

Abstract
Cleavage of proteins in the extracellular milieu, including hormones, growth factors and their receptors, ion channels, and various cell adhesion and extracellular matrix molecules, plays a key role in regulation of cell behavior. Among more than 500 proteolytic enzymes encoded by mammalian genomes, membrane-anchored serine proteases (MASPs), which are expressed on the surface of epithelial cells of all major organs, are excellently suited to mediate signal transduction across the epithelia and are increasingly being recognized as important regulators of epithelial development, function, and disease (1–3). In this minireview, we summarize current knowledge of the in vivo roles of membrane-anchored serine proteases in acquisition and maintenance of some of the defining functions of epithelial tissues, such as barrier formation, ion transport, and sensory perception.
Membrane-Anchored Serine Proteases (MASPs)
The human MASP family currently comprises twenty proteases, with orthologs identified in all vertebrate species analyzed to date (Figure 1). Based on their membrane topology, the majority of the MASPs are classified as type II transmembrane serine proteases (TTSPs) that are anchored to the membrane via a single-pass transmembrane domain located near the amino terminus. This is followed by a variable stem region and an extracellular carboxy-terminal trypsin-like serine protease domain. Based on the sequence similarities between the catalytic domains, TTSPs have been further divided into four distinct sub-families (1). Type I MASPs include three proteases that are composed of an extracellular serine protease domain attached to the membrane via a C terminal transmembrane domain (tryptase γ1) or a glycosylphosphatidylinositol (GPI) anchor (prostasin and testisin).
Figure 1.
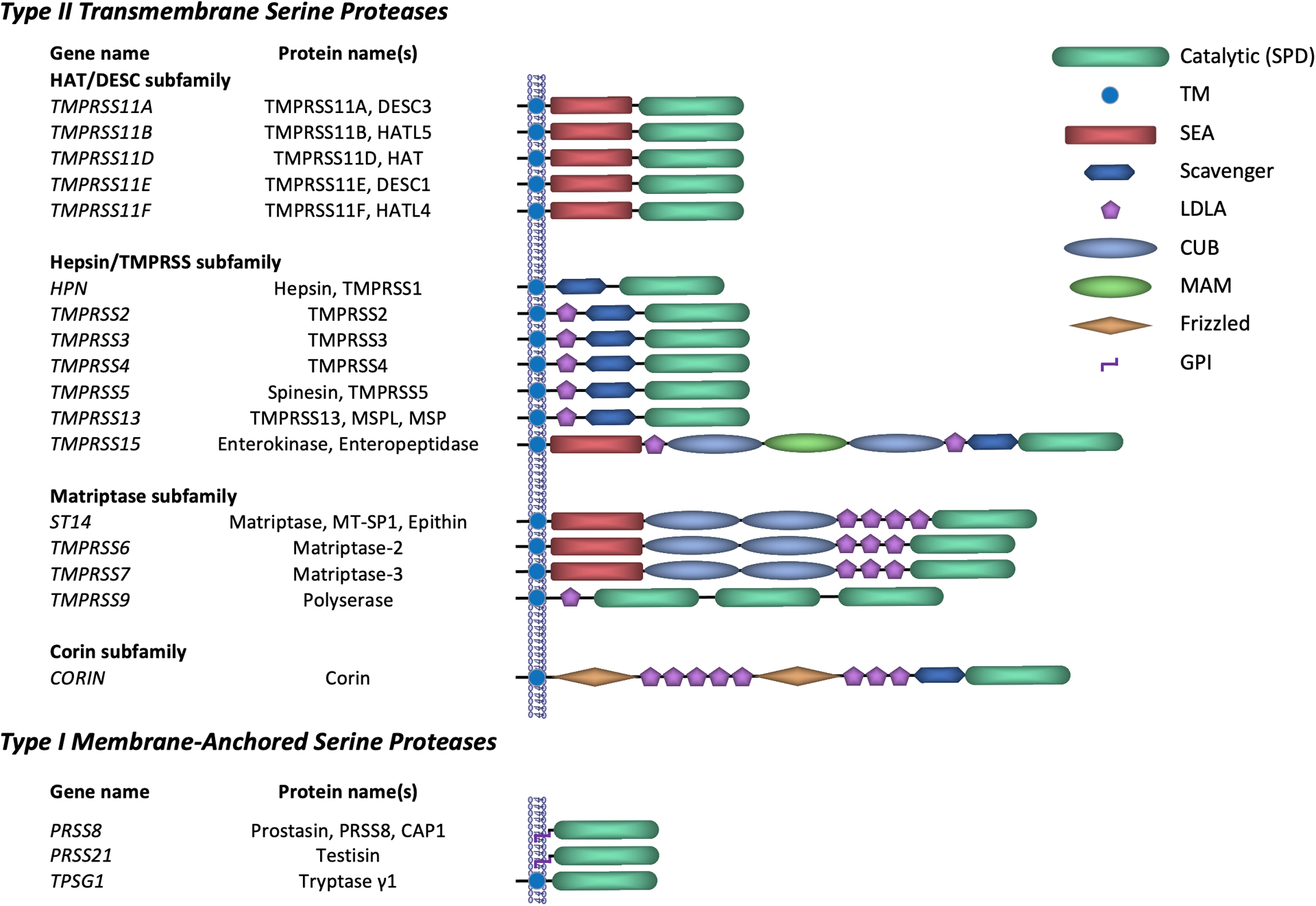
Classification and domain structure of human membrane-anchored serine proteases.
All MASPs belong to the S1 family of serine endoproteases that includes trypsin as the prototypic member and show strong preference for cleavage of substrates after arginine or lysine residues (4). MASPs are synthesized as single-chain zymogens that undergo proteolytic cleavage at a conserved site within the catalytic domain, thus producing a fully active double-chain form (1). While this has long been considered an essential step towards activation of an essentially inactive zymogen, recent findings suggest that at least some MASPs are capable of performing their biological function independent of activation cleavage (5, 6).
MASPs in development and maintenance of epithelial barrier function
Matriptase/prostasin system as general regulator of epithelial barrier function
Epithelia provide barriers that protect tissues from external physical, chemical and microbial insults and maintain fluid homeostasis. The paracellular barrier, which limits permeability between epithelial cells, is critical to maintain transepithelial ion and nutrient gradients, and is a principal defense mechanism preventing entry of pathogens and toxic substances (7). The barrier is primarily regulated at the level of tight junctions (TJs) that are expressed at the apical margin of epithelial junctional complexes and restrict paracellular diffusion (7, 8). The notion that proteolysis may be involved in the regulation of paracellular barrier function was suggested based on the observation that a mild treatment of cultured epithelial cells with endoproteases leads to rapid assembly of TJ strands and strengthening of the barrier (9, 10).
The TTSP matriptase and its proposed activator, prostasin, are expressed in the epithelial compartments of most human and mouse tissues (11). The critical role of the prostasin/matriptase system in epithelial barrier formation was first inferred from the study of genetically-modified mice (Table 1). Mice lacking matriptase develop to term but exhibit abnormal terminal differentiation of epidermal keratinocytes, and loss of epidermal barrier function that results in fatal postnatal dehydration (12, 13). These phenotypes closely match those observed in mice lacking epidermal prostasin, consistent with a proposed functional interaction between the two proteases during skin development (14, 15).
Table 1.
