

Abstract
Here we show intranuclear nanoribbons formed upon dephosphorylation of leucine-rich L- or D-phosphopeptide catalyzed by alkaline phosphatase (ALP) to selectively kill osteosarcoma cells. Being dephosphorylated by ALP, the peptides first transformed into micelles and then convert into nanoribbons. The peptides/assemblies first aggregate on cell membrane, then enter cells via endocytosis, and finally accumulate in nuclei (mainly in nucleoli). Proteomics analysis suggests that the assemblies interact with histone proteins. The peptides kill osteosarcoma cells rapidly and are nontoxic to normal cells. Moreover, the repeated stimulation of the osteosarcoma cells by the peptides sensitizes the cancer cells rather than inducing resistance. This work not only illustrates a novel mechanism for nucleus-targeting, but also may lead a new way to selectively kill osteosarcoma cells and minimize drug resistance.
Keywords: Enzyme instructed self-assembly, Nuclear targeting, Alkaline phosphatase, Histone protein, Osteosarcoma cells
Graphical Abstract
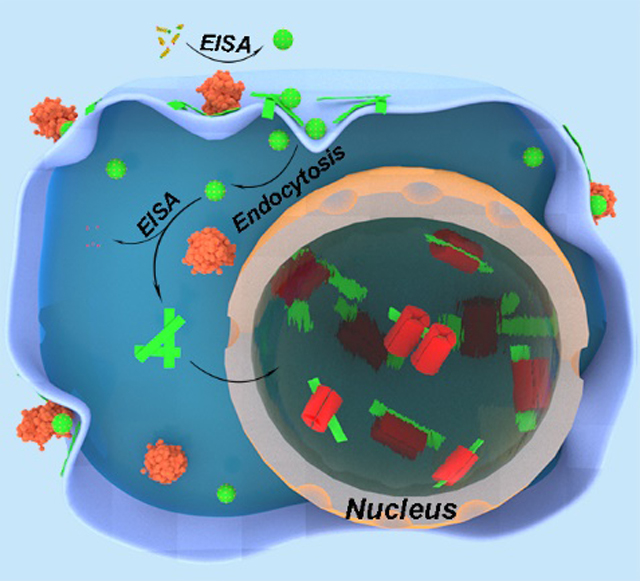
Upon the dephosphorylation of ALP, the leucine-rich phosphopeptides self-assemble into micelles and then convert into nanoribbons. The phosphopeptides/assemblies first aggregate on cell membrane of osteosarcoma cells, and then enter cells via endocytosis, disrupt nuclear membrane, and finally accumulate in nuclei by interaction with histone proteins to induce cell death.
Introduction
Nucleus, being the most sensitive intracellular organelle in eukaryotic cells,[1] performs key cell functions[2]. Consisting of proteins, DNAs and RNAs[3] and acting as the site of ribosome biogenesis[4], nucleolus is a distinct sub-nuclear compartment without membrane. Nucleus-targeting has tremendous potential to inhibit cancer cells[5] because it may lead to effective therapeutic treatment. To target nuclei, usually there are three strategies: passive diffusion of molecules, the active nuclear transport of nuclear localization signal (NLS, ligands that bind nuclear transport receptors[6]), and opening the nuclear envelope[5a]. By using passive diffusion strategies to target nuclei, the small molecules/nanoassemblies need to go through the nuclear pore complexes (NPCs)[7], thus their sizes are usually lower than ~ 10 nm[8] as the passive diffusion is unavailable for nanoassemblies larger than NPCs. One may target nuclei by modifying the nanoassemblies with NLSs, which usually contain positive charges (e.g., lysine or arginine residues) to enhance the cellular and nuclear uptake of nanoparticles and to tightly adhere to anionic DNA[3a, 9]. As to opening the nuclear envelope strategy reported, disturbing the membrane integrity upon light irradiation enhances the nuclear uptake[10]. Besides, other strategies for nucleus targeting were also reported, such as boronated targeting strategy[11].
Recently, we reported a phosphopeptide, consisting of four L-leucine, a N-terminal fluorescent group (NBD), and a C-terminal L-phosphotyrosine, which self-assembles into nanoribbons via enzyme instructed self-assembly (EISA)[12]. The role of NBD is to reveal the location of the peptide assemblies in cells[13]. The dephosphorylated pentapeptide (due to the overexpressed alkaline phosphatase (ALP) by human induced pluripotent stem cells (iPSCs)) self-assembles and accumulates in nuclei of iPSCs and selectively eliminate iPSCs rapidly. Carrying negative charges (a carboxyl group and a phosphate group) and lack any known NLSs, the leucine rich assemblies differ fundamentally from the nuclear targeting strategies reported previously.
The above unexpected result, as the first case of intranuclear assemblies of peptides, stimulates us to use the phosphopeptide (1Lp, NBD-LLLLpY, L = L-leucine, pY = L-phosphotyrosine) to selectively inhibit other cells that overexpress ALP, such as osteosarcoma cells. To study the role of chirality in the peptides, we use D-amino acid residues to replace the L-amino acid residues in 1Lp completely or partially for preparing the D-enantiomers 1Dp (NBD-llllpy, l = D-leucine, py = D-phosphotyrosine) and the diastereomer 1DLp (NBD-lLlLpy). We design 1Dp because the D-peptides are proteolytically resistant and exhibit higher efficacy for cancer cell inhibition[14]. The purpose of 1DLp is to evaluate the self-assembly and cytotoxicity of heterochiral peptides. To study the contribution of dephosphorylation, we synthesized 1L (NBD-LLLLY, Y = L-tyrosine) and 1D (NBD-lllly, y = D-tyrosine) as the controls. To study the mechanism of nucleus-targeting via leucine-rich assembly, we also designed the biotin-modified enzyme substrates 2Lp (NBD-LLLLK(biotin)pY, K = L-lysine) and 2Dp (NBD-llllk(biotin)py, k = D-lysine) to enrich proteins for the proteomics analysis that aims to identify the proteins interacting with the assemblies of the peptides.
Here we show that the phosphopeptide assemblies interact with histone proteins, accumulates in nuclei, and selectively inhibit osteosarcoma cells (Figure 1). Specifically, 1Lp, 1Dp, or 2Lp, upon dephosphorylation catalyzed by ALP, first self-assembles to form micelles and then turns into nanoribbons. Being incubated with osteosarcoma cells, the phosphopeptides first aggregate on cell membrane, then enter cells via endocytosis, disrupt nuclear membrane, and finally accumulate in nuclei (mainly in nucleoli). Proteomics analysis indicates that the assemblies associate with histone proteins in cells, likely contributing of nucleus-targeting. The phosphopeptides show high cytotoxicity against osteosarcoma cells (i.e., SJSA-1 and Saos2), and are nontoxic to normal cells (i.e., HS-5 and HEK293). Surprisingly, the repeated stimulation of the osteosarcoma cells by the phosphopeptides sensitizes the cancer cells to the phosphopeptides rather than inducing acquired drug resistance. This work not only illustrates a novel strategy for nuclei-targeting, but also may lead a new way to selectively kill osteosarcoma cells and minimize acquired drug resistance.
Figure 1.
