

Abstract
Despite the high remission rates achieved using T cells bearing a chimeric antigen receptor (CAR) against hematogical malignancies, there is still a considerable proportion of patients who eventually experience tumor relapse. Clinical studies have established that mechanisms of treatment failure include the down-regulation of target antigen expression and the limited persistence of effective CAR T cells. We hypothesized that dual targeting mediated by a CAR and a chimeric costimulatory receptor (CCR) could simultaneously enhance T cell cytotoxity and improve durability. Indeed, concomitant high affinity engagement of a CD38-binding CCR enhanced the cytotoxicity of BCMA and CD19-CAR T cells by increasing their functional binding avidity. In comparison to second generation BCMA- or CD19-CAR T cells, double targeted CAR+CD38-CCR T cells exhibited increased sensitivity to recognize and lyse tumor variants of multiple myeloma (MM) and acute lymphoblastic leukemia (ALL) with low antigen density in vitro. Additionally, complimentary co-stimulation by 4-1BB and CD28 endodomains provided by the CAR and CCR combination conferred increased cytokine secretion and expansion, and improved persistence in vivo. Notably, the cumulatively improved properties of CAR+CCR T cells enabled the in vivo eradication of antigen-low tumor clones, which were otherwise resistant to treatment with conventional CAR T cells. Therefore, multiplexing targeting and co-stimulation through the combination of a CAR and a CCR is a powerful strategy to improve the clinical outcomes of CAR T cells by enhancing cytotoxic efficacy and persistence, thus preventing relapses of tumor clones with low target-antigen density.
One Sentence Summary:
Combination of a CAR and a chimeric costimulatory receptor augments cytotoxicity and durability of T cells and eliminates of antigen-low tumors.
INTRODUCTION
Adoptive immunotherapy with chimeric antigen receptor (CAR) T cells has emerged as a promising therapeutic tool against cancer. Second generation CARs that provide combined activation and costimulatory signals (1) have been shown to induce impressive clinical responses when targeting CD19 and have been approved for medical use in chemotherapy-resistant B cell leukemias and lymphomas (2, 3). Despite the high rates of complete remissions achieved with CAR T cells, clinical and experimental data of CD22-, B-cell maturation antigen (BCMA)-, and CD19-CAR T cells demonstrate that the emergence of antigen-low variants that evade effector cell mediated killing, as well as the lack of long term in vivo persistence of CAR T cells, are two of the critical mechanisms of disease escape (4).
The expression density of the target-antigen is an important factor that determines the function of engineered T cells since a minimum threshold of antigen expression is needed for preserved T cell activity (5–7). CARs are highly reliant on target-antigen density and, as a consequence, CAR T cells lose their functionality when antigen expression drops below a threshold that depends on the type of the target and the CAR binding properties (7–9). Multi-specific targeting using a pool of CARs, bi-cistronic CARs, or tandem CARs has been proposed as strategies to overcome tumor heterogeneity and antigen expression loss (10–14). These strategies focus on targeting a second tumor-antigen with a second CAR or a second CAR-binding moiety and thus, antigen selection is restricted to antigens with limited off-tumor expression. In addition, the effect of simultaneous CAR activation, when targeting two antigens with two CARs, on the overall function of CAR T cells and the emergence of antigen-loss has not been thoroughly studied.
Furthermore, T cell exhaustion and reduced persistence is another major factor limiting the efficacy of CAR T cells (15, 16). The addition of costimulatory intracellular components has provided second generation CAR T cells with enhanced activation, expansion, and in vivo persistence. The endo-domains of CD28 or 4-1BB are used in the constructs of the two approved CD19 CAR products as well as for the majority of CARs used in clinical studies (17). Extensive research comparing the functional properties of CARs bearing either design has concluded that CD28-based CARs elicit superior and faster cytotoxic capacity allowing for lower antigen density threshold, whereas 4-1BB-based CARs show higher expansion capacity and in vivo persistence (18–21). In physiologic T cell function, CD28 and 4-1BB costimulatory signaling synergize to achieve optimal cell activation and survival (22, 23). Interestingly, CAR T cells engineered to combine signaling of both costimulatory molecules demonstrated greater anti-tumor potency (20, 24, 25).
Overall, experimental and clinical results demonstrate that efficient cytotoxicity and sufficient persistence are key features of an optimal CAR T cell-mediated anti-tumor function. As such, the development of strategies that simultaneously broaden T cell mediated targeting of the tumor population to include clones with low antigen expression and enhance activation, expansion and durability of T cells is critical in order to accelerate the progress of adoptive T cell therapy. Here we propose the concept of double targeting through co-expression of a chimeric costimulatory receptor (CCR) (26) as a means to enhance anti-tumor function of CAR T cell therapy by concomitantly augmenting cytotoxic efficacy and persistence. We show that co-targeting of CD38, a highly expressed antigen in a variety of hematological malignancies (27, 28), using a CCR, results in improved cytotoxicity of CAR engineered T cells by increasing the total functional avidity. Importantly, co-expression of a CCR improved lysis of clonal variants with very low antigen expression, which were otherwise resistant to killing by conventional second-generation CAR T cells. Additionally, the CAR+CD38-CCR combination provided complimentary CD28 and 4-1BB costimulatory signals and improved proliferative capacity, reduced exhaustion, and enhanced in vivo durability of the engineered T cells, resulting in substantial improvement of immunotherapeutic outcomes.
RESULTS
Co-expression of a CCR improves cytotoxicity of CAR-engineered T cells.
In order to test the concept of combinatorial tumor targeting with a CAR and a CCR (Fig. 1A) we focused on hematological malignancies, such as multiple myeloma (MM) and B cell leukemia and lymphoma. We generated first- and second-generation CARs targeting BCMA (fig. S1A). We chose CD38 to serve as a second antigen targeted by the CCR, as CD38 is robustly expressed in almost all hematologic malignancies (27, 28) and therefore a CD38-CCR could be applicable to a variety of diseases. The CD38-CCR constructs were designed either to contain only one of the two most popular costimulatory domains, CD28 or 4-1BB (CD38-28 or CD38-BB), or to combine them (CD38-28BB) (Fig. 1A and fig. S1A). The CAR and the CCR constructs were linked to functionally irrelevant markers [dsRed and low-affinity nerve growth factor receptor (LNGFR), respectively] (fig. S1A). Peripheral blood T cells transduced with CAR+CD38-CCR combinations expressed equal abundance of the CAR on their surface to single CAR-transduced T cells (Fig. 1B and C and fig. S1B). CAR+CD38-CCR T cells showed reduced expression of CD38 on their surface indicating binding (in-cis or in-trans) of the CCR (fig. S1B). However, we did not observe any effect on the differentiation of the produced CAR-, CCR- and CAR+CCR-expressing T cells (Fig. 1D), nor fratricide during the manufacturing of T cells transduced with the CD38-CCR (Fig. 1E). Moreover, compared to mock transduced T cells, CD38-28BB CCR, BCMA-ζ+CD38-28 and BCMA-ζ+CD38-28BB-expressing T cells showed no tonic signaling mediated through the CCR, as evidenced by similar phosphorylation of AKT1 Serine/Threonine Kinase 1 (or AKT1), which reflects CD3ζ and CD28 activation (Fig. 1F).
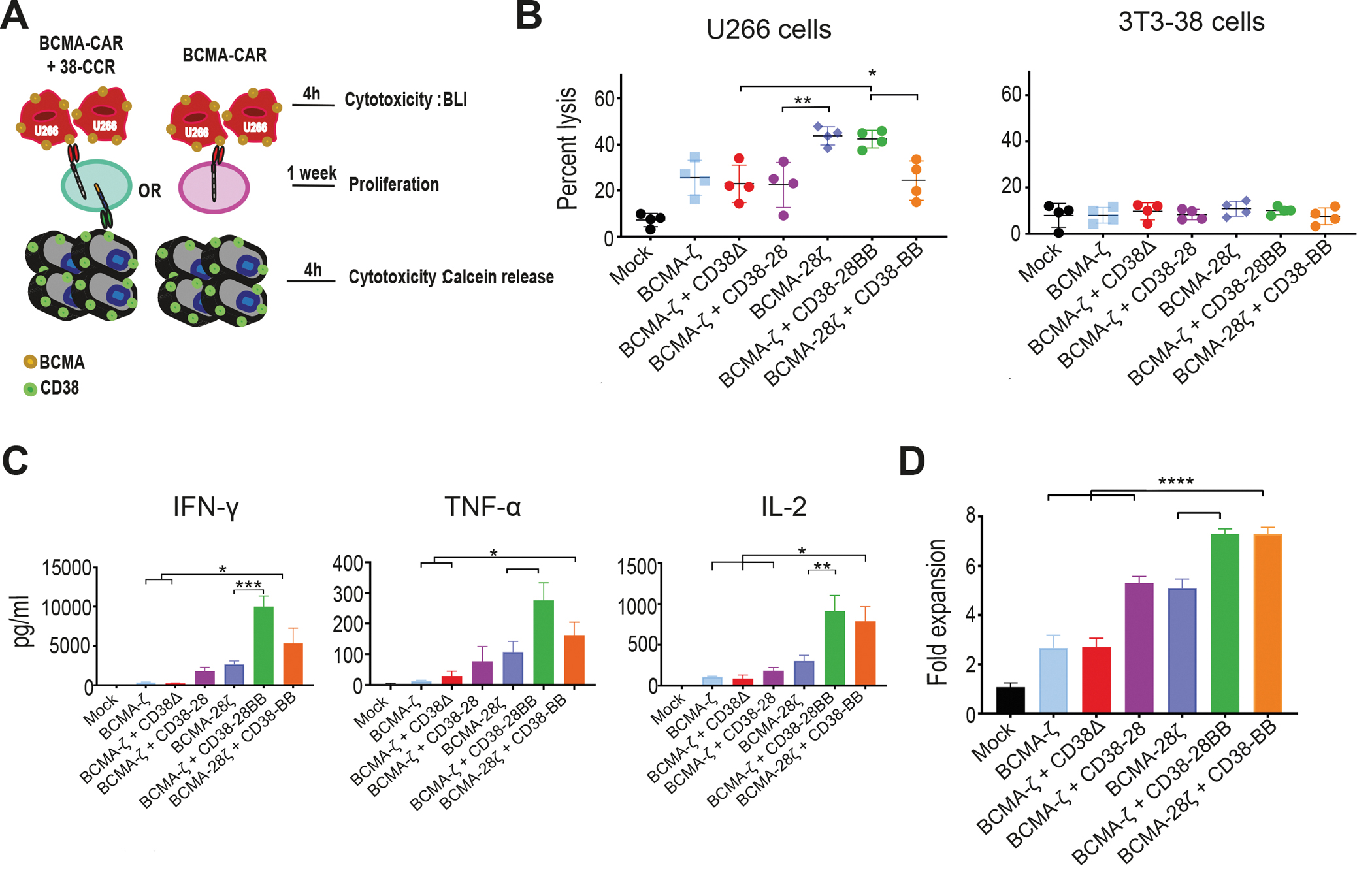
Figure 1. Combinatorial tumor targeting strategy with a CAR and a CCR results in enhanced cytotoxicity.
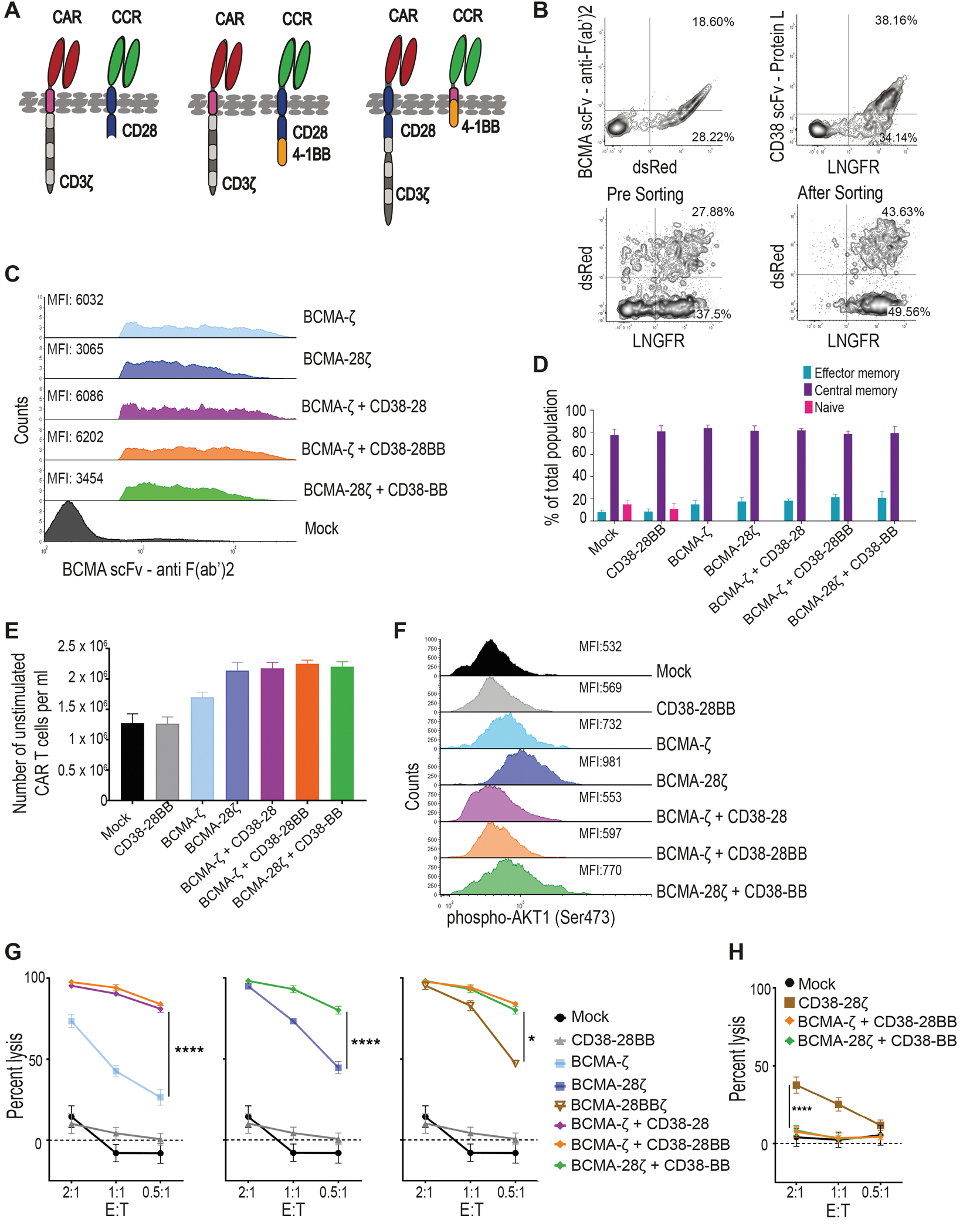
(A) Schematic representations of dual targeted CAR+CCR strategies are shown. First generation CARs were combined with the CD38-CCR construct bearing either CD28 (CD38-28) or both CD28 and 4-1BB costimulatory domains (CD38-28BB). Second generation CARs were combined with the CD38-CCR that contains only the 4-1BB signaling domain (CD38-BB). (B) Representative flow cytometry plots illustrating BCMA-CAR/dsRed and CD38-CCR/LNGFR expression of double transduced T cells are shown. BCMA-CAR expression was measured with an F(ab’)2 Fragment Goat Anti-Mouse antibody. CD38-CCR expression was measured after binding of Protein L. (C) Mean fluorescence intensity (MFI) of BCMA-CAR expression on transduced T cells is shown. MFI values are reported. Mock indicates mock-transduced T cells. (D) Percentage of CAR T cells that have a naïve (CD62L+CD45RA+), central memory (CD62L+CD45RA−), or an effector memory (CD62L−CD45RA−) phenotype are shown (n=4 per group). (E) The absolute number of unstimulated CAR T cells per ml is shown at five days after transduction (n=4 per group). (F) AKT1 phosphorylation (phosho-AKT1) in unstimulated CAR T cells was assessed by flow cytometry. Representative results from one donor are shown. (G) Lysis of luciferase expressing MM1.S (BCMA+CD38+) cells is shown. Tumor cell killing was measured in a 16 hour bioluminescence (BLI) assay (n=4 per group) at the indicated effector:target (E:T) ratios. (H) Calcein-loaded 3T3 cells transduced to express CD38 were co-cultured at indicated E:T ratios with either double-targeting BCMA-CAR+CD38-CCR T cells or single targeting CD38-28ζ CAR T cells. Tumor cell killing was measured after 4 hours (n=4 per group). Statistical analysis in (G) and (H) was performed by two-way ANOVA and subsequent multiple comparison, corrected by Turkey test. *p<0.05, ****p<0.0001.
