

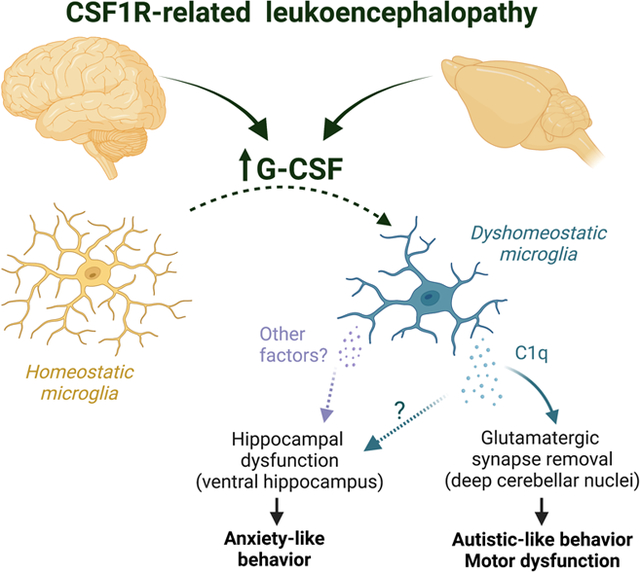
Abstract
Colony stimulating factor (CSF) receptor-1 (CSF-1R)-related leukoencephalopathy (CRL) is an adult-onset, demyelinating and neurodegenerative disease caused by autosomal dominant mutations in CSF1R, modeled by the Csf1r+/− mouse. The expression of Csf2, encoding granulocyte-macrophage CSF (GM-CSF) and of Csf3, encoding granulocyte CSF (G-CSF), are elevated in both mouse and human CRL brains. While monoallelic targeting of Csf2 has been shown to attenuate many behavioral and histological deficits of Csf1r+/− mice, including cognitive dysfunction and demyelination, the contribution of Csf3 has not been explored. In the present study, we investigate the behavioral, electrophysiological and histopathological phenotypes of Csf1r+/− mice following monoallelic targeting of Csf3. We show that Csf3 heterozygosity normalized the Csf3 levels in Csf1r+/− mouse brains and ameliorated anxiety-like behavior, motor coordination and social interaction deficits, but not the cognitive impairment of Csf1r+/− mice. Csf3 heterozygosity failed to prevent callosal demyelination. However, consistent with its effects on behavior, Csf3 heterozygosity normalized microglial morphology in the cerebellum and in the ventral, but not in the dorsal hippocampus. Csf1r+/− mice exhibited altered firing activity in the deep cerebellar nuclei (DCN) associated with increased engulfment of glutamatergic synapses by DCN microglia and increased deposition of the complement factor C1q on glutamatergic synapses. These phenotypes were significantly ameliorated by monoallelic deletion of Csf3. Our current and earlier findings indicate that G-CSF and GM-CSF play largely non-overlapping roles in CRL-like disease development in Csf1r+/− mice.
Keywords: CSF-1 receptor, G-CSF, CRL, HDLS, microglia, leukoencephalopathy, cerebellum
Graphical Abstract
1. INTRODUCTION
CSF1R-related leukoencephalopathy (CRL), previously named adult-onset leukoencephalopathy with axonal spheroids and pigmented glia (ALSP), pigmentary orthochromatic leukodystrophy (POLD) and hereditary diffuse leukoencephalopathy with spheroids (HDLS), is a neurodegenerative disease characterized by progressive cognitive impairment, motor coordination deficits, and psychiatric symptoms (Konno, Kasanuki, Ikeuchi, Dickson, & Wszolek, 2018; Nicholson et al., 2013). CRL is caused by autosomal dominant mutations in the colony stimulating factor-1 receptor gene (CSF1R) that inhibit the kinase activity or abolish the expression of the mutant chain (Rademakers et al., 2011). Based on the finding that haploinsufficiency is sufficient for the development of CRL in humans (Konno et al., 2014; Leng et al., 2019; Miura et al., 2018), we have validated the Csf1r+/− mouse as a model of CRL that reproduces the neurocognitive deficits and histopathological features of the human disease (reviewed in (Chitu, Gokhan, & Stanley, 2021)).
Recent studies indicate that CRL is a primary microgliopathy. Quantitative transcriptomic profiling of autopsied brain samples from patients with CRL revealed the loss of homeostatic microglia (Kempthorne et al., 2020a, 2020b). Genetic studies in mouse showed that Csf1r heterozygosity in Cx3Cr1-expressing mononuclear phagocytes, including microglia, was sufficient to reproduce all aspects of disease produced by global Csf1r+/− heterozygosity (Biundo et al., 2021). Furthermore, consistent with a central role of microglia in disease development, there is no evidence of increased monocytic infiltration in the brains of aged, symptomatic Csf1r+/− mice (Chitu et al., 2020).
In a screen for inflammatory cytokines, chemokines and receptors that could contribute to disease, we found that the mRNAs for Csf2, encoding granulocyte-macrophage CSF (GM-CSF) and for Csf3, encoding granulocyte-CSF (G-CSF), were uniquely elevated in the brains of both presymptomatic and symptomatic Csf1r+/− mice (Chitu et al., 2015). The expression of transcripts for both cytokines is barely detectable in brains at steady-state. However, they can be rapidly induced by a variety of inflammatory stimuli, tissue injury and neurotoxins and signal in microglia to promote functional changes (reviewed in (Chitu, Biundo, & Stanley, 2021)). GM-CSF is a microglial mitogen (Schermer & Humpel, 2002; Xiao, Xu, & Yang, 2002) and promotes a demyelinating phenotype (Smith, 1993), while G-CSF induces a pro-oxidant phenotype (Basso et al., 2017). CSF2 expression is elevated in CRL patient brains (Chitu et al., 2020), which also exhibit gene expression changes consistent with altered G-CSF signaling (Kempthorne et al., 2020a, 2020b). Together, these data suggest that both GM-CSF and G-CSF may contribute to the development of this disease.
In a previous study we showed that GM-CSF contributes to many of the behavioral and histological deficits of Csf1r+/− mice, including microglial expansion and demyelination (Chitu et al., 2020). In the present study, we have explored the role of G-CSF in Csf1r+/− mouse pathology. A conditional G-CSF receptor (Csf3r)-deficient mouse is not available. However, because in the mouse brain, Csf3r expression is almost exclusively found in microglia (Kozareva et al., 2021; Ximerakis et al., 2019), the genetic targeting of the ligand allows the investigation of the G-CSF/G-CSF receptor signaling on microglia function in a similar manner to the lineage-specfic targeting of the receptor. We chose to utilize Csf3 heterozygosity instead of Csf3 deficiency because: 1) Csf3 heterozygosity was sufficient to normalize the level of Csf3 transcripts in the Csf1r+/− mouse brain and 2) in contrast to neutropenic Csf3−/− mice, adult Csf3+/− mice do not exhibit any alterations in peripheral blood parameters (Lieschke et al., 1994) that could potentially confound the interpretation of the data.
We show that Csf3 heterozygosity attenuates the anxiety-like behavior, motor coordination and social novelty deficits of Csf1r+/− mice, but fails to prevent the cognitive deficits, and demyelination. Consistent with these effects, monoallelic targeting of Csf3 reduced microglial morphological changes in the cerebellum and ventral hippocampus, two brain regions involved in motor coordination and anxiety, respectively (Darmohray, Jacobs, Marques, & Carey, 2019; Parfitt et al., 2017), but not in the dorsal hippocampus, or corpus callosum. Csf1r+/− mice exhibited altered electrophysiological responses in the deep cerebellar nuclei (DCN) that were associated with increased expression and deposition of the C1q factor of the complement cascade on glutamatergic synapses and their increased engulfment by DCN microglia. All these phenotypes were attenuated in Csf1r+/−; Csf3+/− mice. Together, our data suggest that in the Csf1r+/− mouse model of CRL, increased G-CSF promotes anxiety and cerebellar dysfunction by activating discrete populations of microglia.
2. MATERIALS AND METHODS
2.1. Ethics statement
All in vivo experiments were performed in accordance with the National Institutes of Health regulations on the care and use of experimental animals and approved by the Institutional Animal Care and Use Committees of Albert Einstein College of Medicine and Hunter College.
2.2. Mouse strains, breeding, and maintenance
Experiments were performed on adult C57BL/6J mice (RRID: IMSR JAX:000664) of the indicated ages and sexes. The generation, maintenance and genotyping of Csf1r+/− mice was described previously (Dai et al., 2002). Csf3+/− mice on a mixed C57BL/6 × 129/Ola genetic background (Lieschke et al., 1994) were obtained from Jackson Laboratories and genotyped by PCR utilizing the following primers: Csf3-Fw (5’- GCACCCTCAGTATCCTTCCA-3’), Csf3-Rev (5’- GCTAGAGCAGCCACTCAGG -3’) and Csf3-Neo (5’-GCTATCAGGACATAGCGTTGG-3’) specific for the neomycin resistance gene. Both lines were backcrossed for more than 10 generations onto the C57BL6/J background. Cohorts were developed from the progeny of matings of Csf1r+/− to Csf3+/− mice, randomized with respect to the litter of origin and maintained on a breeder (PicoLab Rodent Diet 20 5058) rather than a maintenance diet, in order to accelerate symptom development (Chitu, Gokhan, et al., 2021). The age and sex of mice used in each experiment are indicated in the figures.
2.3. Behavioral studies
Male and female mice were tested sequentially for memory, anxiety, motor coordination and social interaction. Data were analyzed for males and females. When similar trends were observed for both sexes the data were pooled. A different cohort was used for active place avoidance. All the experiments were conducted by a blinded experimenter during the light cycle. The animals were allowed to acclimate to the behavior room for one hour before the beginning of each experiment. For each experimental paradigm, mice were randomized and balanced to avoid unwanted effects of confounding factors.
Cognitive flexibility
Cognitive flexibility was evaluated in the active place avoidance paradigm (Burghardt, Park, Hen, & Fenton, 2012) at 7 months of age. The apparatus consisted of a circular 40 cm diameter platform rotating clockwise at 1 rpm. A camera placed above the apparatus recorded the mouse location during each stage of the experiment. The apparatus was controlled by PC-based software (Tracker, Bio-Signal Group Corp., Brooklyn, NY) that tracked the mouse position and delivered a foot shock (500 ms, 60 Hz, 0.2 mA) every time the mouse was inside a 60° stationary shock zone that could be identified by visual cues within the room. The number of entrances into the shock zone was recorded throughout the duration of the experiment.
The task included four steps:
Habituation. Mice were allowed to freely explore the apparatus in the absence of shock for 30 minutes.
Training. For three consecutive days, mice were placed on the apparatus with the shock turned on. Each day, mice were trained with a single 30-minute trial to avoid one shock zone. The location of the shock zone was constant across trials.
Long-term memory test. Mice were returned to the apparatus three days after the last training day with the shock turned on. During this 10-minute trial, the shock zone remained in the same location as the previous training trials.
Cognitive flexibility test. Two hours after the long-term memory test, the location of the shock zone was moved to the opposite side of the arena, which is where mice primarily spent their time on the previous trials. The number of entrances into the new shock zone was recorded over a 20 min period.
Spatial memory
Short-term memory. Mice were assessed at 11.5 months of age for short-term spatial memory in the two-stage version of the Y-maze (Biundo, Ishiwari, Del Prete, & D’Adamio, 2015). In the first training stage, each mouse was introduced into the Y-maze and allowed to explore two of the three arms of the apparatus. In the second testing stage, conducted one hour later, the remaining arm was opened and the mouse returned to the apparatus to freely explore all the three arms. Internal visual cues were placed inside each arm as referential tools to explore the maze. The number of arm entries into each arm was tracked and recorded by Any-maze (Stoelting). The positions of the three arms were randomized within each genotype.
Long-term memory. Long-term spatial recognition memory was evaluated in 11.5-old month mice using the object placement test. Each mouse was allowed to interact with two identical objects placed 10 cm apart parallel to one of the walls of a 40 cm × 40 cm chamber for 10 minutes (training). After 24 hours, one of the objects was displaced into a novel position (15 cm distant, 90° angled) and the mouse returned to explore the objects for 10 minutes (testing). Visual cues were affixed to the walls of the chamber to assist orientation within the arena. Time interacting with the objects was tracked by a blinded experimenter. The discriminatory ratio, calculated as percent time spent exploring the displaced object, was used as an index of preferential interaction with the displaced object.
Anxiety-like behavior
Anxiety was measured by using the elevated zero maze (Ugo Basile Instruments) at the age of 12 months. The apparatus consisted of an elevated ring-shaped apparatus (diameter 50 cm, width 5 cm) including two opposite open zones, and two opposite enclosed zones. Each mouse was allowed to explore the apparatus for 3 minutes. Cumulative time spent in the open zones was tracked by ANY-maze software (ANY-maze, Stoelting), and utilized as a measure inversely related to anxiety (Biundo, Ishiwari, Del Prete, & D’Adamio, 2016).
Motor coordination
Motor coordination was tested in the balance beam test (Gulinello, Chen, & Dobrenis, 2008). The balance beam consisted of a 1-meter-long wooden beam (1.6 cm in diameter) elevated 50 cm above the floor. Each mouse was positioned at one end of the beam and encouraged to cross the beam. The presence of palatable food placed at the opposite end was used as reinforcement to accomplish the task. The number of slips tracked by the experimenter were used as measure of motor coordination.
Social interaction
Sociability was tested in 10–18-month-old mice using the three-chamber sociability test (Kana et al., 2019). This paradigm is based on the natural tendency of mice to preferentially interact with other mice rather than with an inanimate object (social preference), and with a novel mouse rather than with a familiar mouse (social novelty). The three-chamber apparatus consisted of a white Plexiglas box (60 × 40 × 15 cm) divided into three chambers (20 × 40 × 15 cm) by two transparent Plexiglas walls (40 × 15 cm). Entry from the middle chamber to each lateral chamber was made accessible by removable sliding doors (9 × 5.5 cm) (Fig. 5a). The experiment consisted of three 10-minute consecutive stages: habituation, social preference test and social novelty test. Before the start of each stage, the experimental mouse was confined to the middle chamber by the dividing doors. In the habituation, each mouse was allowed to explore the whole empty apparatus. In the social preference test, the test mouse was exposed to an object (a plastic black cube) and to mouse 1 (subsequently designated familiar mouse). After 10 min, the object was replaced by another mouse (novel mouse 2), and the experimental mouse allowed to explore the apparatus and interact with both mice (1 and 2) in the social novelty test. The object, the familiar mouse 1 and the novel mouse 2 were placed under wire mesh pen cups (11.5 cm high, 9.5 cm in diameter) when introduced into the apparatus. The time interacting with each object or mouse was recorded by ANY-maze video tracking system (ANY-maze, Stoelting).
