

Abstract
Purpose:
IL12 promotes adaptive type I immunity and has demonstrated antitumor efficacy, but systemic administration leads to severe adverse events (AE), including death. This pilot trial investigated safety, efficacy, and immunologic activity of intratumoral delivery of IL12 plasmid DNA (tavo) via in vivo electroporation (i.t.-tavo-EP) in patients with Merkel cell carcinoma (MCC), an aggressive virus-associated skin cancer.
Patients and Methods:
Fifteen patients with MCC with superficial injectable tumor(s) received i.t.-tavo-EP on days 1, 5, and 8 of each cycle. Patients with locoregional MCC (cohort A, N = 3) received one cycle before definitive surgery in week 4. Patients with metastatic MCC (cohort B, N = 12) received up to four cycles total, administered at least 6 weeks apart. Serial tumor and blood samples were collected.
Results:
All patients successfully completed at least one cycle with transient, mild (grades 1 and 2) AEs and without significant systemic toxicity. Sustained (day 22) intratumoral expression of IL12 protein was observed along with local inflammation and increased tumor-specific CD8+ T-cell infiltration, which led to systemic immunologic and clinical responses. The overall response rate was 25% (3/12) in cohort B, with 2 patients experiencing durable clinical benefit (16 and 55+ months, respectively). Two cohort A patients (1 with pathologic complete remission) were recurrence-free at 44+ and 75+ months, respectively.
Conclusions:
I.t.-tavo-EP was safe and feasible without systemic toxicity. Sustained local expression of IL12 protein and local inflammation led to systemic immune responses and clinically meaningful benefit in some patients. Gene electrotransfer, specifically i.t.-tavo-EP, warrants further investigation for immunotherapy of cancer.
Introduction
Prior to the success of immune checkpoint inhibitors, immunomodulatory cytokines were the backbone of cancer immunotherapy for decades and provided proof-of-concept for successfully harnessing the immune system against cancer. IL2 and IFN are approved by the FDA for advanced melanoma and renal cell carcinoma. Therapeutic utility of cytokines, however, has been limited by low response rates, significant treatment-associated systemic toxicities, and the paucity of predictive biomarkers. The pursuit of novel cytokines with better efficacy and of novel approaches to deliver cytokines without systemic toxicity remain areas of active investigation in cancer immunotherapy, both as monotherapy and in combination immunotherapy regimens.
IL12 is a 70-kDa heterodimeric, multifunctional protein discovered in 1989 and rapidly recognized as a master regulator of adaptive type I cell-mediated immunity (1, 2). IL12 production by innate immune cells (e.g., monocytes, macrophages, neutrophils, and dendritic cells) polarizes naïve CD4+ T cells toward the Th1 phenotype; these cells then produce high levels of IFNγ upon restimulation (3–5). IL12 also induces the production of IFNγ and increased proliferation and cytotoxicity of natural killer cells and CD8+ T cells (6, 7). In addition, IL12 upregulates the expression of HLA class I and II, and intercellular adhesion molecule-1 on human cancer cells (melanoma), enhancing T-cell extravasation and increasing antigen binding (8). These unique biological properties and promising preclinical data prompted active investigation of recombinant IL12 (rIL12) in the treatment of advanced solid tumors and hematologic malignancies. In early-phase clinical trials using systemic administration of rIL12, encouraging biological and clinical responses were observed, but were tempered by severe multiorgan toxicities that included dose-limiting cytopenias, hepatitis, and even treatment-related deaths in 2 patients (9–11). These observations led to development of localized IL12 delivery directly to the tumor in forms other than recombinant protein. This was attempted through direct injection of IL12 plasmid, viral vectors, or modified fibroblasts, autologous tumor cells, or dendritic cells engineered to secrete IL12 (12–19). These methods showed efficacy in preclinical models, but not as much in clinical trials, likely related to low gene transfer efficiency (20). In 2008, Daud and colleagues reported a clinical trial using intratumoral injection of IL12 plasmid DNA (tavo) followed by in vivo electroporation (i.t.-tavo-EP) to enhance the efficiency of transfection and thus local IL12 protein expression in 24 patients with advanced melanoma (21). Treatment led to robust dose-proportional expression of IL12 protein within injected lesions (21). I.t.-tavo-EP was safe and well-tolerated, with minimal systemic toxicity and no significant systemic IL12 spillage. Four of 19 patients with distant disease, exhibited distant responses in noninjected lesions, including three complete remissions (CR). Six additional patients had stable disease (SD) at distant sites lasting from 4 to 20 months. Responses occurred over a span of 6 to 18 months and were associated with hypopigmentation and gradual regression of abscopal tumors suggesting immune-mediated mechanisms. Encouraged by this data, we investigated this approach in treating another aggressive and highly-immunogenic skin cancer, Merkel cell carcinoma (MCC).
MCC is a rare, highly aggressive skin cancer associated with the Merkel cell polyomavirus (MCPyV) and ultraviolet radiation exposure. MCC is considered immunogenic, with reports of spontaneous regression and responsiveness to immunotherapy (22–27). Strong CD8+ T-cell infiltration is associated with dramatically improved survival, independent of stage at presentation (28). The mechanisms of this immunogenicity, however, have only recently been understood (27). Virus-positive MCC (VP-MCC) accounts for approximately 80% of MCC cases in the United States; MCPyV antigens are persistently expressed in VP-MCC and are immunogenic, which also allows for rigorous study of local and systemic antitumor immune responses in this subset. The remaining 20% of cases are virus-negative (VN-MCC) and are associated with high mutational burden from ultraviolet radiation–induced damage, which likely facilitates expression of neoantigens. These insights have led to multiple successful trials of immunotherapies for VP-MCC and VN-MCC. Programmed death-1 (PD-1) axis checkpoint inhibitors (e.g., avelumab, pembrolizumab, and nivolumab) are now preferred first-line systemic therapy in eligible patients (including VP-MCC and VN-MCC; ref. 29). These agents have impressive frequency, durability, and depth of objective responses, with some of the highest objective response rates (ORR; 40%–60%) reported among solid tumors treated in the first line (22, 30, 31). Unfortunately, approximately half of patients do not experience durable benefit, and 15% to 20% of patients experience severe grade 3–4 immune-related AEs (22, 30, 31), indicating a need for novel therapeutic approaches that target the tumor microenvironment (TME) without excessive systemic immune toxicities and augment responses to checkpoint blockade.
Here, we report the results of a single-arm, open-label, pilot trial of i.t.-tavo-EP for patients with advanced MCC who had at least one superficial (injectable) lesion.
Patients and Methods
Study design
This was a single-arm, open-label, pilot trial of i.t.-tavo-EP in 15 patients with MCC (NCT01440816) evaluating the clinical efficacy, safety, and tolerability, and immunologic changes in the TME, including changes in IL12 protein expression in the TME after treatment. The study was conducted at the University of Washington in accordance with International Conference on Harmonization guidelines for Good Clinical Practice and the code of Federal Regulations and guided by the ethical principles of the Belmont Report. The protocol was approved by the local Institutional Review Board. All patients provided written, informed consent at the time of screening.
Patients
Eligible patients were ≥18 years of age with histologically confirmed MCC and at least one injectable lesion, defined as a superficial (cutaneous, subcutaneous, or nodal) tumor amenable to intratumoral injection in the outpatient setting. MCPyV-positive status was not required. Patients were required to have measurable disease per RECIST v1.1 (32), an Eastern Cooperative Oncology Group (ECOG) performance score of 0 to 2, and adequate hematologic, renal, and hepatic function. Patients were excluded if immunosuppressed. Patients were enrolled onto one of two cohorts: patients with locoregional MCC who were candidates for definitive therapy (surgery ± radiotherapy) were enrolled in cohort A (N = 3) and patients with metastatic MCC in cohort B (N = 12).
Treatment
The treatment schema is provided in Supplementary Fig. S1. Patients received i.t.-tavo-EP on days 1, 5, and 8 of each cycle in an outpatient clinic setting following administration of a local anesthetic and under direct palpation of the tumor mass without radiologic guidance. Patients with locoregional MCC (cohort A) received one cycle before planned definitive surgery in week 4, followed by adjuvant radiotherapy per clinician discretion. Patients with distant metastatic disease (cohort B) could receive up to four cycles total, administered at least 6 weeks apart, with restaging clinical and radiologic evaluation at week 6 of each cycle.
