

Abstract
Mitochondrial permeability transition pore (mPTP)-dependent necrotic cell death is a form of necrotic cell death that is driven by mitochondrial dysfunction by the opening of the mPTP and is triggered by increases in matrix levels of Ca2+ and reactive oxygen species. This form of cell death has been implicated in ischemic injuries of the heart and brain as well as numerous degenerative diseases in the brain and skeletal muscle. This review focuses on the molecular triggers and regulators of mPTP-dependent necrosis in the context of myocardial ischemia reperfusion injury. Research over the past 50 years has led to the identity of regulators and putative pore-forming components of the mPTP. Finally, downstream consequences of activation of the mPTP as well as ongoing questions and areas of research are discussed. These questions pose a particular interest as targeting the mPTP could potentially represent an efficacious therapeutic strategy to reduce infarct size following an ischemic event.
Keywords: MPTP, Necrosis, Ischemia Reperfusion, Mitochondria, Mitochondrial Dysfunction, ANT, BAX, BAK, CypD, ATP Synthase, Calcium, ROS, Permeability Transition
Introduction
The responsibility of cardiac mitochondria is to produce sufficient amounts of ATP to maintain the high energetic demand of the beating heart. Mitochondrial dysfunction is detrimental to this process and has been associated with many cardiomyopathies. Mitochondrial dysfunction is a primary contributor to the cellular damage that occurs during a myocardial infarction (MI). During a MI, the damaged area loses its ability to undergo oxidative phosphorylation due to the lack of O2 and this energy depletion leads to the dysregulation of ions within the myocyte. Following reperfusion of this area, the O2 starved heart becomes replete with nutrients and O2, which leads to oxidative damage. These events are the trigger of a regulated necrotic cell death referred to as mitochondrial permeability transition pore (mPTP)-dependent necrosis. The mPTP is a non-selective pore within the inner mitochondrial membrane that is permeable to solutes up to 1.5 kDa in size [1]. Pore opening leads to the dissipation of the mitochondrial membrane potential, organelle swelling, and eventual rupture [2]. In controlled laboratory settings, pharmacological or genetic desensitization of the mPTP is effective at reducing infarct size following ischemia reperfusion (I/R) injury [3–6]. Unfortunately, the molecular identity of the pore-forming component of the mPTP has evaded investigators since its discovery [2]. In this review, we will discuss the triggers of the mPTP—calcium (Ca2+) and reactive oxygen species (ROS)—matrix regulation of the mPTP by cyclophilin D (CypD), the putative pore-forming inner membrane components, the adenine nucleotide translocator (ANT) family and the F1Fo-ATP synthase, and the outer membrane regulators of the mPTP, the Bcl-2 family. Although the primary focus of this review is the pathological role of the mPTP in the cardiac setting, it bears mentioning that the mPTP also plays an integral physiological role in the heart [7].
Triggers of the mPTP
Numerous studies have focused on the mPTP because it is proposed to be the primary trigger of cell death under many conditions such as I/R injury. The low pH which occurs during ischemia is thought to be inhibitory to mPTP opening, allowing a window of opportunity to deliver drugs to the mitochondria at the beginning of reperfusion to inhibit mPTP-dependent necrosis. Because the identity of the pore-forming components of the pore are still debated, there has been interest in inhibiting mPTP-dependent necrosis by reducing the triggers that activate the mPTP.
Calcium
An increase in mitochondrial Ca2+ is the well-established trigger of the mPTP (Fig. 1). In fact, experimentally Ca2+ is the common trigger used to stimulate mPTP opening in isolated mitochondrial assays such as Ca2+ retention capacity (CRC) and the swelling assay. In CRC, small increments of Ca2+ are added to mitochondria in the presence of an extracellular Ca2+ indicator. The addition of extra-mitochondrial Ca2+ leads to an increase in fluorescence of the Ca2+ sensitive dye which then declines back to baseline as the Ca2+ is taken into the mitochondria. When the mitochondria exceed CRC, the mPTP opens releasing the accumulated Ca2+ which is then detected by the extra-mitochondrial Ca2+ indicator. In the mitochondrial swelling assay, a large bolus of Ca2+ is added to isolated mitochondria inducing mitochondrial swelling, which is measured as a decrease in mitochondrial absorbance at 540 nm. These common mPTP assays rely on Ca2+ to trigger the pore. The primary role for Ca2+ in triggering the mPTP has prompted studies to examine the timing of the rise in Ca2+ during ischemia and reperfusion as well as studies to identify the transporters responsible for the increase in mitochondrial Ca2+ with the goal of inhibiting Ca2+ uptake to block PTP.
Figure 1: Outstanding questions surrounding the regulation, identity, and pathological relevance of the mPTP.
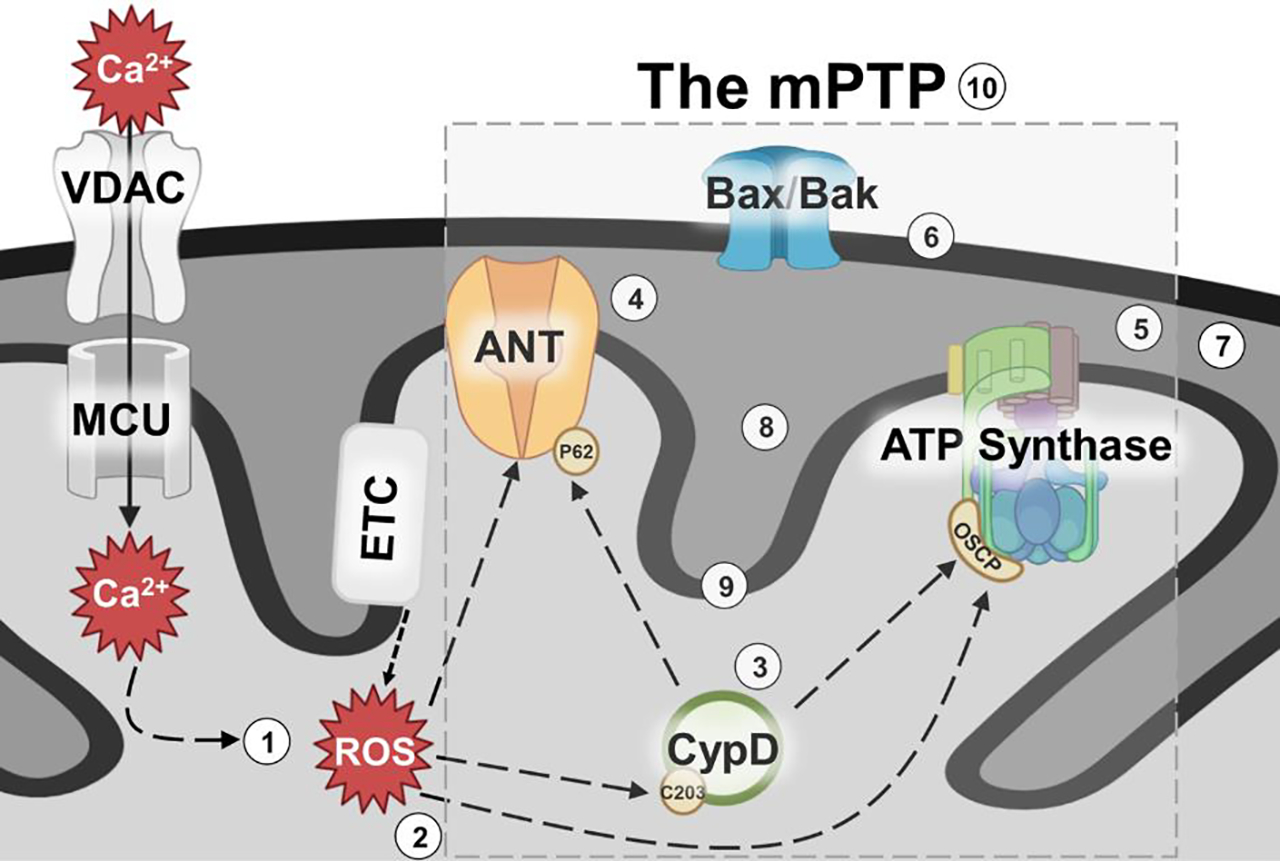
Schematic representation of the current model of the mPTP, its regulators and its triggers. Though it is known that Ca2+ and ROS trigger the mPTP, and various protein components of the mPTP have been proposed, there are essential questions that remain to be addressed: ① How does Ca2+ trigger mPTP? ② How does ROS sensitize mPTP to Ca2+? ③ What is the target of CypD activity? ④ Is ANT a regulator or pore forming component of mPTP? ⑤ Is ATP synthase a regulator or pore forming component of mPTP? ⑥ How does outer mitochondrial membrane permeability influence matrix calcium retention capacity? ⑦ Are contact sites essential for mPTP activity? ⑧ Is the mPTP composed of two pore forming entities (ANT and ATP synthase pores)? ⑨ Are there additional pore forming components of the mPTP? ⑩ Can complete inhibition of mPTP protect humans against myocardial infarct injury?
An increase in cytosolic Ca2+ has been shown to occur during ischemia [8,9]. The mitochondrial Ca2+ uniporter (MCU) is generally thought to be the primary mechanism for Ca2+ uptake into the mitochondria. The driving force for Ca2+ uptake into the mitochondria is the negative mitochondrial membrane potential that is maintained by proton extrusion by the electron transport chain. As electron transport is largely inhibited during ischemia it possible that mitochondrial Ca2+ uptake via MCU would be inhibited during ischemia and would only occur during reperfusion when oxygen is restored and electron transport resumes. Mitochondrial membrane potential has been measured during ischemia and simulated ischemia and it does not appear to fall until late in ischemia after the heart goes into contracture [10,11]. If Ca2+ uptake into mitochondria occurs during ischemia, then inhibition of mitochondrial Ca2+ uptake at the start of reperfusion might not inhibit mPTP opening, depending on the amount of Ca2+ accumulated during ischemia. There are only a few studies examining mitochondrial Ca2+ during simulated ischemia in cells, but these studies suggest some increase in mitochondrial Ca2+ during ischemia.
