

Abstract
Phage research has been vital to fundamental aspects of modern biology. Advances in metagenomics have revealed treasure troves of new, uncharacterized phages that we have yet to understand. However, our ability to find new phages has outpaced our ability to understand phages. Traditional approaches for characterizing phages are limited in scale and face hurdles determining how changes in sequence drive function. In this review, we describe powerful emerging technologies that can be used to clarify sequence-function relationships in phages through high-throughput genome engineering. Using these approaches up to 105 variants can be characterized through pooled selection experiments and deep sequencing. We describe caveats when using these tools and provide examples of basic science and engineering goals pursuable using these approaches.
Keywords: Engineered Bacteriophages, Phage Library, Genome Engineering, Phage Mutagenesis, Phage-Host Interactions, Deep Sequencing
Understanding the interactions between bacteriophages (or ‘phages’) and bacteria has contributed to major advances in modern biology, including the discovery of polymerases, recombinases, and CRISPR-Cas systems [1]. Phages are also increasingly seen as tools for targeted killing of drug-resistant bacteria, precise modulation of the microbiome, gene delivery devices, and diagnostic sensors for pathogens [2–4]. Over the decades, phage biologists have painstakingly isolated and characterized thousands of natural phages. Though invaluable in their scope, these studies often lack a molecular understanding of how sequence changes drive phage function. A major impediment to advancing phage biology from empirical observations to molecular function is the lack of high-throughput methods to systematically and comprehensively profile gene-function or variant-function relationships. Traditional phage assays, such as plaque assays, simply do not scale for large functional genomics studies. As a result, functional studies generally are restricted to one or only a few gene or genome variants which leave large swathes of the sequence-function space unexplored.
In this review, we will describe emerging technologies to elucidate sequence-function relationships in phages through high-throughput genome engineering paired with pooled selection experiments. These approaches enable the functional characterization of up to 105 phage variants and are propelled by a confluence of advances in gene editing, DNA synthesis, and large-scale sequencing. Many functional genomics approaches have been successfully used in bacterial studies and are poised to be implemented in phages. The broad strategy for high throughput characterization of phages involves three essential steps (Figure 1). First, a pool of phage variants, called a phage library (see Glossary), is created. Ideally this library is unbiased, where variants are not lost due to unintended selection during library creation, as those variants might perform well under different conditions. Then, the entire pool of variants is tested in competitive selection experiments. Variants with higher fitness increase in abundance and variants with lower fitness are depleted or lost from the pool of phages. Finally, phage pools from before and after selection are sequenced to score and determine which variants performed better under different selection conditions (Box 1). We explore how these phage libraries can be selected and scored in different contexts to characterize phage-host interactions, understand phage evolution, and uncover gene function. In addition to being a powerful tool to investigate phage biology, these high throughput approaches enable the rational design and engineering of phages with novel properties.
Figure 1. A general outline for library creation, selection, and scoring phage libraries.
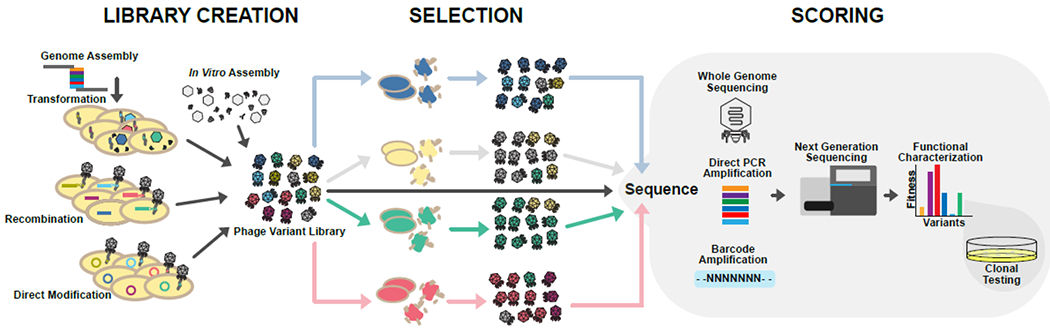
Various approaches can be used to create a phage variant library (represented by different color phages). The phage variant library is selected under different conditions (represented by different color hosts) to select for variants with higher fitness in each condition. Selection can be repeated for multiple replication cycles and may include methods for continual diversification of the phage library during selection. All phage populations, including the original phage variant library, are typically deep sequenced to determine the proportion of each variant in each phage pool. This information is used to score variants and determine which variants were more fit in which condition. Sequencing strategies include whole genome sequencing, direct PCR amplification for targeted mutagenesis, and PCR amplification of barcodes previously mapped to phage variants. Deep sequencing ultimately reveals functional characterization that can be further confirmed in clonal testing.
Box 1. Experimental design for typical pooled selection experiments.
Pooled selection experiments enable researchers to test upwards of tens of thousands of phage variants simultaneously in competitive selection experiments. This pool of variants, called the phage library, can be created using the high throughput approaches described in this review. The phage library is typically contained in a single liquid preparation so all variants can easily be tested at once and contains many copies of each variant to improve experimental consistency. Ideally, the phage library constitutes most or all the phage population with minimal ‘background’ phages that are not part of the intended library. After the library is created, it should be sequenced to check the success of library creation and reveal the abundance of each variant before selection. Many phage preparations can be used directly as template in PCR reactions for deep sequencing. The pre-selection abundance of each variant will be used when calculating the variant score by comparing the abundance of each variant after selection. It is helpful to retain a well-characterized variant or wild type in the phage library to normalize scores. This library can then be easily aliquoted for different selection conditions. A simple experimental approach is passaging the library on several different bacterial hosts or varying the conditions of incubation. It is important to ensure that enough phages are used so the library is fully represented during selection. Controlling the amount of the phage library added or the time the experiment is allowed to proceed allows for estimation of the number of passages. Phage variants more fit will increase in abundance, while those less fit will decrease in abundance. Sequencing the pool of phages after selection reveals the new abundance of each variant which is compared to the pre-selection abundance to score each variant.