I.t.-tavo-EP was applied to specific treatment zones (corresponding to the tumor volume covered by a sterile electroporation applicator comprised of six stainless steel electrodes 1.5 cm long and arranged in a circular array of approximately 1 cm diameter) within the injected tumor(s) that were selected at the beginning of each cycle. Up to four treatment zones (in one or multiple tumors) could be treated within each cycle with no new zones added during a cycle. For patients in cohort B, new treatment zones (in same or different tumors) could be selected at the beginning of each cycle. The pIL12 dose for each treated zone was fixed at 0.5 mL or 0.25 mg (fixed concentration of 0.5 mg/mL). The maximum total dose of plasmid injected in one patient on a given day was capped at 2 mL or 1.0 mg. The plasmid injection in each zone was followed immediately by electroporation before moving onto the next zone. For electroporation, the applicator was inserted into the same zone in which the plasmid was injected, and six electrical pulses at a field strength (E+) of 1,300 volts/cm and pulse width of 100 microseconds at 400-millisecond intervals were administered as described previously (21).
Clinical assessments
Clinical endpoints included safety and tolerability, as well as efficacy, which measured both injected and noninjected lesion regression, recurrence-free survival in cohort A, and objective response by RECIST v1.1 in cohort B. AEs were graded according to NCI Common Terminology Criteria for AEs (v4.03). Clinical assessment of tumor responses was performed for both injected and noninjected (distant) lesions according to RECIST v1.1. For cohort B patients with distant disease, assessment of overall response via radiologic imaging studies was carried out at baseline and at week 6 of each treatment cycle, and then as clinically indicated. After completion of treatment, patients were followed at least annually for relapse, disease progression, and overall survival.
Biopsy and peripheral blood collection
Serial tumor biopsies and peripheral blood samples were collected from all patients to evaluate cellular immunologic changes, as well as changes in IL12 p70 protein expression in treated tumors (Supplementary Fig. S1). Pretreatment tumor biopsy was attempted on day 1 and posttreatment biopsy of the same tumors on day 22; the latter was substituted by surgical specimen collection during week 4 in cohort A patients. Tumor samples were divided for biomarker studies: (i) formaldehyde-fixed paraffin embedded (FFPE) blocks for IHC, multispectral IHC, or gene expression, (ii) flash-frozen for protein expression, and (iii) processed immediately for isolation and culture of tumor-infiltrating lymphocytes (TIL) as described previously (33). Blood samples for immune response analysis were collected at baseline, day 22, and week 6 of each cycle. Peripheral blood mononuclear cells (PBMC) were isolated using routine Ficoll gradient centrifugation and were immediately cryopreserved.
Immune response analyses
Tumor-MCPyV status was assessed using T-antigen IHC (CM2B4 antibody; Santa Cruz Biotechnology) and/or detection of MCPyV oncoprotein antibodies (AMERK test; Table 1) as described previously (34–36).
Table 1.
Patient baseline characteristics.
| Cohort A (N = 3) | Cohort B (N = 12) | Overall (N = 15) | |
|---|---|---|---|
| Age in years | |||
| Median (range) | 59 (50–73) | 69 (53–86) | 66 (50–86) |
| Sex | |||
| Male (%) | 3 (100%) | 9 (75.0%) | 12 (80.0%) |
| Female (%) | 0 (0.0%) | 3 (25.0%) | 3 (20.0%) |
| Ethnicity | |||
| White (%) | 3 (100%) | 11 (92%) | 14 (93%) |
| Other (%) | 1 (8%) | 1 (7%) | |
| ECOG performance status | |||
| Score of 0 (%) | 3 (100%) | 8 (67.7%) | 11 (73.3%) |
| Score of 1 (%) | 0 (0.0%) | 4 (33.3%) | 4 (26.7%) |
| Pathologic stage at enrollment per AJCC 8th edition (43) | |||
| IIIA (%) | 1 (33.3%) | 0 (0.0%) | 1 (6.7%) |
| IIIB (%) | 2 (66.7%) | 1 (8.3%) | 3 (20.0%) |
| IV (%) | 0 (0.0%) | 11 (91.7%) | 11 (73.3%) |
| Prior therapies | |||
| Surgery (%) | 1 (33.3%) | 12 (100%) | 13 (86.7%) |
| Radiation (%) | 1 (33.3%) | 9 (75.0%) | 10 (66.7%) |
| Systemic therapy (%) | 0 (0.0%) | 6 (50.0%) | 6 (40.0%) |
| Chemotherapy | 5 | 5 | |
| Immunotherapya | 6 | 6 | |
| Targeted therapy | 1 | 1 | |
| MCPyV statusb | |||
| Positive (%) | 3 (100%) | 9 (75.0%) | 12 (80.0%) |
| Negative (%) | 0 (0.0%) | 3 (25.0%) | 3 (20.0%) |
Prior immunotherapies included IFNβ, 4-1BB agonistic antibody, and a Toll-like receptor agonist; none had received prior PD-1 blockade.
Viral status was determined by MCPyV oncoprotein serologic status and CM2B4 IHC staining.
IHC analyses
Standard histology was performed on all biopsies. Monochromatic IHC staining with antibodies to CK20 (KS20.8; Dako), CD8 (C8/144B; Dako), CD4 (SP35; Cell Marque), CM2B4 (MCPyV T-antigen, sc-136172; Santa Cruz Biotechnology), MHC class I (EMR8-5; MBL), PD-L1 (E1L3N; Cell Signaling Technology), and FoxP3 (14-5773-82; eBioscience) was performed. Hematoxylin and eosin- and IHC-stained slides were reviewed for evidence of tumor and necrosis prior to performing further biomarker analyses. For multispectral IHC staining, FFPE specimens were deparaffinized and rehydrated, subjected to heat-induced antigen retrieval, and stained as described previously (37). Staining was performed using the following antibodies: PD-1 (EPR4877; Abcam), PD-L1 (SP142; Spring Bio), CD4 (RBT-CD4; BioSB), CD8 (C8/144B; Dako), and CD68 (PG-M1; Dako). Slides were imaged with a Vectra Automated Quantitative Pathology Imaging System (Perkin Elmer). Images were analyzed using inForm Software (Perkin Elmer) and were evaluated by a pathologist.
IL12 p70 protein expression
To determine whether i.t.-tavo-EP led to increased expression of the IL12 protein in the TME, pre- and postbiopsies were collected, lysed, and analyzed for IL12 p70 protein, a heterodimer comprised of p40 and p35 subunits. Pre- and posttreatment frozen paired biopsies were weighed before homogenization in lysis buffer, lysed, and subjected to centrifugation to sediment insoluble cellular material prior to determination of IL12 p70 protein levels using a multianalyte immunoassay platform (MAGPIX; Luminex). Each immunoassay was performed in 96-well plates and according to the MILLIPLEX MAP High Sensitivity Human Cytokine Premixed Magnetic Bead Kit (EMD Millipore Billerica; catalog no. SPRHSCYMG60PX13). Each assay included duplicate 50 μL homogenate samples, which were added to capture bead-treated plates. Detection beads were then added to the plates before quantifying the analyte. For analysis, replicate values below the lower limit of quantification (LLOQ) were adjusted to the run-specific LLOQ value. All values were then adjusted to pg/g of sample and averaged to produce an average expression value. No replicate values were adjusted or excluded from analysis on the basis of the upper limit of quantification.
Gene expression analysis
FFPE- or cell pellet-extracted RNA (approximately 100 ng) was analyzed using the human Immunology V2 panel and the nCounter platform (NanoString Technologies). This panel profiles 594 immunology-related human genes as well as two types of built-in controls: positive controls (spiked RNA at various concentrations to evaluate the overall assay performance) and 15 housekeeping genes (to normalize for differences in total RNA input). Sample preparation and hybridization was carried out according to the manufacturer’s instructions. In brief, 5 μL (100 ng) of total RNA was hybridized at 96°C overnight and then digitally analyzed for frequency of each RNA species. Data were collected using the nCounter Digital Analyzer, and data normalization and analysis were carried out using the nSolver software (V.3). Normalization factors were derived from the geometric mean of housekeeping genes, mean of negative controls, and geometric mean of positive controls.
