

Abstract
Heat shock protein 90 (Hsp90) is a dynamic protein which serves to ensure proper folding of nascent client proteins, regulate transcriptional responses to environmental stress and guide misfolded and damaged proteins to destruction via ubiquitin proteasome pathway. Recent advances in the field of Hsp90 have been made through development of isoform selective inhibitors, Hsp90 C-terminal inhibitors and disruption of protein-protein interactions. These approaches have led to alleviation of adverse off-target effects caused by pan-inhibition of Hsp90 using N-terminal inhibitors. In this review, we provide an overview of relevant advances on targeting the Hsp90 C-terminal Domain (CTD) and the development of Hsp90 C-terminal inhibitors (CTIs) since 2015.
Graphical Abstract
The proper functioning and folding of proteins is necessary for cell survival.1 Cellular stresses such as acidosis, exposure to toxins, viruses, DNA damage, metabolic and oxidative stress, and/or hypoxia lead to the disruption of proteostasis, and can result in a lethal outcome. To maintain proteostasis upon exposure to cellular stresses, cells induce the expression of molecular chaperones such as the heat shock proteins.2 This protein family is divided based on molecular weight and includes Hsp27, Hsp40, Hsp60, Hsp70, Hsp90, as well as other larger members, each of which plays a unique role during the protein folding cycle.3