Defects in epithelial barrier function resulting from loss of matriptase activity
| Organism | Loss of matriptase function | Phenotype | Barrier defect |
|---|---|---|---|
| Human | |||
| ARIH (OMIM#602400) (23) | Expected partial to complete; all tissues | Skin - ichthyosis, hypotrichosis, dysplastic hair Teeth - enamel notching/pitting, conical primary teeth Eye - Comeal opacity, photophobia |
|
| Mouse | |||
| St14−/−(13,14) | Null, all tissues | Perinatal lethality within 48h after birth Skin - Defects in stratum corneum development, ichthyosis, hair follicle hypoplasia, lack of whiskers Thymus - increased thymocyte apoptosis |
Increased transepidermal fluid loss and diffusion of topically or intradermally injected tracers, loss of TJ localization of Z01 |
| St14−/− in PAR2-deficient background (17) | Null, all tissues | Embryonic lethality Placenta - underdevelopment of labyrinth layer |
Increased transplacental diffusion, decreased expression of claudin 1 |
| Vil-Cre;St14fl/fl (14,29) | Null, epithelium of Gl tract | Growth retardation, diarrhea, shortened lifespan Colon - Edema, loss of crypt architecture, severe inflammation | Increased transepithelial diffusion of intestinal luminal biotin into tissue stroma |
| MMTV-Cre;St14fl/fl (14,17) | Null, epithelium of salivary glands | Loss of saliva production | Increased transepithelial electrical conductance in salivary gland, loss of membrane localization of claudin 3 |
| βAct-CreER™;St14fl/fl (14) | Inducible loss of expression, all tissues | Rapid loss of weight and viability Skin - loss of hair, ichthyosis Colon - edema, loss of crypt structure, inflammation |
Skin - decreased expression of TJ proteins claudinl, Z01, and occludin Intestine - increased transepithelial diffusion of intestinal luminal dextran into bloodstream |
| St14hypo/hypo (24,27) | Reduced expression, all tissues | Skin - ichthyosis, epidermal acanthosis, impaired desquamation, delayed hair growth, sparse fur | Skin - increased transepidermal fluid loss Intestine - decreased transepithelial electrical resistance, increased diffusion intestinal luminal dextran into bloodstream, increased expression of claudin 2 |
| Horse | |||
| ST14:c.388G>T (25) | Null, all tissues | Skin - dry and scaly, complete alopecia Thymus - lack of cortico-medullary organization, absence of Hassal corpuscles Spleen, lymph nodes - abnormal or absent T cell zones |
|
| Fruit fly | |||
| NpP6/NpC2 (26) | Null, all tissues | Lack of airway fluid clearance, embryonic lethality | Trachea - increased diffusion of dextran the lumen |
Role of matriptase in other epithelia has been documented using tissue-specific mouse knockouts. Inactivation of the protease results in a loss of saliva production by salivary glands and a disruption of normal tissue architecture within large intestine that results in diarrhea, growth retardation and shortened life span (13, 16). In both organs, elimination of matriptase expression leads to a diminished epithelial barrier function (Table 1) (13, 16). Inactivation of matriptase also significantly increased leakiness of placental epithelia and led to embryonic lethality in mice lacking the G protein-coupled receptor PAR2 (17). Mice and rats carrying hypomorphic mutations in Prss8, encoding prostasin, also exhibit defects in intestinal and placental function, although it is not clear whether any of these defects are related to altered matriptase activity (18–21).
In humans and in horses, loss of function mutations in ST14, encoding matriptase, have been linked to autosomal recessive ichthyosis with hypotrichosis (OMIM#602400) and Naked Foal Syndrome, respectively (22, 23). Both diseases present with skin defects that include mild to moderate ichthyosis, indicative of compromised epidermal barrier function (Table 1). Finally, the matriptase and prostasin homologs, Notopleural and Tracheal-prostasin, are essential for establishment of airway barrier function and embryonic survival in Drosophila (see below) (24). Consistent with the in vivo studies, inhibition or elimination of matriptase activity results in decreased ability of cultured intestinal and kidney epithelial cells to establish and maintain fully functional barriers (25, 26).
Despite the overwhelming evidence for the importance of matriptase for epithelial barrier development, the molecular mechanisms underlying this function remain unknown. Loss of matriptase expression leads to a decreased expression and/or recruitment of several barrier-promoting tight junction-associated proteins, including claudin-1, occludin, and ZO-1, while also increasing expression of permeability-associated protein claudin-2 both in mouse epithelia and in cultured human intestinal epithelial cells (13, 17, 25, 27). Therefore, it is plausible to hypothesize that matriptase promotes epithelial barrier function by regulating assembly and/or stability of barrier-forming junctions. However, as of now, no evidence has been presented to indicate that barrier stability is regulated by a proteolytic cleavage of any of the core components of epithelial junctional complexes.
Although the ability of matriptase to cleave and to regulate the activity of a number of biologically active molecules in vitro is well documented, attempts to validate the involvement of any of these substrates in matriptase-mediated regulation of epithelial development and barrier formation have been unsuccessful. Genetic inactivation of either hepatocyte growth factor (HGF) receptor, cMet, or a G-protein coupled receptor, PAR-2, prevent matriptase-driven tumorigenesis in mouse skin, indicating the ability of matriptase to stimulate HGF- and PAR-2-mediated cell signaling in vivo (28, 29). PAR-2 inactivation also rescued ichthyosis in mice overexpressing prostasin in skin and matriptase-induced defects in epidermal development in zebrafish embryos (30, 31). Likewise, the epithelial sodium channel (ENaC) is activated by prostasin or matriptase in Xenopus oocytes and in mice (see also “MASPs in regulation of transepithelial ion transport“ below) (32). However, loss of PAR-2, HGF or ENaC in mice does not appear to reproduce any of the phenotypes observed in matriptase-deficient mice, indicating that activation of any of these three molecules does not critically contribute to matriptase-mediated formation of epithelial barrier function (33–36).
Recently, epithelial cell adhesion molecule (EpCAM) was proposed to be a pathogenic substrate for matriptase during intestinal development. Inactivating mutations in EPCAM are found in patients with congenital tufting enteropathy (CTE, OMIM #613217), an early-onset severe intestinal insufficiency characterized by epithelial dysplasia and villous atrophy leading to chronic watery diarrhea and failure to thrive (37). EpCAM is efficiently cleaved by matriptase in cell culture systems, and loss of function mutations in SPINT2, encoding the endogenous inhibitor of the matriptase/prostasin pathway, HAI-2, are responsible for the syndromic form of CTE (OMIM #270420) (38, 39). Furthermore, two recently generated HAI-2-deficient mouse models exhibit severe intestinal insufficiency that resembles that of CTE patients, including compromised intestinal epithelial barrier and loss of EpCAM protein expression (40, 41). In one of the models, early onset intestinal defects caused by HAI-2 deficiency were suppressed by genetic inactivation of matriptase, indicating that the intestinal failure in this model of CTE is, in part, matriptase-driven (42). It was therefore suggested that an increased matriptase-mediated cleavage and subsequent degradation of EpCAM in mice and humans lacking functional HAI-2 may be involved in the etiology of the disease. However, whether increased cleavage of EpCAM indeed drives intestinal failure CTE in HAI-2-deficient mice and humans remains to be determined. Furthermore, it is not clear if EpCAM processing has a physiological role in matriptase-mediated barrier acquisition in HAI-2-competent tissues during normal development.
