

Abstract
Splice site variants may lead to transcript alterations, causing exons inclusion, exclusion, truncation, or intron retention. Interpreting the consequences of a specific splice site variant is not straightforward, especially if the variant is located outside of the canonical splice sites. We developed MutSpliceDB: https://brb.nci.nih.gov/splicing, a public resource to facilitate the interpretation of splice sites variants effects on splicing based on manually reviewed RNA-seq BAM files from samples with splice site variants.
Keywords: splice variants, splicing, splice sites, RNA-seq, MutSpliceDB
Introduction
Splice site variants/mutations are one of the well-known classes of genetic alterations playing an important role in biology and diseases. Splice site mutations in cancer are most frequently observed as inactivating alterations in tumor suppressor genes (for example, TP53 (Bouaoun et al., 2016) or RB1 (George et al., 2015)), and to a lesser degree as activating alterations in oncogenes (for example MET (Onozato et al., 2009)). Splice site variants may lead to alterations in mRNA transcripts, causing exons inclusion, exclusion, truncation, or intron retention. Interpreting the consequences of a specific splice site variant is not straightforward, especially if the variant is located outside of the canonical splice sites. Accurate interpretation of the impact of a splice site variant can further our understanding of biology, influence patient treatment, and in cases of germline splice site variants, may have relevance to familial disease predisposition. To facilitate the interpretation of a splice site variants effects, we developed MutSpliceDB (https://brb.nci.nih.gov/splicing), a public resource documenting variants effects on splicing based on manually reviewed publicly available RNA-seq BAM files from samples with splice site variants. MutSpliceDB has stable URLs for each entry and is intended to be a supplemental resource for existing databases, such as ClinVar, ClinGen allele registry, CIViC, LOVD, IARC TP53 DB, OncoKB, etc. Existing databases can point to MutSpliceDB entries; users of such resources would then know that RNA-seq BAM file(s) with the splice variant in question exist, and underlying reads can be easily visualized to review the evidence behind a description of splice variant effect on splicing.
Description
MutSpliceDB is a dynamic resource of RNA based evidence of effects of splice site variants on splicing. Additional entries can be proposed by emailing supporting information, which would be reviewed to verify the existence of clear evidence of splice site mutation effects or lack of splicing defects. Database entries can be edited or removed if problems are identified or new information becomes available. At the time of this publication, MutSpliceDB includes splice site variants for the following genes: APC, ARID1A, ARID1B, ATM, BRCA1, CDKN1A, CDKN1B, CDKN2A, CDKN2C, FANCA, MET, MSH2, NF1, NF2, PALB2, PMS2, POLD1, PTCH1, PTEN, RB1, SMARCA4, SMARCB1, SMARCC1, TP53, TSC1, TSC2, and VHL. Currently, MutSpliceDB contains detailed information for a subset of splice site mutations and their effects on splicing derived from publicly available RNA-seq data from Cancer Cell Lines Encyclopedia (CCLE) (Barretina et al., 2012; Ghandi et al., 2019; Chang, Vural, & Sonkin, 2017) and The Cancer Genome Atlas (TCGA) (Cancer Genome Atlas Research Network et al., 2013).
All entries from MutSpliceDB are in ClinVar or have been submitted to ClinVar to provide functional evidence on effects of splice site variants on splicing (Landrum et al., 2016). For example, the following is a link to one of such ClinVar entries for APC splice site mutation NM_001127511.2:c.250+2T>C https://www.ncbi.nlm.nih.gov/clinvar/variation/619119. Out of 80 unique splice mutations with clear effects on splicing, at the time of entry in MutSpliceDB, 47 had no ClinVar entry, highlighting the importance of MutSpliceDB contributions in providing information on splice cite variants effects on splicing. Out of 33 splice mutations with ClinVar entries, 15 had pathogenic interpretations, 15 had likely pathogenic interpretations, and 3 had a variant of uncertain significance (VUS) interpretations. Eight splice mutations in MutSpliceDB are more than two base pairs (bp) away from an exon-intron junction, including three which are five base pairs (bp) away from an exon-intron junction. Out of these eight mutations, five had no ClinVar entry (NM_000389.5:c.446–3C>G, NM_000314.8:c.635–3C>A, NM_000321.3:c.2211+5G>T, NM_000321.2:c.1389+5G>C, and NM_000548.5:c.3285–3C>G), two had VUS interpretation(s), and one had conflicting interpretations. Figure 1 provides an IGV image snapshot from MutSpliceDB for TP53 NM_000546.5:c.375+5G>A variant, showing clear evidence of intron inclusion between exons 4 and 5 based on RNA-seq data from cell line PK-45H (Robinson et al., 2011; Barretina et al., 2012).
