

Abstract
West Nile viral infection causes severe neuroinvasive disease in less than one percent of infected humans. There are no targeted therapeutics for this serious and potentially fatal disease, and to date no vaccine has been approved for humans. With climate change expected to result in rising incidence of West Nile and other related vector-borne viral infections, there is an increasing need to identify those at risk for serious disease and potential leads for therapeutic and vaccine development. Genetic variation, particularly in genes whose products are either directly or indirectly connected to immune response to infections, is a critical avenue of investigation to identify those at higher risk of clinically apparent West Nile infection. Given the small percent of infections that progress to severe disease and the relatively low numbers of reported infections, it is challenging to conduct well-powered studies to identify genetic factors associated with more severe outcomes. In this chapter, we outline several approaches with the objective to take full advantage of all available data in order to identify genetic factors which lead to increased risk of severe West Nile neuroinvasive disease. These methods are generalizable to other conditions with limited cohort size and rare outcomes.
Keywords: West Nile virus, West Nile Neuroinvasive Disease, candidate gene study, genome-wide association study, imputation, gene-gene interactions, population controls
1. Introduction
Humans infected with West Nile virus (WNV) may be asymptomatic or develop mild or severe disease. West Nile Neuroinvasive Disease (WNND) can be characterized as meningitis, encephalitis, or acute flaccid paralysis, and is estimated to occur in less than 1% of WNV-infected individuals [1, 2]. Given such a small proportion of WNV-infected individuals develop severe disease, genetic factors are likely to play a role in disease pathogenesis, with some genetic variants leading to greater risk of severe disease or in other cases to a reduced risk. With no approved vaccines or disease-specific therapeutics, it is important to understand why some individuals are at increased risk for severe, potentially fatal disease in order to guide development of much-needed vaccines and therapeutics.
To assess genetic predisposition to severe WNND, the most common approaches are case-control studies examining genetic variants, either in specific genes or genome-wide [3]. In the candidate gene studies, analysis is limited to genes suspected to be involved in an aspect of the disease pathogenesis based on a priori knowledge, such as from laboratory experiments or hypotheses from genetic associations identified in similar diseases. Genome-wide association studies (GWAS) are a widely-used genetic tool to analyze all genotyped variants for an association with the phenotype of interest. For example, recent GWAS studies on a large cohort of subjects have identified genetic predisposition to severe COVID-19 infection including 2’−5’-Oligoadenylate Synthetase 1 (OAS1) and tyrosine kinase 2 (TYK2) genes [4, 5]. Importantly, GWAS generally include 1000s of subjects and have a stringent p-value threshold (usually p < 5 × 10−8) to compensate for the large number of statistical analyses. For less common conditions with smaller cohorts of subjects, such as WNV, initial GWAS analysis may be underpowered for definitive identification and thus additional methods may be needed to augment detection of relevant factors.
To date, only one GWAS has been conducted to analyze associations with severe WNND [6]. Set in a North American sample including WNND cases (n = 560 cases) and WNV-positive, mildly ill individuals as the controls (n = 950 controls), this study identified single nucleotide polymorphisms (SNPs) within the replication factor C1 (RFC1), sodium channel neuronal type I α subunit (SCN1A), and ananyl aminopeptidase (ANPEP) genes which may be associated with WNND. At least 12 candidate genes studies have also been conducted, focused on immune-related genes such as C-C chemokine receptor type 5 (CCR5) and interferon regulatory transcription factor 3 (IRF3) [3]. Several of these gene-specific studies have been conducted in Greece, Israel, and Macedonia, and while they include diverse subject demographics, most of the sample sizes were below 500. These studies provide evidence of potential genetic associations with WNND, but additional research is needed to confirm the associations and to describe further the genetic architecture of this disease.