Interestingly, the addition of CD38-28 and CD38-28BB CCR in first-generation (BCMA-ζ) and CD38-BB CCR in second-generation BCMA-CAR T cells (BCMA-28ζ) (Fig. 1A), resulted in remarkable increase of their cytotoxic capacity against MM cell lines and primary CD138+CD38+ patient-derived MM cells (Fig. 1G and fig. S2A and B). The presence of a CD28 transmembrane (TM) and intracellular (IC) domain have been linked to improved cytotoxicity of a CAR and to better response to lower antigen expression (8, 18). We investigated the effect of the positioning of the CD28 TM/IC in the BCMA-CAR instead of in the CD38-CCR. We observed no difference in the cytotoxic capacity between BCMA-28ζ+38-BB and BCMA-ζ+38-28BB T cells (Fig. 1G and fig. S2A and B). Overall, lysis of MM tumor cells by BCMA-CAR+CD38-CCR T cells was significantly higher, in low effector:target (E:T) ratios as compared to lysis by single-targeting second generation BCMA-CAR T cells of both costimulatory designs (BCMA-28ζ or BCMA-BBζ) (P<0.0001, Fig. 1G and fig. S2A and B), or compared to lysis by third generation BCMA-CAR T cells (BCMA-28BBζ) (Fig. 1G). These last findings indicate that the increased cytotoxic potential of BCMA-CAR+CD38-CCR T cells is not solely a result of the CD28/4-1BB costimulatory combination. In addition, T cells bearing the CD38-CCR alone did not induce cell lysis of CD38 expressing targets (Fig. 1G and fig. S2C) and BCMA-CAR+CD38-CCR T cells were unable to induce lysis of NIH-3T3 cells that were transduced to express CD38, but do not express BCMA (3T3-38+ cells) (Fig. 1H). Thus, CD38-CCR expression was not sufficient to induce lysis of tumor by itself or off-tumor lysis in combination with the BCMA-CAR. Notably, the increase in cytotoxicity was completely diminished in the absence of CD38 expression on the target cells when U266 cells (BCMA+CD38−) were used as targets (Fig. 2A). Together, these results demonstrate that it’s the engagement of the co-expressed CD38-CCR with CD38 that results in enhanced cytotoxicity of CAR T cells against tumor targets.
Figure 2. CD38-CCR engagement results in enhanced cytotoxicity of CAR T cells by increasing functional avidity.
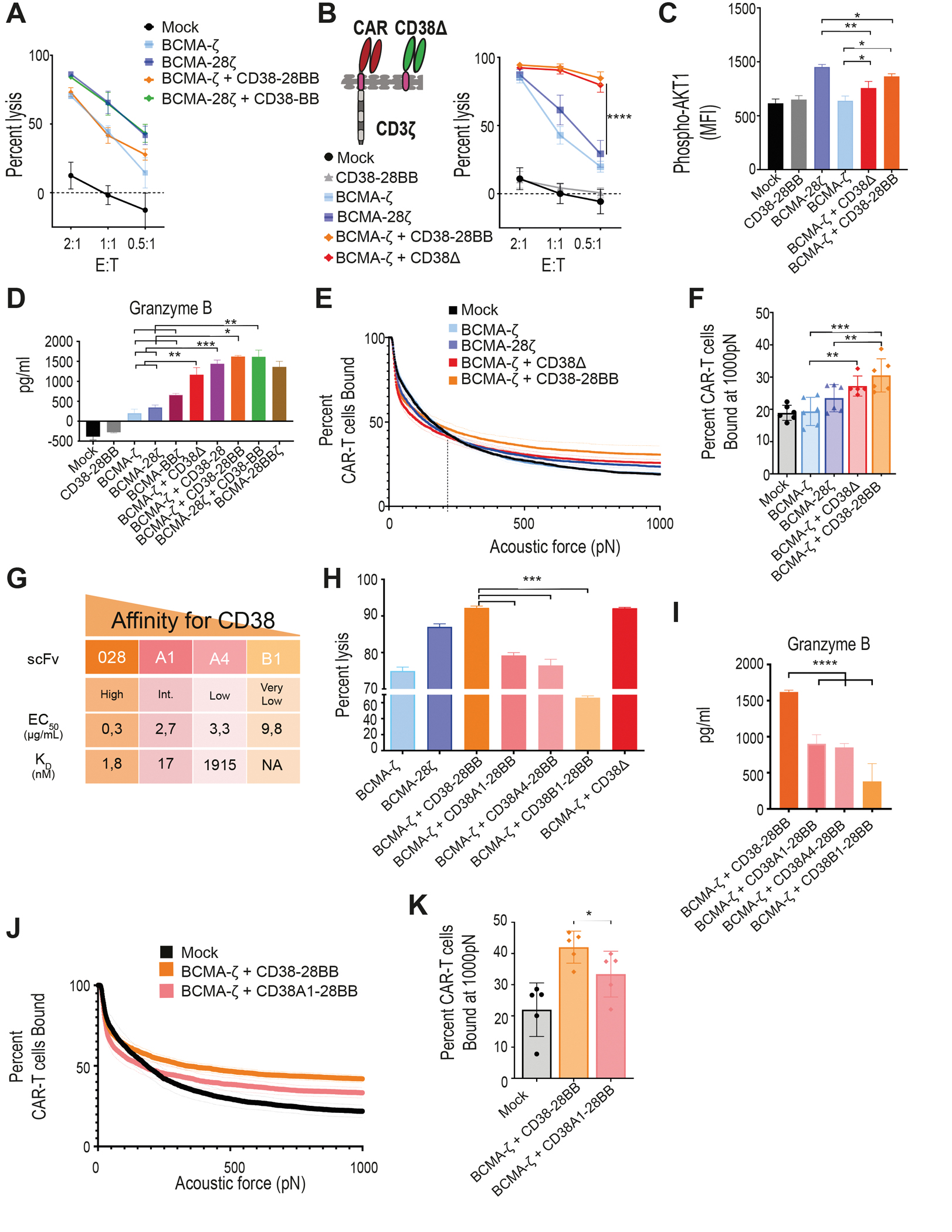
(A) Lysis of luciferase expressing U266 cells (BCMA+CD38−) was measured in a 16 hour BLI assay (n=4 per group) at the indicated E:T ratios. (B) A first generation BCMA-CAR was combined with the CD38Δ construct lacking the intracellular costimulatory tail. BCMA-ζ+CD38Δ T cells and the BCMA-ζ+CD38-28BB T cells were incubated with MM1.S cells and lysis was determined in a 16 hour BLI assay (n=4) at the indicated E:T ratios. Statistical analysis was performed with a two-way ANOVA and subsequent multiple comparison, corrected by Turkey test. ****p<0.0001. (C) MFI of AKT1 phosphorylation (pAKT1) is shown in BCMA-CAR+CD38-CCR T cells activated by stimulation with UM9 cells for 15 minutes as assessed by flow cytometry (n=4 per group). Statistical analysis was performed by one-way ANOVA and subsequent multiple comparison, corrected by Turkey test. *p<0.05, **p<0.01. (D) Granzyme B secretion by CAR T cells co-cultured with ΜΜ1.S (E:T ratio 1:1) for 16 hours is shown (n=4 per group). Statistical analysis was performed by paired t-test. *p<0.05, **p<0.01, ***p<0.001. (E) The strength of interaction between single- and double-targeting T cells and MM1.S target cells was measured. Percentage of total CAR T cells remaining bound to target cells as the acoustic force ramp is applied from 0 to 1000 pN are shown. The dotted line refers to the threshold beyond which all measurement are considered specific binding. (F) The percentage of CAR T cells remaining bound to MM1.S target cells at 1000 pN is shown; data are presented as mean ±SEM from 6 pooled measurements; p-values were calculated using a paired t-test. **p<0.01, ***p<0.001. (G) Affinity characteristics are shown for anti-CD38 antibodies used to generate scFvs. The surface plasmon resonance–determined dissociation constant (KD value, nmol/L) and half-effective concentration (EC50) when titrated on CHO-CD38 cells (μg/mL), described in (29). (H) MM1.S cells were co-cultured at a 1:1 ratio with BCMA-CAR T cells co-expressing CD38-CCRs with gradually lower affinities for CD38 or CD38Δ. Tumor cell lysis was measured in a BLI assay (n=4 per group). Statistical analysis was performed with a paired t-test. (I) Cell supernatants from (H) were harvested to measure granzyme B secretion by ELISA (n=4 per group). Statistical analysis was performed with a paired t-test. ***p<0.001. (J) The strength of interaction between BCMA-ζ + CD38-28BB (EC50 0.3) or BCMA-ζ+CD38A1-28BB (EC50 2.7) T cells and MM1.S target cells is shown as the percentage of total CAR T cells remaining bound to target cells as the acoustic force ramp is applied from 0 to 1000 pN. Dotted line refers to the threshold beyond which all measurement are considered specific binding. (K) The percentage of CAR T cells remaining bound to MM1.S target cells at 1000 pN is shown; data are presented as mean ±SEM from 5 pooled measurements and p-values were calculated using a paired t-test. *p<0.05.
High affinity CCR binding increases CAR T cell functional avidity.
We further aimed to elucidate whether the increase of cytotoxicity observed with CCR co-expression is a result of the CCR binding to the target or of the initiation of downstream costimulatory signals. To investigate this, we constructed a CD38 binding receptor, which lacks the intracellular costimulatory tail (CD38Δ) and therefore is able to bind to the CD38 antigen, with the same affinity as the CD38-CCR, but it is not able to induce activation, nor co-stimulation in the T cells (Fig. 2B and fig. S1A). Surprisingly, the double-targeting BCMA-ζ+CD38Δ T cells lysed MM tumor cells as effective as the BCMA-CAR+CD38-28BB T cells (Fig. 2B). Therefore, it seems that the engagement of the second target-antigen by the CCR, CD38 in this case, can by itself increase the lytic capacity of double-targeting CAR+CCR T cells independent of the addition of costimulatory signaling. Indeed, when we measured the phosphorylation of AKT1 in T cells after target cell encounter, we found that double binding by BCMA-ζ+CD38Δ resulted in increase of pAKT1 mean fluorescence intensity (MFI), compared to single binding by BCMA-ζ (Fig. 2C), indicating that the multivalent binding enhances the downstream signaling of CAR T cell activation. Phosphorylation of AKT1 was further increased in BCMA-ζ+CD38-28BB T cells (Fig. 2C), although that did not translate into better tumor cell lysis in this system. Additionally, we found significantly increased secretion of granzyme B by BCMA-ζ+CD38Δ compared to BCMA-ζ as well as BCMA-28ζ CAR T cells (P=0.0004 and P<0.0001 respectively, Fig. 2D). Moreover, granzyme B secretion by BCMA-ζ+CD38-28 T cells was higher compared to BCMA-28ζ CAR T cells (Fig. 2D), demonstrating once more the effect of multivalent tumor engagement beyond the addition of CD28 costimulation. Further, double targeting and combinatorial costimulation with BCMA-ζ+CD38-28BB and BCMA-28ζ+CD38-BB T cells also showed higher concentrations of secreted granzyme B (Fig. 2D).
We hypothesized that the improved cytotoxic efficacy is a result of an increased functional avidity and the formation of stronger T cell binding due to the multivalent engagement through the BCMA-CAR and the high affinity CD38-CCR. We tested this hypothesis by determining the actual binding strength between tumor cells and CAR T cells with or without CCR co-expression. MM1.S cells were seeded in z-Movi Cell Avidity Analyzer microfluidic chips, following which, mono- and double- targeting CAR T cells were sequentially flushed in and allowed to bind to the target cells. The binding avidity between tumor cells and T cells was quantified by applying acoustic forces (ultrasound waves) of increasing magnitude (pN). We found that the interaction of BCMA-ζ+CD38Δ T cells with tumor cells could withstand the application of higher acoustic forces compared to the interaction of BCMA-ζ with tumor cells, as there were significantly more BCMA-ζ+CD38Δ T cells attached compared to BCMA-ζ cells (P=0.0082, Fig. 2E and F). Addition of costimulatory domains resulted in further slight increase of functional avidity of BCMA-ζ+CD38-28BB T cells, which overall showed significantly higher binding strength compared to both BCMA-ζ and BCMA-28ζ CAR T cells (P= 0.0002 and P= 0.0274, Fig. 2E and F).