The 90kDa Heat Shock Proteins (Hsp90) are ubiquitous and highly-conserved molecular chaperones that are responsible for the activation, stabilization and maturation of ~400 client protein substrates. Hsp90 comprises ~1–2% of all cellular proteins, however, Hsp90 expression is elevated to ~4–6% of total protein content in cancer cells. Hsp90 is a homodimer (Figure 1) with each monomer consisting of three components; an ATP-binding N-terminal domain (NTD), a middle domain (MD) wherein protein-protein interactions occur, and a C-terminal domain (CTD) that is responsible for dimerization.4 The CTD contains a nucleotide binding site that allosterically modulates ATPase activity within the NTD.5 The CTD also possesses a conserved Met-Glu-Glu-Val-Asp (MEEVD) sequence at the terminus to provide interactions with co-chaperones that contain a tetratricopeptide-containing repeat (TPR).6 These interactions with co-chaperones are vital for the regulation and progression of the Hsp90 protein folding cycle.
Figure 1.
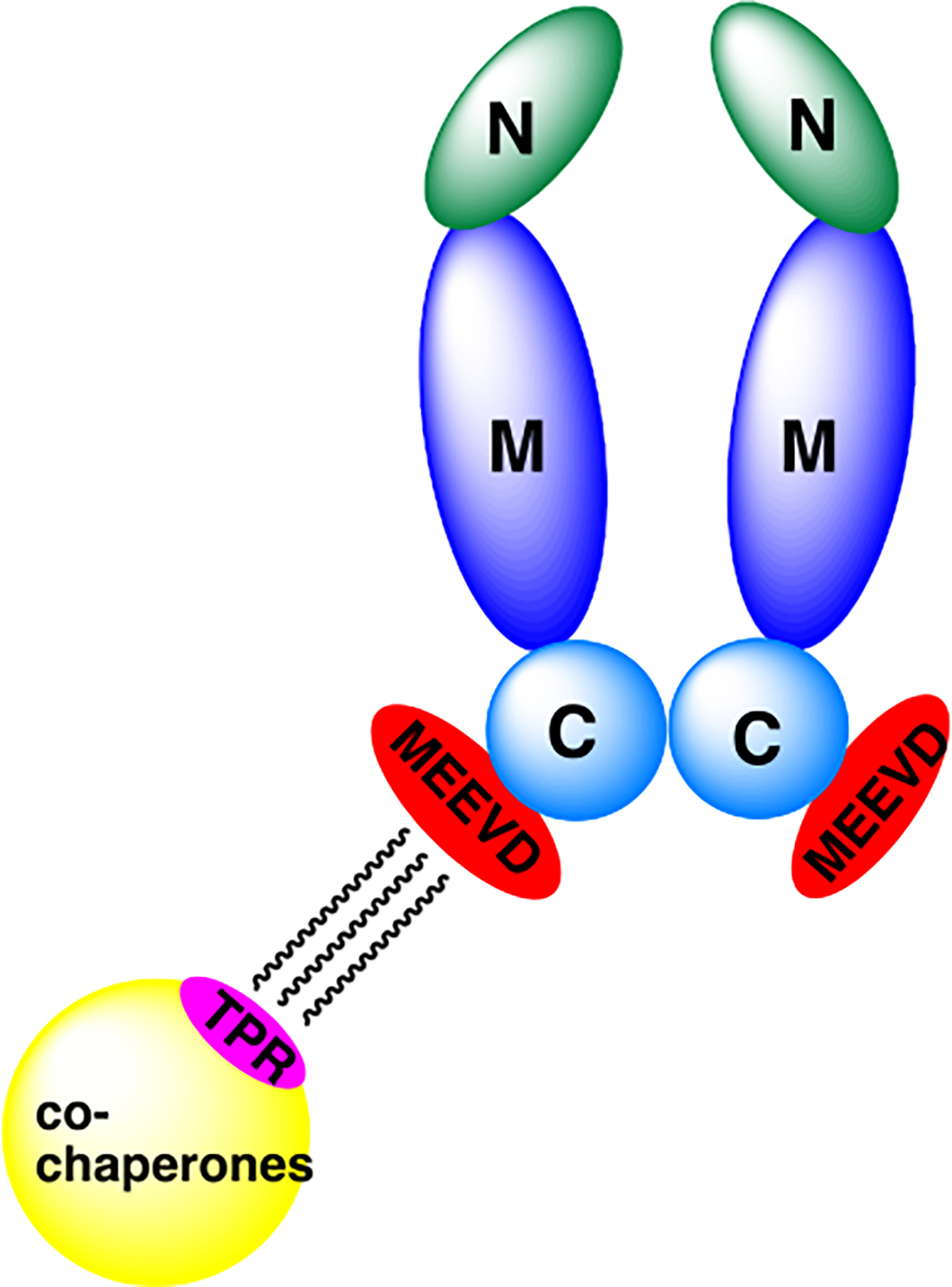
Hsp90 homodimer (N: NTD, M: MD, C: CTD).
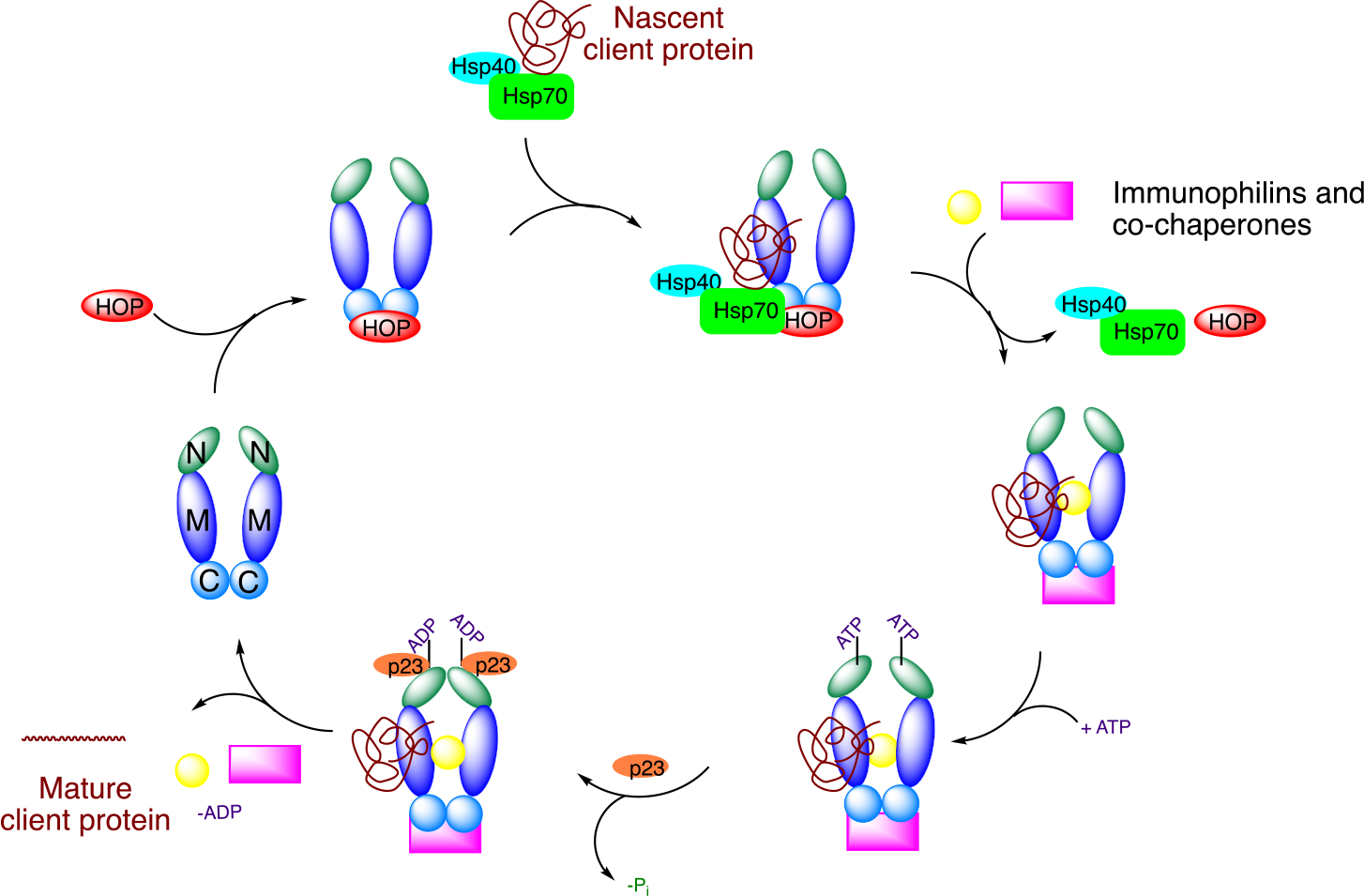
Although the Hsp90 protein folding machinery has been extensively studied, the exact mechanism is not fully understood. Studies suggest that the cycle begins when the Hsp90-Hsp70 Organizing Protein (HOP) transfers a nascent polypeptide from Hsp70 to Hsp90 via binding to the C-terminal domain as displayed in Figure 2.7,8 The nascent client proteins are delivered to Hsp90 through formation of a complex with Hsp70 and Hsp40. Upon delivery, immunophilins and co-chaperones can interact with Hsp90 to form a heteroprotein complex that assembles to fold nascent client proteins into their three-dimensional conformations.9 ATP