Figure 5. Csf3 heterozygosity rescues the altered firing of deep cerebellar nuclei (DCN) cells in Csf1r+/− mice.
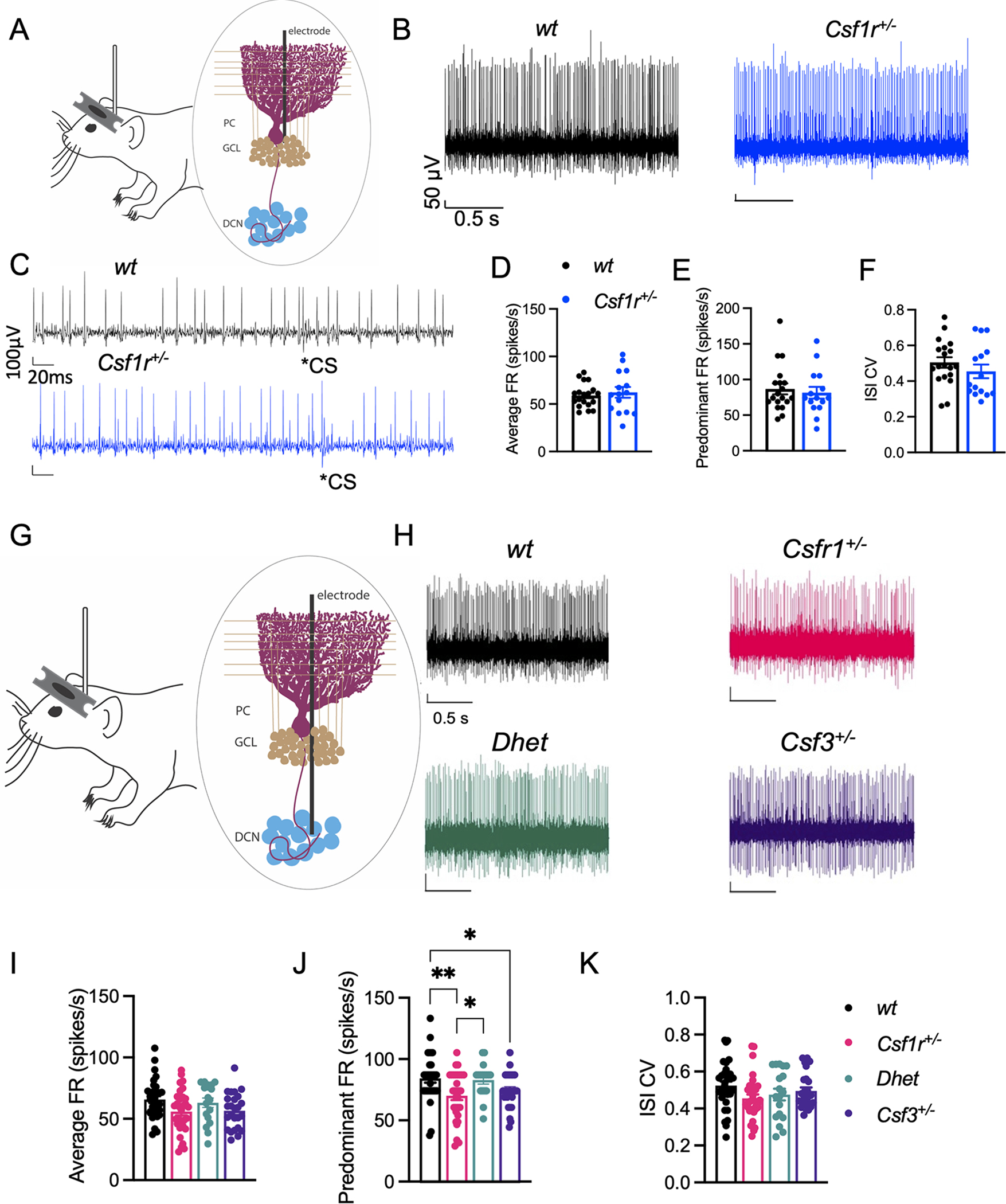
(A-K) 16–22-month-old mice. (A) Schematic of awake head-restrained in vivo single unit electrophysiological recording of cerebellar Purkinje cell (PC) activity. (B) Example recordings of PCs, with (C) examples of complex spikes, from wt and Csf1r+/− mice. (D-F) Quantification of average firing rate (FR) (D), predominant FR (E) and inter-spike interval coefficient of variation (ISI CV) (F) of sorted single units from wt (n = 19 cells, 3 mice) and Csf1r+/− (15 cells, 4 mice). (G) Schematic of in vivo single unit electrophysiological recording of DCN cell activity. (H) Example recordings of DCN cells from wt, Csf1r+/−, Dhet and Csf3+/− mice. (I-K) Quantification of FR (I), predominant FR (J) and ISI CV (K) of sorted single units/cells from wt (n = 32 cells, 5 mice), Csf1r+/− (31 cells, 4 mice), Dhet (17 cells, 3 mice) and Csf3+/− (25 cells, 4 mice). Means ± SEM, significantly different changes are marked by asterisks. *, p < 0.05; **, p < 0.01 (Fisher’s).
2.4. Human studies
Frozen brain tissue blocks containing periventricular white and grey matter were obtained from the Mayo Clinic Brain Bank. Consent for autopsy was obtained from the legal next-of-kin. Information on the CRL patients harboring CSF1R mutations and control cases included in this study is summarized in Supplemental Table S1. Upon removal from the skull according to standard autopsy pathology practices, the brain was divided in the mid-sagittal plane. Half was fixed in 10% neutral buffered formalin, and half was frozen in a −80°C freezer, face down to avoid distortion. The frozen brain was shipped on dry ice to the Neuropathology Laboratory at Mayo Clinic, where it was stored in a −80°C freezer. Frozen tissue was partially thawed before dissection and slabbed in a coronal plane at about 1-cm thickness. Tissue blocks containing the regions of interest were dissected from the frozen slabs and shipped to the research laboratory on dry ice. The area from which the tissue was dissected is indicated in Figure 1A. At all steps, the fresh and frozen tissue was handled with Universal Precautions.
Figure 1. Csf3 heterozygosity attenuates anxiety and motor coordination deficits in CRL mice but fails to improve cognition.
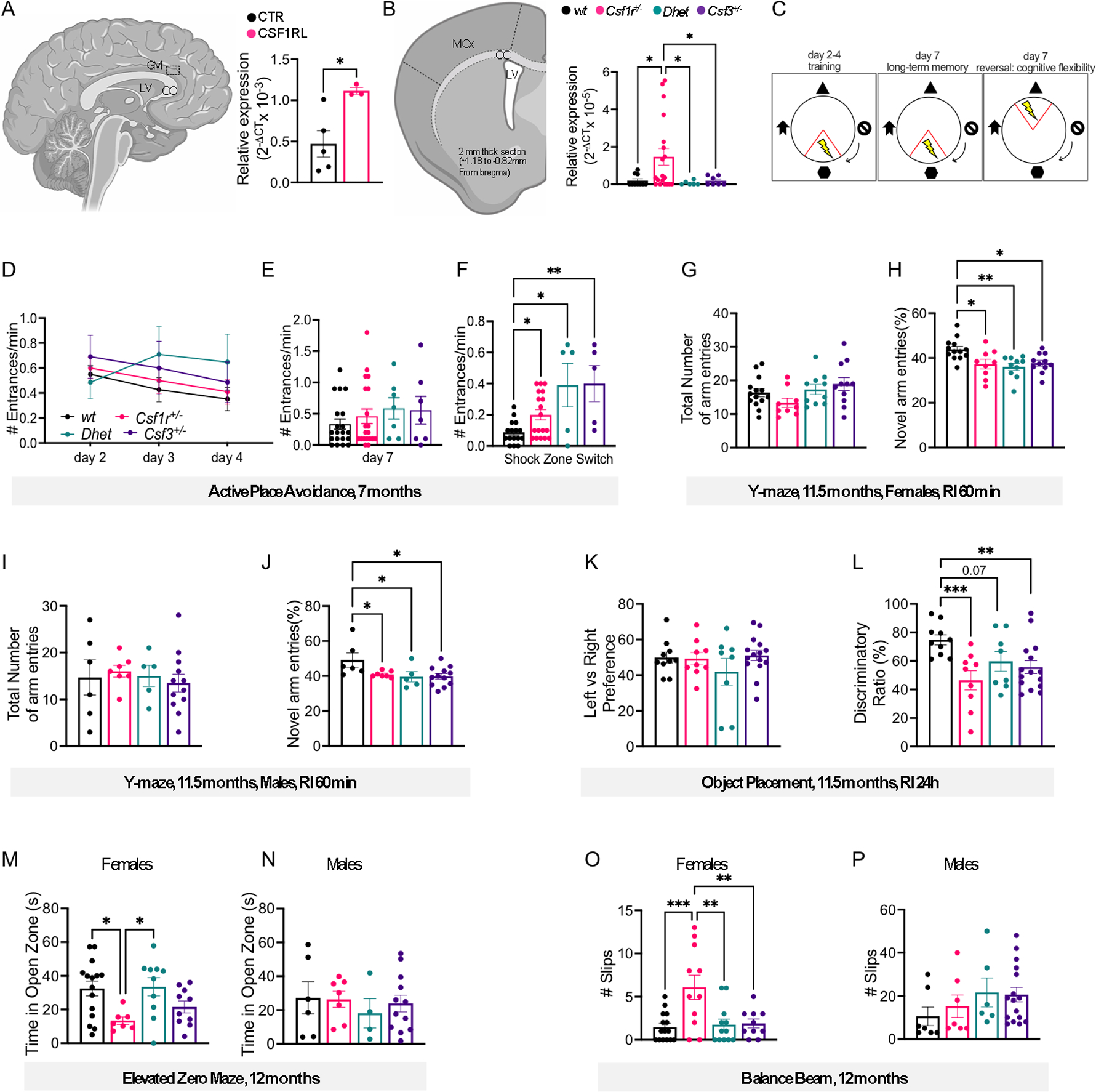
(A) Elevated expression of CSF3 (right panel) in the periventricular grey matter (GM) (left panel) of CRL patients versus healthy controls (Supplemental Table S1) (unpaired t test). LV, lateral ventricle; CC, corpus callosum. (B) Expression of Csf3 (right panel) in the anterior motor cortex (MCx) and corpus callosum (CC) (left panel) of 6-month-old wt and mutant mice (Mann-Whitney test: wt vs Csf1r+/−, Csf1r+/− vs Dhet, Csf1r+/− vs Csf3+/−). The areas marked by dotted lines indicate the region from which RNA was extracted. LV, lateral ventricle. (C-F) Evaluation of cognitive flexibility in 7- month-old mice (females plus males). (C) Schematic of the protocol used for active place avoidance testing. Day 1 (habituation) is not shown. (D) Days 2–4: Training to avoid the initial shock zone location (2-way ANOVA). (E) Evaluation of long-term memory three days after the last training trial. (F) Evaluation of cognitive flexibility after the location of the shock zone was switched at day 7 (E,F; Dunn’s test). (G-J) Assessment of short-term memory at 11.5 months of age in the Y-maze. (G, H), females; (I, J), males. (G, I) Comparable total exploratory activity among genotypes. (H, J) Exploratory preference for the novel arm (G-J; Tukey’s). (K, L) Assessment of long-term memory in the object placement test (females plus males). (K) Ratio of the time exploring the left vs right position of the objects during training. (L) Discriminatory ratio of the time exploring the displaced vs the non-displaced object during testing (K, L; Fisher’s LSD). (M, N) Assessment of anxiety-like behavior for females (M) and males (N) in the elevated zero maze (Bonferroni’s). (O, P) Measurement of motor coordination on the balance beam (Fisher’s). Means ± SEM, significantly different changes are marked by asterisks. Each point in the bar graphs represents an individual patient or mouse. *, p < 0.05; **, p < 0.01; ***, p < 0.001, ****, p < 0.0001. Absence of asterisks in this and all subsequent figures indicates p > 0.05 with reference to the specified post-hoc tests. The post-hoc test used in each panel is indicated in parenthesis in the corresponding description. RI, retention interval.
2.5. Gene expression in CRL patients and mouse brains
RNA was isolated from two 1.5×0.5×0.5cm gray matter fragments of tissue obtained from 3 CRL patients and 5 control patients (see Supplemental Table S1) using Trizol. cDNA was prepared using a Super Script III First Strand Synthesis kit (Invitrogen, Carlsbad, CA). Real time PCR was performed using the PrimePCR CSF3 assay qHsaCED0043218 from BIO-RAD. Human RPL13 (Fw: 5’-AGCCTACAAGAAAGTTTGCCTAT-3’; Rev: 5’-TCTTCTTCCGGTAGTGGATCTTGGC-3’) was used for normalization. Average values from two different blocks of tissue per patient, were used to construct the figure.