We further interrogated the specific affinity requirements of CD38 engagement, which resulted in an increase of cytotoxicity. To this end, we used four single-chain variable fragments (scFvs) binding to the same CD38 epitope with gradually lower affinity, previously described by our group (29) (Fig. 2G). We found that only the high affinity (scFv 028, KD=1.8 nM) binding of the CD38-CCR resulted in superior cytotoxicity (Fig. 2H) of BCMA-CAR+CD38-28BB T cells compared to single targeted BCMA-CAR T cells. Affinity reduction of approximately 10-fold (scFv 38A1, KD=16.5 nM) reduced this effect (Fig. 2H) and, as expected, led to decreased granzyme B secretion (Fig. 2I) and functional avidity (Fig. 2J and K). Lowering the expression of CD38 on the tumor cells also reduced the cytotoxic benefit of BCMA-CAR+CD38-CCR T cells (fig. S3), indicating the requirement for a high expression of the CCR-targeting secondary antigen. Overall, our data demonstrate that a high affinity engagement of a highly expressed secondary antigen, such as CD38, is necessary in order to increase the functional avidity and enhance the cytotoxic capacity of BCMA-CAR T cells.
CAR+CCR combination enables the lysis of low-antigen expressing tumor variants.
A decreased target-antigen density is one of the major mechanisms of tumor escape and relapse after adoptive therapy with CAR T cells. Given the superior cytotoxic capacity of double-targeted CAR+CCR T cells, we examined their efficacy against tumor variants with low antigen density. K562 cell lines (CD38+) were transduced and sorted for the expression of variable concentrations of BCMA. The sorted lines were further characterized as negative (K562-BCMAneg), low (K562-BCMAlow), medium (K562-BCMAmed), and high (K562-BCMAhigh), after quantitative comparison of BCMA expression with MM cell lines and primary MM cells (30) (Fig. 3A). We found that BCMA-CAR+CD38-CCR T cells (BCMA-ζ+CD38-28, BCMA-ζ+CD38-28BB or BCMA-28ζ+CD38-BB) exhibited increased lysis of all K562-BCMA lines compared to BCMA-ζ, BCMA-28ζ and BCMA-28BBζ CAR T cells (Fig. 3B). Notably, K562-BCMAlow cells, which are representative of the median BCMA expression on primary MM, were resistant to lysis by single targeting BCMA-CAR T cells but remained sensitive to BCMA-CAR+CD38-CCR T cells (Fig. 3B). More importantly the lysis of K562-BCMAlow and K562-BCMAmed K562 cells by BCMA-CAR+CD38-CCR T cells was comparable to that of K562-BCMAhigh by BCMA-28ζ CAR T cells (Fig. 3B).
Figure 3. CAR and CCR combinations restore the cytotoxic capacity of T cells against low-antigen expressing tumor variants.
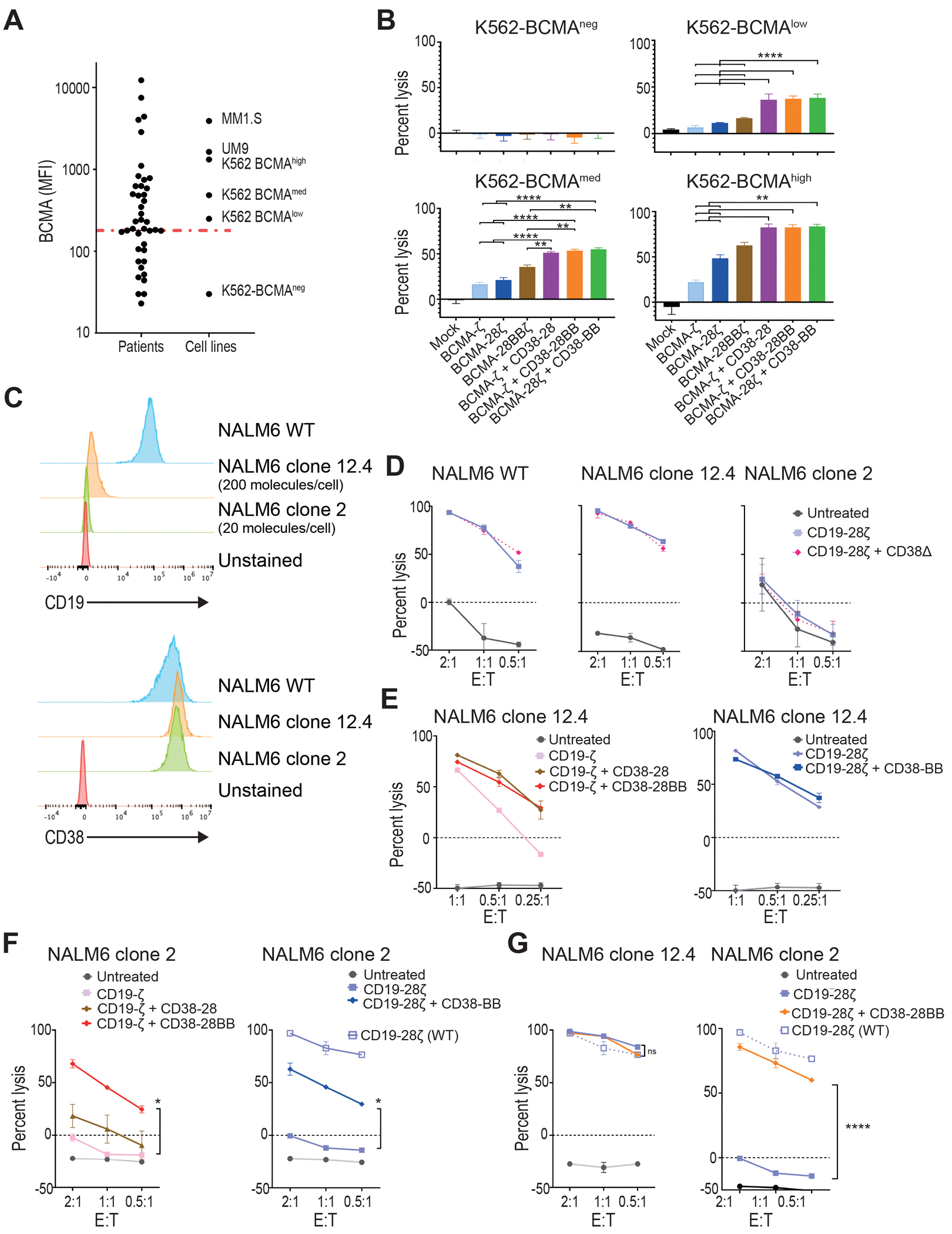
(A) Primary malignant plasma cells and cell lines were analyzed for BCMA expression by flow cytometry. Dot plots depict the MFI of BCMA found on primary MM tumor cells from 49 patients, in comparison with MM cell lines and K562 cells transduced to express BCMA in variant concentrations. The red line depicts median MFI for primary MM cells. (B) K562 cells with different BCMA expression were co-cultured with first, second, or third generation BCMA-CAR T cells and double BCMA-CAR+CD38-CCR T cells (BCMA-ζ+CD38-28BB or BCMA-28ζ+CD38-BB) at 1:1 effector to target ratio. Cell lysis was determined at 16 hours by BLI (n=3 per group). Statistical analysis performed with a two-way ANOVA and subsequent multiple comparison, corrected by Turkey test. **p<0.01, ****p<0.0001. (C) Histograms of CD19 and CD38 expression are shown for the NALM6 acute lymphoblastic leukemia cell line (WT) and on two NALM6 clones (clone 12.4 and clone 2). The red histogram shows the unstained control. (D) Specific lysis of NALM6 WT, clone 12–4 (200 molecules per cell), and NALM6 clone 2 (20 molecules per cell) is shown when cell lines were co-cultured with CD19-28ζ or double CD19-28ζ+CD38Δ T cells. (E) Specific lysis of NALM6 clone 12-4 (200 molecules per cell) is shown when cocultured with CD19-ζ, double CD19-ζ+CD38-28, or double CD19-ζ+CD38-28BB T cells (left) or with CD19-28ζ or double CD19-28ζ+CD38-BB (right) T cells. (F) Specific lysis of NALM6 clone 2 is shown when co-cultured with CD19-ζ, double CD19-ζ+CD38-28, or double CD19-ζ+CD38-28BB T cells (left) or with CD19-28ζ or double CD19-28ζ+CD38-BB (right) T cells. The open squares represent the lysis activity of the CD19 wild type NALM6 cell line when treated with CD19-28ζ CAR T cells. Statistical analysis was performed with using two-way ANOVAs and subsequent multiple comparison, corrected by Turkey test. *p<0.05. (G) Specific cytolysis of NALM6 clone 12-4 (200 molecules per cell) and NALM6 clone 2 (20 molecules per cell) is shown when cells were cocultured with CD19-28ζ or double CD19-28ζ+CD38-28BB T cells at indicated E:T ratios. The open squares represent the lysis activity of the CD19 WT NALM6 cell line when treated with CD19-28ζ CAR T cells. Statistical analysis was performed using two-way ANOVA and subsequent multiple comparison, corrected by Turkey test. ****p<0.0001.
We additionally confirmed this observation in a model of acute lymphoblastic leukemia NALM6 cells engineered and subcloned to express low CD19 antigen density (18). The NALM6 clone 12.4 was generated after biallelic disruption of the Cd19 gene and consecutive lentiviral transduction to express CD19 at low concentrations. NALM6 clone 12.4 carries approximately 200 molecules per cell as determined by flow cytometry-based quantification (Fig. 3C). The NALM6 clone 2 was generated only by monoallelic disruption of the Cd19 gene (18) and displays an ultra-low CD19 surface expression (approximately 20 molecules per cell), which is negative by conventional flow cytometry analysis and only detectable by means of flow cytometry after signal amplification (Fig. 3C). Also, in this system CD19-CAR+CCR T cells specifically recognized CD19 expressing NALM6 targets and not CD19 knockout NALM6 cells (fig. S4A). Interestingly, the addition of CD38Δ did not result in increase of cytotoxicity in this model for the wild type (WT) nor for the CD19-low NALM6 (Fig. 3D). This could be attributed to the fact that the expression of CD38 on NALM6 was 10 times lower than in the MM cell lines (fig. S4B). Clone 12.4 was still sensitive to killing by CD19-ζ and CD19-28ζ CAR T cells (Fig. 3E). The addition of CD38-28 or CD38-28BB CCR equally enhanced the in vitro cytotoxicity of CD19-ζ whereas no difference in lytic capacity was observed between CD19-28ζ and CD19-28ζ+CD38-BB (Fig. 3D and E). Thus, the addition of a CD28 domain was necessary to achieve maximum lysis of clone 12.4. The ultra-low density of CD19 renders NALM6 clone 2 unsusceptible to lysis by both CD19-ζ and second generation CD19-28ζ CAR T cells (Fig. 3D and F). In this case, combination of CD19-CAR and CD38-CCR providing double CD28 and 4-1BB costimulation was necessary to restore lysis (Fig. 3F). CD19-ζ+CD38-28 combination failed to increase lysis, whereas both CD19-ζ+CD38-28BB and CD19-28ζ+CD38-BB T cells elicited significantly higher lysis against the NALM6 clone 2 cells (P=0.0125, Fig. 3F), indicating that in this system the costimulatory design benefits the cytotoxic outcomes, rather than the CD38 engagement. Since we observed that the presence of the CD28 endodomain was important for the optimal lysis of clone 12.4, we interrogated whether the addition of a second CD28 domain would have an additive effect. Interestingly, although CD19-28ζ+CD38-28BB T cells did not elicit a higher lysis of clone 12.4, these cells significantly increased lysis of clone 2 (P<0.0001), which was almost similar to the lysis activity observed when targeting the CD19high wild type NALM6 cell line by conventional CD19-28ζ CAR T cells (Fig. 3G). Altogether, our results suggest that the addition of the CD38-CCR can improve the sensitivity for antigen recognition in different hematological tumor models.
Co-expression of a CCR increases the proliferative capacity of CAR T cells.
Previous studies have elegantly demonstrated that CD28 and 4-1BB intracellular signaling domains equip CAR T cells with distinct kinetics and functions (19, 20), which can be integrated to improve T cell potency. We hypothesized that the combination of CD28 and 4-1BB costimulatory signals delivered by the co-expression of a CCR could enhance not only the cytolytic potency but also the cytokine secretion and the proliferative potential of CAR T cells. Indeed, when challenged with MM cells, BCMA-CAR+CD38-CCR T cells produced significantly higher concentrations of interferon (IFN)-γ (P= 0.0216) and interleukin (IL)-2 (P=0.0433) compared to BCMA-ζ, BCMA-28ζ and BCMA-BBζ CAR T cells, and higher tumor necrosis factor (TNF)-α compared to BCMA-ζ and BCMA-28ζ CAR T cells (Fig. 4A). This was a consequence of the CCR-mediated costimulatory signaling in combination to CAR-mediated activation and not solely of the CCR engagement since no cytokine secretion increase was observed by BCMA-ζ+CD38Δ compared to BCMA-ζ (Fig. 4A). Secretion of cytokines was reduced accordingly, when lowering the affinity of CD38-CCR (fig. S5A). Furthermore, in long-term proliferation assays, double-targeting BCMA-CAR+CD38-CCR T cells (BCMA-ζ+CD38-28, BCMA-ζ+CD38-28BB and BCMA-28ζ+CD38-BB) showed a high expansion potential, outperforming their respective single targeting CARs (BCMA-ζ and BCMA-28ζ) (Fig. 4B). Despite the fact that BCMA-CAR+CD38-CCR T cells showed no difference in cytokine secretion compared to 3rd generation BCMA-28BBζ cells, they clearly expanded better in vitro (Fig. 3B). Notably, the enhanced proliferative capacity of the double-targeting BCMA-CAR+CD38-CCR T cells was similar to BCMA-BBζ T cells and was corroborated with lower surface expression of programmed cell death protein 1 (PD-1), compared to BCMA-ζ, BCMA-28ζ and BCMA-28BBζ CAR T cells (Fig. 4C and fig. S5B). T cells bearing only CD38-BB or CD38-28BB CCR showed no secretion of IFN-γ and TNF-α in response to antigen engagement, and we observed a similar secretion of IL-2 (Fig. 4A and fig. S5C) and expansion (fig. S5D) to mock T cells. However, comparable IL-2 concentrations were secreted by the CD38Δ T cells, indicating that it is not mediated through the costimulatory domains of the CCR; rather it is induced by the engagement of CD38 (fig. S5C) and attributed to enhanced alloreactivity. Interestingly, the highly proliferating BCMA-CAR+CD38-CCR T cells were not characterized by the retention of a central memory phenotype, as observed with BCMA-BBζ T cells, as the majority of BCMA-CAR+CD38-CCR T cells differentiated to a CD45RA−CD62L− effector memory phenotype (Fig. 4D and E and fig. S5E).
