

Summary
The perinatal period represents a critical window for cognitive and immune system development, promoted by maternal and infant gut microbiomes and their metabolites. Here, we tracked the co-development of microbiomes and metabolomes from late pregnancy to one year of age using longitudinal multi-omics data from a cohort of 70 mother-infant dyads. We discovered large-scale mother-to-infant interspecies transfer of mobile genetic elements, frequently involving genes associated with diet-related adaptations. Infant gut metabolomes were less diverse than maternal but featured hundreds of unique metabolites and microbe-metabolite associations not detected in mothers. Metabolomes and serum cytokine signatures of infants who received regular – but not extensively hydrolyzed – formula were distinct from those of exclusively breastfed infants. Taken together, our integrative analysis expands the concept of vertical transmission of the gut microbiome and provides original insights into the development of maternal and infant microbiomes and metabolomes during late pregnancy and early life.
Graphical Abstract
eTOC/In Brief
Maternal gut bacteria that fail to engraft in infants instead influence the assembly and metabolic potential of the infant gut microbiome through horizontal gene transfer.
Introduction
Gut microbiome assembly follows predictable patterns1–9, starting with vertical transmission at birth10,11. Still, infant and maternal microbiomes are shaped by distinctive physiological, dietary, and environmental factors. Breastfeeding provides a competitive advantage to strains that utilize complex sugars in breast milk (human milk oligosaccharides, HMOs), such as Bifidobacterium and Bacteroides. Infants in North America and Western Europe have a comparatively low abundance of bifidobacteria, which coincides with high incidences of allergic and autoimmune conditions12,13. Recently, the early colonizer Bifidobacterium longum subspecies infantis was shown to constrain Th2 and Th17 responses in the infant gut through indole-3-lactic acid production14, providing an important example of the triangular relationship between the gut microbiome, metabolites, and the developing immune system. Early exposure to exogenous proteins through infant formula has also been linked to autoimmune and allergic diseases, and extensively hydrolyzed formula was proposed for infants at risk of these conditions15.
In addition to immune maturation, gut bacteria support cognitive development in part through production of microbial metabolites16–18. A subset of metabolites produced by the maternal gut microbiome, including hippurate and imidazole propionate, promotes axonogenesis in mouse embryos16. Nevertheless, the co-development of microbiomes and metabolomes during the perinatal period and the determinants of this process are not well understood.
In a cohort consisting of 70 mother-infant dyads, we profiled the fecal microbiome and metabolome in late pregnancy and different stages of infancy. Pregnancy was associated with an increase in steroid compounds, including gonadal hormone derivatives and intermediates of bile acid biosynthesis, several of which were independently linked to impaired glucose tolerance. Although infant gut metabolomes were less diverse than maternal, we detected over 2,500 infant-unique metabolomic features. Moreover, we identified numerous infant-specific associations of bacterial species and fecal metabolites, including neurotransmitters and immune modulators.
We used longitudinal sampling to investigate vertical transmission of species, strains, and individual genes. Strikingly, we discovered hundreds of mother-to-infant gene transmission events in the absence of maternal carrier strains in the infant gut. Key maternal donor species in these events belonged to the Bacteroidales order. Although these species were rarely transmitted to infants, their relative abundance in maternal samples exerted a significant influence on infant gut microbial structure and functional potential, including HMO utilization capacity. Hence, apart from classical vertical transmission, the maternal microbiome also shapes the infant gut microbiome through horizontal gene transfer (HGT) events. Together, our large-scale integrative analyses provides a series of high-resolution snapshots of gut colonization dynamics that influence infant development before and after birth.
Results
Infant microbiomes and metabolomes are distinct from maternal and influenced by diet
We analyzed longitudinal fecal and infant serum samples from 74 infants and 137 mothers, including 70 mother-infant dyads, that were part of the EDIA cohort15 to investigate host-microbiome co-development during the first year of life (Fig. 1A). These analyses included deep metagenomic sequencing, untargeted fecal metabolite profiling (Table S1), and assays measuring circulating cytokines, gut permeability, and markers of gut inflammation in infants (Fig. S1A-C). Ordination plots of metagenomic and metabolomic profiles showed distinct separations between infant and maternal samples (Fig. 1B,C).
Figure 1. Multi-omic data from the EDIA cohort.
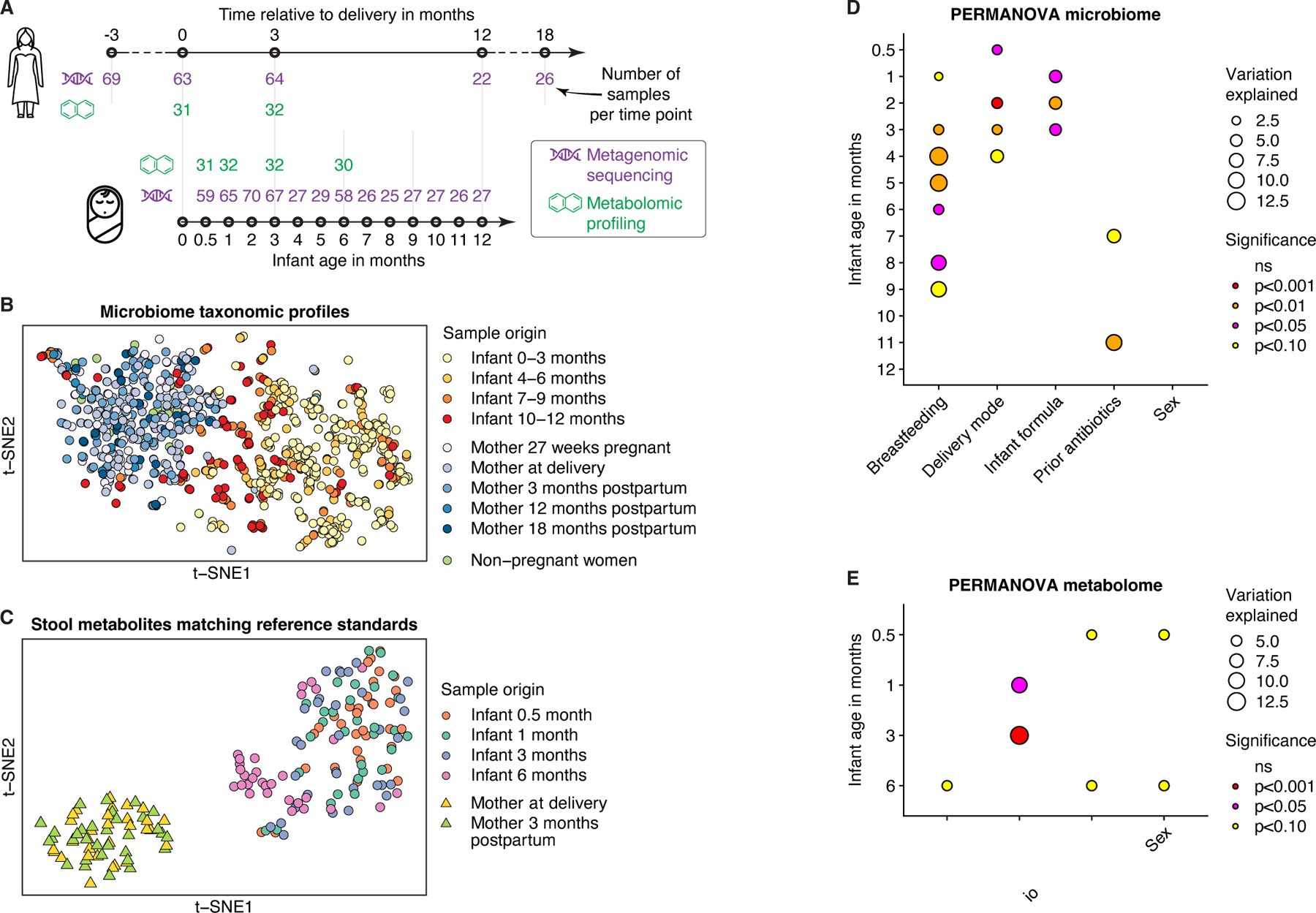
A) Schematic illustration of metagenomic and metabolomic data. Number of samples (n) after quality control at each time point is indicated. B) t-distributed stochastic neighbor embedding (t-SNE) ordination of gut microbiome profiles based on Bray-Curtis dissimilarities of species-level abundances from metagenomic data. C) t-SNE ordination of gut metabolite profiles based on log-scaled and z-score-normalized abundances of n=858 metabolites annotated using reference standards. D-E) Effects of factors on (D) species-level taxonomic composition (Bray-Curtis dissimilarities) and (E) metabolomic profiles (restricted to features annotated using standards) in infants according to cross-sectional PERMANOVA analyses. Factors were assessed in combination, while ordered by individual log10-transformed p-values from initial PERMANOVA analysis of each factor. ‘Infant formula’ comprises three categories: regular, hydrolyzed, and no formula. See also Figure S1; Tables S1-2.
All infants were born between gestational weeks 36 and 42, and eight infants (10.8%) were delivered by cesarean section. Infants underwent a typical transition from exclusive or partial breastfeeding to a diet consisting mostly of solid foods (Fig. S1D; Table S2). Previous investigations identified delivery mode and diet (breastfeeding versus infant formula) as major factors influencing gut microbiome assembly6,12. In our investigation, delivery mode, breastfeeding, use and type of infant formula, and prior antibiotics significantly influenced infant microbiomes or metabolomes for at least one sampling time point (PERMANOVA analysis; Fig. 1D,E). These factors were considered as confounders in subsequent analyses of infant metagenomic and metabolomic profiles.
Microbiome and metabolome shifts during pregnancy may impact maternal metabolic health
The study design allowed us to examine alterations in, and associations between, the metagenomic and metabolomic profiles of mothers and their infants over time. Based on metagenomic analysis of fecal samples from mothers at gestational week 27, delivery, and 3 months postpartum, we observed dynamic patterns in the relative abundances of multiple species during the peripartum period (Fig. 2A). Early infant colonizers Streptococcus thermophilus and Lactobacillus acidophilus transiently expanded around the time of delivery, whereas Anaerotruncus colihominis, a species with potential immunomodulatory properties19,20, was fleetingly reduced. Several species were enriched during pregnancy compared to the postpartum period including Streptococcus salivarius and Streptococcus parasanguinis. Both species are also prevalent members of the infant gut microbiome21,22 detected in 75% and 64% of infant samples, respectively, in our investigation.
Figure 2. Metagenomic and metabolomic signatures during pregnancy included expansion of taurine-conjugated bile acids, Bilophila wadsworthia, and H2S production capacity.
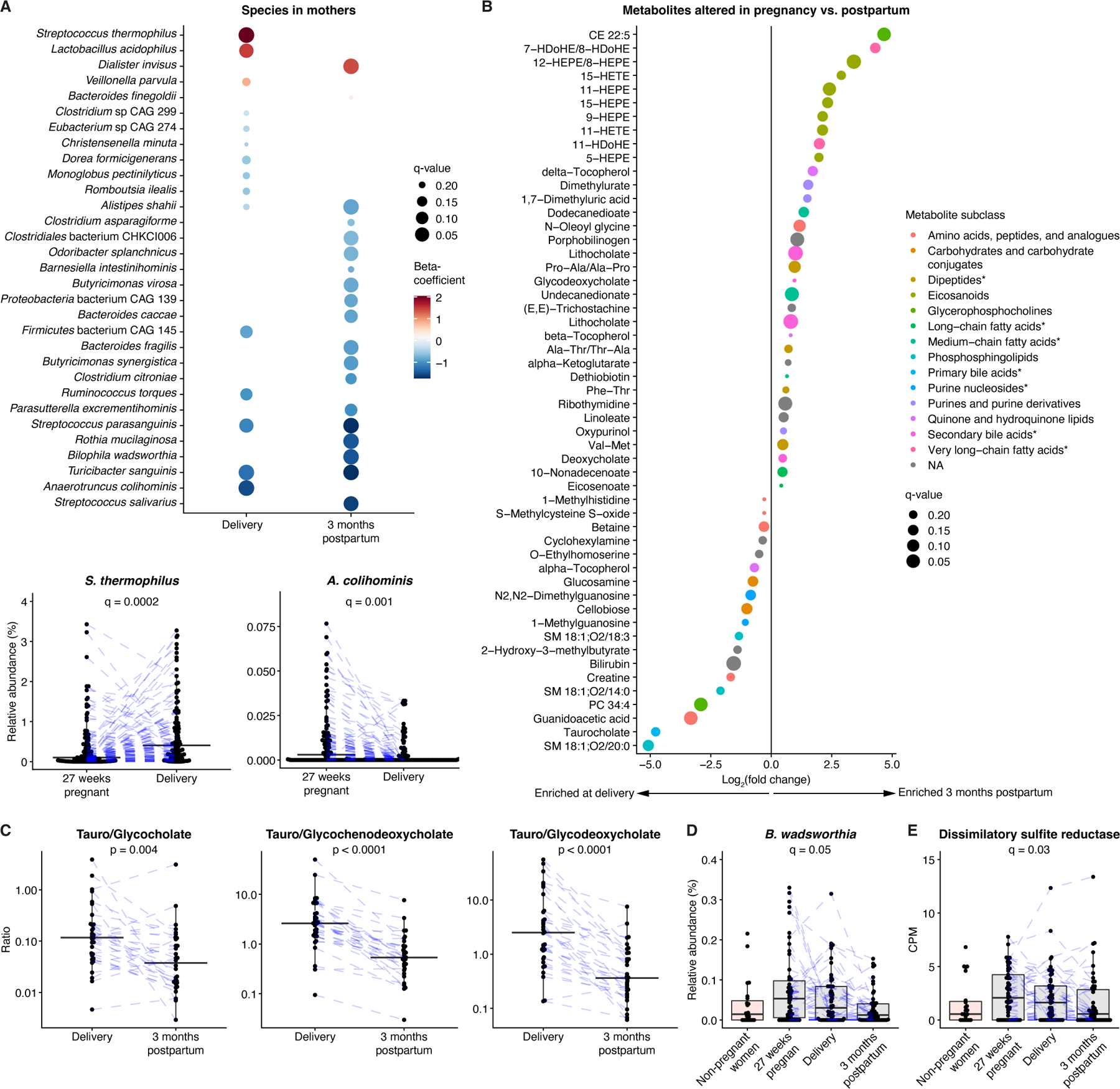
A) Upper panel: Differences in species at delivery and 3 months postpartum compared to gestational week 27. Lower panels: For each boxplot, two high outliers are not shown (both from the delivery time point for S. thermophilus; one from gestational week 27 and one from the delivery time point for A. colihominis). B) Metabolomic differences in paired maternal samples from delivery and 3 months postpartum. Asterisks indicate that subclass identification was replaced by more specific identification. C) Relative expansion of taurine-versus glycine-conjugated bile acids at delivery compared to 3 months postpartum. p-values obtained by the Wilcoxon signed-rank test. D) B. wadsworthia expansion during pregnancy. One high outlier (delivery) is not shown. E) Dissimilatory sulfite reductase expansion during pregnancy. One high outlier (gestational week 27) is not shown. In (D, E) results from a non-pregnant female control group were included as a reference. q-values refer to comparisons of pregnancy and postpartum time points based on paired samples, after correction for longitudinal analysis. For boxplots, midlines represent the median, boxes the interquartile range (25th to 75th percentile), and whiskers the range of data. Blue lines connect data points from the same participant. CPM, copies per million. See also Figure S2; Table S1.
To maximize the metabolomic analysis space in our study, we utilized two approaches: in addition to annotating peaks using reference standards, which yielded 858 metabolomic features, we mapped observed mass-to-charge ratios to the Human Metabolome Database (HMDB; Table S1), identifying 52,669 metabolomic features that represented 8,209 unique compounds23. Metabolomic analysis of paired stool samples from 31 mothers identified alterations in 53 annotated metabolites between delivery and 3 months postpartum (Fig. 2B). We further observed a pronounced expansion of steroids and steroid derivatives such as gonadal hormone metabolites and intermediates of primary bile acid biosynthesis at delivery (Fig. S2A,B). Interestingly, half (n=28/56) of these compounds were independently positively associated with impaired glucose tolerance during pregnancy. These metabolites were consistently associated with higher relative abundance of Methanobrevibacter smithii, which has been linked to improved energy harvest, obesity, impaired glucose tolerance, and diabetes24,25 (Fig. S2B-D).