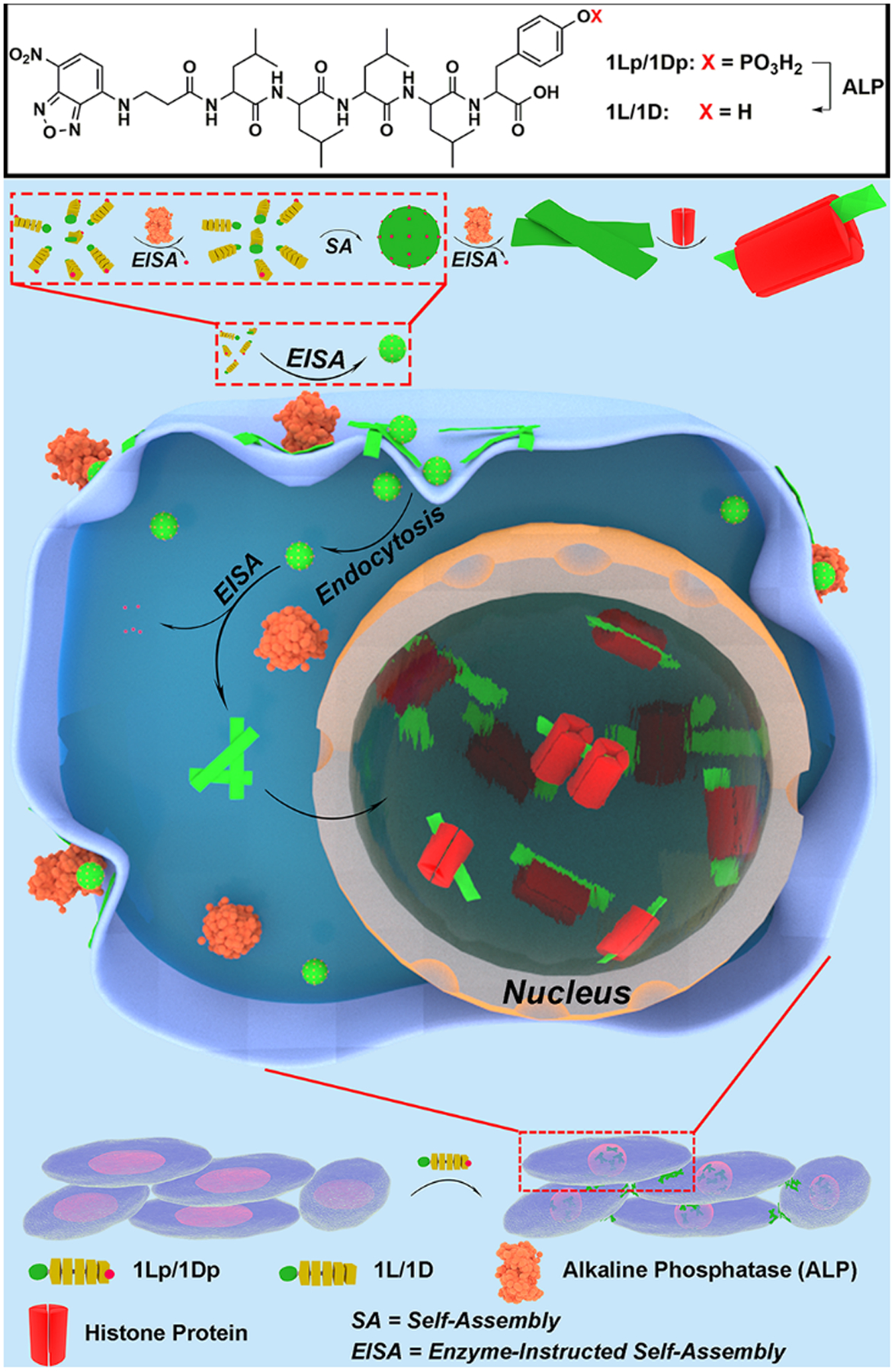
Schematic representation of EISA of 1Lp or 1Dp to result in intranuclear assemblies. After entering the cells, the micelle converts into nanoribbons upon ALP-dephosphorylation and co-assembles with histone proteins in cell nuclei (this illustration only depicts the key assemblies of the dynamic structural continuum generated in the process of EISA).
Results and Discussion
Molecular Synthesis and Characterization.
We used solid phase peptide synthesis (SPPS) to make the peptides (Scheme S1). Briefly, after reacting NBD-Cl and β-alanine to produce NBD-β-alanine and protecting L- or D-phosphotyrosine by 9-fluorenylmethoxycarbonyl (Fmoc), we synthesized the designed peptides via SPPS by using Fmoc-protected amino acids. After the deprotection, the condensation of A or B with biotin produces precursor 2Lp or 2Dp, respectively. After purification by high performance liquid chromatography (HPLC), MS and 1H NMR (Figure S1–S13) confirm the purity and identity of the precursors.
EISA to Form Nanoribbons.
Since the previous study has reported the EISA behaviors of 1Lp[12], we mainly describe that of 1Dp here. 1Dp dissolves in phosphate buffered saline (PBS) to yield a clear orange/yellow solution, even at 10 mM (Figure S14d and e), indicating excellent water solubility of 1Dp. Because of the Tyndall effect of colloids, self-assembly formed nanostructures enable a beam of light to become visible by scattering. Thus, we used the Tyndall test to study the self-assembly by naked eyes[15]. In the Tyndall test of 1Dp solution, beams of light (Figure S14d and e) confirm the self-assembly of 1Dp in PBS. In the solution of 1Dp (200 μM), transmission electron microscopes (TEM) images show only nanoparticles (Figure 2), and the critical micelle concentration (CMC) of 1Dp is 83.3 μM (Figure S18a). After being incubated with ALP (0.5 U/mL, 37°C, 24 h), 1Dp (200 μM) converts to 1D, and yellow precipitate appears. The Tyndall test also shows beam of light, confirming that 1D self-assembles in PBS to form colloids. TEM shows that dephosphorylation of 1Dp results in nanoribbons with the diameters of 85 ± 13 nm, and the CMC of 1D is 10.5 μM (Figure S18b). Interestingly, dephosphorylation of 50 μM of 1Dp by ALP provides nanofibers with diameter of 6 ± 1 nm, while 100 μM and 400 μM of 1D still self-assemble into nanoribbons (Figure S16).
Figure 2.

TEM images of 1Lp and 1Dp (200 μM, PBS) and the corresponding 1L and 1D formed 24 h after adding ALP (0.5 U/mL).
Because cell inhibitory activity of EISA depends the self-assembly supramolecular nanostructures formed by EISA, a key requirement for selectively killing osteosarcoma cells by EISA is that 1Dp undergoes EISA to form nanoribbons on and in the osteosarcoma cells, which overexpress ALP, but not on and in normal cells that do not overexpress ALP. To study the EISA of 1Dp at different ALP concentrations, we incubated 1Dp (200 μM) with different concentrations of ALP (from 0.1 U/mL to 0.8 U/mL) for 1 hour and 4 hours. One hour after the addition of ALP in the solution of 1Dp, TEM shows that micelles with diameter of 434 ± 86 nm form in the presence of 0.1 and 0.2 U/mL of ALP, and nanoribbons form in the presence of 0.4 and 0.8 U/mL of ALP (Figure S15). Four hours after the addition of ALP in the solution of 1Dp, TEM shows that micelles form in the presence of 0.1 U/mL of ALP, and nanoribbons form in the presence of 0.2, 0.4 and 0.8 U/mL of ALP (Figure S15). Besides, in presence of 0.1 U/mL of ALP, 1Dp self-assembles into nanoribbons without other nanostructures after dephosphorylation by ALP for 24 h (Figure S17), indicating that ALP hardly interact with the dephosphorylated pentapeptide (1D). These results indicate that 1Lp[12] and 1Dp share similar self-assembly behaviors, such as ALP triggered nanoparticle-to-nanoribbon transition. Moreover, the 1Lp and 1L share similar CMCs with 1Dp and 1D respectively. However, there are still a few differences: 1Lp shows a higher dephosphorylation rate than that of 1Dp, agreeing with that L-peptides are usually better substrates than D-peptide for enzymatic recognition and catalysis. The self-assembling nanostructures (both of micelle and nanoribbon) from the dephosphorylation of 1Dp has a larger size than that of 1Lp, likely resulting from the different dephosphorylation rates.
To understand the secondary structures of the 1Dp and 1D in the assemblies, we compared the circular dichroism (CD) spectra of 1Dp before and after the addition of ALP with those of 1Lp (Figure S19a and b). Without the addition of ALP, the solution of 1Dp exhibits only weak CD signals, indicating that the phosphate increases the solubility and disfavors forming extensive peptide assemblies. After the addition of ALP in the solution of 1Dp, to produce 1D, CD exhibits two negative bands at 203 and 228 nm. The CD signals of 1Dp and 1D are opposite to those of 1Lp and 1L[12], agreeing with that they are enantiomeric pairs. We measured the CD spectra of 1Dp at different concentrations before and after the addition of ALP. The molar CD intensity decrease as the increase of the concentration of 1Dp (Figure S19c and d), agreeing with the observation that 1D aggregates to form precipitates at high concentrations.
To study the influence of heterochirality, we examined the morphology change of the heterochiral control compound 1DLp (NBD-lLlLpy) before and after adding ALP. At 200 μM, the TEM images only show nanoparticles in both solutions of 1DLp without and with dephosphorylation. In the solution with ALP, we observed more dense nanoparticles, confirming that dephosphorylation enhance the ability of self-assembly (Figure S21). The CD spectra of 1DLp without and with ALP overlay well with each other, exhibiting slight negative bands at 202 nm and 225 nm (Figure S22). These results indicate that heterochirality disfavors self-assembly, agreeing with the our previous report[12].
Intranuclear Self-assembly.
Since the fluorescence of NBD increases significantly in hydrophobic environment, we used confocal laser scanning microscopy (CLSM) to reveal the cellular localization of the pentapeptide assemblies formed upon ALP dephosphorylation. After being incubated with 1Lp or 1Dp (200 μM) for 4 h, the SJSA-1 cells, which overexpress ALP, exhibit strong fluorescence in nuclei and much weaker fluorescence in cytoplasm and on the membrane except for a few puncta (Figure 3a). The bright field images show dark nuclei, which overlay well with the strong fluorescence. In addition, we observed stronger fluorescence and darker region in nucleoli than that in the other part in nuclei. In addition, the sizes of the cells and nuclei appear to be larger of the cells treated by 1Dp than by 1Lp (Figure 3a). This observation, agreeing with the amount of nuclear accumulation (vide infra), likely originates from the proteolytic resistance of D-peptides. The results indicate that the EISA of 1Lp or 1Dp targets and accumulates in nuclei of SJSA-1 cells, especially in nucleoli.