There are unique hurdles for analyzing genetic associations with WNND or other small disease cohorts. Most notably, the potential pool of cases is severely limited given a fairly low incidence of disease and a small fraction of infected individuals which progress to WNND. Studies conducted to date have had small sample sizes and thus limited power to identify associations. Additionally, the majority of studies conducted thus far have been in North American populations of European descent, despite the fact that the virus has been isolated in every continent except Antarctica [7]. With the widespread distribution of WNV and an expected increase in transmission due to climate change [8], it is critical both to conduct new studies and to extract as much information as possible from previously conducted studies. Several innovations in analytic methods can be applied to existing GWAS datasets to elucidate further the role of genetic factors in WNND pathogenesis, and to identify novel associations to this potentially fatal disease. Here we outline methodological approaches for in-depth analysis of existing GWAS data.
2. Materials
The only required material is directly genotyped data for a study of WNND (or similarly rare outcome) cases and controls. Depending on the approach(es) chosen, many software programs are freely available and noted below at the corresponding step.
3. Methods
To conduct a GWAS analysis requires sample collection from cases and appropriate controls, DNA sequencing, quality control steps, and initial univariate analysis [9]. For a rare cohort, where statistical power may be lower due to limited sample size, further analyses following the initial univariate analysis of the dataset can identify novel variants associated with severe WNND or other similarly rare outcomes (see Note 1). Depending on the available data and study aim, researchers may choose to use imputation (3.2), analyze gene-gene interactions (3.3), and/or incorporate population controls (3.4) to identify novel associations (Figure 1).
Figure 1. Overview of Analytical Approaches to Uncover Genetic Associations for Rare Outcomes.
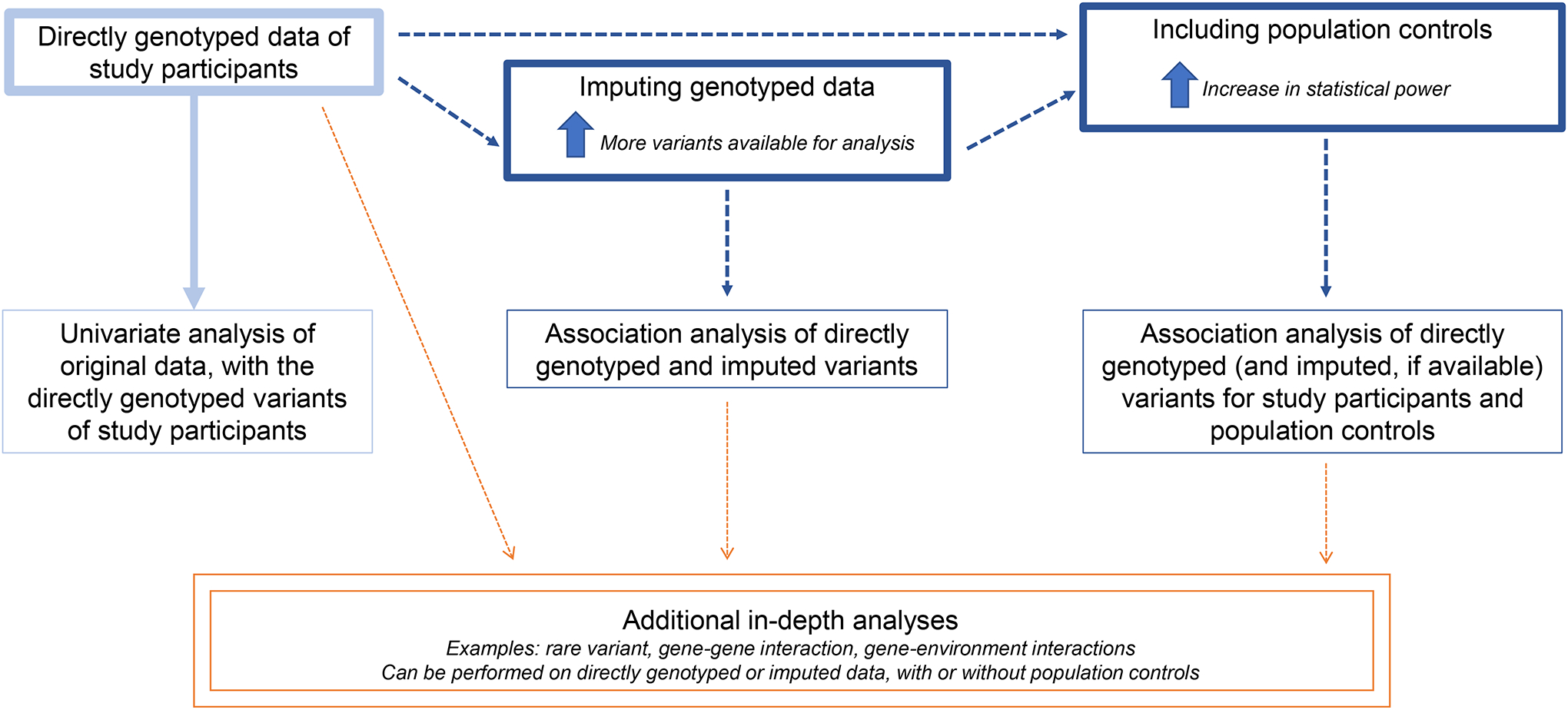
Beginning with directly genotyped data of a study sample, researchers can use imputation to increase the number of variants available for analysis and/or population controls to increase the statistical power. In-depth analysis may also include rare variant, gene-gene interaction, and gene-environment interactions on these datasets.
3.1. Choosing a validated GWAS dataset.