Griffiths et al., examined mitochondrial Ca2+ in isolated adult rat cardiomyocytes (CM) [12]. Indo-1 AM was loaded into the myocytes and manganese was added to quench cytosolic indo-1. Cells were made hypoxic and mitochondrial Ca2+ was measured as a function of time after the myocyte went into rigor. In this model of hypoxia, myocytes stop beating and rigor begins after ~30 min and mitochondrial Ca2+ does not rise until the onset of rigor. Cellular recovery depends on the duration time in rigor. After 30 min of rigor 36% of the myocytes recovered normal rod shape morphology upon re-oxygenation. Mitochondrial Ca2+ prior to ischemia was ~100 nM and by the end of ischemia in cell recovered myocyte mitochondrial Ca2+ rose to ~280 nM versus 743 nM in the myocytes that did not recover. In recovering cells during reoxygenation, mitochondrial Ca2+ initially declined and then rebounded up to 331 nM and then returned to baseline. Surprisingly, Griffiths et al., found that addition of 20 μM ruthenium red (RR) during hypoxia and reoxygenation led to an increase in mitochondrial Ca2+ during hypoxia and a more rapid decrease in mitochondrial Ca2+ upon reoxygenation [13]. Additionally, Griffiths et al., tested the effect of cyclosporin A (CsA), a cyclical peptide immunosuppressant drug that inhibits calcineurin and cyclosporine A, on mitochondrial Ca2+ during anoxia and reoxygenation. The addition of 0.2 μM CsA increased the mitochondrial Ca2+ observed with rigor from 280 nM to 543 nM, whereas 1 μM CsA reduced mitochondrial Ca2+ to 153 nM [12]. With both concentrations of CsA, myocytes recovered compared to only 50% recovery without CsA.
Allen et al., loaded perfused rat heart with fura-2-AM, a fluorescent Ca2+ indicator, and then isolated mitochondria following hypoxic perfusion and hypoxic perfusion followed by reoxygenation [14]. Free Ca2+ was measured in the fura-2-AM isolated mitochondria. Baseline mitochondrial Ca2+ was 156 nM, which increased to 360 nM and 574 nM at 50 and 80 minutes of anoxic perfusion respectively. With 20 minutes of reperfusion after 50 minutes of hypoxia mitochondrial Ca2+ slightly increased to 501 nM, however, after 80 minutes of hypoxia with reperfusion, mitochondrial Ca2+ increased to levels that saturated the Ca2+ indicator (>5000 nM). Addition of 2.5 μM, but not 1 μM RR blocked the rise in mitochondrial Ca2+ upon reperfusion after 80 minutes of hypoxia.
To measure mitochondria Ca2+ in a perfused rat heart Miyamae et al., used the Ca2+ indicator indo-1 with manganese to quench the cytosolic fluorescence of the indo-1 [15]. They measured mitochondrial Ca2+ at baseline prior to ischemia and found mitochondrial Ca2+ to be 230 nM with no difference when RR was added. After 45 min of ischemia and 20 min of reperfusion mitochondrial Ca2+ was 360 nM in hearts without RR and 240 in hearts treated with RR. Mitochondrial Ca2+ was not measured during ischemia.
Ruiz-Meana et al., measured mitochondrial Ca2+ during simulated ischemia and reperfusion in HL-1 cells with and without RR [16]. Mitochondrial Ca2+ levels, measured with Rhod-2, doubled during simulated ischemia; however, when 25 μM RR was added, the rise in Ca2+ was attenuated by 60%. During simulated ischemia, cytosolic Ca2+ showed a steady rise Ca2+ (~2 fold increase) by 60 minute. With addition of RR, cytosolic Ca2+ rose faster and to a higher level (2.5 fold increase). Addition of RR during simulated ischemia resulted in an increase in LDH release.
Taken together the data seem to suggest that mitochondrial Ca2+ increases during ischemia and that RR attenuates this increase. One limitation of these studies is that RR has numerous off target effects and is known to inhibit SR-Ca2+ uptake and other Ca2+ transporters. Ru360, the active species in RR, was purified and found to have few side effects. Studies utilizing Ru360 were done by perfusing rat hearts with 250 nM of Ru360 for 30 minutes prior to 30 min of global ischemia [17]Upon reperfusion, rate pressure product recovered in the Ru360 treated hearts but not in the untreated hearts. Mitochondria were isolated from hearts upon reperfusion and mitochondria Ca2+ was lower in Ru360 treated hearts. Zhang et al., also found that rat hearts perfused with Ru360 (10 μM) had better recovery of post ischemic function [18].
ROS
The importance of ROS and cardiac pathologies was first recognized in the 1970s [19,20]. Upon reperfusion of ischemic tissues, which is overall beneficial when performed in a timely manner, there is additional tissue damage driven by ROS [21–23]. Several groups presented a correlation between the generation of ROS and tissue damage following re-oxygenation [21–25]. This increase in damage post tissue re-oxygenation was coined reperfusion injury and is thought to be driven by toxic levels of ROS as reducing oxidative damage correlates with a reduction in damage [22,26–31].
One potential mechanistic explanation for this correlation is that ROS leads to an increase of membrane permeabilization. It is well established that ROS can peroxidize the lipids in membranes, which can lead to membrane destabilization and the efflux of contents held within. Hicks and Gebicki first established a quantitative relationship between ROS concentration and rate of lipid peroxidation [32]. They demonstrated a proportional relationship between the degree of lipid membrane peroxidation with the amount of solute release from the membranes and the concentration of peroxidizing agents introduced. In addition to direct lipid oxidation, ROS has also been implicated in mPTP opening. Initial investigations revealed that, similar to Ca2+, isolated mitochondria from the heart undergo ROS-induced mPTP opening by treating with t-butylhydroperoxide (Fig. 1) [33]. Further evidence solidified the hypothesis that ROS triggers mPTP opening by demonstrating that ROS accumulation in cells associates with known characteristics of the mPTP. For example, one hallmark of mPTP opening is the depolarization of mitochondrial membrane potential, which occurs when mitochondria are exposed to ROS. Furthermore, bongkrekic acid (BKA) (a known inhibitor of the mPTP) prevents this ROS-dependent depolarization [31]. Since mitochondria are a major source of ROS generation, ROS-dependent mPTP opening may lead to a feed-forward process of additional ROS release into the cytosol of the cell, this has been termed, “ROS-induced ROS release” [31]. Additionally, ROS is able to trigger mPTP-dependent mitochondrial swelling in the presence of low levels of Ca2+, which is inhibited by CsA [31,34,35].
Although ROS can seemingly induce mPTP opening, there are many critical questions addressing this mechanism. One of these fundamental questions is whether or not ROS-mediated mPTP opening is a completely independent event from Ca2+ overload (Fig. 1), or if these two triggers overlap and synergize to lead to mPTP-dependent mitochondrial dysfunction. Another form of necrotic cell death that is dependent on ROS-mediated mitochondrial dysfunction is oxytosis later to be referred to as ferroptosis. This version of cell death leads to a decrease of glutathione peroxidase 4 (GPX4) levels, one of the antioxidant enzymes that reduces oxidized polyunsaturated fatty acids (PUFA), which make up large parts of biological membranes [36]. If GPX4 remains depleted, cells accumulate overwhelming amounts of ROS, which will lead to lipid peroxidation of membranes. This version of cell death has been implicated in I/R injury [37,38]. Following reperfusion of an occlusion, the mechanical and shear stress leads to the lyses of red blood cells. Thus, releasing iron bound hemoglobin, which is taken up by nearby cells. In these cells, the increase of iron leads to the peroxidation of the PUFAs via the Fenton Reaction, in which iron reacts with hydrogen peroxide to form damaging hydroxyl radicals targeting the membrane PUFAs. This causes an ongoing chain of neighboring lipids becoming oxidized eventually leading to membrane collapse. This form of cell death has been reported to occur independently of mPTP activity [39], although inhibition of the mPTP is also protective during I/R injury in numerous animal model systems [3–6]. Potentially, both forms of necrotic cell death may be occurring simultaneously in the heart during I/R injury [40] and ROS levels could be the link between both forms of necrotic cell death. For example, low levels of ROS may facilitate sensitization of Ca2+-mediated mPTP-dependent necrosis, while high levels of ROS may be sufficient to induce ferroptosis independent of the mPTP.
It is clear that there is a relationship between ROS and Ca2+ in mediating mPTP opening. ROS has been established to sensitize the mPTP to Ca2+. It is thought that ROS targets mPTP regulator proteins via posttranslational modifications (PTMs) to facilitate this sensitization. These types of PTMs are referred to as oxidative PTMs, since they occur in an oxidative environment. Indeed, low levels of ROS have been implicated to serve as important precursors in various signal transduction pathways mainly related to cellular redox control, and mitochondrial function [41,42]. For example, certain cysteine proteases that are normally residents of the cytoplasm (a generally reduced environment), are susceptible to PTMs via ROS and become active if the cytoplasm becomes overly oxidative. The presence of the thiol group (-SH) in the side chain render cysteine residues particularly susceptible to oxidative modification [43]. Dysregulation of oxidative PTMs in the mitochondria have been linked with numerous disease pathologies including cardiomyopathies and neurological disorders. The mPTP is composed of numerous proteins and regulators, each of which is potentially susceptible to PTMs, which can affect the sensitivity of the mPTP [44]. S-nitrosylation (SNO/SNOylation) is the addition of a –NO group to thiols of cysteine residues by the donor s-nitrosoglutathione (GSNO) [45]. This modification has been shown to sensitize the mPTP, allowing for the activation of the pore with a lower concentration of Ca2+ by modifying the ANT family and voltage dependent anion channels (VDAC) proteins [46].
CypD has been shown to be modified by SNOylation [40,47]. The specific residue of CypD, cysteine 203, was identified by mass-spectrometric analysis to be SNOylated following treatment with GSNO, and is thought to be the potential site of a triggering PTM to sensitize the mPTP. Subsequent research confirmed that in hearts preconditioned to ischemia, increased SNO-CypD was identified perhaps facilitating the protective effect of ischemic preconditioning [40]. Indeed, GSNO pretreatment inhibited mPTP activation when cells were treated with the H2O2 [40], providing evidence that SNOylation of CypD desensitizes the mPTP. This study also showed that when cysteine 203 of CypD was mutated to serine, cells were resistant to mPTP activation and mitochondria require significantly more Ca2+ to undergo a swelling event. CRC is also increased when liver mitochondria are reconstituted with this mutant version of CypD.
It is clear ROS has the ability to sensitize mitochondria to Ca2+-dependent mPTP opening. Most likely, this is dependent on a ROS-dependent PTM of either a regulator or the actual pore-forming component of the pore. However, high levels of ROS can trigger mitochondrial swelling and dysfunction through lipid peroxidation independently of the mPTP. In the case of I/R injury, additional investigation is needed to determine which form of ROS-dependent mitochondrial dysfunction is dominant. Perhaps it is dependent on ischemic timing or spatial distance from the occlusion.