Figure 3.
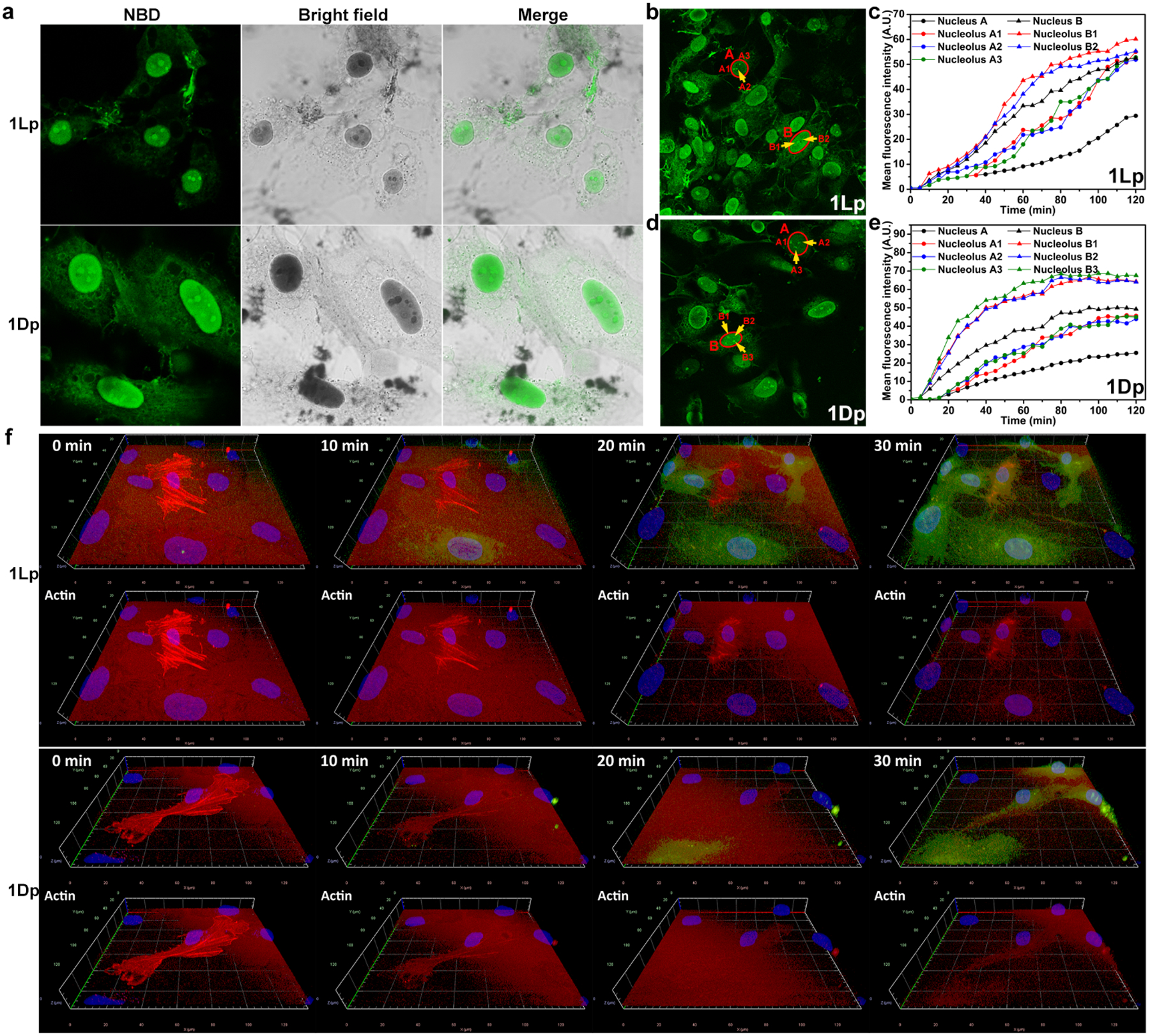
a) CLSM images of SJSA-1 cells treated by 1Lp or 1Dp (200 μM) for 4 h. b) The final fluorescent image of time-dependent CLSM of SJSA-1 cells treated by 1Lp (200 μM) for 2 h (Movie 1). c) The increase of mean fluorescence intensity of the areas in b). d) The final fluorescent image of time-dependent CLSM of SJSA-1 cells treated by 1Dp (200 μM) for 2 h (Movie 2). e) The increase of mean fluorescence intensity of the areas in d). f) 3D CLSM images of SJSA-1 cells (RFP-actin) incubated with 1Lp or 1Dp for 0, 10, 20, and 30 min (Movies 11 and 12).
The nucleus-targeting depends on the concentration of the precursors. When the concentration is 100 μM or lower, there is hardly any fluorescence in the cells (Figure S23 and S24). After incubation with 1Lp or 1Dp for different time (less than 4 h), the fluorescence in cell nuclei increases as the increasing of incubation time (Figure S25, S26, S29 and S30). However, incubating SJSA-1 cells with the heterochiral phosphopeptide, 1DLp, hardly results in any fluorescence after 4 h, even when the concentration is as high as 400 μM (Figure S31). The weaker self-assembly ability of the heterochiral sequence, 1DLp, than that of 1Lp or 1Dp, leads to the absence of nucleus-targeting ability of 1DLp, similar with the case of iPSCs[12].
We also incubated other cells with 1Lp or 1Dp. After being incubated with 1Lp or 1Dp for 4 h, the Saos2 cells, another osteosarcoma cell line, exhibit strong NBD fluorescence in nuclei, like that in SJSA-1 cells (Figure S27 and S28). While only a couple of HeLa cells exhibit NBD fluorescence in nuclei, no fluorescence appears in HepG2 cells, HS-5 cells, and HEK293 cells after incubation with 1Lp or 1Dp for 4 h. The results confirm that the precursors selectively target the nuclei of the osteosarcoma cells that overexpress ALP. Since these cells express different levels of ALP, this observation suggests that 1Lp or 1Dp relies on rapid dephosphorylation catalyzed by ALP for targeting the nuclei.
We used an inhibitor (2,5-dimethoxy-N-(quinolin-3-yl)-benzenesulfonamide (DQB)) to inhibit ALP[16] or treated the cells with phospholipase C (PLC) to remove GPI-anchored ALP on cell membrane[17]. After incubation with 1Lp or 1Dp and DQB for 4 h, SJSA-1 cells exhibit no fluorescence (Figure S32). After the pretreatment of SJSA-1 cells with PLC (0.2 U/mL, 24 h), CLSM shows little fluorescence in the cells incubated 1Lp or 1Dp. SJSA-1 cells incubated with 1L or 1D, which lacks phosphate, only show weak fluorescence on the membrane. These results further support that the nucleus-targeting mainly originates from the ALP-catalyzed dephosphorylation.
Because HEK293 cells has relatively low expression of ALP and no fluorescence in nuclei of HEK293 after incubation with 1Lp or 1Dp (Figure S27 and S28), we used TNAP-RFP transfected HEK293 cells[18], which overexpress and show intracellular distribution TNAP (i.e., tissue-nonspecific ALP), as a control to delineate the role of ALP. The 2D time-lapse CLSM images show the NBD fluorescence appearing in the TNAP-transfected SJSA cells and then in the cell nuclei (Movies 9 and 10, Figure S33 and S34) when the TNAP-RFP transfected HEK293 cells being incubated with 1Lp or 1Dp. These results support with the assumption that nucleus-targeting mainly originates from the ALP-catalyzed EISA.
To trace the dynamics of the EISA of 1Lp and 1Dp inside SJSA-1 cells, we used 2D time-lapse CLSM to image the changes of the fluorescence in the cells being incubated with 1Lp or 1Dp (200 μM) (Movies 1 and 2, Figure S35 and S37). The fluorescence of the assemblies first appears in the cytoplasm and then become intense in nuclei (Figure S36 and S38). The nuclei shrink after the assemblies emerge in the nuclei, and the nuclei show nuclear blebbing (Figure S35 and S37, red arrows). We monitored the increase of fluorescence in several SJSA-1 cells over 2 h (Figure 3b and d). Figure 3c shows the mean fluorescence intensity in the nuclei of the two cells and their nucleoli. The mean fluorescence intensity in the two nuclei keep increasing in 2 h. The fluorescence in the nuclei and nucleoli are synchronous. The SJSA-1 cells, being incubated with 1Dp (200 μM), exhibit similar fluorescence increase to those of SJSA-1 cells being incubated with 1Lp (Figure 3e). We also used 1Lp or 1Dp to treat RFP expressing SJSA-1 cells (Movies 7 and 8). While being incubated with 1Lp or 1Dp, the SJSA-1 cells show the release of RFP, confirming the disruption of cell membrane and release of cytoplasm (Figure S39 and S40, yellow arrows). The nucleus-shrinking, nucleus-blebbing, and the disruption of cell membrane induced by EISA in nuclei contribute to the cell death (vide infra).