Bile acid profiles underwent major alterations during the peripartum period (Fig. 2B-C, S2A). There was a relative enrichment of taurine-conjugated compared to glycine-conjugated and non-conjugated bile acids during pregnancy, accompanied by expansion of taurine respire Bilophila wadsworthia, which has been linked to colitis and glucose dysmetabolism26,27 (Fig. 2A-D). Like other sulfate-reducing bacteria, B. wadsworthia produces hydrogen sulfide (H2S) during respiration through a dissimilatory sulfite reductase28. Gene-level analysis revealed an overall increase of this enzyme at gestational week 27 and delivery compared to paired postpartum samples and samples from a non-pregnant female control group, suggesting increased capacity for microbial H2S production during pregnancy (Fig. 2E). While we cannot account for possible shifts in diet and lifestyle between pregnancy and the postpartum period, our analyses suggest that pregnancy-related alterations of the microbiome and metabolome may impact maternal metabolic health and potentially infant development.
Maternal species influence the infant microbiome through mobile genetic elements
A portion of pioneering infant gut strains originate from the maternal gut and are transmitted at, or soon after, birth11,29,30. With frequent infant metagenomic sampling, we investigated whether strain transmission occurs later in infancy. We analyzed single nucleotide polymorphism (SNP) haplotypes of the dominant strain of each species by StrainPhlAn to identify strains that were identical between mother and infant, then specifically enumerated events of these strains first appearing later in life (Fig. S3A). Lack of the corresponding species earlier in life was confirmed by sensitive detection using MetaPhlAn (Fig. S3B). Maternal gut strains transmitted after the first three months of life included members of the genera Eubacterium, Roseburia, and Blautia.
To further assess the maternal contribution to infant gut microbiome development, we evaluated whether the relative abundance of any maternal gut species had an overall influence on infant gut microbiome structure (PERMANOVA analysis). We found that maternal abundance of certain species, including Bacteroides cellulosilyticus, Bacteroides uniformis, and Alistipes putredinis, was associated with infant gut microbiome structure during the first three months of life (Fig. 3A) despite low rates of stable transmission. The relative abundance of maternal B. cellulosilyticus, a versatile carbohydrate degrader, was positively associated with the overall abundance of microbial glycoside hydrolases in the infant gut, particularly those involved in HMO degradation (Fig. S3C,D). Moreover, this maternal species was inversely correlated with intact HMOs in infant fecal samples and positively associated with HMO-utilizing infant species that are unable to degrade these oligosaccharides, such as Bifidobacterium pseudocatenulatum (Fig. S3D,E). Together, our observations suggest a possible influence of certain rarely-transmitted maternal species on the infant microbiome with marked consequences for infant gut ecology and functional capacity.
Figure 3. Mother-to-infant interspecies gene transmission.
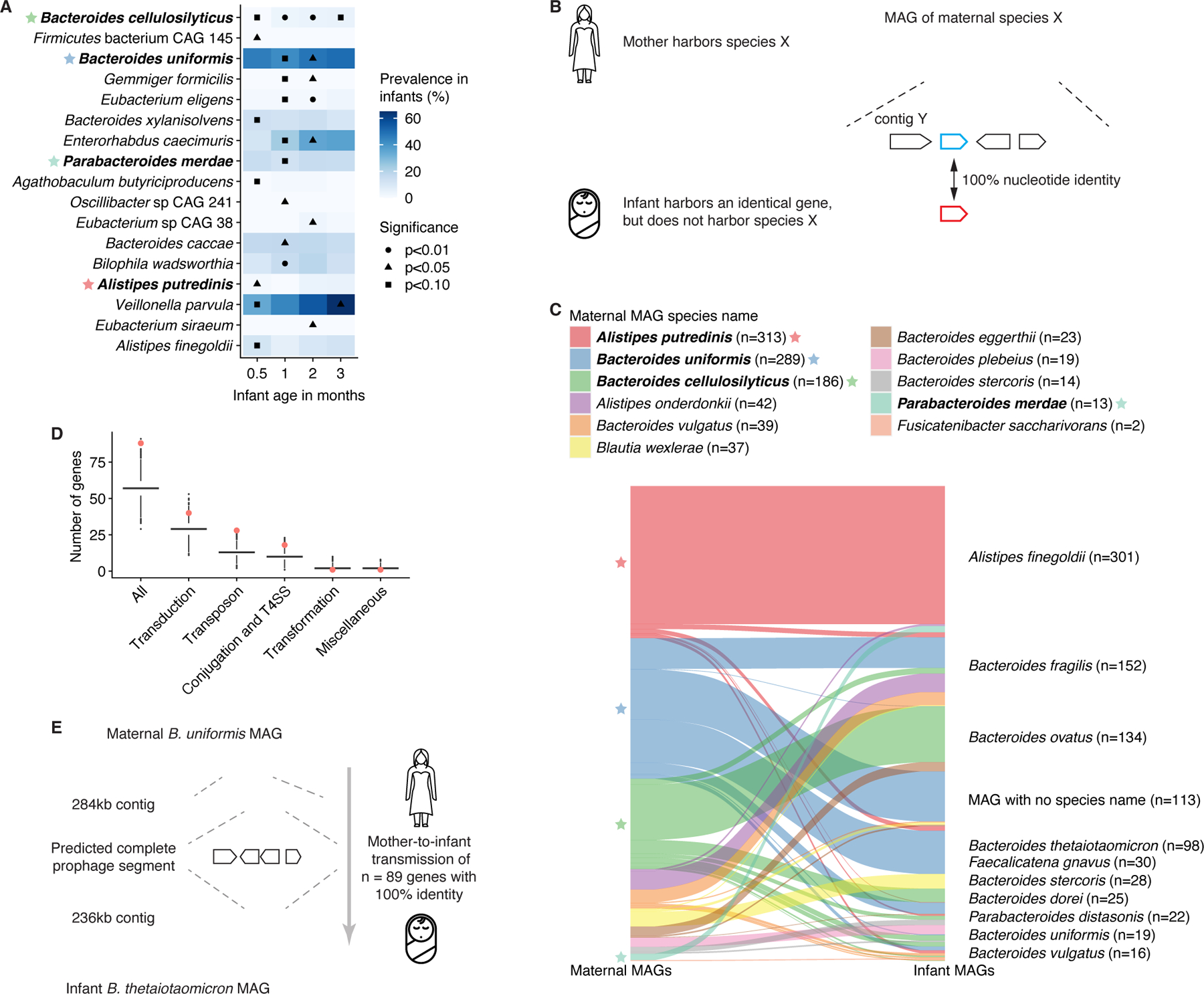
A) Association of prevalent (>50%) maternal species with species-level taxonomic composition (Bray-Curtis dissimilarity) of the gut microbiota in infants aged up to 3 months, based on cross-sectional PERMANOVA analyses. Analyses were corrected for infant sex, delivery mode, breastfeeding, formula use/type, and prior antibiotics; these factors were ordered by relative significance (log10-transformed p-values from individual PERMANOVA analyses) for each time point. Maternal species either significantly (p<0.05) associated with infant species-level taxonomy for ≥1 time points, or explaining >3% of variation, with p-value <0.10 are shown. Color represents species prevalence in infants. Stars indicate species common with (C). B) Schematic illustration of the mother-to-infant interspecies gene transmission hypothesis. C) Graph showing gene flow from maternal (left) to infant (right) species of the 977 gene transmission events identified. Stars indicate species common with (A). D) Numbers of HGT-related genes in 10,000 randomly drawn samples of 977 genes from the assembled gene catalog. Numbers of HGT-related genes among the 977 shared genes identified are highlighted in red. Midlines represent the median, boxes the interquartile range (25th to 75th percentile), and whiskers the range of data. E) Schematic of a transmitted prophage gene segment of n=299 (n=89 identical) genes between maternal B. uniformis and infant B. thetaiotaomicron. See also Figure S3; Tables S3-4.
Based on these observations, we hypothesized that maternal species influence infant gut microbiomes by contributing mobile elements and genes through HGT. To test this, we curated maternal metagenomes for high quality metagenome-assembled genomes (MAGs) and filtered them to species that were not observed in the corresponding infants based on sensitive detection with MetaPhlAn. We then screened the metagenomic assemblies from the infant for exact matches (100% nucleotide identity) to genes harbored by maternal MAGs (Fig. 3B). This approach revealed 977 unique genes that were shared across 22 mother-infant pairs and spanned 11 maternal bacterial species (Fig. 3C; Table S3), representing a significant (permutation test, p=0.01) increase over baseline gene sharing events between unrelated mothers and infants (Fig. S3F). Of these shared genes, 153 (16%) were located in MAGs of different genera and 22 (2%) in MAGs of different phyla in mother and infant (Table S3). Most (81%) of the shared genes were harbored by either B. cellulosilyticus (n=186), B. uniformis (n=289), or A. putredinis (n=313) in the maternal gut, supporting our hypothesis that these species influence the infant gut microbiome. For gene sharing events detected in infants at the first sampling time point (0.5 month), B. cellulosilyticus was the main maternal donor species (contributing n=146/357 genes, 41%; Fig. S3G), consistent with its pronounced influence on infant gut microbiome structure.
To further confirm that the gene transfer occurred between different species in mothers and their infants, we performed pairwise alignment of mother-infant contig pairs harboring shared genes. We identified overlapping regions outside the (near-)identical sequence in 37% of cases (n=56/151 contig pairs with shared genes), with a median length of 2,152 base pairs (bp; Fig. S3H). These sequences exhibited poor alignment between mother and infant with a median nucleotide identity of 47% (Fig. S3I), supporting true interspecies gene transfer events.
The observed gene transmission could be explained by HGT events occurring either in the maternal or infant gut. Of the gene sharing events identified, 30.8% (n=301/977 genes) had tentative mediator genomes in the maternal gut (i.e., the shared gene was detected on more than one maternal MAG representing different bacterial species). For 5.8% (n=57), the mediator genome was of the same species that harbored the gene in the infant. Similarly, 33.3% (n=325) had a tentative mediator genome in the infant gut, but none of these were the same species as the maternal source. We did not identify mediator genomes for 35.9% (n=351) of the gene sharing events; the timing and location of these gene transfers remains an open question.
Horizontal transfer of maternal microbial genes involved in dietary substrate metabolism may affect infant gut ecology
Previous studies have shown that horizontally transferred gene segments are enriched in genes associated with mobilome-related functions, such as transduction and conjugation31. By mapping the 977 shared genes to the KEGG, uniprot and eggNOG databases, we functionally annotated 373 genes (38%). Of these, 80 (21%) were annotated with mobilome-related functions, which represented a significant (permutation test, p=0.0003) enrichment (Fig. 3D, S3J). Genes linked to transduction (n=40; p=0.03), transposons (n=21; p=0.0002), and conjugation (n=17; p=0.01) were all enriched, suggesting that mother-to-infant gene transmission occurs through several mechanisms (Fig. 3D). We identified a high-quality 71,930bp prophage segment on a maternal B. uniformis MAG that was also present in two Bacteroides thetaiotaomicron MAGs from the corresponding infant at 3 and 6 months of age (Fig. 3E; Table S4). The segment from the mother and infant had 98.5% sequence similarity and contained 89 identical genes (100% nucleotide identity); the remaining genes were near-identical. Additionally, we identified 30 tentative, low-quality prophage segments (nine in mothers, 21 in infants; quality classification from checkV32) in metagenomic contigs that harbored genes shared between infants and their mothers (Table S4). These data demonstrate that phages may mediate mother-to-infant gene transfer between bacterial strains in the absence of vertical transmission.
Aside from HGT, predicted functions of the transmitted genes included carbohydrate biosynthesis and utilization (n=56/373 annotated genes, 15%), amino acid metabolism and transport (n=15, 4%), and iron acquisition and storage (n=14, 4%), indicating that gene transfer may have important metabolic consequences for recipient bacteria (Fig. S3J). Annotated genes originating from maternal B. cellulosilyticus were enriched for carbohydrate metabolism and transport relative to other shared genes (n=14/73, 19%; Fisher’s exact test, p=0.007; Fig. S3K), consistent with our previous observations (Fig. S3C-E) and with the extensive capacity for carbohydrate utilization described in this species33. By grouping genes related to carbohydrate utilization by substrate specificity, we found that shared genes linked to metabolism of plant-derived oligo- and polysaccharides appeared only transiently in infants prior to the introduction of solid food, concordant with the absence of these saccharides from a milk-based diet. Genes associated with metabolism of the HMO glycans galactose and fucose, however, were retained for more than one sampling time point (Fig. S3L). These data suggest that maternal species may influence the infant gut microbiome through sharing of genes required for metabolism of dietary substrates, including HMOs.
To further assess the biological relevance of mother-to-infant interspecies gene transfer, we used prevalence and abundance as proxies; biologically relevant microbial genes will be broadly distributed and highly abundant in microbiomes. We estimated the prevalence and abundance of non-redundant gene families (>95% similarity) corresponding to the shared genes and found that they were, on average, more prevalent compared to the null distribution of all genes (n=2.4M) in the metagenomic gene catalog (Fig. S3M). When present, the abundance of the transmitted genes in infants was higher compared to mothers (Kolmogorov-Smirnov test p<10−10; Fig. S3N), further suggesting that genes transferred from mother to infant by HGT have biologically relevant functions in infant gut microbiomes.
A unique infant gut metabolic environment develops in concert with the microbiome
As metabolomic profiles provide a complementary view to microbiome activity, we next compared maternal and infant metabolomes. Of the 858 metabolomic features annotated using reference standards, 395 compounds were more abundant in infants and 227 were more abundant in mothers (linear mixed model, q<0.01; Fig. 4A; Table S5). Metabolites enriched in infants included carnitines, eicosanoids, glycerophosphocholines, and very long-chain fatty acids, whereas secondary bile acids, B-vitamins, and medium-chain fatty acids were more abundant in mothers (Fig. 4B).
Figure 4. Unique metabolomic profiles of the infant gut.
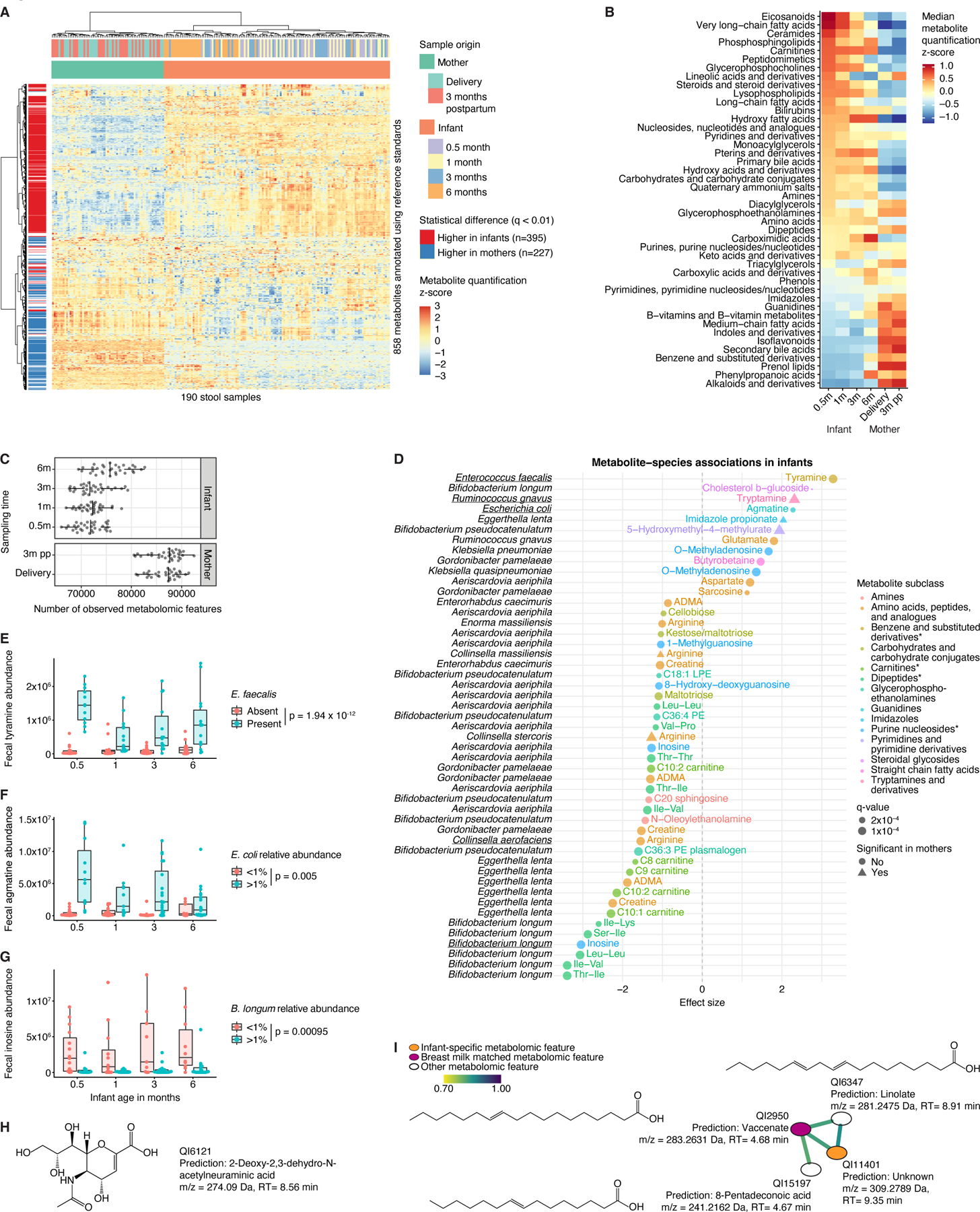
A) Heatmap of n=858 metabolites annotated using reference standards in the infant and maternal gut. B) Median abundance of metabolite classes/subclasses (represented by at least three unique metabolomic features) stratified by sampling time point. C) Metabolomic diversity, measured as the number of observed metabolomic features, in mothers and their infants. D) Significant associations between infant species with >25% prevalence and metabolomic features (annotated by reference standards) after adjustment for longitudinal analysis and correction for infant age, sex, delivery mode, antibiotic usage, and formula use/type. Species-metabolite associations independently confirmed in prior in vitro experiments35 are underscored. Asterisks indicate that subclass identification was replaced by identification on another level. E) Fecal tyramine levels stratified by infant age and presence of Enterococcus faecalis. F) Fecal agmatine levels stratified by age and Escherichia coli relative abundance. G) Fecal inosine levels stratified by age and Bifidobacterium longum relative abundance. H) Chemical structure of infant-specific metabolomic feature QI6121 predicted with SIRIUS. m/z, mass-to-charge ratio; RT, retention time. I) GNPS subnetwork associated with infant-specific metabolomic feature QI11401. Edges connect metabolomic features with cosine similarity >0.7. Chemical structures predicted with SIRIUS. For boxplots, midlines represent the median, boxes the interquartile range (25th to 75th percentile), and whiskers the range of data. See also Figure S4; Tables S1, S5-6.