Matrix Regulation of the mPTP by Cyclophilin D
As discussed previously, it was shown in the 1970s that high matrix Ca2+ leads to mitochondrial swelling due to activation of a non-specific channel and that this swelling could be reversed by chelating Ca2+ [48]. It was later demonstrated that CsA could inhibit the mPTP [49]; the Ca2+ activation of mitochondrial swelling required higher Ca2+ levels in the presence of CsA. It was later demonstrated that the mitochondrial matrix protein CypD was the target of CsA [50,51]. CypD is a peptidyl-prolyl cis/trans isomerases and genetic deletion of CypD directly confirmed that it was an activator of mPTP opening as mitochondria lacking CypD required much larger amounts of Ca2+ to initiate mitochondrial swelling (Fig. 1) [6,52,53]. Although loss or inhibition of CypD increased the amount of Ca2+ needed to activate the mPTP, mPTP opening could still occur with higher levels of Ca2+. This was interpreted as showing that although CypD was an activator of the mPTP, with higher levels of Ca2+, mPTP opening was CypD independent.
The demonstration that CypD could enhance mPTP opening initiated studies to identify targets of CypD. It was originally shown that CypD can bind to ANT leading to the suggestion that the ANT might be the pore-forming unit [54], as discussed below. Studies by Wallace’s group showed that CsA inhibitable mPTP opening can still occur in mitochondria with deletion of several ANT isoforms [55]However, much larger amount of Ca2+ were needed to active mPTP opening. The mitochondrial phosphate carrier (PiC) was also shown to bind to CypD. However genetic deletion of the PiC did not eliminate mPTP activity [56,57]. CypD has also been shown to bind to the ATPO subunit of the F1-ATPase supporting a role for the F1-ATPase as the mPTP or at least a regulator of the mPTP [58]. It should be noted that CypD is a mitochondrial matrix chaperone protein, so it is not surprising that it binds to many proteins.
The mechanism by which CypD activates the mPTP is poorly understood. Inhibitors or genetic ablation of CypD reduce infarct size in animal studies of I/R injury [3–6]. CsA-mediated inhibition of the mPTP and cardioprotection have been demonstrated in cells, isolated hearts, and in vivo models [59]. Furthermore, in a proof of concept, phase II trial, the administration of CsA during acute myocardial infarction decreased infarct size as assessed by MRI [60]; however a subsequent phase III trial showed no benefit to CsA treatment [61]. The potential reasons why CsA has been beneficial in animal studies yet failed to show benefit in the clinical trial have been discussed elsewhere [62,63]. It is worth pointing out that CsA works by binding CypD, which is only a regulator of the pore and robust stimuli can activate mPTP independent of CypD. Alternatively, perhaps a CypD independent mechanism of death (either a second mPTP or an alternative death mechanism) is the primary mechanism of I/R cardiomyocyte death in MI patients.
Amino acids with a proline can exist in either cis or trans conformation and CypD catalyzes this isomerization. The isomerization can occur without CypD but at a slower rate. The role of the CypD prolyl isomerase activity in PTP activation remains unclear. Moreover, other cyclophilin family members are known to have functions that do not involve their isomerase activity [64]. For example, although the drug CsA inhibits the isomerase activity of multiple cyclophilins, mutational analyses have shown that cyclophilin A (CypA) is able to mediate calcineurin inhibition and the immunosuppressive effects of CsA independent its prolyl isomerase activity. There are conflicting data as to whether the isomerase activity is required for CypD activation of PTP. Early inhibitor studies suggested that isomerase activity was not required for mPTP activation [65]. In contrast, Baines et al., expressed a CypD with an R96G mutation, which lacks isomerase activity, in mouse embryonic fibroblasts (MEFs) lacking CypD and showed that the isomerase dead CypD afforded similar protection to CypD-KO when treated with H2O2 [53]. Interestingly, overexpression of CypD in HEK293 and rat glioma C6 cells desensitized the cells to apoptotic stimuli, and this protection required CypD isomerase activity [66]. It is noteworthy that the original studies characterizing the CypD-KO mice showed that loss of CypD inhibited necrotic, but not apoptotic cell death [53]. Additional studies will be needed to clarify the role of the isomerase activity in regulating the mPTP.
It is unclear how CypD activates the mPTP (Fig 1). It has been proposed [67] that CypD functions to regulate the mitochondrial synthasome. ATP production by mitochondria requires the transport of ADP into the mitochondria via the ANT family along with the transport of inorganic phosphate via the PiC. ADP and Pi are then converted to ATP by the F1Fo-ATPase. A complex involving ANT, PiC and the synthase has been shown to form in the mitochondria and it is proposed that this complex more efficiently converts ADP and Pi to ATP. CypD has been proposed to enhance the dissociation of the synthasome into its components (ANT, PiC and F1Fo-ATPase). This would be consistent with data showing that CypD results in a reduction in F1Fo-ATPase activity [58,68], assuming that F1Fo-ATPase activity in the synthasome is higher. Porter and colleagues further propose that neither the F1Fo-ATPase nor ANT are able to facilitate their mPTP modalities while part of the synthasome complex. Therefore, CypD hypothetically promotes mPTP opening by stimulating the dissociation of the synthasome complex into individual components (ANT and the F1Fo-ATPase) that can then form the mPTP. It is also possible that as the synthasome disassembles, it exists in a certain configuration that forms the mPTP.
CypD is known to undergo a number of post translational modifications [69–75] (Table 1). Bernardi’s group showed that phosphorylation of CypD enhanced its activation of the mPTP [76]. Although, the site of phosphorylation was not identified in this study. In another study phosphoproteomics identified an increase in phosphorylation of CypD-S42 in MCU deleted mice [74]. Furthermore, it was shown that mitochondria from MCU null hearts with increased phosphorylation of CypD exhibited increased susceptibility to mPTP opening and required less Ca2+ to trigger the pore. These data suggest that phosphorylation of CypD at S42 can enhance CypD binding to the mPTP exacerbating activation. Other studies showed that phosphorylation of S191 of CypD enhances CypD binding to the oligomycin sensitivity conferring protein (OSCP) of the synthasome and increases mPTP opening [73].
Table 1:
Modifications of Cyclophilin D and its effect on mPTP opening
CypD has been shown to undergo lysine acetylation. SIRT3, a mitochondrial protein that catalyzes the deacetylation of mitochondrial proteins was reported to deacetylate lysine 166 of CypD [72]. Mice lacking SIRT3 showed increased acetylation of CypD at lysine 166 and at 16 months of age mitochondria from SIRT3 null mice showed reduced mitochondrial CRC. Cysteine 202 (C202) of CypD was also shown to play a role in regulating mPTP opening [69,70]. C202 of CypD is highly conserved among species and can undergo redox-sensitive, post-translational modifications [77]. C202 was found to be S-nitrosylated in cardioprotected hearts. It was proposed that oxidation (e.g. perhaps the formation of a disulfide bond with the mPTP) of C202 might target CypD to the mPTP and that SNO of C202 blocks the oxidation that targets CypD to the mPTP. This hypothesis would be consistent with data showing that oxidation targets CypD to the mitochondrial membrane. A knock-in mouse model was developed using CRISPR/Cas9 in which CypD-C202 was mutated to a serine (C202S) [70]. Infarct size was reduced in CypD C202S Langendorff perfused hearts compared to WT. Cardiac mitochondria from CypD-C202S mice also have higher CRC compared to WT. Cysteines can also undergo S-acylation, a reversible post-translational lipid modification involving a thioester bond, and C202 matches a S-acylation motif. S-acylation of CypD-C202 was assessed using resin-assisted capture. WT hearts are abundantly S-acylated on CypD-C202 under baseline conditions indicating that S-acylation on C202 per se does not lead to mPTP opening [70]. CypD C202S knock-in hearts are protected from I/R injury suggesting further that replacing CypD S-acylation at C202 with a serine is not detrimental and does not induce mPTP opening. The data are consistent with the hypothesis that either acylation, SNO or mutation of C202 can block the ability of CypD to activate the mPTP. All of these modifications would block oxidation of this cysteine. Interestingly, ischemia leads to de-acylation of C202 and Ca2+ overload in isolated mitochondria promotes de-acylation of CypD. Taken together, these data suggest that with ischemia CypD is de-acylated at C202 allowing the free cysteine residue to undergo oxidation during the first minutes of reperfusion, which in turn targets it to the mPTP.
These post translational modifications of CypD are consistent with the hypothesis that CypD integrates signaling to regulate activation of the mPTP. It is intriguing to speculate that perhaps these PTMs are important for integrating metabolic signals to regulate F1Fo-ATPase activity and as proposed [67] a by-product of this regulation is alter synthasome formation which regulates PTP activity.
The Inner Membrane Pore:
The ANT Family
The ANT family of proteins are members of the larger mitochondrial solute carrier family and are residents of the inner mitochondrial membrane. The ANTs are the most abundant protein found within this membrane. These proteins transport ATP across the inner membrane from the matrix towards the intermembrane space of mitochondria and ADP towards the matrix from the intermembrane space of the mitochondria in a 1:1 ratio. There are four isoforms of ANT (Ant 1–4) in humans and three expressed in mice (mice lack Ant 3). ANT 1 is most highly expressed in the mitochondria of heart and skeletal muscle. ANT 4 is the predominant isoform in the testis, while ANT 2 and ANT 3 are ubiquitously expressed in other tissues. The first suggestion that ANT may be part of the mPTP came from the observation that Ca2+-dependent mPTP opening is inhibited by ADP [78]. Since ADP binds to the ANT, it was suggested that the specific conformation of this protein can influence the opening of the mPTP [78,79]. Specifically, ADP was found to stabilize the “m” state of ANT, which has the nucleotide-binding site facing the matrix. Additionally, the respiratory toxin, BKA, inhibits the mPTP in the same manner as ADP by stabilizing the ANTs in the m-state [80,81]. Oppositely to the m-state is the “c” state, or cytoplasmic state of the ANTs. Compounds that stabilize the c-state (ATP and carboxyatractyloside (Atr)) are able to sensitize the mPTP to Ca2+. Further, Halestrap and Davidson were first to suggest that the mPTP regulator CypD interacts with ANT [51]. They suggested that the presence of conserved prolines [82] (later identified as proline 62 on the ANTs) in the matrix compartments on this protein may be the site of prolyl-isomerization activity of CypD [51]. Notably, this interaction is ablated with treatments of CsA [51]. Finally, a critical piece of evidence for ANT being the mPTP was demonstrated as ANT reconstituted into lipid bilayers were able to fore pores with similar characteristics as the mPTP (Fig. 1) [82–84].