We also used 3D time-lapse CLSM to image the changes of the fluorescence in SJSA-1 cells being incubated with 1Lp or 1Dp (Movies 3 and 4). The NBD fluorescence first appears on the membrane of cells especially the filopodia contact points among cells (Figure S41 and S42), then the fluorescence appears in the cytoplasm and nuclei. The nucleus-shrinking observed via 2D CLSM imaging is the humping of the nuclei (Movies 3, 4 and 15, side views), which causes the shrinking of intersecting surface. The nucleus-humping usually accompanies cell-humping.
Since actin maintains cell shape[19], the nucleus-humping likely is resulted from the dysregulation of actin dynamics. We transfected actin-RFP fusion construct into SJSA-1 cells, which expresses RFP fused to cell actin. The 3D CLSM imaging shows the disruption of actin filaments after incubation with 1Lp or 1Dp significantly (Figure 3f, Movies 11 and 12). We also used 1Lp or 1Dp to treat another osteosarcoma cell line, Saos2 cell line, the 2D CLSM imaging shows similar cell behaviors observed in SJSA-1 cells (Movies 5 and 6). The 3D CLSM imaging also shows the disruption of actin filaments in cells (Movies 13 and 14). Besides, the Saos2 cells also exhibit the disruption of tubulin (Figure S43). Those results indicate that the EISA in nuclei is resulted from disruption of actin and microtubule dynamics, which lead to the loss of nucleus integrity (evidenced by nucleus-humping and nucleus-blebbing) and the disruption of cell membrane. Such multiple disruptions (of cytoskeletons and membrane integrity) likely contribute to the cell death and minimize acquired drug resistance (vide infra).
Cellular Uptake.
The CLSM image of the SJSA-1 cells with 1Lp or 1Dp at 4°C exhibit no fluorescence, indicating that that the SJSA-1 cells uptake 1Lp and 1Dp upon endocytic process, which slow down at low temperature[14, 20]. To determine the modes of endocytic process being responsible for the uptake, we first seeded the SJSA-1 cells in a 96-well plate, after the co-incubation of several endocytic inhibitors with 1Lp or 1Dp in the culture of the cells, we dissolved the cells in DMSO and measured the fluorescence by a plate reader. Methyl-β-cyclodextrin (M-βCD), filipin III, and cytochalasin D (CytD) significantly decrease the uptake of 1Lp and 1Dp, but 5-(N-ethyl-N-isopropyl)amiloride (EIPA) and chlorpromazine (CPZ) show almost no effects on the uptake (Figure 4c and d). Besides, M-βCD, filipin III, or CytD decreases the cytotoxicity (vide infra) of 1Lp and 1Dp against SJSA-1 cells (Figure 4b). Since M-βCD and filipin III inhibits caveolin-dependent endocytosis, these results suggest that the assemblies enter cells via lipid raft/caveolae-mediated endocytosis[21]. CytD inhibits the endocytosis by inhibiting polymerization of actin[22], but the disruption of actin also affect the nucleus-targeting EISA. Thus, the effect of CytD is inconclusive.
Figure 4.
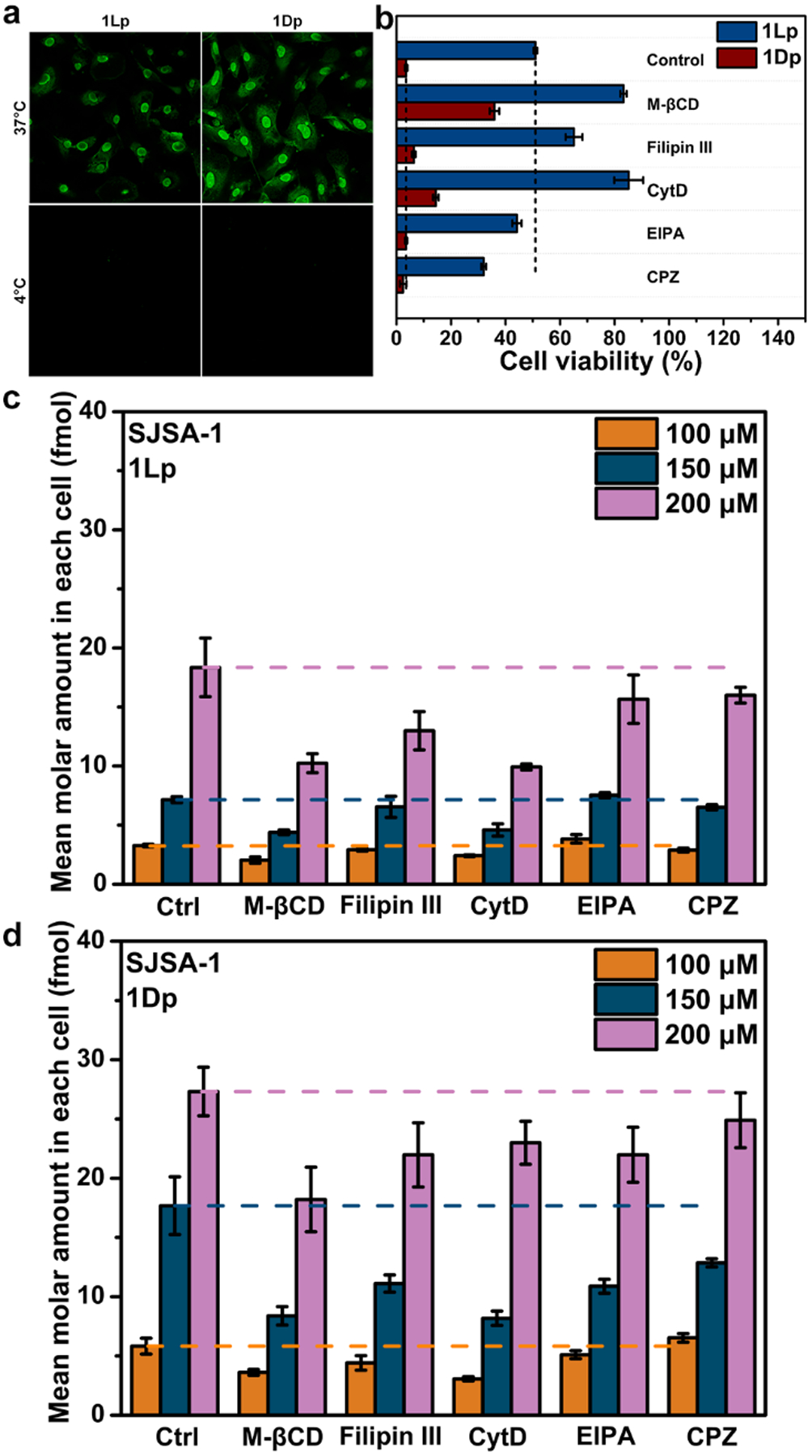
a) CLSM images of SJSA-1 cells after being treated by 1Lp or 1Dp (200 μM) for 4 h at 37°C or 4°C. b) Cell viability of SJSA-1 cells incubated with 1Lp or 1Dp (100 μM) for 24 h in absence (Control) or presence of inhibitors M-βCD (2 mM), Filipin III (2 μg/mL), CytD (2.5 μg/mL), EIPA (1 μM), or CPZ (30 μM). The mean molar amount of uptake in each SJSA-1 cells incubated with c) 1Lp or d) 1Dp (100 μM, 150 μM, and 200 μM) for 2 h in absence (Ctrl) or presence of inhibitors M-βCD (2 mM), Filipin III (2 μg/mL), CytD (2.5 μg/mL), EIPA (1 μM), or CPZ (30 μM).
We also used the CLSM to observe the cells co-incubated with 1Lp or 1Dp and the endocytosis inhibitors. The CLSM of the SJSA-1 cells show that M-βCD and filipin III significantly decrease the fluorescence, while EIPA and CPZ hardly decrease the fluorescence (Figure S44 and S45). CytD decreases the fluorescence in SJSA-1 cells incubated with 1Lp, but hardly decreases the fluorescence in SJSA-1 cells incubated with 1Dp. Besides, 1Dp also causes a shape change of SJSA-1 cells (Figure S45). This difference indicates that 1Lp depends more on actin dynamics than 1Dp for entering the nucleus. The above results indicate that 1Lp and 1Dp enter cells via lipid raft/caveolae-mediated endocytosis.
Nuclear Accumulation.