Figure 4. Co-expression of a CCR enhances the expansion of CAR T cells.
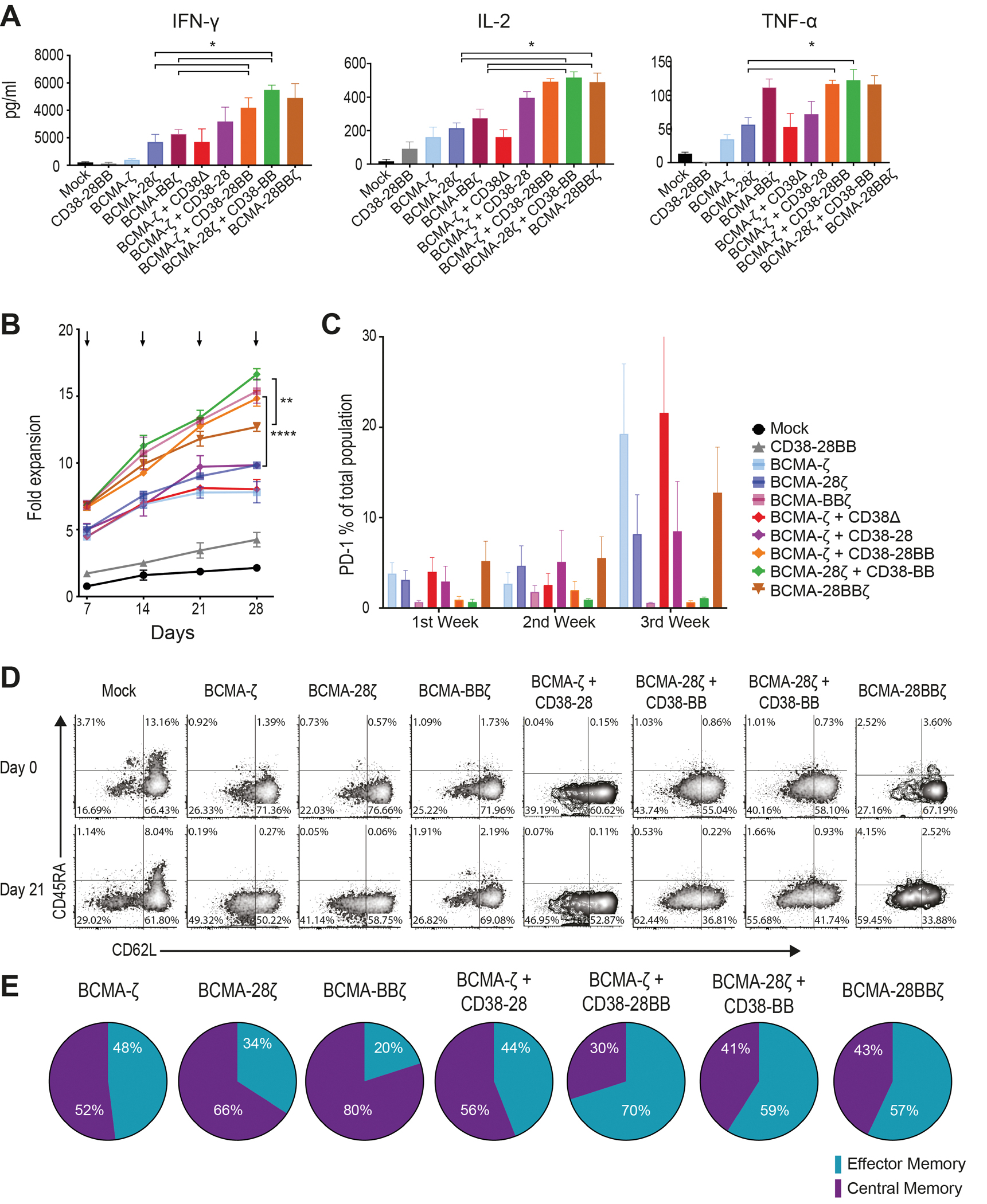
(A) Cytokine secretion is shown in supernatants collected from co-cultures with ΜΜ1.S (E:T ratio 1:1) for 16 hours (n=4 per group). Statistical analysis was performed by paired t-tests. *p<0.05. (B) CAR T cell proliferation is shown following weekly antigen-specific stimulations with irradiated tumor cells. Black arrows indicate addition of irradiated tumor cells. The fold of the expansion of the CAR+ T cells is indicated on the y-axis. The data represent the mean±SEM of 4 experiments with different donors. Statistical analysis was performed using a two-way ANOVA and subsequent multiple comparison, corrected by Turkey test. **p<0.01, ****p<0.0001. (C) PD-1 expression is shown for T cells from the proliferation assay in (B) (n=4 donors). (D) Flow cytometry density plots of phenotypic profile of each group from (B) are shown at day 0 (before expansion) and at day 21. Cells were characterized as naive (CD45RA+/CD62L+), central memory (CD45RA−/CD62L+), effector memory (CD45RA−/CD62L−) or effector (CD45RA+/CD62L−). (E) Percentage of CAR T cells from (B) that have a central memory or an effector memory phenotype at day 21 (n=4 donors).
Since CD38-CCR engagement is a successful strategy to provide co-stimulation to CAR T cells, we interrogated whether it is mandatory that the CCR target is expressed on the tumor cells or whether it is possible that the CCR-mediated costimulatory signaling is initiated after binding in-trans to an antigen expressed on accessory cells. To investigate this, we set up a co-culture cytotoxicity assay where luciferase-positive BCMA+CD38− U266 cells were co-cultured with calcein-loaded 3T3-38+ cells (Fig. 5A). When BCMA-CAR+CD38-CCR T cells were added in the co-culture they preserved their cytotoxicity against the U266 cells while leaving 3T3-38+ cells intact (Fig. 5B). Although, the binding of the CD38-CCR to CD38 expressed on the 3T3 cells did not induce off-tumor toxicity, it was sufficient to induce the costimulatory signaling leading to improved cytokine secretion and expansion of the BCMA-CAR+CD38-CCR T cells compared to respective single-targeting BCMA-CAR T cells (Fig. 5C and D). The BCMA-CAR+CD38-CCR strategies combining both CD28 and 4-1BB costimulation showed higher cytokine secretion and expansion with BCMA-28ζ+CD38-BB T cells outperforming BCMA-ζ+CD38-28BB in IFN-γ and TNF-α secretion (Fig. 5C and D). Thus, the co-expression of a CD38-CCR can enhance the expansion potential of CAR T cells, not only through engagement of the tumor-expressed target-antigen, but also through binding in-trans of the antigen expressed on accessory cells without off-tumor toxicity. When wild type (CD38 negative) 3T3 cells were used in the co-culture (fig. S6A), lysis of U266 was preserved and 3T3 cells remained intact (fig. S6B) but no increase of cytokines and proliferation was observed (fig. S6C and D), indicating that in-cis or in-trans binding of CD38 expressed on the CAR T cells did not contribute to the improved expansion. Since monocytes express CD38 and their activation has been linked to toxicity, we additionally, used primary CD14+ monocytes as accessory cells in the same co-culture system and measured indicators of their activation. We observed that binding of the CD38-CCR still induced the secretion of cytokines by the CAR+CCR T cells but did not induce excessive activation of the monocytes measured by surface expression of CD80, CD86, Human Leukocyte Antigen – DR isotype (HLA-DR) and secretion of IL-6 (fig. S6E and F).
Figure 5. CD38-CCR can signal through in-trans binding of the antigen expressed on accessory cells without off-tumor toxicity.
(A) A schematic representation of the coculture cytotoxicity/proliferation assay is shown. Luciferase-positive BCMA+CD38− U266 cells were co-cultured with calcein-loaded 3T3-38+ cells and single- or double-targeting T cells were added in the co-culture. Lysis of U266 cells was determined by BLI and lysis of 3T3-CD38 cells by calcein release assay. (B) U266 cells and calcein-loaded 3T3-CD38 cells were cocultured at a 3:1 E:T ratio with mock, BCMA-ζ, BCMA-ζ+CD38Δ, BCMA-ζ+CD38-28, BCMA-28ζ, BCMA-28ζ+CD38-BB or BCMA-ζ+CD38-28BB CAR T cells and cell lysis was measured after 4 hours by BLI and a Calcein-AM release assay, respectively (n=4 per group). Statistical analysis was performed by a paired t-test. *p<0.05, **p<0.01. (C) Cell supernatants from (B) were harvested to measure cytokine secretion with a flow cytometry–based assay (n=4 per group). Statistical analysis was performed with paired t-tests. *p<0.05, **p<0.01, ***p<0.001. (D) Irradiated BCMA+CD38− U266 cells were co-cultured with irradiated 3T3-38+ cells. Mock, BCMA-ζ, BCMA-ζ+CD38Δ, BCMA-ζ+CD38-28, BCMA-28ζ, BCMA-28ζ+CD38-BB or BCMA-ζ+CD38-28BB CAR T cells were added in the co-culture and the expansion of CAR T cells was measured 7 days later. Statistical analysis was performed with one-way ANOVAs and subsequent multiple comparison, corrected by Turkey test. ****p<0.0001.
CCR co-expression increases in vivo anti-tumor efficacy of CAR T cells and improves elimination of antigen-low variants.
Our in vitro studies revealed that the combination of BCMA-28ζ+CD38-BB modestly outperformed BCMA-ζ+CD38-28BB in cytokine secretion (through in-cis and in-trans binding) and proliferative potential. Thus, we chose to evaluate the cumulative benefit of tumor co-targeting with BCMA-28ζ+CD38-BB in vivo using a previously described xenograft MM murine model under challenging conditions with low T cell dosing (24, 31). Mice bearing humanized bone marrow (BM)-like scaffold niches were engrafted with luciferase-expressing MM cells and treated 7 days post tumor injection with CD38-28BB, BCMA-28ζ, BCMA-BBζ, BCMA-28ζ+CD38-BB or mock T cells (Fig. 6A). Post-infusion monitoring with bioluminescence imaging (BLI) revealed that treatment with CD38-28BB alone had no effect on tumor progression, confirming in vitro findings that CD38-CCR does not function without an activation signal (fig. S7A). Treatment with BCMA-BBζ and BCMA-28ζ CAR T cells also failed to control tumor growth in this system (Fig. 6B and C and fig. S7B). However, mice treated with double-targeting BCMA-CAR/CD38-CCR T cells (BCMA-28ζ+CD38-BB) showed an impressive delay of tumor growth (Fig. 6B and C and fig. S7B). Post-mortem analysis of the scaffold at 7 weeks after T cell injection revealed lower number of UM9 [Green Fluorescent Protein (GFP)+/CD38+] tumor cells in the BCMA-28ζ+CD38-BB group compared to all other treatment groups (Fig. 6D). No change in the expression of BCMA on the remaining UM9 tumor cells was observed between the evaluable treatment groups (fig. S7C).
Figure 6. BCMA-CAR+CD38-CCR T cells show enhanced in vivo anti-tumor function, improved persistence, and reduced expression of exhaustion markers.