RNA extraction (FFPE or tissue pellets)
FFPE: Total RNA was isolated using the AllPrep Kit (Qiagen) from FFPE specimens after histologic confirmation of evaluable tumor. Tissue pellet: Tumor tissue samples procured by punch biopsy or fine-needle aspirate (FNA) were dissociated in PBS without Ca2+ or Mg2+ (Life Technologies) with protease inhibitors (cOmplete-mini, EDTA-free; Roche Life Science). Clarified supernatants and cell pellets were stored separately at −80°C. Lysates and cell pellets were separated by centrifugation at 18,000g. Total RNA was isolated from cell pellets using TRIzol reagent (Thermo Fisher Scientific) according to the manufacturer’s protocol. Total RNA concentrations were determined using the NanoDrop ND-1000 spectrophotometer (Thermo Fisher Scientific) and quality was assessed using the 2100 Bioanalyzer (Agilent). Samples were excluded from analysis if the Bioanalyzer RIN score was less than 2.0 and smear analysis indicated <30% of RNA were less than 300 bp.
MCPyV-specific tetramer staining
Subjects were HLA-I typed at Bloodworks Northwest. PBMCs and/or TILs from patients with HLA-I types that corresponded to available MCPyV-specific tetramers (A*02:01, A*24:02, B*35:02, B*37:01, B*07:02; n = 13 patients) were incubated with 100 nmol/L of dasatinib for 10 minutes at 37°C. Cells were then stained with appropriate tetramers and analyzed using flow cytometry. At least 2 × 106 PBMCs or TILs were stained with anti–CD8-FITC (clone 3B5; Thermo Fisher Scientific), violet viability dye (Invitrogen), anti-CD14-Pacific Blue (clone M5E2; BioLegend), anti–CD19-Pacific Blue (clone HIB19; BioLegend), anti-CD4 PerCP and either A02/KLLEIAPNC-APC, A24/EWWRSGGFSF-APC, B07/APNCYG-NIPL-PE, B35/FPWEEYGTL-PE tetramer (Immune Monitoring Lab at Fred Hutchinson Cancer Research Center, Seattle, WA) or B37/KEWWRSGGFSF-PE tetramer (Buus Lab) as described previously (33, 38–40). Cells were acquired on an Aria II Cell sorter with BD-FACSDiva software, and were analyzed with FlowJo (v10.0.8) software. Cells of interest were viable and identified as CD8+ tetramer+ cells in the lymphocyte forward/side scatter region with >0.01% of CD8+ T cells costaining with tetramers. Cells staining positively for CD4, CD14, or CD19 were excluded. The frequency of MCPyV-specific CD8 T cells out of the total CD8 T-cell fraction was determined from paired pre- and post-fresh tumor biopsies and PBMCs. The fold change in tetramer-positive T-cell frequency between pre- and posttreatment was calculated as the difference between the posttreatment tetramer-positive T-cell frequency and pretreatment tetramer-positive T-cell frequency over the pretreatment tetramer-positive T-cell frequency. Fold change was used to evaluate changes in tetramer-positive T-cell enrichment following treatment in the periphery and intratumorally.
T-cell receptor sequencing
Total genomic DNA was extracted from whole flash-frozen tumor biopsies using the spin column method and the DNeasy Kit (Qiagen). High-throughput deep sequencing was used to determine T-cell receptor beta locus (TRB) complementarity-determining region 3 (CDR3) with the Illumina Genome Analyzer (Adaptive Biotechnologies) using the immunoSEQ immune-profiling system (41). Read normalization was performed as described previously (42). Identification of the Vβ, Dβ, and Jβ gene segments contributing to each TCRβ CDR3 sequence was performed using the published algorithm (41). The T-cell fraction within MCC tumors was determined by calculating the percentage of a tumor sample that was composed of T cells. To determine whether there was a change in T-cell enrichment between pre- and posttreatment specimens, the fold change in the T-cell fraction was calculated as the difference between the posttreatment T-cell fraction and pretreatment T-cell fraction over the pretreatment T-cell fraction.
Statistical analyses
Descriptive statistics were used to summarize baseline patient characteristics, safety, clinical response, and immunologic response variables. All injected and noninjected tumors were assessed and responses to treatment were classified as complete responses (CR), partial responses (PR), stable disease (SD), or progressive disease (PD) per RECIST v1.1 definitions (32). The overall ORR per standard RECIST v1.1 guidelines is also reported for those patients with distant metastases in cohort B. Progression-free survival and overall survival were calculated from the day of treatment initiation to documented disease progression and/or death, respectively. All analyses and summaries were produced using SAS v9.3 (or higher).
Results
Patient characteristics
Fifteen patients (Pt) with locoregional (cohort A, N = 3) or metastatic (cohort B, N = 12) MCC were enrolled between January 2012 and January 2015; their baseline characteristics are summarized in Table 1. The pathologic stage at enrollment, per AJCC 8th edition (43), for cohort A patients was IIIA (n = 1; T0 pN1b M0) or IIIB (n = 2; T1 pN1b M0 and T1 pN3 M0), and for cohort B patients was unresectable stage IIIB (n = 1; T1 pN3 M0) or stage IV (n = 11; M1). Median patient age at enrollment was 66 years. Patients had an ECOG status of 0 (n = 11) or 1 (n = 4). Most patients (86.7%) had undergone prior surgery, 66% had received prior radiotherapy, 40% had received prior systemic chemotherapy, and 40% had received prior biologic therapy with none receiving prior PD-1/PD-L1 blockade (Table 1). The majority of patients (12/15; 80%) had MCPyV-positive tumors (VP-MCC), correlating well with the expected rate of viral positivity among MCC cases more broadly (44); all 3 patients with VN-MCC were in cohort B.
Safety and tolerability
Overall, i.t.-tavo-EP was safe and well-tolerated (Table 2). In cohort A, all 3 patients successfully completed neoadjuvant i.t.-tavo-EP followed by the planned definitive therapy without any treatment-induced delays. In cohort B, all 12 patients successfully completed at least one treatment cycle (range 1–4 cycles, median 1 cycle) of i.t.-tavo-EP. Treatment-related adverse events (TRAE) were primarily mild to moderate (i.e., grade ≤2); none were grade 3 or higher. There were no deaths or treatment discontinuations due to TRAEs. All patients (15/15; 100%) experienced transient grade 1 pain associated with the electroporation procedure that lasted a few seconds; this occurred despite the use of lidocaine injections, but was manageable with patient education and did not require systemic analgesics or lead to treatment discontinuation. The second most common TRAE was grade 1 injection-site reactions, which did not require medical intervention in most cases. There were no clinically notable systemic toxicities, including laboratory parameters associated with renal and hepatic function. Two patients total (2 of 15; 13.3%) had a serious adverse event (SAE); neither were considered related to the study treatment. One patient developed a grade 3 urinary tract infection that resolved with treatment. Another patient (Pt#13) was diagnosed with hepatocellular carcinoma 9 months after the last i.t.-tavo-EP treatment; this was diagnosed upon elective resection of a preexisting liver lesion that had been presumed to be an MCC metastasis at the time of study entry.
Table 2.
Adverse events.
| All grades | ≥Grade 3 | |
|---|---|---|
| AEsa | n (%) | n (%) |
| Related to study treatment | ||
| Procedural pain | 7 (46.7) | 0 |
| Treatment site inflammation | 3 (20.0) | 0 |
| Treatment site cellulitis | 2 (13.3) | 0 |
| Treatment site bruising | 1 (6.7) | 0 |
| Pain (tumor) | 1 (6.7) | 0 |
| Pain (lymph node) | 1 (6.7) | 0 |
| Peripheral edemab | 1 (6.7) | 0 |
| Pyrexia | 1 (6.7) | 0 |
| Unrelated to study treatment | ||
| Occurring in ≥2 patients | ||
| Fatigue | 3 (20.0) | 0 |
| Severe (grade 3) | ||
| Urinary tract infection (SAE) | — | 1 (6.7) |
| Hepatocellular carcinoma | — | 1 (6.7) |
Abbreviation: SAE, serious adverse event.
Treatment-emergent adverse events were all AEs that began on or after the first administration of study treatment.
Included lymphedema.
Clinical outcomes
Treatment and clinical outcomes are summarized in Table 3. In cohort A, 2 of 3 patients continue to be recurrence-free at 75+ months (Pt#6) and 44+ months (Pt#16) from treatment initiation; the third patient (Pt#9) had disease recurrence at 9 months. Pt#16, who had biopsy-proven recurrent MCC in an enlarged right inguinal lymph node, had pathologic CR after neoadjuvant i.t.-tavo-EP with no detectable residual MCC on histologic review including IHC staining for CK20.
Table 3.