For mouse studies, the RNA was extracted from the anterior motor cortex, corpus callosum or cerebellum of mice as described (Chitu et al., 2015), reverse-transcribed as described above and the qPCR was carried out utilizing SYBR Green in an Eppendorf Realplex II thermocycler. Beta actin was used as a housekeeping gene control. The primers for mouse genes used were as follows: Csf3 (Fw: 5’-GAGCAGTTGTGTGCCACCTA-3’; Rev: 5’-GCTTAGGCACTGTGT CTGCTG-3’), C1qa (Fw: 5’-GGATGGGGCTCCAGGAAATC- 3’; Rev: 5’- CTGATA TTGCCTGGATTGCC- 3’), C1qb (Fw: 5’-TGGCTCTGATGGCCAACCAG-3’; Rev: 5’-GACTTTCTGTGTAGCCCCGT-3’), C1qc (Fw: 5’-AGGACGGGCATGATGGACTC- 3’; Rev: 5’-TGAATACCGACTGGTGCTTC-3’), C3 (Fw: 5’-CGCAACGAACAGGTGGAGATCA- 3’; Rev: 5’-CTGGAAGTAGCGATTCTTGGCG-3’), Itgam (Fw: 5’-CTGAGACTGGAGGCAACCAT-3’; Rev: 5’- GATATCTCCTTCGCGCAGAC-3’), Itgb2 (Fw: 5’-CCCAGGAATGCACCAAGTACA-3’; Rev: 5’- CAGTGAAGTTCAGCTTCTGGCA- 3’), Itgax (Fw: 5’- CTGGATAGCCTTTCTTCTGCTG- 3’; Rev: 5’- GCACACTGTGTCCGAACTCA-3’), Nptx1 (Fw: 5’-ATCACCCCATCAAACCACAG-3’; Rev: 5’ CGATGACATTGCCAGAGAGA-3’), Nptx2 (Fw: 5’-CGGAGCTGGAAGATGAGAAG-3’; Rev: 5’- GGAAGGGACACTTTGAATGC-3’), Hprt (Fw: 5’-CAAACTTTGCTTTCCCTGGT-3’; Rev: CAAGGGCATATCCAACAACA), Actb (Fw: 5’- AGAGGGAAATCGTGCGTGAC-3’; Rev: 5’-CAATAGTGATGACCTGGCCGT-3’).
2.6. Immunofluorescence staining and data analysis
Immunostaining was performed in brain slices prepared as described previously (Biundo et al., 2021). Unless otherwise stated, staining and quantifications were carried out on one section per mouse. To ensure consistency and to avoid bias, tissue sections were chosen from matched anatomical regions. Brain sections were incubated with primary antibodies overnight at 4°C. The primary antibodies used in the study included: Iba1 (1:500) (rabbit IgG; Wako Chemicals RRID: AB_839504 or goat IgG; Abcam RRID:AB_10972670); Calbindin, (1: 500) (mouse IgG, Abcam RRID:AB_1658451); NeuN (1:500) (mouse IgG, Millipore RRID:AB_2149209); GAD67 (1:500) (mouse IgG, Millipore RRID:AB_94905); VGLUT2 (1:500) (polyclonal guinea pig antiserum, Synaptic Systems RRID:AB_887884). Following incubation with primary antibodies, the sections were incubated with secondary antibodies conjugated to either Alexa 488, Alexa 594, or Alexa 647 (1:1000) (Life Technologies) for 1 hour at room temperature. Fluoromyelin staining for myelin (1:350, 30 minutes) was performed according to the manufacturer’s (Molecular Probes, Inc.) instructions.
For C1q staining, slices were blocked with 5% bovine serum albumin (BSA) and 0.2% Triton X-100 solution for 1 h and incubated with primary antibody overnight (1:500) (rabbit IgG, Abcam RRID:AB_2732849). After washing, the secondary antibody was applied and incubation continued for 2 hours at room temperature (Lehrman et al., 2018). Sections were mounted on SuperFrost Plus slides (Thermofisher) using Prolong antifade mountant with DAPI (Thermofisher). Images were captured using a Nikon Eclipse TE300 fluorescence microscope with NIS Elements D4.10.01 software. Cell number quantification was performed manually. Quantification of fluorescent areas was performed using ImageJ. Images were cropped and adjusted for brightness, contrast and color balance using Adobe Photoshop CC.
For confocal microscopy, microscope Z series stacks were obtained by a Leica SP8 Confocal microscope at ×40 magnification with a 0.40 μm interval between stacks. Morphometric analysis of microglia (number of end points and length of cell processes) was performed on maximum intensity projections using FIJI as previously described (Chitu et al., 2020; Young & Morrison, 2018). The extent of microglia-Purkinje cell contacts was examined in confocal 3D surface rendering images using Imaris software (Oxford Instruments Group) as described (Kavetsky et al., 2019).
2.7. Synaptic engulfment and complement deposition
Engulfment of synaptic puncta by microglia was evaluated using a published protocol (Schafer et al., 2012). Briefly, for each animal, a sagittal section of DCN was acquired on SP8 confocal at 40X using 0.4 μm z-steps. ImageJ (NIH) was used to subtract background from all fluorescent channels. Each image was subjected to thresholding for each channel and 3D volume renderings were quantified for microglial engulfment of synaptic puncta (VGLUT2+ and GAD67+) using Volocity (Quorum Technologies, Ontario, Canada). To visualize and measure the volume of engulfed puncta, any synaptic fluorescence not merging with Iba1+ signal was subtracted from the image. To determine the percentage of engulfment, the following calculation was performed: Volume of internalized synaptic puncta (μm3) ×100/volume microglial cell (μm3). To evaluate the extent of C1q deposition into VGLUT2 terminals, each image was similarly subjected to thresholding for each channel and assessed for C1q/VGLUT2 colocalization using Volocity. The percentage of C1q deposition was calculated as follows: number of C1q+ VGLUT2+ puncta × 100/total number of VGLUT2+ puncta.
2.8. In vivo electrophysiology
In vivo single unit recording in the cerebellum was performed in awake, head-restrained mice. Surgeries to implant titanium brackets for head-restraint were conducted as follows: mice were anesthetized with isoflurane (5% induction, 2% maintenance) and mounted on a heated stereotactic frame (Kopf Instruments). The head was shaved, wiped with 70% ethanol and Betadine (Purdue Products) and a midline incision made over the skull and skin reflected to reveal bregma and the interparietal bone overlying the cerebellum. The skull was then lightly scraped and cleaned with ethanol. Recording coordinates were marked (see below) by lightly drilling over the interparietal bone and touching the drilled sites with a marker pen. The skull was then covered with OptiBond (Kerr Corporation, Brea, CA, USA) and cured with ultraviolet light. A titanium bracket was subsequently fixed onto the skull with Charisma (Kulzer GmbH, Germany), centered near bregma, such that the bracket sat entirely anteriorly to the lambdoid suture, enabling later access to the cerebellum, and covered with dental cement (M&S Dental Supply, Jamaica, NY, USA). A recording chamber was simultaneously created with dental cement over the interparietal bone. Once the cement hardened, the chamber was covered with Kwik-Sil silicone elastomer (World Precision Instruments, Sarasota, FL, USA). Mice were administered 10 mL/kg of saline and flunixin analgesic (2.5 mg/kg) after surgery and allowed to recover in a warm cage. The mice were monitored post-surgery for 1 week before neural recordings. During this time, the mice were acclimated to head-restraint in the stereotactic recording apparatus using screws to immobilize the previously implanted bracket. This was done for 1–2h per day for at least 3 days. To control for environmental disturbances, the recordings were carried out in a dimly lit, quiet room without significant background noise. Twenty-four hours prior to recording, a craniotomy was created by removing the Kwik-Sil covering the recording chamber and drilling at coordinates previously marked over the interparietal bone. Purkinje cells were recorded at AP: −6.00 mm, ML: 0 mm and AP: −7.0 mm, ML: 0mm; and deep cerebellar nuclei at AP: −6.2 mm, ML: ± 1.5 mm. The recording chamber was recovered with Kwik-Sil and the mouse was allowed to recover.
For single unit recordings, the mouse was head restrained next to a stereotaxic apparatus on a padded flat air table using the head bracket and screws in a dimly lit, quiet room. The Kwik-Sil was removed from the recording chamber, revealing the craniotomies. A ground electrode was then placed into the recording chamber, a tungsten electrode (2–3 MΩ, Thomas Recording, Giessen, Germany) lowered under microscopic guidance into a craniotomy until it touched the surface of the cerebellum, and the chamber filled with saline. In single unit recording, the electrode was further slowly advanced into the cerebellum until neuronal activity was detected but not more than 3 mm below the surface of the brain. Purkinje cell activity was identified by their characteristic firing rate, location and the brief pauses in firing following complex spikes (Figure 5C). DCN cells were identified based on their location and firing. Cells were recorded for 2–5 mins. Neural signals were bandpass filtered 1 Hz-10 kHz and amplified 2000X on a custom amplifier. Signals were then digitized at 20 kHz sampling rate with a National Instruments BNC-2110 (National Instruments, Austin, TX, USA) analog to digital converter into a PC and visualized with LabView (National Instruments, Austin, TX, USA). Waveforms of recorded single unit activity were sorted offline using Offline Sorter software (Plexon, Dallas, TX, USA) and analyzed using a custom LabView script to obtain the average firing rate, predominant firing rate and the interspike interval coefficient of variation (ISI CV). A histogram showing the distribution of interspike intervals is generated for each unit after sorting and smoothened with a 3-sample running average. The interspike interval corresponding to the peak of this smoothened distribution is the predominant ISI and the predominant firing rate is determined by the inverse of the predominant ISI, i.e. 1/predominant ISI. The ISI CV of a unit is the standard deviation of the ISIs divided by the mean of ISIs and is a measure of regularity of firing. Because the predominant firing rates for all the units for each group of animals were not normally distributed, we used non-parametric tests for post-hoc pairwise comparisons.
2.9. Statistical analyses
Statistical analyses were computed using GraphPad Prism 8 (GraphPad, La Jolla, CA). Data were checked for outliers using the Grubbs’ method. Gaussian distribution was evaluated using the Shapiro-Wilk normality test and the Kolmogorov-Smirnov test. The screened data were analyzed using Student’s t-test (Fig. 1A, 1B only), the Kruskal–Wallis test, or by analysis of variance (one- or two-way ANOVA). When significant effects of the independent variables were detected, single differences between or within genotypes were analyzed by post-hoc multiple comparison tests (Dunnett’s, Bonferroni, Tukey’s, Fisher’s LSD, and the two-stage linear step-up procedure of Benjamini, Krieger and Yekutieli as indicated in the figure legends). Unless otherwise stated, each genotype was compared with the other three. The level of significance was set at p < 0.05. For those comparisons in which no statistical significance is indicated in the figure panels, the p value was > 0.05. Data are presented as means ± SEM. Sample sizes for each experiment are indicated in the figure legends.
3. RESULTS
3.1. Csf3 expression is elevated in mouse and human CRL brains and normalized in mice by Csf3 heterozygosity
In a previous study, we observed an increased expression of Csf3 in brains of pre-symptomatic Csf1r+/− mice. The elevation of Csf3 became more pronounced in aged mice exhibiting behavioral deficits, suggesting a role for G-CSF in their CRL-like phenotype (Chitu et al., 2015). To investigate whether CSF3 expression was increased in CRL patients, we quantified the levels of CSF3 mRNA in brains of CRL patients by qPCR. Consistent with the results in the mouse model, the levels of CSF3 transcripts were significantly higher in CRL than in control brains (Fig. 1A). These results prompted us to generate and characterize an experimental cohort of mice, including mice in which either one allele of Csf1r or of Csf3 was deleted (Csf1r+/−, Csf3+/−), compound heterozygotes (Csf1r+/−;Csf3+/−, referred to as Dhet) and wild type (wt) controls. Measurement of Csf3 expression reproduced the previously reported elevated Csf3 mRNA levels observed in Csf1r+/− mice (Chitu et al., 2015) and showed that levels in Dhet mice did not differ significantly from those of wt mice (Fig. 1B).
3.2. Csf3 heterozygosity fails to prevent the cognitive deficits of Csf1r+/− mice but attenuates anxiety-like behavior and the motor coordination deficits
To assess the contribution of the increased Csf3 expression to the behavioral deficits of Csf1r+/− mice, the experimental cohort was evaluated for cognitive flexibility, spatial memory, anxiety, and motor coordination.
Cognitive flexibility, defined as the ability to change and adapt behavior in response to new environmental stimuli (Armbruster, Ueltzhoffer, Basten, & Fiebach, 2012), is one of the executive functions affected in the early stages of Alzheimer’s disease (Albert, 1996) and deficits were also recently reported in a case of CRL (Zur-Wyrozumska, Kaczmarska, & Mensah-Glanowska, 2021). Cognitive flexibility was evaluated in 7-month-old mice using the active place avoidance test (Fig. 1C) (Burghardt et al., 2012). Regardless of genotype, all mice had the same propensity to avoid the shock zone three days after the last training trial (Fig. 1D, E). These data indicate that at this young age, there are no significant long-term memory deficits associated with Csf1r and/or Csf3 heterozygosity. However, when the location of the shock zone was switched, all mice carrying mutations entered the new shock zone significantly more than wt mice, demonstrating that either single or combined Csf1r and Csf3 deficiencies impair cognitive flexibility (Fig. 1F).
The effects of Csf3 heterozygosity on short- and long-term spatial memory were tested in aged (11.5 month-old) mice, in the Y-maze and object placement tasks, respectively (Fig. 1G–L). All mutant mice (Csf1r+/−, Csf3+/− and Dhet) exhibited a short-term memory deficit as shown by their loss of preference for the novel arm of the Y-maze (Fig. 1H, J). The absence of differences in total number of arm entries indicated that the differences in cognitive performance did not result from different propensities among groups to explore the apparatus (Fig. 1G, I).
Similarly, in the object placement test, all mutant mice showed no preference towards exploring the displaced object versus the non-displaced object, indicative of a long-term memory deficit (Fig. 1L). The absence of differences in time exploring the two initial positions of the objects during training indicated that the cognitive deficits detected during testing were not due to a preferential exploration of one of the sides of the chamber (Fig. 1K).
Mice were tested for anxiety-like behavior in the elevated zero maze at the age of 12 months (Fig. 1M, N). The time spent in the open zone of the circular apparatus was used as an index inversely related to anxiety-like behavior. Deficits observed in female Csf1r+/− mice (Fig. 1M) were prevented by Csf3 heterozygosity, while no significant differences among the genotypes were detected in males (Fig. 1N).
Motor coordination was analyzed at the age of 12 months using the balance beam test (Fig. 1O, P). The deficits observed in female Csf1r+/− mice were rescued by single-allele deletion of Csf3 (Fig. 1O), while no significant differences were observed in males (Fig. 1P). Overall, these behavioral studies show that Csf3 heterozygosity fails to prevent the cognitive deficits of Csf1r+/− mice, but significantly attenuates the anxiety and loss of motor coordination observed in females.