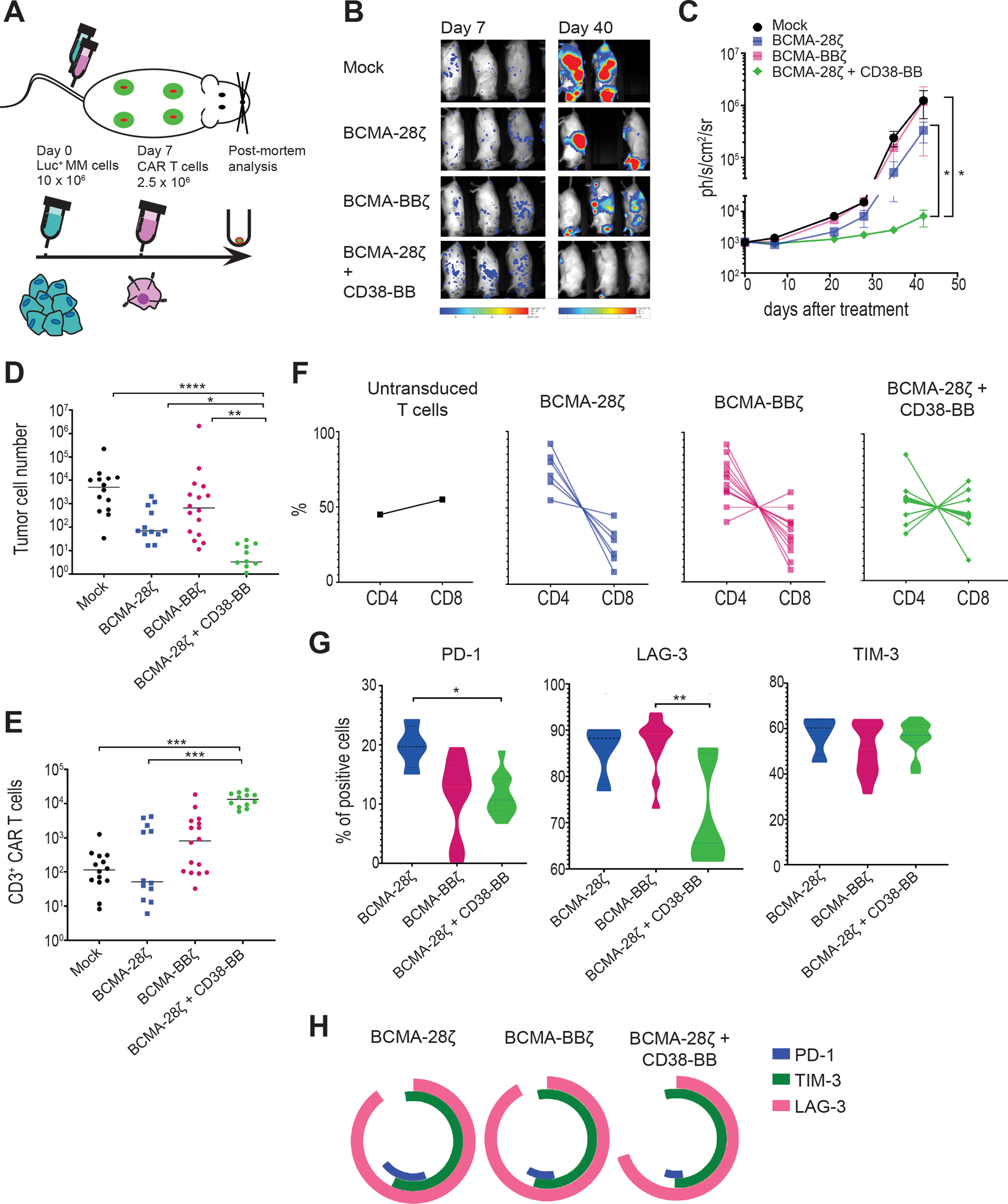
(A) A schematic representation of the MM scaffold-based xenograft murine model is shown. 10 × 106 Luc-GFP UM9 cells were administrated intravenously. Mock-transduced, BCMA-28ζ, BCMA-BBζ or BCMA-28ζ+CD38-BB CAR T cells were infused intravenously 7 days later. Tumor burden was measured weekly by BLI. (B) Representative BLI images of two time points are shown, with the pixel intensity represented in color. (C) Average tumor burden of mice was quantified by BLI and is depicted as units of photons per second per square centimeter per steradian (ph/sec/cm2/sr) (n=4 mice per group). Statistical analysis was performed using a two-way ANOVA and subsequent multiple comparison, corrected by Turkey test. *p<0.05. (D to H) Post-mortem scaffolds were harvested from each mouse and dissociated. Single-cell suspensions were counted, stained, and measured by flow cytometry. (D) Absolute UM9 tumor cell (GFP+/CD38+) numbers in the scaffolds are shown. Each dot represents one scaffold. (E) Absolute CAR T cell numbers in the scaffolds are shown. Each dot represents one scaffold. (F) CD4 and CD8 CAR T cell percentages in each scaffold are shown. (G) Violin plots show the expression of PD-1, LAG-3, and TIM-3 on CAR T cells isolated from scaffolds. (H) Co-expression of inhibitory receptors of (G) are presented. Statistical comparisons in (D), (E), and (G) were performed by Kruskal-Wallis test between the indicated groups; ns, not significant. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
BCMA-28ζ+CD38-BB and BCMA-BBζ T cells showed significantly better persistence than BCMA-28ζ T cells (P= 0.0016, Fig. 6E). Interestingly, there was no difference in the remaining absolute T cell numbers in the scaffolds for the BCMA-28ζ+CD38-BB group and the BCMA-BBζ group, despite the difference in the tumor burden (Fig. 6E), indicating the improved anti-tumor cytotoxic potency of BCMA-28ζ+CD38-BB cells. In addition, we found that an almost 1:1 balanced CD4:CD8 ratio was maintained in the remaining CAR T cells of BCMA-28ζ+CD38-BB group, whereas in the BCMA-28ζ and BCMA-BBζ groups the persisting cells were, to various extents, enriched for CD4+ T cells (Fig. 6F and fig. S7D). Further evaluation of the expression of surface markers related to exhaustion on the CAR T cells revealed that BCMA-28ζ+CD38-BB cells expressed significantly lower concentrations of PD-1 than BCMA-28ζ CAR T cells (P=0.0142) and significantly lower concentrations of lymphocyte-activation gene 3 (LAG-3) compared to BCMA-BBζ (P=0.0015, Fig. 6G). There was no difference in T cell immunoglobulin domain (TIM-3) abundance between the groups (Fig. 6G). Collectively, the BCMA-28ζ+CD38-BB group had lower percentage of triple positive (PD-1+LAG-3+TIM-3+) cells than BCMA-28ζ and BCMA-BBζ (Fig. 6H). These data demonstrate that the combination of a BCMA-CAR and a CD38-CCR not only enhanced cytotoxic anti-tumor function, but also increased persistence and prevented exhaustion of therapeutic T cells, compared to second generation BCMA-targeting CARs. Interestingly, we found that remaining BCMA-BBζ CAR T cells displayed a higher MFI of BCMA expression as compared to persisting BCMA-28ζ+CD38-BB cells (fig. S7E), which was correlated with the co-existence with remaining tumor cells (fig. S7F).
We next interrogated whether the functional boost provided by the CCR co-expression to CAR T cells would also lead to eradication of tumor clones with low antigen expression. In order to investigate that, we employed an in vivo “stress test”, where we treated mice engrafted with the NALM6 clone 12.4 (200 mol of CD19 per cell) or the NALM6 clone 2 (approx. 20 mol per cell) (Fig. 7A). We found that CD19-CAR+CD38-CCR combination delayed tumor growth of both CD19low NALM6 clones and improved the survival of mice compared to both CD19-28ζ and CD19-BBζ CAR T cells. More specifically, although we had not observed differences in the in vitro lysis of NALM6 clone 12.4 between CD19-28ζ and the different CAR+CCR combinations, our in vivo studies revealed that mice treated with CD19-28ζ+CD38-BB and CD19-28ζ+CD38-28BB T cells had significantly lower tumor burden and survived longer than mice treated with CD19-28ζ, CD19-BBζ or CD19-ζ+CD38-28BB T cells (P= 0.0283, Fig. 7B to D and fig. S8A). Most notably, CD19-28ζ+CD38-28BB T cells outperformed all other treatments by inducing long term tumor remissions (Fig. 7B to D and fig. S8A). Importantly, the ultra-low CD19 antigen density NALM6 clone 2 was resistant to killing by both CD19-28ζ and CD19-BBζ CAR T cells as well as CD19-ζ+CD38-28BB and CD19-28ζ+CD38-BB (Fig. 7B, E, and F and fig. S8B). Nevertheless, the addition of a second CD28 domain in CD19-28ζ+CD38-28BB cells effectively delayed tumor progression and significantly prolonged the survival of mice (P<0.0001, Fig. 7B, E, and F and fig. S8B).
Figure 7. The CD19-CAR+CD38-CCR combination elicits improved in vivo anti-tumor function against tumor variants with very low antigen density.
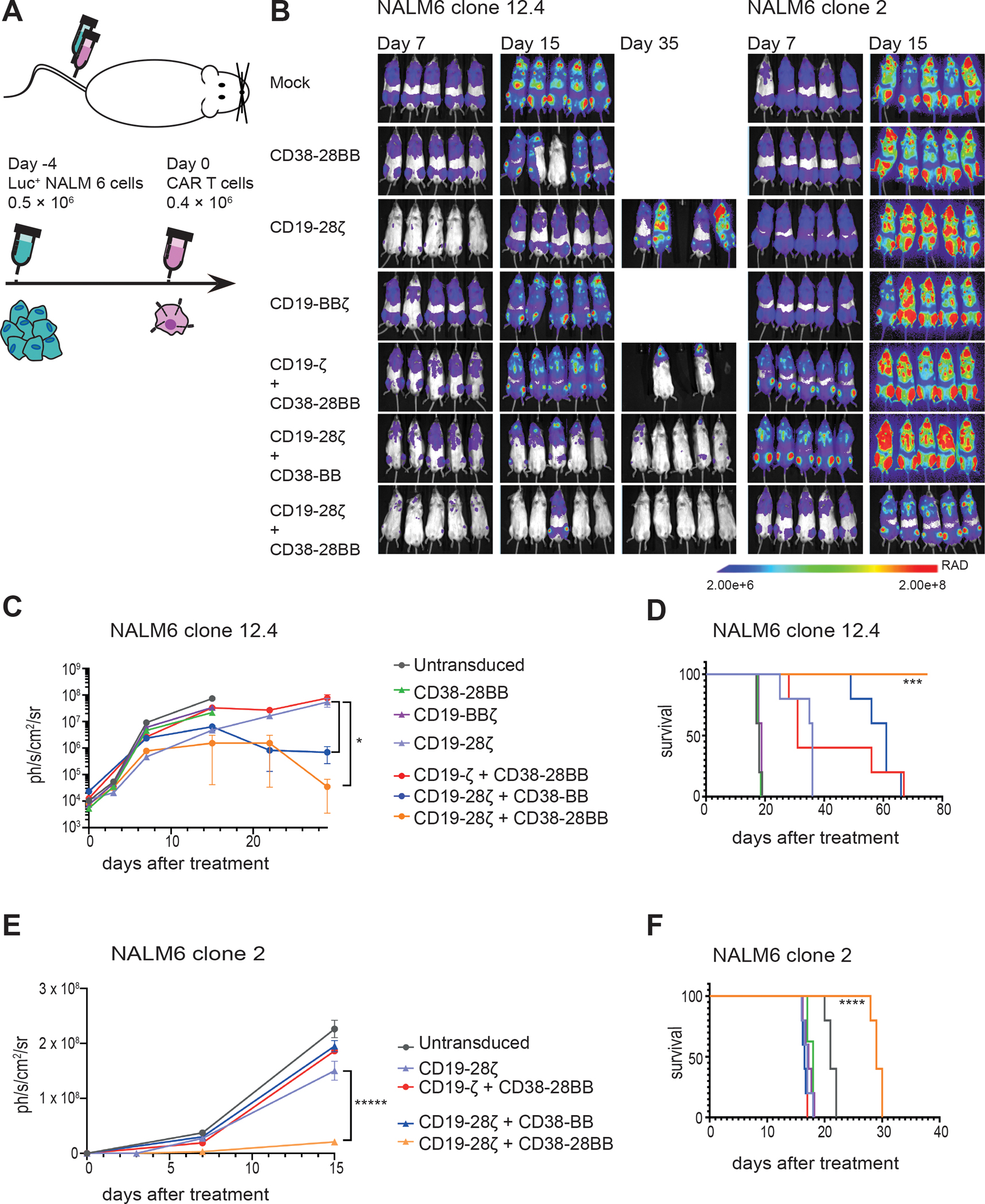
(A) A schematic representation of the ALL in vivo model is shown. 0.5 × 106 FFLuc-GFP NALM6 clone 12.4 or NALM6 clone 2 cells were administrated intravenously. Untransduced, CD38-28BB, CD19-ΒΒζ, CD19-28ζ, CD19-ζ+CD38-28BB, CD19-28ζ+CD38-BB or CD19-28ζ+CD38-28BB CAR T cells were infused intravenously 4 days later. Tumor burden was measured weekly by BLI. (B) Representative images of three time points are shown with the pixel intensity represented in color. (C) Average tumor burden of mice injected with FFLuc-GFP NALM6 clone 12.4 cells is shown over time. Tumor burden is depicted as units of photons per second per square centimeter per steradian (ph/sec/cm2/sr) (n=5 mice per group). (D) Survival of mice injected with NALM6 clone 12-4 is shown. (E) Average tumor burden of mice injected with FFLuc-GFP NALM6 clone 2 cells is shown over time. Tumor burden is depicted as units of photons per second per square centimeter per steradian (ph/sec/cm2/sr); n=5 mice per group. (F) Survival of mice injected with NALM6 clone 2 is shown. Statistical analysis of tumor burden (C and E) was performed with two-way ANOVAs and subsequent multiple comparison, corrected by Turkey test. *p<0.05. Statistical analysis of survival (D and F) was performed with log-rank tests. ***p<0.001, ****p<0.0001.
DISCUSSION
Despite the impressive complete response rates observed with CAR T cells in B-cell acute lymphoblastic leukemia (ALL), lymphomas and MM, there is still a considerable proportion of treatment failure, where the malignancy eventually relapses. Clinical and experimental data have revealed two main mechanisms of disease escape and relapse from adoptive T cell therapy with CAR T cells: the reduction or loss of the target-antigen expression and the limited persistence of the therapeutic T cells (4). Here, we provide evidence that the co-expression of a CCR with a CAR is a successful strategy to concomitantly improve cytotoxicity of the T cells against antigen-low tumor variants and provide optimal co-stimulation to enhance T cell persistence.
Double targeting using a pool of CARs, bi-cistronic CARs, or tandem CARs has been proposed as a strategy to overcome relapses due to antigen loss during CAR T cell therapy (32). In that case the selection of the target combinations is restricted by the requirement of tumor-specificity in order to avoid off-tumor toxicities. Although antigen loss has been documented in relapses after CD19-CAR therapy (13), relapses reported in clinical trials with CD22-CAR or BCMA-CAR T cells have been mostly attributed to the downregulation of the target expression or the selection of tumor clones with very low target expression (5, 33–35). Recent studies suggest that rational design of CAR constructs can improve the cytotoxicity, resulting in better eradication of tumors with low antigen density (8, 18). The combination of a CAR and a CCR has been previously proposed as a strategy to improve specificity of CAR T cell therapy provided that defined affinity requirements are fulfilled for both the CAR and CCR binding domains (36, 37). We demonstrate that the use of a CCR as a secondary binding module in combination with a CAR can be further exploited in order to augment the anti-tumor response. Importantly, since the CCR binding alone does not mediate T cell activation, the selection of the second targeted antigen is not limited by tumor specificity requirements.
Using T cells engineered with two CARs has resulted in improved efficacy in vivo compared to single targeting CAR T cells (11, 13, 18). This has been indirectly attributed to the increase of avidity achieved by the multivalent binding (10). However, detailed investigation of the mechanism resulting in the observed better anti-tumor efficacy of double-targeting CAR T cells and the impact of avidity or co-stimulation has not been performed. Using a highly and uniformly expressed antigen, like CD38, as a CCR target, we were able to observe clear enhancement of the in vitro cytotoxicity and in vivo anti-tumor efficacy of BCMA- and CD19-targeting CARs. With a truncated CD38-engager (CD38Δ) we demonstrate that the engagement of CD38 alone is sufficient to increase the in vitro cytotoxicity, intracellular signalling, and granzyme B secretion and that the binding of the CCR to CD38 could strengthen the synaptic tension between the CAR T cell and the tumor cell. We further investigated the specific requirements of CD38 binding that resulted in increased cytotoxicity. Our collective data from the MM and ALL models demonstrated that the cytotoxicity was increased only when the CCR was binding through a high affinity scFv and when CD38 was expressed at higher densities. Therefore, our data not only provide direct evidence that the engagement of a second receptor along with a CAR enhanced cytotoxic function due to the increase of the functional avidity, but also define specific requirements for the selection of a highly expressed antigen (such as CD38) in combination with a high affinity scFv.
