turkson, Peibin Yue, Francisco Lopez-Tapia, Christine Brotherton-Pleiss, and Marc A. Tius have patent pending to Novella, LLC.
CRediT author statement
Peibin Yue Conceptualization, Investigation, Data curation, Formal analysis, Methodology, Writing- original draft preparation, Writing-Reviewing and Editing; Yinsong Zhu Data curation, Formal analysis, Methodology; Christine Brotherton-Pleiss Conceptualization, Methodology, Formal analysis, Validation, Reviewing and Editing; Wenzhen Fu Data curation, Formal analysis Methodology; Nagendra Verma Data curation, Methodology, Formal analysis; Jasmine Chen Methodology; Kayo Nakamura Methodology, Data curation; Weiliang Chen Methodology, Data curation; Yue Chen Data curation; Felix Alonso-Valenteen Methodology, Data curation, Formal analysis, Writing- original draft preparation; Simoun Mikhael Methodology, Data curation, Formal analysis; Lali Medina-Kauwe Supervision, Formal analysis, Writing-Reviewing and Editing; Kathleen M. Kershaw Methodology, Data curation; Marai Celeridad Methodology, Data curation; Songqin Pan Methodology, Data curation, Formal analysis, Writing- original draft preparation; Allison S. Limpert Methodology, Data curation, Formal analysis, Writing- original draft preparation; Douglas J. Sheffler Methodology, Data curation, Formal analysis, Writing- original draft preparation, Writing- Reviewing, Editing; Nicholas D. P. Cosford Supervision, Formal analysis, Writing- original draft preparation, Writing- Reviewing and Editing; Stephen L. Shiao Supervision, Formal analysis, Writing-original draft preparation; Marc A. Tius Conceptualization, Investigation, Formal analysis, Writing- original draft, Writing- Reviewing and Editing; Francisco Lopez-Tapia Conceptualization, Investigation, Formal analysis, Writing- original draft, Writing-Reviewing and Editing; James Turkson: Conceptualization, Investigation, Formal analysis, Project administration, Resources, Supervision, Writing- original draft preparation, Writing- Reviewing and Editing.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.canlet.2022.215613.
References
- [1].Darnell JE, Validating Stat3 in cancer therapy, Nat. Med 11 (2005) 595–596. [DOI] [PubMed] [Google Scholar]
- [2].Yu H, Herrmann LH,A, Buettner R, Jove R, Revisiting STAT3 signalling in cancer: new and unexpected biological functions, Nat. Rev. Cancer 14 (2014) 736–746. [DOI] [PubMed] [Google Scholar]
- [3].Miklossy G, Hilliard TS, Turkson J, Therapeutic modulators of STAT signaling for human diseases, Nat. Rev. Drug Discov 12 (2013) 611–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Gough DJ, Maríe IJ, Lobry C, Aifantis I, Levy DE, STAT3 supports experimental K-RasG12D-induced murine myeloproliferative neoplasms dependent on serine phosphorylation, Blood 124 (2014) 2252–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Avalle L, Camporeale A, Morciano G, Caroccia N, Ghetti E, Orecchia V, Viavattene D, Giorgi C, Pinton P, Poli V, STAT3 localizes to the ER, acting as a gatekeeper for ER-mitochondrion Ca(2+) fluxes and apoptotic responses, Cell Death Differ. 26 (2019) 932–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Yeh JE, Frank DA, STAT3-Interacting proteins as modulators of transcription factor function: implications to targeted cancer therapy, ChemMedChem 11 (2016) 795–801. [DOI] [PubMed] [Google Scholar]
- [7].Furtek SL, Backos DS, Matheson CJ, Reigan P, Strategies and approaches of targeting STAT3 for cancer treatment, ACS Chem. Biol 11 (2016) 308–318. [DOI] [PubMed] [Google Scholar]