Tools for creating phage libraries frequently have a tradeoff between breadth of genome targeting and programmability of mutations (Figure 2A). For example, chemical mutagenesis and mutagenesis plasmids can introduce mutations across the entire phage genome (higher breadth) but mutations are entirely random (lower programmability). In contrast, specific mutations by oligonucleotide pools (higher programmability) can be inserted into the phage genome but only at a pre-defined locus (lower breadth). The tradeoff between breadth and programmability is an important consideration when selecting an approach for creating and scoring phage libraries (Figure 2). In this review, we describe various tools to introduce point mutations, insertions, and deletions on the phage genome, how they relate in breadth and programmability, and how techniques can introduce bias during library creation which can result in lost opportunities for variants during selection. We motivate the adoption of high-throughput methods by providing examples of basic science questions and engineering goals that can be accomplished using different mutagenesis tools. We conclude with general caveats and considerations when making phage libraries and employing high throughput screens.
Figure 2. Approaches for creating phage libraries have tradeoffs in breadth and programmability.
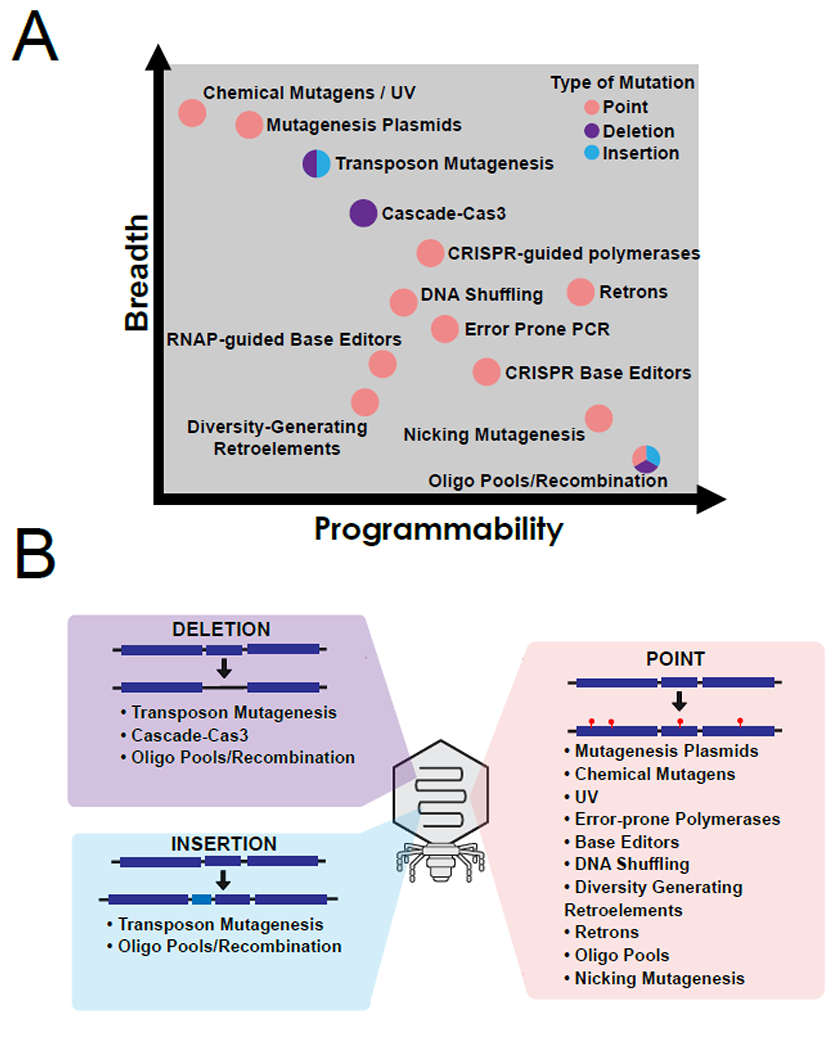
(A) Approaches that can create point mutations (pink), deletions (purple) and insertions (blue) in phage libraries and how they compare in ability to create mutations throughout larger areas of a phage genome (breadth) versus the ability to specify mutations (programmability). (B) Summary of applicable methods for creating deletions, insertions, and point mutations in phages.
Point Mutation Libraries
Methods to introduce point mutations in phages (Figure 2) can be broadly grouped into untargeted and targeted approaches.
Untargeted Phage Mutagenesis
Untargeted mutagenesis is a simple and effective approach for introducing random mutations during continuous evolution experiments and is particularly useful as a screening tool for high-throughput reverse genetics. Point mutations can be introduced randomly into phages using UV radiation or by chemical mutagens such as ethyl methanesulfonate (EMS) or hydroxylamine [5–7]. Radiation and chemical mutagenesis rely on damaging phage DNA, which can cause single nucleotide changes during DNA replication. The frequency of mutagenesis can be tuned in a dose-dependent manner. Chemical mutagenesis, in particular, is relatively easy to implement in any laboratory setting but remains an underutilized tool to study laboratory evolution of phages. Radiation and chemical mutagens can generate unbiased libraries as they do not require passaging in a replication host. Although the mutations are randomly dispersed genome-wide, the nature of mutations is only semi-random as specific transitions or transversion mutations are favored depending on the mutagen [8,9]. UV radiation may also impact the survivability of phages by destabilizing structural proteins by altering protein conformations [10]. An alternative untargeted approach is to use mutagenesis plasmids in the replication host to induce genome-wide mutations through the expression of proteins that damage DNA or reduce DNA replication fidelity [11–13]. However, mutagenesis plasmids require passaging on a replication host, which limits this approach to hosts which can sustain such plasmids and biases phage libraries toward mutations that permit phage variants to grow on that host.