binding and hydrolysis at the N-termini along with the association of co-chaperones (p23) and the activator of Hsp90 ATPase homologue 1 (Aha1) promote client protein maturation.10 After release of the properly folded client protein, the Hsp90 heteroprotein complex disassembles and the Hsp90 homodimer is regenerated to repeat the protein folding cycle.11
Figure 2.
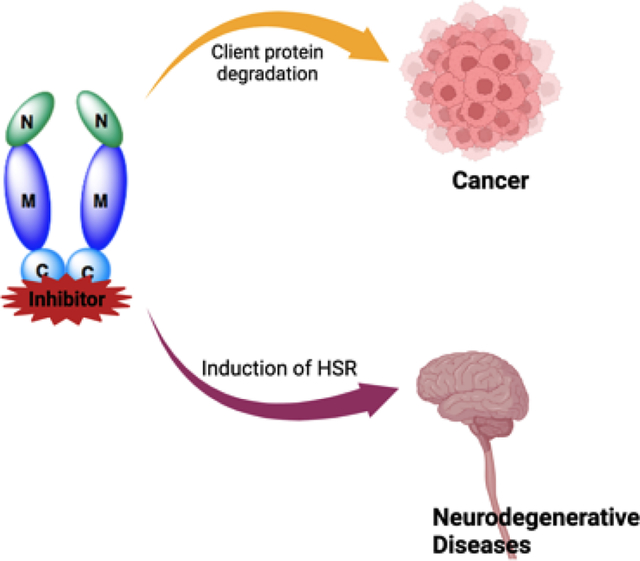
Hsp90 in cancer and neuropathy:
In cancer cells, Hsp90 levels are upregulated due to the excessive need to fold overexpressed and mutated proteins. Hsp90 interacts with more than 400 client proteins, many of which are associated with the ten hallmarks of cancer.1,13,14 These client proteins are often involved with the initiation, progression and/or metastasis of cancer cells. Therefore, inhibition of Hsp90 presents a unique opportunity to selectively target cancer cells and simultaneously disrupt multiple oncogenic pathways, a therapeutic strategy similar to combination therapy. In cancer cells, Hsp90 exists as a heteroprotein complex bound to both client proteins and co-chaperones instead of the inactive homodimer complex that resides within normal tissue. The heteroprotein complex exhibits significantly higher affinity for ATP and ligands that bind competitively versus ATP. Hence, Hsp90 has been shown to exhibit a higher affinity for inhibitors in cancer cells as compared to normal cells.15,16,17 However, when an Hsp90 inhibitor binds to the N- or C- terminus, the client protein does not reach maturation and instead is degraded via the ubiquitin-proteosome pathway.
