

Abstract
Virus emergence may occur through interspecies transmission and recombination of viruses coinfecting a host, with potential to pair novel and adaptive gene combinations. Camels are known to harbor diverse ribonucleic acid viruses with zoonotic and epizootic potential. Among them, astroviruses are of particular interest due to their cross-species transmission potential and endemicity in diverse host species, including humans. We conducted a molecular epidemiological survey of astroviruses in dromedaries from Saudi Arabia and Bactrian camels from Inner Mongolia, China. Herein, we deployed a hybrid sequencing approach coupling deep sequencing with rapid amplification of complementary deoxyribonucleic acid ends to characterize two novel Bactrian and eight dromedary camel astroviruses, including both partial and complete genomes. Our reported sequences expand the known diversity of dromedary camel astroviruses, highlighting potential recombination events among the astroviruses of camelids and other host species. In Bactrian camels, we detected partially conserved gene regions bearing resemblance to human astrovirus types 1, 4, and 8 although we were unable to recover complete reading frames from these samples. Continued surveillance of astroviruses in camelids, particularly Bactrian species and associated livestock, is highly recommended to identify patterns of cross-species transmission and to determine any epizootic threats and zoonotic risks posed to humans. Phylogenomic approaches are needed to investigate complex patterns of recombination among the astroviruses and to infer their evolutionary history across diverse host species.
Keywords: astrovirus, dromedary camel, Bactrian camel, recombination, deep sequencing, RACE, interspecies transmission, virus evolution, molecular surveillance
1. Introduction
Over the past two decades, the world has seen several epidemics and pandemics including Ebola, avian flu, swine flu (pandemic H1N1/2009), severe acute respiratory syndrome, Middle East respiratory syndrome (MERS), Zika, and, most recently, coronavirus disease 2019, which have had devastating impacts on the lives of millions and exhausted even the most advanced health-care systems. In each of these transnational zoonotic emergence events, RNA viruses are the principal etiologic agents involved. It is essential to investigate and characterize the virus diversity in animals closely associated with humans, enabling us to better identify the factors that facilitate the cross-species transmission and adaptation to novel host species.
Astroviruses (AstVs) are of particular interest among the ribonucleic acid (RNA) viruses due to their tendency for recombination, wide host breadth, and association with a range of diseases (Cortez et al. 2017b). Two genera are distinguished within the Astroviridae family, the Mamastrovirus and Avastrovirus, with taxonomy traditionally defined by host affinity (Cortez et al. 2017b). However, the detection of diverse AstVs from a wide breadth of host species poses challenges for this classification, and genetic criteria are increasingly emphasized (Bosch et al. 2011). The cross-species transmission of AstVs has been previously reported (Sun et al. 2014; Pankovics et al. 2015), and the presence of multiple, genetically distinct AstVs within individual hosts may facilitate recombination events and the potential emergence of novel AstVs as well as other RNA viruses (Wohlgemuth, Honce, and Schultz-Cherry 2019).
AstVs are nonenveloped viruses with a positive-sense, single-strand RNA genome of 6.8–7.9 kb in size, generally divided into three open reading frames (ORFs) whose organization differentiates them from other virus families (Méndez and Arias 2013). They are associated with a spectrum of diseases ranging from gastroenteritis to neurological diseases and encephalitis, depending on virus genotype and host factors including species and individual immunity (Cortez et al. 2017b). AstVs have also been found to play a complex role in modulating host interactions with other enteric viruses (Ingle et al. 2019), as well as bacteria, through physiological manipulation of the gut microenvironment (Cortez et al. 2020).
Identifying patterns of virus–host coevolution and host breadth will fill key knowledge gaps in our understanding of RNA virus evolution, allowing us to predict origins of emergence and to better prepare for future outbreaks (Menachery et al. 2015). Dromedary camels have received extensive research focus due to their involvement in the zoonotic emergence of MERS coronavirus (MERS-CoV), and efforts have been made to characterize the virome of these animals (Woo et al. 2014; Sabir et al. 2016; Li et al. 2017). Among the viruses recently discovered and characterized in dromedary camels, dromedary camel astrovirus (DcAstV) has been detected and isolated from fecal samples (Woo et al. 2014, 2015), and short genomic sequences with high similarity to DcAstVs have been detected from dromedary upper respiratory samples in the United Arab Emirates (UAE) (Li et al. 2017).
The virome of Bactrian camels remains largely uncharacterized in comparison to dromedary camels, with the exception of several seroepidemiological and molecular surveys targeting viruses of major public health and agricultural importance (Chan et al. 2015; Adney et al. 2019). The extensive interface between Bactrian camels and humans warrants further surveillance and investigative work in this animal host, as well as other diverse camelid species, given their unique ecology and physiological traits.
In this study, we expand the known diversity of Mamastroviruses (MamAstVs) in camelid hosts and use phylogenomic and recombination analyses to investigate the evolutionary ecology of AstVs in these culturally and economically important animals. Next-generation sequencing techniques are widely used by epidemiologists and evolutionary virologists to study the evolution and transmission of emerging viruses. Here, we describe a rapid amplification of complementary deoxyribonucleic acid (cDNA) ends (RACE)–sequencing technique that facilitates the retrieval of short-length viral genomes from complex samples in which the virus of interest may only be present in low titer. This method may complement targeted detection methods as well as metagenomic methods where genome coverage is incomplete.
2. Materials and methods
2.1. Sample collection
Fresh stool samples (n = 112) were collected from free-ranging Bactrian camels (Camelus bactrianus), kept as pack animals and beasts of burden, in the Inner Mongolia Autonomous Region (Nei Mongol), the People’s Republic of China, during the summers of 2015 (n = 96) and 2016 (n = 16). The fecal material was transported in virus transport medium and stored at −80°C.
Paired nasal and rectal swabs were collected antemortem from dromedary camels (Camelus dromedarius) as part of MERS-CoV surveillance conducted in the Kingdom of Saudi Arabia from May 2014 to April 2015; swabs were transported in viral transport medium and stored at −80°C as described by Sabir et al. (2016). Here, we examined a subset of 198 rectal swabs collected from imported and locally reared male dromedaries at wholesale markets in Jeddah (n = 100) and Riyadh (n = 98) during a single sampling event in June 2014 and July 2014 for each respective location.
2.2. RNA extraction and AstV detection
Viral RNA was extracted from 200 μl fecal homogenate collected from Bactrian camels or from 140 μl rectal swab material collected from dromedaries using the QIAamp Viral RNA Mini Kit (QIAGEN) following the manufacturer’s recommendations. The extracted RNA was used as a template for cDNA synthesis using the PrimeScript™ II 1st Strand cDNA Synthesis Kit (TaKaRa) with random hexamers. Each sample was screened for AstVs using a semi-nested reverse transcription-polymerase chain reaction (PCR) with universal AstV primers targeting a ∼421 base pair (bp) segment of the conserved RNA-dependent RNA polymerase (RdRp) gene region (Chu et al. 2008) using AmpliTaq® Gold DNA Polymerase (Applied Biosystems). Amplicons were purified using the QIAquick Gel Extraction Kit (QIAGEN), and Sanger sequencing was used to confirm AstV detection and to identify viral genogroups through comparison with existing sequences in the GenBank database.
2.3. DNA extraction and host species validation
To validate host species for all Bactrian camel stool samples in which AstVs were detected, DNA was extracted using the QIAamp DNA Stool Kit (QIAGEN). We used conventional PCR to amplify a region of the mitochondrial DNA (mtDNA) encoding the 12S ribosomal RNA (rRNA) as previously described (Xie et al. 2015), with Sanger sequencing used to confirm the host identity through comparison with available sequences in the GenBank database.