- [8].Bai L, Zhou H, Xu R, Zhao Y, Chinnaswamy K, McEachern D, Chen J, Yang CY, Liu Z, Wang M, Liu L, Jiang H, Wen B, Kumar P, Meagher JL, Sun D, Stuckey JA, Wang S, A potent and selective small-molecule degrader of STAT3 achieves complete tumor regression in vivo, Cancer Cell 36 (2019) 498–511, e417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Zhou H, Bai L, Xu R, Zhao Y, Chen J, McEachern D, Chinnaswamy K, Wen B, Dai L, Kumar P, Yang CY, Liu Z, Wang M, Liu L, Meagher JL, Yi H, Sun D, Stuckey JA, Wang S, Structure-based discovery of SD-36 as a potent, selective, and efficacious PROTAC degrader of STAT3 protein, J. Med. Chem 62 (2019) 11280–11300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Siddiquee K, Zhang S, Guida WC, Blaskovich MA, Greedy B, Lawrence HR, Yip ML, Jove R, McLaughlin MM, Lawrence NJ, Sebti SM, Turkson J, Selective chemical probe inhibitor of Stat3, identified through structure-based virtual screening, induces antitumor activity, Proc. Natl. Acad. Sci. U. S. A 104 (2007) 7391–7396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Zhang X, Yue P, Fletcher S, Zhao W, Gunning PT, Turkson J, A novel small-molecule disrupts Stat3 SH2 domain-phosphotyrosine interactions and Stat3-dependent tumor processes, Biochem. Pharmacol 79 (2010) 1398–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Zhang X, Yue P, Page BD, Li T, Zhao W, Namanja AT, Paladino D, Zhao J, Chen Y, Gunning PT, Turkson J, Orally bioavailable small-molecule inhibitor of transcription factor Stat3 regresses human breast and lung cancer xenografts, Proc. Natl. Acad. Sci. U. S. A 109 (2012) 9623–9628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Yue P, Lopez-Tapia F, Paladino D, Li Y, Chen C-H, Hilliard T, Chen Y, Tius M, Turkson J, Hydroxamic acid and benzoic acid-based Stat3 inhibitors suppress human glioma and breast cancer phenotypes in vitro and in vivo, Cancer Res. 76 (2016) 652–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Lopez-Tapia F, Brotherton-Pleiss C, Yue P, Murakami H, Costa Araujo AC, Reis dos Santos B, Ichinotsubo E, Rabkin A, Shah R, Lantz M, Chen S, Tius MA, Turkson J, Linker variation and structure-activity relationship analyses of carboxylic acid-based small molecule STAT3 inhibitors, ACS Med. Chem. Lett 9 (2018) 250–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Brotherton-Pleiss C, Yue P, Zhu Y, Nakamura K, Chen W, Fu W, Kubota C, Chen J, Alonso-Valenteen F, Mikhael S, Medina-Kauwe L, Tius MA, Lopez-Tapia F, Turkson J, Discovery of novel azetidine amides as potent small-molecule STAT3 inhibitors, J. Med. Chem 64 (2021) 695–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Xue J, Patergnani S, Giorgi C, Suarez J, Goto K, Bononi A, Tanji M, Novelli F, Pastorino S, Xu R, Caroccia N, Dogan AU, Pass HI, Tognon M, Pinton P, Gaudino G, Mak TW, Carbone M, Yang H, Asbestos induces mesothelial cell transformation via HMGB1-driven autophagy, Proc. Natl. Acad. Sci. U. S. A 117 (2020) 25543–25552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Crosby EJ, Wei J, Yang XY, Lei G, Wang T, Liu CX, Agarwal P, Korman AJ, Morse MA, Gouin K, Knott SRV, Lyerly HK, Hartman ZC, Complimentary mechanisms of dual checkpoint blockade expand unique T-cell repertoires and activate adaptive anti-tumor immunity in triple-negative breast tumors, OncoImmunology 7 (2018), e1421891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Siddiquee KA, Gunning PT, Glenn M, Katt WP, Zhang S, Schrock C, Sebti SM, Jove R, Hamilton AD, Turkson J, An oxazole-based small-molecule Stat3 inhibitor modulates Stat3 stability and processing and induces antitumor cell effects, ACS Chem. Biol 2 (2007) 787–798. [DOI] [PubMed] [Google Scholar]
- [19].Namanja AT, Wang J, Buettner R, Colson L, Chen Y, Allosteric communication across STAT3 domains associated with STAT3 function and disease-causing mutation, J. Mol. Biol 428 (2016) 579–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Alonso-Valenteen F, Pacheco S, Srinivas D, Rentsendorj A, Chu D, Lubow J, Sims J, Miao T, Mikhael S, Hwang JY, Abrol R, Medina Kauwe LK, HER3-targeted protein chimera forms endosomolytic capsomeres and self-assembles into stealth nucleocapsids for systemic tumor homing of RNA interference in vivo, Nucleic Acids Res. 47 (2019) 11020–11043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].McWhirter C, Chapter One - kinetic mechanisms of covalent inhibition, in: Ward RA, Grimster NP (Eds.), Annual Reports in Medicinal Chemistry, Academic Press, 2021, pp. 1–31. [Google Scholar]