Variant libraries generated by untargeted mutagenesis have high breadth but virtually no programmability (Figure 2A). Libraries are highly diverse but quantifying this diversity will require considerable sequencing depth as the frequency of any particular mutation may be low in the variant population. Despite low initial frequency, mutations that affect a specific trait are likely to exist in the variant population and can rapidly emerge during laboratory selection, making this a powerful approach for continuous evolution. For example, continuous evolution paired with EMS mutagenesis has been used to identify phage variants with improved thermal stability [6]. Due to the preponderance of additional mutations, multiple parallel evolution experiments may be needed to identify mutations that cause a specific phenotype.
Targeted Phage Mutagenesis
Targeted mutagenesis allows deeper exploration of sequence space at a specific genetic locus or loci by concentrating mutations in a defined window. While targeted mutagenesis could be used to discover gene function, it is a particularly effective tool for investigating the effects of sequence variants when the functional role of a gene is known a priori (e.g: genes encoding phage receptor interaction). Targeted genome variants can be created by introducing an externally created library into the phage genome at a specific site or by directly editing the phage genome using accessory proteins.
To generate a library of randomized phage gene variants, we can use common techniques such as error-prone PCR, DNA shuffling, nicking mutagenesis, and degenerate or random primers [14–17]. Alternatively, pre-specified variants can be synthesized using commercially available oligonucleotide pools [18]. These methods are easy-to-implement, guarantee diversity, and generate large variant libraries with high programmability. However, the challenge lies in the efficient integration of the library into the phage genome which can limit breadth (Figure 2A). Site-specific recombinases [18], lambda Red recombineering [19,20], and homologous recombination [15,21,22] have been employed for genome integration.
The phage genome can be directly mutated in vivo using CRISPR systems that localize mutagenic proteins to specific sites. For example, cytidine and adenine base editors can be tethered to or recruited by dead Cas9 or Cas9 nickase to mutate specific loci [23–28]. Cas protein-linked error-prone DNA polymerases with nick translation and/or strand displacement capabilities can be used to introduce every type of point mutation in a defined window [29]. Base editors attached to RNA polymerase can be used to mutate larger regions of a genome engineered with appropriate promoter and terminator sequences flanking the target region [30], although the spectrum of induced mutations is currently limited. Recently, bacterial retroelements (retrons) have been used to genetically engineer T5 phage [31]. Retrons are polycistrons consisting of a reverse transcriptase and covalently linked ssRNA and ssDNA which induce site-specific mutagenesis through a mechanism still yet to be fully elucidated. Libraries of these retroelements can be synthesized in vitro or mutated in vivo for continuous mutagenesis of target sequence [32]. Likewise temperate phage BPP-1 leverages diversity generating retroelements encoded in its genome to introduce targeted hypervariability via mutagenic homing to its receptor binding protein [33]. Such a system could feasibly be adapted to mutate heterologous genes in different phages [34].
Targeted mutagenesis in vivo requires a suitable bacterial host to sustain variant libraries or mutator protein systems. Though versatile, CRISPR approaches are constrained by the location of PAM sites for targeting guide RNA (gRNA), and the difficulty in multiplexing gRNAs to simultaneously edit several loci or to expand the window of activity. The diversity generated by CRISPR guided systems may also vary considerably, with reported rates of 10−4-10−10 mutations/bp/generation [28,29,35]. Recombination efficiency also varies and may require engineering phages to enable recombination, with reported recombination rates ranging from 10−10 to 10−3 [15,18,36]. Lower rates of recombination or mutation incorporation may impact library abundance, decreasing the total number of variants compared to unrecombined phages. One approach to increase library abundance is to use counterselection with CRISPR to remove unrecombined or unmutated phages from the phage population [18,37,38]. Lytic phages may also require multiple passages on the host used to create the library, biasing the library towards variants capable of productively infecting that host. To minimize bias, a ‘helper’ plasmid expressing the wildtype gene was used to mask the effect of variants during library generation in the ORACLE system [18].
Alternatively, some phage genomes can be wholly constructed outside of bacterial hosts. Phage genomes can be assembled in yeast using yeast artificial chromosomes [39,40], or using various ligation cloning methods in vitro [17,41–44]. Assembled genomes or genome fragments can then be transformed into bacterial hosts [39,42,45,46] to ‘reboot’ the phage or packaged into viable phages particles in cell-free systems [41,47,48]. Such ex vivo approaches can maintain high variant abundance and limit library bias as they are not reliant on multiple passages in bacteria. However, these methods are limited to phages that can ‘reboot’ after transformation and by the need for established cell-free systems or bacterial hosts capable of efficient transformation of large DNA constructs, which may limit library size.
Applications of Targeted Phage Mutagenesis Libraries
Targeted mutation libraries can help characterize phage-host interactions and engineer phages at a high resolution by accurately mapping critical functional regions in a phage gene (Figure 3, upper panel). For example, deep mutational scanning (DMS) of the phage receptor-binding protein and subjecting the variant pool to selection on different hosts [18] revealed mutations that play a role in host receptor recognition. Similarly, molecular rules of other phage properties such as capsid assembly, replication fidelity, or host lysis could be probed by DMS of relevant gene(s). This knowledge can be extended to create tailored libraries where variants have been designed for specific purposes, like targeting or avoiding specific hosts, reducing immunogenicity or increasing phage stability in unfavorable physicochemical conditions (e.g: gut environment). Recent advances in machine learning are revolutionizing our ability to decipher the complex relationships between protein sequence, structure, and function [49,50]. Deep learning models enable us to explore vast sequence spaces to predict sequences that are highly optimized for novel function. Large phage mutation-function datasets are a rich resource to train machine learning models to understand the complex relationship between phage sequences and function and to design novel phage variants with desired properties.
Figure 3. Phage libraries can be used to explore questions in fundamental biology and phage engineering.