Epidermal barrier formation – Tmprss13 and Tmprss11f
In addition to matriptase/prostasin pathway, the TTSPs TMPRSS13 and TMPRSS11F contribute to epidermal barrier formation in mouse skin. Both proteases are expressed in a wide array of mouse and human tissues, with high levels detected in keratinized epithelia of skin, oral cavity and upper digestive system (43–46). Both Tmprss13- and Tmprss11f-deficient newborn mice present with significantly increased transepidermal fluid loss, indicative of compromised epidermal barrier function (44, 47, 48). The rate of fluid loss is relatively small compared to that observed in mice lacking either matriptase or prostasin, and it does not affect postnatal development or long-term survival of Tmprss13- or Tmprss11f-deficient mice, suggesting that at least in the absence of additional challenge, the individual contribution of the two proteases to the establishment of epidermal barrier is relatively minor. Loss of TMPRSS11F did not alter the histological appearance of the epidermis, including stratum corneum, or the expression of any of the major components of cornified envelope, whereas newborn Tmprss13-deficient mice exhibit reduced stratum corneum thickness, but no obvious defects in TJ function or profilaggrin processing analogous to the ones observed in matriptase- and prostasin-deficient epidermis (44, 48). It is therefore unclear whether the two proteases contribute to the formation of epidermal barrier function either in parallel or as one of several functionally relevant targets acting downstream of the matriptase/prostasin system.
MASPs in regulation of transepithelial ion transport
Activation of the epithelial sodium channel - prostasin, matriptase, TMPRSS4
Directional transport of ions and water across epithelial barriers is essential for maintaining tissue homeostasis and is a critical function of all polarized epithelia. The epithelial sodium channel (ENaC) is a key component of sodium (Na+) transport across high resistance epithelia and is crucial for salt tasting in tongue epithelium and sodium reabsorption in kidney, lungs and intestines, thus regulating blood pressure, blood potassium levels, and airway and alveolar surface liquid volumes (32). A number of extracellular proteases of serine, cysteine, and metalloproteinase families, including membrane-tethered serine proteases prostasin, matriptase, TMPRSS4, and TMPRSS3, have been shown to activate ENaC in a variety of in vitro and in vivo systems by proteolytic cleavage and removal of an inhibitory tract from its γ subunit (32, 49).
The importance of prostasin-dependent activation of ENaC has been demonstrated using a series of tissue-specific prostasin-deficient mice. In lungs, the level of airway surface liquid (ASL) that covers the apical surface of the airway epithelium is critical for alveolar function and is maintained by moving ASL across the epithelium in a process that is facilitated by ENaC-mediated sodium absorption (50). Mice lacking prostasin in alveolar epithelial cells show 40 % decrease in ENaC-mediated sodium currents and a reduced sodium-driven alveolar clearance, leading to a fluid accumulation in an experimental model of hydrostatic pulmonary edema (51). Aberrant activity of channel-activating proteases may also be involved in the etiology of pulmonary fibrosis. TMPRSS4 and matriptase are upregulated during bleomycin-induced lung fibrosis in mice, and genetic ablation of either of the two proteases or treatment with the serine protease inhibitor, camostat mesilate, attenuates development of the disease (52, 53). Similarly, intratracheal administration of camostat mesilate inhibits ENaC activity in guinea pigs and enhances mucociliary clearance in sheep (54). Furthermore, in cultured primary airway epithelial cells from cystic fibrosis patients, who often suffer from mucus plugging and chronic bacterial infection due to reduced ASL, inhibition of ENaC-activating proteases increased ASL height and improved mucociliary function (55).
Essential functions of ENaC-mediated sodium transport in kidneys have been documented in patients with hypertension associated with inherited forms of hypokalemia (Liddle syndrome, OMIM#177200), or hypotension associated with hyperkalemia (pseudohypoaldosteronism type 1, OMIM#264350), that carry, respectively, gain-of-function and loss-of-function mutations in genes encoding ENaC subunits (56). Although kidney tubule-specific prostasin knockout has not yet been described, the administration of the non-selective serine protease inhibitors, aprotinin and camostat mesilate both led to an increased excretion of sodium in mice and reduced blood pressure and renal injury in a salt-sensitive hypertension rat model, consistent with serine protease-dependent activation of ENaC (57, 58). Reduced ENaC-mediated sodium transport has also been reported in the colon of mice that carry a partial loss-of-function mutation or intestine-specific inactivation of the prostasin gene (18, 20).
Whether regulation of sodium transport plays a role in the barrier-promoting function of the matriptase/prostasin system is not clear. However, barrier acquisition did not appear to be significantly affected in alveolar epithelial cells or colons of prostasin-deficient mice, indicating that two of the proposed physiological functions of prostasin, establishment of epithelial barrier and regulation of transepithelial ion transport, may not be directly linked (20, 51).
Regulation of blood pressure and kidney function – corin, hepsin
The TTSP corin is essential for proteolytic activation of pro-atrial natriuretic peptide (pro-ANP), a pro-form of a cardiac hormone ANP that regulates water-salt balance and blood pressure by promoting excretion of both sodium and water by the kidneys (59). Loss-of-function mutations in CORIN are associated with hypertension, edema and heart failure in humans, and with impaired renal sodium excretion, salt-sensitive hypertension, water retention and cardiac hypertrophy in mice (59–61). While natriuretic peptides and corin are mainly produced by the heart, both pro-ANP and corin are expressed in several non-cardiac tissues involved in blood pressure regulation, including placenta and kidney (62, 63). In the kidney, corin is expressed at the apical membrane of epithelial cells along the entire length of the nephron, where it is believed to process filtered and/or locally produced pro-ANP (63). Mature ANP decreases sodium reabsorption and urine-concentrating capacity by inhibiting several sodium transport systems, most notably the Na-K-ATPase, Na/H exchanger, and type IIa Na-Pi cotransporter in the proximal tubule, and by inhibiting Cl- transport via decreasing apical membrane expression of Na-K-2Cl cotransporter (NKCC2) in the distal tubule (Figure 2) (64). Natriuretic and diuretic effects of ANP also involve suppression of both ENaC-mediated sodium reabsorption and vasopressin-stimulated water reabsorption in collecting ducts of the kidney (65). Consistent with this, impaired sodium excretion and increased water retention in corin-deficient mice were suppressed by treatment with an ENaC inhibitor, amiloride (60).
Figure 2.