Summary of clinical outcomesa.
| Progression-free survivalb | Median, in months (range) |
|---|---|
|
| |
| Cohort A | NRc (9–75+) |
| Cohort B (all; N = 12) | 1.5 (1.5–55+) |
| Cohort B responders (N = 3) | 7 (3–55+) |
| ORR (Cohort B only; N = 12) | n (%) |
| PR | 3 (25.0) |
| SD | 1 (8.3) |
| PD | 8 (66.7) |
| Local lesion response rated | n (%) |
| (Response-evaluable local lesions; N = 27c) | |
| Major regression (≥30%) | 12 (44.4) |
| Distant lesion response rate | n (%) |
| (Response-evaluable cohort B patientse; N = 10) | |
| Patients with regression of ≥1 distant lesion(s)f | 3 (30.0) |
Clinical outcomes in patients with potential clinical benefit are described in detail in the manuscript text.
Interval is defined as the duration between the date of treatment initiation (Study Day 1) to the date of documented objective or clinical disease progression (cohort B)/relapse (cohort A).
NR, not reported.
Proportion of treated lesions with major (≥30%) regression.
These patients had at least one distant tumor that was defined as a noninjected MCC tumor, clearly distinct from treated lesions.
Proportion of patients with at least one lesion left untreated that reduced in size by ≥30% suggesting systemic immune response.
In cohort B, 3 of 12 (25%) patients had an objective response (all 3 were PR), 1 patient had SD, and 8 patients had PD as the best response. The median time to progression for cohort B was 1.5 months overall and was 7 months for the 3 responders. Two (Pt#2 and Pt#13) of the 3 responders had clinically meaningful benefit lasting 16 months and 55+ months, respectively. Pt#2 was a 55-year-old man with primary VP-MCC on the left ankle and multiple recurrences in the left lower extremity and regional lymph nodes over 2 years despite several prior therapies (including surgery, radiotherapy, chemotherapy, and intratumoral IFN). He received two cycles of i.t.-tavo-EP treatment and had an impressive PR (>70% regression) associated with regression of distant untreated lesions; this clinical response was accompanied by evidence for induction of a systemic immune response. He was progression-free for 7 months and then received another 2 cycles of i.t.-tavo-EP with an additional progression-free period lasting 9 months. Pt#13 was a 82-year-old man with primary MCC on the right proximal elbow and subsequent distant metastatic disease with two large abdominal wall subcutaneous nodules and a suspicious liver lesion. Prior therapies included surgery, chemotherapy (carboplatin plus etoposide), and intratumoral injections of a toll-like receptor 4 agonist on a clinical trial. He received two cycles of i.t.-tavo-EP with complete resolution of his bulky treated lesions over the next several months (NOTE: Only a small proportion of the two lesions was treated and the majority of the bulky lesions were untreated); the suspicious liver lesion remained unchanged, was subsequently resected, and found to be hepatocellular carcinoma. The patient has been MCC progression-free for 55+ months after starting study treatment. Pt#14 also had a PR lasting 3 months, but developed PD with a new brain metastasis. He received stereotactic radiosurgery to the brain lesion followed by pembrolizumab, resulting in a CR lasting 44 months.
A high proportion (12/27; 44.4%) of measurable injected lesions exhibited major (defined as ≥30%) regression. Ten of 12 patients in cohort B had distant untreated lesions that were response-evaluable; 3 of these 10 patients (30%) had major regression in at least one distant lesion, which suggests systemic antitumor immune responses from local therapy in these patients. These patients included Pt#2 and Pt#14 who had overall objective responses too. As mentioned above, both of the measurable lesions in Pt#13 were treated and hence he did not have measurable noninjected distant lesions. Another patient with distant regression of a noninjected lesion had overall PD.
Intratumoral IL12 gene and protein expression
Elevated intratumoral IL12 gene and protein expression were observed in posttreatment specimens (Fig. 1A and B). Specifically, increased expression (by 2-fold or more) of IL12 p70 protein was observed posttreatment (on day 22) versus baseline (day 1) in 6 of 10 (60%) patients with evaluable paired tumor biopsies (Fig. 1B). Although we did not measure systemic IL12 levels in the peripheral blood in this study, there was no evidence of clinically significant systemic spillage of IL12 beyond the local TME, based on a complete lack of systemic TRAEs in this study and similar to results reported by Daud and colleagues in the phase I study (21).
Figure 1.
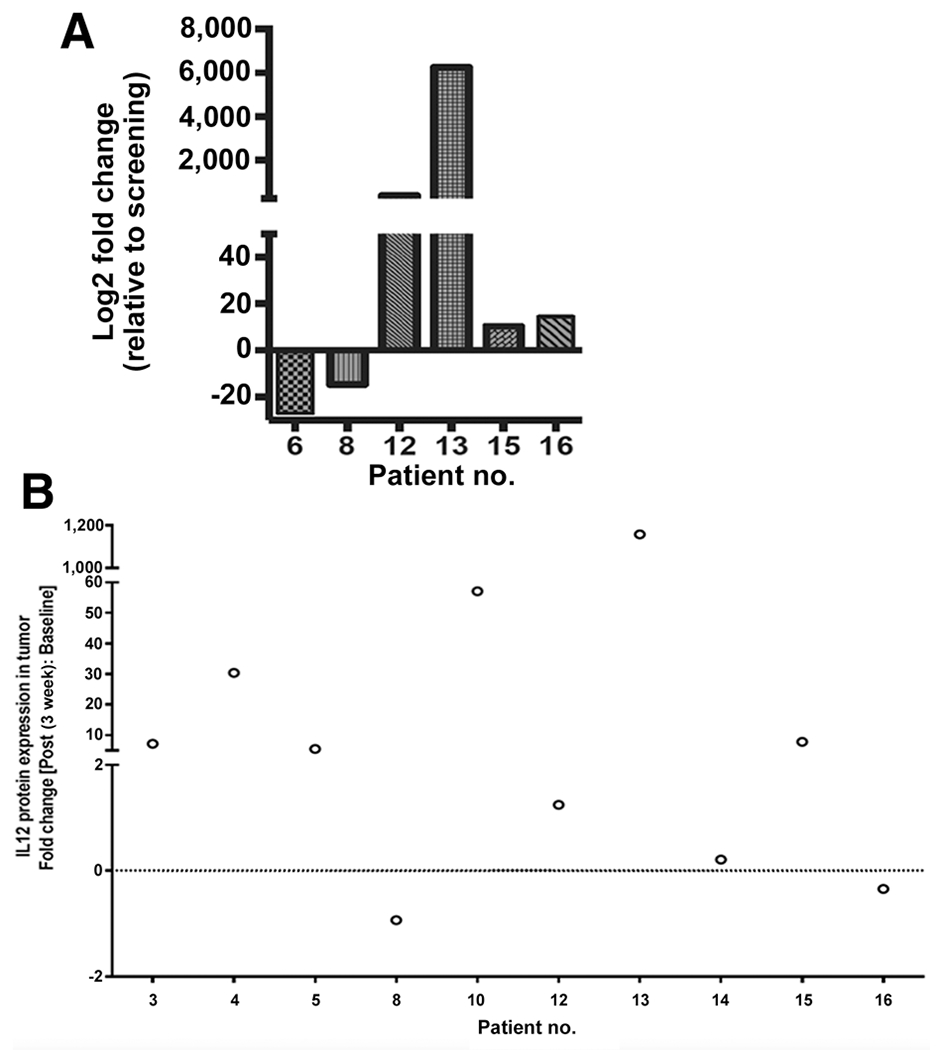
Increased IL12 gene and protein expression posttherapy in electroporated lesions. A, IL12 gene expression (IL12A subunit) was elevated in several tumors posttreatment compared with pretreatment. Changes in gene expression were analyzed using NanoString nCounter platform. B, Paired pre- and post-biopsy samples from 10 patients were evaluable for IL12 p70 protein levels by MAGPIX. Data are presented as fold change (post-/pretreatment). The majority (6 of 10; 60%) had greater than 2-fold increase in IL12 protein levels suggesting sustained expression (at day 22) after treatment on days 1, 5, and 8. no., number.