2.4. RACE-Seq assays and library preparation
RACE coupled with deep sequencing of amplicons (RACE-Seq) was adapted to separately amplify both the 5ʹ and 3ʹ regions of the Bactrian camel AstV (BcAstV) and DcAstV genomes. RACE assays were designed with nested, gene-specific sense or antisense primers to limit off-target amplification, with amplicons subsequently sequenced on the MiSeq benchtop sequencer (Illumina). 3ʹ RACE-Seq was attempted for all dromedary and Bactrian camel samples in which AstVs were detected, while 5ʹ RACE-Seq was only performed for samples with successful recovery of the 3ʹ region.
RACE assays were carried out with cDNA synthesized from the extracted RNA using SuperScript® IV Reverse Transcriptase (Invitrogen) and spiked with Oligo d(T) in preparation for 3ʹ RACE or a gene-specific antisense primer in preparation for 5ʹ RACE. The cDNA prepared for 5ʹ RACE was treated with Ribonuclease H (Invitrogen) according to the manufacturer’s instructions to digest residual RNA in preparation for second-strand DNA synthesis. Double-stranded DNA (dsDNA) was prepared for 5ʹ RACE using Sequenase™ Version 2.0 DNA Polymerase (Applied Biosystems) to append an adaptor to the 5ʹ end. The QIAquick PCR Purification Kit (QIAGEN) was used to purify dsDNA, which was then used as a template for 5ʹ RACE.
In 3ʹ RACE assays, cDNA was used directly as a template for PCR amplification using AccuPrime™ Taq DNA Polymerase (Invitrogen) with nested gene-specific sense primers and an anchor primer fixed to the poly-A tail. In 5ʹ RACE assays, the purified dsDNA was used as a template for PCR amplification using AccuPrime Taq with a sense primer targeting the adaptor region and nested gene-specific antisense primers. The first-round RACE assays were conducted using gene-specific primers designed within the RdRp region and based on the sequences obtained during virus detection, while primers used in the subsequent RACE assays were designed further upstream or downstream based on sequencing results obtained (primers available on request).
In preparation for Illumina sequencing, RACE products were purified using Agencourt AMPure XP magnetic beads (Beckman Coulter, Inc.), and samples were uniformly diluted to a concentration of 1 ng/μl. Fragmentation and indexing were performed using the TruePrep™ DNA Library Prep Kit V2 or V3 for Illumina (Vazyme™). Library purification was performed using AMPure XP magnetic beads to select for an even size distribution of fragments at approximately 200 bp. Sequencing was performed on the Illumina MiSeq benchtop sequencer using V3 chemistry to generate paired-end reads (150 bp).
2.5. Genome assembly
Preprocessing of the raw reads was done using Trimmomatic 0.39 (Bolger, Lohse, and Usadel 2014) and an in-house bioinformatics pipeline with built-in filters to remove indexes and perform quality checks on sequencing data. De novo assembly of all high-quality Illumina reads into contigs was performed using Trinity v2.8.5 (Grabherr et al. 2011). Taxonomic classification of all high-quality reads was performed with BLASTn using the National Center for Biotechnology Information (NCBI) virus nucleotide (nt) database with an E-value cut-off of 1e-10, and the classification of de novo assembled contigs was performed with DIAMOND BLASTx v0.9.24 (Buchfink, Xie, and Huson 2015) using the NCBI non-redundant protein database with an E-value cut-off of 1e-5.
Genome assembly of all contigs and high-quality reads matching to the family Astroviridae was performed using the Geneious assembler (Kearse et al. 2012) as implemented in Geneious Prime® 2019.2.1 (Biomatters, Ltd). Assembled genomes were examined for completeness of the expected ORFs and were further evaluated by mapping all trimmed, high-quality reads back to the assembled genomes using Bowtie2 v2.3.5 (Langmead and Salzberg 2012), with post-processing of mapped reads performed with SAMtools 1.9 (Li et al. 2009).
2.6. Genome characterization
The ORFs of each BcAstV and DcAstV were predicted by the DNASTAR ORF search tool and further verified with NCBI ORF finder (https://www.ncbi.nlm.nih.gov/orffinder/). ORF1b was identified by a conserved heptanucleotide frameshift signal common to AstVs (Jiang et al. 1993). Conserved motifs were identified using Pfam (Finn et al. 2006). RNA secondary structures were evaluated using RNAstructure 6.1 (https://rna.urmc.rochester.edu/RNAstructure.html), while the 3′ untranslated region (UTR), 5′ UTR, and the putative ribosomal frameshift signal were predicted with m-fold (Zuker 2003). The prediction of transmembrane domains and nuclear localization signals (NLS) were analyzed by a transmembrane protein topology prediction method with a hidden Markov model; program (https://www.cbs.dtu.dk/services/TMHMM/) and a computer program for prediction of the classical importin-alpha/beta pathway-specific NLSs (cNLS Mapper) (Kosugi et al. 2009), respectively. The pairwise mean amino acid (aa) genetic distance (p-dist) analysis between viruses characterized in this study and representative AstV sequences were estimated by bootstrap method (N = 1,000 replicates) and partial deletion (95 per cent) with MEGA10.0.5 (Kumar et al. 2018). Per cent identity between genomic sequences and between coding regions was determined using Geneious Prime. Visualization of pairwise comparisons of per cent identity and p-dist was created with Morpheus (https://software.broadinstitute.org/morpheus).
2.7. Phylogenetic analysis
To infer the evolutionary relationships of our DcAstV and BcAstV viruses, a detailed phylogenetic analysis was carried out using nt sequences of the ORFs: ORF1a, ORF1b (including RdRp), and ORF2. The available MamAstV sequences were retrieved from the GenBank database. Geneious Prime was used for sequence assembly and alignment using MAFFT v7.450 (Katoh and Standley 2013). Initial RdRp and capsid region phylogenies were produced to refine the reference sequences used in later analysis. Maximum-likelihood (ML) phylogenies were inferred from the nt alignments of individual gene regions. ML phylogenies were generated using RAxML v8.2.12 (Stamatakis 2014), picking the GTRGAMMA or PROTGAMMAAUTO substitution models for nt or protein alignments, respectively. Branch support on the best ML tree was estimated using 1,000 rapid bootstrap replicates.
2.8. Recombination analysis
AstV genomic sequences were screened for potential recombination breakpoints using a suite of detection methods implemented in recombination detection program (RDP4) (Martin et al. 2015), including RDP, GENECONV, Bootscan, MaxChi, Chimaera, and SiScan. Sequences in which recombination signals were detected by more than two methods were analyzed in more detail. Simplot v3.5.1 (Lole et al. 1999) was used to perform similarity comparisons using sequences with recombination break points identified by RDP4 as query sequences. ML phylogenies were constructed for genome segments and coding regions on either side of recombination breakpoints to examine sequence movements within the tree topology.
3. Results
3.1. AstV detection and host species confirmation
AstVs were detected in sixteen Bactrian camel stool samples (16/112) from Inner Mongolia, with complete genomes successfully retrieved from two samples. All Bactrian camel samples in which AstVs were detected were collected during the summer of 2015 (16/96) and were not detected in samples collected during the summer of 2016 (0/16) (Table 1). The amplification of mtDNA-encoded 12S rRNA showed complete homology to Bactrian camel mtDNA, confirming fecal material originated from Bactrian camels and discounting the possibility of misidentification during the collection of stool samples.
Table 1.