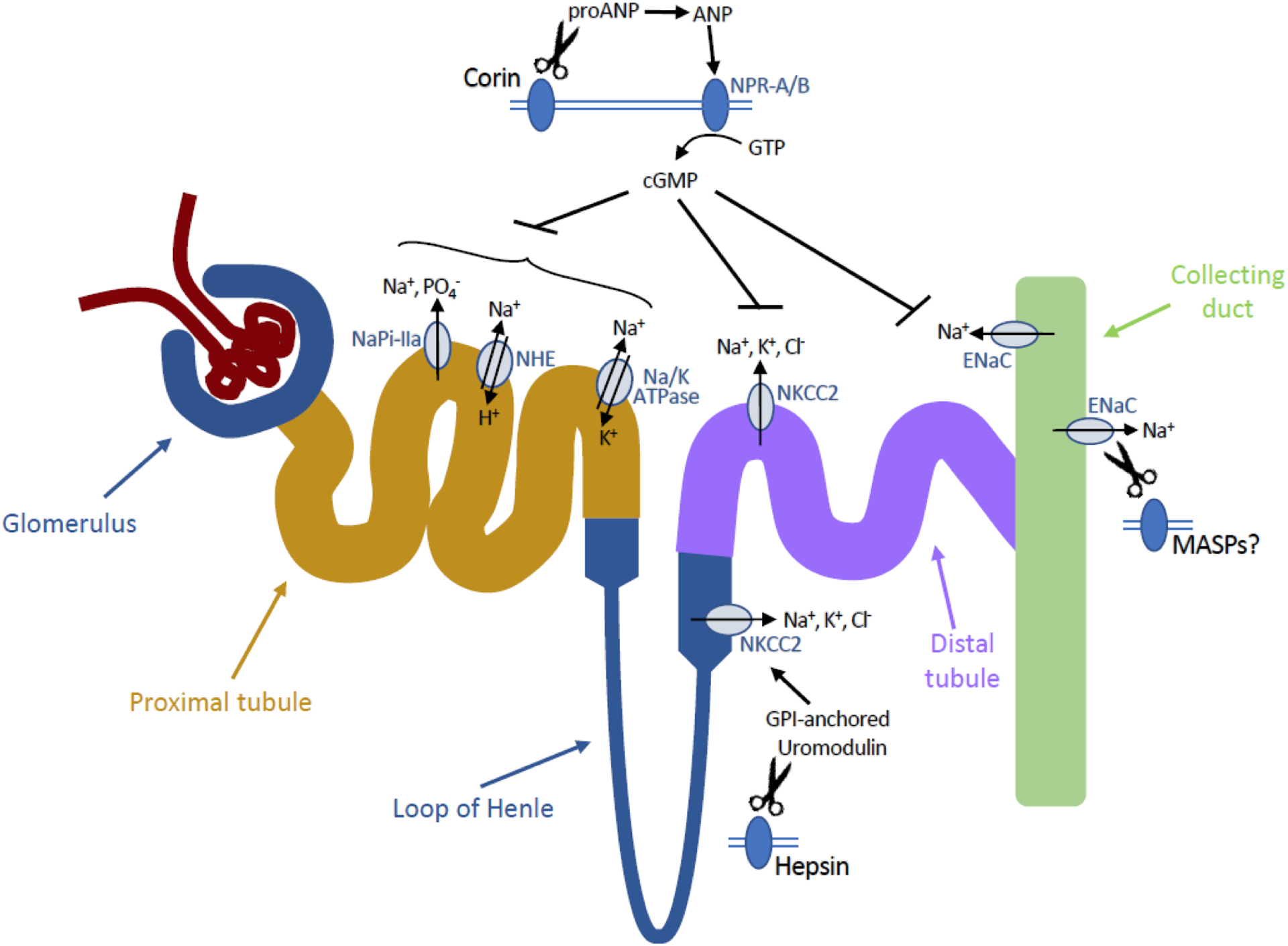
Proposed roles of MASPs in regulation of ion transport in the kidneys. Corin-generated ANP binds to its receptor NPR-A and enhances its guanylyl cyclase activity. CyclicGMP prevents sodium reabsorption by inhibiting activity of several ion transport proteins in different segments of nephron. Hepsin mediates shedding of the GPI-anchored protein uromodulin, leading to a decreased activity of Na+, K+, Cl− cotransporter NKCC2 in the thick ascending limb. Finally, several MASPs, including prostasin, matriptase, and TMPRSS4, have been proposed to activate the epithelial sodium channel ENaC, suggesting their possible involvement in sodium reabsorption in the distal tubules and collecting ducts. Abbreviations: ANP, atrial natriuretic peptide; cGMP, cyclic guanosine monophosphate; ENaC, epithelial sodium channel; GPI, glycosylphosphatidylinositol, GTP, guanosine triphosphate; Na/K ATPase, sodium-potassium adenosine triphosphatase; NaPi-IIa, sodium-phosphate cotransporter IIa; NHE, sodium-proton exchanger; NKCC2, sodium-potassium-chloride cotransporter; NPR, natriuretic peptide receptor.
Corin mutations that impair the natriuretic peptide-processing activity have also been identified in patients with pregnancy-associated hypertension (preeclampsia) (62). Corin expression was detected in decidual cells in pregnant uterus in mice and in humans, and within late secretory endometrium and in villous cytotrophoblasts from first trimester placenta, and uterine corin expression was significantly lower in individuals with pre-eclampsia, compared to normal pregnancies (62, 66, 67). Histological analysis of placentas from corin-deficient mice and patients with pre-eclampsia revealed delayed trophoblast invasion and impaired spiral artery remodeling in the uterus (62, 68, 69). Pregnant mice lacking corin or ANP develop high blood pressure and proteinuria that is not prevented by heart-specific re-expression of corin that restored pro-ANP processing in the heart and normalized blood pressure in non-pregnant mice, suggesting that decreased local uterine, rather than cardiac, corin activity is critical for development of pre-eclampsia (62, 70).
Hepsin regulates sodium transport and homeostasis in the kidney via processing of the GPI-anchored, zona pellucida-type protein, uromodulin, that is expressed on the apical surface of kidney epithelial cells within the thick ascending limb of loop of Henle (71, 72). Mutations in the uromodulin gene cause medullary cystic kidney disease 2 (OMIM #603860) and familial juvenile hyperuricemic nephropathy (OMIM#162000) and have been associated with increased risk of chronic kidney disease, calcium stones, and hypertension (73–75). Uromodulin appears to modulate ion reabsorption by regulating the surface abundance and activity of the sodium, potassium, chloride cotransporter, NKCC2, and the potassium channel, ROMK2 (76, 77). Hepsin mediates proteolytic cleavage and subsequent shedding of uromodulin from the cell surface, and loss of hepsin function leads to hyperactivation of the NKCC2 transporter and increased sodium and water reabsorption (Figure 2) (71, 72). Prolonged exposure of hepsin-deficient mice to high-salt diet resulted in severe deterioration of renal function with salt wasting and subsequent rise in plasma osmolarity, hypocalcemia and hypomagnesemia, underscoring the importance of hepsin-mediated processing of uromodulin in the regulation of salt homeostasis and kidney function (72).
Development of hearing - TMPRSS3, hepsin
The ability to perceive sound depends on proper functioning of the cochlea, a spiral organ within the inner ear with a specialized epithelium that contains electro conducting hair cells for detecting vibrations from the outer and middle ear and converting them into nerve impulses (78). Loss-of-function mutations in TMPRSS3 causes non-syndromic autosomal recessive deafness (DFNB8/10, OMIM#601072), characterized by early onset, progressive, bilateral hearing loss (79). Similarly, mice lacking a functional Tmprss3 protein exhibit severe loss of hearing associated with rapid degeneration of cochlear and vestibular hair cells (80). Prior to degeneration, loss of Tmprss3 reduced the number of Ca2+-activated K+ (BK) channels and decreased expression of several intracellular calcium-binding proteins in cultured primary hair cells, and decreased expression of the α subunit of the BK channel and a loss of BK-dependent fast K+ conductance within the cochlea of Tmprss3-deficient mice (81, 82). Furthermore, activation of the wildtype, but not the deafness-causing variants of Tmprss3 increases amiloride-sensitive sodium transport in a Xenopus oocyte expression system, indicating that the protease could potentially be involved in activation of inner ear-expressed ENaC (49). However, pseudohypoaldosteronism type 1 patients carrying inactivating mutations in the alpha subunit of ENaC have normal hearing, thus challenging the notion that ENaC activation is crucial for auditory system development (83). Interestingly, deficiency in hepsin also leads to loss of hearing function in mice and is associated with reduced levels of BK channels in the sensory hair cells, indicating that the two proteases may regulate development of hearing function via overlapping mechanisms, possibly even as parts of the same proteolytic cascade regulating ion transport in the inner ear (84).