Although higher avidity resulted in better cytotoxicity, there was no benefit in the cytokine secretion and expansion of CAR T cells. Experimental data from our group and others have pointed out both the synergy of CD28 and 4-1BB costimulatory signaling and the superior anti-tumor function of CAR T cells combining both modalities (18, 20, 24). We hypothesized that the co-expression of a CCR allows not only for double targeting, but also complimentary co-stimulation by the addition of both the CD28 and 4-1BB intracellular domains, either in the CCR alone or in the CAR and CCR respectively. More specifically, double co-stimulation provided CAR+CCR T cells with improved cytokine and granzyme B production and proliferative capacity in vitro, as well as impressive persistence and reduced expression of exhaustion-related markers in vivo, compared to both second generation CAR T cell designs. Therefore, double CD28/4-1BB co-stimulation and the achievement of high effector-to-tumor ratios is a second mechanism of improving anti-tumor efficacy by using a CAR+CCR combination. Importantly, the CCR mediated co-stimulation could also be mediated through in-trans binding to CD38 expressed by accessory cells without increasing off-tumor lysis or activation of monocytes, meaning that the costimulatory effects of a CCR could be harnessed by using targets present in the tumor environment and not necessarily on the tumor. We observed no difference in cytotoxicity and only slight differences in cytokine secretion and proliferation between the combinations having different positioning of CD28 costimulatory domain (ζ+CD38-28BB or 28ζ+CD38-BB) in our BCMA/CD38 co-targeting system. However, when targeting NALM6 cells with low CD19 expression we did observe that the selection of costimulatory domains had an effect on the anti-tumor function. Although, the ζ+CD38-28BB and 28ζ+CD38-BB cells showed again the same lytic capacity, we found that the addition of a second CD28 transmembrane and intracellular domain in a 28ζ+CD38-28BB design improved in vitro cytotoxicity, as well as in vivo tumor eradication. Thus, the 28ζ+CD38-28BB combination seems superior when targeting very low target densities. This could be attributed not only to the amplification of CD28 intracellular signaling but also to the duplication of the CD28 transmembrane domain, which has been shown recently to benefit the cytotoxic function of CAR T cells (8) potentially through interactions with endogenous partners (38).
Interestingly, although in vitro and in vivo expansion of CAR+CCR cells was similar to 4-1BB-based CAR T cells, it was not correlated with the retention of a CD62L+CD45RA+ central memory phenotype as previously described for 4-1BB-based CAR T cells (16, 19), but rather with an effector memory phenotype combined with lower expression of inhibitory receptors PD-1 and LAG-3. A similar phenotype has been observed in another study when costimulation was achieved by the combination of a CD19-28ζ CAR and co-expression of 4-1BB ligand (20). In addition, we found that BCMA-28ζ+CD38-BB T cells maintained a balanced (1:1) CD4:CD8 ratio in vivo, which has been associated with optimal anti-tumor function and high rate of clinical remissions (39, 40). Although, post-mortem analysis of the MM in vivo model revealed no difference in BCMA expression in the remaining tumor cells of the BCMA-BBζ group, we found an increased BCMA expression in the persisting BCMA-BBζ CAR T cells of some scaffolds, but not in the BCMA-28ζ+CD38-BB T cells, which correlated with the co-existence with tumor cells. Similar finding has been reported previously and correlated to the inefficient anti-tumor cytotoxic efficacy of the BCMA-BBζ CAR T cells (18).
The antigen density threshold required for efficient cytotoxicity from CAR T cells can vary between different targets and CAR constructs. Investigations on CD19-targeting CARs established that the cytotoxicity of 4-1BB-based CARs is more sensitive to a decrease in antigen density than CD28-based CARs (8, 18). Roughly the efficient cytotoxicity threshold for 4-1BB-based CD19-CARs has been described to be around 1,000 target molecules per cell (8, 18) whereas cytotoxicity of CD28-based CD20CARs was reduced at 200 molecules per cell (9). Super resolution microscopy has showed that CD19-targeting CARs bearing a CD28 transmembrane domain and a 4-1BB signaling domain eliminated MM cells with fewer than 100 CD19 molecules per cell in short-term in vitro assays (41). Modeling the BCMA expression of MM patients and using engineered NALM6 cell lines bearing very low CD19 (200 and 20 molecules per cell) we show that multi-targeting with a CAR and a CCR can improve lysis of tumor variants with low target-antigen expression and even restore lysis of resistant ultra-low expressing tumor cells in vitro and in vivo. In a previous study using a similar ALL model, improving only the cytotoxic capacity of 4-1BB-based CD19-CAR T cells by fine tuning the CAR design delayed tumor growth but was not sufficient to induce sustained anti-tumor responses in vivo (8). Notably, using the CAR+CCR combination CD19-28ζ+CD38-28BB, we observed complete eradication of CD19+ tumors with target density as low as 200 molecules per cell and delay of tumor growth for CD19+ tumors with 20 molecules per cell.
We chose CD38 as target for the CCR as CD38 is widely expressed in various hematological malignancies. Clinical experience of using anti-CD38 monoclonal antibodies underscores the potential downregulation of CD38 expression on the tumor (42, 43). Although, in this case the benefit of CCR expression on cytotoxicity would be absent, we show that a CD38-CCR could at least provide in-trans co-stimulation to improve persistence by engaging on surrounding cells expressing CD38, for example natural killer (NK) cells or monocytes. Further, T cells themselves express CD38 in lower abundance and T cells bearing a CD38-CCR or CD38Δ showed decrease of CD38 expression on their surface due to either in-cis or in-trans engagement of endogenous CD38. Since CD38-CCR expression does not exert any cytotoxic capacity or tonic signaling, we observed no negative effect on CAR T cell viability upon CD38-CCR expression.
Our study has limitations. In the present study we focused on demonstrating how the CAR+CCR combination can improve therapeutic outcomes and the construction requirements. Thus we chose to limit the combination of costimulatory domains to CD28 and 4-1BB, two domains that are already known to work synergistically. It remains to be investigated if other costimulatory domains, such as the TNF Receptor Superfamily Member 4 (TNFRSF4 or OX40) or the Inducible T-Cell Co-Stimulator (ICOS), could be used to provide similar results and how they could be combined and split between the two receptors. In addition, here we studied only the combinatorial targeting of BCMA+CD38 and CD19+CD38 using specific scFvs. Using different target combinations or different scFvs may differently impact the effect of avidity or costimulation in the anti-tumor function. Finally, although we show enhancement of T cell efficacy against tumor clones with very low antigen density, our study was limited by the lack of models of relapse with antigen-deficient or antigen-low tumor cells in order to investigate the effect on the actual incidence of relapse.
Our results demonstrate that the use of a CD38-CCR in combination with any CAR would benefit the immunotherapeutic outcomes against hematological malignancies such as MM and B-cell lymphomas. In addition, the increase of cytotoxicity of CD19-CAR+CD38-CCR T cells against tumor cells with ultra-low CD19 antigen density advocates that the co-expression of a CD38-CCR could potentially improve the unsatisfactory clinical outcomes of CD19-CAR T cell therapy for MM (44). Furthermore, beyond hematological malignancies, our study provides the requirements of CCR target selection and CCR construction and thus, the CAR+CCR strategy could be also applicable and be used to improve CAR T cell therapy of solid tumors. Using a 1st generation CAR construct with a low affinity scFv could allow also for selective tumor killing as previously described (36). Collectively our data show that the combination of a CAR and a CCR allows for multiplexed targeting, as well as co-stimulation, generating potent and durable effectors with enhanced anti-tumor function including tumor variants with ultra-low antigen density. Thus, the application of this strategy could improve clinical outcomes and accelerate the progress of CAR T cell therapy for various malignancies.
MATERIALS AND METHODS
Study design
The purpose of this study was to investigate the hypothesis that dual targeting of tumor with a CAR and a CCR would improve the therapeutic outcomes of engineered T cells. We chose to test our strategy in hematological tumor models of MM and ALL and thus, selected CD38 as a CCR target, a molecule expressed in various hematologic tumors. At first, we investigated the cytotoxic potential of CAR+CCR T cells compared to conventional single targeted BCMA- or CD19- CAR T cells. We interrogated the mechanism of the observed increase of cytolytic capacity and the contribution of the extracellular and intracellular part of the CCR. Next, we used MM and ALL tumor cell lines with low BCMA and CD19 density to investigate the sensitivity of recognition from CAR+CCR T cells compared to CAR T cells. A second objective was the assessment of the cytokine production, expansion potential and differentiation of CAR+CCR T cells and the contribution of the extracellular and intracellular part of the CCR. For the in vivo models, mice (with a minimum of n = 4 per group) were randomly assigned to treatment groups and we analyzed the anti-tumor efficacy and persistence of CAR+CCR T cells and their efficacy against tumor variants with low target-antigen density. The animal experiments were performed under the approval of the central authority for scientific procedures on animals (CCD). All primary samples (healthy donor and patient material) were obtained after informed consent and approval by the institutional medical ethical committee. Each experiment was performed multiple times using T cells from at least four different donors and we present pooled data. There were no outliers in our experimental data.
Cell lines
Human cell lines MM1.S (kind gift of C. Mitsiadis), UM9 (obtained from UMC Utrecht), K562 (American Type Culture Collection, ATCC), U266 (ATCC) and NALM6 (ATCC) (unmodified or expressing luciferase) were cultured in RPMI-1640 (Thermo Fisher Scientific) with 10% HyClone Fetal Clone I (Thermo Fisher Scientific) and antibiotics (penicillin; 100 U/mL, streptomycin; 100 mg/mL). K562 cells were lentivirally transduced to express human CD38 and human BCMA and sorted for gradually increasing concentrations of BCMA expression. The mouse fibroblast NIH/3T3 cell line (ATCC) and Phoenix Ampho cells (ATCC) were maintained in Dulbecco’s Modified Eagle Medium (DMEM) GlutaMAX with 10% fetal bovine serum (FBS) (Invitrogen) and antibiotics (penicillin 10,000 U/ml; streptomycin 10,000 μg/ml). NIH/3T3 cells were lentivirally transduced to express human CD38 (3T3-CD38). Banks of all cell lines were checked with Short Tandem Repeat (STR) analysis and were regularly tested for mycoplasma.
Primary cells from MM patients and healthy individuals.
Healthy donor peripheral blood mononuclear cells (PBMCs) from buffy coats (Sanquin blood-bank) or bone marrow mononuclear cells (BM-MNCs) from MM patient’s bone marrow aspirates [10 to 40% malignant cells, determined by flow cytometry (CD138+/CD38+)], were isolated by Ficoll-Paque (GE Healthcare Life Sciences) density centrifugation. Isolated cells were directly used in cytotoxicity assays or cryopreserved in liquid nitrogen until use. All primary samples were obtained after informed consent and approval by the institutional medical ethical committee.
CAR and CCR vector constructs
CAR and CCR constructs were cloned into SFG γ-retroviral vectors (45) using standard molecular biology techniques. BCMA-CAR constructs used an extracellular single chain variable fragment (scFv) published and described in WO2016/094304 A2 (BCMA02, drug product name bb2121). The scFv was followed by a CD8α transmembrane domain and CD3ζ signaling domain (BCMA-ζ) or by a CD8α transmembrane domain and the 4-1BB and CD3ζ signaling domains (BCMA-BBζ) or a CD28 transmembrane and intracellular sequence fused to CD3ζ intracellular domain (BCMA-28ζ). The CAR sequences were linked by a P2A sequence to a dsRed fluorescent expression marker or to a truncated LNGFR sequence. The CD19-CAR constructs used the SJ25C1 scFv and have been previously described (1). For the CCR construct the CD38-specific scFvs, previously described from our group (46), were followed by a CD28 transmembrane and intracellular sequence and the 4-1BB intracellular domain or by a CD8α transmembrane domain and the 4-1BB signaling domain. The CCR sequences were linked by a P2A element to a truncated LNGFR sequence.
Generation of Retroviral Particles and Transduction of T Cells
Phoenix-Ampho packaging cells were calcium phosphate transfected with 10μg CAR constructs. 16 hours post-transfection, complete medium (DMEM with 10% FBS) was refreshed. Two and three days after transfection, cell free supernatants containing retroviral particles were collected and directly used for transduction. Peripheral blood mononuclear cells (PBMCs) from healthy donors (3×106 per well) were stimulated with lectin-like phytohemagglutinin (PHA-L) (Roche) or with Dynabeads Human T-Activator CD3/CD28 (Thermo Fisher Scientific) in a 6-well plate (Greiner Bio-One) in culture medium (RPMI-1640 with 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin). After 48 hours, 3×106 cells per ml were transferred to retronectin coated (15 μg/ml) (Takara) 6-well plates (Falcon). Retroviral transduction was performed by addition of 2 ml virus per well followed by spinoculation (1500g for 1 hour at room temperature) in the presence of 4 μg/ml Polybrene. A second transduction was conducted after 16 hours, replacing two-thirds of the cell supernatant with freshly obtained virus (2 ml). Six to eight hours after the second transduction, half of the cell supernatant was replaced by fresh RPMI-1640 with 10% FBS and 50 IU/ml recombinant human (rh) IL-2 (Proleukin, Novartis). Transduction efficiencies were determined 72 hours later by flow cytometric detection of LNGFR (CD271) or dsRed expression. In order to isolate double-transduced CAR+CCR T cells, an EasySep allophycocyanin (APC) Positive Selection Kit II (Stemcell Technologies) was used per manufacturer’s instructions to isolate T cells labeled with an CD271 (NGFR)-APC antibody (CD271; clone ME20.4, BioLegend) (staining with 3 μg per mL of sample, incubation for 15 minutes at room temperature), by positive selection. Sorted T cells bearing the CD38-CCR (a mix of double CAR+CCR transduced and CCR-transduced) were further used in assays, assuming that CD38-CCR-transduced T cells would remain non-functional upon antigen engagement.
Bioluminescent Imaging based cytotoxicity assay
Seven to ten days after transduction, serial dilutions (effector:target 2:1, 1:1, 1:2) of CAR T cells were incubated with luciferase-expressing cell lines. The luciferase signal produced by surviving cells was determined after 16 to 24 hours with a GloMax 96 Microplate Luminometer (Promega) within 15 minutes after the addition of 125 μg/mL beetle luciferin (Promega). Percent lysis was calculated as: % lysis = 1 − (BLI signal in treated wells / BLI signal in untreated wells) × 100%.