Infant gut metabolomes consistently displayed less diversity, with on average slightly over 70,000 observed metabolomic features compared to nearly 90,000 in mothers (Fig. 4C). Lower taxonomic and metabolomic diversity could result in increased significance of specific species-metabolite associations—not only in terms of statistical power for detection but also due to a lack of biological redundancy. Indeed, out of 163 significant associations between bacterial species and annotated metabolites in infants, only 5 (3%) were observed in mothers (Fig. 4D; Table S6). Unique infant microbe-metabolite associations included positive correlations of Enterococcus faecalis and Escherichia coli with neurotransmitters tyramine and agmatine, respectively (Fig. 4E-F, S4A-B). There was an infant-specific inverse association of Collinsella aerofaciens and fecal arginine, possibly mediated by arginine deiminase (arcA; Fig. S4C). Arginine is an essential amino acid for premature and low-birth weight infants and modulates immune system development, partially as a substrate for polyamine and nitric oxide biosynthesis. B. longum abundance was inversely correlated with inosine, a purine metabolite with immunoregulatory properties34 (Fig. 4G, S4D). These four species-metabolite associations were independently observed in prior in vitro metabolic profiling of 178 gut microbe strains35. Additionally, there was an infant-specific inverse association between B. longum and dipeptides containing branched-chain amino acids (BCAAs; Fig. 4D). Cyclic BCAA-containing dipeptides were recently identified as a bifidogenic factor produced by certain Bacilli36.
We also detected 2,555 metabolomic features unique to infant fecal samples, including many unknown features. Of these, 433 were present in at least 50% of samples, including 131 (30%) features that were annotated based on mapping to the HMDB. These prominently included organooxygen compounds (mostly carbohydrates) and flavonoids, particularly flavonoid glycosides (Fig. S4E). Twenty-eight common (present in >33% of samples) annotated infant-specific metabolites were associated with serum cytokine levels at 6 months of age (p<0.01; Fig. S4F). In particular, sialic acid levels inversely correlated with several proinflammatory cytokines, notably IFNγ. Sialic acids are HMO components that may dampen immune responses by signaling through Siglec receptors37,38. Forty-one (of 433, 9%) of the highly prevalent infant-specific metabolomic features were significantly associated with 27 bacterial species, some of which were also unique to infant samples (Fig. S4G). These included Klebsiella michiganensis (present in 125 infant versus 3 maternal samples), which lacks the pathogenic potential of other Klebsiella species and may contribute to colonization resistance during antibiotic perturbation39. Interestingly, K. michiganensis was more prevalent in samples from infants who previously received systemic antibiotics (37.1% versus 21.1%; Fisher’s exact test, p=0.002). Thus, this species may contribute to stabilization of the fragile infant gut ecosystem.
We next investigated longitudinal trends in infant-specific metabolomic features and found statistically significant shifts in 287 (11.2%) features (Kruskal-Wallis test, FDR corrected p-value<0.01; Fig. S4H). Most (n=253) were highly abundant around birth and declined over time, whereas only 34 increased longitudinally. The observed decrease tended to be exacerbated by the introduction of solid foods or infant formula (Fig. S4I), suggesting that decreasing molecules originated from breast milk. Indeed, among 59 such features that could be annotated by mapping to the HMDB, we identified known breast milk components including arachidonic, eicosadienoic, and eicosapentaenoic fatty acids as well as lactic acid, which is also a product of, and substrate for, microbial metabolism.
To further investigate potential links between infant-specific metabolomic features and breast milk, we applied tandem mass spectrometry (MS2) characterization to 10 infant samples that contained a high level of a diverse repertoire of the common infant-specific metabolomic features, successfully acquiring spectra for 77. SIRIUS40 predicted molecular formulas and structures for 30 (Table S1), including the sialic acid derivative 2-deoxy-2,3-dehydro-N-acetylneuraminic acid (Fig. 4H). We further used molecular networking on MS2 spectra with GNPS41 and identified a subnetwork with four SIRIUS-predicted eicosenoic acid derivatives: vaccenate, linolate, 8-pentadeconoic acid, and QI11401—an infant-specific feature inversely correlated with TNFa (Fig. 4I). Consistent with vaccenate being an important lipid component of breast milk42, this metabolomic peak was also found in human breast milk MS2 spectra that we generated from 40 pooled samples collected as part of the OriGiN study29. Interestingly, we observed numerous infant-specific metabolomic features that networked (GNPS cosine>0.7) with breast milk metabolomic features or reference standards, such as retinol (Fig. S4J-L).
Dietary modulations of infant gut metagenomes and metabolomes influence immune maturation
Gut microbiome and metabolomic profiles are influenced by diet and modulate immune responses and inflammation43,44. The 74 infants in our analyses were part of a randomized clinical trial to evaluate the effects of extensively hydrolyzed formula on intestinal inflammation and permeability in infants with HLA-conferred susceptibility to type 1 diabetes15. Despite this genetic risk, the baseline microbiome composition of infants in our investigation largely resembled previous observations1–9. Given that infant diet influenced metagenomic and metabolomic profiles (Fig. 1D,E), we further examined specific effects of breastfeeding as well as formula use and type on infant metagenomes, metabolomes, and markers of intestinal and systemic inflammation.
Forty (54%) infants were randomized to regular and 34 (46%) to hydrolyzed formula; however, regular formula use was reported for 27 infants (111 samples) and hydrolyzed formula use for 23 infants (107 samples; Table S2). Hydrolyzed formula was associated with expansion of Ruminococcus gnavus, whereas regular formula was linked to enrichment of E. faecalis, Actinomyces radingae, and S. thermophilus (Fig. 5A). Serum levels of proinflammatory cytokines (IL-12, IL-6, IL-9, IL-13) increased in infants on regular but not hydrolyzed formula compared to non-formula-fed infants (Fig. 5B). Moreover, regular formula was associated with metabolomic shifts, including a depletion of lysophosphatidylcholines (Fig. 5C,D) that may modulate immune responses by various leukocyte subsets45,46 and were inversely correlated with proinflammatory cytokines (Fig. S5).
Figure 5. Formula types were associated with distinct metagenomic and metabolic profiles.
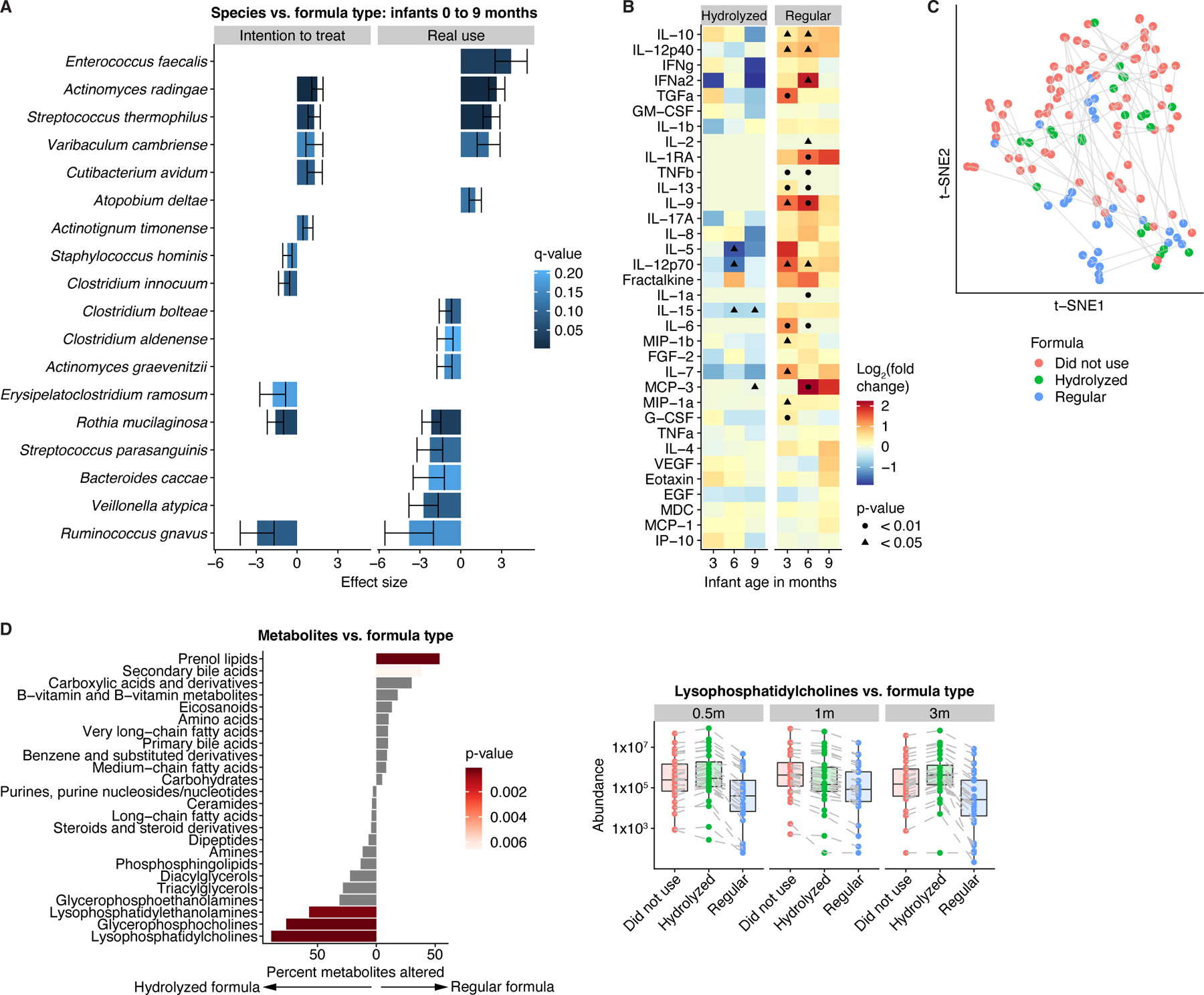
A) Species differences (q<0.25) between infants who were randomized to (left) or received (right) regular versus hydrolyzed formula. Results obtained from general linear models, adjusted for longitudinal analysis and corrected for age, sex, delivery mode, antibiotic usage, and breastfeeding. Error bars represent standard error. Positive effect sizes indicate species enriched in infants who were randomized to or received regular formula, while negative effect sizes indicate species enriched in infants who were randomized to or received hydrolyzed formula. B) Enrichment of proinflammatory cytokines in infants given regular versus no formula. p-values obtained by the Mann-Whitney U-test. C) t-SNE ordination of fecal metabolomics profiles in infants, colored by formula use/type. D) Left panel: Percentage of metabolites per subclass/category that were altered (q<0.25) between infants on hydrolyzed and regular formula for at least one time point. p-values obtained through Fisher’s exact test. Right panel: Median levels of lysophosphatidylcholines stratified by age and formula use/type. Lines connect identical metabolites. Midlines represent the median, boxes the interquartile range (25th to 75th percentile), and whiskers the range of data. See also Figure S5; Tables S1-2.
Baseline measures of intestinal inflammation are typically increased in infants compared to adults; however, the implications of this and the connection with systemic inflammation are poorly understood47,48. Investigating correlations with proinflammatory serum cytokines, we observed a striking, age-dependent dichotomy: markers of intestinal and systemic inflammation positively correlated in one-year-olds and inversely correlated in younger infants (Fig. 6A). Calprotectin and beta-defensin 2, which tended to be elevated in breastfed infants, exhibited positive associations with beneficial species, including B. longum and B. breve, and inverse correlations with linoleoyl ethanolamide, which promotes aberrant microbiome shifts49 (Fig. 6B, S6A-C). We also observed positive correlations between calprotectin and eicosanoids, particularly arachidonic acid derivatives, which are critical for early immune and neuro-development50 (Fig. 6C). Eicosanoids were positively associated with previously identified breast milk microbiome members, including Streptococcus species21,22 and Corynebacterium kroppenstedtii (Fig. 6D). Collectively, our observations suggest that intestinal inflammation markers in young infants may reflect immune maturation, potentially facilitated by breastfeeding, rather than underlying pathology.
Figure 6. Associations of inflammation and permeability markers with infant fecal metagenomes and metabolomes.