- [22].Strelow JM, A perspective on the kinetics of covalent and irreversible inhibition, SLAS Discov 22 (2017) 3–20. [DOI] [PubMed] [Google Scholar]
- [23].Pagadala NS, Syed K, Tuszynski J, Software for molecular docking: a review, Biophys Rev 9 (2017) 91–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Schust J, Berg T, A high-throughput fluorescence polarization assay for signal transducer and activator of transcription 3, Anal. Biochem 330 (2004) 114–118. [DOI] [PubMed] [Google Scholar]
- [25].Turkson J, Ryan D, Kim JS, Zhang Y, Chen Z, Haura E, Laudano A, Sebti S, Hamilton AD, Jove R, Phosphotyrosyl peptides block Stat3-mediated DNA binding activity, gene regulation, and cell transformation, J. Biol. Chem 276 (2001) 45443–45455. [DOI] [PubMed] [Google Scholar]
- [26].Zhao W, Jaganathan S, Turkson J, A cell-permeable Stat3 SH2 domain mimetic inhibits Stat3 activation and induces antitumor cell effects in vitro, J. Biol. Chem 285 (2010) 35855–35865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ren Z, Cabell LA, Schaefer TS, McMurray JS, Identification of a high-affinity phosphopeptide inhibitor of stat3, Bioorg. Med. Chem. Lett 13 (2003) 633–636. [DOI] [PubMed] [Google Scholar]
- [28].Raptis L, Arulanandam R, Vultur A, Geletu M, Chevalier S, Feracci H, Beyond structure, to survival: activation of Stat3 by cadherin engagement, Biochem. Cell. Biol 87 (2009) 835–843. [DOI] [PubMed] [Google Scholar]
- [29].Vultur A, Cao J, Arulanandam R, Turkson J, Jove R, Greer P, Craig A, Elliott B, Raptis L, Cell-to-cell adhesion modulates Stat3 activity in normal and breast carcinoma cells, Oncogene 23 (2004) 2600–2616. [DOI] [PubMed] [Google Scholar]
- [30].Turkson J, Bowman T, Garcia R, Caldenhoven E, De Groot RP, Jove R, Stat3 activation by Src induces specific gene regulation and is required for cell transformation, Mol. Cell Biol 18 (1998) 2545–2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Sharma P, López-Tarruella S, García-Saenz JA, Ward C, Connor CS, Gómez HL, Prat A, Moreno F, Jerez-Gilarranz Y, Barnadas A, Picornell AC, Del Monte-Millán M, Gonzalez-Rivera M, Massarrah T, Pelaez-Lorenzo B, Palomero MI, González Del Val R, Cortes J, Fuentes Rivera H, Bretel Morales D, Máarquez-Rodas I, Perou CM, Wagner JL, Mammen JM, McGinness MK, Klemp JR, Amin AL, Fabian CJ, Heldstab J, Godwin AK, Jensen RA, Kimler BF, Khan QJ, Martin M, Efficacy of neoadjuvant carboplatin plus docetaxel in triple-negative breast cancer: combined analysis of two cohorts, Clin. Cancer Res 23 (2017) 649–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Hu XC, Zhang J, Xu BH, Cai L, Ragaz J, Wang ZH, Wang BY, Teng YE, Tong ZS, Pan YY, Yin YM, Wu CP, Jiang ZF, Wang XJ, Lou GY, Liu DG, Feng JF, Luo JF, Sun K, Gu YJ, Wu J, Shao ZM, Cisplatin plus gemcitabine versus paclitaxel plus gemcitabine as first-line therapy for metastatic triple-negative breast cancer (CBCSG006): a randomised, open-label, multicentre, phase 3 trial, the Lancet, Oncology 16 (2015) 436–446. [DOI] [PubMed] [Google Scholar]
- [33].Yang PL, Liu LX, Li EM, Xu LY, STAT3, the challenge for chemotherapeutic and radiotherapeutic efficacy, Cancers 12 (2020) 2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Yue P, Zhang X, Paladino D, Sengupta B, Ahmad S, Holloway RW, Ingersoll SB, Turkson J, Hyperactive EGF receptor, Jaks and Stat3 signaling promote enhanced colony-forming ability, motility and migration of cisplatin-resistant ovarian cancer cells, Oncogene 31 (2012) 2309–2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Wang X, Zhang X, Qiu C, Yang N, STAT3 contributes to radioresistance in cancer, Front. Oncol 10 (2020) 1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Wang Y, Hu Z, Ma W, Niu Y, Su J, Zhang L, Zhao P, Signal transducer and activator of transcription 3 inhibition alleviates resistance to BRAF inhibition in anaplastic thyroid cancer, Invest. N. Drugs 39 (2021) 764–774. [DOI] [PubMed] [Google Scholar]