AstV surveillance conducted in dromedary and Bactrian camels from Saudi Arabia and Inner Mongolia, China, respectively.
| Camel species | Sampling locality and year | Individuals sampled (AstV positive) |
|---|---|---|
| Dromedary camel (Camelus dromedarius) | Jeddah, 2014 | 100 (8) |
| Riyadh, 2014 | 98 (0) | |
| Total | 198 (8) | |
| Bactrian camel (C. bactrianus) | Nei Mongol, 2015 | 96 (16) |
| Nei Mongol, 2016 | 16 (0) | |
| Total | 112 (16) |
Additionally, AstVs were detected in eight dromedary camel rectal swab samples (8/198). All AstV-positive dromedary samples were collected at a wholesale market in Jeddah during June 2014 (8/100), while none were detected in samples collected from a wholesale market in Riyadh during July 2014 (0/98). AstV-positive dromedaries were locally reared animals with a median age of 4.5 months (Supplementary Table S1).
3.2. RACE-Seq assays
Successful sequencing runs generated an average of 157,890 (SD 59,189) quality-trimmed reads for 3ʹ RACE assays, with an average of 33.1 per cent reads mapped to the assembled AstV genome with an average depth of 3,982 (SD 2,129) reads per covered base (Supplementary Table S2). An average of 89,656 (SD 47,307) quality-trimmed reads were generated for 5ʹ RACE assays, with an average of 9.36 per cent reads mapped to the assembled AstV genome with an average depth of 750 (SD 1,152) reads per covered base. Successive RACE assays were used to extend the genomes in either the 3ʹ or 5ʹ directions (see Fig. 1A).
Figure 1.

Genome structures of the BcAstVs and DcAstVs. (A) A sampling graphical illustration showing aggregate coverage of high-quality reads achieved through successive rounds of RACE-Seq. Here, two 3ʹ and three 5ʹ RACE assays were executed to cover the full BcAstV/NMG31 genome (read coverage across genome ranged from a minimum coverage of 212 reads to a maximum coverage of 23,782, with an average of 10,632.2 reads per nucleotide position). (B) Genome maps of the two novel BcAstVs and a representative DcAstV from this study as determined through sequence comparison with already characterized dromedary AstVs. The tentative designation and estimated complete size of camelid AstV genomes are shaded in gray. The viral ORFs identified through homology mapping to known MamAstV gene sets are depicted by successive forward directional handles that are distinctively colored (sky and hazy blue, respectively, indicate BcAstV/NMG31 and BcAstV/NMG59, while brown stains indicate DcAstV/JD53 genome) to mark individual genomes. The underlying conserved domains are shown in black arrow bars. Functional motifs detected using various web-based tools as stated in Section 2 are indicated by their putative genome location. The length of viral ORFs (nucleotide and amino acid) is mentioned in red text on top of individual ORFs.
3.3. Genome organization
Two complete BcAstV genomes, BcAstV-NMG31 (6,342 nt) and NMG59 (6,368 nt), were recovered along with four complete and four partial DcAstV genomes, DcAstV-Jd53 (6,317 nt), Jd57 (6,326 nt), Jd60 (3,112 nt), Jd67 (6,350 nt), Jd70 (5,450 nt), Jd73 (3,639 nt), Jd74 (6,337 nt), and Jd76 (5,431 nt). All complete camelid AstV genomes exhibited a standard ensemble of four MamAstV-specific ORFs (ORF1a, ORF1b, ORFX, and ORF2), a 5ʹ and 3ʹ UTR, and a polyadenylated tail (Fig. 1B). Polyproteins resulting from post-translational processing of the viral ORFs were predicted to include a trypsin-like peptidase, the RdRp, and an AstV capsid protein. A viral protein genome-linked motif was also identified.
The ORF1a encoded a nonstructural polyprotein with a predicted trypsin-like peptidase domain. Structural projections indicated potential transmembrane helices as well as motifs typical of serine proteases ‘GMSG’ and a bipartite NLS (KGKNKRGRGARLMRLAGAKKRKQK). The cis-regulatory heptanucleotide frameshift signal (AAAAAAC), which facilitates the translation of the viral RNA polymerase encoded within the ORF1b, was positioned near the end of ORF1a followed by a stem-loop structure potentially forming a pseudoknot with the ensuing sequence (Jiang et al. 1993; Méndez and Arias 2013).
The ORF1b was generated by a −1 ribosomal frameshift and encoded the replicase polyprotein, which contained the highly conserved RdRp. The RdRp was verified by the presence of signature motifs including ‘DWTRYD’, ‘GNPSG’, ‘YGDD’, and ‘FGMWVK’ (Zhang et al. 2017). A stretch of highly conserved mammalian subgenomic RNA (sgRNA) promotor sequence (Méndez and Arias 2013) was observed at the 3ʹ end of the ORF1b, just upstream of the ORF2 start codon. The end of ORF1b and the beginning of ORF2 shared an overlap of 8 nt, with ORF2 maintaining the same reading frame as ORF1a (De Benedictis et al. 2011; Woo et al. 2015).
The ORF2 encoded a large structural polyprotein, which is proteolytically processed to generate a bipartite capsid consisting of a conserved N-terminal and a hyper variable and potentially receptor interacting C-terminal domain (Méndez and Arias 2013). The stem-loop II-like motif (s2m) predicted to be located at the 3ʹ end of the genomic RNA of several AstVs was found in these camelid AstVs (Luo et al. 2011). This motif is also present in some coronaviruses and equine rhinovirus serotype 2 (Jonassen, Jonassen, and Grinde 1998).
In addition to the ORFs shared by all members of the MamAstV genus, the DcAstV and BcAstV genomes contained an additional seventy-one to seventy-five codon ORFX, which overlaps with the 5ʹ end of ORF2 as previously described in DcAstVs (Woo et al. 2015). The ORFX start codon was located fifty-nine nt downstream from that of ORF2 and translated in the +1 ribosomal frameshift through a leaky scanning mechanism (Méndez and Arias 2013).
3.4. Evolutionary relationships of the camelid AstVs
Preliminary phylogenetic investigation of the conserved RdRp region, obtained through the direct sequencing of the amplicons from AstV detection, found that two of the sixteen AstVs detected in Bactrian camels (BcAstV-NMG13 and BcAstV-NMG16) fell within the classical human AstV clade pairing human AstV4 and AstV1, respectively. However, attempts to obtain longer genomic sequences for these human-like AstVs through RACE-Seq were unsuccessful. The remaining fourteen BcAstV RdRp sequences are represented by the two novel BcAstVs (BcAstV-NMG31 and BcAstV-NMG59) based on homology. The novel BcAstVs and DcAstVs described in this study, as well as the previously reported DcAstVs (NCBI Accession Nos. KR868721–KR868724), are distributed across two basal clusters alongside strains of PoAstV-2 and a porcupine AstV (Fig. 2A).
Figure 2.

Phylogenetic trees of RdRp and capsid genes of camelid AstVs. Maximum-likelihood phylogenetic trees of the (A) RdRp (n = 110; 486 nt) and (B) complete capsid gene (n = 95; >2,000 nt) were inferred for the two novel Bactrian and eight dromedary AstVs from this study along with representative members of the genus Mamastrovirus. The difference in ‘n’ between the estimated phylogenies is due to the differential availability of RdRp and ORF2 sequences in GenBank. Camelid AstVs reported in this study are color-marked (taxa in red represent dromedary, while those in sky blue indicate Bactrian AstVs). The human AstV-like viruses exclusively found in Bactrian species and for whom only RdRp regions could be amplified are stated in green. Branch lengths reflect the number of nucleotide substitutions per site and the trees were rooted by avian AstV (Y15936).