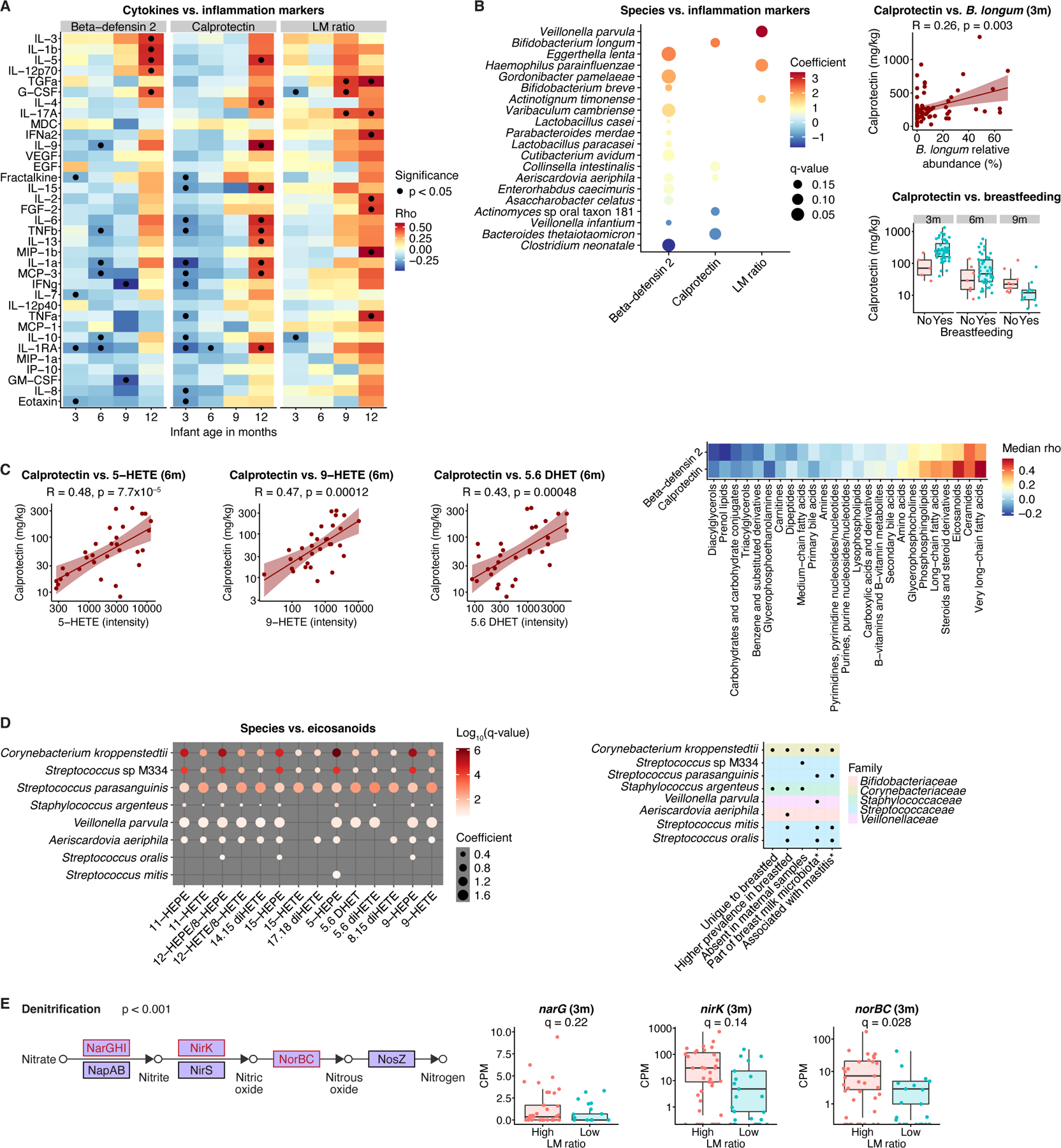
A) Markers of intestinal permeability and inflammation were inversely correlated with proinflammatory serum cytokines during the first months of life but positively correlated at one year of age. B) Left panel: Calprotectin and beta-defensin 2 were positively associated with beneficial species in infants up to 6 months old. Effects are from general linear models, corrected for longitudinal sampling, age, sex, delivery mode, antibiotics, breastfeeding, and formula use/type. Right panels: Calprotectin levels were higher in breastfed infants and correlated with Bifidobacterium longum relative abundance. C) Calprotectin levels were positively correlated with fecal eicosanoids, particularly arachidonic acid metabolites in 6-month-old infants. R values for scatter plots in (B, C) are Kendall’s tau. D) Levels of infant fecal eicosanoid metabolites were positively associated with relative abundances of species previously linked to the breast milk microbiome, including causative agents of mastitis. *based on previous literature21,22,55. Results (left) are from general linear models, corrected for longitudinal sampling, age, sex, delivery mode, antibiotics, and formula use/type. E) LM ratio was positively associated with microbial genes linked to denitrification via nitric oxide production. The cut-off of 0.03 for high versus low LM ratio was determined a priori. Overall p-value for denitrification enrichment was obtained by Fisher’s exact test. q-values derived from a general linear model including infants aged up to one year, corrected for longitudinal sampling, age, sex, delivery mode, antibiotics, breastfeeding, and formula use/type. For boxplots, midlines represent the median, boxes the interquartile range (25th to 75th percentile), and whiskers the range of data. See also Figure S6; Table S1.
Intestinal permeability (measured by lactulose-mannitol (LM) ratio) was positively associated with relative abundances of the nitrate-reducing species Veillonella parvula and Haemophilus parainfluenzae (Fig. 6B). Gene-level analysis of infants with high LM ratios identified an enrichment of the denitrification pathway (Fisher’s exact test, p<0.001), which reduces nitrate to nitrogen via nitric oxide (Fig. 6E). Together, our observations offer insight into how diet and the gut microbiome modulate immune maturation.
Discussion
By examining the development of maternal and infant gut microbiomes, we discovered an additional mode of vertical microbiome transmission, where maternal gut bacterial strains shared genes with infant gut strains in the absence of persistent transmission of the bacterial genomes themselves. Besides functions related to mobile genetic elements, the 977 transmitted genes encoded functions related to carbohydrate utilization, amino acid metabolism, and iron acquisition and storage. Thus, mother-to-infant HGT influences the metabolic potential of the infant gut microbiome, with possible consequences for immune system development. As our analysis only covered identical genes harbored by high-quality MAGs, the full magnitude and functional ramifications of HGT in gut microbiome assembly during infancy remain to be expanded by future studies.
Many bacterial species involved in gene transmission events belonged to the Bacteroidales order, concordant with previous reports of extensive interspecies gene sharing between Bacteroidales members in the human intestine51, although we also observed gene sharing events between different bacterial orders and even phyla. Gene sharing events among dyads were significantly more common than between unrelated mothers and infants, with a significant enrichment of genes related to different modes of HGT. One interesting possibility is that the distinct environment of the infant gut triggers prophage induction in vertically transmitted strains that are unable to engraft.
We previously reported that in Finnish infants HMO degradation was mainly performed by Bacteroides rather than Bifidobacterium species12. Not all Bacteroides strains, however, possess the required glycoside hydrolases for HMO utilization, and horizontal transfer of these genes between Bacteroides could confer major advantages in the infant gut ecosystem. In our analysis, maternal B. cellulosilyticus – a major donor species in gene sharing events – was positively associated with HMO-metabolizing glycoside hydrolases and species that scavenge liberated HMO glycans in infant samples, and inversely correlated with intact fecal HMOs, indicating that maternal B. cellulosilyticus may indirectly influence the repertoire of carbohydrate-active enzymes in the infant microbiome in the absence of species transmission.
We tracked the longitudinal development of infant microbiomes and metabolomes and mapped their relationships to diet, systemic inflammatory responses, and gut permeability. The use of infant formula containing intact exogenous proteins was linked to a global increase of proinflammatory serum cytokines relative to exclusive breastfeeding, consistent with data indicating that formula-fed infants have a higher risk of developing autoimmune disease52. Intestinal permeability was associated with Veillonella parvula, a genus previously linked to type 1 diabetes53.
Breastfeeding has previously been associated with beneficial effects on immune system development through modulation of the infant microbiome and provision of antibodies and antimicrobial proteins54. We linked breastfeeding to an early increase in intestinal inflammation markers, including fecal calprotectin and beta-defensin 2. These markers correlated inversely with proinflammatory serum cytokines in young infants, suggesting that their levels may reflect immune maturation rather than underlying pathology in this age group. Moreover, we observed a positive association of infant intestinal inflammatory mediators, such as eicosanoids, with previously identified members of the breast milk microbiome and with C. kroppenstedtii, a species isolated almost exclusively from clinical mastitis samples55 and uniquely present in stool from breastfed infants in our analysis. Thus, early exposure to microbial pathobionts and inflammatory mediators could expedite immune system education, potentially resulting in more tolerogenic responses to subsequent challenges.
Gut bacteria influence human physiology and development through the production of bioactive compounds. Our analysis revealed over 2,500 infant-specific metabolomic features that, despite varying proportions of redundancies and artifacts in untargeted LC-MS metabolomics56, likely reflect several hundred unique compounds. Using paired metagenomic and metabolomic data, we identified numerous microbe-metabolite relationships that were statistically significant in infants but not mothers, including an inverse association between B. longum and inosine. Inosine has been reported to have neuroprotective and immunomodulatory properties34,57. Thus, utilization of inosine by B. longum, potentially with subspecies-specific variations, may also influence immunological and neurological development in infants.
Pregnancy was associated with alterations of fecal bile acids, with a relative increase in taurine conjugation. These changes were mirrored by a gut microbiome shift, with expansion of the taurine-degrading, sulfate-reducing species B. wadsworthia and of microbial H2S production capacity. Endogenous taurine production increases after intestinal infections and promotes resistance to pathogen expansion through Bilophila-derived H2S58. Consequently, a taurine-induced increase of Bilophila species during pregnancy and the peripartum may provide critical protection against infectious complications in this vulnerable period. However, B. wadsworthia has also been linked to unfavorable metabolic effects, including host glucose dysmetabolism26, and the transient increase of this species may therefore contribute to the deterioration of metabolic parameters commonly observed during pregnancy.
Our investigation represents a unique perspective into the co-development of infant gut microbiomes and metabolomes under the influence of known maternal and dietary factors, with potentially profound implications for immune and neuro-development. The discovery that mother-to-infant interspecies HGT events shape infant microbial metabolic activities expands our understanding of maternal influences on the infant gut microbiome. Moreover, the identification of distinctive metabolomic profiles and microbe–metabolite interactions in the infant gut constitutes a platform for further study of microbial contributions to development. Together, these observations open new prospects for targeted interventions to ensure optimal opportunities for growth and development in infancy.
Limitations of the study
Aspects of our study merit consideration. First, assembling metagenomic contigs from a complex mixture of closely-related genomes could result in assembly and/or binning errors. We attempted to minimize this risk by only investigating genes harbored by near-complete MAGs. Second, we did not consider diet and lifestyle changes between pregnancy and the postpartum period, which may have affected microbiomes and metabolomes. Third, the proportion of metabolomic features that can be annotated using reference standards represents a general conundrum; continued work to expand the database of annotated features reported here will provide an opportunity to discover additional microbe-metabolite-host relationships.
Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Ramnik J. Xavier (xavier@molbio.mgh.harvard.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
Metagenomic data have been deposited at NCBI Sequence Read Archive. Metabolomic data have been deposited at Metabolomics Workbench. Data are publicly available as of the date of publication. Accession numbers and DOIs are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the Lead Contact upon request.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| Maternal and infant stool samples | Tampere Center for Child Health Research, Tampere University Hospital, Tampere, Finland | EDIA |
| Infant serum samples | Tampere Center for Child Health Research, Tampere University Hospital, Tampere, Finland | EDIA |
| Breast milk samples | Vincent Obstetrics & Gynecology Department, Massachusetts General Hospital, Boston, MA, USA | OriGiN |
| Critical commercial assays | ||
| PowerSoil DNA Isolation Kit | MoBio Laboratories | Cat#12888-100 |
| Nextera XT DNA Library Preparation kit | Illumina | Cat#FC-131-1096 |
| CalproLab ELISA (ALP) | Calpro AS | Cat#CALP0170 |
| β-Defensin 2 ELISA | Immundiagnostik | Cat#KR6500 |
| MILLIPLEX MAP Human Cytokine/Chemokine Magnetic Bead Panel | Millipore | Cat#HCYTMAG-60K-PX38 |
| Deposited data | ||
| Metagenomic sequencing data | This paper | SRA: PRJNA821542 |
| Stool metabolomic profiles | This paper | http://dx.doi.org/10.21228/M8C70Q |
| Software and algorithms | ||
| KneadData v0.7.2 | Curtis Huttenhower laboratory | https://huttenhower.sph.harvard.edu/kneaddata/ |
| MetaPhlAn2/StrainPhlAn v2.9.21 | Beghini et al. 70 | https://huttenhower.sph.harvard.edu/metaphlan/ |
| HUMANn2 v0.11.2 | Franzosa et al. 71 | https://huttenhower.sph.harvard.edu/humann/ |
| megaHIT v1.1.4-2-gd1998a1 | Li et al. 72 | https://github.com/voutcn/megahit |
| Prodigal v2.6.3 | Hyatt et al. 73 | https://github.com/hyattpd/Prodigal |
| metaBAT2 v2.15-3-g367a7ef | Kang et al. 74 | https://bitbucket.org/berkeleylab/metabat |
| checkM v1.1.2 | Parks et al. 75 | https://ecogenomics.github.io/CheckM/ |
| GTDB-Tk v1.0.2 | Chaumeil et al. 76 | https://github.com/Ecogenomics/GTDBTk |
| CD-HIT v4.7 | Fu et al. 77 | http://weizhong-lab.ucsd.edu/cd-hit/ |
| eggNOG-mapper v2.0.1 | Huerta-Cepas et al. 78 | http://eggnog-mapper.embl.de/ |
| KMA | Clausen et al. 79 | https://bitbucket.org/genomicepidemiology/kma |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cohort recruitment.
Healthy pregnant women were recruited at the fetal ultrasonography visit around gestational week 20, from January 2013 to February 201515. Written informed consent was signed by the parents to analyze the offspring’s HLA genotype and to collect and analyze infant and maternal stool and blood samples. Inclusion criteria for the intervention study were informed consent signed by the parents and an eligible HLA genotype of the newborn conferring increased risk for type 1 diabetes59. The study protocol was approved by the Ethical Committee of the Joint Municipal Authority of the Pirkanmaa Hospital District.
Trial design and randomization.
Eligible pregnant women came to their first clinical study visit at the beginning of the last trimester. Randomization for the study formula was performed during the 35th gestational week, and families received the first batch of the study formula before delivery. Cord blood samples were used for HLA genotyping in the newborn infants, and the families were informed about the genotyping results within 10 days after delivery. Eligible infants visited the study center at the age of 3, 6, 9, and 12 months. During the visits, blood samples were taken and the lactulose-mannitol (LM) ratio test was performed in order to assess intestinal permeability. Stool samples were collected at the age of 2 weeks and monthly between the age of 1 and 12 months. The recruited mothers were encouraged to breastfeed. Study formulas were used as a part of the diet until the age of 9 months, whenever infants needed additional feeding. During the intervention period, the infants were on a diet free of cow-based proteins, while their mothers’ diets remained unrestricted. Study formula use, food consumption frequency, and compliance to the intervention diet were monitored with frequent dietary questionnaires and interviews (at the age of 2 weeks, monthly between the age of 1 and 9 months, and at 12 months of age). Follow-up ended when the infant reached the age of 12 months, but participants were offered the possibility to continue in the DIPP study follow-up59. The primary outcome of this study was the LM ratio at 9 months of age. By the protocol, at that age the intended exposure to study formula could have reached 90 days, even if the infants were exclusively breastfed for the first 6 months. The secondary outcomes were LM ratios at 3, 6, and 12 months, and the levels of fecal calprotectin and beta-defensin 2 at the age of 3, 6, 9, and 12 months. In addition, the study protocol included exploratory analyses on all analyzed data types: gut metagenomes and metabolomes, circulating cytokines and intestinal inflammation markers.
The infants were randomized to one of four color-coded, blinded formulas, two of which contained the extensively hydrolyzed formula (EHF) and two the control formula. A data handling software (BC CLIN version 3.6, Biocomputing Platforms Ltd., Espoo, Finland) was used for the randomization (1:1 block permutation). The manufacturer of the formulas (Mead Johnson Nutrition, Glenview, IN) kindly provided packing and labeling of the study formulas as well as guarded the randomization codes during the intervention period. Participating families and all study personnel remained blinded until the last participant’s 12-month visit had been completed. Detailed trial design and clinical findings were described previously15.
Sample collection and DNA extractions.
The participating women collected stool samples at home or in the delivery hospital. Infant stool samples were collected by the mothers at home and stored in the household freezer (−20°C) until the next visit to the study center. The samples were then shipped on dry ice to the EDIA Core Laboratory in Helsinki, where the samples were stored at −80°C until shipping to Tampere University for DNA extraction. DNA extractions from 0.2 grams of stool were carried out using the vacuum protocol of PowerSoil DNA Isolation Kit (MoBio Laboratories, Inc., Carlsbad, CA, USA) according to the manufacturer’s standard protocol. The extracted DNA was stored at −80°C.
Breast milk cohort information and collection.
Human patient research in the OriGiN cohort29 was reviewed and approved by the Partners Human Research Committee (ref.2015P000460/PHS). Each mother signed informed consent forms prior to participation. Breast milk was collected between day 13–29 after delivery and stored at 1:1 volume in 100% ethanol. Samples were returned at room temperature within 24h of collection and were stored at −80°C until 40 samples were pooled for metabolomic analysis.
METHOD DETAILS
Metagenome library construction and sequencing.
DNA samples were quantified by Quant-iT PicoGreen dsDNA Assay (Life Technologies) and normalized to a concentration of 50 pg/mL. Illumina sequencing libraries were prepared from 100–250 pg DNA using the Nextera XT DNA Library Preparation kit (Illumina) according to the manufacturer’s recommended protocol, with reaction volumes scaled accordingly. Metagenomic libraries were sequenced on the Illumina HiSeq 2500 platform, targeting 2.5 Gb of sequence per sample with 101 bp paired end reads.