- [37].Martinez MN, Amidon GL, A mechanistic approach to understanding the factors affecting drug absorption: a review of fundamentals, J. Clin. Pharmacol 42 (2002) 620–643. [DOI] [PubMed] [Google Scholar]
- [38].Di L, Kerns E, Concepts, Structure Design and Methods from ADME to Toxicity Optimization, second ed., Elsevier, 2016. [Google Scholar]
- [39].Chaturvedi PR, Decker CJ, Odinecs A, Prediction of pharmacokinetic properties using experimental approaches during early drug discovery, Curr. Opin. Chem. Biol 5 (2001) 452–463. [DOI] [PubMed] [Google Scholar]
- [40].Kerns EH, Di L, Carter GT, In vitro solubility assays in drug discovery, Curr. Drug Metabol 9 (2008) 879–885. [DOI] [PubMed] [Google Scholar]
- [41].Di L, Kerns EH, Ma XJ, Huang Y, Carter GT, Applications of high throughput microsomal stability assay in drug discovery, Comb. Chem. High Throughput Screen 11 (2008) 469–476. [DOI] [PubMed] [Google Scholar]
- [42].Copeland RA, Evaluation of enzyme inhibitors in drug discovery: a Guide for medicinal chemists and pharmacologists, in: Chapter 10: Quantitative Biochemistry in the Pharmacological Evaluation of Drugs, second ed., Wiley, 2013, pp. 383–469, 2013. [Google Scholar]
- [43].Houston JB, Utility of in vitro drug metabolism data in predicting in vivo metabolic clearance, Biochem. Pharmacol 47 (1994) 1469–1479. [DOI] [PubMed] [Google Scholar]
- [44].Turkson J, Zhang S, Palmer J, Kay H, Stanko J, Mora LB, Sebti S, Yu H, Jove R, Inhibition of constitutive signal transducer and activator of transcription 3 activation by novel platinum complexes with potent antitumor activity, Mol. Cancer Therapeut 3 (2004) 1533–1542. [PubMed] [Google Scholar]
- [45].Song H, Wang R, Wang S, Lin J, A low-molecular-weight compound discovered through virtual database screening inhibits Stat3 function in breast cancer cells, Proc. Natl. Acad. Sci. U. S. A 102 (2005) 4700–4705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Lin L, Deangelis S, Foust E, Fuchs J, Li C, Li PK, Schwartz EB, Lesinski GB, Benson D, Lu J, Hoyt D, Lin J, A novel small molecule inhibits STAT3 phosphorylation and DNA binding activity and exhibits potent growth suppressive activity in human cancer cells, Mol. Cancer 9 (2010) 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Chen J, Bai L, Bernard D, Nikolovska-Coleska Z, Gomez C, Zhang J, Yi H, Wang S, Structure-based design of conformationally constrained, cell-permeable STAT3 inhibitors, ACS Med. Chem. Lett 1 (2010) 85–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Bhasin D, Cisek K, Pandharkar T, Regan N, Li C, Pandit B, Lin J, Li P, Design, synthesis, and studies of small molecule STAT3 inhibitors, Bioorg. Med. Chem. Lett 18 (2008) 391–395. [DOI] [PubMed] [Google Scholar]
- [49].Hopper M, Gururaja T, Kinoshita T, Dean JP, Hill RJ, Mongan A, Relative selectivity of covalent inhibitors requires assessment of inactivation kinetics and cellular occupancy: a case study of ibrutinib and acalabrutinib, J. Pharmacol. Exp. Therapeut 372 (2020) 331–338. [DOI] [PubMed] [Google Scholar]
- [50].Salgia R, Pharaon R, Mambetsariev I, Nam A, Sattler M, The improbable targeted therapy: KRAS as an emerging target in non-small cell lung cancer (NSCLC), Cell Rep Med 2 (2021) 100186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Canon J, Rex K, Saiki AY, Mohr C, Cooke K, Bagal D, Gaida K, Holt T, Knutson CG, Koppada N, Lanman BA, Werner J, Rapaport AS, San Miguel T, Ortiz R, Osgood T, Sun JR, Zhu X, McCarter JD, Volak LP, Houk BE, Fakih MG, O’Neil BH, Price TJ, Falchook GS, Desai J, Kuo J, Govindan R, Hong DS, Ouyang W, Henary H, Arvedson T, Cee VJ, Lipford JR, The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity, Nature 575 (2019) 217–223. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.