Although there is substantial evidence to suggest that the ANT family is a pore-forming component or a critical regulator of the mPTP, reports have challenged this hypothesis [85]. Genetic removal of the two major isoforms of the ANT family, Ant1 and Ant2, from mouse liver did not inhibit mPTP opening, however, the mitochondria required significantly more Ca2+ to open the pore. The treatment of CsA further desensitized these mitochondria to mPTP opening, suggesting that CypD may function independently of the ANT family in regulating the pore. One caveat to this study was that Ant4 could potentially compensate for the loss of the other isoforms of Ants in the liver. Recently, this caveat was addressed when researchers deleted all isoforms of Ants from the mouse liver. Surprisingly, these mice survived into adulthood with no obvious liver dysfunction [86]. Therefore, an unidentified compensatory mechanism must have been responsible for the transport of ADP/ATP across the inner mitochondrial membrane in these livers. Mitochondria isolated from these livers were extremely resistant to Ca2+-induced mPTP opening, although mPTP opening could still occur with very high levels of Ca2+. However, when CypD was inhibited or deleted on the Ant null background, Ca2+-induced mPTP opening is completely inhibited, suggesting, that together the ANT family and CypD are essential regulators of the mPTP or that there may be the presence of two mPTP-like pores. One, consisting of the ANT family and the other requiring CypD activity. Therefore, genetic evidence has confirmed that the ANT family is an important regulatory or partial pore-forming component of the mPTP, however, the family is not essential for Ca2+ induced mPTP opening, similarly to CypD.
Perhaps the unknown compensatory mechanism that is responsible for the physiological function of the ANT family that engages in their absence is also responsible for the remaining mPTP activity. Additionally, both of these genetic loss-of-function approaches relied on the same Cre/LoxP approach for the deletion of Ant2 in the liver, since the complete loss of Ant2 is embryonically lethal. These systems are inherently problematic when deleting essential genes in tissues that are highly regenerative, such as the liver. There remains a possibility of incomplete deletion of Ant2, which may give these hepatocytes a competitive proliferative advantage to maintain liver functionality and may explain the remaining mPTP activity. Additionally, these data put into question the relationship between CypD and the ANT family and further investigation is needed to determine if their interaction has a functional consequence on mPTP sensitivity.
F1Fo-ATPase
The F1Fo-ATPase has been proposed by two groups to be the pore-forming unit of the mPTP (Fig. 1). Briefly, recent data have suggested that the F1Fo-ATPase can, under certain circumstances, form the mPTP. Bernardi’s group has proposed that dimers of the F1Fo-ATPase can form the pore-forming unit of the mPTP [87]. Jonas’ group has suggested that de-lipidation of the c-ring of the F1Fo-ATPase can form the mPTP [88]. Although there are strong data in support for both hypotheses, there are also data in opposition [89,90]. Data from Walker’s group has shown a CsA-inhibitable mPTP-like swelling is still present in cells lacking the c-ring and other components of the F1Fo-ATPase that are required for dimerization. Furthermore, patch clamping results from the Jonas lab indicating similar conductance activity as the mPTP to the c-subunit ring pore showed that this pore is oligomycin (F1Fo-ATPase inhibitor) sensitive [91], however; Ca2+ induced mPTP opening by the mitochondrial swelling assay was not inhibited by oligomycin treatment [92].
One explanation for the discrepancy is that there are two or more CsA-inhibitable pores [86]. As discussed above data from over 30 years ago proposed a role for ANT as a key component of the mPTP. CypD was shown to bind to ANT and adenine nucleotides were known inhibitors of the mPTP. BKA and Atr, two inhibitors of ANT that lock ANT in different conformations, were shown to regulate mPTP opening. BKA locks ANT in the matrix facing conformation and inhibits mPTP opening whereas Ca2+ binds to the cytosolic facing conformation and activates PTP. The hypothesis that ANT was the pore-forming component of the mPTP fell out of favor in 2005 when Wallace’s group reported that a CsA-inhibitable PTP, albeit requiring higher Ca2+ for activation, was present when Ant1 and Ant2 were deleted [93]. Recent studies have revisited a role for ANT as a pore component of the mPTP. Karch et al., showed that mitochondria with deletion of Ant1, Ant2 and Ant4 were desensitized to Ca2+-induced PTP opening, and PTP opening was completely inhibited by the further addition of CsA [86]. They proposed that there are two distinct pore-forming components of the mPTP: consisting of the ANT family and a second pore (Fig. 1). Both components may be activated by CypD, although the non-ANT component has a strict requirement for CypD.
Another feature of the mPTP that may also suggest two independent channels or regulatory components is its bistable conductance states, high and low [94]. Low conductance mPTP is thought to occur in more physiological settings due to the ability for the membrane potential to remain intact during this transient opening and closing. The high conductance state is considered to be pathological due to the depletion of membrane potential. Perhaps the ANT family or the F1Fo-ATPase favors a high vs low mPTP channel. Neginskaya et al., reported that mitochondria deficient for the c-subunit of the F1Fo-ATPase lack the high conductance mPTP channel, while a low conductance CsA sensitive channel still persists [95]. More insight is required on determining how the ANT family or the F1Fo-ATPase influences the high vs low conductance mPTP channels and how this may affect physio- and pathological conditions.
Outer Membrane Regulators of the mPTP
The mPTP is defined as an inner mitochondrial membrane pore that is triggered mainly by increases in matrix Ca2+ levels. Although the pore is regulated from the matrix and takes place in the inner membrane, evidence suggests that the outer mitochondrial membrane plays a significant role in its sensitivity. In the early models of the mPTP, it was hypothesized that the mPTP spanned the inner mitochondrial space and occurred at inner and outer membrane contact sites. Some early evidence of this originated when it was demonstrated that ANT was isolated and identified at contact sites between the inner and outer membranes [96,97]. Furthermore, at these sites it has been shown that there is a pore that has similar conductance characteristics to that of the mPTP [97]. Together, these results provided evidence for an outer membrane component of the mPTP and research began into potential identities of this pore.
One such protein that became a suitable candidate is the voltage dependent anion channel (VDAC). VDAC is the most abundant protein on the mitochondrial outer membrane. This protein family form the main channel that regulate ion and metabolite flux between the outer mitochondrial membrane and the cytosol. Early evidence that the VDAC family may be part of the mPTP came from patch clamping experiments that showed that opening of the mPTP was voltage dependent [98]. The conductance characteristic of the mPTP was then compared to that of bilayers with reconstituted VDAC and similar conductance patterns were observed [99]. Later evidence showed affinity capture experiments that identified VDAC and ANT as binding partners of a GST-CypD fusion protein [100]. Additionally, reconstitution of CypD, ANT, and VDAC produced the characteristic trace of mPTP opening in the presence of Ca2+, which could be inhibited by pretreatment with CsA [100].
Though there is significant evidence to suggest VDAC is the outer membrane regulator of the mPTP, genetic evidence challenged this hypothesis. Baines et. al, utilized a dual deletion approach of two of the three isoforms of the VDAC family expressed in the heart. (Vdac1 and Vdac3) to demonstrate that mitochondria isolated from cardiomyocytes were still susceptible to Ca2+ induced mPTP-dependent swelling [101]. Unlike the deletion of CypD or the ANT family, the deletion of these isoforms did not alter the sensitivity of the mPTP to Ca2+ [101]. Utilizing Vdac1 and Vdac3 double deleted MEFs and an siRNA approach to abolish the expression of Vdac2, they found that VDAC family null MEFs were not protected against necrotic or apoptotic stimuli, suggesting that they are dispensable for mPTP activity [101].
In addition to the VDAC family, the Bcl-2 family is another set of proteins that can greatly affect the permeability of the outer mitochondrial membrane and has been implicated in regulating the mPTP (Fig. 1). The Bcl-2 family consists of over 25 family members characterized by containing at least one Bcl-2 homology (BH) domain and consist of both pro-apoptotic and anti-apoptotic members [102]. During homeostasis, the pro-apoptotic BH3-only family members are in balance with the anti-apoptotic BH1-4 containing members [102–104]. The downstream ramification of these family members is the inactivation or activation of BAX and BAK (BH1-3, effector Bcl-2 family members) [102]. Upon activation, BAX and BAK form homo-/hetero oligomers, which leads to increased permeability of the outer mitochondrial membrane allowing molecules up to 100 kDa to freely cross the outer membrane vs the normal 5 kDa molecules [105–107].
The first evidence that BAX may be a component of the mPTP was determined when depletion of BAX from cells inhibited the ability to undergo permeability transition [108]. This same study showed that BAX and ANT interact in a yeast two-hybrid system and cells with reconstituted BAX could not induce cell death in cells lacking ANT [108]. It was also shown that recombinant BAX could lead to a dissipation of the inner membrane potential, which is associated with mPTP opening [108,109]. Strong supportive evidence for the role of BAX and BAK in the mPTP came from showing that these proteins are necessary for cells to engage regulated necrosis. These studies demonstrated that cells lacking both Bax and Bak1 are resistant apoptotic cell death as well as multiple forms of regulated necrotic cell death, including mPTP-dependent necrosis [110–113]. Further, mitochondria isolated from these cells were unable to undergo Ca2+ -induced swelling and displayed significantly increased CRC [112,114]. During apoptosis BAX/BAK form large pores within the outer mitochondrial membrane, however, this role of BAX and BAK is not required for mPTP-dependent cell death. Indeed, double deleted cells reconstituted with oligomeric-dead mutant versions of BAX restore the ability to undergo some forms of regulated necrotic cell death, but not apoptosis [110–112]. Notably, the inner membrane pore is still intact and measurable by patch clamping mitoplasts isolated from Bax and Bak1 deleted cells. Thus suggesting, that BAX and BAK are eliciting their mPTP sensitizing effects by increasing outer mitochondrial membrane permeability.
In a recent paper, it was shown that cells are more susceptible to Ca2+-induced necrosis when treated with BH3-mimetic compounds targeting and inhibiting various members of the anti-apoptotic Bcl-2 family (Bcl-2, Mcl-1, and Bcl-xL) [114]. Further, isolated mitochondria treated with the same BH3-mimetics displayed decreases in CRC due to the sensitization of the mPTP [114]. Indeed, mitochondria lacking both BAX and BAK remained unaffected by the treatment of the various BH3-mimetics [114]. Together, these data support the hypothesis that upstream Bcl-2 family members are able to affect mPTP sensitivity by influencing the activation status of BAX and BAK, which alters outer mitochondrial membrane permeability. Mechanistic understanding of how outer mitochondrial membrane permeability directly affects matrix Ca2+ capacity and mPTP sensitivity is required. Potentially, active BAX and BAK may lead to the release of a critical regulator of the mPTP found within the inner membrane space, similarly to cytochrome-c release during apoptosis.
Conclusion
MPTP-dependent necrosis is dependent on mitochondrial dysfunction via prolonged mPTP opening. If the heart does not produce sufficient levels of ATP, it cannot maintain contractibility or ion homeostasis, which can lead to osmotically driven cellular rupture and necrosis. Although mPTP-dependent necrotic cell death is a primary mode of cell death during I/R injury, the process of permeability transition can also lead to other forms of cell death as well (Fig. 2). For example, if a limited population of mitochondria undergo mPTP opening and lose membrane integrity, this can lead to cytochrome-c (Cyt-C) release [115,116] or mtDNA release, which can initiate apoptosis or pyroptosis respectively [117]. Additionally, these dysfunctional mitochondria need to be processed through mitophagy and the energetic deficit caused by their dysfunction can lead to an increase in macroautophagy, which may lead to autophagy or lysosomal-dependent forms of cell death [118,119]. Furthermore, compensation for the loss of oxidative phosphorylation through glycolysis leads to a decrease in NAD+ pools within the cell, which are required for PARP-dependent DNA repair, which may sensitize a cell to undergo parthanatos, another form of necrotic cell death [120].