Nineteen Hsp90 N-terminal inhibitors have entered clinical trials, but most have failed due to toxicities resulting from pan-inhibitory activity and their ability to induce the heat shock response (HSR).18 The HSR is a pro-survival mechanism that leads to the upregulation of Hsp90 and other heat shock proteins as a method to overcome cellular stress. Increased concentrations of inhibitors are necessary to inhibit the increased Hsp90 levels and unfortunately, results in dose-escalating toxicities. Therefore, C-terminal inhibition offers a valuable alternative to target cancer cells without induction of the HSR.19
The HSR is regulated by the transcription factor, heat shock factor-1 (HSF-1). Under normal cellular conditions, HSF-1 remains bound to Hsp90 (Figure 3). However, during cellular stress or elevated temperatures, HSF-1 dissociates from Hsp90 and trimerizes before entering the nucleus, which leads to the transcriptional upregulation of antioxidant genes and molecular chaperones.12,20 Studies have shown that the upregulation of Hsp can lead to the rematuration of denatured and/or damaged proteins.21 An increase in Hsp70 therefore, can result in a significant reduction in Aβ-induced cell death as well as a reduction in hyperphosphorylated Tau proteins.15,22 In addition, the upregulation of Hsp70 has been shown to reverse diabetic peripheral neuropathy (DPN) and improve mitochondrial bioenergetics.12,22,23,24 Thus, while induction of the HSR poses detrimental activities for anticancer agents, it is advantageous for the treatment of neurodegenerative diseases.
Figure 3.
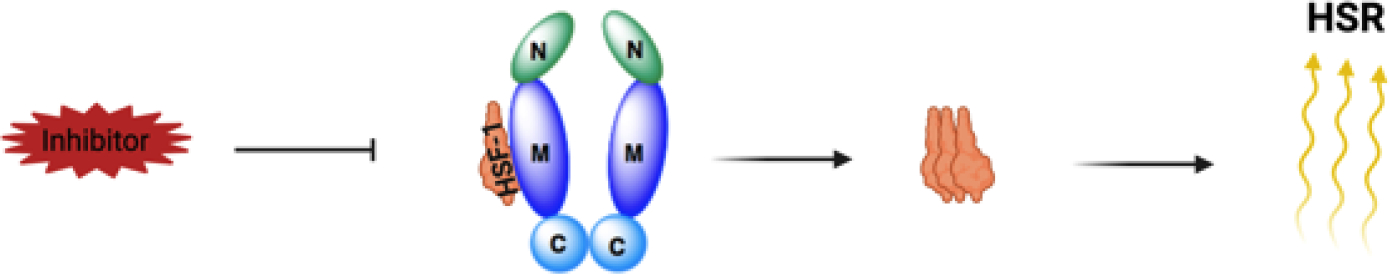
Mechanism of the heat shock response (HSR).