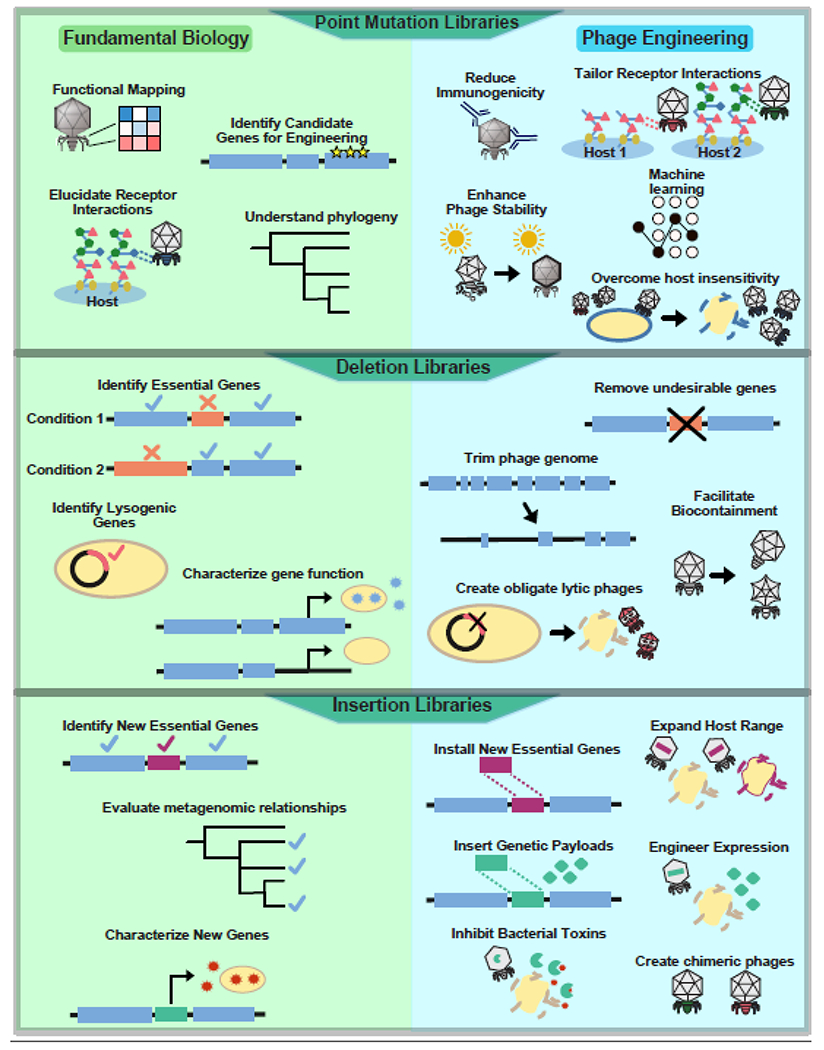
Phage libraries can be used to explore diverse applications in fundamental biology (left) and phage engineering (right). Point mutation libraries (top panel) can be used to map regions relevant for phage function and identify specific residues responsible for host receptor recognition. These approaches can further be leveraged to identify candidate genes for engineering and identify conserved regions to trace phylogeny. This information can be used to engineer phages to reduce host immunogenicity, enhance stability in challenging environments or tailor phages for different receptor interactions. Testing libraries against insensitive hosts may reveal avenues to overcome insensitivity and restore activity. Deletion libraries (middle panel) can be used to identify essential genes, reveal genes responsible for lysogeny and characterize function by exploring the effects on bacterial hosts. Undesirable genes can be removed, trimming the phage genome to make the phage more efficient, while lysogenic genes can be deleted to create new obligate lytic phages. Alternatively essential genes can be removed to create fully bio-contained phages. Insertion libraries (bottom panel) can be used to explore roles for metagenomic proteins and identify genes that enable function in new contexts or characterize the role of genes in specific hosts. Insertion of new essential genes can expand host range, or genetic payloads like fluorescent markers, enzymes to degrade bacterial toxins, or other desired genetic cassettes can be added to the phage genome to tailor expression. Alternatively chimeric phages can be constructed by inserting new structural genes into the genome.
Phage Insertion and Deletion Libraries
Targeted deletions or insertions in phage genomes evaluates the functional impact of larger genomic perturbations ranging from tens of bases to kilobases. Smaller deletions of tens to hundreds of bases can be created in vivo by expressing Cas9 in the replication host and larger deletions up to several kilobases can be generated in vivo using Cascade-Cas3 which degrades DNA using dual helicase-nuclease activity [51] (Figure 2B). Transposon mutagenesis with site-specific mariner class transposons or random insertion transposons such as Tn5, which have been used extensively to create genome-wide loss-of-function libraries in bacteria [52–55], could be implemented in phages in vivo to create gene deletion libraries. Alternatively, transposon mutagenesis can be used to generate more programmable single, double, and triple residue deletions in specific genes (Figure 2A). These insertion or deletion libraries can be synthesized in oligonucleotide pools and recombined into the phage genome [56] using site-specific recombinases or by homologous recombination [53,57]. Cas-based editing requires the user to direct insertions or deletions to defined regions of the genome while transposon-based editing is broader and can cover the entire genome. Barcodes may be inserted into the donor or transposon sequence to score the genome perturbations using short read sequencing [58]. Insertion of sequences encoding sequence motifs to small protein domains can be created but currently suffer tradeoffs between library size, genome insertion efficiency, and cost. Large, diverse libraries can be created from oligonucleotide pools at relatively low cost, but the efficiency of genome insertion into the phage genome is the limiting factor.