Figure 2: mPTP dependent mitochondrial dysfunction can potentially feed into other cell death pathways.
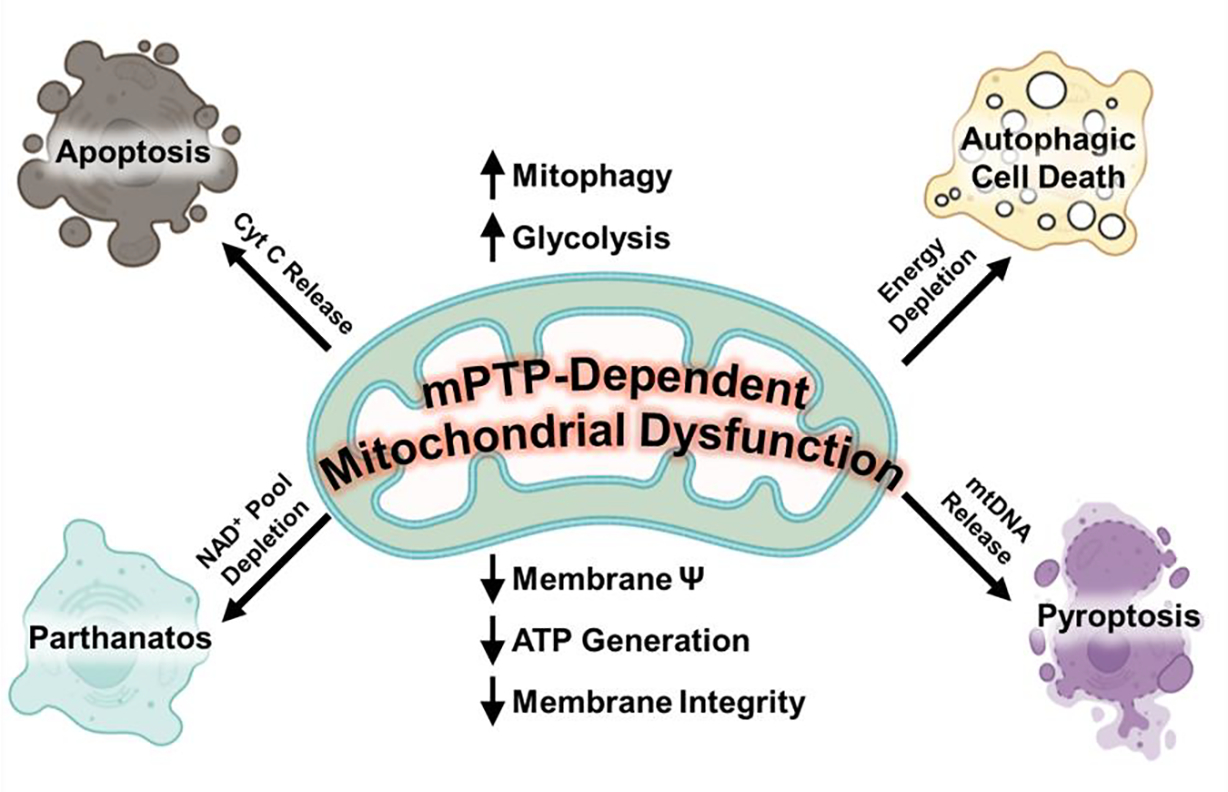
Following mPTP opening, mitochondrial membrane potential and eventually integrity decreases. This leads to a decrease in mitochondrial ATP production, which can lead to an increase in glycolysis as a compensatory mechanism to maintain the energetic demand of the cell. These events culminate in the reduction of the NAD+ pools which can sensitize a cell to parthanatos. Energetic depletion may also result in the over activation of autophagy, which may result in autophagic or lysosomal dependent cell death. In addition, mitochondrial membrane rupture can trigger other cell death modalities. For example, rupture can lead to Cytochrome C (Cyt C) release, which may initiate apoptosis or mtDNA release, which can initiate pyroptosis.
The lack of molecular characterization of the mPTP has hampered the progress and impact of this cell death mechanism. Potentially, pinning down the identification has challenged researchers due to the possibility of being composed of more than one pore-forming component. Many ongoing questions concerning the regulation of the mPTP have been highlighted throughout this review (Fig. 1). Identifying the molecular composition of this pore will be the catalyst for answering most of these remaining questions. Regardless, it is clear that the mPTP is a significant contributing factor to the diseases of the heart and identifying novel strategies to target mPTP regulation will be therapeutically beneficial.
Highlights.
Ischemia reperfusion injury is a consequence of mPTP-dependent necrosis
Prolonged mPTP opening leads to mitochondrial dysfunction
mPTP opening is triggered by calcium and reactive oxygen species
The mPTP may consist of multiple pore-forming entities
Outer mitochondrial membrane permeability through the Bcl-2 family alters mPTP sensitivity to calcium
Acknowledgment
This work was supported by National Institute of Health grants R01HL150031 (to JK) and by the NHLBI intramural program (to EM)
Footnotes
Figures were generated using Biorender.com and Microsoft PowerPoint
Disclosures of all authors
None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature Cited
- [1].Haworth RA, Hunter DR, The Ca2+-induced membrane transition in mitochondria, Arch Biochem Biophys. 195 (1979) 460–467. 10.1016/0003-9861(79)90372-2. [DOI] [PubMed] [Google Scholar]
- [2].Halestrap AP, What is the mitochondrial permeability transition pore?, J Mol Cell Cardiol. 46 (2009) 821–831. 10.1016/j.yjmcc.2009.02.021. [DOI] [PubMed] [Google Scholar]
- [3].Skyschally A, Schulz R, Heusch G, Cyclosporine A at Reperfusion Reduces Infarct Size in Pigs, Cardiovasc Drugs Ther. 24 (2010) 85–87. 10.1007/s10557-010-6219-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Argaud L, Gateau O, Muntean D, Chalabreysse L, Loufouat J, Robert D, Ovize M, Specific inhibition of the mitochondrial permeability transition prevents lethal reperfusion injury, J Mol Cell Cardiol. 38 (2005) 367–374. 10.1016/j.yjmcc.2004.12.001. [DOI] [PubMed] [Google Scholar]
- [5].Zhang C, Cheng Y, Liu D, Liu M, Cui H, Zhang B, Mei Q, Zhou S, Mitochondria-targeted cyclosporin A delivery system to treat myocardial ischemia reperfusion injury of rats, J Nanobiotechnology. 17 (2019) 18. 10.1186/s12951-019-0451-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, Inohara H, Kubo T, Tsujimoto Y, Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death, Nature. 434 (2005) 652–658. 10.1038/nature03317. [DOI] [PubMed] [Google Scholar]
- [7].Kwong JQ, Molkentin JD, Physiological and Pathological Roles of the Mitochondrial Permeability Transition Pore in the Heart, Cell Metab. 21 (2015) 206–214. 10.1016/j.cmet.2014.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Steenbergen C, Murphy E, Levy L, London RE, Elevation in cytosolic free calcium concentration early in myocardial ischemia in perfused rat heart., Circ Res. 60 (1987) 700–707. 10.1161/01.RES.60.5.700. [DOI] [PubMed] [Google Scholar]
- [9].Marban E, Kitakaze M, Kusuoka H, Porterfield JK, Yue DT, Chacko VP, Intracellular free calcium concentration measured with 19F NMR spectroscopy in intact ferret hearts., Proceedings of the National Academy of Sciences. 84 (1987) 6005–6009. 10.1073/pnas.84.16.6005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Bauer TM, Giles A. v., Sun J, Femnou A, Covian R, Murphy E, Balaban RS, Perfused murine heart optical transmission spectroscopy using optical catheter and integrating sphere: Effects of ischemia/reperfusion, Anal Biochem. 586 (2019) 113443. 10.1016/j.ab.2019.113443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].di Lisa F, Blank PS, Colonna R, Gambassi G, Silverman HS, Stern MD, Hansford RG, Mitochondrial membrane potential in single living adult rat cardiac myocytes exposed to anoxia or metabolic inhibition., J Physiol. 486 (1995) 1–13. 10.1113/jphysiol.1995.sp020786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Griffiths EJ, Ocampo CJ, Savage JS, Stern MD, Silverman HS, Protective effects of low and high doses of cyclosporin A against reoxygenation injury in isolated rat cardiomyocytes are associated with differential effects on mitochondrial calcium levels, Cell Calcium. 27 (2000) 87–95. 10.1054/ceca.1999.0094. [DOI] [PubMed] [Google Scholar]
- [13].Griffiths E, Ocampo CJ, Rutter GA, Hansford RG, Stern MD, Silverman HS, Mitochondrial calcium transporting pathways during hypoxia and reoxygenation in single rat cardiomyocytes, Cardiovasc Res. 39 (1998) 423–433. 10.1016/S0008-6363(98)00104-7. [DOI] [PubMed] [Google Scholar]
- [14].Allen SP, Darley-Usmar VM, McCormack JG, Stone D, Changes in Mitochondrial Matrix Free Calcium in Perfused Rat Hearts Subjected to Hypoxia-Reoxygenation, J Mol Cell Cardiol. 25 (1993) 949–958. 10.1006/jmcc.1993.1107. [DOI] [PubMed] [Google Scholar]
- [15].Miyamae M, Camacho SA, Weiner MW, Figueredo VM, Attenuation of postischemic reperfusion injury is related to prevention of [Ca2+]m overload in rat hearts, American Journal of Physiology-Heart and Circulatory Physiology. 271 (1996) H2145–H2153. 10.1152/ajpheart.1996.271.5.H2145. [DOI] [PubMed] [Google Scholar]
- [16].Ruizmeana M, Garciadorado D, Mirocasas E, Abellan A, Solersoler J, Mitochondrial Ca2+ uptake during simulated ischemia does not affect permeability transition pore opening upon simulated reperfusion☆, Cardiovasc Res. 71 (2006) 715–724. 10.1016/j.cardiores.2006.06.019. [DOI] [PubMed] [Google Scholar]
- [17].de Jesús García-Rivas G, Guerrero-Hernández A, Guerrero-Serna G, Rodríguez-Zavala JS, Zazueta C, Inhibition of the mitochondrial calcium uniporter by the oxo-bridged dinuclear ruthenium amine complex (Ru360) prevents from irreversible injury in postischemic rat heart, FEBS Journal. 272 (2005) 3477–3488. 10.1111/j.1742-4658.2005.04771.x. [DOI] [PubMed] [Google Scholar]
- [18].Zhang S, Gao Q, Cao C, Bruce IC, Xia Q, Involvement of the mitochondrial calcium uniporter in cardioprotection by ischemic preconditioning, Life Sci. 78 (2006) 738–745. 10.1016/j.lfs.2005.05.076. [DOI] [PubMed] [Google Scholar]
- [19].Hearse D, Humphrey S, Bullock G, The oxygen paradox and the calcium paradox: Two facets of the same problem?, J Mol Cell Cardiol. 10 (1978) 641–668. 10.1016/S0022-2828(78)80004-2. [DOI] [PubMed] [Google Scholar]
- [20].Hearse D, Abrupt reoxygenation of the anoxic potassium-arrested perfused rat heart: A study of myocardial enzyme release, J Mol Cell Cardiol. 5 (1973) 395–407. 10.1016/0022-2828(73)90030-8. [DOI] [PubMed] [Google Scholar]
- [21].Guarnieri C, Flamigni F, Caldarera CM, Role of Oxygen in the Cellular Damage Induced by Re-oxygenation of Hypoxic Heart*, 1980. [DOI] [PubMed]
- [22].Granger DN, Rutili G, McCord JM, Superoxide radicals in feline intestinal ischemia, Gastroenterology. 81 (1981) 22–29. 10.1016/0016-5085(81)90648-x. [DOI] [PubMed] [Google Scholar]
- [23].Granger DN, Kvietys PR, Reperfusion injury and reactive oxygen species: The evolution of a concept, Redox Biol. 6 (2015) 524–551. 10.1016/j.redox.2015.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Toescu EC, Normal brain ageing: models and mechanisms, Philosophical Transactions of the Royal Society B: Biological Sciences. 360 (2005) 2347–2354. 10.1098/rstb.2005.1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Downey JM, Free Radicals and Their Involvement During Long-Term Myocardial Ischemia and Reperfusion, Annu Rev Physiol. 52 (1990) 487–504. 10.1146/annurev.ph.52.030190.002415. [DOI] [PubMed] [Google Scholar]
- [26].FISCHER EG, Ames A, Studies on Mechanisms of Impairment of Cerebral Circulation Following Ischemia: Effect of Hemodilution and Perfusion Pressure, Stroke. 3 (1972) 538–542. 10.1161/01.STR.3.5.538. [DOI] [PubMed] [Google Scholar]
- [27].Ames A, Wright RL, Kowada M, Thurston JM, Majno G, Cerebral ischemia. II. The no-reflow phenomenon., Am J Pathol. 52 (1968) 437–53. [PMC free article] [PubMed] [Google Scholar]