Figure 1.
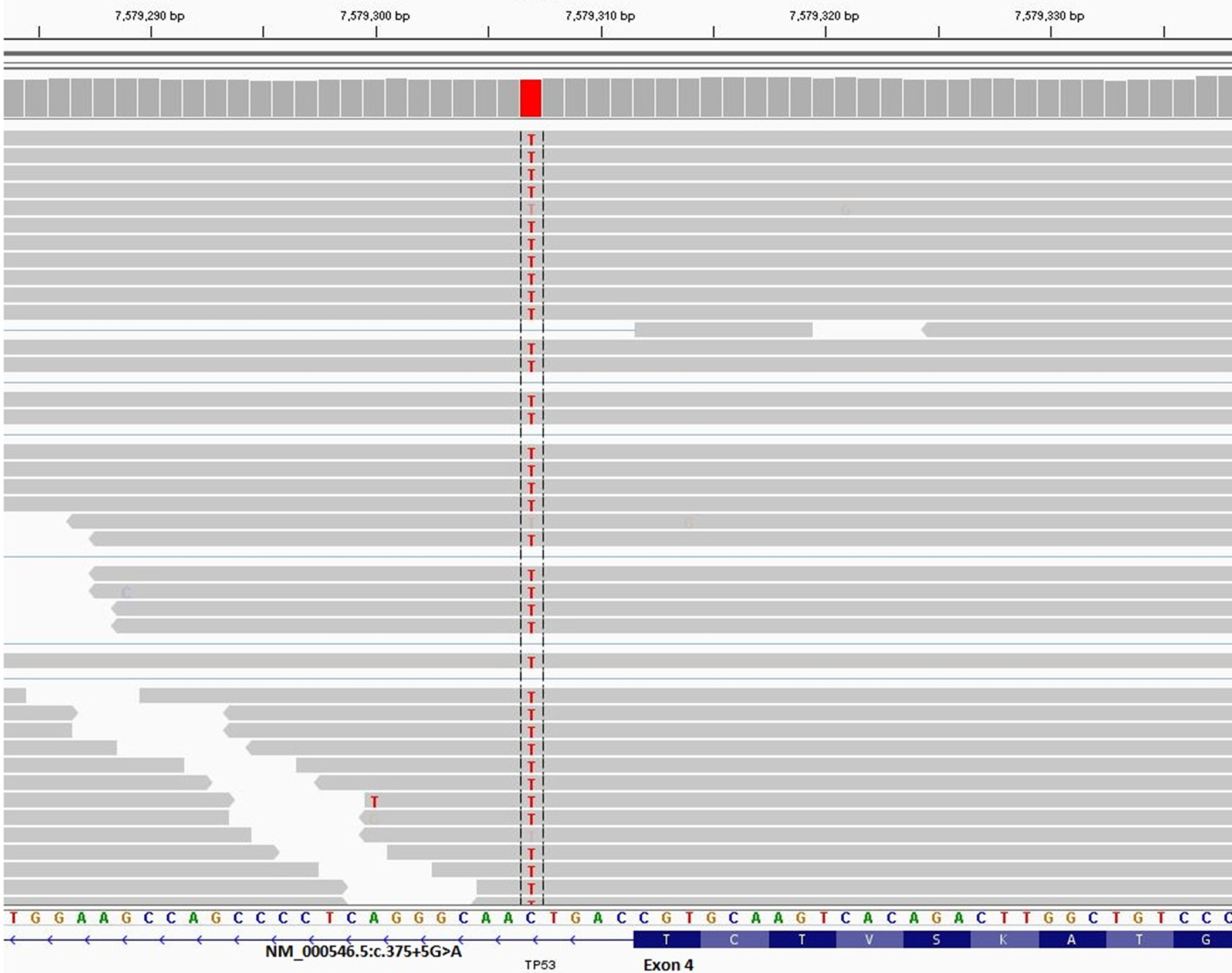
IGV image snapshot from MutSpliceDB for TP53 NM_000546.5:c.375+5G>A variant
MutSpliceDB documents splice site variant effect(s) on splicing based on manual review of RNA-seq BAM files from samples with splice site variant. Manual review of RNA-seq BAM files confirms at least 20X coverage for relevant region(s), at least 10 reads with a splice site variant, and no evidence of alternative explanation(s) for observed splicing effects. In cases of exon skipping, RNA-seq BAM files from other samples without exon skipping should have at least 20X coverage for the exon of interest to rule out a drop of sequencing depth as a technical artifact. Additionally, if a splice site variant is not observed in the RNA-seq data due to exon skipping, DNA sequencing data is reviewed to confirm the presence of a variant and rule out potential alternative explanations for the observed exon skipping. Splice site variants (especially non-canonical) in gene regions, which are known to have well supported alternative splicing isoforms expressed at a reasonable level, are included only if it is possible to differentiate between an alternative splicing isoform and the effects of splice site variant. RNA-seq BAM files from other samples without a variant are always checked during the manual review process; however it is possible that technical artifacts, such as DNA contamination, may be specific to a particular sample and appear as an intron retention event. Additionally, the presence of a variant in a sufficient fraction in DNA sequencing data is confirmed to avoid complications due to the possible subclonal nature of a variant or potentially high level of inclusion of normal tissue in tumors biopsies/resections.