Hsp90 C-terminal binding site:
Given the therapeutic advantages of targeting the Hsp90 CTD, several researchers have strived to develop small molecule inhibitors. However, one of the key challenges for the rational development of improved analogs is the lack of a co-crystal structure of inhibitors bound to the Hsp90 CTD.25 The CTD exhibits four biochemical properties; 1) binds ligands, 2) forms an Hsp90 homodimer, 3) maintains interactions with co-chaperones, and 4) provides resistance to proteolysis in the ligand-bound state.26,27,28 The Hsp90 CTD contains several binding regions; a nucleotide binding site, the MEEVD motif located at the termini, and the region between the MD and dimerization interface.29 In addition, computational studies have identified at least four pockets in the CTD as putative binding sites for Hsp90 inhibitors.30
Despite identification of these putative binding pockets, the exact location of the nucleotide binding site remains unknown. An overview of approaches used to elucidate the ATP binding site is presented in Figure 4.31 The presence of a nucleotide binding site in the CTD was first proposed by Matts and co-workers when they found that molybdate inhibited the binding of geldanamycin (a N-terminal inhibitor) to the N-terminus, and simultaneously protected the C-terminus from proteolysis.32,33 Subsequent studies by Neckers and coworkers demonstrated that ATP and novobiocin bound to the same C-terminal binding site, and within amino acids 657–677.34,35 Further studies revealed that the C-terminal ATP binding site is only accessible upon occupancy of the N-terminal ATP binding site. Moreover, the binding of novobiocin at the CTD produces a conformational change that allosterically modulates the ATPase activity within the NTD.5 The CTD binds both purine and pyrimidine nucleotides, whereas NTD specifically binds purine nucleotides. These functional differences highlight the importance of obtaining a co-crystal structure and exact location of the C-terminal nucleotide binding site.36
Figure 4.
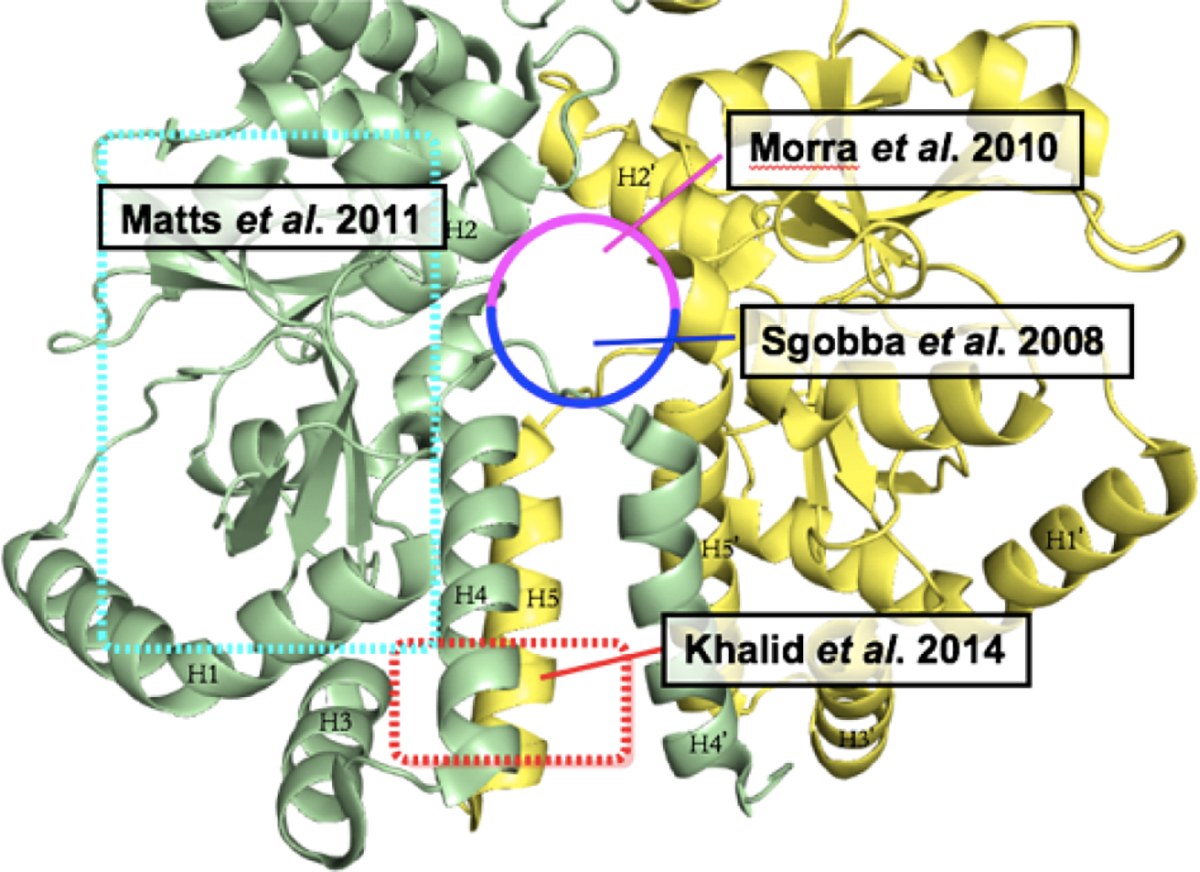
Potential CTD binding sites.31