- [28].Murry CE, Jennings RB, Reimer KA, Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium., Circulation. 74 (1986) 1124–1136. 10.1161/01.CIR.74.5.1124. [DOI] [PubMed] [Google Scholar]
- [29].Jennings RB, Sommers HM, Smyth GA, Flack HA, Linn H, Myocardial necrosis induced by temporary occlusion of a coronary artery in the dog., Arch Pathol (Chic). 70 (1960) 68–78. [PubMed] [Google Scholar]
- [30].Sewell WH, Koth DR, Huggins CE, Ventricular fibrillation in dogs after sudden return of flow to the coronary artery., Surgery. 38 (1955) 1050–3. [PubMed] [Google Scholar]
- [31].Zorov DB, Filburn CR, Klotz L-O, Zweier JL, Sollott SJ, Reactive Oxygen Species (Ros-Induced) Ros Release, Journal of Experimental Medicine. 192 (2000) 1001–1014. 10.1084/jem.192.7.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Hicks M, Gebicki JM, A quantitative relationship between permeability and the degree of peroxidation in ufasome membranes, Biochem Biophys Res Commun. 80 (1978) 704–708. 10.1016/0006-291X(78)91301-3. [DOI] [PubMed] [Google Scholar]
- [33].Crompton M, Costi A, Hayat L, Evidence for the presence of a reversible Ca2+-dependent pore activated by oxidative stress in heart mitochondria, Biochemical Journal. 245 (1987) 915–918. 10.1042/bj2450915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Seidlmayer LK, Juettner V. v., Kettlewell S, Pavlov E. v., Blatter LA, Dedkova EN, Distinct mPTP activation mechanisms in ischaemia–reperfusion: contributions of Ca2+, ROS, pH, and inorganic polyphosphate, Cardiovasc Res. 106 (2015) 237–248. 10.1093/cvr/cvv097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Červinková Z, Lotková H, Křivaková P, Roušar T, Kučera O, Tichý L, Červinka M, Drahota Z, Evaluation of Mitochondrial Function in Isolated Rat Hepatocytes and Mitochondria during Oxidative Stress, Alternatives to Laboratory Animals. 35 (2007) 353–361. 10.1177/026119290703500303. [DOI] [PubMed] [Google Scholar]
- [36].Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji AF, Clish CB, Brown LM, Girotti AW, Cornish VW, Schreiber SL, Stockwell BR, Regulation of Ferroptotic Cancer Cell Death by GPX4, Cell. 156 (2014) 317–331. 10.1016/j.cell.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Fang X-L, Ding S-Y, Du X-Z, Wang J-H, Li X-L, Ferroptosis—A Novel Mechanism With Multifaceted Actions on Stroke, Front Neurol. 13 (2022). 10.3389/fneur.2022.881809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Yan H, Zou T, Tuo Q, Xu S, Li H, Belaidi AA, Lei P, Ferroptosis: mechanisms and links with diseases, Signal Transduct Target Ther. 6 (2021) 49. 10.1038/s41392-020-00428-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, Morrison B, Stockwell BR, Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death, Cell. 149 (2012) 1060–1072. 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Nguyen TT, Stevens M. v., Kohr M, Steenbergen C, Sack MN, Murphy E, Cysteine 203 of Cyclophilin D Is Critical for Cyclophilin D Activation of the Mitochondrial Permeability Transition Pore, Journal of Biological Chemistry. 286 (2011) 40184–40192. 10.1074/jbc.M111.243469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Scherz-Shouval R, Shvets E, Elazar Z, Oxidation as a Post-Translational Modification that Regulates Autophagy, Autophagy. 3 (2007) 371–373. 10.4161/auto.4214. [DOI] [PubMed] [Google Scholar]
- [42].Fratelli M, Goodwin LO, Ørom UA, Lombardi S, Tonelli R, Mengozzi M, Ghezzi P, Gene expression profiling reveals a signaling role of glutathione in redox regulation, Proceedings of the National Academy of Sciences. 102 (2005) 13998–14003. 10.1073/pnas.0504398102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Chung HS, Wang S-B, Venkatraman V, Murray CI, van Eyk JE, Cysteine Oxidative Posttranslational Modifications, Circ Res. 112 (2013) 382–392. 10.1161/CIRCRESAHA.112.268680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Alves-Figueiredo H, Silva-Platas C, Lozano O, Vázquez-Garza E, Guerrero-Beltrán CE, Zarain-Herzberg A, García-Rivas G, A systematic review of post-translational modifications in the mitochondrial permeability transition pore complex associated with cardiac diseases, Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 1867 (2021) 165992. 10.1016/j.bbadis.2020.165992. [DOI] [PubMed] [Google Scholar]
- [45].van der Vliet A, Chr. t Hoen PA, Wong PS-Y, Bast A, Cross CE, Formation of S-Nitrosothiols via Direct Nucleophilic Nitrosation of Thiols by Peroxynitrite with Elimination of Hydrogen Peroxide, Journal of Biological Chemistry. 273 (1998) 30255–30262. 10.1074/jbc.273.46.30255. [DOI] [PubMed] [Google Scholar]
- [46].Chang AHK, Sancheti H, Garcia J, Kaplowitz N, Cadenas E, Han D, Respiratory Substrates Regulate S-Nitrosylation of Mitochondrial Proteins through a Thiol-Dependent Pathway, Chem Res Toxicol. 27 (2014) 794–804. 10.1021/tx400462r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Kohr MJ, Aponte AM, Sun J, Wang G, Murphy E, Gucek M, Steenbergen C, Characterization of potential S - nitrosylation sites in the myocardium, American Journal of Physiology-Heart and Circulatory Physiology. 300 (2011) H1327–H1335. 10.1152/ajpheart.00997.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Hunter DR, Haworth RA, Hunter DR, The Ca2+-induced membrane transition in mitochondria, Arch Biochem Biophys. 195 (1979). [DOI] [PubMed] [Google Scholar]
- [49].Crompton M, Ellinger H, Costi A, Inhibition by cyclosporin A of a Ca2+-dependent pore in heart mitochondria activated by inorganic phosphate and oxidative stress., Biochem J. 255 (1988) 357–60. [PMC free article] [PubMed] [Google Scholar]
- [50].Crompton M, Virji S, Ward JM, Cyclophilin-D binds strongly to complexes of the voltage-dependent anion channel and the adenine nucleotide translocase to form the permeability transition pore, Eur J Biochem. 258 (1998) 729–735. 10.1046/j.1432-1327.1998.2580729.x. [DOI] [PubMed] [Google Scholar]
- [51].Woodfield K, Rück A, Brdiczka D, Halestrap AP, Direct demonstration of a specific interaction between cyclophilin-D and the adenine nucleotide translocase confirms their role in the mitochondrial permeability transition, Biochemical Journal. 336 (1998) 287–290. 10.1042/bj3360287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Basso E, Fante L, Fowlkes J, Petronilli V, Forte MA, Bernardi P, Properties of the Permeability Transition Pore in Mitochondria Devoid of Cyclophilin D, Journal of Biological Chemistry. 280 (2005) 18558–18561. 10.1074/jbc.C500089200. [DOI] [PubMed] [Google Scholar]
- [53].Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, Robbins J, Molkentin JD, Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death, Nature. 434 (2005) 658–662. 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- [54].Halestrap A, Brenner C, The Adenine Nucleotide Translocase: A Central Component of the Mitochondrial Permeability Transition Pore and Key Player in Cell Death, Curr Med Chem. 10 (2003) 1507–1525. 10.2174/0929867033457278. [DOI] [PubMed] [Google Scholar]
- [55].Kokoszka JE, Waymire KG, Levy SE, Sligh JE, Cai J, Jones DP, MacGregor GR, Wallace DC, The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore, Nature. 427 (2004) 461–465. 10.1038/nature02229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Gutiérrez-Aguilar M, Douglas DL, Gibson AK, Domeier TL, Molkentin JD, Baines CP, Genetic manipulation of the cardiac mitochondrial phosphate carrier does not affect permeability transition, J Mol Cell Cardiol. 72 (2014) 316–325. 10.1016/j.yjmcc.2014.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Kwong JQ, Davis J, Baines CP, Sargent MA, Karch J, Wang X, Huang T, Molkentin JD, Genetic deletion of the mitochondrial phosphate carrier desensitizes the mitochondrial permeability transition pore and causes cardiomyopathy, Cell Death Differ. 21 (2014) 1209–1217. 10.1038/cdd.2014.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Giorgio V, Bisetto E, Soriano ME, Dabbeni-Sala F, Basso E, Petronilli V, Forte MA, Bernardi P, Lippe G, Cyclophilin D Modulates Mitochondrial F0F1-ATP Synthase by Interacting with the Lateral Stalk of the Complex, Journal of Biological Chemistry. 284 (2009) 33982–33988. 10.1074/jbc.M109.020115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Bauer TM, Murphy E, Role of Mitochondrial Calcium and the Permeability Transition Pore in Regulating Cell Death, Circ Res. 126 (2020) 280–293. 10.1161/CIRCRESAHA.119.316306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Piot C, Croisille P, Staat P, Thibault H, Rioufol G, Mewton N, Elbelghiti R, Cung TT, Bonnefoy E, Angoulvant D, Macia C, Raczka F, Sportouch C, Gahide G, Finet G, André-Fouët X, Revel D, Kirkorian G, Monassier J-P, Derumeaux G, Ovize M, Effect of Cyclosporine on Reperfusion Injury in Acute Myocardial Infarction, New England Journal of Medicine. 359 (2008) 473–481. 10.1056/NEJMoa071142. [DOI] [PubMed] [Google Scholar]