Increased peripheral and intratumoral MCPyV-specific T cells are associated with response
Using our library of MCPyV-specific HLA class-I tetramers (33, 38–40), we tracked MCPyV-specific T-cell responses, both locally in the treated lesions (Fig. 2A), and also systemically, in the peripheral blood (Fig. 2B) and in untreated lesions (Fig. 3A and B). Thirteen of 15 patients expressed suitable HLA class I alleles for tetramer screening (Supplementary Table S1). We performed MCPyV-specific tetramer staining on paired pre- and posttherapy TIL from 5 patients with MCC-VP tumors (Fig. 2A). The frequency of MCPyV-specific tetramer+ TIL significantly increased (≥1.5-fold change in at least one MCPyV-specific T-cell population) in 3 of 5 patients (Pt#2, Pt#8, Pt#13) following treatment, and decreased in 1 patient (Pt#7), with 1 patient (Pt#10) not having any detectable MCPyV-specific T cells in pre- or postspecimens. Interestingly, all 3 patients with increased frequency of MCPyV-specific TIL-experienced clinical responses.
Figure 2.
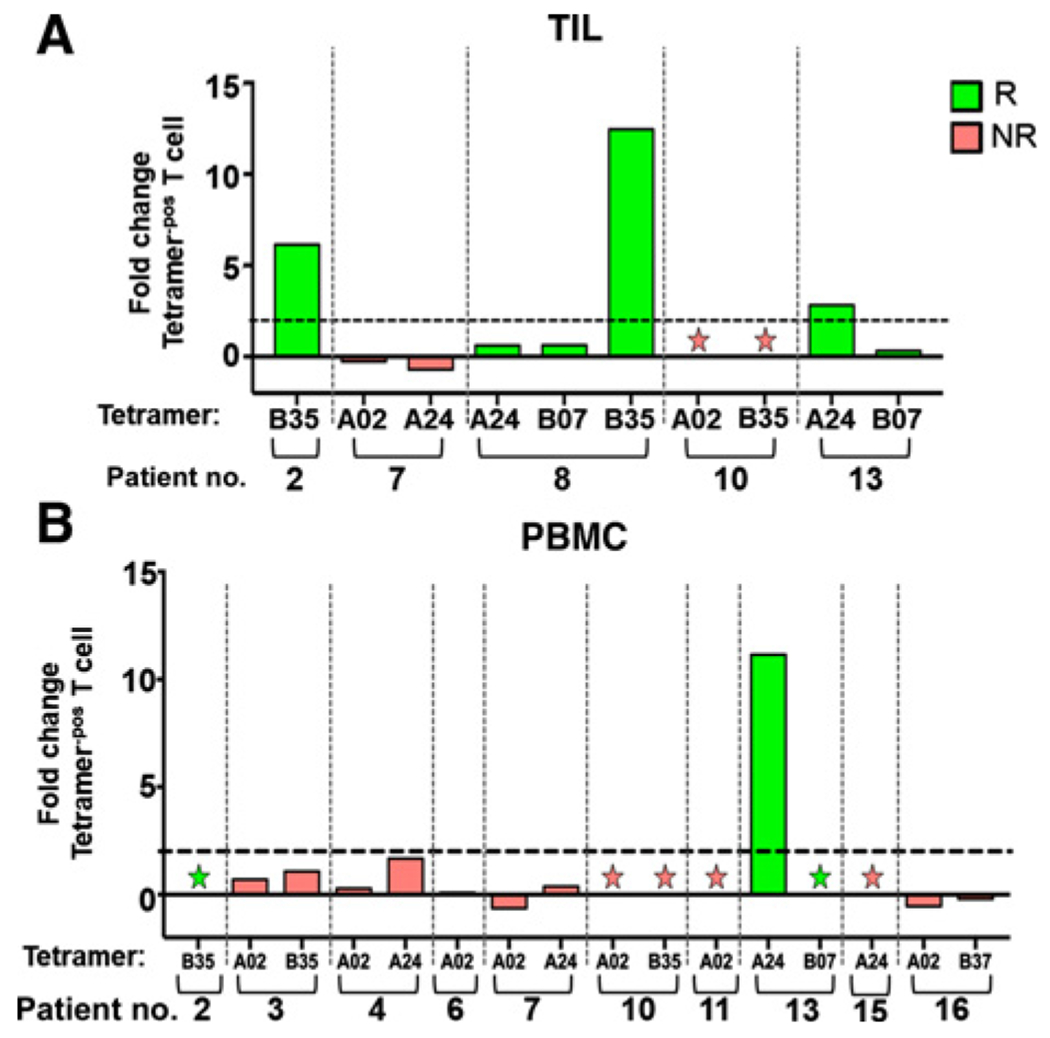
I.t.-tavo-EP induces intratumoral infiltration and systemic MCPyV-specific T-cell expansion. MCPyV-specific CD8 T cells were evaluated intratumorally (A) and in PBMCs (B), using MCPyV-specific tetramers. A, Paired pre- and post-fresh tumor biopsies were cultured in the presence of IL2 and IL15 to obtain TILs. B, Systemic evaluation of MCPyV-specific CD8 T cells from PBMCs was performed as in A. Fold changes, post-IL12 versus pre-IL12 therapy, in the MCPyV-specific tetramer-positive population, as described in Materials and Methods, are shown (the dotted line indicates a 2-fold change). Responders (R) are in green; nonresponders (NR) are in red. Tetramer HLA type is denoted on the x-axis, and stars denote samples that were negative for tetramer positive cells at both pre- and post-time points. no., number.
Figure 3.
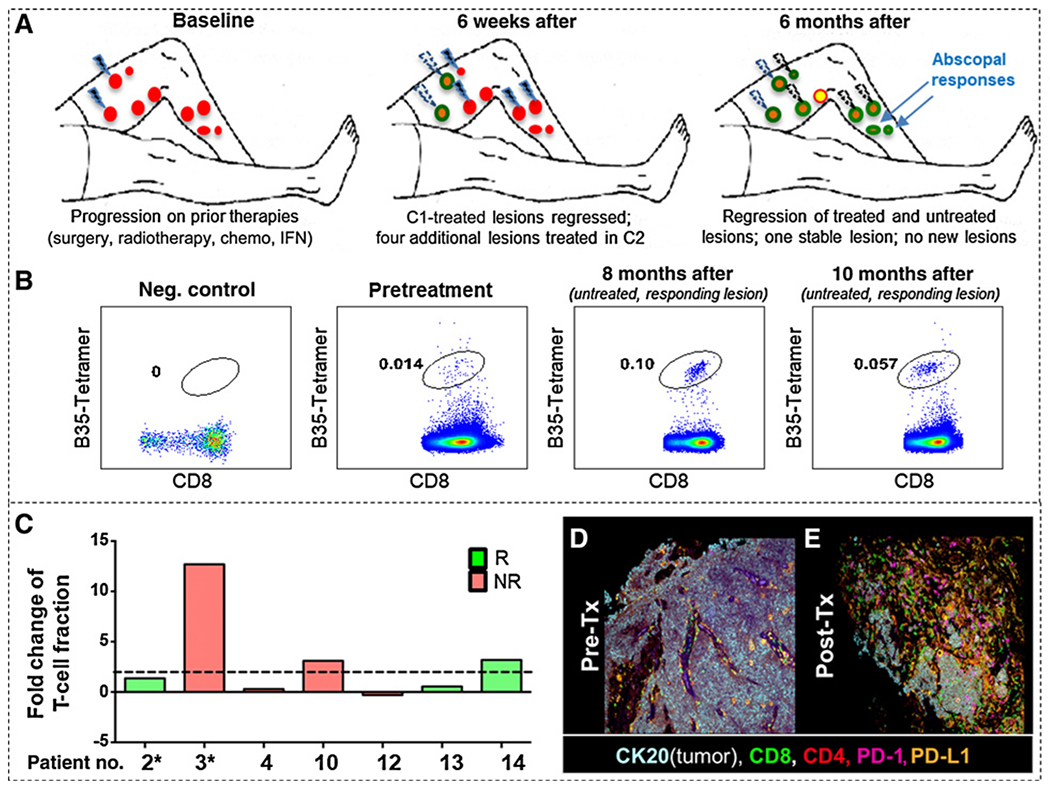
Regression of treated and untreated lesions is associated with robust T-cell responses and infiltration. A, Schema of treatment and response in Pt#2 with baseline (red circles), regressed lesions (green circles), a persistent lesion (yellow circle). Lesions treated with i.t.-tavo-EP are shown as lightning bolts (blue: current cycle; white: prior cycles). Two untreated lesions that regressed are shown in top right panel (blue arrows). B, Tetramer staining of MCPyV-specific CD8 T cells from TIL from pre- and posttherapy from Pt#2. Tetramer-positive CD8 T cells are circled in each panel. Negative control is an HLA mismatched donor. C, T-cell receptor (TCR) beta chain sequencing was used to determine TCR variable beta chain diversity using ImmunoSeq V3 (Adaptive Biotechnology) from pre- and posttreatment tumor specimens (patient number is shown on x-axis, and * denotes patients in whom the pre- and posttreatment samples were from different lesions). Data are presented as fold change (a ratio of the difference between the posttreatment T-cell fraction and pretreatment T-cell fraction over the pretreatment T-cell fraction). Bars are colored in correlation with clinical response: responder (R, green) and nonresponder (NR, red). The dotted line denotes a greater than or equal to 2-fold change. D and E, Multispectral IHC images of pretreatment show an MCC tumor (CK20, white/light blue) with very few infiltrating T cells (CD8 in green and CD4 in red; D). Three weeks after treatment (E), there was a large infiltrate of CD8 T cells (green), accompanied by increased PD-1 (magenta) and PD-L1 (orange) and loss of tumor (CK20, white/light blue). C1, cycle 1; C2, cycle 2; no., number; RT, radiotherapy; Tx, treatment.