Matts and coworkers recently reported the co-crystal structure of a coumarin based compound, 7-diethylamino-3-[N-(2-maleimidoethyl)carbamoyl]coumarin (MDCC) bound to the Hsp90α middle and C-terminal (MC) domain interface. In this binding site, there are 3 loops; a catalytic loop that is important for ATP hydrolysis, an Src loop and a CTD loop for client binding, which reorients upon ligand binding to the Hsp90αMC domain. Hsp90α adopts a unique conformation upon binding to MDCC, which leads to the inhibition of client protein binding to the CTD and Src loops. In addition, the Hsp90 dimer is required for client protein maturation, however, upon MDCC binding to Hsp90, the protein is locked into an inactive hexamer, which prevents ATP hydrolysis. A florescence competition assay was used to demonstrate that other coumarin-based compounds such as novobiocin also compete with the binding of MDCC. In the co-crystal structure presented in this study, the coumarin core is sequestered into a hydrophobic binding pocket, which is consistent with prior SAR studies on novobiocin.37
Challenges associated with the binding of modulators to the CTD in the absence of a co-crystal structure has been partially circumvented by the use of saturation transfer difference (STD) NMR spectroscopy.38,39,40 Consistent with prior observations, this study recognized that the inhibitory activity manifested by CTD modulators is caused by long range allosteric structural rearrangements within the NTD, which is facilitated through movement of the MD or disruption of co-chaperone interactions. Upon the binding of novobiocin (1) or KU32 (2) (a first-generation novobiocin analog) to the CTD, a strong allosteric cross-talk was observed between the CTD and NTD, which reduced Hsp90’s affinity for geldanamycin (a N-terminal inhibitor) by >100 fold (Figure 5).38 Given the importance of the allosteric binding site, several small molecules have emerged as CTD modulators and are briefly highlighted below.
Figure 5.
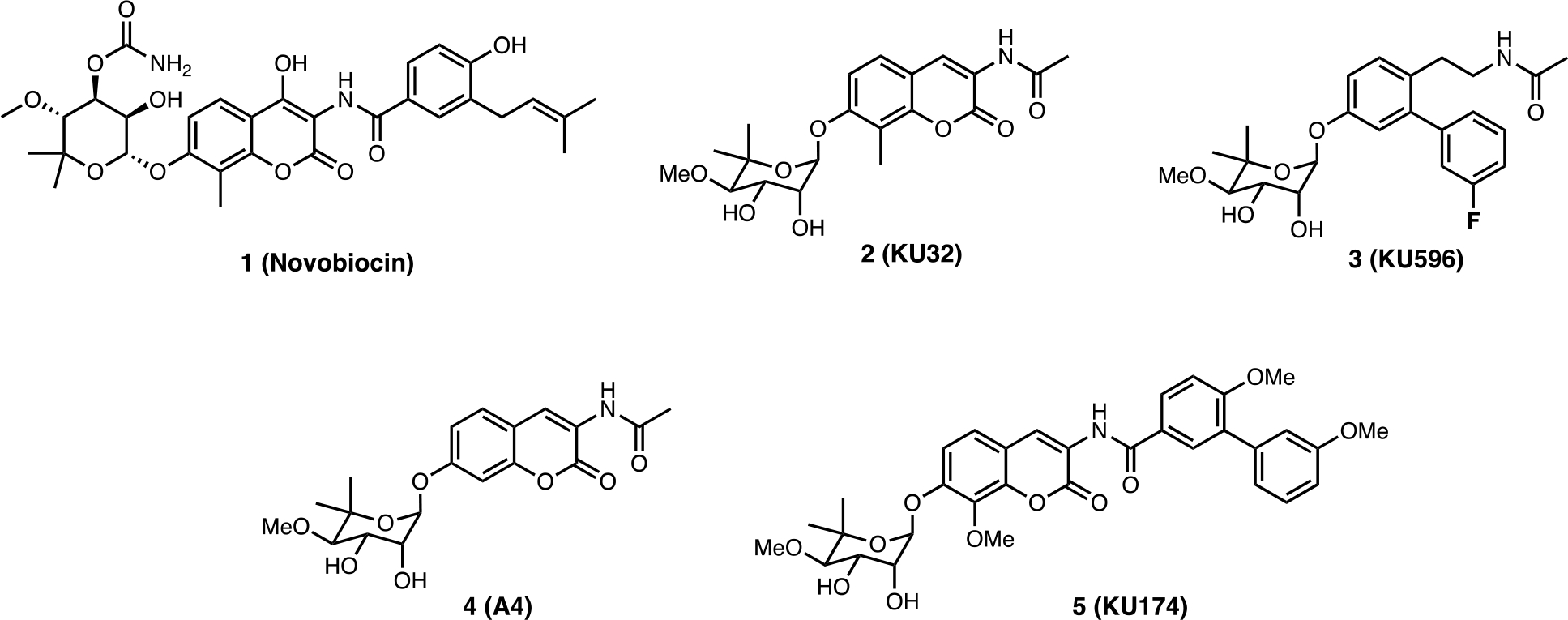
Novobiocin and its analogs.
Novobiocin-based CTIs:
Since the discovery of novobiocin as the first CTI, many analogs have been prepared and some structure-activity relationships (SAR) elucidated, which led to improved modulators of the CTD. A4 (4) was the first novobiocin analog reported and incorporated an acetamide side chain in lieu of the prenylated benzamide side chain found in novobiocin. Interestingly, replacement of the side chain with a smaller acetamide led to induction of the HSR at a concentration 1000-fold lower than that needed for client protein degradation.41 Consequently, an SAR study on the amide side chain was pursued and demonstrated that a five carbon appendage on the amide side chain diverts a C-terminal modulator from a neuroprotective agent to an antiproliferative agent. In fact, molecules were found to manifest an increase in antiproliferative activity as the amide side chain got larger.42 Exploration of the bulkiness on the amide side chain led to KU174 (5), which includes both a noviose sugar and a benzamide side chain. In contrast to 4, 5 disrupted the Hsp90α/Aha1 complex and exhibited significant anti-proliferative activity against MCF-7 cells.42,43 On the other hand, optimization of 4 led to KU32 (2) and eventually KU596 (3) as neuroprotective agents. Both 2 and 3 can induce the HSR without client protein degradation, whereas 5 led to the degradation of client proteins without an induction of HSR, clearly highlighting mechanistic activities that differ from N-terminal inhibitors.24 Therefore, C-terminal modulators are promising agents that can display either antiproliferative or neuroprotective activity.
Following this study, a series of novobiocin analogs were prepared that focused on optimization and exploration of the coumarin core, the benzamide side chain, and the noviose sugar present in 1. Replacement of the noviose sugar with noviomimetics 6 (a cyclohexyl derivative containing a 4-benzyl ether) solved many synthetic challenges associated with the 10-step preparation of noviose, while maintaining the ability to induce the HSR (Figure 6), which is a key mechanistic feature required for neuroprotective activity.44 Based on 6, 3’- and 4’-substituted cyclohexyl scaffolds were identified as good noviomimetics that increased mitochondrial bioenergetics as needed for cytoprotective activity. 7 exhibited a significant shift in the ATP index towards OxPhos, also indicating cytoprotection.45
Figure 6.