Applications of Phage Deletion and Insertion Libraries
Phage deletion libraries are helpful to study gene essentiality in different environmental and host conditions (Figure 3, middle panel). When the phage deletion library is subjected to selection under a certain condition, variants with deletions in genes or regulatory elements essential in that condition will deplete. For phage genomes that contain overlapping genetic elements, high-resolution deletion libraries with smaller windows of deletions minimize disruption of overlapping genomic regions. Small windows of deletions may reveal the presence of essential regulatory sequences found in non-essential genes. Temperate phages which are disfavored for therapeutic use could be converted into obligate lytic phages by the systematic removal of recombinases, transcription factors or other unknown mechanisms that regulate lysogeny [43]. Deletions may also improve the activity of phage in different conditions, as some genetic elements may only be beneficial in specific contexts and are otherwise disadvantageous to the phage, such as gene 1.7 in T7 phage which confers sensitivity to dideoxythymidine [59].
Deletion libraries are helpful for phage engineering. Mapping essential genes can be used to create synthetic phages with a minimal genome [60], whose activity may be higher than wildtype. Host range can be customized in engineered phages by removing essential genes for one host but not another. Targeted deletions of key genes can be used as a strategy to improve the biocontainment of phages, as deletion of essential genes restricts phage replication.
Larger-sized insertions can be used to characterize metagenomic gene function (Figure 3, bottom panel). While phage genome databases are rapidly growing, tools to characterize functions of these sequences has lagged far behind. Libraries of metagenomic sequences can be rapidly tested by inserting them into a well characterized phage genome. For example, exchanging receptor binding proteins between different phages can show evolutionary relationships between phages or identify new phage chimeras that are able to infect novel hosts [15,39]. Exchanging or inserting phage genes predicted to deter host defenses, like Cas inhibitors, and passaging this library on the relevant host would allow identification of metagenomic genes that are effective against that particular host defense [61].
Phage genomes could be augmented with new capabilities (e.g: new lysins) by inserting characterized genes into non-essential regions (Figure 3, bottom panel). Addition of host genes that are essential to the phage, like trxA for T7 phage, removes reliance on the host, while insertion of genes that deter host defenses like Cas inhibitors can improve the ability of engineered phages to target and/or eliminate new hosts [37,61,62]. The insertion of larger sequences can enable tailored protein production during the phage lifecycle, such as fluorescent markers, or can be used to inactivate undesirable proteins made by the bacterial host, such as toxins [63,64]. Gene cassettes that facilitate host killing may be incorporated into phages, such as Crispr-Cas elements that target the host genome, ensuring host cell destruction [65].
Considerations for Creating and Screening Phage Libraries
Any approach used to create phage libraries should account for library bias. Bias is the reduction or elimination of phage variants due to selection pressure during library formation. Bias frequently occurs during passage on a replication host. Variants lost to library bias may be active on different hosts or in different conditions. These variants are likely to be the most relevant to understanding or engineering that phage, as they necessarily can affect the activity and host range of that phage. Bias can be attenuated by limiting rounds of replication or by using a helper plasmid for libraries of specific genes. In vitro assembly of phage libraries avoids selection bias from replication hosts but could introduce bias if altered structural proteins assemble differently in vivo versus in vitro. Methods for scoring the phage library should be able to take the extent of bias into account. If the library is sequenced prior to selection, it is critical that the library be assessed after any bias may have altered library distribution (Figure 1). For example, sequencing a plasmid library before integration into a phage genome may produce dramatically different results than sequencing the phage library after integration. If the library is not assessed prior to selection, possible uncharacterized bias should be considered when analyzing results.
Library abundance should be evaluated to ensure that an experimentally appropriate number of unique phage variants are used during selection. Experimental consistency is likely to be improved if there are hundreds or thousands of a particular variant during selection. Reduced phage-host ratios during selection may result in complete attenuation of some variants in the phage pool. If a library has been biased during creation, some members may have much lower proportional representation than other members. Some methods such as homologous recombination result in an extreme overabundance of wild type phages. This can dramatically reduce the number of relevant variants in the phage pool and add significant noise to the experiment. The proportion of wild type or other background phages that have not been engineered can be reduced by using counterselection strategies like CRISPR during passage or by incorporating essential genes during library creation that can be used to select for engineered phages [18,37,38].
Variants that dramatically outperform other library members may overtake the phage population over a few rounds of replication during selection. This effect can semi-randomly mask variants with less pronounced effects that are still be valuable for understanding phage host interactions or engineering the phage. Comparison of biological replicates can reveal this effect, which can be reduced by lowering the number of passages used for selection. Selection also does not necessarily require host lysis or productive phage replication. For example, evaluating the efficiency of different integrases or efficiency of packaging frequency using phagemids are viable selection schemes that do not require productive phage replication.
Libraries are sequenced to score phage variants, ideally before and after selection (Figure 1). The sequencing depth necessary should be based on the ability to detect the lowest abundant variants. Low sequencing coverage or low initial abundance can dramatically alter the limit of detection for the assay and this effect must be kept in mind when analyzing results. A clonally validated reference, typically the wild type, can be used to normalize scoring to an active phage. Sequencing and scoring variants can rely on whole genome sequencing or sequencing of specific target regions. Long read sequencing such as PacBio or Oxford Nanopore and short read sequencing such as Illumina platforms are viable options. Longer reads come at the cost of depth, as short read sequencing can sequence millions of times [66]. Targeted mutation libraries or short, ~500 bp windows are easiest to sequence and score as they can be directly sequenced using abundant short reads. For larger windows and changes which surpass ~500 bp, more costly long read sequencing is generally necessary. Alternatively, short nucleotide barcodes can be integrated in tandem with the variant sequence. Strategies include integrating barcodes in a known region alongside the variant [41], or barcodes could replace deleted regions [58]. This barcode can be sequenced as a proxy readout for the mutated region after mapping unique barcodes to individual variants using long read sequencing, after which mutants can be scored using less expensive short read sequencing. These strategies which use targeted deep sequencing of variants enable researchers to obtain quantitative fitness data for thousands, if not millions, of phage variants, exceeding the throughput of arrayed assays by several orders of magnitude.