Paired baseline and posttreatment peripheral blood samples from patients with MCPyV-positive tumors (n = 10 evaluable patients) were stained with the same panel of five MCPyV tetramers as used to evaluate TILs. Only 1 patient (1/10; 10%) exhibited ≥1.5-fold increase of MCPyV-specific T cells (Fig. 2B); this patient (Pt#13) did have a durable clinical response (PR). Five patients did not have tetramer+ T cells for one or two tetramers at baseline or posttreatment. Taken together, these data suggest that i.t.-tavo-EP therapy can induce systemic and intratumoral expansion of MCPyV-specific T cells, which appear to be associated with a clinical response to treatment.
Abscopal responses and increased intratumoral T-cell infiltration following i.t.-tavo-EP therapy
Additional evidence of systemic, abscopal immune responses was observed in Pt#2, who experienced an objective PR with regression of several treated as well as nontreated lesions (Fig. 3A). Expanded TILs were analyzed from biopsies obtained pretherapy from a treated lesion and 8 months posttherapy from an untreated lesion. TILs from the untreated lesion showed greater MCPyV-specific tetramer staining (6-fold) relative to the baseline pretreatment TILs isolated from a separate treated lesion; Fig. 3B), suggesting abscopal induction of substantial MCPyV-specific T-cell recruitment in untreated lesions by i.t.-tavo-EP.
To evaluate global changes in intratumoral T-cell responses (not restricted to MCPyV-specific T cells), sequencing of TRB chains was performed on pre- and posttreatment tumor specimens. In 6 of 7 patients, the fraction of T cells (relative to the total tumor cell number) increased in posttreatment samples independent of clinical response (Fig. 3C), suggesting that i.t.-tavo-EP can increase infiltration or expansion of T cells.
Multispectral IHC staining was used to evaluate spatial changes in intratumoral T-cell infiltration and their expression of immunoregulatory molecules PD-1 and PD-L1 in a pre- and posttreatment lesion from Pt#10 (Fig. 3D and E). I.t.-tavo-EP induced increased infiltration of CD8+ and CD4+ T cells, and also a concomitant increase in PD-1 and PD-L1 expression in this lesion. Despite evidence of increased T-cell infiltration, this patient did not respond to i.t.-tavo-EP therapy. Taken together, these data indicate that i.t.-tavo-EP therapy can increase intratumoral T cells, including those specific for MCPyV epitopes, but that additional mechanisms of local immune evasion may limit clinical responses.
Discussion
This pilot trial investigated i.t.-tavo-EP in 15 patients with advanced MCC. I.t.-tavo-EP administered on days 1, 5, and 8 resulted in sustained (day 22) intratumoral IL12 expression without any systemic or severe AEs. Resultant local inflammation led to systemic immune responses, suggested by regression of noninjected MCC lesions in 3 of 10 (30%) patients and an overall response rate of 25% (3 of 12) in cohort B patients with metastatic MCC. Neoadjuvant administration before surgery and radiotherapy in locoregional MCC was feasible; one of 3 cohort A patients experienced a pathologic CR.
This trial validates the previously reported findings of the phase I trial of i.t.-tavo-EP in patients with melanoma (21). It highlights the immunogenicity of this approach that evokes clinically meaningful and durable responses in a subset of patients. Similar to the phase I trial, this intratumoral treatment was safe and well tolerated; there were no severe TRAEs, suggesting lack of systemic spillage of IL12 protein. This is remarkable because systemic delivery of IL12, a promising cytokine that facilitates Th1 responses, has previously been associated with severe AEs, including death. Sustained local expression of IL12 in the TME on day 22 with treatment only on days 1, 5, and 8 highlights the potential of gene electrotransfer (GET) to overcome a limitation of most intratumoral immunotherapy approaches, the requirement for frequent repeated injections due to limited intratumoral persistence of the injected drugs. The platform for GET is relatively flexible for inducing local intratumoral expression of plasmid DNA of choice and also mitigates neutralizing antibodies and the risk of viral integration observed with gene delivery via viral vectors (20).
Because of shared viral antigens expressed by VP-MCC (12 of 15 patients within this study), study of cancer-specific T-cell responses against conserved viral epitopes was possible. Using 5 MCPyV-specific HLA-peptide tetramers (33, 38–40), we found detectable increases in MCPyV-specific T cells in the MCC TME following i.t.-tavo-EP in patients with clinical responses, with a concomitant increase of tetramer-positive T cells in the PBMC of 1 patient. In addition, we found that i.t.-tavo-EP induced increased intratumoral infiltration or expansion of T cells independent of response to therapy. This suggests that additional mechanisms of immune evasion are involved and indeed, i.t.-tavo-EP treatment in 1 evaluable patient was associated with increased expression of PD-1 and PD-L1 within the TME (Fig. 3D). Notably, IL12 has been shown to significantly induce expression of IFNγ, which can drive potent antitumor effects via enhanced immunogenicity and direct tumoral-static mechanisms, but this cytokine can also trigger upregulation of PD-L1 and IDO-1 in the TME (45). Consequently, combining i.t.-tavo-EP with PD-1/PD-L1 pathway blockade may provide an appealing therapeutic combination. Indeed, tumor regression after therapeutic PD-1 blockade may require preexisting cancer-specific immune response (46). Therefore, the observation that i.t.-tavo-EP induces intratumoral infiltration and systemic expansion of T cells, and may increase PD-L1 expression suggests a potential for therapeutic synergy. Interestingly, intratumoral PD-L1+ tumor-associated macrophages can also contribute to “adaptive resistance,” yet IL12 has been reported to reduce the frequency of MDSC suppressor subsets (47). Further investigation into the role and potential modulation of this innate immune subset during i.t.-tavo-EP therapy is warranted. Several studies using IL12 delivery in combination with PD-1 blockade have shown promising efficacy in murine models (48, 49). Intriguingly, 5 patients with MCC in this trial received PD-1 blocking therapies (as monotherapy or in combination with other immunotherapies) at some point in their treatment course after i.t.-tavo-EP; all 5 had an objective response to PD-1/PD-L1 axis blockade (Supplementary Table S2). On the basis of these observations and other clinical trials in melanoma and metastatic triple-negative breast cancer, combined with the excellent safety profile of this approach, several clinical trials are currently investigating the combination of i.t.-tavo-EP with PD-1 blocking therapies (NCT03132675, NCT03567720).
In vivo electroporation of tumors also has several limitations, many of which are shared with other intratumoral approaches. It has primarily been investigated in patients with superficial, clinically accessible tumors; administration to deeper-seated (e.g., visceral) lesions, while possible and in development, will have additional challenges. I.t.-tavo-EP monotherapy is less suitable for patients with bulky visceral lesions and/or rapidly progressive disease due to the relatively low proportion of treated tumor burden that is unlikely to overcome the immune evasion mechanisms in rapidly growing distant lesions. It will be ideal to combine this approach with other systemic therapies in such patients. Nevertheless, this study provides proof-of-concept of this approach in enhancing immunogenicity in MCC and is worthy of further exploration in MCC and other cancers.
In conclusion, i.t.-tavo-EP was safe and feasible in patients with MCC without severe systemic toxicities. It led to sustained local expression of IL12 protein, which facilitated local and systemic immunity and clinically meaningful responses in a subset of patients. GET warrants further investigation for immunotherapy of cancer, either as monotherapy or in combination with agents such as PD-1 blockade therapy.