MASPs in extracellular matrix remodeling – lessons from Drosophila
Drosophila Notopleural and Trp, which encode proteases homologous to, respectively, human matriptase and prostasin, have recently been shown to be essential for degradation of intraluminal chitin in the trachea of Drosophila, leading to a defect in barrier function, impaired tracheal liquid clearance and death before hatching (24). Notopleural mutant and Trp knockdown embryos show defects in cleavage of apical ECM zona pellucida (ZP) protein Dumpy, which plays an important role in attachment of the epithelium to the exoskeleton at later stages of Drosophila development (85). Interestingly, depletion of Stubble (Sb-sbd), encoding a TTSP homologous to Notopleural, impairs degradation of Dumpy in epithelial cells within imaginal discs during morphogenesis, and therefore inhibits elongation and expansion of wings and legs (86). Although Dumpy does not have a homolog in mammalian systems, the recent discovery of hepsin-mediated cleavage of another ZP protein, uromodulin, in mammalian kidney (see above) indicates that cleavage of ZP domain-containing proteins may present an evolutionary-conserved means of regulating epithelial function by cell surface-linked proteolysis.
MASPs in viral processing
Respiratory epithelium is a point of entry into the organism for a number of pathogens, including viruses. Many enveloped viruses, including influenza A and B, depend on a cleavage of hemaglutinin (HA)-type surface glycoproteins for viral entry into the target cell (87, 88). Several trypsin-like serine proteases, including airway epithelium-expressed TTSPs TMPRSS2, TMPRSS4, TMPRSS13, TMPRSS11A, TMPRSS11D/HAT, TMPRSS11E/DESC1, and matriptase, activate influenza HA and promote infectivity in cell culture systems (88, 89). Recently, TMPRSS2 was identified as a susceptibility gene for Severe 2009 Pandemic A(H1N1) and A(H7N9) Influenza, and inactivation of either TMPRSS2 or TMPRSS4 confers partial to complete resistance to the spread and pathogenesis of influenza virus in mice (90–95). Surface proteins of several other types of viruses, including Middle East respiratory syndrome coronavirus (MERS-CoV), severe acute respiratory syndrome (SARS-CoV), parainfluenza viruses, human metapneumovirus (HMPV), and Sendai virus (SeV), undergo TTSP-mediated activation cleavage in culture, indicating a potential widespread role of membrane-anchored serine proteases in viral respiratory illnesses that makes these enzymes potential targets for new antiviral therapies (96–102).
Perspectives
Importance of the field: Membrane-anchored serine proteases are critical regulators of epithelial physiology. Changes in MASP expression and/or activity underlie the etiology of numerous diseases and developmental defects in both humans and in animal models.
Current thinking: Localization to the cell surface makes MASPs ideal instruments of regulation of cell behavior by extracellular stimuli. Significant progress has been made in recent years to uncover some of the molecular mechanisms of MASP-mediated regulation of epithelial function, including activation of proANP in kidney and placenta, processing of zona pellucida proteins in kidney and tracheal system in Drosophila, or activation cleavage of viral surface proteins.
Future directions: Imaging and analyzing the activity of individual proteases in living tissues using highly selective antibodies and activity probes will be crucial to provide further insight into physiological and pathological roles of MASPs, as well as into the mechanisms that govern their expression, cell localization, zymogen activation, and modulation of their proteolytic activity by auxiliary proteins. Development of selective inhibitors of matriptase and other MASPs shown to contribute to disease development may open alternative new strategies to treat conditions such as Congenital Tufting Enteropathy or viral respiratory infections.
Acknowledgements
This work was supported by the Intramural Research Program at the National Institute of Dental and Craniofacial Research.
References
- 1.Szabo R, Bugge TH. Membrane-anchored serine proteases in vertebrate cell and developmental biology. Annu Rev Cell Dev Biol. 2011;27:213–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Antalis TM, Conway GD, Peroutka RJ, Buzza MS. Membrane-anchored proteases in endothelial cell biology. Curr Opin Hematol. 2016;23(3):243–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Martin CE, List K. Cell surface-anchored serine proteases in cancer progression and metastasis. Cancer Metastasis Rev. 2019;38(3):357–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Netzel-Arnett S, Hooper JD, Szabo R, Madison EL, Quigley JP, Bugge TH, et al. Membrane anchored serine proteases: a rapidly expanding group of cell surface proteolytic enzymes with potential roles in cancer. Cancer Metastasis Rev. 2003;22(2–3):237–58. [DOI] [PubMed] [Google Scholar]
- 5.Friis S, Madsen DH, Bugge TH. Distinct Developmental Functions of Prostasin (CAP1/PRSS8) Zymogen and Activated Prostasin. J Biol Chem. 2016;291(6):2577–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Friis S, Tadeo D, Le-Gall SM, Jurgensen HJ, Sales KU, Camerer E, et al. Matriptase zymogen supports epithelial development, homeostasis and regeneration. BMC Biol. 2017;15(1):46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marchiando AM, Graham WV, Turner JR. Epithelial barriers in homeostasis and disease. Annu Rev Pathol. 2010;5:119–44. [DOI] [PubMed] [Google Scholar]
- 8.Garcia MA, Nelson WJ, Chavez N. Cell-Cell Junctions Organize Structural and Signaling Networks. Cold Spring Harb Perspect Biol. 2018;10(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cohen E, Talmon A, Faff O, Bacher A, Ben-Shaul Y. Formation of tight junctions in epithelial cells. I. Induction by proteases in a human colon carcinoma cell line. Exp Cell Res. 1985;156(1):103–16. [DOI] [PubMed] [Google Scholar]
- 10.Ronaghan NJ, Shang J, Iablokov V, Zaheer R, Colarusso P, Dion S, et al. The serine protease-mediated increase in intestinal epithelial barrier function is dependent on occludin and requires an intact tight junction. Am J Physiol Gastrointest Liver Physiol. 2016;311(3):G466–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.List K, Hobson JP, Molinolo A, Bugge TH. Co-localization of the channel activating protease prostasin/(CAP1/PRSS8) with its candidate activator, matriptase. J Cell Physiol. 2007;213(1):237–45. [DOI] [PubMed] [Google Scholar]
- 12.List K, Haudenschild CC, Szabo R, Chen W, Wahl SM, Swaim W, et al. Matriptase/MT-SP1 is required for postnatal survival, epidermal barrier function, hair follicle development, and thymic homeostasis. Oncogene. 2002;21(23):3765–79. [DOI] [PubMed] [Google Scholar]