Metabolomic analysis.
Metabolomic profiles of a subset of 192 stool samples were generated using a combination of four liquid chromatography–mass spectrometry (LC–MS) methods comprised of a Shimadzu Nexera X2 U-HPLC (Shimadzu Corp.; Marlborough, MA) coupled to a Q Exactive Hydro Quadrupole Orbitrap or Exactive Plus Mass Spectrometer (Thermo Fisher Scientific). These methods measure the following complementary metabolite classes: 1) HILIC-pos: positive ion mode MS analyses of polar metabolites, 2) HILIC-neg: negative ion mode MS analyses of polar metabolites, 3) C8-pos: polar and nonpolar lipids, and 4) C18-neg: negative ion mode analyses of metabolites of intermediate polarity. Method-specific protocols were as follows44. HILIC-pos: Metabolites were extracted from samples (10 μL) using 90 μL of acetonitrile/methanol/formic acid (74.9:24.9:0.2 v/v/v) containing stable isotope-labeled internal standards (valine-d8, Sigma-Aldrich; St. Louis, MO and phenylalanine-d8, Cambridge Isotope Laboratories; Andover, MA). The samples were centrifuged (10 min, 9,000 × g, 4°C), and the supernatants were injected directly onto a 150 × 2 mm, 3 μm Atlantis HILIC column (Waters; Milford, MA). The column was eluted isocratically at a flow rate of 250 μL/min with 5% mobile phase A (10 mM ammonium formate and 0.1% formic acid in water) for 0.5 minute followed by a linear gradient to 40% mobile phase B (acetonitrile with 0.1% formic acid) over 10 minutes. MS analyses were carried out using electrospray ionization in the positive ion mode using full scan analysis over 70–800 mass-to-charge ratio (m/z) at 70,000 resolution and 3 Hz data acquisition rate. HILIC-neg: Metabolites were extracted from samples (30 μL) using 120 μL of 80% methanol containing inosine-15N4, thymine-d4 and glycocholate-d4 internal standards (Cambridge Isotope Laboratories; Andover, MA). The samples were centrifuged (10 min, 9,000 x g, 4°C), and the supernatants were injected directly onto a 150 × 2.0 mm Luna NH2 column (Phenomenex; Torrance, CA). The column was eluted at a flow rate of 400 μL/min with initial conditions of 10% mobile phase A (20 mM ammonium acetate and 20 mM ammonium hydroxide in water) and 90% mobile phase B (10 mM ammonium hydroxide in 75:25 v/v acetonitrile/methanol) followed by a 10 min linear gradient to 100% mobile phase A. MS analyses were carried out using electrospray ionization in the negative ion mode using full scan analysis over 70–750 m/z at 70,000 resolution and 3 Hz data acquisition rate. C8-pos: Lipids were extracted from samples (10 μL) using 190 μL of isopropanol containing 1,2-didodecanoyl-sn-glycero-3-phosphocholine (Avanti Polar Lipids; Alabaster, AL). After centrifugation, supernatants were injected directly onto a 100 × 2.1 mm, 1.7 μm ACQUITY BEH C8 column (Waters; Milford, MA). The column was eluted isocratically with 80% mobile phase A (95:5:0.1 vol/vol/vol 10mM ammonium acetate/methanol/formic acid) for 1 minute followed by a linear gradient to 80% mobile phase B (99.9:0.1 vol/vol methanol/formic acid) over 2 minutes, a linear gradient to 100% mobile phase B over 7 minutes, then 3 minutes at 100% mobile phase B. MS analyses were carried out using electrospray ionization in the positive ion mode using full scan analysis over 200–1100 m/z at 70,000 resolution and 3 Hz data acquisition rate. C18-neg: Samples (30 μL) were extracted using 90 μL of methanol containing PGE2-d4 as an internal standard (Cayman Chemical Co.; Ann Arbor, MI) and centrifuged (10 min, 9,000 x g, 4°C). The supernatants (10 μL) were injected onto a 150 × 2.1 mm ACQUITY BEH C18 column (Waters; Milford, MA). The column was eluted isocratically at a flow rate of 450 μL/min with 20% mobile phase A (0.01% formic acid in water) for 3 minutes followed by a linear gradient to 100% mobile phase B (0.01% acetic acid in acetonitril) over 12 minutes. MS analyses were carried out using electrospray ionization in the negative ion mode using full scan analysis over 70–850 m/z at 70,000 resolution and 3 Hz data acquisition rate.
Raw data were processed using TraceFinder 3.3 software (Thermo Fisher Scientific; Waltham, MA) and Progenesis QI (Nonlinear Dynamics; Newcastle upon Tyne, UK). For each method, metabolite identities were confirmed using authentic reference standards or reference samples.
Metabolite classification was based on the ClassyFire software (v1.0) using the Human Metabolome database (HMDB, v4.0)60,61. The set of peaks spanning unique m/z and retention time (RT) values are referred to as metabolomic features.
Metabolomic features not matching the internal standards were matched approximately via adduct subtraction and molecular formula matching to compounds downloaded from the HMDB on Sep 16, 2020. The measured m/z values were adjusted for method-specific adducts and molecular formulas matching to within 5 parts per million (ppm) were selected as candidate identifiers. When multiple molecular formulas matched the adduct-adjusted mass (as a result of multiple potential adducts), one with minimal ppm difference was selected. The following common adducts are assumed for different LC-MS methods: [M - H](−) for negative mode methods (C18-neg, HILIC-neg) and [M + H](+), [M + NH4](+), [M + Na](+), [M-H2O+H](+) for positive mode methods (C8-pos, HILIC-pos). This yielded molecular formula matches for 52,669 (41%) metabolomic features, with a median of one (range 1–4) candidate annotation per annotated peak.
MS2 data generation.
MS2 data generation was carried out on a Thermo ID-X mass spectrometer (Thermo Fisher Scientific; Waltham, MA) using the targeted MS2 acquisition on features of interest or the AcquireX method, a modified data-dependent MS2 acquisition method provided with the ID-X instrument designed to generate a comprehensive library of MS2 scans for all the features detected in a sample. The AcquireX pipeline as well as the targeted MS2 data generation were applied for the C8-pos method.
Stool–breast milk analyses.
To identify only milk-derived lipids detected in stool, a human milk pooled sample (n=40 subjects) was analyzed alongside the stool study pool in the C8-pos method. The breast milk sample was titrated, and features with abundances that did not correlate with the titration (Pearson<0.8) were removed. The resulting features shared by milk and stool were then targeted to generate MS2 data to confirm the identity of the match and assist in further compound identification efforts. Finally, the stool pool analyzed with milk was used to adjust m/z and RT deviations and align the resulting matching features to the original study data.
MS2 data processing.
MS2-containing raw files were converted to *.mzML format files using MSConvert62. MS2 runs were processed in ProgenesisQI and aligned to the original study data to create a mapping of the features originally annotated in the study to the RT and m/z observed in the MS2 run. All MS2 parsing was conducted using the R package MSnbase v. 3.1263 and in-house scripts for producing extracted ion chromatograms and MS2 spectra visualizations. MS2 data were extracted by matching the precursor ion for each scan to the detected MS1 peak m/z within a RT window of ±0.1 minute and a ±0.2 atomic mass unit (amu). Matching MS2 peaks within 5ppm across MS2 scans spanning the range were aggregated whenever more than one MS2 scan was mapped to each individual feature. The resulting peak height for aggregated peaks was determined as the average of the aggregated peak intensities. When more than one MS2 scan was obtained for a peak, MS2 fragments only detected in one scan were removed from the aggregate. Additionally, an electronic noise fragment detected in the MS2 of low abundance peaks in the m/z range 173.46–174.49 was removed from the parsed data. Parsed MS2 data were formatted as an input for molecular structure predictions (*.ms) or MS2-based similarity networks (*.MGF).
Compound predictions and molecular networking.
Parsed MS2 data were analyzed using SIRIUS CSI-Finger ID version 4.7.2 to generate MS2-based compound predictions40 and chemical classes with CANOPUS64. Molecular formula predictions were generated with Orbitrap-specific settings (MS2 isotope scorer: ignore, mass deviation: 5 ppm, candidates: 10, candidates per ion: 1, possible ionizations: [M+H]+, [M+K]+, [M+Na]+). Structure elucidations were done using all included databases and the adducts ([M+H]+, [M+K]+, [M+Na]+, [M-H2O+H]+, [M+H3N+H]+H). Predictions were exported containing the top structure elucidations parsed for each feature. MS2-based networks were built using the Global Natural Products Social Molecular Networking (GNPS)41, and resulting networks were visualized with Cytoscape v. 3.8.265.
Gut biomarker analysis.
Fecal calprotectin and beta-defensin 2 levels were analyzed in stool samples from infants aged 3, 6, 9 and 12 months with commercial ELISA kits according to the manufacturer’s instructions (Calpro AS, Lysaker, Norway and β-Defensin 2 ELISA Kit, Immundiagnostik, Bensheim, Germany)66,67. Briefly, approximately 100 mg of feces was obtained from each frozen sample. Extraction buffer was then added at a dilution of 1:50 for both beta-defensin 2 and calprotectin detection. Fecal material with the extraction buffer was vortexed for 30 seconds and mixing was continued in a shaker at 1,000 rpm for 3 minutes or until solid particles had dissolved. Samples were then centrifuged for 10 minutes at 10,000g at room temperature, and the supernatants were collected and stored at −20°C until analyzed.
Gut permeability test.
The infants were given an oral dose of 2 ml/kg of a lactulose-mannitol solution containing 5 g lactulose and 2 g mannitol per 100 ml after fasting for a minimum of 4 hours. Urine was collected with special urine collection bags for 5 hours. After measuring the total collected urine volume, the sample was stored in plastic tubes at −20°C. Lactulose concentrations were measured as described previously68 with the following modifications: sample and enzyme volumes 25 μl of sample or standard, 12.5 μl of β-galactosidase, 680 μl enzyme cocktail, and 20 μl of PGI. The final concentrations in the solutions were 500 U/mL for β-galactosidase, 10 mM for ATP, 14.9 mM for NADP, 3.64 U/ml for HK/G6P-DH, and 350 U/mL for PGI. Enzyme reactions were performed in plastic tubes, and absorbance measurements were taken in 96-well plates. VICTOR Wallac 1420 workstation (Perkin Elmer, Waltham, MA) was used for measuring absorbance levels. Mannitol concentrations were determined as described previously69 with the following modifications: sample and enzyme volumes 5 μl of sample or standard, 250 μl of enzyme cocktail, and 12.5 μl of mannitol dehydrogenase. The final concentrations in the solutions were 6.25 mM for NAD+, 6.55 mM for ATP, 3.64 U/mL for HK/G6P-DH, and 133 U/mL for mannitol dehydrogenase. Samples were analyzed in 96-well plates. Megazyme (Wicklow, Ireland) was the provider of mannitol dehydrogenase, whereas all other reagents were purchased from Sigma-Aldrich (Darmstadt, Germany). After calculating the proportions of the excreted lactulose and mannitol, LM ratio was calculated by dividing the lactulose value by the mannitol value.
Circulating cytokine measurements.
Unthawed serum samples were used for the cytokine analysis, as repeated freezing and thawing of the samples decreases the concentrations of detectable analytes. Cytokines detected were: EGF, Eotaxin, FGF-2, Flt-3L, Fractalkine, G-CSF, GM-CSF, GRO, IFNa2, IFNg, IL-1RA, IL-1a, IL-1b, IL-2, IL-3, IL-4, IL-5, IL-6, IL-7, IL-8, IL-9, IL-10, IL-12p40, IL-12p70, IL-13, IL-15, IL-17A, IP-10 (CXCL10), MCP-1, MCP-3, MDC (CCL22), MIP-1a, MIP-1b (CCL4), TGFa, TNFa, TNFb, sCD40L, and VEGF. Cytokine concentrations were assessed as pg/ml using multiplex ELISA (MILLIPLEX MAP Human Cytokine/Chemokine Magnetic bead 38-plex Panel, Millipore, Billerica, MA) according to the manufacturer’s instructions, except for adding a third wash with washing buffer to the plates and replacing sheath fluid with phosphate-buffered saline (PBS) to the samples for Luminex reading. The Bio-Rad Bio-Plex 200 System (Bio-Rad Laboratories, Hercules, CA) instrument was used with the Bio-Plex Manager 5.0 program to run plates and generate quantitative data.
Taxonomic microbiome profiling.
Quality control of metagenomic sequencing reads involved removal of adaptor sequences using Trim Galore! v0.4.4 followed by trimming and/or removal of low quality reads and human sequences with KneadData v0.7.2. Samples with at least 5 million sequencing reads after quality control were included in the analysis. Species-level taxonomic profiles were generated by MetaPhlAn2 v2.9.21 (21 Aug 2019) using species-specific marker database v2.94 (Jan 2019)70. Functional metagenomic profiles were generated using HUMAnN2 v0.11.271.
Metagenomic assembly.
Quality-controlled sequencing reads were assembled using megaHIT v1.1.4–2-gd1998a172, independently for each sample, and open reading frames were predicted using Prodigal v2.6.373. Adaptive contig binning in metaBAT2 v2.15–3-g367a7ef74 was used to reconstruct metagenome-assembled genomes (MAGs) followed by MAG quality and contamination estimation by checkM v1.1.275 and taxonomic assignment by GTDB-Tk v1.0.2 (database release 89)76. A non-redundant gene catalog from all assembled genes (not only the genes harbored by MAGs) was constructed by clustering genes based on sequence similarity (95% identity, 90% coverage) using CD-HIT v4.777. The genes in the catalog were functionally annotated by eggNOG-mapper v2.0.178 and their abundance was estimated by mapping sequencing reads from each sample against the gene catalog with k-mer alignment in KMA79.
Mother-to-infant strain transmission.
SNP-haplotypes of the dominant bacterial strains of a given species per sample were determined by StrainPhlAn v2.7.780 by requiring a minimum coverage of 5 bases for SNP calling (‘–min_read_depth 5’ command line parameter for sample2markers.py Python script). Following previous mother-to-infant transmission analysis10, SNP haplotype similarities for a given species were first median-normalized, and values less than 0.25 were treated as identical strains.
Mother-to-infant gene transmission.
To survey microbial genes shared between mother and infant independently from the harboring species, we first identified maternal MAGs present at delivery of species that were not observed in the MetaPhlAn taxonomic profiles of the offspring. Only samples predating the first observation of any given species in an infant were included in the analysis. We then identified microbial genes from the infant sample that shared 100% sequence identity with genes on the subset of maternal MAGs: first, we collected non-redundant gene clusters (from the gene catalog described above under Metagenomic assembly) that included genes from both the maternal MAGs and the corresponding infant samples, then conducted multiple sequence alignment of these gene clusters using Clustal Omega v1.2.481 (with Kimura distance correction) and identified mother-infant gene pairs with 100% nucleotide identity. We filtered these down to a list of non-redundant gene transmission events (Table S3) by removing any repeated observations of the same gene over time. Contigs harboring shared genes were analyzed using checkV v0.7.032 to identify prophage segments. The Basic Local Alignment Search Tool (BLAST)82 was used for the alignment of contigs harboring genes shared between mother and infant, and to identify non-orthologous, overlapping sequences. Global alignment of these was performed with EMBOSS Stretcher83.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis of metagenomic data.
All statistical analyses were performed using R version 4.1.1 (2021–08-10), and figure panels were created using ggplot2 (v.3.3.5). Ordination plots were generated using t-distributed stochastic neighbor embedding (t-SNE, package Rtsne v. 0.15)84. The influence of metadata variables and maternal species’ relative abundance on infant microbiome composition was quantified using PERMANOVA analysis with the adonis2 function from the vegan package (v. 2.5–7). Differences in maternal species-level taxonomy and gene-level composition between gestational week 27, delivery, and 3 months postpartum were quantified using MaAsLin2 (v.1.7.2)85, with participant identity included in the model as a random effect to account for the longitudinal analysis. Alterations of infant microbial species, genes, and glycoside hydrolase families that were associated with maternal species’ relative abundance, abundance of different fecal metabolites, use and type of infant formula, and markers of intestinal inflammation and permeability were also assessed using MaAsLin2. For these analyses, participant identity was included in the model as a random effect, and infant age, sex, delivery mode, formula use and type, prior antibiotic treatment, and breastfeeding were included as fixed effects, as indicated in the figure legends. For the MaAsLin2 analyses, a prevalence filter of 10% was imposed. The significance threshold for these analyses was a false discovery rate (FDR)-corrected p-value (q-value) of 0.25, unless otherwise stated.