Supplementary Material
Translational Relevance.
Intratumoral immunotherapy is gaining momentum, but its applicability (and likely efficacy too) is limited by the need for repeated injections because of limited intratumoral persistence of the injected drugs. We utilized plasmid IL12 DNA (pIL12 or tavokinogene telseplasmid; “tavo”) and in vivo intratumoral electroporation (i.t.-tavo-EP) to achieve prolonged intratumoral IL12 expression in 15 patients with Merkel cell carcinoma (MCC), an aggressive, immunogenic cancer. Systemic delivery of IL12, a cytokine that facilitates Th1 responses, had previously been associated with severe adverse events (AE), including death. I.t.-tavo-EP administered on days 1, 5, and 8 resulted in sustained (day 22) intratumoral IL12 expression without any systemic or severe AEs. Resultant local inflammation led to systemic immune responses, with regression of noninjected MCC lesions in 3 of 10 (30%) patients and an overall response rate of 25% (3 of 12) in patients with metastatic MCC. Gene electrotransfer, specifically i.t.-tavo-EP, warrants further investigation for immunotherapy of cancer.
Acknowledgments
This study was funded by the NIH/NCI Cancer Center Support Grant P30 CA015704, R01-CA162522 (to N. Longino, N. Miller, R. Kulikauskas, D. Ibrani, and P. Nghiem), NIH-K24-CA139052 (to N. Longino, N. Miller, and P. Nghiem), Merkel patient gift funds, and OncoSec Medical Inc. The authors would like to acknowledge the patients and families; Seattle Cancer Care Alliance clinical care team (including RNs Jon Smith, Sharon Rockwell, and Debra Martin); Nichole Pelz for research coordination, and Jennifer Kahle, PhD (BPS International) for medical writing assistance. The results of an interim analysis of this study were presented previously at the 2015 ESMO congress (50).
Disclosure of Potential Conflicts of Interest
S. Bhatia has served as a paid consultant for Bristol-Myers Squibb, Sanofi Genzyme, and EMD Serono, and reports receiving commercial research grants from Oncosec, Bristol-Myers Squibb, EMD Serono, Merck, Immune Design, Novartis, Nektar, and NantKwest. N.J. Miller is listed as a co-inventor on a patent regarding Merkel Cell Polyomavirus T Antigen-Specific TCRS and Uses thereof, co-owned by the University of Washington and Fred Hutch Cancer Research Center. C.G. Twitty holds ownership interest (including patents) in Oncosec. J.S. Campbell is an employee/paid consultant for Oncosec Medical and is listed as a co-inventor on two patents: one regarding systems and methods for improved tissue-sensing based electroporation and one regarding plasmid constructs for heterologous protein expression and methods of use, both of which are owned by OncoSec Medical. M.H. Le is an employee/paid consultant for OncoSec Medical, Inc. and Symvivo, and holds ownership interest (including patents) in OncoSec Medical, Inc. S. Gargosky is an employee/paid consultant for OncoSec. R.H. Pierce is an employee/paid consultant for OncoSec Medical and is an unpaid consultant/advisory board member for SymVivo. R. Heller holds ownership interest (including patents) in and is an unpaid consultant/advisory board member for OncoSec Medical, Inc. P. Nghiem has served as a paid consultant for Merck and EMD Serono. No potential conflicts of interest were disclosed by the other authors.
Footnotes
Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
References
- 1.Kobayashi M, Fitz L, Ryan M, Hewick RM, Clark SC, Chan S, et al. Identification and purification of natural killer cell stimulatory factor (NKSF), a cytokine with multiple biologic effects on human lymphocytes. J Exp Med 1989;170:827–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stern AS, Podlaski FJ, Hulmes JD, Pan YC, Quinn PM, Wolitzky AG, et al. Purification to homogeneity and partial characterization of cytotoxic lymphocyte maturation factor from human B-lymphoblastoid cells. Proc Natl Acad Sci U S A 1990;87:6808–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.D’Andrea A, Rengaraju M, Valiante NM, Chehimi J, Kubin M, Aste M, et al. Production of natural killer cell stimulatory factor (interleukin 12) by peripheral blood mononuclear cells. J Exp Med 1992;176:1387–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Macatonia SE, Hosken NA, Litton M, Vieira P, Hsieh CS, Culpepper JA, et al. Dendritic cells produce IL-12 and direct the development of Th1 cells from naive CD4+ T cells. J Immunol 1995;154:5071–9. [PubMed] [Google Scholar]
- 5.Hsieh CS, Macatonia SE, Tripp CS, Wolf SF, O’Garra A, Murphy KM. Development of TH1 CD4+ T cells through IL-12 produced by Listeria-induced macrophages. Science 1993;260:547–9. [DOI] [PubMed] [Google Scholar]
- 6.Brunda MJ, Luistro L, Warrier RR, Wright RB, Hubbard BR, Murphy M, et al. Antitumor and antimetastatic activity of interleukin 12 against murine tumors. J Exp Med 1993;178:1223–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ma X, Trinchieri G. Regulation of interleukin-12 production in antigen-presenting cells. Adv Immunol 2001;79:55–92. [DOI] [PubMed] [Google Scholar]
- 8.Yue FY, Geertsen R, Hemmi S, Burg G, Pavlovic J, Laine E, et al. IL-12 directly upregulates the expression of HLA class I, HLA class II and ICAM-1 on human melanoma cells: a mechanism for its antitumor activity? Eur J Immunol 1999;29:1762–73. [DOI] [PubMed] [Google Scholar]
- 9.Atkins MB, Robertson MJ, Gordon M, Lotze MT, DeCoste M, DuBois JS, et al. Phase I evaluation of intravenous recombinant human interleukin 12 in patients with advanced malignancies. Clin Cancer Res 1997;3:409–17. [PubMed] [Google Scholar]
- 10.Cohen J. IL-12 deaths: explanation and a puzzle. Science 1995;270:908. [DOI] [PubMed] [Google Scholar]
- 11.Robertson MJ, Cameron C, Atkins MB, Gordon MS, Lotze MT, Sherman ML, et al. Immunological effects of interleukin 12 administered by bolus intravenous injection to patients with cancer. Clin Cancer Res 1999;5:9–16. [PubMed] [Google Scholar]
- 12.Mendiratta SK, Quezada A, Matar M, Wang J, Hebel HL, Long S, et al. Intratumoral delivery of IL-12 gene by polyvinyl polymeric vector system to murine renal and colon carcinoma results in potent antitumor immunity. Gene Ther 1999;6:833–9. [DOI] [PubMed] [Google Scholar]
- 13.Lohr F, Lo DY, Zaharoff DA, Hu K, Zhang X, Li Y, et al. Effective tumor therapy with plasmid-encoded cytokines combined with in vivo electroporation. Cancer Res 2001;61:3281–4. [PubMed] [Google Scholar]
- 14.Lucas ML, Heller L, Coppola D, Heller R. IL-12 plasmid delivery by in vivo electroporation for the successful treatment of established subcutaneous B16.F10 melanoma. Mol Ther 2002;5:668–75. [DOI] [PubMed] [Google Scholar]
- 15.Mahvi DM, Henry MB, Albertini MR, Weber S, Meredith K, Schalch H, et al. Intratumoral injection of IL-12 plasmid DNA—results of a phase I/IB clinical trial. Cancer Gene Ther 2007;14:717–23. [DOI] [PubMed] [Google Scholar]
- 16.Mazzolini G, Alfaro C, Sangro B, Feijoo E, Ruiz J, Benito A, et al. Intratumoral injection of dendritic cells engineered to secrete interleukin-12 by recombinant adenovirus in patients with metastatic gastrointestinal carcinomas. J Clin Oncol 2005;23:999–1010. [DOI] [PubMed] [Google Scholar]
- 17.Triozzi PL, Allen KO, Carlisle RR, Craig M, LoBuglio AF, Conry RM. Phase I study of the intratumoral administration of recombinant canarypox viruses expressing B7.1 and interleukin 12 in patients with metastatic melanoma. Clin Cancer Res 2005;11:4168–75. [DOI] [PubMed] [Google Scholar]
- 18.Heinzerling L, Burg G, Dummer R, Maier T, Oberholzer PA, Schultz J, et al. Intratumoral injection of DNA encoding human interleukin 12 into patients with metastatic melanoma: clinical efficacy. Hum Gene Ther 2005;16:35–48. [DOI] [PubMed] [Google Scholar]
- 19.Sun Y, Jurgovsky K, Moller P, Alijagic S, Dorbic T, Georgieva J, et al. Vaccination with IL-12 gene-modified autologous melanoma cells: preclinical results and a first clinical phase I study. Gene Ther 1998;5:481–90. [DOI] [PubMed] [Google Scholar]