The complete ORF2 region ML phylogeny continued placing camelid AstVs in two clades stacked below a bat-borne AstV (NCBI Accession No. KX645667). All the DcAstVs from this study closely aligned with known dromedary strains extending prior observations on the diversity of AstVs in dromedaries, while the two novel Bactrian AstVs bifurcated, each becoming part of a separate homology group. The BcAstV-NMG59 along with the dromedary-based AstVs formed a basal cluster adjacent to a PoAstV-2 subset that belongs to MamAstV-32. The imputed pairwise aa identities among members of this group ranged from 62 to 86 per cent and with a group mean genetic distance of 0.221. In contrast, the BcAstV-NMG31 assembled with PoAstV-2 strains that correspond to MamAstV-31 sharing an intragroup mean genetic distance of 0.306 (pairwise distance values: 0.257–0.358). This phylogenetic cluster included isolates from a broad range of host species including swine, porcupine, and diverse domestic livestock and wild ruminant species. The high genetic similarity of an individual Bactrian AstV strain to porcine-origin AstVs may also hint at a possible historic interspecies virus spillover event.
To fully resolve AstV genetic diversity in dromedary and Bactrian camels, additional gene trees were built comparing complete ORF1a, 1b, and 2 regions of camelid AstVs and those of related MamAstVs. The resulting phylogenies replicated topological patterns as obtained in previous RdRp and ORF2 trees. However, the position of some camelid AstVs differed in their position between gene trees, hinting at potential recombinant strains and supporting further investigation (Fig. 3A).
Figure 3.
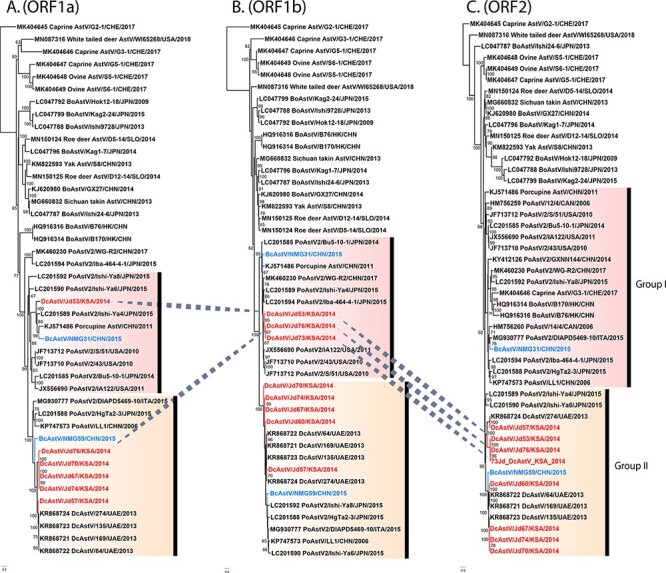
Phylogenetic trees of ORF1 and ORF2 regions of camelid AstVs. Maximum-likelihood phylogenetic trees of the (A) ORF1a (n = 43; >2,000 nt), (B) ORF1b (n = 45; >2,000 nt), and (C) the complete ORF2 nucleotide sequence (n = 48; >2,000 nt) were inferred for the two novel Bactrian and eight dromedary AstVs from this study along with representative members of the genus Mamastrovirus. The difference in ‘n’ is due to the differential availability of sequences in GenBank. Camelid AstVs are mentioned in red (dromedary) and sky blue (Bactrian) text. The dotted lines indicate the movement of taxa along different phylogenetic groups in the compared gene trees. Branch lengths reflect the number of nucleotide substitutions per site, and the trees were rooted by a distantly related MK404646 caprine AstV.
The classification scheme adapted by the Astrovirus Study Group of the International Committee for Taxonomy of Viruses sees conspecific MamAstVs as (1) being isolated from the same hosts and (2) having a p-dist within a range of 0.000–0.318, considering the complete capsid region at the aa level (Bosch et al. 2011; Guix, Bosch, and Pintó 2012). The pairwise aa distances (p-dist) among all DcAstV sequences ranged from 0.000 to 0.378 with an overall mean distance of 0.239 in the ORF2 region (Fig. 4). These figures indicate that all DcAstVs described in this study form a single species along with previously isolated DcAstVs.
Figure 4.

Sequence similarity between the camelid AstVs and representative members of Mamastrovirus. Maximum-likelihood phylogenetic trees of the complete ORF2 based on (A) nucleotide (n = 27; >2,000 nt) and (C) aa (n = 27; >750 aa) sequences were inferred for the two novel Bactrian and eight dromedary AstVs from this study along with representative members of the genus Mamastrovirus. The taxa names highlighted in red mark dromedary AstV variants, while those shaded in sky blue represent Bactrian AstV species. Branch lengths reflect the number of nucleotide and aa substitutions per site, and both trees were rooted by a distantly related MK404646 caprine AstV. (B) The color-coded matrix representing pairwise nucleotide per cent identities (lower left) and aa p-distance estimates (upper right) are, respectively, generated from complete ORF2 nucleotide and aa sequences of the reported Bactrian and dromedary AstVs along with representative members of the genus Mamastrovirus. Each colored cell stands for per cent identity or genetic distance score between a given set of two sequences. A color key indicates the correspondence between pairwise identities or distances and the colors displayed in the matrix.
Furthermore, we have identified four discreet variant groups (V1–V4) within the DcAstV (Fig. 4A), based on the criteria that intraspecific variants share more than 75 per cent homology in the capsid protein gene (Bosch et al. 2011) and that nt homology between groups be less than 93 per cent (De Benedictis et al. 2011). Intragroup pairwise identity values ranged from 94 to 100 per cent for the ORF2 region, while intergroup pairwise identity values ranged from 62 to 87 per cent. Variant group 1 (V1) contained two DcAstVs, Jd57 from this study and KR868724, sharing 94.6 per cent identity. Variant group 2 (V2) contained three DcAstVs all from this study, Jd53, Jd73, and Jd76, sharing 92–96 per cent identity and all containing putative recombination breakpoints (see details later). Variant group 3 (V3) contained one DcAstV from this study, Jd60, and two previously characterized viruses, KR868721 and KR868722, sharing 94–99 per cent identity. Variant group 4 (V4) was most closely related to V3 and contained three dromedary viruses from this study, Jd67, Jd70, and Jd74, as well as the previously described KR868723, sharing 96–100 per cent identity.
3.5. Recombination analysis
Phylogenetic incongruence between the tree topologies of the ORF1a, ORF1b, and ORF2 coding regions was observed for DcAstV isolates Jd53, Jd73, and Jd76 (Fig. 3), suggesting a complex history of multiple recombination events between members of this clade. Interestingly, all three of these putative recombinant DcAstVs belong to variant group 2 (Fig. 4). In each isolate, the putative recombination breakpoints identified are generally positioned at or around the junctions between ORFs although some fall within coding regions (see Supplementary Table S3 for details of the recombination signals detected by each method incorporated in RDP4). Recombination hotspots have been hypothesized at the junction of ORF1b and ORF2, influenced by the presence of an sgRNA promotor at this junction (Walter et al. 2001; Pantin-Jackwood, Spackman, and Woolcock 2006; Wohlgemuth, Honce, and Schultz-Cherry 2019).
DcAstV-Jd53 appears to consist of PoAstV-2-like nonstructural proteins with a DcAstV capsid protein. A putative recombination breakpoint was identified at nt position 1,242 (99 per cent Confidence interval, CI: 1,036–1,442), approximately in the middle of the ORF1a, sharing the highest similarity to BcAstV-NMG31 in the 5ʹ region, but most similar to PoAstV-2 (JF713712) within the 3ʹ region of the ORF1a (Fig. 5). Throughout the ORF1b, Jd53 showed high similarity to NMG31, PoAstV-2, and related viruses including porcupine AstV. A second recombination breakpoint was detected at nt position 4,094 (99 per cent CI: 3,863–4,167), covering the ORF1b-ORF2 junction and most of ORFX (Fig. 5). Here, we observed a notable change in the position of Jd53 within the tree topology, moving from the PoAstV-2 clade within the ORF1b to the DcAstV clade in ORF2 (Figs 3 and 5). A third recombination breakpoint was identified at nt position 5,811 (99 per cent CI: 5,241–5,938) (Fig. 5).