Genes were annotated by the Kyoto Encyclopedia of Genes and Genomes (KEGG) database – including a separate database for enzymes under the Enzyme Nomenclature (EC) system – and with eggNOG (evolutionary genealogy of genes: Non-supervised Orthologous Groups)78 and UniProt databases86. Carbohydrate-active enzymes were separately annotated using the Carbohydrate Active Enzymes (CAZy) database (http://www.cazy.org/). For permutation analysis, genes associated with different modes of HGT were identified by filtering for the following eggNOG free text description terms: 1) Transduction: antitermination, baseplate, capsid, DUF4102, excisionase, head, hmm pf00665, KilA-N, ORF11CD3, phage, portal, tail, terminase, tape, T5orf172, viral, virion; 2) Conjugation and type IV secretion systems: conjugal, conjugation, conjugative, DotD, IV secretory, IV secretion, MobA, mobilisation, mobilization, MobL, Pfam:DUF955, plasmid, relaxase, TcpE family, TraG, TraL, TraM; and 3) Transposable elements: DDE, hmm pf01609, IS66, IstB-like ATP binding, transposase, transposon, transposition. Genes associated with transformation were identified by filtering for the following KEGG Orthology ids: K02237, K02238, K02242, K02243, K02244, K02245, K02246, K12296, K04096, K06198, K07343, K22299. Certain designations that may be related to different modes of HGT were categorized as ‘miscellaneous’; these included the following terms from eggNOG free text descriptions: anti-restriction, antirestriction, DNA integrase, DNA integration, integrase core domain, PFAM integrase catalytic. The terms used for identification of genes associated with different mobilome-related functions were selected based on thorough analysis of KEGG, eggNOG and UniProt annotations from 20 randomly drawn samples of 977 genes (19,540 genes in total) from the gene catalog.
The Wilcoxon signed-rank test and the Mann-Whitney U-test were used for comparisons of paired and unpaired continuous data, respectively, whereas the Fisher’s exact test was used for categorical data, with corrections for multiple comparisons performed with either the Benjamini-Hochberg or the Bonferroni method as indicated in the figure legends.
Statistical analysis of metabolomic data.
Before statistical analysis, missing intensity values were imputed with half of the minimum value per metabolomic feature. Ordination plots and heatmaps were generated using intensity values that had been standardized (z-scores) across the dataset. All statistical analyses were performed using R version 4.1.1 (2021–08-10), and figure panels were created using ggplot2 (v.3.3.5). Ordination plots were generated using t-SNE (package Rtsne v. 0.15). The influence of metadata variables on infant metabolomes was quantified using PERMANOVA analysis with the adonis function from the vegan package (v. 2.5–7). The Wilcoxon signed-rank test and the Mann-Whitney U-test were used for comparisons of paired and unpaired continuous data, respectively, whereas the Fisher’s exact test or the chi-square test with Yates’ correction were used for categorical data, with corrections for multiple comparisons performed with either the Benjamini-Hochberg or the Bonferroni method as indicated in the figure legends.
Supplementary Material
Figure S1. Age-dependent trajectories of serum cytokines, intestinal permeability and inflammation markers, and diet in infants, Related to Figure 1. A) Median serum cytokine levels stratified by infant age. Included are infants with data from all four time points. Levels of EGF, eotaxin, G-CSF, MDC, MIP-1a and MIP-1b differed significantly (Friedman test, p<0.05) between time points. B) Fecal inflammation markers calprotectin and beta-defensin 2 are elevated in young infants. Blue lines connect data points from the same infant. p-values obtained by the Friedman test. C) Lactulose-mannitol ratio by age. D) Proportion of infants who were breastfed, formula-fed, and/or received solid food for each time point. Number of samples per time point varies, explaining the increased proportion of breastfed infants at age six months. For boxplots, midlines represent the median, boxes the interquartile range (25th to 75th percentile), and whiskers the range of data.
Figure S2. Steroids and derivatives were enriched in the maternal gut at delivery compared to three months postpartum, Related to Figure 2. A) Metabolite subclasses altered between delivery and 3 months postpartum based on altered metabolomic features (Benjamini-Hochberg-corrected p-value <0.25, Wilcoxon signed-rank test). Metabolomic features were grouped by predicted nominal identity (with/without common adducts). B) Steroids and derivatives altered in pregnancy (Benjamini-Hochberg-corrected p-value <0.25) that were also independently positively associated with glucose intolerance (p<0.01, Mann-Whitney U-test, delivery time point only). GD, gestational diabetes; OGTT, oral glucose tolerance test. C) Examples of steroids enriched in pregnancy and impaired glucose tolerance. Lower row shows results from the delivery time point. Midlines represent the median, boxes the interquartile range (25th to 75th percentile), and whiskers the range of data. One high outlier (for tetrahydrocorticosterone under impaired glucose tolerance) was excluded. 3m pp, 3 months postpartum. D) Associations between species and selected steroids/steroid derivatives that were increased in pregnancy and positively associated with impaired glucose tolerance. Metabolomic features were annotated by mapping observed mass-to-charge ratios to the HMDB (Methods).
Figure S3. Maternal Bacteroides cellulosilyticus expands the carbohydrate utilization capacity of the infant gut microbiome through HGT, Related to Figure 3. A) Number of maternally-derived strains in infant gut metagenomes at each sampling time point. B) Maternally-derived gut strains obtained at 3 months or older, stratified by first detection month. C) Significant (q<0.25) association of maternal B. cellulosilyticus with infant abundances of microbial glycoside hydrolases. D) Left panel: Associations (q<0.5) of maternal B. cellulosilyticus with microbial glycoside hydrolases in infants. Right panels: Negative correlation of maternal B. cellulosilyticus and certain infant fecal HMOs (annotated by mapping observed mass-to-charge ratios to the HMDB) at 0.5 (3-Fucosyllactose) and 1 month (3’-Sialyllactose) of age. R represents Spearman’s rho. E) Associations (q<0.25) of maternal B. cellulosilyticus with species in infants up to 3 months old. Results in (C-E) are from general linear models, adjusted for longitudinal sampling and corrected for infant age, sex, delivery mode, breastfeeding, antibiotics, and formula use/type. Results in (C) are also corrected for infant Bifidobacterium and Bacteroides species with >10% prevalence. Error bars represent standard error. F) Number of gene transmission events in a permutation test where infants were assigned to a random mother 100 times (mean gene transfer events per permutation=443, median=438). Red dot indicates the 977 observed gene transfer events. G) Mother-to-infant gene transmission events, stratified by donor species and infant age at first detection of the gene. H) Illustration of average nucleotide identity estimation between maternal and infant contigs. Case 2 represents a scenario with an overlapping, non-orthologous region. I) Overall nucleotide identity of non-orthologous (<95% nucleotide identity), overlapping sequences of contigs harboring genes shared between mother and infant, stratified by the top three maternal donor species. Midlines represent the median, boxes the interquartile range (25th to 75th percentile), and whiskers the range. J) A high proportion of mother-to-infant gene transmission events was linked to the processes of HGT and carbohydrate utilization and biosynthesis. K) Genes linked to carbohydrate metabolism and transport were commonly transmitted from maternal species to different infant species. Events were stratified by the top three maternal donor species. Point size indicates the percentage of total mapped gene transmission events where the gene was linked to a particular functional category. Genes associated with HGT (e.g., transduction, conjugation, T4SS, transformation, transposon) or generic processes (e.g., replication and repair, translation and transcriptional regulation) not shown. L) Predicted substrates for transmitted genes associated with carbohydrate metabolism and transport that were detected in infants 0.5 to 2 months old, stratified by retention (present at 1 versus ≥2 time points). M) Prevalence of non-redundant genes representing genes involved in mother-to-infant HGT (teal, n=977) and all observed non-redundant genes (red, n=2,358,086). Mean±standard deviation shown. N) Abundance distributions of non-redundant genes representing the 977 genes involved in mother-to-infant HGT and all 2,358,086 genes in the catalog of metagenomically assembled genes.
Figure S4. Infant-specific metabolites and microbe-metabolite associations, Related to Figure 4. A-D) Correlations of (A) Enterococcus faecalis versus tyramine (age 0.5 month) and tyrosine decarboxylase versus tyramine (age 1 month), (B) Escherichia coli versus agmatine (age 3 months), C) Collinsella aerofaciens versus arginine (age 6 months) and arginine deiminase versus arginine (age 3 months), and D) Bifidobacterium longum versus inosine (age 6 months). R values are Kendall’s tau. E) Class distribution of infant-specific metabolites present in ≥50% of infant samples. Red color indicates enriched metabolite classes; p-values obtained by chi-square test with Yates’ correction. F) Associations of infant-specific metabolites (present in ≥⅓ of infant samples) and serum cytokines in 6-month-old infants. Metabolomic features in (E, F) were annotated by mapping observed mass-to-charge ratios to the HMDB. G) Species significantly associated with infant-specific metabolites (present in ≥50% of infant samples and no maternal samples) after adjustment for longitudinal analysis and correction for infant age, sex, delivery mode, antibiotics, and formula use/type. p-values comparing species prevalence in adults and infants obtained by Fisher’s exact test; colors represent prevalence. H) Mean abundance of n=253 infant-specific metabolomic features (both annotated and unknown) with longitudinal trends (Kruskal-Wallis test, FDR corrected p<0.01). Each point shows mean peak intensity value per time point and metabolomic feature. Lines connect points corresponding to metabolomic features. I) Mean abundance of infant-specific metabolomic features in (H) stratified by breastfeeding status: teal, exclusive breastfeeding; red, formula and/or solid foods introduced. Midlines represent the median, boxes the interquartile range (25th to 75th percentile), and whiskers the range. J) Infant-specific metabolomic features connect in GNPS networks (cosine>0.7) with human breast milk metabolomic features. K) GNPS network with densely interconnected infant-specific and breast milk matched metabolomic features. L) Subnetworks built using GNPS with MS2 data for stool metabolomics peaks, infant-specific peaks, breast milk peaks, and MS2 for reference standards. Visualization limited to first and second neighbors of infant-specific peaks that included reference standards.
Figure S5. Correlations of metabolite categories and serum cytokines in infants, Related to Figure 5. Heatmap of correlations between metabolite categories and serum cytokines in 3-month-old infants.
Figure S6. Fecal inflammation marker associations in infants, Related to Figure 6. A) Beta-defensin 2 levels tended to be higher in breastfed infants. Midlines represent the median, boxes the interquartile range (25th to 75th percentile), and whiskers the range of data. B) Positive correlation of Bifidobacterium breve and beta-defensin 2 in 3-month-old infants. C) Inverse correlation between linoleoyl ethanolamide (LEA) and intestinal inflammation markers in 6-month-old infants.
Table S1. Metabolomic features observed in this study, Related to Figures 1, 2, 4, 5 and 6. The sheet named ‘Reference standards’ lists 858 metabolites annotated using reference standards, while the sheet named ‘HMDB based’ lists 52,669 metabolomic features with putative annotations based on molecular formula matching against the HMDB. A single representative candidate representing the best match is indicated in the column NominalHit along with adduct and ppm delta information. The sheet named ‘MSMS’ lists molecular formulas and structures as predicted by SIRIUS for 30 infant-specific metabolomic features characterized by MS2 of 10 infant samples.
Table S2. Demographic, dietary, and clinical characteristics of infants in the study, stratified by age, Related to Figures 1 and 5. Data in the table are restricted to samples with conclusive results from metagenomic analysis. Infant age is shown in months (m). abx, antibiotic; RF, regular formula; HF, hydrolyzed formula. 1variable with missing data for certain time points.
Table S3. Mother-to-infant gene transmission events, Related to Figure 3. The sheet ‘Transmitted genes’ shows n=977 genes that were identical (100% nucleotide identity) between mother and infant and were harbored by MAGs of discordant bacterial species in mother and infant. Subsequent sheets tabulate the gene transmission events between MAGs of different genera, families, and phyla, respectively.
Table S4. Predicted prophage segments of MAG contigs harboring mother-to-infant interspecies transmitted genes, Related to Figure 3. Prophage predictions by checkV.
Table S5. Differentially abundant metabolites in mothers and infants, Related to Figure 4. List of n=368 metabolites annotated using reference standards that were more abundant in infants (q<0.01) and n=189 metabolites that were more abundant in mothers (q<0.01).
Table S6. Associations between species and metabolites in the infant gut, Related to Figure 4. The table shows associations between the relative abundances of bacterial species and the abundances of metabolites annotated by reference standards in infant fecal samples. The associations were derived from general linear models adjusted for longitudinal analysis and corrected for infant age, sex, delivery mode, antibiotics, and formula use and type. Included are associations that were statistically significant (q<0.25) following Bonferroni correction for multiple comparisons. Associations were marked as “significant in mothers” in the event of a congruous association in maternal samples with an uncorrected q-value <0.25. The column marked as “confirmed in prior in vitro experiment” refers to results of a study investigating the metabolic signatures of 178 strains from the gut microbiome [S1].
Highlights.
Mobile genetic elements from maternal bacteria shape offspring gut microbiomes
Microbiome and metabolome shifts in pregnancy may impact maternal metabolic health
The infant gut harbors unique metabolites and species-metabolite relationships
Diet modulates metabolomic profiles and immune system maturation in infants
Acknowledgements
We thank Tiffany Poon, Luke Besse, and Timothy Arthur for sample and data management, and Theresa Reimels for editorial assistance and figure preparation. This work was funded by the National Institutes of Health (P30 DK043351 to RJX), Juvenile Diabetes Research Foundation (2-SRA-2016-247-S-B), Center for Microbiome Informatics and Therapeutics, and Wallenberg Foundations (to KSJ). MY is the Rosalind, Paul and Robin Berlin Faculty Development Chair in Perinatal Research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
RJX is co-founder of Jnana Therapeutics and Celsius Therapeutics, Board Director at MoonLake Immunotherapeutics, and consultant to Nestlé; these organizations had no role in the study. All other authors declare no competing interests.