We also examined the accumulation of the assemblies quantitatively. After using 1Lp or 1Dp to incubate the cells, we lysed the SJSA-1 cells and used centrifugation[13, 23] to fraction the cells. We mainly obtained two samples: Sample N consisting of nuclei; and Sample M consisting of mitochondria, lysosomes and peroxisomes (Figure 5). We measured the NBD fluorescence to quantify the mean amount of 1Lp (or 1L) or 1Dp (or 1D) in each cell. The SJSA-1 incubated with 1Lp and 1Dp show similar amounts of assemblies in mitochondria, lysosomes and peroxisomes of each cell, which are (5.5 ± 0.096) × 10−16 and (5.6 ± 0.091) × 10−16 mol, respectively. However, the amounts of the assemblies of the L- and D-peptides in nuclei differ: (3.8 ± 0.041) × 10−16 and (6.7 ± 0.071) × 10−16 mol of 1Lp (or 1L) and 1Dp (or 1D), respectively accumulate in the nucleus. The volume of an osteosarcoma cell is about 4000 μm3, and nucleus is about 10% of cell volume. Thus, the estimated concentrations of 1Lp (or 1L) and 1Dp (or 1D) in each nucleus are 950 ± 10 and 1675 ± 18 μM, respectively (ignore the size change of nuclei). which are much higher than the incubation concentration (200 μM). This result confirms that the peptide assemblies accumulate in nuclei. The concentration of L-assemblies is slightly lower than that of D-assemblies, agreeing with that L-peptides are more proteolytic than D-peptides.
Figure 5.
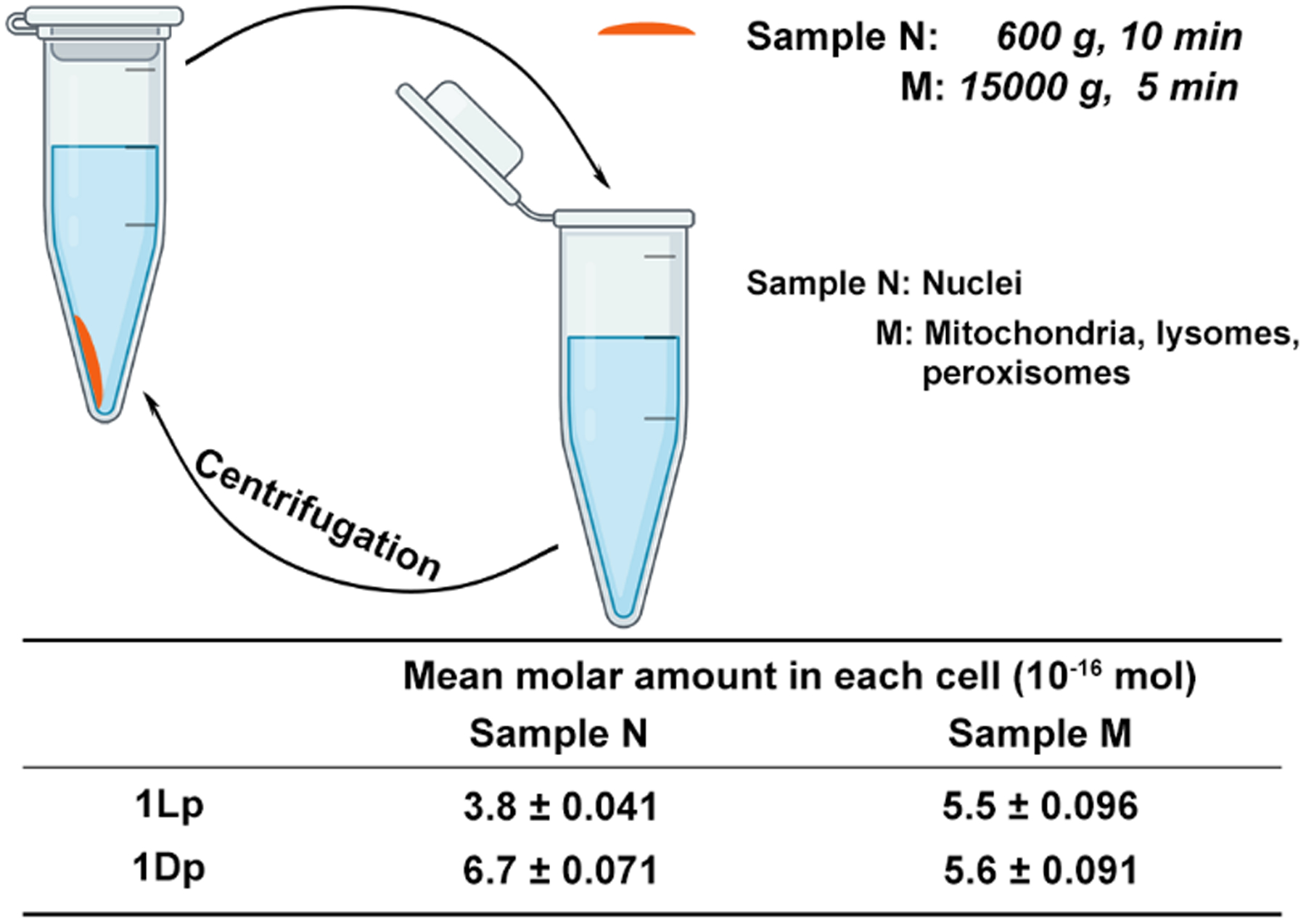
The fractionation of assemblies in SJSA-1 cells incubated with 1Lp or 1Dp (200 μM) for 4 h.
Interactions with Histone Proteins
To study the mechanism for intranuclear self-assembly, we examined the interaction of assemblies with the proteins in the SJSA-1 cells. We used RayBio Biotin beads, magnetic beads modified with streptavidin, to enrich the proteins co-assembling with the assemblies from 1Lp or 1Dp because streptavidin shows high-affinity for biotin binding[24]. We synthesized 2Lp and 2Dp by attaching biotin to 1Lp and 1Dp for targeting nuclei, co-assembling, and enriching proteins that interact with the peptide assemblies. Before dephosphorylation, 2Lp and 2Dp (400 μM) self-assemble to form short nanofibers (Figure S46). After ALP-dephosphorylation (0.5 U/mL, 24 h), TEM images of 2Lp (or 2Dp) exhibit long nanofibers (Figure S46), differing from those of 1Lp and 1Dp (nanoribbons). The CD spectra of 2Lp and 2Dp after dephosphorylation exhibit weak CD signals (Figure S47). The CD spectra exhibit both α-helix component and β-sheet component, but the CD spectrum of dephosphorylated-2Lp (2L) exhibits higher α-helix component ratio than that of 2D.
The SJSA-1 cells, being treated by 2Lp or 2Dp, exhibit significant NBD-fluorescence in nuclei in the case of 2Lp, but but not in the case of 2Dp (Figure S48). The difference of the nucleus-targeting ability is consistent with the weak self-assembling ability of the diastereomer that consists of biotin and D-peptide and agrees with our previous result show that a diastereomer (NBD-llllpY) possesses weak nuclear-targeting ability[12].
Based on the above results, we chose 2Lp as the probe to target the cell nuclei and enrich proteins. We made two samples, PC (Proteomics Cell) and PL (Proteomics Lysate), for proteomics analysis (Figure 6a). The LC-MS data were analyzed with the PEAKS proteomics software[25]. 187 proteins were detected in Sample PC (Table S1), while only 22 proteins were detected in Sample PL (Table S2). Figure 6b lists the proteins found in Sample PC but not found in Sample PL with a coverage higher than 10%. Those proteins likely involve the nucleus-targeting by the peptides (2Lp). The protein with the highest coverage (62%) is histone H4 (H4C1), which is a core component of nucleosome. Another two histone proteins also present: Histone H3.1 (H3C1), a core component of nucleosome, reaches a coverage of 38%; Histone H1.5 (H1–5), which binds to linker DNA between nucleosomes and chromatin fibers, exhibits a coverage of 19%. The TEM images also show that 1Lp and 1Dp can co-assemble with Histone H4 (Figure S49d and e). The results suggest that the EISA of 1Lp and 1Dp prefer to co-assemble with histone proteins in nuclei and imply that some of 1Lp or 1Dp remain phosphorylated prior entering nucleus. Since nucleolus is composed of ribosomal RNA and proteins[26], the results agree with the above observations that the EISA molecules prefer to accumulate in nucleoli.
Figure 6.