Figure 5.

Genomic recombination in the camelid astrovirus DcAstV-Jd53. (A) Partial genome structure of DcAstV-Jd53 with ML trees constructed before and after each putative breakpoint corresponding to the alternating white- and gray-shaded regions. (B) Similarity analysis performed using the partial genome of DcAstV-Jd53R as the query sequence. (C) Bootscan analysis plots for DcAstV-Jd53R. Putative recombination breakpoints in DcAstV-Jd53R was identified at 1,242 nt (99 per cent CI: 1,036–1,442) through comparison with sequences identified as putative parental sequences in RDP4. Nucleotide position within the multiple alignments is represented on the x-axis, while the y-axis represents pairwise bootstrap support (%). Pairwise comparison of PoAstV2 (JF713712) and BcAstV-NMG31 represented in yellow; PoAstV2 (JF713712) and DcAstV-Jd53R represented in green; and BcAstV-NMG31 and DcAstV-Jd53R represented in purple. The red box masks genome regions used for further analysis and identification of recombination breakpoints by additional means. (D) Additional putative recombination breakpoints were identified through further analysis at 4,094 nt (99 per cent CI: 3,863–4,167) and with an uncertain ending breakpoint at 5,811 nt (99 per cent CI: 5,241–5,938). Pairwise comparison of DcAstV-64 (KR868722) and BcAstV-NMG31 represented in yellow; DcAstV-64 (KR868722) and DcAstV-Jd53R represented in green; and BcAstV-NMG31 and DcAstV-Jd53R represented in purple. The red box masks the recombination breakpoint region identified in the ORF1ab coding sequence (CDS).
DcAstV-Jd76 appears to have a chimeric DcAstV and PoAstV-like nonstructural protein and a DcAstV capsid protein although we were unable to obtain the 5ʹ region of the ORF1a. DcAstV-Jd76 had recombination breakpoints identified at nt position 1,200 (99 per cent CI: 1,168–1,245) [nt position 2,088 (99 per cent CI: 2,056–2,133) based on homologous positions in Jd53 genome] within the ORF1a (Fig. 5). Prior to this breakpoint, Jd76 shares high similarity with Jd67 and other previously described DcAstVs. Following this breakpoint, Jd76 has a short region that shares high similarity with BcAstV-NMG31 until a second recombination breakpoint at nt position 1,617 (99 per cent CI: 1,489–1,691) [nt position 2,505 (99 per cent CI: 2,377–2,579) based on Jd53] covering the frameshift region within the ORF1ab. Throughout the ORF1b, Jd76 shares the highest similarity with Jd53, as well as NMG31 and PoAstV-2 viruses (Figs 3 and 6). A third recombination breakpoint was identified at the ORF1b-ORF2 junction at nt position 3,227 (99 per cent CI: 3,200–3,344) [nt position 4,115 (99 per cent CI: 4,088–4,232) based on Jd53]. This breakpoint was positioned at the end of ORFX and similar to the breakpoint observed in Jd53, with the ORF2 gene region most similar to Jd53 and clustering with other DcAstVs (Fig. 3).
Figure 6.

Genomic recombination in the camelid astrovirus DcAstV-Jd76. (A) Near-complete genome structure for DcAstV-Jd76 with ML trees constructed before and after each putative breakpoint corresponding to the alternating white- and gray-shaded regions. (B) Similarity analysis performed using the partial genome of DcAstV-Jd76R as the query sequence. Note that ‘*’ indicates a sequence containing putative recombination breakpoints. (C) Bootscan analysis plot for DcAstV-Jd76R. Putative recombination breakpoints in DcAstV-Jd76R were identified at 1,200 nt (99 per cent CI: 1,168–1,245); at 1,617 nt (99 per cent CI: 1,489–1,691); and at 3,227 nt (99 per cent CI: 3,200–3,344) through comparison with sequences identified as putative parental sequences in RDP4. Nucleotide position within the multiple alignment is represented on the x-axis, while the y-axis represents pairwise bootstrap support (%). Pairwise comparison of PoAstV2 (JF713712) and DcAstV-Jd67R represented in yellow; PoAstV2 (JF713712) and DcAstV-Jd76R represented in green; and DcAstV-Jd67R and DcAstV-Jd76R represented in purple.
DcAstV-Jd73 shares high similarity to both Jd67 and Jd76, with evidence of a similar recombination breakpoint at the ORF1b-ORF2 junction as identified in Jd53 and Jd76 (Figs 3 and 7). A putative recombination breakpoint was identified at nt position 214 (99 per cent CI: 204–219) [nt position 2,878 (99 per cent CI: 2,868–2,883) based on Jd53], sharing high similarity to Jd67 and other DcAstVs prior to this position, but following this point, Jd73 shares high similarity to the recombinant Jd76 as well as PoAstV-2 and BcAstV-NMG31 throughout most of the ORF1b regions. A second recombination breakpoint was identified at nt position 1,237 (99 per cent CI: 1,209–1,260) [nt position 3,901 (99 per cent CI: 3,873–3,924) based on Jd53], just upstream of the ORF1b-ORF2 junction. Here, Jd73 shares the highest similarity to Jd67 and other DcAstVs. A third recombination breakpoint was identified within the ORF2 at nt position 1,718 (99 per cent CI: 1,708–1,733) [nt position 4,391 (99 per cent CI: 4,381–4,406) based on Jd53], at which point Jd73 returns to sharing the highest similarity with recombinant Jd76. Within the ORF2 phylogeny, Jd73 groups with Jd76 and Jd53 in DcAstV variant group 2 (Figs 3 and 4).
Figure 7.

Genomic recombination in the camelid astrovirus DcAstV-Jd73. (A) Partial genome structure for DcAstV-Jd73 with ML trees constructed before and after each putative breakpoint corresponding to the alternating white- and gray-shaded regions. (B) Similarity analysis performed using the partial genome of DcAstV-Jd73R as the query sequence. Note that ‘*’ indicates sequences containing putative recombination breakpoints. (C) Bootscan analysis plot for DcAstV-Jd73R. Putative recombination breakpoints in DcAstV-Jd73R were identified at 214 nt (99 per cent CI: 204–219); at 1,237 nt (99 per cent CI: 1,209–1,260); and at 1,718 nt (99 per cent CI: 1,708–1,733) through comparison with sequences identified as putative parental sequences in RDP4. Nucleotide position within the multiple alignments is represented on the x-axis, while the y-axis represents pairwise bootstrap support (%). Pairwise comparison of DcAstV-Jd76R and DcAstV-Jd67R represented in yellow; DcAstV-Jd76R and DcAstV-Jd73R represented in green; and DcAstV-Jd67R and DcAstV-Jd73R represented in purple.
4. Discussion
Our survey of AstVs in Bactrian camels from the Inner Mongolian region of China and in dromedaries from Saudi Arabia revealed the presence of diverse and novel AstVs sharing gene regions with multiple viruses within the putative species clade including MamAstV-29, 30, 31, 32, and 33. Previous reports have suggested the potential for interspecies transmission events within this lineage containing AstVs of dromedaries, swine, porcupine, and diverse ruminant host species (Smits et al. 2010; Hu et al. 2014; Donato and Vijaykrishna 2017; Guan et al. 2018).