- 13.List K, Kosa P, Szabo R, Bey AL, Wang CB, Molinolo A, et al. Epithelial integrity is maintained by a matriptase-dependent proteolytic pathway. Am J Pathol. 2009;175(4):1453–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leyvraz C, Charles RP, Rubera I, Guitard M, Rotman S, Breiden B, et al. The epidermal barrier function is dependent on the serine protease CAP1/Prss8. J Cell Biol. 2005;170(3):487–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peters DE, Szabo R, Friis S, Shylo NA, Uzzun Sales K, Holmbeck K, et al. The membrane-anchored serine protease prostasin (CAP1/PRSS8) supports epidermal development and postnatal homeostasis independent of its enzymatic activity. J Biol Chem. 2014;289(21):14740–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yin H, Kosa P, Liu X, Swaim WD, Lai Z, Cabrera-Perez J, et al. Matriptase deletion initiates a Sjogren’s syndrome-like disease in mice. PLoS One. 2014;9(2):e82852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Szabo R, Peters DE, Kosa P, Camerer E, Bugge TH. Regulation of feto-maternal barrier by matriptase- and PAR-2-mediated signaling is required for placental morphogenesis and mouse embryonic survival. PLoS Genet. 2014;10(7):e1004470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Frateschi S, Keppner A, Malsure S, Iwaszkiewicz J, Sergi C, Merillat AM, et al. Mutations of the serine protease CAP1/Prss8 lead to reduced embryonic viability, skin defects, and decreased ENaC activity. Am J Pathol. 2012;181(2):605–15. [DOI] [PubMed] [Google Scholar]
- 19.Hummler E, Dousse A, Rieder A, Stehle JC, Rubera I, Osterheld MC, et al. The channel-activating protease CAP1/Prss8 is required for placental labyrinth maturation. PLoS One. 2013;8(2):e55796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Malsure S, Wang Q, Charles RP, Sergi C, Perrier R, Christensen BM, et al. Colon-specific deletion of epithelial sodium channel causes sodium loss and aldosterone resistance. J Am Soc Nephrol. 2014;25(7):1453–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Keppner A, Malsure S, Nobile A, Auberson M, Bonny O, Hummler E. Altered Prostasin (CAP1/Prss8) Expression Favors Inflammation and Tissue Remodeling in DSS-induced Colitis. Inflamm Bowel Dis. 2016;22(12):2824–39. [DOI] [PubMed] [Google Scholar]
- 22.Bauer A, Hiemesch T, Jagannathan V, Neuditschko M, Bachmann I, Rieder S, et al. A Nonsense Variant in the ST14 Gene in Akhal-Teke Horses with Naked Foal Syndrome. G3 (Bethesda). 2017;7(4):1315–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Basel-Vanagaite L, Attia R, Ishida-Yamamoto A, Rainshtein L, Ben Amitai D, Lurie R, et al. Autosomal recessive ichthyosis with hypotrichosis caused by a mutation in ST14, encoding type II transmembrane serine protease matriptase. American Journal of Human Genetics. 2007;80(3):467–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Drees L, Konigsmann T, Jaspers MHJ, Pflanz R, Riedel D, Schuh R. Conserved function of the matriptase-prostasin proteolytic cascade during epithelial morphogenesis. PLoS Genet. 2019;15(1):e1007882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Buzza MS, Netzel-Arnett S, Shea-Donohue T, Zhao A, Lin CY, List K, et al. Membrane-anchored serine protease matriptase regulates epithelial barrier formation and permeability in the intestine. Proc Natl Acad Sci U S A. 2010;107(9):4200–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gray K, Elghadban S, Thongyoo P, Owen KA, Szabo R, Bugge TH, et al. Potent and specific inhibition of the biological activity of the type-II transmembrane serine protease matriptase by the cyclic microprotein MCoTI-II. Thromb Haemost. 2014;112(2):402–11. [DOI] [PubMed] [Google Scholar]
- 27.Kosa P, Szabo R, Molinolo AA, Bugge TH. Suppression of Tumorigenicity-14, encoding matriptase, is a critical suppressor of colitis and colitis-associated colon carcinogenesis. Oncogene. 2012;31(32):3679–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Szabo R, Rasmussen AL, Moyer AB, Kosa P, Schafer JM, Molinolo AA, et al. c-Met-induced epithelial carcinogenesis is initiated by the serine protease matriptase. Oncogene. 2011;30(17):2003–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sales KU, Friis S, Konkel JE, Godiksen S, Hatakeyama M, Hansen KK, et al. Non-hematopoietic PAR-2 is essential for matriptase-driven pre-malignant progression and potentiation of ras-mediated squamous cell carcinogenesis. Oncogene. 2015;34(3):346–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schepis A, Barker A, Srinivasan Y, Balouch E, Zheng Y, Lam I, et al. Protease signaling regulates apical cell extrusion, cell contacts, and proliferation in epithelia. J Cell Biol. 2018;217(3):1097–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Frateschi S, Camerer E, Crisante G, Rieser S, Membrez M, Charles RP, et al. PAR2 absence completely rescues inflammation and ichthyosis caused by altered CAP1/Prss8 expression in mouse skin. Nat Commun. 2011;2:161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kleyman TR, Eaton DC. Regulating ENaC’s gate. Am J Physiol Cell Physiol. 2020;318(1):C150–C62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Adams MN, Ramachandran R, Yau MK, Suen JY, Fairlie DP, Hollenberg MD, et al. Structure, function and pathophysiology of protease activated receptors. Pharmacol Ther. 2011;130(3):248–82. [DOI] [PubMed] [Google Scholar]
- 34.Hummler E, Barker P, Gatzy J, Beermann F, Verdumo C, Schmidt A, et al. Early death due to defective neonatal lung liquid clearance in alpha-ENaC-deficient mice. Nat Genet. 1996;12(3):325–8. [DOI] [PubMed] [Google Scholar]
- 35.Schmidt C, Bladt F, Goedecke S, Brinkmann V, Zschiesche W, Sharpe M, et al. Scatter factor/hepatocyte growth factor is essential for liver development. Nature. 1995;373(6516):699–702. [DOI] [PubMed] [Google Scholar]
- 36.Uehara Y, Minowa O, Mori C, Shiota K, Kuno J, Noda T, et al. Placental defect and embryonic lethality in mice lacking hepatocyte growth factor/scatter factor. Nature. 1995;373(6516):702–5. [DOI] [PubMed] [Google Scholar]
- 37.Sivagnanam M, Mueller JL, Lee H, Chen Z, Nelson SF, Turner D, et al. Identification of EpCAM as the gene for congenital tufting enteropathy. Gastroenterology. 2008;135(2):429–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu CJ, Feng X, Lu M, Morimura S, Udey MC. Matriptase-mediated cleavage of EpCAM destabilizes claudins and dysregulates intestinal epithelial homeostasis. J Clin Invest. 2017;127(2):623–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Heinz-Erian P, Muller T, Krabichler B, Schranz M, Becker C, Ruschendorf F, et al. Mutations in SPINT2 cause a syndromic form of congenital sodium diarrhea. Am J Hum Genet. 2009;84(2):188–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Szabo R, Bugge TH. Loss of HAI-2 in mice with decreased prostasin activity leads to an early-onset intestinal failure resembling congenital tufting enteropathy. PLoS One. 2018;13(4):e0194660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kawaguchi M, Yamamoto K, Takeda N, Fukushima T, Yamashita F, Sato K, et al. Hepatocyte growth factor activator inhibitor-2 stabilizes Epcam and maintains epithelial organization in the mouse intestine. Commun Biol. 2019;2:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Szabo R, Callies LK, Bugge TH. Matriptase drives early-onset intestinal failure in a mouse model of congenital tufting enteropathy. Development. 2019;146(22). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim DR, Sharmin S, Inoue M, Kido H. Cloning and expression of novel mosaic serine proteases with and without a transmembrane domain from human lung. Biochim Biophys Acta. 2001;1518(1–2):204–9. [DOI] [PubMed] [Google Scholar]