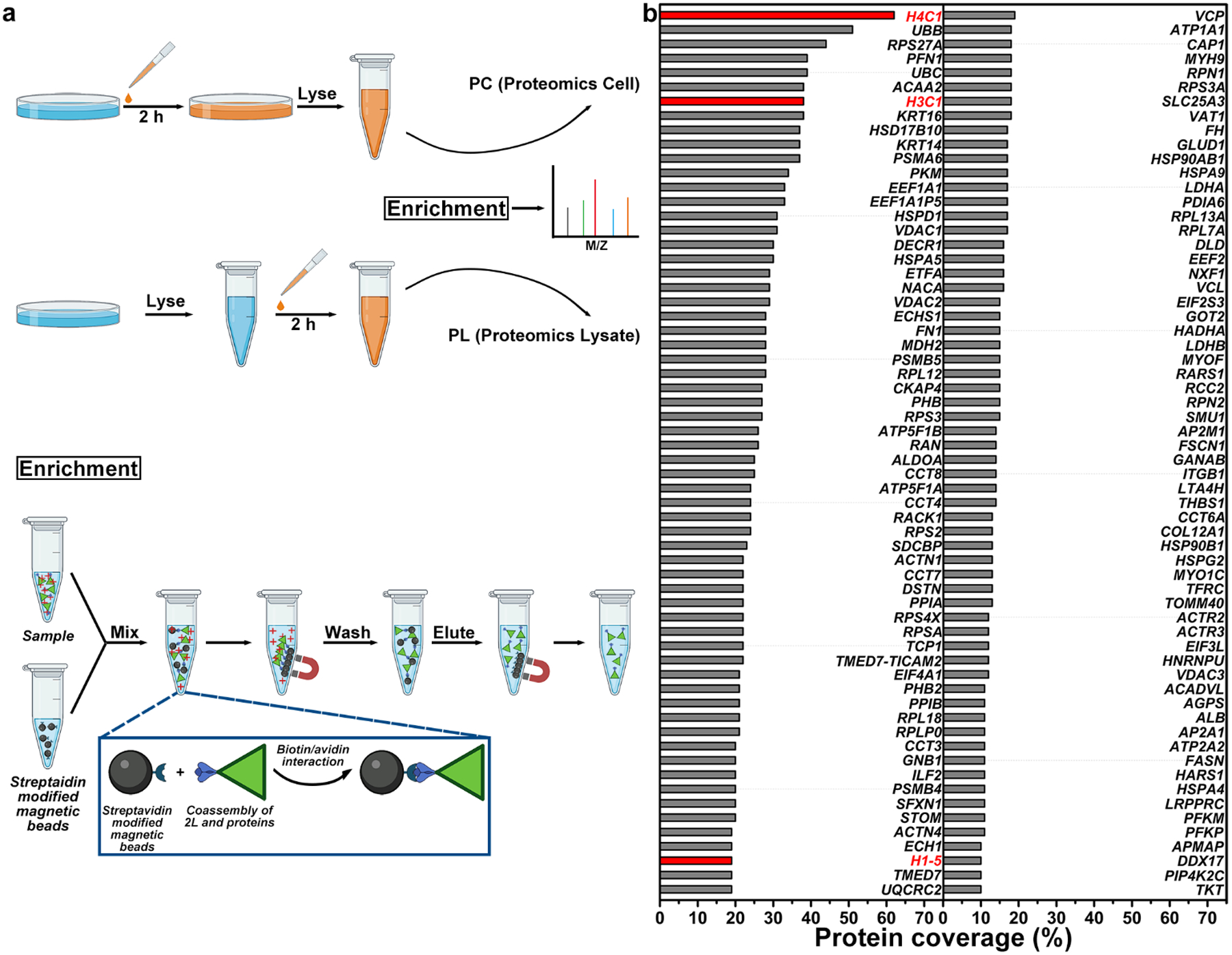
a) The protocol for the preparation of samples PC and PL. b) The coverage of the proteins found in Sample PC but not found in Sample PL with a coverage higher than 10%. The red color shows the histone proteins, which may relate to nuclear targeting.
We also examine the ability of 1Lp and 1Dp to aggregate with DNA. We incubated the mixture of 1Lp or 1Dp, DNA and ALP at 37°C for 24 h. After centrifugation, we measured the DNA concentration of the supernatant as the concentration of remain DNA. The result shows that the EISA of 1Lp induces the co-precipitation of 33.4% DNA and that of 1Dp induces the co-precipitation of 24.7% DNA (Figure S50). TEM images of the mixture of DNA and 1L or 1D (Figure S51) hardly show any new nanostructure of the co-assembles or morphological changes that usually associate with strong binding, indicating that 1L or 1D weakly interact with DNA moderately. However, the direct or indirect interactions of the nanoribbons with DNA inside cells remains to be verified.
Selectively Killing Osteosarcoma Cells
We used 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay[27] to examine the viability of SJSA-1 cells incubated with the precursors (25 μM to 200 μM) for 24 h. After incubation of 24 h, 200 μM of 1Lp or 1Dp kills more than 96% or 99% of SJSA-1 cells (Figure 7a), respectively. 1Lp and 1Dp exhibits IC50 of 101 and 69 μM (24 h), respectively. Interestingly, the two precursors kill the SJSA-1 cells rapidly (Figure S52b and c). After 3 h incubation, 200 μM of 1Lp or 1Dp kills more than 94% or 97% of SJSA-1 cells, respectively. Since cell death occurs fast, the release of the phosphopeptide and peptide from the cytosol make it difficult to precisely determine the percentage of dephosphorylation of the intracellular assemblies. Besides, the mixture of 1Lp and 1Dp hardly effects their cytotoxicity (Figure S52a). We also examined the viability of Saos2 cells cultured with 1Lp and 1Dp (Figure S53) and found the IC50 to 122 μM and 99 μM (24 h), respectively. Against both the osteosarcoma cells, 1Dp exhibit higher cytotoxicity than 1Lp. As a control, we examined the viability of normal cells (e.g., HS5 and HEK293) and other cancer cells (e.g., HeLa, HepG2, OVSAHO, A2780 and A2780cis). After incubation of 24 h, all the cells exhibit viability higher than 60% (Figure 7b). The results indicate that the two precursors selectively inhibit the osteosarcoma cells, agreeing with that the two precursors selectively target the nuclei of the osteosarcoma cells overexpressing ALP.
Figure 7.
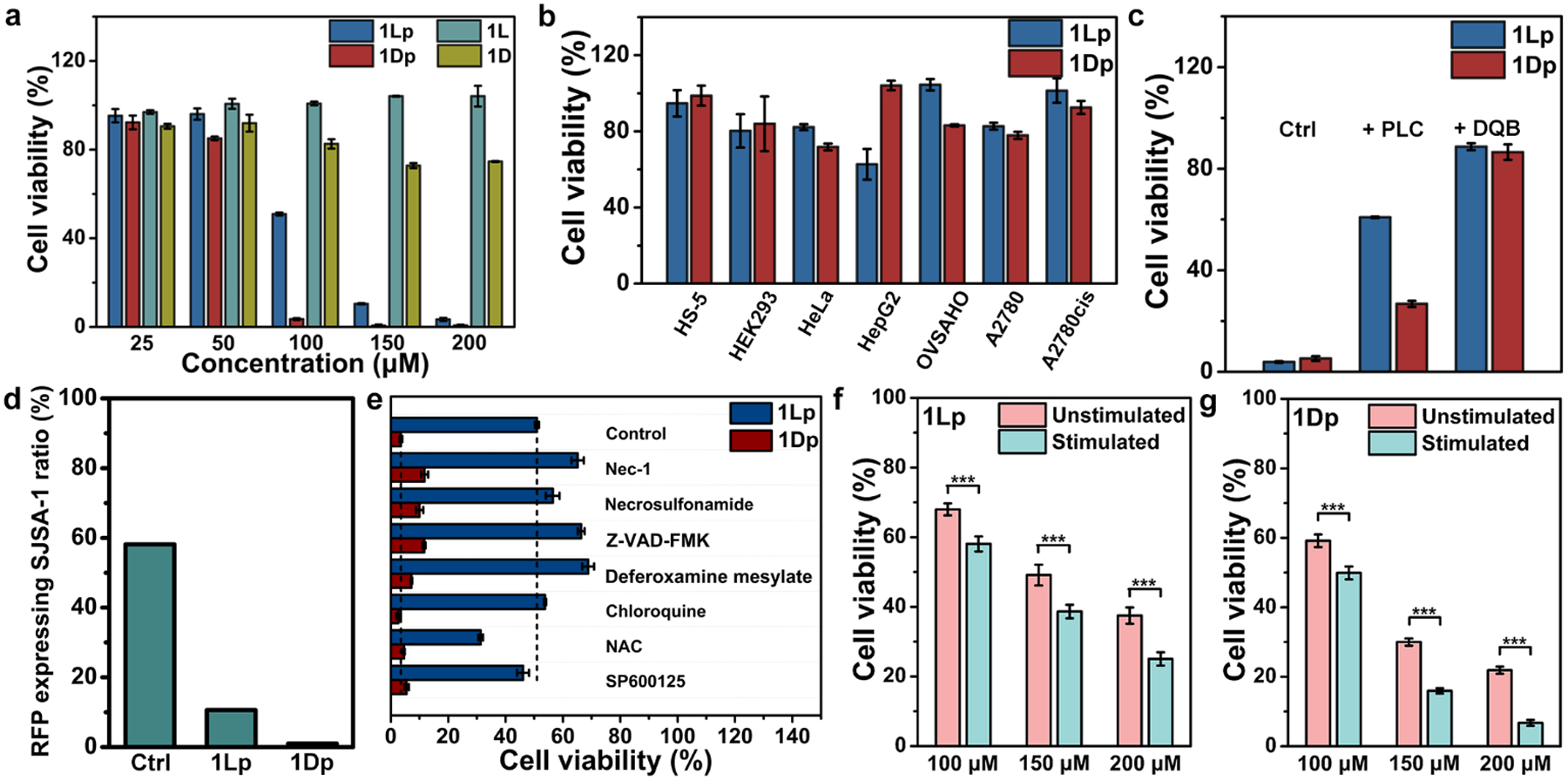
a) Cell viability of SJSA-1 cells incubated with 1Lp, 1Dp, 1L, or 1D for 24 h. b) The cell viability of HS-5, HEK293, HeLa, HepG2, OVSAHO, A2780, and A2780cis cells incubated with 1Lp or 1Dp (200 μM) for 24 h. c) Cell viability of SJSA-1 cells incubated with 1Lp or 1Dp (200 μM) for 24 h in absence (Ctrl) or presence of PLC (0.2 U/mL, pretreat for 24 h) or DQB (5 μM). d) The remain ratio of RFP expressing SJSA-1 in the mixture of RFP expressing SJSA-1 and HS-5 incubated with 1Lp or 1Dp for 4 h and subculture. The starting ratio of RFP expressing SJSA-1 in the mixture is 50%. e) Cell viability of SJSA-1 cells incubated with 1Lp or 1Dp (100 μM) for 24 h in absence (Control) or presence of death signaling inhibitors Nec-1 (50 μM), Necrosulfonamide (1 μM), Z-VAD-FMK (50 μM), Deferoxamine mesylate (200 μM), Chloroquine (10 μM), NAC (1 mM), or SP600125 (25 μM). Cell viability of unstimulated SJSA-1 cells or stimulated SJSA-1 cells (after 5 weeks’ treatment of the precursors with gradually increase concentrations) incubated with f) 1Lp or g) 1Dp (100 μM, 150μM and 200 μM) for 24 h (n= 3, ***, p < 0.001).