- [61].Cung T-T, Morel O, Cayla G, Rioufol G, Garcia-Dorado D, Angoulvant D, Bonnefoy-Cudraz E, Guérin P, Elbaz M, Delarche N, Coste P, Vanzetto G, Metge M, Aupetit J-F, Jouve B, Motreff P, Tron C, Labeque J-N, Steg PG, Cottin Y, Range G, Clerc J, Claeys MJ, Coussement P, Prunier F, Moulin F, Roth O, Belle L, Dubois P, Barragan P, Gilard M, Piot C, Colin P, de Poli F, Morice M-C, Ider O, Dubois-Randé J-L, Unterseeh T, le Breton H, Béard T, Blanchard D, Grollier G, Malquarti V, Staat P, Sudre A, Elmer E, Hansson MJ, Bergerot C, Boussaha I, Jossan C, Derumeaux G, Mewton N, Ovize M, Cyclosporine before PCI in Patients with Acute Myocardial Infarction, New England Journal of Medicine. 373 (2015) 1021–1031. 10.1056/NEJMoa1505489. [DOI] [PubMed] [Google Scholar]
- [62].Bernardi P, di Lisa P, Cyclosporine before PCI in Acute Myocardial Infarction, New England Journal of Medicine. 374 (2016) 88–90. 10.1056/NEJMc1514192. [DOI] [PubMed] [Google Scholar]
- [63].Linkermann A, Konstantinidis K, Kitsis RN, Catch me if you can: targeting the mitochondrial permeability transition pore in myocardial infarction, Cell Death Differ. 23 (2016) 1–2. 10.1038/cdd.2015.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Zydowsky LD, Etzkorn FA, Chang HY, Ferguson SB, Stolz LA, Ho SI, Walsh CT, Active site mutants of human cyclophilin A separate peptidyl-prolyl isomerase activity from cyclosporin A binding and calcineurin inhibition, Protein Science. 1 (1992) 1092–1099. 10.1002/pro.5560010903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Scorrano L, Nicolli A, Basso E, Petronilli V, Bernardi P, Two modes of activation of the permeability transition pore: the role of mitochondrial cyclophilin, Mol Cell Biochem. 174 (1997) 181–184. [PubMed] [Google Scholar]
- [66].Lin D-T, Lechleiter JD, Mitochondrial Targeted Cyclophilin D Protects Cells from Cell Death by Peptidyl Prolyl Isomerization, Journal of Biological Chemistry. 277 (2002) 31134–31141. 10.1074/jbc.M112035200. [DOI] [PubMed] [Google Scholar]
- [67].Beutner G, Alanzalon RE, Porter GA, Cyclophilin D regulates the dynamic assembly of mitochondrial ATP synthase into synthasomes, Sci Rep. 7 (2017) 14488. 10.1038/s41598-017-14795-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Chinopoulos C, Konràd C, Kiss G, Metelkin E, Töröcsik B, Zhang SF, Starkov AA, Modulation of F 0 F 1 -ATP synthase activity by cyclophilin D regulates matrix adenine nucleotide levels, FEBS Journal. 278 (2011) 1112–1125. 10.1111/j.1742-4658.2011.08026.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Nguyen TT, Stevens M. v., Kohr M, Steenbergen C, Sack MN, Murphy E, Cysteine 203 of Cyclophilin D Is Critical for Cyclophilin D Activation of the Mitochondrial Permeability Transition Pore, Journal of Biological Chemistry. 286 (2011) 40184–40192. 10.1074/jbc.M111.243469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Amanakis G, Sun J, Fergusson MM, McGinty S, Liu C, Molkentin JD, Murphy E, Cysteine 202 of cyclophilin D is a site of multiple post-translational modifications and plays a role in cardioprotection, Cardiovasc Res. 117 (2021) 212–223. 10.1093/cvr/cvaa053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Lv B, Peng H, Qiu B, Zhang L, Ge M, Bu D, Li K, Yu X, Du J, Yang L, Tang C, Huang Y, Du J, Jin H, Sulphenylation of CypD at Cysteine 104: A Novel Mechanism by Which SO2 Inhibits Cardiomyocyte Apoptosis, Front Cell Dev Biol. 9 (2022). 10.3389/fcell.2021.784799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Hafner A. v., Dai J, Gomes AP, Xiao C-Y, Palmeira CM, Rosenzweig A, Sinclair DA, Regulation of the mPTP by SIRT3-mediated deacetylation of CypD at lysine 166 suppresses age-related cardiac hypertrophy, Aging. 2 (2010) 914–923. 10.18632/aging.100252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Hurst S, Gonnot F, Dia M, Crola Da Silva C, Gomez L, Sheu S-S, Phosphorylation of cyclophilin D at serine 191 regulates mitochondrial permeability transition pore opening and cell death after ischemia-reperfusion, Cell Death Dis. 11 (2020) 661. 10.1038/s41419-020-02864-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Parks RJ, Menazza S, Holmström KM, Amanakis G, Fergusson M, Ma H, Aponte AM, Bernardi P, Finkel T, Murphy E, Cyclophilin D-mediated regulation of the permeability transition pore is altered in mice lacking the mitochondrial calcium uniporter, Cardiovasc Res. 115 (2019) 385–394. 10.1093/cvr/cvy218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Ghosh JC, Siegelin MD, Vaira V, Faversani A, Tavecchio M, Chae YC, Lisanti S, Rampini P, Giroda M, Caino MC, Seo JH, Kossenkov A. v., Michalek RD, Schultz DC, Bosari S, Languino LR, Altieri DC, Adaptive Mitochondrial Reprogramming and Resistance to PI3K Therapy, JNCI: Journal of the National Cancer Institute. 107 (2015). 10.1093/jnci/dju502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Rasola A, Sciacovelli M, Chiara F, Pantic B, Brusilow WS, Bernardi P, Activation of mitochondrial ERK protects cancer cells from death through inhibition of the permeability transition, Proceedings of the National Academy of Sciences. 107 (2010) 726–731. 10.1073/pnas.0912742107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Linard D, Kandlbinder A, Degand H, Morsomme P, Dietz K-J, Knoops B, Redox characterization of human cyclophilin D: Identification of a new mammalian mitochondrial redox sensor?, Arch Biochem Biophys. 491 (2009) 39–45. 10.1016/j.abb.2009.09.002. [DOI] [PubMed] [Google Scholar]
- [78].Halestrap AP, Woodfield K-Y, Connern CP, Oxidative Stress, Thiol Reagents, and Membrane Potential Modulate the Mitochondrial Permeability Transition by Affecting Nucleotide Binding to the Adenine Nucleotide Translocase, Journal of Biological Chemistry. 272 (1997) 3346–3354. 10.1074/jbc.272.6.3346. [DOI] [PubMed] [Google Scholar]
- [79].Halestrap AP, Davidson AM, Inhibition of Ca2+-induced large-amplitude swelling of liver and heart mitochondria by cyclosporin is probably caused by the inhibitor binding to mitochondrial-matrix peptidyl-prolyl cis-trans isomerase and preventing it interacting with the adenine nucleotide translocase, Biochemical Journal. 268 (1990) 153–160. 10.1042/bj2680153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Hendersoni PJF, Lardy HA, Bongkrekic Acid an Inhibitor of the Adenine Nucleotide Translocase of Mitochondria, n.d. [PubMed] [Google Scholar]
- [81].Malekova L, Kominkova V, Ferko M, Stefanik P, Krizanova O, Ziegelhöffer A, Szewczyk A, Ondrias K, Bongkrekic acid and atractyloside inhibits chloride channels from mitochondrial membranes of rat heart, Biochimica et Biophysica Acta (BBA) - Bioenergetics. 1767 (2007) 31–44. 10.1016/j.bbabio.2006.10.004. [DOI] [PubMed] [Google Scholar]
- [82].Halestrap A, Brenner C, The Adenine Nucleotide Translocase: A Central Component of the Mitochondrial Permeability Transition Pore and Key Player in Cell Death, Curr Med Chem. 10 (2003) 1507–1525. 10.2174/0929867033457278. [DOI] [PubMed] [Google Scholar]
- [83].Dierks T, Salentin A, Krämer R, Pore-like and carrier-like properties of the mitochondrial aspartate/glutamate carrier after modification by SH-reagents: evidence for a preformed channel as a structural requirement of carrier-mediated transport, Biochimica et Biophysica Acta (BBA) - Biomembranes. 1028 (1990) 281–288. 10.1016/0005-2736(90)90177-P. [DOI] [PubMed] [Google Scholar]
- [84].Dierks T, Salentin A, Heberger C, Krämer R, The mitochondrial aspartate/glutamate and ADP/ATP carrier switch from obligate counterexchange to unidirectional transport after modification by SH-reagents, Biochimica et Biophysica Acta (BBA) - Biomembranes. 1028 (1990) 268–280. 10.1016/0005-2736(90)90176-O. [DOI] [PubMed] [Google Scholar]