3.3. Csf3 heterozygosity attenuates microglial activation in the ventral but not dorsal hippocampus of Csf1r+/− mice
The dorsal hippocampus plays a critical role in cognition while the ventral hippocampus relates to emotions and stress (Fanselow & Dong, 2010). Administration of recombinant G-CSF has been reported to increase the number of microglia and their activation in vivo (Basso et al., 2017; Sanchez-Ramos et al., 2009) and during aging, G-CSF expression is elevated in the ventral, but not the dorsal hippocampus (Porcher et al., 2021). Previous studies have shown that dysregulation of Csf1r signaling in microglia of Csf1r+/− mice leads to low grade microgliosis in multiple regions of the brain, including the dorsal hippocampus (Arreola et al., 2021; Biundo et al., 2021; Chitu et al., 2020). To investigate whether G-CSF regulates microglial changes in the hippocampus, we analyzed microglia densities and morphology in 16-month-old mice (Fig. 2). Iba1 staining revealed that microglial densities were significantly increased in the dorsal hippocampi of Csf1r+/− mice, and that Csf3 heterozygosity failed to attenuate this increase (Fig. 2A - upper panels and Fig. 2B), yet there were no significant differences in microglial densities in the ventral hippocampus (Fig. 2A - lower panels, Fig. 2C). Interestingly, while the branching and length of microglial processes did not vary with genotype in the dorsal hippocampus (Fig. 2D – left panels, Fig. 2E), both the branching and length of microglial processes were decreased in the ventral hippocampus of Csf1r+/− mice (Fig. 2D – right panels, Fig. 2F). Monoallelic deletion of Csf3 restored process branching, albeit it had no significant effect on process length (Fig. 2F). These data are consistent with a contribution of G-CSF to the development of anxiety-like behavior, but not to the cognitive deficits in Csf1r+/− mice through regulation of ventral but not dorsal hippocampus microglia (Fig. 1C–M).
Figure 2. Csf3 heterozygosity reduces microglial morphological alterations in the ventral hippocampus of Csf1r+/− mice.
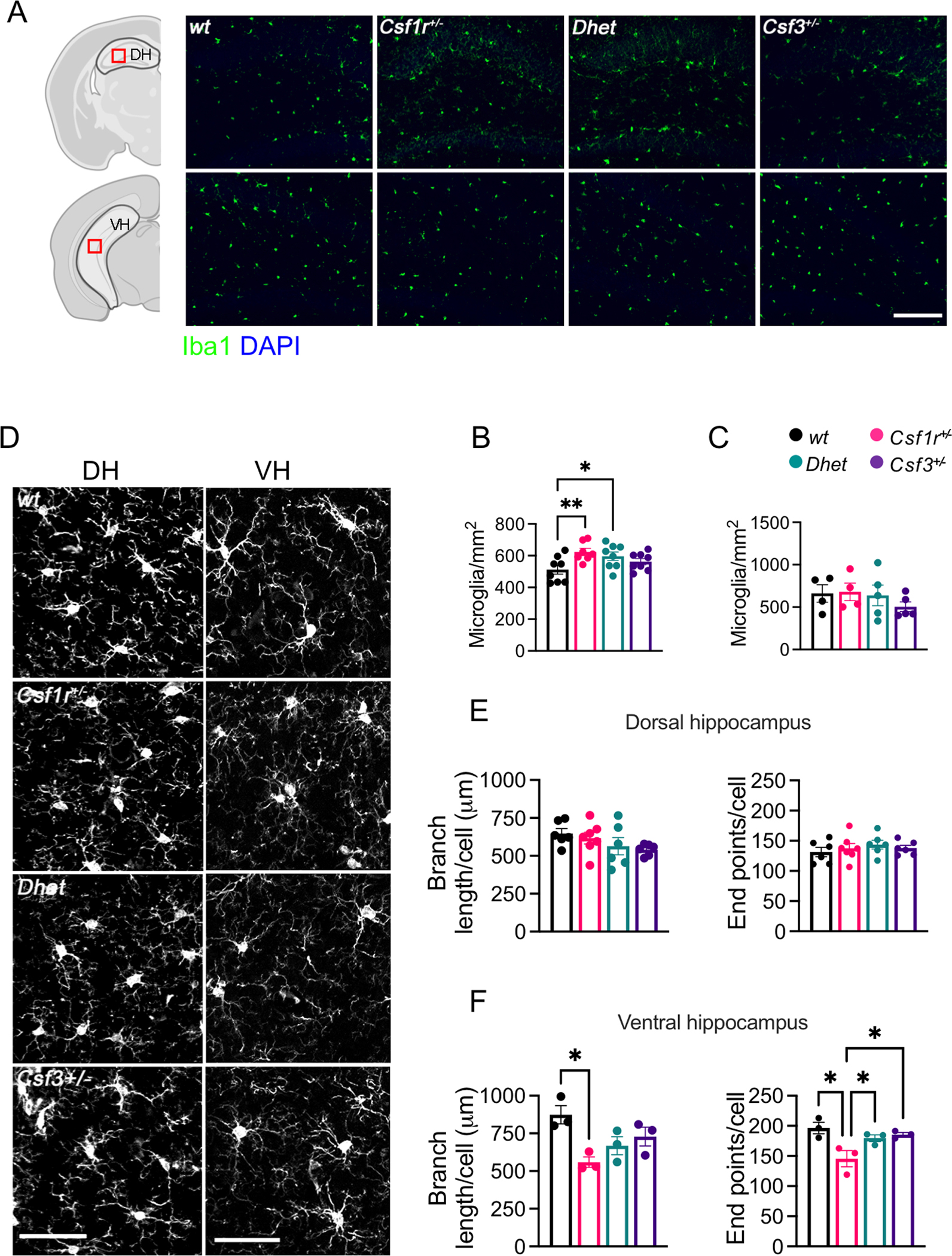
Examination of medial-lateral sagittal sections of dorsal hippocampus (DH) and coronal sections of ventral hippocampus (VH) of 16-month-old female mice. (A) Iba1+ cell densities in the DH and VH (green; scale bar, 100 μm). The areas in which microglia were counted are delineated by the grey outlines (left panels). Red squares indicate the positions of the microscopic fields subjected to morphometric analysis. (B, C) Quantification of microglial densities in the DH (B) and VH (C) (4–8 mice per genotype, 1 section per mouse, Fisher’s LSD). (D) Morphology of the microglial ramifications in the DH and VH (scale bar, 50 μm). (E, F) Quantitation of the ramifications in DH (E) and VH (F) (3–6 mice per genotype, 1 section per mouse, average cell numbers per DH section: wt: 26.83±1.48, Csf1r+/−: 23.43±1.39, Dhet: 26.17±3.56, Csf3+/−: 23.33±0.98 and per VH section: wt: 17.00±1.00, Csf1r+/−: 24.67±4.63, Dhet: 19.00±0.57, Csf3+/−: 19.67±1.86 (two-stage linear step-up procedure of Benjamini, Krieger and Yekutieli). Each point in the bar graphs represents a mouse. Means ± SEM, significantly different changes are marked by asterisks. *, p < 0.05; **, p < 0.01; ****, p < 0.0001.
3.4. Csf3 heterozygosity fails to prevent callosal demyelination and neurodegeneration in the motor cortex of Csf1r+/− mice
Previous studies have shown that dysregulation of CSF-1R signaling in microglia of Csf1r+/− mice promotes callosal demyelination and the loss of layer V neurons in the motor cortex (Chitu et al., 2020). To test whether G-CSF regulates microglial changes in the corpus callosum, we analyzed microglia density and morphology in 16-month-old Csf1r+/− and wt control mice. Consistent with the previously published data (Chitu et al., 2020), analysis of multiple sagittal brain sections detected patches of high microglial density as well as a significant elevation in the total number of microglia in the corpus callosum of Csf1r+/− compared to wt mice (Supplementary Figure S2A, C). Although Csf3 heterozygosity reduced the total number of microglia in the white matter of Csf1r+/− mice, it did not significantly reduce their propensity to cluster (Supplementary Figure S2C, right panel) and failed to prevent the shortening of their processes and the loss of process branching (Supplementary Figure S2D, E). The presence of clusters of activated microglia has recently been correlated with the active clearing of degenerated myelin (Safaiyan et al., 2021). Consistent with its inability to reduce white matter microglia dyshomeostasis, Csf3 heterozygosity also failed to attenuate callosal demyelination (Supplementary Figure S2F, G). Histological evaluation of the motor cortex revealed that Csf3 heterozygosity failed to attenuate microglia expansion in the motor cortex (Supplementary Figure S2H, I), although it attenuated their morphological alteration (Supplementary Figure S2 J, K). Csf3 heterozygosity also failed to prevent the loss of Layer V neurons (Supplementary Figure S2L, M). These data indicate that, although G-CSF may regulate some phenotypic aspects of callosal and cortical microglia, its actions do not contribute significantly to callosal demyelination or to neurodegeneration in the motor cortex.
3.5. Csf3 heterozygosity prevents microglial morphological alterations in the cerebellum
In addition to the corpus callosum and the motor cortex, the cerebellum plays an important role in motor coordination. This prompted us to analyze the impact of Csf3 heterozygosity on microglia density and morphology in the cerebellum. Histological analysis revealed that either Csf1r or Csf3 heterozygosity increased the density of Iba1+ microglia in the cerebellar cortex, while double heterozygosity had no effect, suggesting that balanced actions of Csf1r and Csf3 are required to maintain normal microglial densities in the cerebellar cortex of aged mice. In contrast, no differences in microglia densities were detected in the dorsal protuberance of the medial cerebellar nucleus (MedDL), and the interposed cerebellar nuclei (Int) (Fig. 3B, C). Morphometric analysis revealed morphological alterations of microglia in all the areas of the Csf1r+/− cerebelli, that were prevented by Csf3 monoallelic deletion (Fig. 3D, E and Fig. 3G–I). However, neither Csf1r or Csf3 heterozygosity altered the extent of microglia contacts with the Purkinje cells (Fig. 3D, F). These data indicate that G-CSF contributes to cerebellar microglia dyshomeostasis in Csf1r+/− mice.
Figure 3. Csf3 heterozygosity reduces microglial morphological alterations in the cerebella of 16-month-old female Csf1r+/− mice.
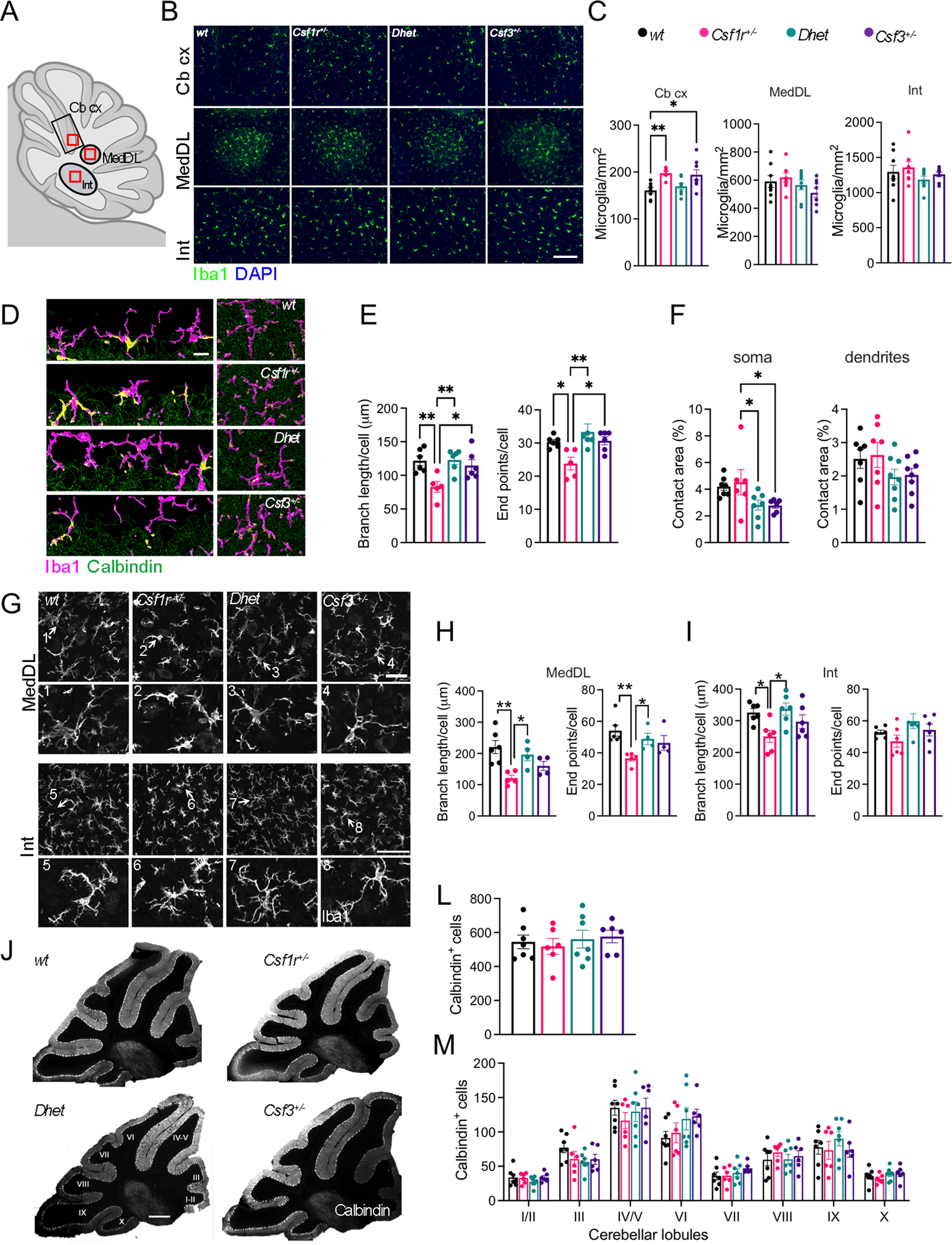
(A) Diagram of a sagittal section of mouse cerebellum. Outlined black regions indicate the areas examined for microglia density analysis. Outlined red squares indicate the subregions selected for morphometric analysis. (B) Iba1+ cell densities (green) in the cerebellar cortex (Cb cx) and in the deep cerebellar nuclei, the dorsal protuberance of the medial cerebellar nucleus (MedDL) and the interposed nucleus (Int) (scale bar, 100 μm). (C) Quantification of data in (B) (6–8 mice per genotype, 1 section per mouse, Bonferroni’s). (D) Imaging of microglia (magenta) and their contacts (yellow) with the Purkinje cell (green) somas (left panels) and dendrites (right panels). Scale bar, 15 μm. (E) Morphometry of microglia in the cerebellar cortex (4–5 mice per genotype, average cell number per Cb cx section: wt: 26.17±4.85, Csf1r+/−: 20.60±3.19, Dhet: 14.67±0.98, Csf3+/−: 14.33±2.15. Dunn’s). (F) Quantification of microglia contact with Purkinje cell somas and dendrites (% of total Calbindin+ area). (G) Morphology of microglia in the deep cerebellar nuclei (scale bars, 50 μm and 70 μm respectively). The arrows and numbers in each upper panel designate the cells shown 2x enlarged in the numbered panels below. (H, I) Morphometry of microglia in the deep cerebellar nuclei (4–5 mice per genotype, average cell number per MedDL section: wt: 25.17±1.17, Csf1r+/−: 24.40±2.11, Dhet: 22.25±2.14, Csf3+/−: 24.50±3.88; cell number per Int section: wt: 20.33±0.98, Csf1r+/−: 16.60±1.16, Dhet: 18.25±1.31, Csf3+/−: 24.00±1.83. Dunn’s, and Tukey’s). (J) Representative images of Calbindin+ Purkinje cells (PC) distributed in the cerebellar lobules (scale bar, 500 μm) (L) Quantification of the total number PC per section and (M) Quantification of the number of Calbindin+ PCs in each lobule (4–5 mice per genotype, 1 section per mouse). Each point in the bar graphs represents a mouse. Means ± SEM, significantly different changes are marked by asterisks. *, p < 0.05; **, p < 0.01.