The approaches outlined below should be conducted on existing cleaned genotype data, either directly prepared by study researchers or requested through data repositories (see section 3.4.1). GWAS datasets contain sensitive personal information and are often large files, and a secure, robust computing environment that meets laws and institutional policies is required. Prior to analysis, researchers should clean the dataset through quality control (QC) steps, such as application of thresholds for individual and variant call rates [10]. For WNV, we worked with the sole WNV GWAS dataset, and used a high-performance computing cluster with a Linux operating system to execute the below analyses. QC steps ensured analyses was on SNPs and individuals with < 5% missing data and we excluded any SNPs that deviated significantly from Hardy Weinberg Equilibrium [11].
3.2. Imputation
Imputation estimates variants that were not genotyped in the original study based on comparison to larger reference datasets, which consist of directly genotyped samples. Imputation increases the ability to detect novel loci for an association with WNND or another phenotype of interest. With imputation, the number of common variants available for analysis may increase by 8-fold or greater [11, 12]. The steps below outline a hands-on and versatile approach for imputing data. Researchers with limited coding experience may choose to use the Michigan Imputation Server, which provides a free and user-friendly online platform to impute study data [13, 14].
3.2.1. Build Conversion and Pre-Phasing.
If the samples were collected or processed using older technology, the data may need to be converted to a more recent build for imputation and analysis (see Note 2). University of California, Santa Cruz provides the Batch Coordinate Conversion (LiftOver) tool to quickly and easily convert data [15, 16].
Pre-phasing, or estimating haplotypes, improves the accuracy and efficiency of imputation. ShapeIt is a popular tool for pre-phasing and provides output that can then be used in Impute2 for the imputation step [17–20].
3.2.2. Imputation, using software such as Impute2.
Additional software such as Beagle and Minimac are well-used [21, 22], with comparable accuracy depending on the ancestry of the study participants [23, 24].
Imputation relies on a reference panel to estimate ungenotyped variants, and the 1000 Genomes is a validated and well-established option [25–27]. Reference panels should match the ancestry of the study participants as closely as possible for accurate imputation [28].
Impute2 recommends imputing chunks of chromosomes to increase computational efficiency and imputation accuracy [29]. One strategy is to divide the chromosomes in equal sized chunks, however, some chunks may have too few SNPs for accurate analysis and/or crossover the centromere. Research groups from the University of Michigan Center for Statistical Genetics and University of California, Santa Cruz Genome Browser have provided detailed instructions on chromosome chunk intervals and centromere locations, respectively [30, 31].
Impute2 assesses imputed quality for each variant with an INFO score, ranging between zero and 1.0, with a higher score indicating increased certainty [32]. To ensure only variants imputed with high certainty are included in the analysis, a cutoff threshold such as 0.7 should be applied to the INFO scores.