For each splice site variant, MutSpliceDB contains the following key information: description of the splicing effect; IGV image snapshot of the splicing effect; and mini BAM file (if there are no restrictions on nucleotide level data distribution) with reads only for relevant genes. The mini BAM file can be explored using web based IGV, without the need to download the file or use any additional software. In addition, each splice site variant in MutSpliceDB also contains: gene symbol, Entrez gene ID, HGVS compliant transcript based variant notation, ClinGen allele registry ID, sample name, sample source, name of RNA-seq BAM file, and the name of BAM file with DNA sequencing data, if the RNA-seq BAM file does not contain reads with splice site variant (e.g., due to exon skipping).. The ClinGen Allele Registry contains direct links from the Allele Registry entry to corresponding entry in MutSpliceDB (Pawliczek et al., 2018). Allele Registry entries contain links to ClinVar, Exome Aggregation Consortium (ExAC), Genome Aggregation Database (gnomAD), Catalogue of Somatic Mutations in Cancer (COSMIC), and other resources if they contain information for variant described by Allele Registry entry (Lek et al., 2016; Karczewski et al., 2019; Tate et al., 2019). Figure 2 illustrates a page with RNA-seq details on splice site variant for TP53 NM_000546.5:c.375+5G>A variant. Documentation on how to use MutSpliceDB website and how to submit evidence for review can be found at https://brb.nci.nih.gov/splicing/documentation.html.
Figure 2.

Splice site variant RNA-seq evidence details page for TP53 NM_000546.5:c.375+5G>A variant
As mentioned above, entries in MutSpliceDB are fully integrated with ClinVar and ClinGen allele registry. For example, the TP53 variant NM_000546.5:c.375+5G>A MutSpliceDB entry: https://brb.nci.nih.gov/cgi-bin/splicing/splicing_evidence.cgi?caid=CA645589233 contains a link to ClinGen allele registry: http://reg.clinicalgenome.org/redmine/projects/registry/genboree_registry/by_caid?caid=CA645589233, which in turn contains a link to ClinVar: https://www.ncbi.nlm.nih.gov/clinvar/variation/481015, containing accession: SCV000925744.1 from MutSpliceDB with a link back to MutSpliceDB entry.