Deletion of Csf1 in the Nestin+ neural lineage, resulting in CSF1R signaling deficiency in the cerebellum, was associated, not only with alterations of cerebellar microglia homeostasis, but also with decreased Purkinje cell (PC) numbers (Kana et al., 2019). Furthermore, a reduction of PC number was also documented in Csf1op/op mice with global Csf1 deficiency (Nandi et al., 2012). These findings, together with our observation that G-CSF may regulate the function of cerebellar microglia, prompted us to explore how Csf1r and/or Csf3 heterozygosities impact PC number (Fig. 3J–M). Neither the total numbers of Calbindin+ PC cells (Fig. 3L) nor their distribution in each lobule of the cerebellar cortex (Fig. 3M) were significantly different in mice carrying single or compound mutations in Csf1r and Csf3.
Together, these data indicate that, although Csf1r heterozygosity does not cause the loss of Purkinje cells, it promotes a dyshomeostatic phenotype in cerebellar microglia in a G-CSF-dependent manner.
3.6. Csf3 heterozygosity prevents deficits in social behavior
Aside from its role in motor coordination, the cerebellum is involved in regulation of aspects of social behavior (Carta, Chen, Schott, Dorizan, & Khodakhah, 2019). Both direct genetic disruption of Purkinje cell activity (Tsai et al., 2012) and disruption of cerebellar microglia homeostasis in mice with neural-lineage specific deletion of Csf1 (Kana et al., 2019) cause autistic-like behavior manifested as a loss of social novelty preference. This prompted us to investigate how decreased CSF-1R signaling impacts social behavior and to evaluate the contribution of G-CSF. Using the three-chamber sociability paradigm for social preference and social novelty (Fig. 4A), we found that aged Csf1r+/− mice showed clear deficits in social preference (Fig. 4B) and social novelty (Fig. 4C). This phenotype was prevented by monoallelic targeting of Csf3 (Fig. 4B, C). These data suggest that G-CSF may contribute to the development of autistic-like behavior in Csf1r+/− mice by impairing cerebellar function.
Figure 4. Evaluation of social interaction in the three-chamber sociability paradigm.
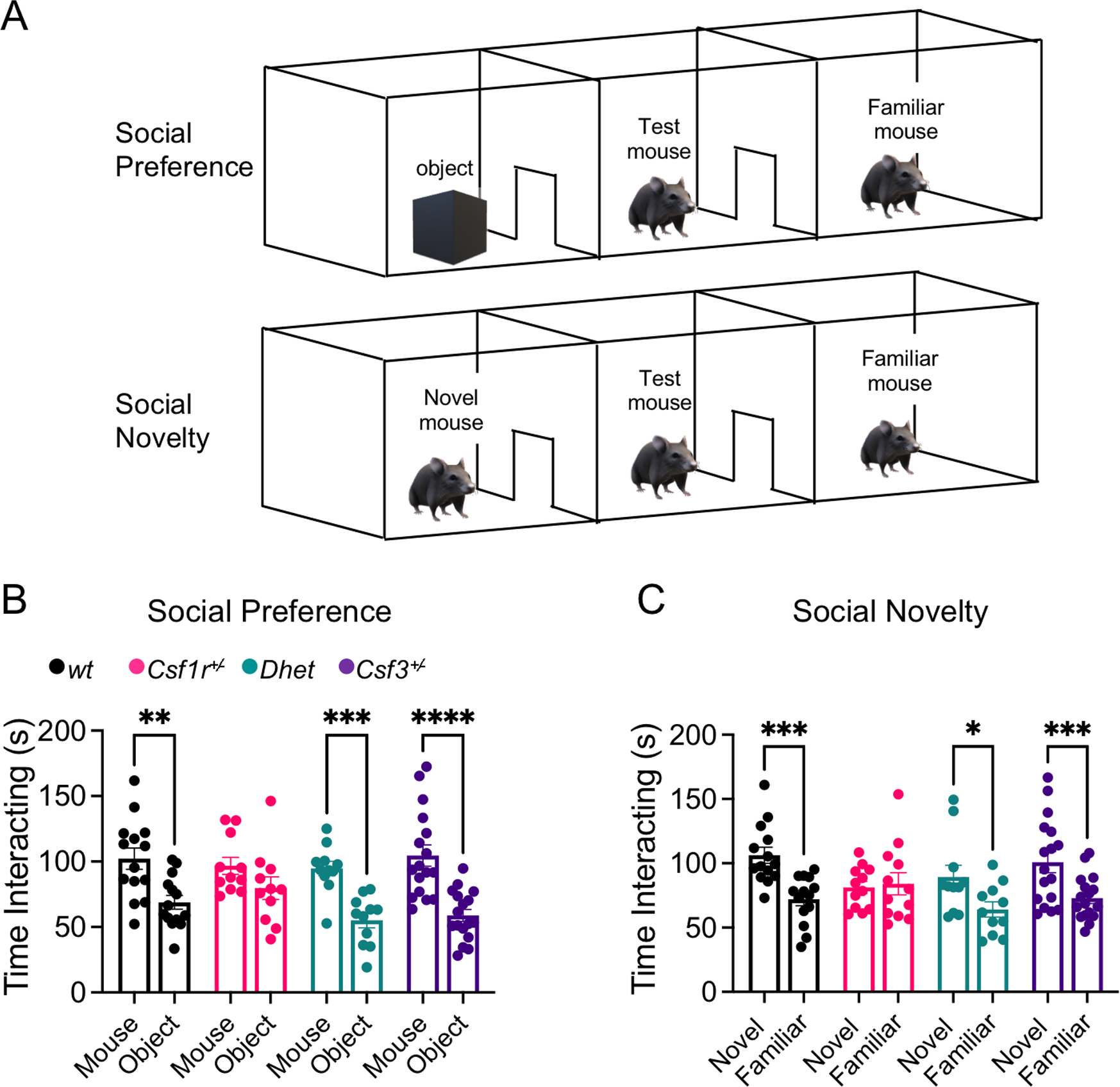
(A) Schematic of the testing apparatus for in the three-chamber sociability paradigm. (B) Social preference. Preferential exploration of mouse compared to object. (C) Social novelty. Preference of novel mouse over familiar mouse. Combined female and male data from 13–16-month-old mice. Means ± SEM, significantly different changes are marked by asterisks. *, p < 0.05; ****, p < 0.0001, Fisher’s).
3.7. Csf3 heterozygosity prevents the development of electrophysiological alterations in the deep cerebellar nuclei of Csf1r+/− mice
The attenuation of cerebellar microglial dyshomeostasis (Fig. 3) together with the correction of the cerebellum-dependent behaviors by Csf3 heterozygosity (Fig. 1O and Fig. 4) suggested that G-CSF may play a role in cerebellar physiology. To test this hypothesis, we analyzed the firing properties of PC and DCN cells in vivo. Electrophysiological recordings of the activity of Purkinje cells in awake, head-restrained mice revealed no difference in average firing rate (FR), predominant FR, or inter-spike interval coefficient of variation (ISI CV) between wt and Csf1r+/− mice (Fig. 5A–F). In contrast, recordings in the DCN revealed a significant decrease in the predominant FR in Csf1r+/− mice that was normalized when one allele of Csf3 was removed (Fig. 5G–K). These data indicate that G-CSF mediates the disruption of DCN firing properties in Csf1r+/− mice.
3.8. Csf3 heterozygosity prevents the excessive elimination of glutamatergic synapses in the deep cerebellar nuclei of Csf1r+/− mice
Single cell transcriptome profiling experiments of mouse cerebelli detect Csf3r transcripts in microglia while the expression in neural lineage cells, including Purkinje cells, is sporadic, at best (Kozareva et al., 2021; Ximerakis et al., 2019). Microglia play a pivotal role in remodeling neuronal networks by pruning or eliminating synapses during development and in adult life (Schafer & Stevens, 2013; Sierra et al., 2010). To investigate whether G-CSF-activated microglia may contribute to aberrant synapse pruning in the DCN of Csf1r+/− mice, we quantified the presence of markers of glutamatergic (VGLUT2+) and of GABAergic (GAD67+) synapses (Baumel, Jacobson, & Cohen, 2009) in DCN microglia (Fig. 6A). While the amount of GAD67+ synaptic material present within Iba1+ microglia was comparable among all genotypes (Fig. 6A, B), Csf1r+/− DCN microglia contained more VGLUT2+ synaptic material (Fig. 6A, C). This phenotype was reversed in Csf1r+/−; Csf3+/− compound heterozygous mice, suggesting that G-CSF stimulates the pruning of glutamatergic synapses by Csf1r+/− microglia in the DCN.
Figure 6. G-CSF mediates excessive complement-mediated engulfment of DCN glutamatergic synapses by microglia in Csf1r+/− mice.
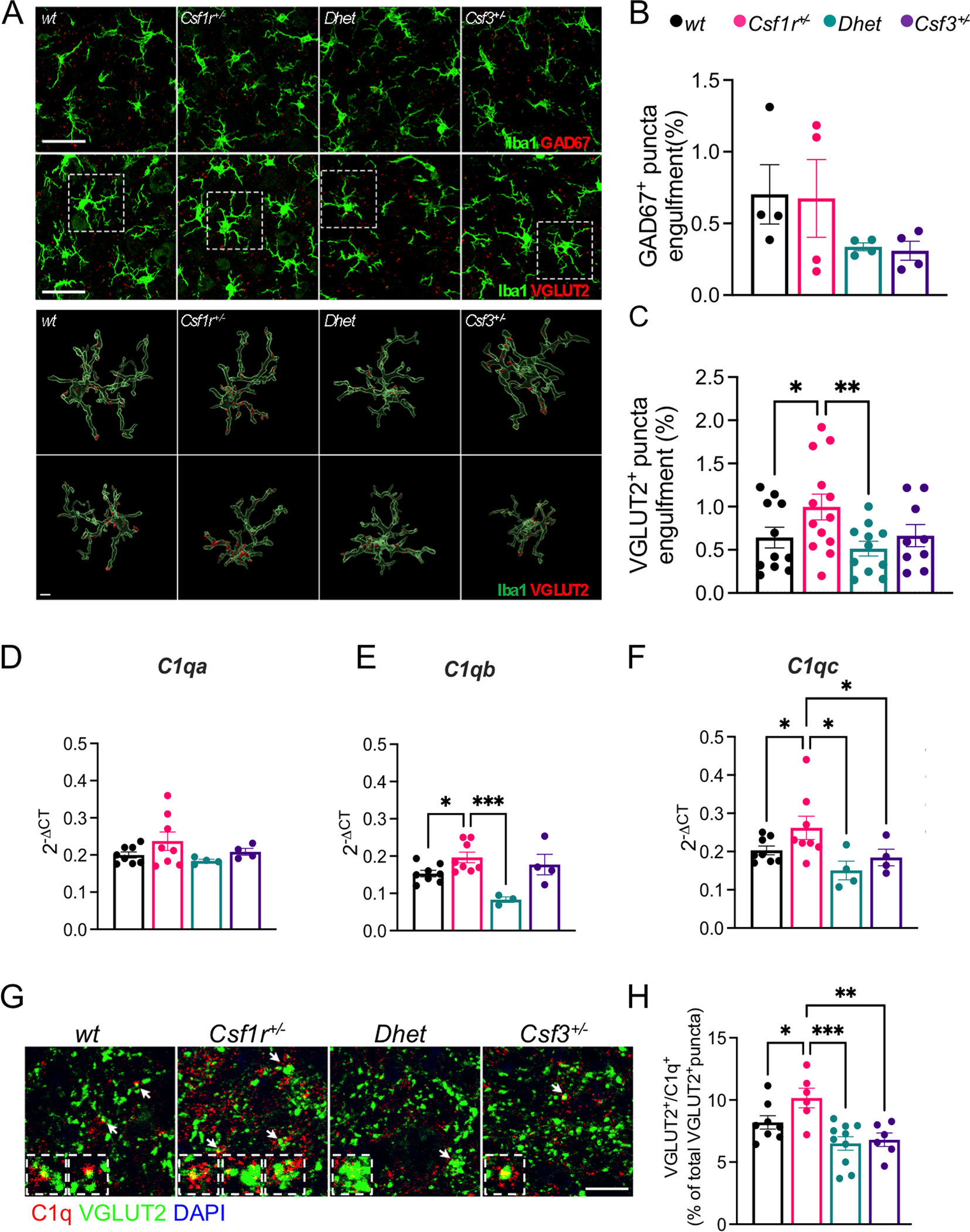
(A-C) 16-month-old female mice. (A) Upper panels, immunofluorescence staining showing the engulfment of GAD67+ and VGLUT2+ puncta (red) by Iba1+ microglia (green) in the DCN. Lower panels, 3D reconstruction with surface rendering showing VGLUT2+ puncta inside the branches and the cell bodies of single microglia, confirming the engulfment of synaptic material. Microglia are shown in their original position (top) and rotated 180 degrees along the z axis (bottom). Scale bars: 30 μm (upper panels), 5 μm (lower panels). (B-C) Quantification of the percentage of engulfment of GAD67+ and VGLUT2+ puncta by Iba1+ cells (4–8 mice per genotype, Newman-Keuls). (D-F) Quantification of the expression of transcripts of C1q genes C1qa, C1qb, and C1qc in the cerebella of 16–24-month-old male and female mice (4–8 mice per genotype; two-stage linear step-up procedure of Benjamini, Krieger and Yekutieli). (G) Co-localization of VGLUT2+ puncta (green) and C1q (red) indicated by the arrows in the DCN of 16-month-old female mice. Inserts show enlarged representative examples. (H) Quantification of data from G (2 sections per mouse, 3–5 mice per genotype; Holm-Sidak’s). Each point in the bar graphs represents a mouse. Means ± SEM, significant differences are marked by asterisks. *, p < .05; **, p < 0.01, ***, p < 0.001.