3.2.3. Univariate analysis in PLINK.
The directly genotyped and high quality imputed variants can be analyzed for an association with WNND in PLINK or a similar software, with adjustments as needed for any covariates of interest [33].
3.3. Gene-gene Interactions
Another valuable tool to identify genetic influences in a rare disease or small cohort is assessing gene-gene interactions, also called epistasis. This method adds to the analysis by identifying pairs of genes which together may play a role in the outcome under study [34]. Identifying the combined effects of variation at two loci of interest can provide additional insight into the genetic architecture of WNND.
3.1.1. Minor Allele Frequency (MAF) cutoff.
To assess gene-gene interactions, the dataset should only include common variants with minor allele frequency (MAF) above a threshold such as 10%. MAF is the estimated frequency of the second most common allele for a variant in a population of matching ancestry; more common variants have a higher MAF while rare variants have low MAF values [35]. For this analysis, only common variants with MAF above a threshold such as 10% should be included. Limiting the dataset to variants ≥ 10% MAF can be done using the --maf flag in PLINK [33].
3.3.1. Fast-epistasis using genotype count tables in PLINK.
As an initial step to filter the variants, PLINK’s fast-epistasis function can quickly test interactions by comparing genotype count tables between all SNPs with MAF ≥10%.
Variants with an interaction p-value below a cutoff (such as 5 × 10−8) can then be included in further, more precise analysis of interactions.
3.3.2. Regular epistasis function in PLINK.
The regular epistasis function, which employs a logistic regression approach, can then be conducted between SNPs that met the threshold from the fast-epistasis step. Alternatively, interactions can be assessed between one SNP that met the fast-epistasis cutoff and all other genotyped variants.
3.4. Population Controls
Including population controls is another relevant option to enhance the analysis of the role of genetic variation on rare outcomes. The addition of relevant genotype datasets to serve as population controls may increase the study size and statistical power to detect associations. Given the low incidence of WNND, many studies are limited in size and underpowered to examine genome-wide associations. An appropriate population control dataset can be used to increase a study’s power to identity potential variants associated with WNND.
3.4.1. Selection of population controls.
Population controls should closely match the study’s population in ancestry, as well as any additional limitations such as age, location, and comorbidities. Possible datasets can be searched through databases like dbGaP [36, 37], which requires an application detailing rationale and proposed use for access to individual-level data.
3.4.2. Matching quality control measures.
Data from the population controls should be processed similarly to data from the cases and controls of the original study. Both datasets should have the same process to assess and apply thresholds for missing data at the individual and variant level, minor allele frequency, and deviation from Hardy-Weinberg Equilibrium. Both datasets need to be of the same build, with software like Batch Coordinate Conversion (LiftOver) used to update build as needed.
3.4.3. Principal components and matching minor allele frequency.
To adjust for heterogeneity and differences in ancestry of the population controls and study sample, principal components can be calculated and outliers removed using software such as Eigenstrat [38, 39] (see Note 3).
Principal components should be plotted (e.g., Principal Component 1 by Principal Component 2 for each individual, using distinct colors to differentiate population controls from study cases and controls) to visually assess similarity of the datasets.
To further reduce heterogeneity between the samples, analysis should be limited to variants with MAF within 5 percent between the population controls and the study controls.
3.4.4. Univariate analysis.
Analysis of the association of variants with WNND can be conducted in PLINK or similar software. Adjustments should be made for principal components and study membership (either population control or WNND study), in addition to any other covariates of interest (such as genetic sex and age).
3.5. Discussion
WNV can cause severe, fatal illness in some infected individuals, with no approved vaccines or targeted therapeutics. Given the difficulty of identifying sufficient cases for a well-powered GWAS, it is necessary to consider novel approaches both for future studies and in order to extract further information to fully utilize previously conducted studies. There are many challenges of working with existing datasets, as some data such as type of neuroinvasive disease or covariates may be unavailable and older studies may have utilized now-discontinued chips or processing methods. With rapid improvement in technology used in genetic research, there may be a need to update the datasets in order to work with current software but methods such as the ones we outlined above make this feasible. Here we have identified additional approaches, such as analysis of rare variants or pathways for association with severe disease [40, 41], that can also be utilized on either novel or existing datasets to strengthen the ability to detect genetic variants relevant to the disease cohort (see Note 4). Gene-environment interactions may also be of value [42, 43], but use may be limited for datasets from previously conducted studies depending on the covariates collected at the time.