Concluding Remarks
Over the decades, our understanding of how phages and bacteria interact has continued to evolve. These studies have already revealed a wealth of novel biologic functions that are fundamental to many fields like polymerases, recombinases, and Crispr-Cas systems. Our increasing understanding of phages has positioned phages as potent tools for precision editing of complex microbial communities and as treatments for drug-resistant bacterial infections. Still, advances in metagenomics have revealed many new phages that remain uncharacterized, and many fundamental rules of phage-host interactions remain to be investigated. New techniques have been introduced recently to study phenotypic effects of host perturbations at scale [53,67].
Here we describe complementary high throughput approaches that enable systematic characterization of phages to establish sequence-function relationships. Screening large libraries of phage variants in pooled selection experiments enables characterization of any attribute which affects phage fitness (e.g., adsorption, entry, avoidance of host restriction-modification systems, replication, host lysis, structural stability, etc.). Still, many of the techniques highlighted in this review have been developed only in model hosts or phages. While techniques such as chemical mutagenesis and CRISPR tools have been shown to be generalizable to non-model phages and hosts, this has yet to be demonstrated for other approaches [68]. While many questions remain as to the best strategy to implement these techniques in phages (see Outstanding Questions), these approaches present exciting new avenues for research to enhance our understanding of these viruses so essential to our ecosystem and our study of biology.
Outstanding Questions Box.
Continuous evolution experiments can frequently introduce variants that are immediately selected against. These variants are difficult to detect and score. How can those variants that do poorly be identified in these experiments?
Many techniques have been developed in model bacteria like E. coli. How productive are these techniques in non-model bacteria and how can we extend these approaches to these new frontiers?
Reducing the prevalence of wild type or other ‘background’ phages is an important consideration for library abundance. Beyond CRISPR counterselection or inclusion of essential genes what other strategies can be incorporated into library creation to ensure phages are efficiently engineered?
How can we link the activity of specific phage variants to different members of complex microbial communities?
How can information from screens be used to create machine learning models to tailor phage function?
Can deep sequencing be used to mine for related phages in complex metagenomic databases?
What generalizable approaches can be developed to limit bias when creating phage libraries?
How do rates of mutagenesis and recombination rates differ for phage genomes compared to bacterial genomes?
How can mutagenesis and recombination rates be optimized within the setting of an active phage infection?
Highlights.
High throughput approaches can generate large phage libraries that can be screened simultaneously in pooled selection experiments and scored using deep sequencing.
Approaches for creating phage libraries frequently have a tradeoff between creating mutations throughout larger areas of a phage genome (breadth) versus ability to specify mutations (programmability).
Untargeted mutagenesis is a simple and effective approach for introducing random mutations and is a useful tool for high-throughput reverse genetics.
Targeted mutagenesis can characterize phage-host interactions at a high resolution by accurately mapping critical functional regions in a phage genome.
Phage deletion libraries can be used to study gene essentiality in different environmental and host conditions while insertion libraries can be used to characterize metagenomic gene function.
Acknowledgements
This work was supported by NIAID grant R21AI156785 (to S.R.) and the National Institute of General Medical Sciences of the National Institute of Health under Award Number T32GM135066 (to J.C.).
Glossary
- Base Editors:
proteins which introduce single nucleotide alteration without forming double-stranded breaks, typically by damaging DNA through deamination.
- Breadth:
The ability of an approach to create mutations throughout larger areas of a phage genome. High breadth indicates mutations can be incorporated over much of the phage genome, while low breadth indicates an approach is restricted to smaller regions.
- Continuous Evolution:
An experimental approach whereby phage diversification and selection occur uninterrupted over multiple rounds of replication. The approach for mutating phages may be included in every round of replication to allow for increased diversification.
- DNA Shuffling:
Approach for generated diversity in a gene by treatment with restriction enzymes and reassembly by PCR without primers or through nonhomologues random recombination.
- Library Abundance:
The proportion of phages in a phage population that are the actual library variants. For example, a library is 1% abundant if 99% of a phage population is wild type and 1% of phages are the library phage variants.
- Library Bias:
The degree to which phage variants have been proportionally reduced or eliminated in a phage library due to selection pressure during library formation, skewing the phage library towards variants more fit to the condition in which the library was created. Bias can eliminate variants that are viable in other conditions and alter interpretation of results if unaccounted for.
- Mutagenic homing:
Transposition of a mutated genetic element into a targeted region, where the mutations found in the transposed element are inherited by the target sequence.
- Nickase:
A Cas9 variant with one of the two nuclease domains inactivated. Instead of generating double-stranded breaks at the target site, these enzymes introduce a single-stranded nick.
- Nicking mutagenesis:
PCR-based method which generates diversity at target loci on a dsDNA plasmid using mutagenic oligos where wildtype DNA strands are successively degraded with nicking enzymes and exonuclease treatment.
- ORACLE:
A method (Optimized Recombination, Accumulation and Library Expression) designed to create unbiased phage libraries using a combination of site-specific recombination, CRISPR counterselection and helper plasmids.
- Phage Library:
A pool of phages which contains all the phage variants to be evaluated in a mixed population.
- Programmability:
The ability of an approach to create specific, desired variants in a phage library. High programmability indicates precise control over which mutations are incorporated into a library, while low programmability indicates no control over which mutations are incorporated.
- Rebooting:
Process by which phage genomes are directly transformed into compatible hosts to initiate infection and create viable phage particles.
- Sequencing Depth:
The number of times a given nucleotide position is read during deep sequencing when scoring libraries. Every different read is presumed to be a different sequence from a different phage. For example, if a position has a sequencing depth of 1000 it is presumed 1000 different phages have been sequenced at that position.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declarations of Interest
No interests are declared.