3.9. Csf3 heterozygosity prevents the overexpression of C1q genes and attenuates C1q deposition on glutamatergic synapses in Csf1r+/− mice
The complement cascade of the innate immune system has emerged as an important mediator of synapse pruning during both brain development and disease (Hong et al., 2016; Stevens et al., 2007). The C1q factor, comprising 6 C1qA, 6 C1qB and 6 C1qC chains, is the initiating protein of the classical complement cascade and was reported to associate with synapses to promote their removal by microglia (Hong et al., 2016). These findings prompted us to analyze the expression of C1q genes in the cerebella and the extent of C1q deposition on glutamatergic synapses of DCN, in Csf1r+/− mice. Quantitative RT-PCR revealed significantly increased expression of C1qb and C1qc transcripts in Csf1r+/− mice, that was normalized by monoallelic targeting of Csf3 (Fig. 6D–F). The increased expression of C1q genes in Csf1r+/− mice was associated with increased C1q deposition on glutamatergic synapses, evidenced by the increased colocalization of C1q with VGLUT2+ puncta (Fig. 6G, H) and was normalized by monoallelic deletion of Csf3. In contrast, the expression of genes encoding other components of the complement cascade (C3), complement receptors (Itgam, Itgax, Itgb2), of neuronal proteins that mediate the synaptic deposition of C1q (Nptx1, Nptx2) (Kovacs et al., 2020) and of Trem2, a microglial receptor involved in synapse removal (Filipello et al., 2018) was unchanged (Supplementary Fig. S1). Given that Csf3r expression in the brain is restricted to microglia (Kozareva et al., 2021; Ximerakis et al., 2019), which are also the dominant source of C1q in the mouse brain (Fonseca et al., 2017), our data suggest that the elevated G-CSF signaling in Csf1r+/− mice causes an excessive removal of glutamatergic synapses in the DCN by regulating C1q production by microglia.
4. DISCUSSION
Csf1r heterozygosity in mice reproduces the hallmark features of CRL (Chitu et al., 2015). The cognitive, emotional and motor deficits are accompanied by histological alterations, including elevated brain microglial density, callosal demyelination, cortical neuronal loss, and callosal axonal spheroids. These phenotypes were associated with increased brain expression of Csf2, and of Csf3 (Chitu et al., 2015). Subsequent studies in autopsied brain tissue of CRL patients showed increased expression of CSF2 (Chitu et al., 2020) and provided evidence of dysregulated G-CSF signaling (Kempthorne et al., 2020a, 2020b) suggesting an important role for these factors in CRL. Since the CSF-1R, as well as the receptors for both G- and GM-CSF, are predominantly expressed in microglia and regulate their activation (reviewed in (Chitu, Biundo, et al., 2021)), it was suggested that CRL could be a primary microgliopathy. Indeed, monoallelic deletion of Csf1r in the microglial lineage recapitulated the phenotype observed in Csf1r+/− mice, indicating that CRL is a primary microgliopathy (Biundo et al., 2021)
Targeting Csf2, that encodes a microglial mitogen, rescued some behavioral defects (spatial memory, depression and olfactory) and the histological alterations observed in the forebrain of Csf1r+/− mice (microgliosis, callosal demyelination, decreased callosal volume), but did not attenuate the elevated microglial density in the cerebellum (Chitu et al., 2020). While G-CSF is not a microglial mitogen (Giulian & Ingeman, 1988), its administration was reported to induce the expansion of microglia in vivo (Chen et al., 2013), to activate a Cathepsin S-CX3CR1-inducible NOS pathway in microglia and to induce the production of factors that promote neuronal excitability (Basso et al., 2017). Therefore, we explored whether G-CSF may contribute to the CRL-like pathogenesis in Csf1r+/− mice.
A hallmark feature of CRL is the loss of callosal white matter (reviewed in (Chitu, Gokhan, et al., 2021; Konno et al., 2018)). The primary function of the corpus callosum is to integrate information by joining both cerebral hemispheres to process motor, sensory, and cognitive signals. Disruption of myelination could potentially impact all these functions. Indeed, a study employing advanced MRI techniques revealed altered functional connectivity between the cerebral hemispheres in CRL patients and highlighted an association between their symptoms and the disconnection of the two cerebral hemispheres, due to the loss of connection fibers in the corpus callosum (Zhan et al., 2020). Furthermore, studies in both autopsied tissue from CRL patients and the Csf1r+/− mouse model suggest that microglial activation contributes to the loss of callosal white matter (Biundo et al., 2021; Chitu et al., 2020; E. J. Kim et al., 2015; Kinoshita et al., 2021). Intriguingly, although targeting Csf3 reduced the density of microglia in the corpus callosum, in contrast to Csf2 reduction (Chitu et al., 2020), it did not prevent their morphological alterations and clustering, nor did it prevent demyelination. Together, these data indicate that in contrast to GM-CSF, G-CSF does not play a major role in promoting demyelination in the Csf1r+/− mouse model of CRL. Interestingly however, as observed with Csf2 targeting, targeting Csf3 did not prevent the loss of layer V neurons, a population that is uniquely dependent on trophic support from microglia (Ueno et al., 2013), suggesting that the loss of these neurons may require the targeting of both factors together, or is caused by other mechanisms.
The memory deficits of Csf1r+/− mice developed independently of the level Csf3 expression. Furthermore, consistent with a previously published study in Csf3−/− mice (Diederich et al., 2009), we show that monoallelic deletion of Csf3 was sufficient to cause cognitive dysfunctions in wild type mice, a phenotype that could be related to the reduction of its neurogenic actions in the dorsal hippocampus. Nevertheless, monoallelic targeting of Csf3 in female Csf1r+/− mice reduced microglial activation in the ventral hippocampus and ameliorated anxiety-like behavior. Of interest in this respect, is the observation that a female CRL patient had an unusual response to G-CSF infusions following hematopoietic stem cell transplantation, becoming more agitated (Dr. Troy C. Lund, personal communication). The factors contributing to the differential effects of G-CSF in different areas of the hippocampus remain to be elucidated. The finding that G-CSF is selectively elevated with age in the ventral but not dorsal hippocampus (Porcher et al., 2021), could explain the apparently region-specific response of hippocampal microglia to the monoallelic targeting of Csf3. The rescue of two cerebellum-dependent functions, i.e. the motor and social interaction deficits (Carta et al., 2019; Mateen et al., 2010) by Csf3 heterozygosity suggests that G-CSF mediates cerebellar dysfunction in Csf1r+/− mice, a hypothesis supported by the attenuation of microglial morphological alterations in all areas of the cerebellum following monoallelic targeting of Csf3 in Csf1r+/− mice. Csf3 heterozygosity also decreased the elimination of glutamatergic synapses and restored electrophysiological activity in the deep cerebellar nuclei. Furthermore, these effects of Csf3 heterozygosity were associated with normalization of the expression of C1q genes and of C1q deposition on glutamatergic synapses. The lack of a conditional allele of the Csf3r precludes further mechanistic investigations. However, since within the brain Csf3r expression is restricted to microglia (Kozareva et al., 2021; Ximerakis et al., 2019), we suggest that G-CSF mediated alterations of microglia in specific brain regions promotes the development of anxiety-like behavior, motor coordination and social interaction deficits in Csf1r+/− mice.
Females tend to be more severely affected by CRL and exhibit a higher prevalence of gait disorders (Chitu, Gokhan, et al., 2021). Furthermore, ataxia and cerebellar involvement have also been reported predominantly in female CRL patients (13 out of 15 documented cases) (Bonvegna et al., 2020; Guerreiro et al., 2013; Karle et al., 2013; E. J. Kim et al., 2015; S. I. Kim et al., 2019; Lynch et al., 2016; Lynch et al., 2017; Mateen et al., 2010; Meyer-Ohlendorf et al., 2015; Riku et al., 2014; Sundal et al., 2015). These findings raise the possibility that estrogens and/or androgens might specifically regulate subpopulations of microglia such as the cerebellar microglia, or those interacting with motor neurons. Remarkably, targeting Csf3 selectively rescued the motor coordination deficits of female Csf1r+/− mice, while it also tended to worsen motor function in males (Fig. 1P). This finding is not unique to the CRL mouse model. Administration of G-CSF has also produced sex-specific effects in both preclinical and clinical trials for amyotrophic lateral sclerosis, providing protection in males by attenuating inflammation and exacerbating the loss of motor function in females (Amirzagar et al., 2015; Naumenko et al., 2011; Pollari et al., 2011). These data suggest an interaction of G-CSF with sex-specific factors, likely hormonal, in the control of neuroinflammation, an aspect that deserves further exploration.
The Csf1r+/− model of CRL used in this study (Chitu et al., 2015) was based on the finding that CSF-1R haploinsufficiency causes CRL (Konno et al., 2014). Since this initial report, 2 additional CRL mutations have been shown to cause CSF1R haploinsufficiency (Leng et al., 2019; Miura et al., 2018). For the mouse model, the C57BL6/J background was chosen primarily because Csf1r−/− mice on this background exhibited the most severe phenotype observed (perinatal mortality) in several mouse strains (Chitu, Gokhan, Nandi, Mehler, & Stanley, 2016). These mice phenocopy the cognitive deficits, motor coordination deficits, depression- and anxiety-like behavior (Biundo et al., 2021; Chitu et al., 2020; Chitu et al., 2015), as well as the age-dependent disease penetrance, of patients with CRL mutations (Konno et al., 2018). However, recent reports show that neither Csf1r haploinsufficiency in rats (Patkar et al., 2021), nor heterozygosity of a missense mutation (E631K corresponding to the E633K CRL-associated mutation) in mice (Stables et al., 2022) reproduce characteristics of CRL or of Csf1r+/− C57BL6/J mice, such as the presence of motor coordination deficits, or the increase in microglial densities. Based on their findings, the authors argue against Csf1r haploinsufficiency as a model of CRL (Patkar et al., 2021). This argument is premature. First, the behavioral experiments in Csf1r E631K/+ mice and Csf1r+/− rats were carried out under sub-optimal conditions. Csf1r E631K/+ mice were evaluated at 10 months of age with mixed groups of males and females, while the experiments in the Csf1r+/− rats involved small groups of male mice. As reproducibly shown in the present study (Fig. 1 O, P) and in a previous publication (Chitu et al., 2020), the motor coordination deficits preferentially occur in females and develop after 13 months of age. In addition, patients with mutations causing haploinsufficiency (early truncations and nonsense-mediated RNA decay) develop symptoms significantly earlier than those with missense mutations (Chitu, Gokhan, et al., 2021), suggesting that animal models based on loss-of-function missense mutations should be characterized at ages at least as advanced as those studied in Csf1r+/− mice. Furthermore, given the penetrance of the disease in Csf1r+/− mice (Chitu, Gokhan, et al., 2021), large rodent numbers are required for the behavioral experiments. Second, the consequences of Csf1r deficiency tend to be less severe in rats (reviewed in (Chitu, Gokhan, et al., 2021)). In contrast to the perinatal lethality of homozygous-null Csf1r C57BL6/J mice, most Csf1r−/− dark agouti rats survive to 7–8 weeks of age. Consequently, Csf1r heterozygosity in rats may also not be severe enough for symptom development.
Another argument against Csf1r haploinsufficiency as a model of CRL is based on the discrepancy between the small increase in microglial numbers documented in C57BL6/J Csf1r+/− mice (Arreola et al., 2021; Chitu et al., 2015) and the decrease or lack of change in microglia densities respectively observed in Csf1r E631K/+ mice (Stables et al., 2022) and Csf1r+/− rats (Patkar et al., 2021). However, the increase in microglial densities observed in Csf1r+/− mice is established during development (Arreola et al., 2021) and, unless accompanied by other features such as morphological alterations or cell clustering, may not reflect a disease-related, reactive state. In fact, studies in autopsied tissue from CRL patients have established that microglial densities change during disease progression, peaking at early stages and declining thereafter (Kinoshita et al., 2021; Oyanagi et al., 2017). Nevertheless, the reduction in microglia processes, indicative of changes in their activation state, is a common feature of both Csf1r+/− and Csf1r E631K/+ mice (Figs. 2F, 3E,K and 4E,H,I and (Stables et al., 2022)), that was also described in the human disease (Kinoshita et al., 2021). We conclude that at present, the Csf1r+/− C57BL6/J mouse is the only validated model of CRL that reproduces most symptoms of CRL.