When conducting future studies, an emphasis should be placed on greater diversity of study samples to match the global spread of this virus. WNV has been isolated worldwide, and an increasing number of outbreaks have occurred in recent years in Europe and the Middle East[7, 44]. There has been a movement to expand the diversity of GWAS beyond the initial disproportionate concentration on samples of individuals with European-descent [45–47], and there are many tools and reference datasets now available to accurately analyze non-European and admixed populations [26, 48]. With better powered and more diverse studies, researchers will be better equipped to identify host factors that place individuals at increased risk for severe disease.
While WNV has been isolated worldwide and incidence is expected to continue to rise due to climate change, the number of affected individuals remain small compared to some other diseases. With an estimated 80% of infected individuals remaining asymptomatic [2], there are challenges to identifying and enrolling a sufficient number of cases for a well-powered GWAS. Creative approaches, such as testing the blood of individuals enrolled in other studies for antibodies to WNV or collaborating with blood banks to find current asymptomatic infections [49, 50], can be used to increase sample sizes and to provide asymptomatic controls.
Genetic studies have great potential to guide the development of vaccines and targeted therapeutics, as well as potentially identifying individuals at elevated risk for severe neuroinvasive disease. Continued research through novel analyses of existing datasets using approaches like the ones we outlined above, as well as conducting new, well-powered, and diverse studies are expected to provide further evidence of genetic factors associated with severe outcomes and novel avenues of investigation for developing methods to prevent and treat WNND. It is challenging to research the role of genetic variation in the development of WNND and other rare conditions given the obstacles noted above, such as case identification and statistical power. Through the use of innovative technology and pooling of samples and resources, significant progress continues to be made towards identifying those at risk and developing preventative and therapeutic measures to protect populations against WNND and other public health threats [51–54].
4. Notes
Use the most recent approaches. Methodologic and analytical approaches will depend on the existing knowledge at the time of the study, the research question (e.g., whether whole genome or focused on specific genes), and the available study population. Genomic technology advances rapidly, and it is recommended that researchers review recent publications for new approaches prior to designing a study.
Navigate reference builds and program versions. If using an older dataset, be sure to check that it is in the right version and format for a software, as discussed under 3.2.1. Older datasets may have been sequenced on a now-outdated reference build. Different software programs will have different formatting requirements; check the program’s website for the latest versions.
Address population stratification. Cases and controls need to be as similar as possible for all factors other than the disease of interest; population stratification will lead to inaccurate or unusable results. The careful selection of controls – whether within the study or as supplemental population controls – is critical. Principal component analysis, outlined in 3.4.3 above and more extensively in other resources [10, 55, 56], is an important step to identifying and adjusting heterogeneity.
Compare data from closely related cohorts. Similar phenotypes can be analyzed together for greater insight, especially for rare and related outcomes. Like WNV, dengue is a mosquito-borne flavivirus that causes severe disease in some infected individuals; we analyzed dengue and WNV GWAS datasets together to identify variants associated with both outcomes. This approach has also been used for more common outcomes, particularly psychiatric conditions [3, 57, 58].
Acknowledgements
This work was supported in part by the US National Institutes of Health (NIH)/National Institute of Allergy and Infectious Diseases (NIAID) Human Immunology Project Consortium (HIPC) award U19 AI 089992. The authors are grateful to Dr. Andrew Dewan for expert guidance and Ms. Xiaomei Wang for valuable support.