Bibliography
- 1.Salmond GPC and Fineran PC (2015) A century of the phage: past, present and future. Nat. Rev. Microbiol 13, 777–786 [DOI] [PubMed] [Google Scholar]
- 2.Mutalik VK and Arkin AP (2022) A Phage Foundry Framework to Systematically Develop Viral Countermeasures to Combat Antibiotic-Resistant Bacterial Pathogens. iScience 25, 104121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eskenazi A et al. (2022) Combination of pre-adapted bacteriophage therapy and antibiotics for treatment of fracture-related infection due to pandrug-resistant Klebsiella pneumoniae. Nat. Commun 13, 302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hatfull GF et al. (2022) Phage Therapy for Antibiotic-Resistant Bacterial Infections. Annu. Rev. Med 73, 197–211 [DOI] [PubMed] [Google Scholar]
- 5.Yosef I. et al. (2017) Extending the Host Range of Bacteriophage Particles for DNA Transduction. Mol. Cell 66, 721–728.e3 [DOI] [PubMed] [Google Scholar]
- 6.Favor AH et al. (2020) Optimizing bacteriophage engineering through an accelerated evolution platform. Sci. Rep 10, 13981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arimoto-Kobayashi S. et al. (2000) Oxidative damage and induced mutations in M13mp2 phage DNA exposed to N-nitrosopyrrolidine with UVA radiation. Mutagenesis 15, 473–477 [DOI] [PubMed] [Google Scholar]
- 8.Shibai A. et al. (2017) Mutation accumulation under UV radiation in Escherichia coli. Sci. Rep 7, 14531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li C-LF et al. (2016) Gene discovery by chemical mutagenesis and whole-genome sequencing in Dictyostelium. Genome Res. 26, 1268–1276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Santos AL et al. (2013) Effects of UV Radiation on the Lipids and Proteins of Bacteria Studied by Mid-Infrared Spectroscopy. Environ. Sci. Technol 47, 6306–6315 [DOI] [PubMed] [Google Scholar]
- 11.Badran AH and Liu DR (2015) Development of potent in vivo mutagenesis plasmids with broad mutational spectra. Nat. Commun 6, 8425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Esvelt KM et al. (2011) A System for the Continuous Directed Evolution of Biomolecules. Nature 472, 499–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miller SM et al. (2020) Phage-assisted continuous and non-continuous evolution. Nat. Protoc 15, 4101–4127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wrenbeck EE et al. (2016) Plasmid-based one-pot saturation mutagenesis. Nat. Methods 13, 928–930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yehl K. et al. (2019) Engineering Phage Host-Range and Suppressing Bacterial Resistance through Phage Tail Fiber Mutagenesis. Cell 179, 459–469.e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stemmer WPC (1994) Rapid evolution of a protein in vitro by DNA shuffling. Nature 370, 389–391 [DOI] [PubMed] [Google Scholar]
- 17.Dunne M. et al. (2019) Reprogramming Bacteriophage Host Range through Structure-Guided Design of Chimeric Receptor Binding Proteins. Cell Rep. 29, 1336–1350.e4 [DOI] [PubMed] [Google Scholar]
- 18.Huss P. et al. (2021) Mapping the functional landscape of the receptor binding domain of T7 bacteriophage by deep mutational scanning. eLife 10, e63775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jensen JD et al. (2020) λ Recombineering Used to Engineer the Genome of Phage T7. Antibiotics 9, 805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Oppenheim AB et al. (2004) In vivo recombineering of bacteriophage lambda by PCR fragments and single-strand oligonucleotides. Virology 319, 185–189 [DOI] [PubMed] [Google Scholar]
- 21.Hoshiga F. et al. (2019) Modification of T2 phage infectivity toward Escherichia coli O157:H7 via using CRISPR/Cas9. FEMS Microbiol. Lett 366, fnz041. [DOI] [PubMed] [Google Scholar]
- 22.Marinelli LJ et al. (2019) Genetic Manipulation of Lytic Bacteriophages with BRED: Bacteriophage Recombineering of Electroporated DNA. Methods Mol. Biol. Clifton NJ 1898, 69–80 [DOI] [PubMed] [Google Scholar]
- 23.Moore CL et al. (2018) A Processive Protein Chimera Introduces Mutations across Defined DNA Regions In Vivo. J. Am. Chem. Soc 140, 11560–11564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Álvarez B. et al. (2020) In vivo diversification of target genomic sites using processive base deaminase fusions blocked by dCas9. Nat. Commun 11, 6436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hess GT et al. (2016) Directed evolution using dCas9-targeted somatic hypermutation in mammalian cells. Nat. Methods 13, 1036–1042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Komor AC et al. (2016) Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gaudelli NM et al. (2017) Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 551, 464–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zheng K. et al. (2018) Highly efficient base editing in bacteria using a Cas9-cytidine deaminase fusion. Commun. Biol 1, 1–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Halperin SO et al. (2018) CRISPR-guided DNA polymerases enable diversification of all nucleotides in a tunable window. Nature 560, 248–252 [DOI] [PubMed] [Google Scholar]
- 30.Cravens A. et al. (2021) Polymerase-guided base editing enables in vivo mutagenesis and rapid protein engineering. Nat. Commun 12, 1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ramirez-Chamorro L. et al. (2021) Strategies for Bacteriophage T5 Mutagenesis: Expanding the Toolbox for Phage Genome Engineering. Front. Microbiol 12, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Simon AJ et al. (2018) Retroelement-Based Genome Editing and Evolution. ACS Synth. Biol. 7, 2600–2611 [DOI] [PubMed] [Google Scholar]