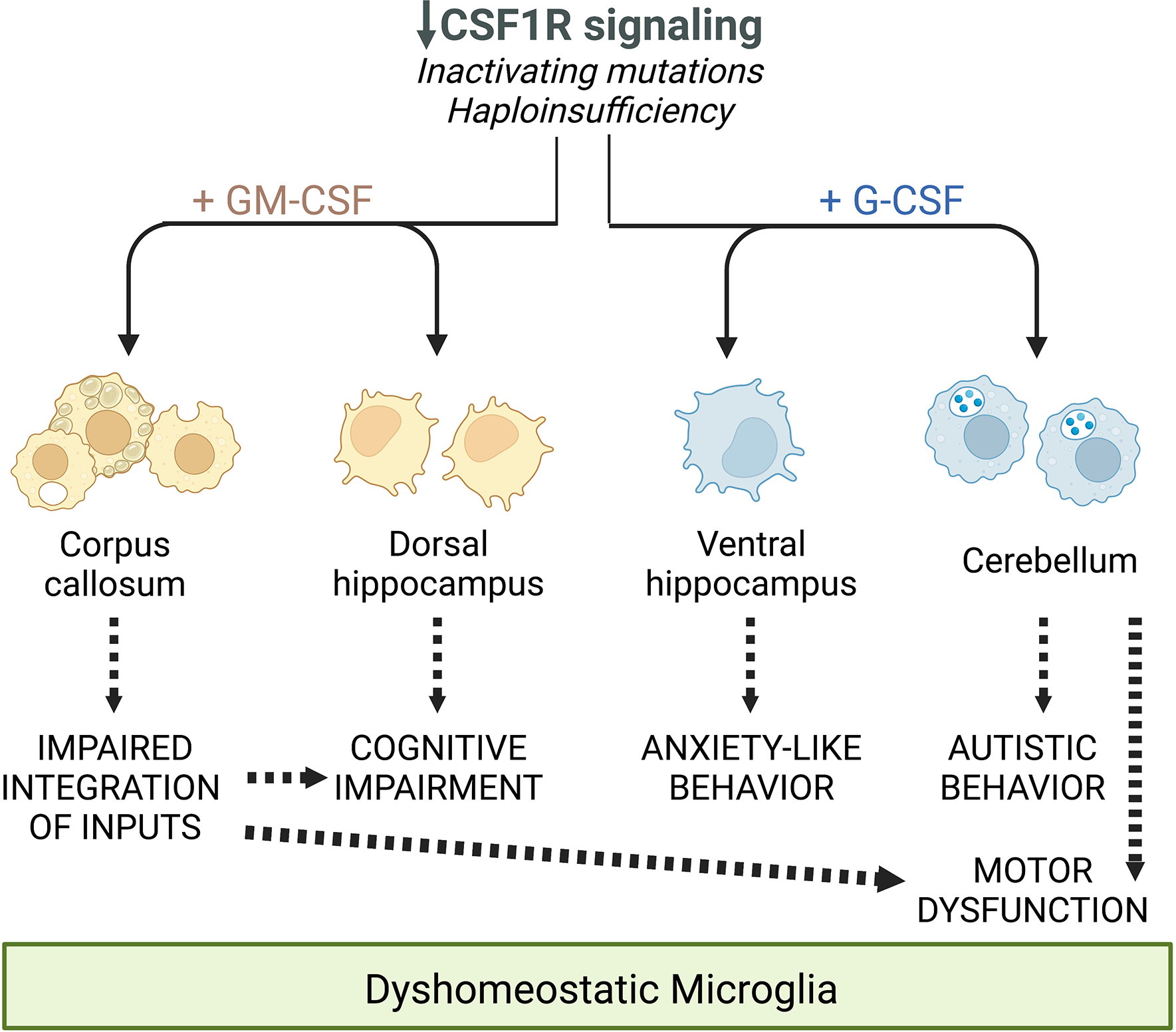
In conclusion, this study identifies elevated G-CSF as the main factor driving anxiety and cerebellar dysfunctions contributing to the motor coordination and social preference deficits in Csf1r+/− mice. Apart from their overlapping contributions to the motor coordination deficit, the effects of elevated G-CSF and GM-CSF in Csf1r+/− mice are non-redundant (Chitu et al., 2020) (Fig. 7). Further investigations are necessary to establish the benefit of combined, G-CSF and GM-CSF, genetic and pharmacological targeting.
Figure 7. Schematic summarizing the differential and overlapping contributions of the increased expression of GM-CSF and G-CSF to the deficits observed in CRL mice, mediated through their regulation of microglia.
Supplementary Material
Acknowledgements
The authors thank Hillary Guzik, Andrea Briceno and Dr. Vera Des-Marais of the Einstein Analytical Imaging Facility for help with imaging and histomorphometry, Dr. Daniel Wilton of Dr. Beth Stevens laboratory for sharing their protocol for C1q staining and Christopher Fernandes and Jude Oppong-Asare for technical assistance.
FUNDING INFORMATION
This work was supported by grants from the National Institutes of Health: Grant R01NS091519 (to E. R. S.), R01NS105470 (to K.K.), R21MH114182 (N.S.B), U54 HD090260 (support for the Rose F. Kennedy IDDRC), the P30CA013330 NCI Cancer Center Grant and a gift from David and Ruth Levine. ZKW is partially supported by the NIH/NIA and NIH/NINDS (1U19AG063911, FAIN: U19AG063911), Mayo Clinic Center for Regenerative Medicine, Mayo Clinic in Florida Focused Research Team Program, the gifts from The Sol Goldman Charitable Trust, and the Donald G. and Jodi P. Heeringa Family, the Haworth Family Professorship in Neurodegenerative Diseases fund, and The Albertson Parkinson’s Research Foundation.
Footnotes
CONFLICT OF INTEREST
The authors declare that they have no competing interests.
DATA AVAILABILITY STATEMENT
The data sets used and analyzed during the current study are available from the corresponding author on reasonable request.
REFERENCES:
- Albert MS (1996). Cognitive and neurobiologic markers of early Alzheimer disease. Proc Natl Acad Sci U S A, 93(24), 13547–13551. doi: 10.1073/pnas.93.24.13547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amirzagar N, Nafissi S, Tafakhori A, Modabbernia A, Amirzargar A, Ghaffarpour M, … Harirchian MH. (2015). Granulocyte colony-stimulating factor for amyotrophic lateral sclerosis: a randomized, double-blind, placebo-controlled study of Iranian patients. J Clin Neurol, 11(2), 164–171. doi: 10.3988/jcn.2015.11.2.164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armbruster DJ, Ueltzhoffer K, Basten U, & Fiebach CJ (2012). Prefrontal cortical mechanisms underlying individual differences in cognitive flexibility and stability. J Cogn Neurosci, 24(12), 2385–2399. doi: 10.1162/jocn_a_00286 [DOI] [PubMed] [Google Scholar]
- Arreola MA, Soni N, Crapser JD, Hohsfield LA, Elmore MRP, Matheos DP, … Green KN. (2021). Microglial dyshomeostasis drives perineuronal net and synaptic loss in a CSF1R(+/−) mouse model of ALSP, which can be rescued via CSF1R inhibitors. Sci Adv, 7(35). doi: 10.1126/sciadv.abg1601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basso L, Lapointe TK, Iftinca M, Marsters C, Hollenberg MD, Kurrasch DM, & Altier C (2017). Granulocyte-colony-stimulating factor (G-CSF) signaling in spinal microglia drives visceral sensitization following colitis. Proc Natl Acad Sci U S A, 114(42), 11235–11240. doi: 10.1073/pnas.1706053114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumel Y, Jacobson GA, & Cohen D (2009). Implications of functional anatomy on information processing in the deep cerebellar nuclei. Front Cell Neurosci, 3, 14. doi: 10.3389/neuro.03.014.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biundo F, Chitu V, Shlager GGL, Park ES, Gulinello ME, Saha K, … Stanley ER. (2021). Microglial reduction of colony stimulating factor-1 receptor expression is sufficient to confer adult onset leukodystrophy. Glia, 69, 779–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biundo F, Ishiwari K, Del Prete D, & D’Adamio L (2015). Interaction of ApoE3 and ApoE4 isoforms with an ITM2b/BRI2 mutation linked to the Alzheimer disease-like Danish dementia: Effects on learning and memory. Neurobiol Learn Mem, 126, 18–30. doi: 10.1016/j.nlm.2015.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biundo F, Ishiwari K, Del Prete D, & D’Adamio L (2016). Deletion of the gamma-secretase subunits Aph1B/C impairs memory and worsens the deficits of knock-in mice modeling the Alzheimer-like familial Danish dementia. Oncotarget, 7(11), 11923–11944. doi: 10.18632/oncotarget.7389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonvegna S, Straccia G, Golfre Andreasi N, Elia AE, Marucci G, Di Bella D, … Eleopra R. (2020). Parkinsonism and Nigrostriatal Damage Secondary to CSF1R-Related Primary Microgliopathy. Mov Disord, 35(12), 2360–2362. doi: 10.1002/mds.28290 [DOI] [PubMed] [Google Scholar]
- Burghardt NS, Park EH, Hen R, & Fenton AA (2012). Adult-born hippocampal neurons promote cognitive flexibility in mice. Hippocampus, 22(9), 1795–1808. doi: 10.1002/hipo.22013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carta I, Chen CH, Schott AL, Dorizan S, & Khodakhah K (2019). Cerebellar modulation of the reward circuitry and social behavior. Science, 363(6424). doi: 10.1126/science.aav0581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CH, Huang SY, Chen NF, Feng CW, Hung HC, Sung CS, … Chen WF. (2013). Intrathecal granulocyte colony-stimulating factor modulate glial cell line-derived neurotrophic factor and vascular endothelial growth factor A expression in glial cells after experimental spinal cord ischemia. Neuroscience, 242, 39–52. doi: 10.1016/j.neuroscience.2013.02.017 [DOI] [PubMed] [Google Scholar]
- Chitu V, Biundo F, Shlager GGL, Park ES, Wang P, Gulinello ME, … Stanley ER. (2020). Microglial Homeostasis Requires Balanced CSF-1/CSF-2 Receptor Signaling. Cell Rep, 30(9), 3004–3019 e3005. doi: 10.1016/j.celrep.2020.02.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chitu V, Biundo F, & Stanley ER (2021). Colony stimulating factors in the nervous system. Semin Immunol, 101511. doi: 10.1016/j.smim.2021.101511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chitu V, Gokhan S, Gulinello M, Branch CA, Patil M, Basu R, … Stanley ER. (2015). Phenotypic characterization of a Csf1r haploinsufficient mouse model of adult-onset leukodystrophy with axonal spheroids and pigmented glia (ALSP). Neurobiol Dis, 74, 219–228. doi:S0969-9961(14)00372-6 [pii] 10.1016/j.nbd.2014.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chitu V, Gokhan S, Nandi S, Mehler MF, & Stanley ER (2016). Emerging Roles for CSF-1 Receptor and its Ligands in the Nervous System. Trends Neurosci, 39(6), 378–393. doi: 10.1016/j.tins.2016.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chitu V, Gokhan S, & Stanley ER (2021). Modeling CSF-1 receptor deficiency diseases - how close are we? The FEBS journal. doi: 10.1111/febs.16085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai XM, Ryan GR, Hapel AJ, Dominguez MG, Russell RG, Kapp S, … Stanley ER. (2002). Targeted disruption of the mouse colony-stimulating factor 1 receptor gene results in osteopetrosis, mononuclear phagocyte deficiency, increased primitive progenitor cell frequencies, and reproductive defects. Blood, 99(1), 111–120. [DOI] [PubMed] [Google Scholar]
- Darmohray DM, Jacobs JR, Marques HG, & Carey MR (2019). Spatial and Temporal Locomotor Learning in Mouse Cerebellum. Neuron, 102(1), 217–231 e214. doi: 10.1016/j.neuron.2019.01.038 [DOI] [PubMed] [Google Scholar]
- Diederich K, Sevimli S, Dorr H, Kosters E, Hoppen M, Lewejohann L, … Schabitz WR. (2009). The role of granulocyte-colony stimulating factor (G-CSF) in the healthy brain: a characterization of G-CSF-deficient mice. J Neurosci, 29(37), 11572–11581. doi: 10.1523/JNEUROSCI.0453-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanselow MS, & Dong HW (2010). Are the dorsal and ventral hippocampus functionally distinct structures? Neuron, 65(1), 7–19. doi: 10.1016/j.neuron.2009.11.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipello F, Morini R, Corradini I, Zerbi V, Canzi A, Michalski B, … Matteoli M. (2018). The Microglial Innate Immune Receptor TREM2 Is Required for Synapse Elimination and Normal Brain Connectivity. Immunity, 48(5), 979–991 e978. doi: 10.1016/j.immuni.2018.04.016 [DOI] [PubMed] [Google Scholar]
- Fonseca MI, Chu SH, Hernandez MX, Fang MJ, Modarresi L, Selvan P, … Tenner AJ. (2017). Cell-specific deletion of C1qa identifies microglia as the dominant source of C1q in mouse brain. J Neuroinflammation, 14(1), 48. doi: 10.1186/s12974-017-0814-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giulian D, & Ingeman JE (1988). Colony-stimulating factors as promoters of ameboid microglia. J. Neurosci, 8, 4707–4717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerreiro R, Kara E, Le Ber I, Bras J, Rohrer JD, Taipa R, … Houlden H. (2013). Genetic analysis of inherited leukodystrophies: genotype-phenotype correlations in the CSF1R gene. JAMA Neurol, 70(7), 875–882. doi:1684866 [pii] 10.1001/jamaneurol.2013.698 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulinello M, Chen F, & Dobrenis K (2008). Early deficits in motor coordination and cognitive dysfunction in a mouse model of the neurodegenerative lysosomal storage disorder, Sandhoff disease. Behavioural brain research, 193(2), 315–319. doi:S0166-4328(08)00311-2 [pii] 10.1016/j.bbr.2008.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, … Stevens B. (2016). Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science, 352(6286), 712–716. doi: 10.1126/science.aad8373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kana V, Desland FA, Casanova-Acebes M, Ayata P, Badimon A, Nabel E, … Merad M. (2019). CSF-1 controls cerebellar microglia and is required for motor function and social interaction. J Exp Med, 216(10), 2265–2281. doi: 10.1084/jem.20182037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karle KN, Biskup S, Schule R, Schweitzer KJ, Kruger R, Bauer P, … Schols L. (2013). De novo mutations in hereditary diffuse leukoencephalopathy with axonal spheroids (HDLS). Neurology, 81(23), 2039–2044. doi:01.wnl.0000436945.01023.ac [pii] 10.1212/01.wnl.0000436945.01023.ac [DOI] [PubMed] [Google Scholar]
- Kavetsky L, Green KK, Boyle BR, Yousufzai FAK, Padron ZM, Melli SE, … Soto I. (2019). Increased interactions and engulfment of dendrites by microglia precede Purkinje cell degeneration in a mouse model of Niemann Pick Type-C. Sci Rep, 9(1), 14722. doi: 10.1038/s41598-019-51246-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kempthorne L, Yoon H, Madore C, Smith S, Wszolek ZK, Rademakers R, … Dickson DW. (2020a). Correction to: Loss of homeostatic microglial phenotype in CSF1R-related Leukoencephalopathy. Acta Neuropathol Commun, 8(1), 90. doi: 10.1186/s40478-020-00970-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kempthorne L, Yoon H, Madore C, Smith S, Wszolek ZK, Rademakers R, … Dickson DW. (2020b). Loss of homeostatic microglial phenotype in CSF1R-related Leukoencephalopathy. Acta Neuropathol Commun, 8(1), 72. doi: 10.1186/s40478-020-00947-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EJ, Shin JH, Lee JH, Kim JH, Na DL, Suh YL, … Huh GY. (2015). Adult-onset leukoencephalopathy with axonal spheroids and pigmented glia linked CSF1R mutation: Report of four Korean cases. J Neurol Sci, 349(1–2), 232–238. doi:S0022-510X(14)00786-2 [pii] 10.1016/j.jns.2014.12.021 [doi] [DOI] [PubMed] [Google Scholar]