- 44.Madsen DH, Szabo R, Molinolo AA, Bugge TH. TMPRSS13 deficiency impairs stratum corneum formation and epidermal barrier acquisition. Biochem J. 2014;461(3):487–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sales KU, Hobson JP, Wagenaar-Miller R, Szabo R, Rasmussen AL, Bey A, et al. Expression and genetic loss of function analysis of the HAT/DESC cluster proteases TMPRSS11A and HAT. PLoS One. 2011;6(8):e23261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fagerberg L, Hallstrom BM, Oksvold P, Kampf C, Djureinovic D, Odeberg J, et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol Cell Proteomics. 2014;13(2):397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang Z, Hu Y, Yan R, Dong L, Jiang Y, Zhou Z, et al. The Transmembrane Serine Protease HAT-like 4 Is Important for Epidermal Barrier Function to Prevent Body Fluid Loss. Sci Rep. 2017;7:45262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Callies LK, Tadeo D, Simper J, Bugge TH, Szabo R. Iterative, multiplexed CRISPR-mediated gene editing for functional analysis of complex protease gene clusters. J Biol Chem. 2019;294(44):15987–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guipponi M, Vuagniaux G, Wattenhofer M, Shibuya K, Vazquez M, Dougherty L, et al. The transmembrane serine protease (TMPRSS3) mutated in deafness DFNB8/10 activates the epithelial sodium channel (ENaC) in vitro. Hum Mol Genet. 2002;11(23):2829–36. [DOI] [PubMed] [Google Scholar]
- 50.Gaillard EA, Kota P, Gentzsch M, Dokholyan NV, Stutts MJ, Tarran R. Regulation of the epithelial Na+ channel and airway surface liquid volume by serine proteases. Pflugers Arch. 2010;460(1):1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Planes C, Randrianarison NH, Charles RP, Frateschi S, Cluzeaud F, Vuagniaux G, et al. ENaC-mediated alveolar fluid clearance and lung fluid balance depend on the channel-activating protease 1. EMBO Mol Med. 2010;2(1):26–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Valero-Jimenez A, Zuniga J, Cisneros J, Becerril C, Salgado A, Checa M, et al. Transmembrane protease, serine 4 (TMPRSS4) is upregulated in IPF lungs and increases the fibrotic response in bleomycin-induced lung injury. PLoS One. 2018;13(3):e0192963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bardou O, Menou A, Francois C, Duitman JW, von der Thusen JH, Borie R, et al. Membrane-anchored Serine Protease Matriptase Is a Trigger of Pulmonary Fibrogenesis. Am J Respir Crit Care Med. 2016;193(8):847–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Coote K, Atherton-Watson HC, Sugar R, Young A, MacKenzie-Beevor A, Gosling M, et al. Camostat attenuates airway epithelial sodium channel function in vivo through the inhibition of a channel-activating protease. J Pharmacol Exp Ther. 2009;329(2):764–74. [DOI] [PubMed] [Google Scholar]
- 55.Reihill JA, Walker B, Hamilton RA, Ferguson TE, Elborn JS, Stutts MJ, et al. Inhibition of Protease-Epithelial Sodium Channel Signaling Improves Mucociliary Function in Cystic Fibrosis Airways. Am J Respir Crit Care Med. 2016;194(6):701–10. [DOI] [PubMed] [Google Scholar]
- 56.Rossier BC, Staub O, Hummler E. Genetic dissection of sodium and potassium transport along the aldosterone-sensitive distal nephron: importance in the control of blood pressure and hypertension. FEBS Lett. 2013;587(13):1929–41. [DOI] [PubMed] [Google Scholar]
- 57.Bohnert BN, Menacher M, Janessa A, Worn M, Schork A, Daiminger S, et al. Aprotinin prevents proteolytic epithelial sodium channel (ENaC) activation and volume retention in nephrotic syndrome. Kidney Int. 2018;93(1):159–72. [DOI] [PubMed] [Google Scholar]
- 58.Maekawa A, Kakizoe Y, Miyoshi T, Wakida N, Ko T, Shiraishi N, et al. Camostat mesilate inhibits prostasin activity and reduces blood pressure and renal injury in salt-sensitive hypertension. J Hypertens. 2009;27(1):181–9. [DOI] [PubMed] [Google Scholar]
- 59.Zhou Y, Wu Q. Corin in natriuretic peptide processing and hypertension. Curr Hypertens Rep. 2014;16(2):415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang W, Shen J, Cui Y, Jiang J, Chen S, Peng J, et al. Impaired sodium excretion and salt-sensitive hypertension in corin-deficient mice. Kidney Int. 2012;82(1):26–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chan JC, Knudson O, Wu F, Morser J, Dole WP, Wu Q. Hypertension in mice lacking the proatrial natriuretic peptide convertase corin. Proc Natl Acad Sci U S A. 2005;102(3):785–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cui Y, Wang W, Dong N, Lou J, Srinivasan DK, Cheng W, et al. Role of corin in trophoblast invasion and uterine spiral artery remodelling in pregnancy. Nature. 2012;484(7393):246–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Polzin D, Kaminski HJ, Kastner C, Wang W, Kramer S, Gambaryan S, et al. Decreased renal corin expression contributes to sodium retention in proteinuric kidney diseases. Kidney Int. 2010;78(7):650–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Theilig F, Wu Q. ANP-induced signaling cascade and its implications in renal pathophysiology. Am J Physiol Renal Physiol. 2015;308(10):F1047–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Guo LJ, Alli AA, Eaton DC, Bao HF. ENaC is regulated by natriuretic peptide receptor-dependent cGMP signaling. Am J Physiol Renal Physiol. 2013;304(7):F930–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yan W, Sheng N, Seto M, Morser J, Wu Q. Corin, a mosaic transmembrane serine protease encoded by a novel cDNA from human heart. J Biol Chem. 1999;274(21):14926–35. [DOI] [PubMed] [Google Scholar]
- 67.Kaitu’u-Lino TJ, Ye L, Tuohey L, Dimitriadis E, Bulmer J, Rogers P, et al. Corin, an enzyme with a putative role in spiral artery remodeling, is up-regulated in late secretory endometrium and first trimester decidua. Hum Reprod. 2013;28(5):1172–80. [DOI] [PubMed] [Google Scholar]
- 68.Red-Horse K, Zhou Y, Genbacev O, Prakobphol A, Foulk R, McMaster M, et al. Trophoblast differentiation during embryo implantation and formation of the maternal-fetal interface. J Clin Invest. 2004;114(6):744–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pijnenborg R, Vercruysse L, Hanssens M. The uterine spiral arteries in human pregnancy: facts and controversies. Placenta. 2006;27(9–10):939–58. [DOI] [PubMed] [Google Scholar]
- 70.Baird RC, Li S, Wang H, Naga Prasad SV, Majdalany D, Perni U, et al. Pregnancy-Associated Cardiac Hypertrophy in Corin-Deficient Mice: Observations in a Transgenic Model of Preeclampsia. Can J Cardiol. 2019;35(1):68–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Brunati M, Perucca S, Han L, Cattaneo A, Consolato F, Andolfo A, et al. The serine protease hepsin mediates urinary secretion and polymerisation of Zona Pellucida domain protein uromodulin. Elife. 2015;4:e08887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Olinger E, Lake J, Sheehan S, Schiano G, Takata T, Tokonami N, et al. Hepsin-mediated Processing of Uromodulin is Crucial for Salt-sensitivity and Thick Ascending Limb Homeostasis. Sci Rep. 2019;9(1):12287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hart TC, Gorry MC, Hart PS, Woodard AS, Shihabi Z, Sandhu J, et al. Mutations of the UMOD gene are responsible for medullary cystic kidney disease 2 and familial juvenile hyperuricaemic nephropathy. J Med Genet. 