- 33.Medhekar B. and Miller JF (2007) Diversity-generating retroelements. Curr. Opin. Microbiol 10, 388–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guo H. et al. (2011) Target Site Recognition by a Diversity-Generating Retroelement. PLOS Genet. 7, e1002414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kantor A. et al. (2020) CRISPR-Cas9 DNA Base-Editing and Prime-Editing. Int. J. Mol. Sci 21, 6240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pires DP et al. (2016) Genetically Engineered Phages: a Review of Advances over the Last Decade. Microbiol. Mol. Biol. Rev 80, 523–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grigonyte AM et al. (2020) Comparison of CRISPR and Marker-Based Methods for the Engineering of Phage T7. Viruses 12, 193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nayeemul Bari SM et al. (2017) Strategies for Editing Virulent Staphylococcal Phages Using CRISPR-Cas10. ACS Synth. Biol 6, 2316–2325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ando H. et al. (2015) Engineering Modular Viral Scaffolds for Targeted Bacterial Population Editing. Cell Syst. 1, 187–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jaschke PR et al. (2012) A fully decompressed synthetic bacteriophage øX174 genome assembled and archived in yeast. Virology 434, 278–284 [DOI] [PubMed] [Google Scholar]
- 41.Andrews B and Fields S (2021) Balance between promiscuity and specificity in phage λ host range. ISME J. 15, 2195–2205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pulkkinen EM et al. (2019) Utilizing in vitro DNA assembly to engineer a synthetic T7 Nanoluc reporter phage for Escherichia coli detection. Integr. Biol 11, 63–68 [DOI] [PubMed] [Google Scholar]
- 43.Kilcher S. et al. (2018) Cross-genus rebooting of custom-made, synthetic bacteriophage genomes in L-form bacteria. Proc. Natl. Acad. Sci 115, 567–572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pryor JM et al. (2022) Rapid 40 kb Genome Construction from 52 Parts through Data-optimized Assembly Design. ACS Synth. Biol DOI: 10.1021/acssynbio.1c00525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Faber MS et al. (2020) Saturation Mutagenesis Genome Engineering of Infective ΦX174 Bacteriophage via Unamplified Oligo Pools and Golden Gate Assembly. ACS Synth. Biol 9, 125–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Assad-Garcia N. et al. Cross-Genus “Boot-Up” of Synthetic Bacteriophage in Staphylococcus aureus by Using a New and Efficient DNA Transformation Method. Appl. Environ. Microbiol 88, e01486–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rustad M. et al. (2017) Synthesis of Infectious Bacteriophages in an E. coli-based Cell-free Expression System. JoVE J. Vis. Exp DOI: 10.3791/56144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shin J. et al. (2012) Genome replication, synthesis, and assembly of the bacteriophage T7 in a single cell-free reaction. ACS Synth. Biol 1, 408–413 [DOI] [PubMed] [Google Scholar]
- 49.Wu Z. et al. (2019) Machine learning-assisted directed protein evolution with combinatorial libraries. Proc. Natl. Acad. Sci 116, 8852–8858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gelman S. et al. (2021) Neural networks to learn protein sequence–function relationships from deep mutational scanning data. Proc. Natl. Acad. Sci 118, e2104878118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Csörgő B. et al. (2020) A compact Cascade–Cas3 system for targeted genome engineering. Nat. Methods 17, 1183–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.van Opijnen T. et al. (2009) Tn-seq; high-throughput parallel sequencing for fitness and genetic interaction studies in microorganisms. Nat. Methods 6, 767–772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mutalik VK et al. (2020) High-throughput mapping of the phage resistance landscape in E. coli. PLOS Biol. 18, e3000877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Langridge GC et al. (2009) Simultaneous assay of every Salmonella Typhi gene using one million transposon mutants. Genome Res. 19, 2308–2316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Goodman AL et al. (2009) Identifying genetic determinants needed to establish a human gut symbiont in its habitat. Cell Host Microbe 6, 279–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Emond S. et al. (2020) Accessing unexplored regions of sequence space in directed enzyme evolution via insertion/deletion mutagenesis. Nat. Commun 11, 3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jones GM et al. (2008) A systematic library for comprehensive overexpression screens in Saccharomyces cerevisiae. Nat. Methods 5, 239–241 [DOI] [PubMed] [Google Scholar]
- 58.Wetmore KM et al. (2015) Rapid Quantification of Mutant Fitness in Diverse Bacteria by Sequencing Randomly Bar-Coded Transposons. mBio 6, e00306–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tran NQ et al. (2008) Gene 1.7 of bacteriophage T7 confers sensitivity of phage growth to dideoxythymidine. Proc. Natl. Acad. Sci. U. S. A 105, 9373–9378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pires DP et al. (2021) Designing P. aeruginosa synthetic phages with reduced genomes. Sci. Rep 11, 2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Landsberger M. et al. (2018) Anti-CRISPR Phages Cooperate to Overcome CRISPR-Cas Immunity. Cell 174, 908–916.e12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Qimron U. et al. (2006) Genomewide screens for Escherichia coli genes affecting growth of T7 bacteriophage. Proc. Natl. Acad. Sci. U. S. A 103, 19039–19044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vinay M. et al. (2015) Phage-Based Fluorescent Biosensor Prototypes to Specifically Detect Enteric Bacteria Such as E. coli and Salmonella enterica Typhimurium. PLOS ONE 10, e0131466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Song S and Wood TK (2020) A Primary Physiological Role of Toxin/Antitoxin Systems Is Phage Inhibition. Front. Microbiol 11, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Selle K. et al. In Vivo Targeting of Clostridioides difficile Using Phage-Delivered CRISPR-Cas3 Antimicrobials. mBio 11, e00019–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hu T. et al. (2021) Next-generation sequencing technologies: An overview. Hum. Immunol 82, 801–811 [DOI] [PubMed] [Google Scholar]
- 67.Kortright KE et al. (2020) High-throughput discovery of phage receptors using transposon insertion sequencing of bacteria. Proc. Natl. Acad. Sci. U. S. A 117, 18670–18679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vento JM et al. (2019) Barriers to genome editing with CRISPR in bacteria. J. Ind. Microbiol. Biotechnol 46, 1327–1341 [DOI] [PMC free article] [PubMed] [Google Scholar]