Through incorporating a hybrid RACE-Seq workflow, we managed to expand the known diversity of MamAstVs and provide evidence for recombination among members of this lineage. To our knowledge, this is the first report of AstV detection in Bactrian camels, which we propose to formally describe as Bactrian camel astrovirus 1 (BcAstV-1), corresponding to BcAstV-NMG31, and Bactrian camel astrovirus 2 (BcAstV-2), represented by BcAstV-NMG59. The BcAstV isolates were highly divergent, with one (BcAstV-NMG59) falling within the dromedary AstV clade and another, more distantly related isolate (BcAstV-NMG31), nested within the PoAstV-2 clade (MamAstV-32). BcAstV-NMG31 may be a bona fide camelid virus capable of infecting and replicating within the camel gastrointestinal tract that had evaded the detection until now although it may also represent an instance of cross-species transmission from other livestock species.
We also detected short RdRp fragments of human or human-like AstVs in the Bactrian camel samples, but we were unable to obtain longer-length genomic sequences for these viruses using RACE-Seq. Whether these represent cross-species transmission between humans and camels or if this may be attributed to environmental contamination requires further investigation. We cannot discount the possibility that these virus particles were present in camel feed or otherwise entered their diet through behaviors such as coprophagy, passing through the camel digestive tract but not necessarily causing active infection. The AstVs we have isolated from dromedaries formed a clade with previously described dromedary AstV isolates from the UAE (Woo et al. 2015), based on the capsid protein or ORF2 phylogeny. The close relatedness among the dromedary AstV strains points to a shared common ancestry of DcAstVs.
We provide evidence for the occurrence of recombination among camelid AstVs and their relatives, indicating that cross-species transmission of viruses within this clade may be especially common and facilitated through recombination. Coinfection of AstV species or strains is a prerequisite for recombination (De Benedictis et al. 2011), with reports of coinfection in pediatric patients (Cortez et al. 2017a) and of genetically distinct AstVs in porcine and bovine hosts (Luo et al. 2011; Tse et al. 2011; Xiao et al. 2013). The frequency of AstV coinfection that underlies the potential for recombination is necessarily dependent on viral, host, and environmental factors as recorded for other RNA viruses (Simon-Loriere and Holmes 2011). Recombinant AstVs have been previously described from human, bovine, porcine, and avian hosts (De Benedictis et al. 2011; Wohlgemuth, Honce, and Schultz-Cherry 2019), but the absence of widespread genomic surveillance in diverse host species presents a challenge for the detection of recombination events and likely leads to underreporting of this phenomenon.
Earlier, we reported the substantial presence of respiratory viruses such as alpha- and MERS-CoVs in juvenile dromedaries at about twice the rate detected in older camels (Sabir et al. 2016). Similarly, in this study, most AstVs were observed in camel calves under 6 months old. Among them, individuals Jd53, Jd73, and Jd76 were found to harbor recombinant DcAstVs in rectal swabs and camel alphacoronaviruses in their paired nasal swabs (Sabir et al. 2016) (Supplementary Table S1). With the small sample size in this study, we cannot confidently say whether this was just a coincidence or if there were any associations among the presence of recombinant AstV, the coinfections of AstV and CoV, and the age of camels. More investigations are needed to explore patterns of virus–virus interactions and coinfection dynamics.
The MamAstV diversity we have characterized from the dromedary and Bactrian camels appears to represent globally distributed viruses of livestock and domestically reared animals, indicating potential origins in other livestock or farmed animal species. Based on evidence for recombination between diverse AstVs in this lineage, it may also be possible that these camelid-associated AstVs have originated in other wild ungulate or livestock species, and these recombination events may have occurred following interspecies transmission from an as-yet-unknown host to camels. We also hypothesize that novel combinations of nonstructural and capsid proteins may prove adaptive in novel host environments.
The characterization of AstVs from old world and new world camelids may provide further evidence for the existence of a clade of camelid-associated AstVs and may provide a clearer understanding of the evolutionary history of these viruses. Given the wide breadth of host species from which AstVs have been described, interspecies transmission clearly plays an important role in the evolutionary history of this virus (Zhu et al. 2009; De Grazia et al. 2012; Hu et al. 2014; Meliopoulos et al. 2014; Sun et al. 2014; Mendenhall, Smith, and Vijaykrishna 2015; Pankovics et al. 2015). Our findings warrant further characterization of these viruses from a broader geographic region and from diverse host species in the context of virus evolution and emergence. Further work is needed to investigate the epidemiology and pathogenesis of AstV infection in camelids and to establish the role of AstVs as etiologic agents of neurologic or diarrheal diseases. Future directions in AstV research should prioritize (1) surveillance for AstVs in diverse host species across different geographic regions and seasons, aiming to better understand AstV ecology, evolution, and emergence; (2) characterization of host transmission networks for particular AstV subtypes, especially those subtypes containing viruses with zoonotic or epizootic potential; and (3) investigation of recombination events within and across AstV lineages to uncover patterns of interspecific transmission.
Supplementary Material
Acknowledgements
We thank our colleagues, collaborators, and laboratory support staff at the State Key Laboratory of Emerging Infectious Diseases (HKU), the Joint Institute of Virology (STU-HKU), and King Abdulaziz University.
Contributor Information
Muhammad I Qureshi, Guangdong-Hong Kong Joint Laboratory of Emerging Infectious Diseases/MOE Joint Laboratory for International Collaboration in Virology and Emerging Infectious Diseases, Joint Institute of Virology (Shantou University/The University of Hong Kong), Shantou University, 243 Daxue Road, Shantou, Guangdong 515063, China.
Brian M Worthington, Guangdong-Hong Kong Joint Laboratory of Emerging Infectious Diseases/MOE Joint Laboratory for International Collaboration in Virology and Emerging Infectious Diseases, Joint Institute of Virology (Shantou University/The University of Hong Kong), Shantou University, 243 Daxue Road, Shantou, Guangdong 515063, China; State Key Laboratory of Emerging Infectious Diseases, School of Public Health, Li Ka Shing Faculty of Medicine, The University of Hong Kong, 5/F, Lab Block, 21 Sassoon Road, Pokfulam, Hong Kong SAR 000, China; Exploration, Knowledge, Intelligence and Health, Gewuzhikang (EKIH) Pathogen Research Institute, 13/F, Building 3, 3 Binglang Road, Futian District, Shenzhen, Guangdong 518045, China.
Yongmei Liu, Guangdong-Hong Kong Joint Laboratory of Emerging Infectious Diseases/MOE Joint Laboratory for International Collaboration in Virology and Emerging Infectious Diseases, Joint Institute of Virology (Shantou University/The University of Hong Kong), Shantou University, 243 Daxue Road, Shantou, Guangdong 515063, China; State Key Laboratory of Emerging Infectious Diseases, School of Public Health, Li Ka Shing Faculty of Medicine, The University of Hong Kong, 5/F, Lab Block, 21 Sassoon Road, Pokfulam, Hong Kong SAR 000, China; Exploration, Knowledge, Intelligence and Health, Gewuzhikang (EKIH) Pathogen Research Institute, 13/F, Building 3, 3 Binglang Road, Futian District, Shenzhen, Guangdong 518045, China.
William Y.-M Cheung, Guangdong-Hong Kong Joint Laboratory of Emerging Infectious Diseases/MOE Joint Laboratory for International Collaboration in Virology and Emerging Infectious Diseases, Joint Institute of Virology (Shantou University/The University of Hong Kong), Shantou University, 243 Daxue Road, Shantou, Guangdong 515063, China; State Key Laboratory of Emerging Infectious Diseases, School of Public Health, Li Ka Shing Faculty of Medicine, The University of Hong Kong, 5/F, Lab Block, 21 Sassoon Road, Pokfulam, Hong Kong SAR 000, China; Exploration, Knowledge, Intelligence and Health, Gewuzhikang (EKIH) Pathogen Research Institute, 13/F, Building 3, 3 Binglang Road, Futian District, Shenzhen, Guangdong 518045, China.