Conclusions
MutSpliceDB provides a place for the genetics community to submit RNA-seq based evidence of splice site variants effects on splicing which allows the preservation of the complex details needed for full examination of effects of splice sites variants. ACMG/AMP variant interpretation guidelines recommend usage of RNA-seq data as one of evidence types for the interpretation of splice site variants (Richards et al., 2015). MutSpliceDB can be an especially valuable resource for curators who are following ACMG guidelines to interpret pathogenicity of splice site variants.
Acknowledgements
The authors would like to thank the anonymous reviewers and the journal editorial board for providing valuable feedback on earlier versions of this manuscript. We would also like to thank the NIH Library Writing Center for manuscript editing assistance.
Footnotes
Conflict of Interest: authors declare no conflicts of interest
Web Resources: https://brb.nci.nih.gov/splicing
Data Availability Statement:
all data is publicly available at https://brb.nci.nih.gov/splicing
References
- Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, ... Garraway LA (2012). The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature, 483(7391), 603–607. 10.1038/nature11003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouaoun L, Sonkin D, Ardin M, Hollstein M, Byrnes G, Zavadil J, & Olivier M (2016). TP53 Variations in Human Cancers: New Lessons from the IARC TP53 Database and Genomics Data. Human Mutation. 10.1002/humu.23035 [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network, Weinstein JN, Collisson EA, Mills GB, Shaw KRM, Ozenberger BA, ... Stuart JM (2013). The Cancer Genome Atlas Pan-Cancer analysis project. Nature Genetics, 45(10), 1113–1120. 10.1038/ng.2764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang L-C, Vural S, & Sonkin D (2017). Detection of homozygous deletions in tumor-suppressor genes ranging from dozen to hundreds nucleotides in cancer models. Human Mutation. 10.1002/humu.23308 [DOI] [PubMed] [Google Scholar]
- George J, Lim JS, Jang SJ, Cun Y, Ozreti? L, Kong G, ... Thomas RK (2015). Comprehensive genomic profiles of small cell lung cancer. Nature, 524(7563), 47–53. 10.1038/nature14664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghandi M, Huang FW, Jané-Valbuena J, Kryukov GV, Lo CC, McDonald ER, ... Sellers WR (2019). Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature. 10.1038/s41586-019-1186-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, ... MacArthur DG (2019). Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes [Preprint]. Genomics. 10.1101/531210 [DOI] [Google Scholar]
- Landrum MJ, Lee JM, Benson M, Brown G, Chao C, Chitipiralla S, ... Maglott DR (2016). ClinVar: Public archive of interpretations of clinically relevant variants. Nucleic Acids Research, 44(D1), D862–868. 10.1093/nar/gkv1222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, ... Exome Aggregation Consortium. (2016). Analysis of protein-coding genetic variation in 60,706 humans. Nature, 536(7616), 285–291. 10.1038/nature19057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onozato R, Kosaka T, Kuwano H, Sekido Y, Yatabe Y, & Mitsudomi T (2009). Activation of MET by gene amplification or by splice mutations deleting the juxtamembrane domain in primary resected lung cancers. Journal of Thoracic Oncology: Official Publication of the International Association for the Study of Lung Cancer, 4(1), 5–11. 10.1097/JTO.0b013e3181913e0e [DOI] [PubMed] [Google Scholar]
- Pawliczek P, Patel RY, Ashmore LR, Jackson AR, Bizon C, Nelson T, ... on behalf of the Clinical Genome (ClinGen) Resource. (2018). ClinGen Allele Registry links information about genetic variants. Human Mutation, 39(11), 1690–1701. 10.1002/humu.23637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, ... ACMG Laboratory Quality Assurance Committee. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine: Official Journal of the American College of Medical Genetics, 17(5), 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, & Mesirov JP (2011). Integrative genomics viewer. Nature Biotechnology, 29(1), 24–26. 10.1038/nbt.1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N, ... Forbes SA (2019). COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Research, 47(D1), D941–D947. 10.1093/nar/gky1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
all data is publicly available at https://brb.nci.nih.gov/splicing