Calcein-AM release assay
Target cells (3T3-CD38) were suspended in complete medium at a final concentration of 106 per ml and labeled with Calcein-AM, purchased from Molecular Probes. CAR T cells were incubated together with labeled target cells at E:T ratios ranging from 2:1 to 0.5:1 for 4 hours. Controls included wells for spontaneous (mock transduced T cells from the same donor) and maximum release (only target cells in medium plus 2% Triton X-100). After incubation, clarified supernatant was sampled for Calcein-AM detection in a SpectraMax Gemini dual-scanning microplate spectrofluorimeter (Molecular Devices) with an excitation filter of 485 ± 9 nm and a band-pass filter of 530 ± 9 nm. Data are expressed as arbitrary fluorescent units (AFU). Percent lysis was calculated as: Specific Lysis (%) = (AFU in treated wells − AFU in spontaneous release)/(AFU in maximum release − AFU in spontaneous release) × 100%.
Flow cytometry–based cytotoxicity assay
CAR T cells were incubated in serial dilutions (effector:target 2:1, 1:1, or 0.5:1) with CellTrace Violet kit Thermo Fisher Scientific)-labeled BM-MNC for 16 hours. Cell subsets were identified after staining with different markers (CD3, CD14, CD19, CD38, BCMA, CD56, and CD138 (BD Biosciences), as described below. Flow-Count Fluorospheres (Beckman 7547053) were added and cells were quantitatively analyzed through Flow-Count–equalized measurements. Percentage cell lysis was calculated as: % lysis = 1 − ((# viable target cells in treated wells/# of beads)/(# viable target cells in untreated wells/# of beads)) × 100%.
Avidity measurement
Cellular avidity of single or double targeting CAR T cells to MM1.S tumor cells was investigated as described previously (47, 48). In brief, MM1.S cells were allowed to adhere to the surface of a temperature-controlled microfluidic chip (LUMICKS CA B.V.) for a total of 2.5 hours. Chips were placed into the z-Movi Cell Avidity Analyzer (LUMICKS) where experiments were performed at 37°C. Indicated CAR T cells were subsequently flowed into the microfluidics chip and allowed to interact with the MM1.S monolayer for 5 minutes, after which an acoustic force ramp was applied (1000 pN relative force over 150 seconds). All experiments were performed in triplicate.
Proliferation assay
BCMA-CAR T cells were counted and stimulated weekly with irradiated (60 Gy) BCMA+CD38+ MM1.S cells. Starting 7 days after transduction, 0.5 × 106 CAR T cells were seeded in a 24-well plate containing 0.25 × 106 MM1.S, to final volume of 1mL. No additional cytokines were added.
Intracellular phosho-AKT1 assessment
Stimulation and staining of cells were performed in 5mL Round Bottom Polystyrene FACS Tubes. CAR T cells (0.5×106) were co-cultured together with UM9 cells (0.5×106) for 15 minutes at room temperature in complete RPMI-1640 medium with the recommended amount of APC-conjugated anti-CD271 antibody (CD271; clone ME20.4, BioLegend). At the end of the incubation time, tubes were transferred on ice and cells were washed twice with FACs buffer (phosphate-buffered saline with 0.5 to 1% bovine serum albumin or 5 to 10% FBS and 0.1% sodium azide). The eBioscience Intracellular Fixation & Permeabilization Buffer Set was used according to manufacturer’s protocol. After permeabilization and without washing, the recommended amount of the Phospho-AKT11 (Ser473) Monoclonal Antibody (clone SDRNR, eBioscience) was added and the cells were incubated for 30 minutes at room temperature, protected from light. Cells were washed twice with Permeabilization Buffer and flow cytometry was performed on BD LSR Fortessa.
Monocyte activation
CD14+ monocytes were isolated from PBMCs by positive selection. Isolated monocytes (10,000 cells) were plated in a 96-wells flat-bottom plate. 24 hours later, 10,000 CAR T cells and 10,000 tumor cells (MM1.S) were added in each well and were incubated for 24 hours. After incubation, the wells were washed with phosphate-buffered saline twice and detached from the wells with Trypsin/EDTA solution (Lonza) for 20 minutes. Monocytes were stained with CD3 (clone SK7, BD Biosciences), CD14 (clone HCD14, BioLegend), HLA-DR (clone G46-6, BD Biosciences), CD86 (clone HA5.2B7, Beckman Coulter), and CD80 (clone 16-10A1, BioLegend) (staining with 1 μg per mL of sample, incubation for 30 minutes at 4°C in PBS with 1% BSA, 10% FBS and 0.1% NaN3 sodium azide). Flow cytometry was performed on BD LSR Fortessa.
Flow cytometry
Flow cytometry was performed on BD LSR Fortessa. Transduction efficiency was measured with an APC-conjugated antibody toward NGFR (CD271; clone ME20.4, BioLegend) and Protein L for CCR, whereas for CARs it was measured in the phycoerythrin (PE) channel to detect dsRed and AffiniPure F(ab’)2 Fragment Goat Anti-Mouse IgG (Jackson ImmunoResearch). The following antibodies were used for flow cytometry staining: Anti-Human CD3 (clone UCHT1, BD Biosciences), Anti-Human CD4 (clone L200, BD Biosciences), Anti-Mouse CD45 (clone 30-F11, BD Biosciences), Anti-Human CD8 (clone SK1, BD Biosciences), Anti-Human CD45 (clone HI30, BD Biosciences), Anti-human CD366 (TIM-3) (clone F38-2E2, BioLegend or clone F38-2E2, Thermo Fisher Scientific), Anti-human CD279 (PD-1) (clone EH12.2H7, BioLegend or clone eBioJ105, Thermo Fisher Scientific), Anti-human CD62L (clone DREG-56, BioLegend), Anti-human CD269 (BCMA) (clone 19F2, BioLegend), Anti-human CD38 (clone HIT2, BioLegend), CD45RA (clone HI100, Thermo Fisher Scientific), LAG-3 (CD223; clone 3DS223H, Thermo Fisher Scientific) (staining 1 μg per mL of sample, incubation for 30 minutes at 4°C in PBS with 1% BSA, 10% FBS and 0.1% NaN3 sodium azide). For the quantification of the CD38, the Quantibrite Phycoerythrin (PE) Fluorescence Quantitation Kit (BD) was used. Flow cytometry data analysis was performed with FCS Express 6 flow software.
Cytokine and Granzyme B measurements
To determine cytokine production, cell supernatants were harvested 16 hours after co-culture with target cells. The Cytokine Bead Array (CBA) Human Th1/Th2/Th17 cytokine Kit (BD Biosciences) was used according to the manufacturer’s protocol. Human granzyme B was quantitatively determined by the Human Granzyme B enzyme-linked immunosorbent assay (ELISA) development kit (Mabtech, 3485-1H-6) according to the manufacturer’s guidelines.
In vivo xenograft studies
For the MM in vivo model, RAG-2−/−γc−/− female mice were bred and maintained at the Amsterdam Animal Research Center (Universitair proefdiercentrum, AARC-UPC). The animal experiments were performed under the approval of the central authority for scientific procedures on animals (protocol number AVD114002015345). Animal welfare was monitored, and euthanasia was induced in strict accordance with the Dutch Animal Experimentation Act. The scaffold-based xenograft murine model was prepared as previously described (31). Mice were subcutaneously implanted with hybrid scaffolds, each consisting of three 2- to 3-mm3 biphasic calcium phosphate particles, coated in vitro with human bone marrow mesenchymal stromal cells (BM-MSC; 2 × 105 cells/scaffold). Eight to twelve weeks after implantation, mice were injected with bisulvex intraperitoneally (18 mg/kg in 500 μl). Next day, they were intravenously injected with luciferase-transduced multiple myeloma cells (10 × 106 UM9 or 2.5 × 106 MM1S). Seven days after injection of tumor cells, mice received transduced T cells (2.5 × 106 cells/mice) intravenously. Tumor growth was monitored by weekly bioluminescence (BLI) measurements using a PhotonIMAGER (Biospace Lab) (intraperitoneal injection of 100 μl D-luciferin). Postmortem BM and scaffolds were harvested from each mouse, dissociated (scaffolds) and filtered through a 70 μm filter. Single-cell suspensions were counted, stained, and measured by flow cytometry.
For the ALL in vivo model, female 8–12-week-old NOD.Cg-PrkdcscidIl2rgtmWjl/SzJ (NSG) mice (Jackson Laboratory) were used, under a protocol approved by the MSKCC Institutional Animal Care and Use Committee. A total of 0.5 × 106 FFLuc-GFP NALM6 cells were administered intravenously. Four days later, 0.2 × 106 CAR or CAR+CCR T cells were administered. Tumor burden was measured, after intraperitoneal injection of 100 μl D-luciferine, by bioluminescence imaging using the Xenogen IVIS Imaging System (Xenogen).
Statistical analysis
Raw, individual level data are shown in data file S1. Data analysis and visualization was performed using GraphPad Prism 8.2.1 (GraphPad Prism, RRID:SCR_002798) software. No pre-specified effect size was used to determine sample sizes. Graphs represent individual values ± standard error of the mean (SEM), for in vitro and in vivo experiments. Normality of the data was confirmed with the Kolmogorov-Smirnov test. The statistical tests that were used to calculate the p values are described in the relevant figure legends. Differences were considered significant at p <0.05 and p values are denoted with asterisks as follows: p>0.5, not significant (ns), *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
Supplementary Material
Acknowledgments
The authors would like to thank Constantine Mitsiades (Dana Farber Cancer Institute) for providing the MM1.S cell line.
Funding:
This work was supported by Stichting VUmc CCA grant CCA2014-1-17 to AK and MT, Multiple Myeloma Research Foundation Immunotherapy Network of Excellence grant to M. Sjöstrand, DS, DA, TM, M. Sadelain and MT, the Swedish Research Council grant to M. Sjöstrand, and the National Institutes of Health grant R01 CA196664 to RWJG.
Footnotes
Competing interests: J.M-S. holds unrelated patent on CAR T cell technologies. N.W.C.J.v.d.D. has received research support from Janssen Pharmaceuticals, AMGEN, Celgene, Novartis, and BMS, and serves on advisory boards for Janssen Pharmaceuticals, AMGEN, Celgene, BMS, Novartis, Takeda, Bayer, and Servier. S.Z. has received research support from Celgene, Takeda and Janssen Pharmaceuticals and serves in advisory boards for Celgene, Takeda, Janssen and Amgen. T.M. has received research support from Janssen Research and Development, Celgene, Onkimmune and Genmab. I.S., Y.R.P., E.M., and R.M.R. are employed by LUMICKS. H.Y. and J.d.B. are employed by Kuros Biosciences BV. D.A. has received research support from Celgene, Pharmacyslics and Kite Pharma and serves in advisory boards for Celgene, Juno, Partners Tx, Karyopharm, BMS, Aviv MedTech Ltd., Takeda, Legend BioTech, and Chugai. M.S. receives research funding from Fate Therapeutics, Takeda Pharmaceuticals and Atara Biotherapeutics and holds patents on CAR T cell technologies. R.W.J.G. has received research support from Janssen Research and Development. M.T. holds unrelated patent on CAR T cell technologies. The rest of the authors have no conflicts of interest to declare.
Data and materials availability:
All data associated with this study are in the paper or supplementary materials.