References
- 1.Debiasi RL. West nile virus neuroinvasive disease. Curr Infect Dis Rep. 2011;13(4):350–9. doi: 10.1007/s11908-011-0193-9. [DOI] [PubMed] [Google Scholar]
- 2.Prevention CfDCa: West Nile virus: Symptoms, Diagnosis, & Treatment. https://www.cdc.gov/westnile/symptoms/index.html (2018). Accessed 2021.
- 3.Cahill ME, Conley S, DeWan AT, et al. Identification of genetic variants associated with dengue or West Nile virus disease: a systematic review and meta-analysis. BMC Infect Dis. 2018;18(1):282. doi: 10.1186/s12879-018-3186-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Initiative C-HG. Mapping the human genetic architecture of COVID-19. Nature. 2021;600(7889):472–7. doi: 10.1038/s41586-021-03767-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pairo-Castineira E, Clohisey S, Klaric L, et al. Genetic mechanisms of critical illness in COVID-19. Nature. 2021;591(7848):92–8. doi: 10.1038/s41586-020-03065-y. [DOI] [PubMed] [Google Scholar]
- 6.Loeb M, Eskandarian S, Rupp M, et al. Genetic variants and susceptibility to neurological complications following West Nile virus infection. J Infect Dis. 2011;204(7):1031–7. doi: 10.1093/infdis/jir493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chancey C, Grinev A, Volkova E, et al. The global ecology and epidemiology of West Nile virus. BioMed research international. 2015;2015:376230. doi: 10.1155/2015/376230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Paz S Climate change impacts on West Nile virus transmission in a global context. Philosophical transactions of the Royal Society of London Series B, Biological sciences. 2015;370(1665). doi: 10.1098/rstb.2013.0561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marees AT, de Kluiver H, Stringer S, et al. A tutorial on conducting genome-wide association studies: Quality control and statistical analysis. Int J Methods Psychiatr Res. 2018;27(2):e1608. doi: 10.1002/mpr.1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Turner S, Armstrong LL, Bradford Y, et al. Quality control procedures for genome-wide association studies. Curr Protoc Hum Genet. 2011;Chapter 1:Unit1 19. doi: 10.1002/0471142905.hg0119s68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cahill ME, Loeb M, Dewan AT, et al. In-Depth Analysis of Genetic Variation Associated with Severe West Nile Viral Disease. Vaccines (Basel). 2020;8(4). doi: 10.3390/vaccines8040744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wood AR, Perry JR, Tanaka T, et al. Imputation of variants from the 1000 Genomes Project modestly improves known associations and can identify low-frequency variant-phenotype associations undetected by HapMap based imputation. PLoS One. 2013;8(5):e64343. doi: 10.1371/journal.pone.0064343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Das S, Forer L, Schonherr S, et al. Next-generation genotype imputation service and methods. Nat Genet. 2016;48(10):1284–7. doi: 10.1038/ng.3656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Christian Fuchsberger LF, Sebastian Schoenherr, Sayantan Das, Gonçalo Abecasis: Michigan Imputation Server: Free Next-Generation Genotype Imputation Service. https://imputationserver.sph.umich.edu/ (2021). Accessed 2021.
- 15.Kent WJ, Sugnet CW, Furey TS, et al. The human genome browser at UCSC. Genome Res. 2002;12(6):996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.University of California SCGBG: Lift Genome Annotations. https://genome.ucsc.edu/cgi-bin/hgLiftOver Accessed 2021.
- 17.Delaneau O, Marchini J, Zagury JF. A linear complexity phasing method for thousands of genomes. Nat Methods. 2011;9(2):179–81. doi: 10.1038/nmeth.1785. [DOI] [PubMed] [Google Scholar]
- 18.Delaneau O, Zagury JF, Marchini J. Improved whole-chromosome phasing for disease and population genetic studies. Nat Methods. 2013;10(1):5–6. doi: 10.1038/nmeth.2307. [DOI] [PubMed] [Google Scholar]
- 19.Delaneau O: SHAPEIT. https://mathgen.stats.ox.ac.uk/genetics_software/shapeit/ Accessed 2021.