Shuo Su, Ministry of Education (MOE), Joint International Research Laboratory of Animal Health and Food Safety, Jiangsu Engineering Laboratory of Animal Immunity, Institute of Immunology, College of Veterinary Medicine, Academy for Advanced Interdisciplinary Studies, Nanjing Agricultural University, 1 Weigang Road, Nanjing, Jiangsu 210095, China.
Zuoyi Zheng, Guangdong-Hong Kong Joint Laboratory of Emerging Infectious Diseases/MOE Joint Laboratory for International Collaboration in Virology and Emerging Infectious Diseases, Joint Institute of Virology (Shantou University/The University of Hong Kong), Shantou University, 243 Daxue Road, Shantou, Guangdong 515063, China.
Lifeng Li, Guangdong-Hong Kong Joint Laboratory of Emerging Infectious Diseases/MOE Joint Laboratory for International Collaboration in Virology and Emerging Infectious Diseases, Joint Institute of Virology (Shantou University/The University of Hong Kong), Shantou University, 243 Daxue Road, Shantou, Guangdong 515063, China; State Key Laboratory of Emerging Infectious Diseases, School of Public Health, Li Ka Shing Faculty of Medicine, The University of Hong Kong, 5/F, Lab Block, 21 Sassoon Road, Pokfulam, Hong Kong SAR 000, China; Exploration, Knowledge, Intelligence and Health, Gewuzhikang (EKIH) Pathogen Research Institute, 13/F, Building 3, 3 Binglang Road, Futian District, Shenzhen, Guangdong 518045, China.
Tommy T -Y Lam, Guangdong-Hong Kong Joint Laboratory of Emerging Infectious Diseases/MOE Joint Laboratory for International Collaboration in Virology and Emerging Infectious Diseases, Joint Institute of Virology (Shantou University/The University of Hong Kong), Shantou University, 243 Daxue Road, Shantou, Guangdong 515063, China; State Key Laboratory of Emerging Infectious Diseases, School of Public Health, Li Ka Shing Faculty of Medicine, The University of Hong Kong, 5/F, Lab Block, 21 Sassoon Road, Pokfulam, Hong Kong SAR 000, China; Exploration, Knowledge, Intelligence and Health, Gewuzhikang (EKIH) Pathogen Research Institute, 13/F, Building 3, 3 Binglang Road, Futian District, Shenzhen, Guangdong 518045, China; Laboratory of Data Discovery for Health Limited, 12/F, Building 19W, 19 Science Park West Avenue, Hong Kong Science Park, Pak Shek Kok, New Territories, Hong Kong SAR 000, China.
Yi Guan, Guangdong-Hong Kong Joint Laboratory of Emerging Infectious Diseases/MOE Joint Laboratory for International Collaboration in Virology and Emerging Infectious Diseases, Joint Institute of Virology (Shantou University/The University of Hong Kong), Shantou University, 243 Daxue Road, Shantou, Guangdong 515063, China; State Key Laboratory of Emerging Infectious Diseases, School of Public Health, Li Ka Shing Faculty of Medicine, The University of Hong Kong, 5/F, Lab Block, 21 Sassoon Road, Pokfulam, Hong Kong SAR 000, China; Exploration, Knowledge, Intelligence and Health, Gewuzhikang (EKIH) Pathogen Research Institute, 13/F, Building 3, 3 Binglang Road, Futian District, Shenzhen, Guangdong 518045, China; Laboratory of Data Discovery for Health Limited, 12/F, Building 19W, 19 Science Park West Avenue, Hong Kong Science Park, Pak Shek Kok, New Territories, Hong Kong SAR 000, China.
Huachen Zhu, Guangdong-Hong Kong Joint Laboratory of Emerging Infectious Diseases/MOE Joint Laboratory for International Collaboration in Virology and Emerging Infectious Diseases, Joint Institute of Virology (Shantou University/The University of Hong Kong), Shantou University, 243 Daxue Road, Shantou, Guangdong 515063, China; State Key Laboratory of Emerging Infectious Diseases, School of Public Health, Li Ka Shing Faculty of Medicine, The University of Hong Kong, 5/F, Lab Block, 21 Sassoon Road, Pokfulam, Hong Kong SAR 000, China; Exploration, Knowledge, Intelligence and Health, Gewuzhikang (EKIH) Pathogen Research Institute, 13/F, Building 3, 3 Binglang Road, Futian District, Shenzhen, Guangdong 518045, China; Laboratory of Data Discovery for Health Limited, 12/F, Building 19W, 19 Science Park West Avenue, Hong Kong Science Park, Pak Shek Kok, New Territories, Hong Kong SAR 000, China.
Data availability
GenBank accession numbers for complete and partial genomes sequenced in this study are OP076743-OP076766.
Supplementary data
Supplementary data are available at Virus Evolution online.
Funding
This work was supported by the Shenzhen (HZQB-KCZYZ-2021014) and Guangdong government (2019B121205009 and 190824215544727), research grants from the National Natural Science Foundation of China (31270435/C030102), the National Center for International Research of Genomics (2017B01012), the National Key Plan for Scientific Research and Development of China (2016YFD0500302 and 2017YFE0190800), and the Li Ka Shing Foundation.
Conflict of interest:
No potential conflicts of interest were disclosed.
References
- Adney D. R. et al. (2019) ‘Bactrian Camels Shed Large Quantities of Middle East Respiratory Syndrome Coronavirus (MERS-CoV) after Experimental Infection’, Emerging Microbes & Infections, 8: 717–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger A. M., Lohse M., and Usadel B. (2014) ‘Trimmomatic: A Flexible Trimmer for Illumina Sequence Data’, Bioinformatics, 30: 2114–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch A. et al. (2011) ‘Family Astroviridae’. In: King A. M. Q., Adams M. J., Carstens E. B. and Lefkowitz E. J. (eds) Virus Taxonomy: Classification and Nomenclature of Viruses (Ninth Report of the International Committee on the Taxonomy of Viruses), pp. 953–9. Elsevier Academic Press: New York. [Google Scholar]
- Buchfink B., Xie C., and Huson D. H. (2015) ‘Fast and Sensitive Protein Alignment Using DIAMOND’, Nature Methods, 12: 59–60. [DOI] [PubMed] [Google Scholar]
- Chan S. M. S. et al. (2015) ‘Absence of MERS-Coronavirus in Bactrian Camels, Southern Mongolia, November 2014’, Emerging Infectious Diseases, 21: 1269–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu D. K. W. et al. (2008) ‘Novel Astroviruses in Insectivorous Bats’, Journal of Virology, 82: 9107–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortez V. et al. (2017a) ‘Persistent Infections with Diverse Co-Circulating Astroviruses in Pediatric Oncology Patients, Memphis, Tennessee, USA’, Emerging Infectious Diseases, 23: 288–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— et al. (2017b) ‘Astrovirus Biology and Pathogenesis’, Annual Review of Virology, 4: 327–48. [DOI] [PubMed] [Google Scholar]
- ——— et al. (2020) ‘Astrovirus Infects Actively Secreting Goblet Cells and Alters the Gut Mucus Barrier’, Nature Communications, 11: 2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Benedictis P. et al. (2011) ‘Astrovirus Infections in Humans and Animals – Molecular Biology, Genetic Diversity, and Interspecies Transmissions’, Infection, Genetics and Evolution, 11: 1529–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Grazia S. et al. (2012) ‘Genetic Heterogeneity and Recombination in Human Type 2 Astroviruses’, Journal of Clinical Microbiology, 50: 3760–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donato C., and Vijaykrishna D. (2017) ‘The Broad Host Range and Genetic Diversity of Mammalian and Avian Astroviruses’, Viruses, 9: 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn R. D. et al. (2006) ‘Pfam: Clans, Web Tools and Services’, Nucleic Acids Research, 34: D247–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabherr M. G. et al. (2011) ‘Full-Length Transcriptome Assembly from RNA-Seq Data without a Reference Genome’, Nature Biotechnology, 29: 644–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan T.-P. et al. (2018) ‘Metagenomic Analysis of Sichuan Takin Fecal Sample Viromes Reveals Novel Enterovirus and Astrovirus’, Virology, 521: 77–91. [DOI] [PubMed] [Google Scholar]