References and Notes
- 1.van der Stegen SJ, Hamieh M, Sadelain M, The pharmacology of second-generation chimeric antigen receptors. Nat Rev Drug Discov 14, 499–509 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, Bader P, Verneris MR, Stefanski HE, Myers GD, Qayed M, De Moerloose B, Hiramatsu H, Schlis K, Davis KL, Martin PL, Nemecek ER, Yanik GA, Peters C, Baruchel A, Boissel N, Mechinaud F, Balduzzi A, Krueger J, June CH, Levine BL, Wood P, Taran T, Leung M, Mueller KT, Zhang Y, Sen K, Lebwohl D, Pulsipher MA, Grupp SA, Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N Engl J Med 378, 439–448 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Park JH, Riviere I, Gonen M, Wang X, Senechal B, Curran KJ, Sauter C, Wang Y, Santomasso B, Mead E, Roshal M, Maslak P, Davila M, Brentjens RJ, Sadelain M, Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N Engl J Med 378, 449–459 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Majzner RG, Mackall CL, Clinical lessons learned from the first leg of the CAR T cell journey. Nat Med 25, 1341–1355 (2019). [DOI] [PubMed] [Google Scholar]
- 5.Fry TJ, Shah NN, Orentas RJ, Stetler-Stevenson M, Yuan CM, Ramakrishna S, Wolters P, Martin S, Delbrook C, Yates B, Shalabi H, Fountaine TJ, Shern JF, Majzner RG, Stroncek DF, Sabatino M, Feng Y, Dimitrov DS, Zhang L, Nguyen S, Qin H, Dropulic B, Lee DW, Mackall CL, CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat Med 24, 20–28 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Walker AJ, Majzner RG, Zhang L, Wanhainen K, Long AH, Nguyen SM, Lopomo P, Vigny M, Fry TJ, Orentas RJ, Mackall CL, Tumor Antigen and Receptor Densities Regulate Efficacy of a Chimeric Antigen Receptor Targeting Anaplastic Lymphoma Kinase. Mol Ther 25, 2189–2201 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ramakrishna S, Highfill SL, Walsh Z, Nguyen SM, Lei H, Shern JF, Qin H, Kraft IL, Stetler-Stevenson M, Yuan CM, Hwang JD, Feng Y, Zhu Z, Dimitrov D, Shah NN, Fry TJ, Modulation of Target Antigen Density Improves CAR T-cell Functionality and Persistence. Clin Cancer Res 25, 5329–5341 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Majzner RG, Rietberg SP, Sotillo E, Dong R, Vachharajani VT, Labanieh L, Myklebust JH, Kadapakkam M, Weber EW, Tousley AM, Richards RM, Heitzeneder S, Nguyen SM, Wiebking V, Theruvath J, Lynn RC, Xu P, Dunn AR, Vale RD, Mackall CL, Tuning the Antigen Density Requirement for CAR T-cell Activity. Cancer Discov 10, 702–723 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Watanabe K, Terakura S, Martens AC, van Meerten T, Uchiyama S, Imai M, Sakemura R, Goto T, Hanajiri R, Imahashi N, Shimada K, Tomita A, Kiyoi H, Nishida T, Naoe T, Murata M, Target antigen density governs the efficacy of anti-CD20-CD28-CD3 zeta chimeric antigen receptor-modified effector CD8+ T cells. J Immunol 194, 911–920 (2015). [DOI] [PubMed] [Google Scholar]
- 10.de Larrea CF, Staehr M, Lopez AV, Ng KY, Chen Y, Godfrey WD, Purdon TJ, Ponomarev V, Wendel HG, Brentjens RJ, Smith EL, Defining an Optimal Dual-Targeted CAR T-cell Therapy Approach Simultaneously Targeting BCMA and GPRC5D to Prevent BCMA Escape-Driven Relapse in Multiple Myeloma. Blood Cancer Discov 1, 146–154 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hegde M, Corder A, Chow KK, Mukherjee M, Ashoori A, Kew Y, Zhang YJ, Baskin DS, Merchant FA, Brawley VS, Byrd TT, Krebs S, Wu MF, Liu H, Heslop HE, Gottschalk S, Yvon E, Ahmed N, Combinational targeting offsets antigen escape and enhances effector functions of adoptively transferred T cells in glioblastoma. Mol Ther 21, 2087–2101 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Qin H, Cho M, Haso W, Zhang L, Tasian SK, Oo HZ, Negri GL, Lin Y, Zou J, Mallon BS, Maude S, Teachey DT, Barrett DM, Orentas RJ, Daugaard M, Sorensen PH, Grupp SA, Fry TJ, Eradication of B-ALL using chimeric antigen receptor-expressing T cells targeting the TSLPR oncoprotein. Blood 126, 629–639 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ruella M, Barrett DM, Kenderian SS, Shestova O, Hofmann TJ, Perazzelli J, Klichinsky M, Aikawa V, Nazimuddin F, Kozlowski M, Scholler J, Lacey SF, Melenhorst JJ, Morrissette JJ, Christian DA, Hunter CA, Kalos M, Porter DL, June CH, Grupp SA, Gill S, Dual CD19 and CD123 targeting prevents antigen-loss relapses after CD19-directed immunotherapies. J Clin Invest 126, 3814–3826 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schneider D, Xiong Y, Wu D, Nlle V, Schmitz S, Haso W, Kaiser A, Dropulic B, Orentas RJ, A tandem CD19/CD20 CAR lentiviral vector drives on-target and off-target antigen modulation in leukemia cell lines. J Immunother Cancer 5, 42 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fraietta JA, Lacey SF, Orlando EJ, Pruteanu-Malinici I, Gohil M, Lundh S, Boesteanu AC, Wang Y, O’Connor RS, Hwang WT, Pequignot E, Ambrose DE, Zhang C, Wilcox N, Bedoya F, Dorfmeier C, Chen F, Tian L, Parakandi H, Gupta M, Young RM, Johnson FB, Kulikovskaya I, Liu L, Xu J, Kassim SH, Davis MM, Levine BL, Frey NV, Siegel DL, Huang AC, Wherry EJ, Bitter H, Brogdon JL, Porter DL, June CH, Melenhorst JJ, Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat Med 24, 563–571 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, Smith JP, Walker AJ, Kohler ME, Venkateshwara VR, Kaplan RN, Patterson GH, Fry TJ, Orentas RJ, Mackall CL, 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med 21, 581–590 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guedan S, Calderon H, Posey AD Jr., Maus MV, Engineering and Design of Chimeric Antigen Receptors. Mol Ther Methods Clin Dev 12, 145–156 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hamieh M, Dobrin A, Cabriolu A, van der Stegen SJC, Giavridis T, Mansilla-Soto J, Eyquem J, Zhao Z, Whitlock BM, Miele MM, Li Z, Cunanan KM, Huse M, Hendrickson RC, Wang X, Riviere I, Sadelain M, CAR T cell trogocytosis and cooperative killing regulate tumour antigen escape. Nature 568, 112–116 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kawalekar OU, RS OC, Fraietta JA, Guo L, McGettigan SE, Posey AD Jr., Patel PR, Guedan S, Scholler J, Keith B, Snyder NW, Blair IA, Milone MC, June CH, Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity 44, 712 (2016). [DOI] [PubMed] [Google Scholar]
- 20.Zhao Z, Condomines M, van der Stegen SJC, Perna F, Kloss CC, Gunset G, Plotkin J, Sadelain M, Structural Design of Engineered Costimulation Determines Tumor Rejection Kinetics and Persistence of CAR T Cells. Cancer Cell 28, 415–428 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Feucht J, Sadelain M, Function and evolution of the prototypic CD28ζ and 4-1BBζ chimeric antigen receptors. Immuno-Oncology Technology, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Melero I, Bach N, Hellstrom KE, Aruffo A, Mittler RS, Chen L, Amplification of tumor immunity by gene transfer of the co-stimulatory 4-1BB ligand: synergy with the CD28 co-stimulatory pathway. Eur J Immunol 28, 1116–1121 (1998). [DOI] [PubMed] [Google Scholar]
- 23.Rudolf D, Silberzahn T, Walter S, Maurer D, Engelhard J, Wernet D, Buhring HJ, Jung G, Kwon BS, Rammensee HG, Stevanovic S, Potent costimulation of human CD8 T cells by anti-4-1BB and anti-CD28 on synthetic artificial antigen presenting cells. Cancer Immunol Immunother 57, 175–183 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Drent E, Poels R, Ruiter R, van de Donk N, Zweegman S, Yuan H, de Bruijn J, Sadelain M, Lokhorst HM, Groen RWJ, Mutis T, Themeli M, Combined CD28 and 4-1BB Costimulation Potentiates Affinity-tuned Chimeric Antigen Receptor-engineered T Cells. Clin Cancer Res 25, 4014–4025 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhong XS, Matsushita M, Plotkin J, Riviere I, Sadelain M, Chimeric antigen receptors combining 4-1BB and CD28 signaling domains augment PI3kinase/AKT/Bcl-XL activation and CD8+ T cell-mediated tumor eradication. Mol Ther 18, 413–420 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Krause A, Guo HF, Latouche JB, Tan C, Cheung NK, Sadelain M, Antigen-dependent CD28 signaling selectively enhances survival and proliferation in genetically modified activated human primary T lymphocytes. J Exp Med 188, 619–626 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Naik J, Themeli M, de Jong-Korlaar R, Ruiter RWJ, Poddighe PJ, Yuan H, de Bruijn JD, Ossenkoppele GJ, Zweegman S, Smit L, Mutis T, Martens ACM, van de Donk N, Groen RWJ, CD38 as a therapeutic target for adult acute myeloid leukemia and T-cell acute lymphoblastic leukemia. Haematologica 104, e100–e103 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Krejcik J, Casneuf T, Nijhof IS, Verbist B, Bald J, Plesner T, Syed K, Liu K, van de Donk NW, Weiss BM, Ahmadi T, Lokhorst HM, Mutis T, Sasser AK, Daratumumab depletes CD38+ immune regulatory cells, promotes T-cell expansion, and skews T-cell repertoire in multiple myeloma. Blood 128, 384–394 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Drent E, Themeli M, Poels R, de Jong-Korlaar R, Yuan H, de Bruijn J, Martens ACM, Zweegman S, van de Donk N, Groen RWJ, Lokhorst HM, Mutis T, A Rational Strategy for Reducing On-Target Off-Tumor Effects of CD38-Chimeric Antigen Receptors by Affinity Optimization. Mol Ther 25, 1946–1958 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Frerichs KA, Broekmans MEC, Marin Soto JA, van Kessel B, Heymans MW, Holthof LC, Verkleij CPM, Boominathan R, Vaidya B, Sendecki J, Axel A, Gaudet F, Pillarisetti K, Zweegman S, Adams HC 3rd, Mutis T, van de Donk N, Preclinical Activity of JNJ-7957, a Novel BCMAxCD3 Bispecific Antibody for the Treatment of Multiple Myeloma, Is Potentiated by Daratumumab. Clin Cancer Res 26, 2203–2215 (2020). [DOI] [PubMed] [Google Scholar]
- 31.Groen RWJ, Noort WA, Raymakers RA, Prins H-J, Aalders L, Hofhuis FM, Moerer P, van Velzen JF, Bloem AC, van Kessel B, Rozemuller H, van Binsbergen E, Buijs A, Yuan H, de Bruijn JD, de Weers M, Parren PWHI, Schuringa JJ, Lokhorst HM, Mutis T, Martens ACM, Reconstructing the human hematopoietic niche in immunodeficient mice: opportunities for studying primary multiple myeloma. Blood 120, e9–e16 (2012). [DOI] [PubMed] [Google Scholar]
- 32.Rafiq S, Hackett CS, Brentjens RJ, Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat Rev Clin Oncol 17, 147–167 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brudno JN, Maric I, Hartman SD, Rose JJ, Wang M, Lam N, Stetler-Stevenson M, Salem D, Yuan C, Pavletic S, Kanakry JA, Ali SA, Mikkilineni L, Feldman SA, Stroncek DF, Hansen BG, Lawrence J, Patel R, Hakim F, Gress RE, Kochenderfer JN, T Cells Genetically Modified to Express an Anti-B-Cell Maturation Antigen Chimeric Antigen Receptor Cause Remissions of Poor-Prognosis Relapsed Multiple Myeloma. J Clin Oncol 36, 2267–2280 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cohen AD, Garfall AL, Stadtmauer EA, Melenhorst JJ, Lacey SF, Lancaster E, Vogl DT, Weiss BM, Dengel K, Nelson A, Plesa G, Chen F, Davis MM, Hwang WT, Young RM, Brogdon JL, Isaacs R, Pruteanu-Malinici I, Siegel DL, Levine BL, June CH, Milone MC, B cell maturation antigen-specific CAR T cells are clinically active in multiple myeloma. J Clin Invest 129, 2210–2221 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Green DJ, O’Steen S, Lin Y, Comstock ML, Kenoyer AL, Hamlin DK, Wilbur DS, Fisher DR, Nartea M, Hylarides MD, Gopal AK, Gooley TA, Orozco JJ, Till BG, Orcutt KD, Wittrup KD, Press OW, CD38-bispecific antibody pretargeted radioimmunotherapy for multiple myeloma and other B-cell malignancies. Blood 131, 611–620 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M, Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol 31, 71–75 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wilkie S, van Schalkwyk MC, Hobbs S, Davies DM, van der Stegen SJ, Pereira AC, Burbridge SE, Box C, Eccles SA, Maher J, Dual targeting of ErbB2 and MUC1 in breast cancer using chimeric antigen receptors engineered to provide complementary signaling. J Clin Immunol 32, 1059–1070 (2012). [DOI] [PubMed] [Google Scholar]
- 38.Muller YD, Nguyen DP, Ferreira LMR, Ho P, Raffin C, Valencia RVB, Congrave-Wilson Z, Roth TL, Eyquem J, Van Gool F, Marson A, Perez L, Wells JA, Bluestone JA, Tang Q, The CD28-Transmembrane Domain Mediates Chimeric Antigen Receptor Heterodimerization With CD28. Front Immunol 12, 639818 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sommermeyer D, Hudecek M, Kosasih PL, Gogishvili T, Maloney DG, Turtle CJ, Riddell SR, Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia 30, 492–500 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Turtle CJ, Hanafi LA, Berger C, Gooley TA, Cherian S, Hudecek M, Sommermeyer D, Melville K, Pender B, Budiarto TM, Robinson E, Steevens NN, Chaney C, Soma L, Chen X, Yeung C, Wood B, Li D, Cao J, Heimfeld S, Jensen MC, Riddell SR, Maloney DG, CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest 126, 2123–2138 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nerreter T, Letschert S, Gotz R, Doose S, Danhof S, Einsele H, Sauer M, Hudecek M, Super-resolution microscopy reveals ultra-low CD19 expression on myeloma cells that triggers elimination by CD19 CAR-T. Nat Commun 10, 3137 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nijhof IS, Casneuf T, van Velzen J, van Kessel B, Axel AE, Syed K, Groen RW, van Duin M, Sonneveld P, Minnema MC, Zweegman S, Chiu C, Bloem AC, Mutis T, Lokhorst HM, Sasser AK, van de Donk NW, CD38 expression and complement inhibitors affect response and resistance to daratumumab therapy in myeloma. Blood 128, 959–970 (2016). [DOI] [PubMed] [Google Scholar]
- 43.Krejcik J, Frerichs KA, Nijhof IS, van Kessel B, van Velzen JF, Bloem AC, Broekmans MEC, Zweegman S, van Meerloo J, Musters RJP, Poddighe PJ, Groen RWJ, Chiu C, Plesner T, Lokhorst HM, Sasser AK, Mutis T, van de Donk N, Monocytes and Granulocytes Reduce CD38 Expression Levels on Myeloma Cells in Patients Treated with Daratumumab. Clin Cancer Res 23, 7498–7511 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Garfall AL, Stadtmauer EA, Hwang WT, Lacey SF, Melenhorst JJ, Krevvata M, Carroll MP, Matsui WH, Wang Q, Dhodapkar MV, Dhodapkar K, Das R, Vogl DT, Weiss BM, Cohen AD, Mangan PA, Ayers EC, Nunez-Cruz S, Kulikovskaya I, Davis MM, Lamontagne A, Dengel K, Kerr ND, Young RM, Siegel DL, Levine BL, Milone MC, Maus MV, June CH, Anti-CD19 CAR T cells with high-dose melphalan and autologous stem cell transplantation for refractory multiple myeloma. JCI Insight 4, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rivière I, Brose K, Mulligan RC, Effects of retroviral vector design on expression of human adenosine deaminase in murine bone marrow transplant recipients engrafted with genetically modified cells. Proceedings of the National Academy of Sciences 92, 6733 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Drent E, Groen RWJ, Noort WA, Themeli M, Lammerts van Bueren JJ, Parren PWHI, Kuball J, Sebestyen Z, Yuan H, de Bruijn J, van de Donk NWCJ, Martens ACM, Lokhorst HM, Mutis T, Pre-clinical evaluation of CD38 chimeric antigen receptor engineered T cells for the treatment of multiple myeloma. Haematologica 101, 616 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kamsma D, Bochet P, Oswald F, Alblas N, Goyard S, Wuite GJL, Peterman EJG, Rose T, Single-Cell Acoustic Force Spectroscopy: Resolving Kinetics and Strength of T Cell Adhesion to Fibronectin. Cell Rep 24, 3008–3016 (2018). [DOI] [PubMed] [Google Scholar]
- 48.Sitters G, Kamsma D, Thalhammer G, Ritsch-Marte M, Peterman EJ, Wuite GJ, Acoustic force spectroscopy. Nat Methods 12, 47–50 (2015). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data associated with this study are in the paper or supplementary materials.