- [85].Kokoszka JE, Waymire KG, Levy SE, Sligh JE, Cai J, Jones DP, MacGregor GR, Wallace DC, The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore, Nature. 427 (2004) 461–465. 10.1038/nature02229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Karch J, Bround MJ, Khalil H, Sargent MA, Latchman N, Terada N, Peixoto PM, Molkentin JD, Inhibition of mitochondrial permeability transition by deletion of the ANT family and CypD, Sci Adv. 5 (2019). 10.1126/sciadv.aaw4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Giorgio V, von Stockum S, Antoniel M, Fabbro A, Fogolari F, Forte M, Glick GD, Petronilli V, Zoratti M, Szabó I, Lippe G, Bernardi P, Dimers of mitochondrial ATP synthase form the permeability transition pore, Proceedings of the National Academy of Sciences. 110 (2013) 5887–5892. 10.1073/pnas.1217823110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Alavian KN, Beutner G, Lazrove E, Sacchetti S, Park H-A, Licznerski P, Li H, Nabili P, Hockensmith K, Graham M, Porter GA, Jonas EA, An uncoupling channel within the c-subunit ring of the F 1 F O ATP synthase is the mitochondrial permeability transition pore, Proceedings of the National Academy of Sciences. 111 (2014) 10580–10585. 10.1073/pnas.1401591111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].He J, Carroll J, Ding S, Fearnley IM, Walker JE, Permeability transition in human mitochondria persists in the absence of peripheral stalk subunits of ATP synthase, Proceedings of the National Academy of Sciences. 114 (2017) 9086–9091. 10.1073/pnas.1711201114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Carroll J, He J, Ding S, Fearnley IM, Walker JE, Persistence of the permeability transition pore in human mitochondria devoid of an assembled ATP synthase, Proceedings of the National Academy of Sciences. 116 (2019) 12816–12821. 10.1073/pnas.1904005116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Mnatsakanyan N, Llaguno MC, Yang Y, Yan Y, Weber J, Sigworth FJ, Jonas EA, A mitochondrial megachannel resides in monomeric F1FO ATP synthase, Nat Commun. 10 (2019) 5823. 10.1038/s41467-019-13766-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Chávez E, Rodríguez J, García G, García N, Correa F, Oligomycin strengthens the effect of cyclosporin A on mitochondrial permeability transition by inducing phosphate uptake, Cell Biol Int. 29 (2005) 551–558. 10.1016/j.cellbi.2005.03.009. [DOI] [PubMed] [Google Scholar]
- [93].Kokoszka JE, Waymire KG, Levy SE, Sligh JE, Cai J, Jones DP, MacGregor GR, Wallace DC, The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore, Nature. 427 (2004) 461–465. 10.1038/nature02229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Pérez MJ, Quintanilla RA, Development or disease: duality of the mitochondrial permeability transition pore, Dev Biol. 426 (2017) 1–7. 10.1016/j.ydbio.2017.04.018. [DOI] [PubMed] [Google Scholar]
- [95].Neginskaya MA, Solesio ME, Berezhnaya E. v., Amodeo GF, Mnatsakanyan N, Jonas EA, Pavlov E. v., ATP Synthase C-Subunit-Deficient Mitochondria Have a Small Cyclosporine A-Sensitive Channel, but Lack the Permeability Transition Pore, Cell Rep. 26 (2019) 11–17.e2. 10.1016/j.celrep.2018.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Bücheler K, Adams V, Brdiczka D, Localization of the ATP/ADP translocator in the inner membrane and regulation of contact sites between mitochondrial envelope membranes by ADP. A study on freeze-fractured isolated liver mitochondria, Biochimica et Biophysica Acta (BBA) - Bioenergetics. 1056 (1991) 233–242. 10.1016/S0005-2728(05)80054-4. [DOI] [PubMed] [Google Scholar]
- [97].Brdiczka D, Contact sites between mitochondrial envelope membranes. Structure and function in energy- and protein-transfer, Biochimica et Biophysica Acta (BBA) - Reviews on Biomembranes. 1071 (1991) 291–312. 10.1016/0304-4157(91)90018-R. [DOI] [PubMed] [Google Scholar]
- [98].Szabó I, Zoratti M, The mitochondrial permeability transition pore may comprise VDAC molecules I. Binary structure and voltage dependence of the pore, FEBS Lett. 330 (1993) 201–205. 10.1016/0014-5793(93)80273-W. [DOI] [PubMed] [Google Scholar]
- [99].Szabó I, de Pinto V, Zoratti M, The mitochondrial permeability transition pore may comprise VDAC molecules II. The electrophysiological properties of VDAC are compatible with those of the mitochondrial megachannel, FEBS Lett. 330 (1993) 206–210. 10.1016/0014-5793(93)80274-X. [DOI] [PubMed] [Google Scholar]
- [100].Crompton M, Virji S, Ward JM, Cyclophilin-D binds strongly to complexes of the voltage-dependent anion channel and the adenine nucleotide translocase to form the permeability transition pore, Eur J Biochem. 258 (1998) 729–735. 10.1046/j.1432-1327.1998.2580729.x. [DOI] [PubMed] [Google Scholar]
- [101].Baines CP, Kaiser RA, Sheiko T, Craigen WJ, Molkentin JD, Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death, Nat Cell Biol. 9 (2007) 550–555. 10.1038/ncb1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Shamas-Din A, Kale J, Leber B, Andrews DW, Mechanisms of action of Bcl-2 family proteins., Cold Spring Harb Perspect Biol. 5 (2013) a008714. 10.1101/cshperspect.a008714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Oltval ZN, Milliman CL, Korsmeyer SJ, Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programed cell death, Cell. 74 (1993) 609–619. 10.1016/0092-8674(93)90509-O. [DOI] [PubMed] [Google Scholar]
- [104].Chittenden T, Harrington EA, O’Connor R, Remington C, Lutz RJ, Evan GI, Guild BC, Induction of apoptosis by the Bcl-2 homologue Bak, Nature. 374 (1995) 733–736. 10.1038/374733a0. [DOI] [PubMed] [Google Scholar]
- [105].Tait SWG, Green DR, Mitochondria and cell death: outer membrane permeabilization and beyond, Nat Rev Mol Cell Biol. 11 (2010) 621–632. 10.1038/nrm2952. [DOI] [PubMed] [Google Scholar]
- [106].Walther DM, Rapaport D, Biogenesis of mitochondrial outer membrane proteins, Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 1793 (2009) 42–51. 10.1016/j.bbamcr.2008.04.013. [DOI] [PubMed] [Google Scholar]
- [107].Kalkavan H, Green DR, MOMP, cell suicide as a BCL-2 family business, Cell Death Differ. 25 (2018) 46–55. 10.1038/cdd.2017.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Marzo I, Brenner C, Zamzami N, Jürgensmeier JM, Susin SA, Vieira HLA, Prévost M-C, Xie Z, Matsuyama S, Reed JC, Kroemer G, Bax and Adenine Nucleotide Translocator Cooperate in the Mitochondrial Control of Apoptosis, Science (1979). 281 (1998) 2027–2031. 10.1126/science.281.5385.2027. [DOI] [PubMed] [Google Scholar]
- [109].Narita M, Shimizu S, Ito T, Chittenden T, Lutz RJ, Matsuda H, Tsujimoto Y, Bax interacts with the permeability transition pore to induce permeability transition and cytochrome c release in isolated mitochondria, Proceedings of the National Academy of Sciences. 95 (1998) 14681–14686. 10.1073/pnas.95.25.14681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Karch J, Schips TG, Maliken BD, Brody MJ, Sargent MA, Kanisicak O, Molkentin JD, Autophagic cell death is dependent on lysosomal membrane permeability through Bax and Bak, (2017). 10.7554/eLife.30543.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Karch J, Kanisicak O, Brody MJ, Sargent MA, Michael DM, Molkentin JD, Necroptosis Interfaces with MOMP and the MPTP in Mediating Cell Death, PLoS One. 10 (2015) e0130520. 10.1371/journal.pone.0130520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Karch J, Kwong JQ, Burr AR, Sargent MA, Elrod JW, Peixoto PM, Martinez-Caballero S, Osinska H, Cheng EH-Y, Robbins J, Kinnally KW, Molkentin JD, Bax and Bak function as the outer membrane component of the mitochondrial permeability pore in regulating necrotic cell death in mice, Elife. 2 (2013). 10.7554/eLife.00772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Martens MD, Karch J, Gordon JW, The molecular mosaic of regulated cell death in the cardiovascular system, Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 1868 (2022) 166297. 10.1016/j.bbadis.2021.166297. [DOI] [PubMed] [Google Scholar]
- [114].Patel P, Mendoza A, Robichaux DJ, Wang MC, Wehrens XHT, Karch J, Inhibition of the Anti-Apoptotic Bcl-2 Family by BH3 Mimetics Sensitize the Mitochondrial Permeability Transition Pore Through Bax and Bak, Front Cell Dev Biol. 9 (2021). 10.3389/fcell.2021.765973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Bustamante J, Nutt L, Orrenius S, Gogvadze V, Arsenic stimulates release of cytochrome from isolated mitochondria via induction of mitochondrial permeability transition, Toxicol Appl Pharmacol. 207 (2005) 110–116. 10.1016/j.taap.2005.01.024. [DOI] [PubMed] [Google Scholar]
- [116].Zhang D, Lu C, Whiteman M, Chance B, Armstrong JS, The Mitochondrial Permeability Transition Regulates Cytochrome c Release for Apoptosis during Endoplasmic Reticulum Stress by Remodeling the Cristae Junction, Journal of Biological Chemistry. 283 (2008) 3476–3486. 10.1074/jbc.M707528200. [DOI] [PubMed] [Google Scholar]
- [117].Yu C-H, Davidson S, Harapas CR, Hilton JB, Mlodzianoski MJ, Laohamonthonkul P, Louis C, Low RRJ, Moecking J, de Nardo D, Balka KR, Calleja DJ, Moghaddas F, Ni E, McLean CA, Samson AL, Tyebji S, Tonkin CJ, Bye CR, Turner BJ, Pepin G, Gantier MP, Rogers KL, McArthur K, Crouch PJ, Masters SL, TDP-43 Triggers Mitochondrial DNA Release via mPTP to Activate cGAS/STING in ALS, Cell. 183 (2020) 636–649.e18. 10.1016/j.cell.2020.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Elmore SP, Qian T, Grissom SF, Lemasters JJ, The mitochondrial permeability transition initiates autophagy in rat hepatocytes, The FASEB Journal. 15 (2001) 1–17. 10.1096/fj.01-0206fje. [DOI] [PubMed] [Google Scholar]
- [119].Carreira RS, Lee Y, Ghochani M, Gustafsson ÅB, Gottlieb RA, Cyclophilin D is required for mitochondrial removal by autophagy in cardiac cells, Autophagy. 6 (2010) 462–472. 10.4161/auto.6.4.11553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Karch J, Molkentin JD, Regulated Necrotic Cell Death, Circ Res. 116 (2015) 1800–1809. 10.1161/CIRCRESAHA.116.305421. [DOI] [PMC free article] [PubMed] [Google Scholar]