- Guix S.Bosch A., and Pintó R. M. (2012) ‘Astrovirus Taxonomy’. In: Schultz-Cherry S. (ed.) Astrovirus Research, pp. 97–118. Springer: New York, NY. [Google Scholar]
- Hu B. et al. (2014) ‘Detection of Diverse Novel Astroviruses from Small Mammals in China’, Journal of General Virology, 95: 2442–9. [DOI] [PubMed] [Google Scholar]
- Ingle H. et al. (2019) ‘Viral Complementation of Immunodeficiency Confers Protection against Enteric Pathogens via Interferon-λ’, Nature Microbiology, 4: 1120–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang B. et al. (1993) ‘RNA Sequence of Astrovirus: Distinctive Genomic Organization and a Putative Retrovirus-Like Ribosomal Frameshifting Signal That Directs the Viral Replicase Synthesis’, Proceedings of the National Academy of Sciences, 90: 10539–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonassen C. M., Jonassen T. Ø., and Grinde B. (1998) ‘A Common RNA Motif in the 3h End of the Genomes of Astroviruses, Avian Infectious Bronchitis Virus and an Equine Rhinovirus’, Journal of General Virology, 79: 715–8. [DOI] [PubMed] [Google Scholar]
- Katoh K., and Standley D. M. (2013) ‘MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability’, Molecular Biology and Evolution, 30: 772–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearse M. et al. (2012) ‘Geneious Basic: An Integrated and Extendable Desktop Software Platform for the Organization and Analysis of Sequence Data’, Bioinformatics, 28: 1647–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosugi S. et al. (2009) ‘Systematic Identification of Cell Cycle-Dependent Yeast Nucleocytoplasmic Shuttling Proteins by Prediction of Composite Motifs’, Proceedings of the National Academy of Sciences, 106: 10171–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S. et al. (2018) ‘MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms’, Molecular Biology and Evolution, 35: 1547–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B., and Salzberg S. L. (2012) ‘Fast Gapped-Read Alignment with Bowtie 2’, Nature Methods, 9: 357–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. et al. (2009) ‘The Sequence Alignment/Map Format and SAMtools’, Bioinformatics, 25: 2078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y. et al. (2017) ‘Identification of Diverse Viruses in Upper Respiratory Samples in Dromedary Camels from United Arab Emirates’, PLoS ONE, 12: e0184718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lole K. S. et al. (1999) ‘Full-Length Human Immunodeficiency Virus Type 1 Genomes from Subtype C-Infected Seroconverters in India, with Evidence of Intersubtype Recombination’, Journal of Virology, 73: 152–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Z. et al. (2011) ‘Multiple Novel and Prevalent Astroviruses in Pigs’, Veterinary Microbiology, 149: 316–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin D. P. et al. (2015) ‘RDP4: Detection and Analysis of Recombination Patterns in Virus Genomes’, Virus Evolution, 1: vev003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meliopoulos V. A. et al. (2014) ‘Detection of Antibodies against Turkey Astrovirus in Humans’, PLoS ONE, 9: e96934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menachery V. D. et al. (2015) ‘A SARS-Like Cluster of Circulating Bat Coronaviruses Shows Potential for Human Emergence’, Nature Medicine, 21: 1508–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendenhall I. H., Smith G. J. D., and Vijaykrishna D. (2015) ‘Ecological Drivers of Virus Evolution: Astrovirus as a Case Study’, Journal of Virology, 89: 6978–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Méndez E., and Arias C. F. (2013) ‘Astroviruses’. In: Knipe, D. M., and Howley, P. M. (eds) Fields Virology, Vol. 1. 6th edn, pp. 609–28. Lippincott Williams & Wilkins: Philadelphia. [Google Scholar]
- Pankovics P. et al. (2015) ‘Detection of a Mammalian-Like Astrovirus in Bird, European Roller (Coracias garrulus)’, Infection, Genetics and Evolution, 34: 114–21. [DOI] [PubMed] [Google Scholar]
- Pantin-Jackwood M. J., Spackman E., and Woolcock P. R. (2006) ‘Phylogenetic Analysis of Turkey Astroviruses Reveals Evidence of Recombination’, Virus Genes, 32: 187–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabir J. S. M. et al. (2016) ‘Co-Circulation of Three Camel Coronavirus Species and Recombination of MERS-CoVs in Saudi Arabia’, Science, 351: 81–4. [DOI] [PubMed] [Google Scholar]
- Simon-Loriere E., and Holmes E. C. (2011) ‘Why Do RNA Viruses Recombine?’, Nature Reviews. Microbiology, 9: 617–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smits S. L. et al. (2010) ‘Identification and Characterization of Deer Astroviruses’, Journal of General Virology, 91: 2719–22. [DOI] [PubMed] [Google Scholar]
- Stamatakis A. (2014) ‘RAxML Version 8: A Tool for Phylogenetic Analysis and Post-Analysis of Large Phylogenies’, Bioinformatics, 30: 1312–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun N. et al. (2014) ‘Detection and Characterization of Avastrovirus Associated with Diarrhea Isolated from Minks in China’, Food and Environmental Virology, 6: 169–74. [DOI] [PubMed] [Google Scholar]
- Tse H. et al. (2011) ‘Rediscovery and Genomic Characterization of Bovine Astroviruses’, Journal of General Virology, 92: 1888–98. [DOI] [PubMed] [Google Scholar]
- Walter J. E. et al. (2001) ‘Molecular Characterization of a Novel Recombinant Strain of Human Astrovirus Associated with Gastroenteritis in Children’, Archives of Virology, 146: 2357–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohlgemuth N., Honce R., and Schultz-Cherry S. (2019) ‘Astrovirus Evolution and Emergence’, Infection, Genetics and Evolution, 69: 30–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo P. C. Y. et al. (2014) ‘Metagenomic Analysis of Viromes of Dromedary Camel Fecal Samples Reveals Large Number and High Diversity of Circoviruses and Picobirnaviruses’, Virology, 471–473: 117–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— et al. (2015) ‘A Novel Astrovirus from Dromedaries in the Middle East’, Journal of General Virology, 96: 2697–707. [DOI] [PubMed] [Google Scholar]
- Xiao C.-T. et al. (2013) ‘Identification and Characterization of Novel Porcine Astroviruses (PAstVs) with High Prevalence and Frequent Co-Infection of Individual Pigs with Multiple PAstV Types’, Journal of General Virology, 94: 570–82. [DOI] [PubMed] [Google Scholar]
- Xie J. et al. (2015) ‘Identification of Mammalian Species Using the Short and Highly Variable Regions of Mitochondrial DNA’, Mitochondrial DNA, 26: 550–4. [DOI] [PubMed] [Google Scholar]
- Zhang Y. et al. (2017) ‘Complete Genome Sequence of a Novel Avastrovirus in Goose’, Archives of Virology, 162: 2135–9. [DOI] [PubMed] [Google Scholar]
- Zhu H. et al. (2009) ‘Detection of Diverse Astroviruses from Bats in China’, Journal of General Virology, 90: 883–7. [DOI] [PubMed] [Google Scholar]
- Zuker M. (2003) ‘Mfold Web Server for Nucleic Acid Folding and Hybridization Prediction’, Nucleic Acids Research, 31: 3406–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
GenBank accession numbers for complete and partial genomes sequenced in this study are OP076743-OP076766.