- 20.Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 2009;5(6):e1000529. doi: 10.1371/journal.pgen.1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Browning SR, Browning BL. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am J Hum Genet. 2007;81(5):1084–97. doi: 10.1086/521987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Howie B, Fuchsberger C, Stephens M, et al. Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nat Genet. 2012;44(8):955–9. doi: 10.1038/ng.2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shi S, Yuan N, Yang M, et al. Comprehensive Assessment of Genotype Imputation Performance. Hum Hered. 2018;83(3):107–16. doi: 10.1159/000489758. [DOI] [PubMed] [Google Scholar]
- 24.Roshyara NR, Horn K, Kirsten H, et al. Comparing performance of modern genotype imputation methods in different ethnicities. Sci Rep. 2016;6:34386. doi: 10.1038/srep34386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Clarke L, Fairley S, Zheng-Bradley X, et al. The international Genome sample resource (IGSR): A worldwide collection of genome variation incorporating the 1000 Genomes Project data. Nucleic Acids Res. 2017;45(D1):D854–D9. doi: 10.1093/nar/gkw829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Genomes Project C, Auton A, Brooks LD, et al. A global reference for human genetic variation. Nature. 2015;526(7571):68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Resource IGS: Data Portal. https://www.internationalgenome.org/data (2021). Accessed 2021.
- 28.Huang GH, Tseng YC. Genotype imputation accuracy with different reference panels in admixed populations. BMC Proc. 2014;8(Suppl 1 Genetic Analysis Workshop 18Vanessa Olmo):S64. doi: 10.1186/1753-6561-8-S1-S64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Howie B, Marchini J: Impute2: Analyzing Whole Chromosomes. https://mathgen.stats.ox.ac.uk/impute/impute_v2.html#whole_chroms Accessed 2021.
- 30.Luan JT A; Zhao J; Fuchsberger C; Willer C: IMPUTE2: 1000 Genomes Imputation Cookbook. https://genome.sph.umich.edu/wiki/IMPUTE2:_1000_Genomes_Imputation_Cookbook (2012). Accessed 2021.
- 31.University of California SCGBG: Cytoband. http://hgdownload.cse.ucsc.edu/goldenPath/hg19/database/cytoBand.txt.gz Accessed 2021.
- 32.Howie B, Marchini J: Impute2: Details about ‘info’ metric. https://mathgen.stats.ox.ac.uk/impute/impute_v2.html#info_metric_details Accessed 2021.
- 33.Purcell S: PLINK 1.9 Input Filtering. https://www.cog-genomics.org/plink/1.9/filter (2021). Accessed.
- 34.Cordell HJ. Epistasis: what it means, what it doesn’t mean, and statistical methods to detect it in humans. Hum Mol Genet. 2002;11(20):2463–8. doi: 10.1093/hmg/11.20.2463. [DOI] [PubMed] [Google Scholar]
- 35.Panagiotou OA, Evangelou E, Ioannidis JP. Genome-wide significant associations for variants with minor allele frequency of 5% or less--an overview: A HuGE review. Am J Epidemiol. 2010;172(8):869–89. doi: 10.1093/aje/kwq234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tryka KA, Hao L, Sturcke A, et al. NCBI’s Database of Genotypes and Phenotypes: dbGaP. Nucleic Acids Res. 2014;42(Database issue):D975–9. doi: 10.1093/nar/gkt1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.National Center for Biotechnology Information USNLoM: Database of Genotypes and Phenotypes (dbGaP). https://www.ncbi.nlm.nih.gov/gap/ Accessed.