To mimic tumor microenvironment that the cancer cells mix with normal cells, we mixed the same amount of RFP expressing SJSA-1 cells and HS-5 cells. Then, we used 1Lp or 1Dp to treat the mixed cells for 4 h, refreshed culture media and removed dead cells. Then, we counted the cells and calculated the ratio of RFP expressing SJSA-1 cells by using CLSM, after Hoechst staining (Figure 7d). After above treatment, the ratio of RFP expressing SJSA-1 cells in the control group is 58%, which is higher than 50% and indicates slightly faster growth SJSA-1 over HS-5 cells. After the treatment of 1Lp, the ratio of RFP expressing SJSA-1 cells in the mixture is 11%, and after the treatment of 1Dp, the ratio of RFP expressing SJSA-1 cells in the mixture is 0.98%. The observation indicates that 1Lp or 1Dp significantly decreases the ratio of RFP expressing SJSA-1 cells in the co-culture. The higher efficacy of 1Dp agrees with that 1Dp exhibit higher cytotoxicity than 1Lp against osteosarcoma cells.
After 24 h incubation, both 1L and 1D (200 μM) hardly exhibit cytotoxicity against SJSA-1 cells (Figure 7a). We also used DQB to inhibit ALP or used PLC to remove GPI-anchored ALP on cell membrane during cell viability experiment (Figure 7c). Co-incubation of DQB (5 μM) or pretreatment of PLC (0.2 U/mL, 24 h) significantly rescue SJSA-1 cells incubated with 1Lp or 1Dp (200 μM) for 24 h, agreeing with the CLSM imaging results (Figure S32). Thees results reveal that the ALP-dephosphorylation is essential for inhibition of osteosarcoma cells, agreeing with the results that ALP-dephosphorylation is essential for 1Lp or 1Dp to target the nuclei of the osteosarcoma cells.
Modes of Cell Death.
To evaluate the modality of cell death induced by 1Lp and 1Dp, we co-incubated cell death inhibitors[28] (necrostatin-1 (Nec-1), necrosulfonamide, Z-VAD-FMK, deferoxamine mesylate, chloroquine, N-acetyl-L-cysteine (NAC), and SP600125) with the 1Lp or 1Dp in the culture of the SJSA-1 cells (Figure 7e). Nec-1 and necrosulfonamide are necroptosis inhibitors[29], and they both rescue the SJSA-1 cells incubated with 1Lp or 1Dp. Z-VAD-FMK is a pan-caspase inbibitor[30], which is able to reduce the cytotoxicity of 1Lp and 1Dp. Deferoxamine mesylate, a ferroptosis inhibitor[31], also rescues SJSA-1 cells when being co-incubated with 1Lp or 1Dp. On the other hand, chloroquine (an inhibitor of autophagy-dependent cell death[28), NAC (an inhibitor of alkaliptosis, oxeiptosis and lysosome-dependent cell death[28]) or SP600125 (an inhibitor of lysosome permeation-induced cell death[32) hardly reduce the cytotoxicity of 1Lp and 1Dp. Interestingly, the co-incubation with NAC results in more cell death. The results indicate that the cell death induced by the nucleus-targeting EISA of 1Lp or 1Dp involves multiple cell death modality (e.g., necroptosis, apoptosis, and ferroptosis).
Minimizing Acquired Drug Resistance.
Based on the above results, the cell death induced by the nucleus-targeting EISA relates to accumulation of assemblies in nuclei, we reckon that cancer cells unlikely would acquire drug resistance. To test this assumption, we incubated 1Lp or 1Dp with SJSA-1 cells by gradually increasing the concentrations of the precursors from 20 μM to IC50 (101 and 69 μM for 1Lp 1Dp, respectively) for 5 weeks and selected the cells that survive the treatment. Then we examined the viability of the stimulated SJSA-1 cells incubated with 1Lp or 1Dp by MTT assay (Figure 7f and g). Surprisingly, the repeated stimulation significantly sensitizes the SJSA-1 cells to 1Lp and 1Dp. The observation confirms that 1Lp and 1Dp selectively inhibit osteosarcoma cells without evolving acquired drug resistance[33].
Conclusion
In summary, we report a pair of L- and D-leucine-rich phosphopeptide (1Lp and 1Dp) that selectively kills osteosarcoma cells by intranuclear peptide assemblies formed by EISA. The assemblies accumulate in cell nuclei and likely interact with histone proteins in nuclei. The high selectivity of the phosphopeptides towards to SJSA-1 originates from that the highest expression of ALP (Figure S54) in SJSA-1 results in fast generation of nanoribbons and the activity of EISA depends on the rate of EISA process[34]. The cell death induced by the nucleus-targeting EISA involve multiple cell death modality (e.g., necroptosis, apoptosis, and ferroptosis), and the cells hardly acquire drug resistance after repeated treatment of the phosphopeptides. Although the concentration of the phosphopeptide is relatively high, this result indicates that a nucleus-targeting EISA minimizing acquiring drug resistance, which promises a development of anti-tumor method[35] via subcellular targeting EISA. The nucleus-targeting relates to co-assembly with histone proteins in nuclei, which may provide a novel strategy to disrupt interorganelle communication for inducing cell death. Although this work focuses on pentapeptides, the insights obtained from this work may be useful for design other peptide assemblies[36] or for EISA triggered by other enzymes[37].
Supplementary Material
Acknowledgements
We acknowledge the National Institutes of Health (CA142746 and CA262920) and National Science Foundation (DMR-2011846).
Footnotes
Conflict of interest
The authors declare no conflict of interest.