2002;39(12):882–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Padmanabhan S, Melander O, Johnson T, Di Blasio AM, Lee WK, Gentilini D, et al. Genome-wide association study of blood pressure extremes identifies variant near UMOD associated with hypertension. PLoS Genet. 2010;6(10):e1001177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gudbjartsson DF, Holm H, Indridason OS, Thorleifsson G, Edvardsson V, Sulem P, et al. Association of variants at UMOD with chronic kidney disease and kidney stones-role of age and comorbid diseases. PLoS Genet. 2010;6(7):e1001039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mutig K, Kahl T, Saritas T, Godes M, Persson P, Bates J, et al. Activation of the bumetanide-sensitive Na+,K+,2Cl- cotransporter (NKCC2) is facilitated by Tamm-Horsfall protein in a chloride-sensitive manner. J Biol Chem. 2011;286(34):30200–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Renigunta A, Renigunta V, Saritas T, Decher N, Mutig K, Waldegger S. Tamm-Horsfall glycoprotein interacts with renal outer medullary potassium channel ROMK2 and regulates its function. J Biol Chem. 2011;286(3):2224–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mittal R, Aranke M, Debs LH, Nguyen D, Patel AP, Grati M, et al. Indispensable Role of Ion Channels and Transporters in the Auditory System. J Cell Physiol. 2017;232(4):743–58. [DOI] [PubMed] [Google Scholar]
- 79.Guipponi M, Antonarakis SE, Scott HS. TMPRSS3, a type II transmembrane serine protease mutated in non-syndromic autosomal recessive deafness. Front Biosci. 2008;13:1557–67. [DOI] [PubMed] [Google Scholar]
- 80.Fasquelle L, Scott HS, Lenoir M, Wang J, Rebillard G, Gaboyard S, et al. Tmprss3, a transmembrane serine protease deficient in human DFNB8/10 deafness, is critical for cochlear hair cell survival at the onset of hearing. J Biol Chem. 2011;286(19):17383–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Molina L, Fasquelle L, Nouvian R, Salvetat N, Scott HS, Guipponi M, et al. Tmprss3 loss of function impairs cochlear inner hair cell Kcnma1 channel membrane expression. Hum Mol Genet. 2013;22(7):1289–99. [DOI] [PubMed] [Google Scholar]
- 82.Tang PC, Alex AL, Nie J, Lee J, Roth AA, Booth KT, et al. Defective Tmprss3-Associated Hair Cell Degeneration in Inner Ear Organoids. Stem Cell Reports. 2019;13(1):147–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Peters TA, Levtchenko E, Cremers CW, Curfs JH, Monnens LA. No evidence of hearing loss in pseudohypoaldosteronism type 1 patients. Acta Otolaryngol. 2006;126(3):237–9. [DOI] [PubMed] [Google Scholar]
- 84.Guipponi M, Tan J, Cannon PZ, Donley L, Crewther P, Clarke M, et al. Mice deficient for the type II transmembrane serine protease, TMPRSS1/hepsin, exhibit profound hearing loss. Am J Pathol. 2007;171(2):608–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ray RP, Matamoro-Vidal A, Ribeiro PS, Tapon N, Houle D, Salazar-Ciudad I, et al. Patterned Anchorage to the Apical Extracellular Matrix Defines Tissue Shape in the Developing Appendages of Drosophila. Dev Cell. 2015;34(3):310–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Diaz-de-la-Loza MD, Ray RP, Ganguly PS, Alt S, Davis JR, Hoppe A, et al. Apical and Basal Matrix Remodeling Control Epithelial Morphogenesis. Dev Cell. 2018;46(1):23–39 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bertram S, Glowacka I, Blazejewska P, Soilleux E, Allen P, Danisch S, et al. TMPRSS2 and TMPRSS4 facilitate trypsin-independent spread of influenza virus in Caco-2 cells. J Virol. 2010;84(19):10016–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Shin WJ, Seong BL. Type II transmembrane serine proteases as potential target for anti-influenza drug discovery. Expert Opin Drug Discov. 2017;12(11):1139–52. [DOI] [PubMed] [Google Scholar]
- 89.Bottcher-Friebertshauser E, Klenk HD, Garten W. Activation of influenza viruses by proteases from host cells and bacteria in the human airway epithelium. Pathog Dis. 2013;69(2):87–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cheng Z, Zhou J, To KK, Chu H, Li C, Wang D, et al. Identification of TMPRSS2 as a Susceptibility Gene for Severe 2009 Pandemic A(H1N1) Influenza and A(H7N9) Influenza. J Infect Dis. 2015;212(8):1214–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Keppner A, Andreasen D, Merillat AM, Bapst J, Ansermet C, Wang Q, et al. Epithelial Sodium Channel-Mediated Sodium Transport Is Not Dependent on the Membrane-Bound Serine Protease CAP2/Tmprss4. PLoS One. 2015;10(8):e0135224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lambertz RLO, Gerhauser I, Nehlmeier I, Leist SR, Kollmus H, Pohlmann S, et al. Tmprss2 knock-out mice are resistant to H10 influenza A virus pathogenesis. J Gen Virol. 2019;100(7):1073–8. [DOI] [PubMed] [Google Scholar]
- 93.Kuhn N, Bergmann S, Kosterke N, Lambertz RLO, Keppner A, van den Brand JMA, et al. The Proteolytic Activation of (H3N2) Influenza A Virus Hemagglutinin Is Facilitated by Different Type II Transmembrane Serine Proteases. J Virol. 2016;90(9):4298–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sakai K, Ami Y, Tahara M, Kubota T, Anraku M, Abe M, et al. The host protease TMPRSS2 plays a major role in in vivo replication of emerging H7N9 and seasonal influenza viruses. J Virol. 2014;88(10):5608–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hatesuer B, Bertram S, Mehnert N, Bahgat MM, Nelson PS, Pohlmann S, et al. Tmprss2 is essential for influenza H1N1 virus pathogenesis in mice. PLoS Pathog. 2013;9(12):e1003774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bertram S, Glowacka I, Muller MA, Lavender H, Gnirss K, Nehlmeier I, et al. Cleavage and activation of the severe acute respiratory syndrome coronavirus spike protein by human airway trypsin-like protease. J Virol. 2011;85(24):13363–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Abe M, Tahara M, Sakai K, Yamaguchi H, Kanou K, Shirato K, et al. TMPRSS2 is an activating protease for respiratory parainfluenza viruses. J Virol. 2013;87(21):11930–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zmora P, Blazejewska P, Moldenhauer AS, Welsch K, Nehlmeier I, Wu Q, et al. DESC1 and MSPL activate influenza A viruses and emerging coronaviruses for host cell entry. J Virol. 2014;88(20):12087–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zmora P, Hoffmann M, Kollmus H, Moldenhauer AS, Danov O, Braun A, et al. TMPRSS11A activates the influenza A virus hemagglutinin and the MERS coronavirus spike protein and is insensitive against blockade by HAI-1. J Biol Chem. 2018;293(36):13863–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Reinke LM, Spiegel M, Plegge T, Hartleib A, Nehlmeier I, Gierer S, et al. Different residues in the SARS-CoV spike protein determine cleavage and activation by the host cell protease TMPRSS2. PLoS One. 2017;12(6):e0179177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kam YW, Okumura Y, Kido H, Ng LF, Bruzzone R, Altmeyer R. Cleavage of the SARS coronavirus spike glycoprotein by airway proteases enhances virus entry into human bronchial epithelial cells in vitro. PLoS One. 2009;4(11):e7870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Straus MR, Kinder JT, Segall M, Dutch RE, Whittaker GR. SPINT2 inhibits proteases involved in activation of both influenza viruses and metapneumoviruses. Virology. 2020;543:43–53. [DOI] [PMC free article] [PubMed] [Google Scholar]