- 38.Patterson N, Price AL, Reich D. Population structure and eigenanalysis. PLoS Genet. 2006;2(12):e190. doi: 10.1371/journal.pgen.0020190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Price AL, Patterson NJ, Plenge RM, et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38(8):904–9. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 40.Auer PL, Lettre G. Rare variant association studies: considerations, challenges and opportunities. Genome Med. 2015;7(1):16. doi: 10.1186/s13073-015-0138-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cirillo E, Parnell LD, Evelo CT. A Review of Pathway-Based Analysis Tools That Visualize Genetic Variants. Front Genet. 2017;8:174. doi: 10.3389/fgene.2017.00174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cooley PCC, R. F; Folsom RE Assessing Gene-Environment Interactions in Genome-Wide Association Studies: Statistical Approaches. RTI Press Research Report Series Research Triangle Park (NC): Research Triangle Institute; 2014. [PubMed] [Google Scholar]
- 43.Lin WY, Huang CC, Liu YL, et al. Genome-Wide Gene-Environment Interaction Analysis Using Set-Based Association Tests. Front Genet. 2018;9:715. doi: 10.3389/fgene.2018.00715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Control. ECfDPa. West Nile virus infection. Annual Epidemiological Report for 2019 Stockholm: ECDC; 2021. [Google Scholar]
- 45.Wojcik GL, Graff M, Nishimura KK, et al. Genetic analyses of diverse populations improves discovery for complex traits. Nature. 2019;570(7762):514–8. doi: 10.1038/s41586-019-1310-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mills MC, Rahal C. The GWAS Diversity Monitor tracks diversity by disease in real time. Nat Genet. 2020;52(3):242–3. doi: 10.1038/s41588-020-0580-y. [DOI] [PubMed] [Google Scholar]
- 47.Peterson RE, Kuchenbaecker K, Walters RK, et al. Genome-wide Association Studies in Ancestrally Diverse Populations: Opportunities, Methods, Pitfalls, and Recommendations. Cell. 2019;179(3):589–603. doi: 10.1016/j.cell.2019.08.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Atkinson EG, Maihofer AX, Kanai M, et al. Tractor uses local ancestry to enable the inclusion of admixed individuals in GWAS and to boost power. Nat Genet. 2021;53(2):195–204. doi: 10.1038/s41588-020-00766-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cahill ME, Yao Y, Nock D, et al. West Nile Virus Seroprevalence, Connecticut, USA, 2000–2014. Emerg Infect Dis. 2017;23(4):708–10. doi: 10.3201/eid2304.161669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Garcia MN, Hause AM, Walker CM, et al. Evaluation of prolonged fatigue post-West Nile virus infection and association of fatigue with elevated antiviral and proinflammatory cytokines. Viral Immunol. 2014;27(7):327–33. doi: 10.1089/vim.2014.0035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Beloor J, Maes N, Ullah I, et al. Small Interfering RNA-Mediated Control of Virus Replication in the CNS Is Therapeutic and Enables Natural Immunity to West Nile Virus. Cell host & microbe. 2018;23(4):549–56 e3. doi: 10.1016/j.chom.2018.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Diamond MS. Progress on the development of therapeutics against West Nile virus. Antiviral Res. 2009;83(3):214–27. doi: 10.1016/j.antiviral.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ulbert S West Nile virus vaccines - current situation and future directions. Hum Vaccin Immunother. 2019;15(10):2337–42. doi: 10.1080/21645515.2019.1621149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bai F, Thompson EA, Vig PJS, et al. Current Understanding of West Nile Virus Clinical Manifestations, Immune Responses, Neuroinvasion, and Immunotherapeutic Implications. Pathogens. 2019;8(4). doi: 10.3390/pathogens8040193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bouaziz M, Ambroise C, Guedj M. Accounting for population stratification in practice: a comparison of the main strategies dedicated to genome-wide association studies. PLoS One. 2011;6(12):e28845. doi: 10.1371/journal.pone.0028845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhao H, Mitra N, Kanetsky PA, et al. A practical approach to adjusting for population stratification in genome-wide association studies: principal components and propensity scores (PCAPS). Stat Appl Genet Mol Biol. 2018;17(6). doi: 10.1515/sagmb-2017-0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cross-Disorder Group of the Psychiatric Genomics Consortium. Electronic address pmhe, Cross-Disorder Group of the Psychiatric Genomics C. Genomic Relationships, Novel Loci, and Pleiotropic Mechanisms across Eight Psychiatric Disorders. Cell. 2019;179(7):1469–82 e11. doi: 10.1016/j.cell.2019.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wellcome Trust Case Control C. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447(7145):661–78. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]