ADDITIONAL RESOURCES
Clinicaltrials.gov Identifier: NCT01735123
References
- 1.Charbonneau MR, Blanton LV, DiGiulio DB, Relman DA, Lebrilla CB, Mills DA, and Gordon JI (2016). A microbial perspective of human developmental biology. Nature 535, 48–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rao C, Coyte KZ, Bainter W, Geha RS, Martin CR, and Rakoff-Nahoum S (2021). Multi-kingdom ecological drivers of microbiota assembly in preterm infants. Nature 591, 633–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Derrien M, Alvarez A-S, and de Vos WM (2019). The Gut Microbiota in the First Decade of Life. Trends Microbiol 27, 997–1010. [DOI] [PubMed] [Google Scholar]
- 4.Stewart CJ, Ajami NJ, O’Brien JL, Hutchinson DS, Smith DP, Wong MC, Ross MC, Lloyd RE, Doddapaneni H, Metcalf GA, et al. (2018). Temporal development of the gut microbiome in early childhood from the TEDDY study. Nature 562, 583–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M, Magris M, Hidalgo G, Baldassano RN, Anokhin AP, et al. (2012). Human gut microbiome viewed across age and geography. Nature 486, 222–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bäckhed F, Roswall J, Peng Y, Feng Q, Jia H, Kovatcheva-Datchary P, Li Y, Xia Y, Xie H, Zhong H, et al. (2015). Dynamics and Stabilization of the Human Gut Microbiome during the First Year of Life. Cell Host Microbe 17, 852. [DOI] [PubMed] [Google Scholar]
- 7.Lim ES, Zhou Y, Zhao G, Bauer IK, Droit L, Ndao IM, Warner BB, Tarr PI, Wang D, and Holtz LR (2015). Early life dynamics of the human gut virome and bacterial microbiome in infants. Nat. Med 21, 1228–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Palmer C, Bik EM, DiGiulio DB, Relman DA, and Brown PO (2007). Development of the Human Infant Intestinal Microbiota. PLoS Biol 5, e177–e177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vatanen T, Plichta DR, Somani J, Münch PC, Arthur TD, Hall AB, Rudolf S, Oakeley EJ, Ke X, Young RA, et al. (2019). Genomic variation and strain-specific functional adaptation in the human gut microbiome during early life. Nature Microbiology 4, 470–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ferretti P, Pasolli E, Tett A, Asnicar F, Gorfer V, Fedi S, Armanini F, Truong DT, Manara S, Zolfo M, et al. (2018). Mother-to-Infant Microbial Transmission from Different Body Sites Shapes the Developing Infant Gut Microbiome. Cell Host Microbe 24, 133–145.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yassour M, Jason E, Hogstrom LJ, Arthur TD, Tripathi S, Siljander H, Selvenius J, Oikarinen S, Hyöty H, Virtanen SM, et al. (2018). Strain-Level Analysis of Mother-to-Child Bacterial Transmission during the First Few Months of Life. Cell Host Microbe 24, 146–154.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vatanen T, Kostic AD, d’Hennezel E, Siljander H, Franzosa EA, Yassour M, Kolde R, Vlamakis H, Arthur TD, Hämäläinen A-M, et al. (2016). Variation in Microbiome LPS Immunogenicity Contributes to Autoimmunity in Humans. Cell 165, 1551. [DOI] [PubMed] [Google Scholar]
- 13.Lewis ZT, Totten SM, Smilowitz JT, Popovic M, Parker E, Lemay DG, Van Tassell ML, Miller MJ, Jin Y-S, German JB, et al. (2015). Maternal fucosyltransferase 2 status affects the gut bifidobacterial communities of breastfed infants. Microbiome 3, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Henrick BM, Rodriguez L, Lakshmikanth T, Pou C, Henckel E, Arzoomand A, Olin A, Wang J, Mikes J, Tan Z, et al. (2021). Bifidobacteria-mediated immune system imprinting early in life. Cell 10.1016/j.cell.2021.05.030. [DOI] [PubMed]
- 15.Siljander H, Jason E, Ruohtula T, Selvenius J, Koivusaari K, Salonen M, Ahonen S, Honkanen J, Ilonen J, Vaarala O, et al. (2021). Effect of Early Feeding on Intestinal Permeability and Inflammation Markers in Infants with Genetic Susceptibility to Type 1 Diabetes: A Randomized Clinical Trial. J. Pediatr 10.1016/j.jpeds.2021.07.042. [DOI] [PubMed]
- 16.Vuong HE, Pronovost GN, Williams DW, Coley EJL, Siegler EL, Qiu A, Kazantsev M, Wilson CJ, Rendon T, and Hsiao EY (2020). The maternal microbiome modulates fetal neurodevelopment in mice. Nature 586, 281–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Luk B, Veeraragavan S, Engevik M, Balderas M, Major A, Runge J, Luna RA, and Versalovic J (2018). Postnatal colonization with human “infant-type” Bifidobacterium species alters behavior of adult gnotobiotic mice. PLoS One 13, e0196510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Olin A, Henckel E, Chen Y, Lakshmikanth T, Pou C, Mikes J, Gustafsson A, Bernhardsson AK, Zhang C, Bohlin K, et al. (2018). Stereotypic Immune System Development in Newborn Children. Cell 174, 1277–1292.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lau SKP (2006). Bacteraemia caused by Anaerotruncus colihominis and emended description of the species. Journal of Clinical Pathology 59, 748–752. 10.1136/jcp.2005.031773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bianchimano P, Britton GJ, Wallach DS, Smith EM, Cox LM, Liu S, Weiner HL, Faith JJ, Clemente JC, and Tankou SK (2021). Human gut derived Anaerotruncus colihominis ameliorates experimental autoimmune encephalomyelitis. bioRxiv, 2021.11.10.468120. 10.1101/2021.11.10.468120. [DOI]
- 21.Fehr K, Moossavi S, Sbihi H, Boutin RCT, Bode L, Robertson B, Yonemitsu C, Field CJ, Becker AB, Mandhane PJ, et al. (2020). Breastmilk Feeding Practices Are Associated with the Co-Occurrence of Bacteria in Mothers’ Milk and the Infant Gut: the CHILD Cohort Study. Cell Host Microbe 28, 285–297.e4. [DOI] [PubMed] [Google Scholar]
- 22.Martín V, Mediano P, Del Campo R, Rodríguez JM, and Marín M (2016). Streptococcal Diversity of Human Milk and Comparison of Different Methods for the Taxonomic Identification of Streptococci. J. Hum. Lact 32, NP84–NP94. [DOI] [PubMed] [Google Scholar]
- 23.Gauglitz JM, West KA, Bittremieux W, Williams CL, Weldon KC, Panitchpakdi M, Di Ottavio F, Aceves CM, Brown E, Sikora NC, et al. (2022). Enhancing untargeted metabolomics using metadata-based source annotation. Nat. Biotechnol 10.1038/s41587-022-01368-1. [DOI] [PMC free article] [PubMed]
- 24.Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, and Gordon JI (2006). An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444, 1027–1031. [DOI] [PubMed] [Google Scholar]
- 25.Mathur R, Chua KS, Mamelak M, Morales W, Barlow GM, Thomas R, Stefanovski D, Weitsman S, Marsh Z, Bergman RN, et al. (2016). Metabolic effects of eradicating breath methane using antibiotics in prediabetic subjects with obesity. Obesity 24, 576–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Natividad JM, Lamas B, Pham HP, Michel M-L, Rainteau D, Bridonneau C, da Costa G, van Hylckama Vlieg J, Sovran B, Chamignon C, et al. (2018). Bilophila wadsworthia aggravates high fat diet induced metabolic dysfunctions in mice. Nat. Commun 9, 2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Devkota S, Wang Y, Musch MW, Leone V, Fehlner-Peach H, Nadimpalli A, Antonopoulos DA, Jabri B, and Chang EB (2012). Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10−/− mice. Nature 487, 104–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Peck SC, Denger K, Burrichter A, Irwin SM, Balskus EP, and Schleheck D (2019). A glycyl radical enzyme enables hydrogen sulfide production by the human intestinal bacterium Bilophila wadsworthia. Proc. Natl. Acad. Sci. U. S. A 116, 3171–3176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mitchell CM, Mazzoni C, Hogstrom L, Bryant A, Bergerat A, Cher A, Pochan S, Herman P, Carrigan M, Sharp K, et al. (2020). Delivery Mode Affects Stability of Early Infant Gut Microbiota. Cell Rep Med 1, 100156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Asnicar F, Manara S, Zolfo M, Truong DT, Scholz M, Armanini F, Ferretti P, Gorfer V, Pedrotti A, Tett A, et al. (2017). Studying Vertical Microbiome Transmission from Mothers to Infants by Strain-Level Metagenomic Profiling. mSystems 2. 10.1128/mSystems.00164-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Groussin M, Poyet M, Sistiaga A, Kearney SM, Moniz K, Noel M, Hooker J, Gibbons SM, Segurel L, Froment A, et al. (2021). Elevated rates of horizontal gene transfer in the industrialized human microbiome. Cell 184, 2053–2067.e18. [DOI] [PubMed] [Google Scholar]
- 32.Nayfach S, Camargo AP, Schulz F, Eloe-Fadrosh E, Roux S, and Kyrpides NC (2021). CheckV assesses the quality and completeness of metagenome-assembled viral genomes. Nat. Biotechnol 39, 578–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McNulty NP, Wu M, Erickson AR, Pan C, Erickson BK, Martens EC, Pudlo NA, Muegge BD, Henrissat B, Hettich RL, et al. (2013). Effects of diet on resource utilization by a model human gut microbiota containing Bacteroides cellulosilyticus WH2, a symbiont with an extensive glycobiome. PLoS Biol 11, e1001637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mager LF, Burkhard R, Pett N, Cooke NCA, Brown K, Ramay H, Paik S, Stagg J, Groves RA, Gallo M, et al. (2020). Microbiome-derived inosine modulates response to checkpoint inhibitor immunotherapy. Science 369, 1481–1489. [DOI] [PubMed] [Google Scholar]
- 35.Han S, Van Treuren W, Fischer CR, Merrill BD, DeFelice BC, Sanchez JM, Higginbottom SK, Guthrie L, Fall LA, Dodd D, et al. (2021). A metabolomics pipeline for the mechanistic interrogation of the gut microbiome. Nature 595, 415–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hatanaka M, Morita H, Aoyagi Y, Sasaki K, Sasaki D, Kondo A, and Nakamura T (2020). Effective bifidogenic growth factors cyclo-Val-Leu and cyclo-Val-Ile produced by Bacillus subtilis C-3102 in the human colonic microbiota model. Sci. Rep 10, 7591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Duan S, and Paulson JC (2020). Siglecs as Immune Cell Checkpoints in Disease. Annu. Rev. Immunol 38, 365–395. [DOI] [PubMed] [Google Scholar]
- 38.Charbonneau MR, O’Donnell D, Blanton LV, Totten SM, Davis JCC, Barratt MJ, Cheng J, Guruge J, Talcott M, Bain JR, et al. (2016). Sialylated Milk Oligosaccharides Promote Microbiota-Dependent Growth in Models of Infant Undernutrition. Cell 164, 859–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Oliveira RA, Ng KM, Correia MB, Cabral V, Shi H, Sonnenburg JL, Huang KC, and Xavier KB (2020). Klebsiella michiganensis transmission enhances resistance to Enterobacteriaceae gut invasion by nutrition competition. Nat Microbiol 5, 630–641. [DOI] [PubMed] [Google Scholar]
- 40.Dührkop K, Fleischauer M, Ludwig M, Aksenov AA, Melnik AV, Meusel M, Dorrestein PC, Rousu J, and Böcker S (2019). SIRIUS 4: a rapid tool for turning tandem mass spectra into metabolite structure information. Nat. Methods 16, 299–302. [DOI] [PubMed] [Google Scholar]
- 41.Wang M, Carver JJ, Phelan VV, Sanchez LM, Garg N, Peng Y, Nguyen DD, Watrous J, Kapono CA, Luzzatto-Knaan T, et al. (2016). Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat. Biotechnol 34, 828–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Floris LM, Stahl B, Abrahamse-Berkeveld M, and Teller IC (2020). Human milk fatty acid profile across lactational stages after term and preterm delivery: A pooled data analysis. Prostaglandins Leukot. Essent. Fatty Acids 156, 102023. [DOI] [PubMed] [Google Scholar]
- 43.Schirmer M, Smeekens SP, Vlamakis H, Jaeger M, Oosting M, Franzosa EA, Horst RT, Jansen T, Jacobs L, Bonder MJ, et al. (2016). Linking the Human Gut Microbiome to Inflammatory Cytokine Production Capacity. Cell 167, 1897. [DOI] [PubMed] [Google Scholar]
- 44.Franzosa EA, Sirota-Madi A, Avila-Pacheco J, Fornelos N, Haiser HJ, Reinker S, Vatanen T, Hall AB, Mallick H, McIver LJ, et al. (2019). Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat Microbiol 4, 293–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gräler MH, and Goetzl EJ (2002). Lysophospholipids and their G protein-coupled receptors in inflammation and immunity. Biochim. Biophys. Acta 1582, 168–174. [DOI] [PubMed] [Google Scholar]
- 46.Knuplez E, and Marsche G (2020). An Updated Review of Pro- and Anti-Inflammatory Properties of Plasma Lysophosphatidylcholines in the Vascular System. Int. J. Mol. Sci 21. 10.3390/ijms21124501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li F, Ma J, Geng S, Wang J, Liu J, Zhang J, and Sheng X (2015). Fecal calprotectin concentrations in healthy children aged 1–18 months. PLoS One 10, e0119574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Campeotto F, Baldassarre M, Laforgia N, Viallon V, Kalach N, Amati L, Butel MJ, Dupont C, and Kapel N (2010). Fecal expression of human β-defensin-2 following birth. Neonatology 98, 365–369. [DOI] [PubMed] [Google Scholar]
- 49.Fornelos N, Franzosa EA, Bishai J, Annand JW, Oka A, Lloyd-Price J, Arthur TD, Garner A, Avila-Pacheco J, Haiser HJ, et al. (2020). Growth effects of N-acylethanolamines on gut bacteria reflect altered bacterial abundances in inflammatory bowel disease. Nat Microbiol 5, 486–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hadley KB, Ryan AS, Forsyth S, Gautier S, and Salem N, Jr (2016). The Essentiality of Arachidonic Acid in Infant Development. Nutrients 8, 216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Coyne MJ, Zitomersky NL, McGuire AM, Earl AM, and Comstock LE (2014). Evidence of extensive DNA transfer between bacteroidales species within the human gut. MBio 5, e01305–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Garcia-Larsen V, Ierodiakonou D, Jarrold K, Cunha S, Chivinge J, Robinson Z, Geoghegan N, Ruparelia A, Devani P, Trivella M, et al. (2018). Diet during pregnancy and infancy and risk of allergic or autoimmune disease: A systematic review and meta-analysis. PLoS Med 15, e1002507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Leiva-Gea I, Sánchez-Alcoholado L, Martín-Tejedor B, Castellano-Castillo D, Moreno-Indias I, Urda-Cardona A, Tinahones FJ, Fernández-García JC, and Queipo-Ortuño MI (2018). Gut Microbiota Differs in Composition and Functionality Between Children With Type 1 Diabetes and MODY2 and Healthy Control Subjects: A Case-Control Study. Diabetes Care 41, 2385–2395. [DOI] [PubMed] [Google Scholar]
- 54.Walker WA, and Iyengar RS (2015). Breast milk, microbiota, and intestinal immune homeostasis. Pediatr. Res 77, 220–228. [DOI] [PubMed] [Google Scholar]
- 55.Tauch A, Fernández-Natal I, and Soriano F (2016). A microbiological and clinical review on Corynebacterium kroppenstedtii. Int. J. Infect. Dis 48, 33–39. [DOI] [PubMed] [Google Scholar]
- 56.Chen L, Lu W, Wang L, Xing X, Chen Z, Teng X, Zeng X, Muscarella AD, Shen Y, Cowan A, et al. (2021). Metabolite discovery through global annotation of untargeted metabolomics data. Nat. Methods 18, 1377–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Benowitz LI, Goldberg DE, Madsen JR, Soni D, and Irwin N (1999). Inosine stimulates extensive axon collateral growth in the rat corticospinal tract after injury. Proc. Natl. Acad. Sci. U. S. A 96, 13486–13490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stacy A, Andrade-Oliveira V, McCulloch JA, Hild B, Oh JH, Juliana Perez-Chaparro P, Sim CK, Lim AI, Link VM, Enamorado M, et al. (2021). Infection trains the host for microbiota-enhanced resistance to pathogens. Cell 184, 615–627.e17. 10.1016/j.cell.2020.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ilonen J, Hammais A, Laine A-P, Lempainen J, Vaarala O, Veijola R, Simell O, and Knip M (2013). Patterns of β-cell autoantibody appearance and genetic associations during the first years of life. Diabetes 62, 3636–3640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Djoumbou Feunang Y, Eisner R, Knox C, Chepelev L, Hastings J, Owen G, Fahy E, Steinbeck C, Subramanian S, Bolton E, et al. (2016). ClassyFire: automated chemical classification with a comprehensive, computable taxonomy. J. Cheminform 8, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wishart DS, Jewison T, Guo AC, Wilson M, Knox C, Liu Y, Djoumbou Y, Mandal R, Aziat F, Dong E, et al. (2012). HMDB 3.0—The Human Metabolome Database in 2013. Nucleic Acids Research 41, D801–D807. 10.1093/nar/gks1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chambers MC, Maclean B, Burke R, Amodei D, Ruderman DL, Neumann S, Gatto L, Fischer B, Pratt B, Egertson J, et al. (2012). A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol 30, 918–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gatto L, Gibb S, and Rainer J (2021). MSnbase, Efficient and Elegant R-Based Processing and Visualization of Raw Mass Spectrometry Data. J. Proteome Res 20, 1063–1069. [DOI] [PubMed] [Google Scholar]
- 64.Dührkop K, Nothias L-F, Fleischauer M, Reher R, Ludwig M, Hoffmann MA, Petras D, Gerwick WH, Rousu J, Dorrestein PC, et al. (2021). Systematic classification of unknown metabolites using high-resolution fragmentation mass spectra. Nat. Biotechnol 39, 462–471. [DOI] [PubMed] [Google Scholar]
- 65.Otasek D, Morris JH, Bouças J, Pico AR, and Demchak B (2019). Cytoscape Automation: empowering workflow-based network analysis. Genome Biol 20, 185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tøn H, Brandsnes S. Dale, Holtlund J, Skuibina E, Schjønsby H, and Johne B(2000). Improved assay for fecal calprotectin. Clin. Chim. Acta 292, 41–54. [DOI] [PubMed] [Google Scholar]
- 67.Wehkamp J, Fellermann K, Herrlinger KR, Baxmann S, Schmidt K, Schwind B, Duchrow M, Wohlschläger C, Feller AC, and Stange EF (2002). Human β-defensin 2 but not β-defensin 1 is expressed preferentially in colonic mucosa of inflammatory bowel disease. Eur. J. Gastroenterol. Hepatol 14, 745. [DOI] [PubMed] [Google Scholar]
- 68.Northrop CA, Lunn PG, and Behrens RH (1990). Automated enzymatic assays for the determination of intestinal permeability probes in urine. 1. Lactulose and lactose. Clin. Chim. Acta 187, 79–87. [DOI] [PubMed] [Google Scholar]
- 69.Blood J, Ingle AR, Allison N, Davies GR, and Hill PG (1991). Rapid enzymatic method for the measurement of mannitol in urine. Ann. Clin. Biochem 28 (Pt 4), 401–406. [DOI] [PubMed] [Google Scholar]
- 70.Beghini F, McIver LJ, Blanco-Míguez A, Dubois L, Asnicar F, Maharjan S, Mailyan A, Manghi P, Scholz M, Thomas AM, et al. (2021). Integrating taxonomic, functional, and strain-level profiling of diverse microbial communities with bioBakery 3. Elife 10. 10.7554/eLife.65088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Franzosa EA, McIver LJ, Rahnavard G, Thompson LR, Schirmer M, Weingart G, Lipson KS, Knight R, Caporaso JG, Segata N, et al. (2018). Species-level functional profiling of metagenomes and metatranscriptomes. Nat. Methods 15, 962–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Li D, Liu C-M, Luo R, Sadakane K, and Lam T-W (2015). MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676. [DOI] [PubMed] [Google Scholar]
- 73.Hyatt D, Chen G-L, Locascio PF, Land ML, Larimer FW, and Hauser LJ (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kang DD, Li F, Kirton E, Thomas A, Egan R, An H, and Wang Z (2019). MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 7, e7359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, and Tyson GW (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res 25, 1043–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chaumeil P-A, Mussig AJ, Hugenholtz P, and Parks DH (2019). GTDB-Tk: a toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 10.1093/bioinformatics/btz848. [DOI] [PMC free article] [PubMed]
- 77.Fu L, Niu B, Zhu Z, Wu S, and Li W (2012). CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Huerta-Cepas J, Szklarczyk D, Heller D, Hernández-Plaza A, Forslund SK, Cook H, Mende DR, Letunic I, Rattei T, Jensen LJ, et al. (2019). eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res 47, D309–D314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Clausen PTLC, Aarestrup FM, and Lund O (2018). Rapid and precise alignment of raw reads against redundant databases with KMA. BMC Bioinformatics 19, 307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Truong DT, Tett A, Pasolli E, Huttenhower C, and Segata N (2017). Microbial strain-level population structure and genetic diversity from metagenomes. Genome Res, gr.216242.116–gr.216242.116. [DOI] [PMC free article] [PubMed]
- 81.Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, et al. (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol 7, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Altschul SF, Gish W, Miller W, Myers EW, and Lipman DJ (1990). Basic local alignment search tool. J. Mol. Biol 215, 403–410. [DOI] [PubMed] [Google Scholar]
- 83.Myers EW, and Miller W (1988). Optimal alignments in linear space. Comput. Appl. Biosci 4, 11–17. [DOI] [PubMed] [Google Scholar]
- 84.Van der Maaten L, and Hinton G (2008). Visualizing data using t-SNE. J. Mach. Learn. Res 9. [Google Scholar]
- 85.Mallick H, Rahnavard A, McIver LJ, Ma S, Zhang Y, Nguyen LH, Tickle TL, Weingart G, Ren B, Schwager EH, et al. (2021). Multivariable association discovery in population-scale meta-omics studies. PLoS Comput. Biol 17, e1009442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Consortium UniProt (2019). UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res 47, D506–D515. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Age-dependent trajectories of serum cytokines, intestinal permeability and inflammation markers, and diet in infants, Related to Figure 1. A) Median serum cytokine levels stratified by infant age. Included are infants with data from all four time points. Levels of EGF, eotaxin, G-CSF, MDC, MIP-1a and MIP-1b differed significantly (Friedman test, p<0.05) between time points. B) Fecal inflammation markers calprotectin and beta-defensin 2 are elevated in young infants. Blue lines connect data points from the same infant. p-values obtained by the Friedman test. C) Lactulose-mannitol ratio by age. D) Proportion of infants who were breastfed, formula-fed, and/or received solid food for each time point. Number of samples per time point varies, explaining the increased proportion of breastfed infants at age six months. For boxplots, midlines represent the median, boxes the interquartile range (25th to 75th percentile), and whiskers the range of data.