- 20.Cha E, Daud A. Plasmid IL-12 electroporation in melanoma. Hum Vaccin Immunother 2012;8:1734–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Daud AI, DeConti RC, Andrews S, Urbas P, Riker AI, Sondak VK, et al. Phase I trial of interleukin-12 plasmid electroporation in patients with metastatic melanoma. J Clin Oncol 2008;26:5896–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nghiem PT, Bhatia S, Lipson EJ, Kudchadkar RR, Miller NJ, Annamalai L, et al. PD-1 blockade with pembrolizumab in advanced Merkel-cell carcinoma. N Engl J Med 2016;374:2542–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kaufman HL, Russell JS, Hamid O, Bhatia S, Terheyden P, D’Angelo SP, et al. Updated efficacy of avelumab in patients with previously treated metastatic Merkel cell carcinoma after >/=1 year of follow-up: JAVELIN Merkel 200, a phase 2 clinical trial. J Immunother Cancer 2018;6:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bhatia S, Miller NJ, Lu H, Longino NV, Ibrani D, Shinohara MM, et al. Intratumoral G100, a TLR4 agonist, induces antitumor immune responses and tumor regression in patients with Merkel cell carcinoma. Clin Cancer Res 2019;25:1185–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pang C, Sharma D, Sankar T. Spontaneous regression of Merkel cell carcinoma: a case report and review of the literature. Int J Surg Case Rep 2015;7C:104–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sugamata A, Goya K, Yoshizawa N. A case of complete spontaneous regression of extremely advanced Merkel cell carcinoma. J Surg Case Rep 2011;2011:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schadendorf D, Nghiem P, Bhatia S, Hauschild A, Saiag P, Mahnke L, et al. Immune evasion mechanisms and immune checkpoint inhibition in advanced merkel cell carcinoma. Oncoimmunology 2017;6:e1338237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Paulson KG, Iyer JG, Tegeder AR, Thibodeau R, Schelter J, Koba S, et al. Transcriptome-wide studies of merkel cell carcinoma and validation of intratumoral CD8+ lymphocyte invasion as an independent predictor of survival. J Clin Oncol 2011;29:1539–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Paulson KG, Bhatia S. Advances in immunotherapy for metastatic merkel cell carcinoma: a clinician’s guide. J Natl Compr Canc Netw 2018;16:782–90. [DOI] [PubMed] [Google Scholar]
- 30.D’Angelo SP, Russell J, Lebbe C, Chmielowski B, Gambichler T, Grob JJ, et al. Efficacy and safety of first-line avelumab treatment in patients with stage IV metastatic Merkel cell carcinoma: a preplanned interim analysis of a clinical trial. JAMA Oncol 2018;4:e180077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Topalian S, Bhatia S, Hollebecque A, Awada A, De Boer J, Kudchadkar R, et al. Non-comparative, open-label, multiple cohort, phase 1/2 study to evaluate nivolumab (NIVO) in patients with virus-associated tumors (CheckMate 358): efficacy and safety in Merkel cell carcinoma (MCC) [abstract]. In: Proceedings of the American Association for Cancer Research Annual Meeting 2017; 2017 Apr 1–5; Washington, DC. Philadelphia (PA): AACR; 2017. Abstract nr CT074. [Google Scholar]
- 32.Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 2009;45:228–47. [DOI] [PubMed] [Google Scholar]
- 33.Iyer JG, Afanasiev OK, McClurkan C, Paulson K, Nagase K, Jing L, et al. Merkel cell polyomavirus-specific CD8(+) and CD4(+) T-cell responses identified in Merkel cell carcinomas and blood. Clin Cancer Res 2011;17:6671–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Busam KJ, Jungbluth AA, Rekthman N, Coit D, Pulitzer M, Bini J, et al. Merkel cell polyomavirus expression in merkel cell carcinomas and its absence in combined tumors and pulmonary neuroendocrine carcinomas. Am J Surg Pathol 2009;33:1378–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Paulson KG, Carter JJ, Johnson LG, Cahill KW, Iyer JG, Schrama D, et al. Antibodies to merkel cell polyomavirus T antigen oncoproteins reflect tumor burden in merkel cell carcinoma patients. Cancer Res 2010;70:8388–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moshiri AS, Doumani R, Yelistratova L, Blom A, Lachance K, Shinohara MM, et al. Polyomavirus-negative Merkel cell carcinoma: a more aggressive subtype based on analysis of 282 cases using multimodal tumor virus detection. J Invest Dermatol 2017;137:819–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med 2016;375:819–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chapuis AG, Afanasiev OK, Iyer JG, Paulson KG, Parvathaneni U, Hwang JH, et al. Regression of metastatic Merkel cell carcinoma following transfer of polyomavirus-specific T cells and therapies capable of re-inducing HLA class-I. Cancer Immunol Res 2014;2:27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ibrani D, Iyer J, Miller N, Vandeven N, Afanasiev O, Koelle D, et al. Identifying Merkel polyomavirus-specific CD4+ and CD8+ T-cells in Merkel cell carcinoma patients’ tumor-infiltrating lymphocytes [abstract]. In: Proceedings of the 2015 Society for Investigative Dermatology Annual Meeting; 2015 May 6–9; Atlanta, GA. Cleveland (OH):SID; 2015. Poster #015. [Google Scholar]
- 40.Lyngaa R, Pedersen NW, Schrama D, Thrue CA, Ibrani D, Met O, et al. T-cell responses to oncogenic merkel cell polyomavirus proteins distinguish patients with merkel cell carcinoma from healthy donors. Clin Cancer Res 2014;20:1768–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Robins HS, Campregher PV, Srivastava SK, Wacher A, Turtle CJ, Kahsai O, et al. Comprehensive assessment of T-cell receptor beta-chain diversity in alphabeta T cells. Blood 2009;114:4099–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Miller NJ, Church CD, Dong L, Crispin D, Fitzgibbon MP, Lachance K, et al. Tumor-infiltrating Merkel cell polyomavirus-specific T cells are diverse and associated with improved patient survival. Cancer Immunol Res 2017;5:137–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Harms KL, Healy MA, Nghiem P, Sober AJ, Johnson TM, Bichakjian CK, et al. Analysis of prognostic factors from 9387 Merkel cell carcinoma cases forms the basis for the New 8th Edition AJCC Staging System. Ann Surg Oncol 2016;23:3564–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Feng H, Shuda M, Chang Y, Moore PS. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science 2008;319:1096–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Berraondo P, Etxeberria I, Ponz-Sarvise M, Melero I. Revisiting interleukin-12 as a cancer immunotherapy agent. Clin Cancer Res 2018;24:2716–8. [DOI] [PubMed] [Google Scholar]
- 46.Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature 2017;541:321–30. [DOI] [PubMed] [Google Scholar]
- 47.Kerkar SP, Goldszmid RS, Muranski P, Chinnasamy D, Yu Z, Reger RN, et al. IL-12 triggers a programmatic change in dysfunctional myeloid-derived cells within mouse tumors. J Clin Invest 2011;121:4746–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Quetglas JI, Labiano S, Aznar MA, Bolanos E, Azpilikueta A, Rodriguez I, et al. Virotherapy with a Semliki Forest virus-based vector encoding IL12 synergizes with PD-1/PD-L1 blockade. Cancer Immunol Res 2015;3:449–54. [DOI] [PubMed] [Google Scholar]
- 49.Fallon JK, Vandeveer AJ, Schlom J, Greiner JW. Enhanced antitumor effects by combining an IL-12/anti-DNA fusion protein with avelumab, an anti-PD-L1 antibody. Oncotarget 2017;8:20558–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bhatia S, Iyer J, Ibrani D, Blom A, Byrd D, Parvathaneni U, et al. Intratumoral delivery of Interleukin-12 DNA via in vivo electroporation leads to regression of injected and non-injected tumors in Merkel cell carcinoma: final results of a phase 2 study [abstract]. In: Proceedings of the 40th European Society for Medical Oncology (ESMO) Congress; 2015 Sept 25–29; Vienna, Austria; Lugano, Switzerland: ESMO; 2015. Abstract nr 504. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.