References
- [1].a) Lamond AI, Earnshaw WC, Science 1998, 280, 547–553; [DOI] [PubMed] [Google Scholar]; b) Vankayala R, Kuo C-L, Nuthalapati K, Chiang C-S, Hwang KC, Adv. Funct. Mater 2015, 25, 5934–5945. [Google Scholar]
- [2].Dundr M, Misteli T, Biochem. J 2001, 356, 297–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].a) Wang H, Feng Z, Tan W, Xu B, Bioconjugate Chem. 2019, 30, 2528–2532; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) O’Sullivan JM, Pai DA, Cridge AG, Engelke DR, Ganley ARD, Biomol. Concepts 2013, 4, 277–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Carotenuto P, Pecoraro A, Palma G, Russo G, Russo A, Cells 2019, 8, 1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].a) Pan L, Liu J, Shi J, Chem. Soc. Rev 2018, 47, 6930–6946; [DOI] [PubMed] [Google Scholar]; b) Pouton CW, Wagstaff KM, Roth DM, Moseley GW, Jans DA, Adv. Drug Deliver. Rev 2007, 59, 698–717. [DOI] [PubMed] [Google Scholar]
- [6].a) Jeang KT, Xiao H, Rich EA, J. Biol. Chem 1999, 274, 28837–28840; [DOI] [PubMed] [Google Scholar]; b) Pan L, He Q, Liu J, Chen Y, Ma M, Zhang L, Shi J, J. Am. Chem. Soc 2012, 134, 5722–5725. [DOI] [PubMed] [Google Scholar]
- [7].Fontana P, Dong Y, Pi X, Tong AB, Hecksel CW, Wang L, Fu T-M, Bustamante C, Wu H, Science 2022, 376, eabm9326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].a) Huo S, Jin S, Ma X, Xue X, Yang K, Kumar A, Wang PC, Zhang J, Hu Z, Liang X-J, ACS Nano 2014, 8, 5852–5862; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hinde E, Thammasiraphop K, Duong HTT, Yeow J, Karagoz B, Boyer C, Gooding JJ, Gaus K, Nat. Nanotechnol 2017, 12, 81–89. [DOI] [PubMed] [Google Scholar]
- [9].a) Arisaka A, Mogaki R, Okuro K, Aida T, J. Am. Chem. Soc 2018, 140, 2687–2692; [DOI] [PubMed] [Google Scholar]; b) Cheng Y, Sun C, Liu R, Yang J, Dai J, Zhai T, Lou X, Xia F, Angew. Chem. Int. Ed 2019, 58, 5049–5053; [DOI] [PubMed] [Google Scholar]; c) Kong J, Wang Y, Zhang J, Qi W, Su R, He Z, Angew. Chem. Int. Ed 2018, 57, 14032–14036. [DOI] [PubMed] [Google Scholar]
- [10].Zhu Y-X, Jia H-R, Pan G-Y, Ulrich NW, Chen Z, Wu F-G, J. Am. Chem. Soc 2018, 140, 4062–4070. [DOI] [PubMed] [Google Scholar]
- [11].Tang R, Wang M, Ray M, Jiang Y, Jiang Z, Xu Q, Rotello VM, J. Am. Chem. Soc 2017, 139, 8547–8551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Liu S, Zhang Q, Shy AN, Yi M, He H, Lu S, Xu B, J. Am. Chem. Soc 2021, 143, 15852–15862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Gao Y, Shi J, Yuan D, Xu B, Nat. Commun 2012, 3, 1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Wang H, Feng Z, Wang Y, Zhou R, Yang Z, Xu B, J. Am. Chem. Soc 2016, 138, 16046–16055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kraemer EO, Dexter ST, J. Phys. Chem 2002, 31, 764–782. [Google Scholar]
- [16].Sekaran S, Vimalraj S, Thangavelu L, Biomolecules 2021, 11, 1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Hutchinson TE, Rastogi A, Prasad R, Pereira BMJ, Anim. Prod. Sci 2005, 88, 271–286. [DOI] [PubMed] [Google Scholar]
- [18].a) He H, Guo J, Xu J, Wang J, Liu S, Xu B, Nano Lett. 2021, 21, 4078–4085; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Backliwal G, Hildinger M, Hasija V, Wurm FM, Biotechnol. Bioeng 2008, 99, 721–727. [DOI] [PubMed] [Google Scholar]
- [19].Pollard TD, Cooper JA, Science 2009, 326, 1208–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Gump JM, Dowdy SF, Trends Mol. Med 2007, 13, 443–448. [DOI] [PubMed] [Google Scholar]
- [21].a) Ros-Baró A, López-Iglesias C, Peiró S, Bellido D, Palacín M, Zorzano A, Camps M, Proc. Natl. Acad. Sci 2001, 98, 12050–12055; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Rejman J, Oberle V, Zuhorn IS, Hoekstra D, Biochem. J 2004, 377, 159–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Casella JF, Flanagan MD, Lin S, Nature 1981, 293, 302–305. [DOI] [PubMed] [Google Scholar]
- [23].Dopp E, von Recklinghausen U, Hartmann LM, Stueckradt I, Pollok I, Rabieh S, Hao L, Nussler A, Katier C, Hirner AV, Rettenmeier AW, Drug Metab. Dispos 2008, 36, 971–979. [DOI] [PubMed] [Google Scholar]
- [24].Weber PC, Ohlendorf DH, Wendoloski JJ, Salemme FR, Science 1989, 243, 85–88. [DOI] [PubMed] [Google Scholar]
- [25].Ma B, Zhang K, Hendrie C, Liang C, Li M, Doherty-Kirby A, Lajoie G, Rapid. Commun. Mass. Sp 2003, 17, 2337–2342. [DOI] [PubMed] [Google Scholar]
- [26].a) Hernandez-Verdun D, Roussel P, Thiry M, Sirri V, Lafontaine DLJ, WIRES RNA 2010, 1, 415–431; [DOI] [PubMed] [Google Scholar]; b) Scheer U, Hock R, Curr. Opin. Cell Biol 1999, 11, 385–390. [DOI] [PubMed] [Google Scholar]
- [27].Gerlier D, Thomasset N, Immuno J. Methods 1986, 94, 57–63. [DOI] [PubMed] [Google Scholar]
- [28].Tang D, Kang R, Berghe TV, Vandenabeele P, Kroemer G, Cell Res. 2019, 29, 347–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].a) Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA, Yuan J, Nat. Chem. Bio 2005, 1, 112–119; [DOI] [PubMed] [Google Scholar]; b) Liao D, Sun L, Liu W, He S, Wang X, Lei X, MedChemComm 2014, 5, 333–337. [Google Scholar]
- [30].Slee EA, Zhu H, Chow SC, MacFarlane M, Nicholson DW, Cohen GM, Biochem. J 1996, 315, 21–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Ouyang S, You J, Zhi C, Li P, Lin X, Tan X, Ma W, Li L, Xie W, Cell Death Dis. 2021, 12, 782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Werneburg NW, Guicciardi ME, Bronk SF, Kaufmann SH, Gores GJ, J. Biol. Chem 2007, 282, 28960–28970. [DOI] [PubMed] [Google Scholar]
- [33].Housman G, Byler S, Heerboth S, Lapinska K, Longacre M, Snyder N, Sarkar S, Cancers 2014, 6, 1769–1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Zhou J, Du X, Xu B, Angew. Chem. Int. Ed 2016, 55, 5770–5775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Chen P, Ma Y, Zheng Z, Wu C, Wang Y, Liang G, Nat. Commun 2019, 10, 1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].a) Wu D, Sinha N, Lee J, Sutherland BP, Halaszynski NI, Tian Y, Caplan J, Zhang HV, Saven JG, Kloxin CJ, Pochan DJ, Nature 2019, 574, 658–662; [DOI] [PubMed] [Google Scholar]; b) Wang F, Su H, Xu D, Dai W, Zhang W, Wang Z, Anderson CF, Zheng M, Oh R, Wan F, Cui H, Nat. Biomed. Eng 2020, 4, 1090–1101; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Majumder P, Singh A, Wang Z, Dutta K, Pahwa R, Liang C, Andrews C, Patel NL, Shi J, de Val N, Walsh STR, Jeon AB, Karim B, Hoang CD, Schneider JP, Nat. Nanotechnol 2021, 16, 1251–1259; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Zhong Y, Zhan J, Xu G, Chen Y, Qin Q, Liao X, Ma S, Yang Z, Cai Y, Angew. Chem. Int. Ed 2021, 60, 8121–8129; [DOI] [PubMed] [Google Scholar]; e) Yamamoto S, Nishimura K, Morita K, Kanemitsu S, Nishida Y, Morimoto T, Aoi T, Tamura A, Maruyama T, Biomacromolecules 2021, 22, 2524–2531; [DOI] [PubMed] [Google Scholar]; f) Wang F, Gnewou O, Modlin C, Beltran LC, Xu C, Su Z, Juneja P, Grigoryan G, Egelman EH, Conticello VP, Nat. Commun 2021, 12, 407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].a) Zhao X-X, Li L-L, Zhao Y, An H-W, Cai Q, Lang J-Y, Han X-X, Peng B, Fei Y, Liu H, Qin H, Nie G, Wang H, Angew. Chem. Int. Ed 2019, 58, 15287–15294; [DOI] [PubMed] [Google Scholar]; b) Wang C, Du W, Wu C, Dan S, Sun M, Zhang T, Wang B, Yuan Y, Liang G, Angew. Chem. Int. Ed 2022, 61, e202114766; [DOI] [PubMed] [Google Scholar]; c) Chen Z, Chen M, Zhou K, Rao J, Angew. Chem. Int. Ed 2020, 59, 7864–7870; [DOI] [PubMed] [Google Scholar]; d) Yang L, Peltier R, Zhang M, Song D, Huang H, Chen G, Chen Y, Zhou F, Hao Q, Bian L, He M.-l., Wang Z, Hu Y, Sun H, J. Am. Chem. Soc 2020, 142, 18150–18159. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.