Figure S2. Steroids and derivatives were enriched in the maternal gut at delivery compared to three months postpartum, Related to Figure 2. A) Metabolite subclasses altered between delivery and 3 months postpartum based on altered metabolomic features (Benjamini-Hochberg-corrected p-value <0.25, Wilcoxon signed-rank test). Metabolomic features were grouped by predicted nominal identity (with/without common adducts). B) Steroids and derivatives altered in pregnancy (Benjamini-Hochberg-corrected p-value <0.25) that were also independently positively associated with glucose intolerance (p<0.01, Mann-Whitney U-test, delivery time point only). GD, gestational diabetes; OGTT, oral glucose tolerance test. C) Examples of steroids enriched in pregnancy and impaired glucose tolerance. Lower row shows results from the delivery time point. Midlines represent the median, boxes the interquartile range (25th to 75th percentile), and whiskers the range of data. One high outlier (for tetrahydrocorticosterone under impaired glucose tolerance) was excluded. 3m pp, 3 months postpartum. D) Associations between species and selected steroids/steroid derivatives that were increased in pregnancy and positively associated with impaired glucose tolerance. Metabolomic features were annotated by mapping observed mass-to-charge ratios to the HMDB (Methods).
Figure S3. Maternal Bacteroides cellulosilyticus expands the carbohydrate utilization capacity of the infant gut microbiome through HGT, Related to Figure 3. A) Number of maternally-derived strains in infant gut metagenomes at each sampling time point. B) Maternally-derived gut strains obtained at 3 months or older, stratified by first detection month. C) Significant (q<0.25) association of maternal B. cellulosilyticus with infant abundances of microbial glycoside hydrolases. D) Left panel: Associations (q<0.5) of maternal B. cellulosilyticus with microbial glycoside hydrolases in infants. Right panels: Negative correlation of maternal B. cellulosilyticus and certain infant fecal HMOs (annotated by mapping observed mass-to-charge ratios to the HMDB) at 0.5 (3-Fucosyllactose) and 1 month (3’-Sialyllactose) of age. R represents Spearman’s rho. E) Associations (q<0.25) of maternal B. cellulosilyticus with species in infants up to 3 months old. Results in (C-E) are from general linear models, adjusted for longitudinal sampling and corrected for infant age, sex, delivery mode, breastfeeding, antibiotics, and formula use/type. Results in (C) are also corrected for infant Bifidobacterium and Bacteroides species with >10% prevalence. Error bars represent standard error. F) Number of gene transmission events in a permutation test where infants were assigned to a random mother 100 times (mean gene transfer events per permutation=443, median=438). Red dot indicates the 977 observed gene transfer events. G) Mother-to-infant gene transmission events, stratified by donor species and infant age at first detection of the gene. H) Illustration of average nucleotide identity estimation between maternal and infant contigs. Case 2 represents a scenario with an overlapping, non-orthologous region. I) Overall nucleotide identity of non-orthologous (<95% nucleotide identity), overlapping sequences of contigs harboring genes shared between mother and infant, stratified by the top three maternal donor species. Midlines represent the median, boxes the interquartile range (25th to 75th percentile), and whiskers the range. J) A high proportion of mother-to-infant gene transmission events was linked to the processes of HGT and carbohydrate utilization and biosynthesis. K) Genes linked to carbohydrate metabolism and transport were commonly transmitted from maternal species to different infant species. Events were stratified by the top three maternal donor species. Point size indicates the percentage of total mapped gene transmission events where the gene was linked to a particular functional category. Genes associated with HGT (e.g., transduction, conjugation, T4SS, transformation, transposon) or generic processes (e.g., replication and repair, translation and transcriptional regulation) not shown. L) Predicted substrates for transmitted genes associated with carbohydrate metabolism and transport that were detected in infants 0.5 to 2 months old, stratified by retention (present at 1 versus ≥2 time points). M) Prevalence of non-redundant genes representing genes involved in mother-to-infant HGT (teal, n=977) and all observed non-redundant genes (red, n=2,358,086). Mean±standard deviation shown. N) Abundance distributions of non-redundant genes representing the 977 genes involved in mother-to-infant HGT and all 2,358,086 genes in the catalog of metagenomically assembled genes.
Figure S4. Infant-specific metabolites and microbe-metabolite associations, Related to Figure 4. A-D) Correlations of (A) Enterococcus faecalis versus tyramine (age 0.5 month) and tyrosine decarboxylase versus tyramine (age 1 month), (B) Escherichia coli versus agmatine (age 3 months), C) Collinsella aerofaciens versus arginine (age 6 months) and arginine deiminase versus arginine (age 3 months), and D) Bifidobacterium longum versus inosine (age 6 months). R values are Kendall’s tau. E) Class distribution of infant-specific metabolites present in ≥50% of infant samples. Red color indicates enriched metabolite classes; p-values obtained by chi-square test with Yates’ correction. F) Associations of infant-specific metabolites (present in ≥⅓ of infant samples) and serum cytokines in 6-month-old infants. Metabolomic features in (E, F) were annotated by mapping observed mass-to-charge ratios to the HMDB. G) Species significantly associated with infant-specific metabolites (present in ≥50% of infant samples and no maternal samples) after adjustment for longitudinal analysis and correction for infant age, sex, delivery mode, antibiotics, and formula use/type. p-values comparing species prevalence in adults and infants obtained by Fisher’s exact test; colors represent prevalence. H) Mean abundance of n=253 infant-specific metabolomic features (both annotated and unknown) with longitudinal trends (Kruskal-Wallis test, FDR corrected p<0.01). Each point shows mean peak intensity value per time point and metabolomic feature. Lines connect points corresponding to metabolomic features. I) Mean abundance of infant-specific metabolomic features in (H) stratified by breastfeeding status: teal, exclusive breastfeeding; red, formula and/or solid foods introduced. Midlines represent the median, boxes the interquartile range (25th to 75th percentile), and whiskers the range. J) Infant-specific metabolomic features connect in GNPS networks (cosine>0.7) with human breast milk metabolomic features. K) GNPS network with densely interconnected infant-specific and breast milk matched metabolomic features. L) Subnetworks built using GNPS with MS2 data for stool metabolomics peaks, infant-specific peaks, breast milk peaks, and MS2 for reference standards. Visualization limited to first and second neighbors of infant-specific peaks that included reference standards.
Figure S5. Correlations of metabolite categories and serum cytokines in infants, Related to Figure 5. Heatmap of correlations between metabolite categories and serum cytokines in 3-month-old infants.
Figure S6. Fecal inflammation marker associations in infants, Related to Figure 6. A) Beta-defensin 2 levels tended to be higher in breastfed infants. Midlines represent the median, boxes the interquartile range (25th to 75th percentile), and whiskers the range of data. B) Positive correlation of Bifidobacterium breve and beta-defensin 2 in 3-month-old infants. C) Inverse correlation between linoleoyl ethanolamide (LEA) and intestinal inflammation markers in 6-month-old infants.
Table S1. Metabolomic features observed in this study, Related to Figures 1, 2, 4, 5 and 6. The sheet named ‘Reference standards’ lists 858 metabolites annotated using reference standards, while the sheet named ‘HMDB based’ lists 52,669 metabolomic features with putative annotations based on molecular formula matching against the HMDB. A single representative candidate representing the best match is indicated in the column NominalHit along with adduct and ppm delta information. The sheet named ‘MSMS’ lists molecular formulas and structures as predicted by SIRIUS for 30 infant-specific metabolomic features characterized by MS2 of 10 infant samples.
Table S2. Demographic, dietary, and clinical characteristics of infants in the study, stratified by age, Related to Figures 1 and 5. Data in the table are restricted to samples with conclusive results from metagenomic analysis. Infant age is shown in months (m). abx, antibiotic; RF, regular formula; HF, hydrolyzed formula. 1variable with missing data for certain time points.
Table S3. Mother-to-infant gene transmission events, Related to Figure 3. The sheet ‘Transmitted genes’ shows n=977 genes that were identical (100% nucleotide identity) between mother and infant and were harbored by MAGs of discordant bacterial species in mother and infant. Subsequent sheets tabulate the gene transmission events between MAGs of different genera, families, and phyla, respectively.
Table S4. Predicted prophage segments of MAG contigs harboring mother-to-infant interspecies transmitted genes, Related to Figure 3. Prophage predictions by checkV.
Table S5. Differentially abundant metabolites in mothers and infants, Related to Figure 4. List of n=368 metabolites annotated using reference standards that were more abundant in infants (q<0.01) and n=189 metabolites that were more abundant in mothers (q<0.01).
Table S6. Associations between species and metabolites in the infant gut, Related to Figure 4. The table shows associations between the relative abundances of bacterial species and the abundances of metabolites annotated by reference standards in infant fecal samples. The associations were derived from general linear models adjusted for longitudinal analysis and corrected for infant age, sex, delivery mode, antibiotics, and formula use and type. Included are associations that were statistically significant (q<0.25) following Bonferroni correction for multiple comparisons. Associations were marked as “significant in mothers” in the event of a congruous association in maternal samples with an uncorrected q-value <0.25. The column marked as “confirmed in prior in vitro experiment” refers to results of a study investigating the metabolic signatures of 178 strains from the gut microbiome [S1].
Data Availability Statement
Metagenomic data have been deposited at NCBI Sequence Read Archive. Metabolomic data have been deposited at Metabolomics Workbench. Data are publicly available as of the date of publication. Accession numbers and DOIs are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the Lead Contact upon request.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| Maternal and infant stool samples | Tampere Center for Child Health Research, Tampere University Hospital, Tampere, Finland | EDIA |
| Infant serum samples | Tampere Center for Child Health Research, Tampere University Hospital, Tampere, Finland | EDIA |
| Breast milk samples | Vincent Obstetrics & Gynecology Department, Massachusetts General Hospital, Boston, MA, USA | OriGiN |
| Critical commercial assays | ||
| PowerSoil DNA Isolation Kit | MoBio Laboratories | Cat#12888-100 |
| Nextera XT DNA Library Preparation kit | Illumina | Cat#FC-131-1096 |
| CalproLab ELISA (ALP) | Calpro AS | Cat#CALP0170 |
| β-Defensin 2 ELISA | Immundiagnostik | Cat#KR6500 |
| MILLIPLEX MAP Human Cytokine/Chemokine Magnetic Bead Panel | Millipore | Cat#HCYTMAG-60K-PX38 |
| Deposited data | ||
| Metagenomic sequencing data | This paper | SRA: PRJNA821542 |
| Stool metabolomic profiles | This paper | http://dx.doi.org/10.21228/M8C70Q |
| Software and algorithms | ||
| KneadData v0.7.2 | Curtis Huttenhower laboratory | https://huttenhower.sph.harvard.edu/kneaddata/ |
| MetaPhlAn2/StrainPhlAn v2.9.21 | Beghini et al. 70 | https://huttenhower.sph.harvard.edu/metaphlan/ |
| HUMANn2 v0.11.2 | Franzosa et al. 71 | https://huttenhower.sph.harvard.edu/humann/ |
| megaHIT v1.1.4-2-gd1998a1 | Li et al. 72 | https://github.com/voutcn/megahit |
| Prodigal v2.6.3 | Hyatt et al. 73 | https://github.com/hyattpd/Prodigal |
| metaBAT2 v2.15-3-g367a7ef | Kang et al. 74 | https://bitbucket.org/berkeleylab/metabat |
| checkM v1.1.2 | Parks et al. 75 | https://ecogenomics.github.io/CheckM/ |
| GTDB-Tk v1.0.2 | Chaumeil et al. 76 | https://github.com/Ecogenomics/GTDBTk |
| CD-HIT v4.7 | Fu et al. 77 | http://weizhong-lab.ucsd.edu/cd-hit/ |
| eggNOG-mapper v2.0.1 | Huerta-Cepas et al. 78 | http://eggnog-mapper.embl.de/ |
| KMA | Clausen et al. 79 | https://bitbucket.org/genomicepidemiology/kma |