- Kim SI, Jeon B, Bae J, Won JK, Kim HJ, Yim J, … Park SH. (2019). An Autopsy Proven Case of CSF1R-mutant Adult-onset Leukoencephalopathy with Axonal Spheroids and Pigmented Glia (ALSP) with Premature Ovarian Failure. Exp Neurobiol, 28(1), 119–129. doi: 10.5607/en.2019.28.1.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita M, Oyanagi K, Kondo Y, Ishizawa K, Ishihara K, Yoshida M, … Ikeda SI. (2021). Pathologic basis of the preferential thinning of thecorpus callosum in adult-onset leukoencephalopathy with axonal spheroids and pigmented glia (ALSP). eNeurologicalSci, 22, 100310. doi: 10.1016/j.ensci.2021.100310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konno T, Kasanuki K, Ikeuchi T, Dickson DW, & Wszolek ZK (2018). CSF1R-related leukoencephalopathy: A major player in primary microgliopathies. Neurology, 91(24), 1092–1104. doi: 10.1212/WNL.0000000000006642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konno T, Tada M, Tada M, Koyama A, Nozaki H, Harigaya Y, … Ikeuchi T. (2014). Haploinsufficiency of CSF-1R and clinicopathologic characterization in patients with HDLS. Neurology, 82(2), 139–148. doi:WNL.0000000000000046 [pii] 10.1212/WNL.0000000000000046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs RA, Vadaszi H, Bulyaki E, Torok G, Toth V, Matyas D, … Kardos J. (2020). Identification of Neuronal Pentraxins as Synaptic Binding Partners of C1q and the Involvement of NP1 in Synaptic Pruning in Adult Mice. Front Immunol, 11, 599771. doi: 10.3389/fimmu.2020.599771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozareva V, Martin C, Osorno T, Rudolph S, Guo C, Vanderburg C, … Macosko E. (2021). A transcriptomic atlas of mouse cerebellar cortex comprehensively defines cell types. Nature, 598(7879), 214–219. doi: 10.1038/s41586-021-03220-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehrman EK, Wilton DK, Litvina EY, Welsh CA, Chang ST, Frouin A, … Stevens B. (2018). CD47 Protects Synapses from Excess Microglia-Mediated Pruning during Development. Neuron, 100(1), 120–134 e126. doi: 10.1016/j.neuron.2018.09.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leng C, Lu L, Wang G, Zhang Y, Xu Y, Lin X, … Ge W. (2019). A novel dominant-negative mutation of the CSF1R gene causes adult-onset leukoencephalopathy with axonal spheroids and pigmented glia. Am J Transl Res, 11(9), 6093–6101. [PMC free article] [PubMed] [Google Scholar]
- Lieschke GJ, Grail D, Hodgson G, Metcalf D, Stanley E, Cheers C, … Dunn AR. (1994). Mice Lacking Granulocyte-Colony-Stimulating Factor Have Chronic Neutropenia, Granulocyte and Macrophage Progenitor-Cell Deficiency, and Impaired Neutrophil Mobilization. Blood, 84(6), 1737–1746. [PubMed] [Google Scholar]
- Lynch DS, Jaunmuktane Z, Sheerin UM, Phadke R, Brandner S, Milonas I, … Houlden H. (2016). Hereditary leukoencephalopathy with axonal spheroids: a spectrum of phenotypes from CNS vasculitis to parkinsonism in an adult onset leukodystrophy series. J Neurol Neurosurg Psychiatry, 87(5), 512–519. doi: 10.1136/jnnp-2015-310788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch DS, Rodrigues Brandao de Paiva A, Zhang WJ, Bugiardini E, Freua F, Tavares Lucato L, … Houlden H. (2017). Clinical and genetic characterization of leukoencephalopathies in adults. Brain, 140(5), 1204–1211. doi: 10.1093/brain/awx045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateen FJ, Keegan BM, Krecke K, Parisi JE, Trenerry MR, & Pittock SJ (2010). Sporadic leucodystrophy with neuroaxonal spheroids: persistence of DWI changes and neurocognitive profiles: a case study. J Neurol Neurosurg Psychiatry, 81(6), 619–622. doi: 10.1136/jnnp.2008.169243 [DOI] [PubMed] [Google Scholar]
- Meyer-Ohlendorf M, Braczynski A, Al-Qaisi O, Gessler F, Biskup S, Weise L, … Bahr O. (2015). Comprehensive diagnostics in a case of hereditary diffuse leukodystrophy with spheroids. BMC Neurol, 15, 103. doi: 10.1186/s12883-015-0368-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura T, Mezaki N, Konno T, Iwasaki A, Hara N, Miura M, … Ikeuchi T. (2018). Identification and functional characterization of novel mutations including frameshift mutation in exon 4 of CSF1R in patients with adult-onset leukoencephalopathy with axonal spheroids and pigmented glia. J Neurol, 265(10), 2415–2424. doi: 10.1007/s00415-018-9017-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nandi S, Gokhan S, Dai XM, Wei S, Enikolopov G, Lin H, … Stanley ER. (2012). The CSF-1 receptor ligands IL-34 and CSF-1 exhibit distinct developmental brain expression patterns and regulate neural progenitor cell maintenance and maturation. Dev Biol, 367(2), 100–113. doi: 10.1016/j.ydbio.2012.03.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naumenko N, Pollari E, Kurronen A, Giniatullina R, Shakirzyanova A, Magga J, … Giniatullin R. (2011). Gender-Specific Mechanism of Synaptic Impairment and Its Prevention by GCSF in a Mouse Model of ALS. Front Cell Neurosci, 5, 26. doi: 10.3389/fncel.2011.00026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson AM, Baker MC, Finch NA, Rutherford NJ, Wider C, Graff-Radford NR, … Rademakers R. (2013). CSF1R mutations link POLD and HDLS as a single disease entity. Neurology, 80(11), 1033–1040. doi:Doi 10.1212/Wnl.0b013e31828726a7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyanagi K, Kinoshita M, Suzuki-Kouyama E, Inoue T, Nakahara A, Tokiwai M, … Ikeda SI. (2017). Adult onset leukoencephalopathy with axonal spheroids and pigmented glia (ALSP) and Nasu-Hakola disease: lesion staging and dynamic changes of axons and microglial subsets. Brain Pathol, 27(6), 748–769. doi: 10.1111/bpa.12443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parfitt GM, Nguyen R, Bang JY, Aqrabawi AJ, Tran MM, Seo DK, … Kim JC. (2017). Bidirectional Control of Anxiety-Related Behaviors in Mice: Role of Inputs Arising from the Ventral Hippocampus to the Lateral Septum and Medial Prefrontal Cortex. Neuropsychopharmacology, 42(8), 1715–1728. doi: 10.1038/npp.2017.56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patkar OL, Caruso M, Teakle N, Keshvari S, Bush SJ, Pridans C, … Hume DA. (2021). Analysis of homozygous and heterozygous Csf1r knockout in the rat as a model for understanding microglial function in brain development and the impacts of human CSF1R mutations. Neurobiol Dis, 151, 105268. doi: 10.1016/j.nbd.2021.105268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollari E, Savchenko E, Jaronen M, Kanninen K, Malm T, Wojciechowski S, … Magga J. (2011). Granulocyte colony stimulating factor attenuates inflammation in a mouse model of amyotrophic lateral sclerosis. J Neuroinflammation, 8, 74. doi: 10.1186/1742-2094-8-74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porcher L, Bruckmeier S, Burbano SD, Finnell JE, Gorny N, Klett J, … Kelly MP. (2021). Aging triggers an upregulation of a multitude of cytokines in the male and especially the female rodent hippocampus but more discrete changes in other brain regions. J Neuroinflammation, 18(1), 219. doi: 10.1186/s12974-021-02252-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rademakers R, Baker M, Nicholson AM, Rutherford NJ, Finch N, Soto-Ortolaza A, … Wszolek ZK. (2011). Mutations in the colony stimulating factor 1 receptor (CSF1R) gene cause hereditary diffuse leukoencephalopathy with spheroids. Nat Genet, 44(2), 200–205. doi:ng.1027 [pii] 10.1038/ng.1027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riku Y, Ando T, Goto Y, Mano K, Iwasaki Y, Sobue G, & Yoshida M (2014). Early pathologic changes in hereditary diffuse leukoencephalopathy with spheroids. J Neuropathol Exp Neurol, 73(12), 1183–1190. doi: 10.1097/NEN.0000000000000139 [DOI] [PubMed] [Google Scholar]
- Safaiyan S, Besson-Girard S, Kaya T, Cantuti-Castelvetri L, Liu L, Ji H, … Simons M. (2021). White matter aging drives microglial diversity. Neuron, 109(7), 1100–1117 e1110. doi: 10.1016/j.neuron.2021.01.027 [DOI] [PubMed] [Google Scholar]
- Sanchez-Ramos J, Song S, Sava V, Catlow B, Lin X, Mori T, … Arendash GW. (2009). Granulocyte colony stimulating factor decreases brain amyloid burden and reverses cognitive impairment in Alzheimer’s mice. Neuroscience, 163(1), 55–72. doi: 10.1016/j.neuroscience.2009.05.071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, … Stevens B. (2012). Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron, 74(4), 691–705. doi: 10.1016/j.neuron.2012.03.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer DP, & Stevens B (2013). Phagocytic glial cells: sculpting synaptic circuits in the developing nervous system. Curr Opin Neurobiol, 23(6), 1034–1040. doi: 10.1016/j.conb.2013.09.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schermer C, & Humpel C (2002). Granulocyte macrophage-colony stimulating factor activates microglia in rat cortex organotypic brain slices. Neurosci Lett, 328(2), 180–184. [DOI] [PubMed] [Google Scholar]
- Sierra A, Encinas JM, Deudero JJ, Chancey JH, Enikolopov G, Overstreet-Wadiche LS, … Maletic-Savatic M. (2010). Microglia shape adult hippocampal neurogenesis through apoptosis-coupled phagocytosis. Cell Stem Cell, 7(4), 483–495. doi:S1934-5909(10)00437-6 [pii] 10.1016/j.stem.2010.08.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith ME (1993). Phagocytosis of myelin by microglia in vitro. J Neurosci Res, 35(5), 480–487. doi: 10.1002/jnr.490350504 [DOI] [PubMed] [Google Scholar]
- Stables J, Green EK, Sehgal A, Patkar OL, Keshvari S, Taylor I, … Hume DA. (2022). A kinase-dead Csf1r mutation associated with adult-onset leukoencephalopathy has a dominant inhibitory impact on CSF1R signalling. Development, 149(8). doi: 10.1242/dev.200237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, … Barres BA. (2007). The classical complement cascade mediates CNS synapse elimination. Cell, 131(6), 1164–1178. doi: 10.1016/j.cell.2007.10.036 [DOI] [PubMed] [Google Scholar]
- Sundal C, Baker M, Karrenbauer V, Gustavsen M, Bedri S, Glaser A, … Andersen O. (2015). Hereditary diffuse leukoencephalopathy with spheroids with phenotype of primary progressive multiple sclerosis. Eur J Neurol, 22(2), 328–333. doi: 10.1111/ene.12572 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai PT, Hull C, Chu Y, Greene-Colozzi E, Sadowski AR, Leech JM, … Sahin M. (2012). Autistic-like behaviour and cerebellar dysfunction in Purkinje cell Tsc1 mutant mice. Nature, 488(7413), 647–651. doi: 10.1038/nature11310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno M, Fujita Y, Tanaka T, Nakamura Y, Kikuta J, Ishii M, & Yamashita T (2013). Layer V cortical neurons require microglial support for survival during postnatal development. Nat Neurosci, 16(5), 543–551. doi:nn.3358 [pii] 10.1038/nn.3358 [DOI] [PubMed] [Google Scholar]
- Xiao BG, Xu LY, & Yang JS (2002). TGF-beta 1 synergizes with GM-CSF to promote the generation of glial cell-derived dendriform cells in vitro. Brain Behav Immun, 16(6), 685–697. doi:S088915910200020X [pii] [DOI] [PubMed] [Google Scholar]
- Ximerakis M, Lipnick SL, Innes BT, Simmons SK, Adiconis X, Dionne D, … Rubin LL. (2019). Single-cell transcriptomic profiling of the aging mouse brain. Nat Neurosci, 22(10), 1696–1708. doi: 10.1038/s41593-019-0491-3 [DOI] [PubMed] [Google Scholar]
- Young K, & Morrison H (2018). Quantifying Microglia Morphology from Photomicrographs of Immunohistochemistry Prepared Tissue Using ImageJ. J Vis Exp(136). doi: 10.3791/57648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan FX, Zhu ZY, Liu Q, Zhou HY, Luan XH, Huang XJ, … Cao L. (2020). Altered structural and functional connectivity in CSF1R-related leukoencephalopathy. Brain Imaging Behav. doi: 10.1007/s11682-020-00360-0 [DOI] [PubMed] [Google Scholar]
- Zur-Wyrozumska K, Kaczmarska P, & Mensah-Glanowska P (2021). Adult-onset leukoencephalopathy with axonal spheroids and pigmented glia associated with an A792D mutation in the CSF1R gene in a Polish patient. Neurol Neurochir Pol, 53, 322–324. doi: 10.5603/PJNNS.a2021.0012 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data sets used and analyzed during the current study are available from the corresponding author on reasonable request.