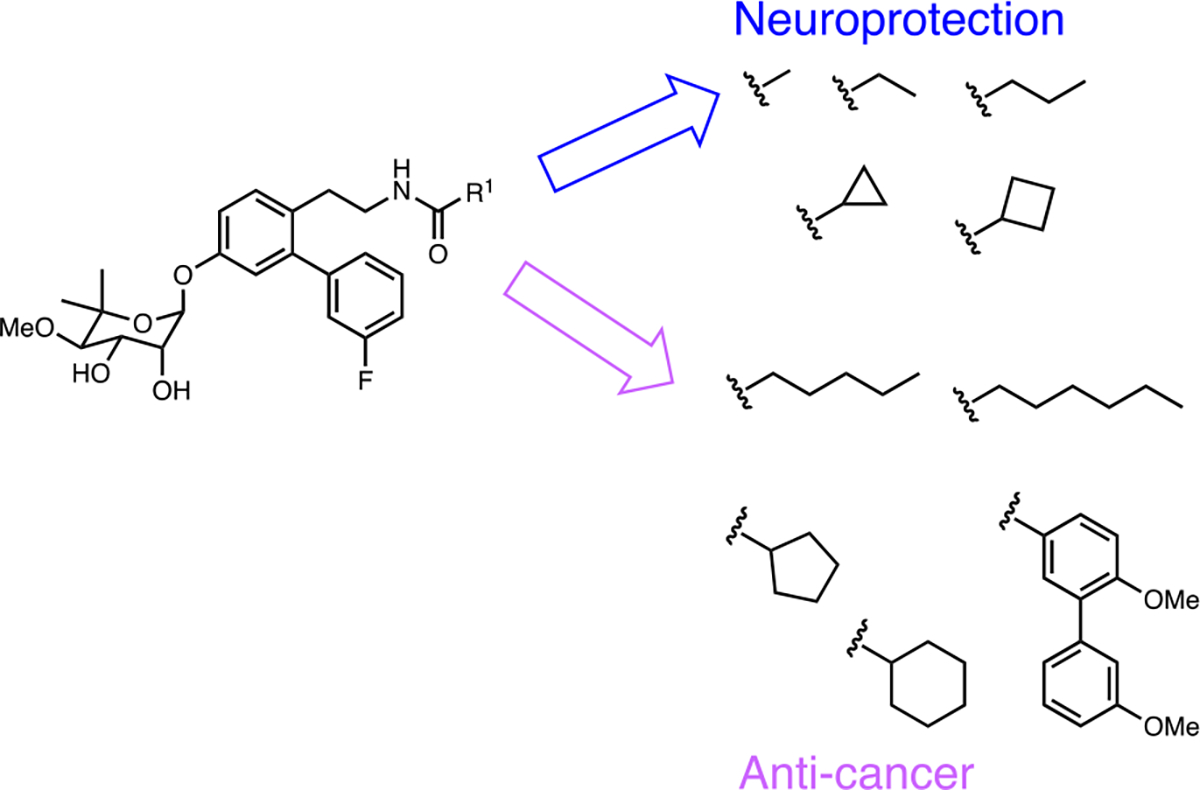
Differentiation of side chain moiety for neuroprotection vs anticancer.42
Previous SAR studies indicated that replacement of the coumarin core with a biphenyl moiety led enhanced antiproliferative activity. Therefore, a series of biphenylamides (8) were synthesized and contained a hydrophilic region, which improved potency against both MCF-7 and SKBr3 cells as confirmed through western blot analyses and luciferase refolding assays. Amines attached to a three carbon linker provided enhanced inhibitory activity.46 Furthermore, a saturated ring that allowed the molecule to adopt a three-dimensional conformation also appeared to improve binding as an increase in anti-proliferative activity was observed. Such studies eventually led to the discovery of phenyl cyclohexyl carboxamides 9, which manifested improved antiproliferative activity against SKBr3 cells.47
Prior studies on the length of the amide side chain provided additional insights into the ability to target different diseases. In the presence of an acetamide (eg. KU32, KU598), Hsp90-Aha1 interactions are stabilized and are important for Hsp90-mediated protein folding activity, whereas the benzamide containing compounds (eg. KU174) destabilized interactions between Hsp90 and Aha1, which led to the inhibition of Hsp90 protein folding activity (Figure 6).43 After identification of the optimal sugar surrogates, the amide was investigated. SAR studies led to identification of thiourea and urea based derivatives, KU711 (10) and KU757 (12), which displayed potent activity against head and neck squamous cell carcinoma (HNSCC). 10 and 12 significantly decreased the rate migration, invasion, self-renewal, and epithelial to mesenchymal transition (EMT) of several HNSCC cell lines at a low μM concentrations (Figure 7).48 Furthermore, 12 displayed potency against two thyroid cancer cell lines and exhibited 4- to 10-fold differential selectivity over normal fibroblasts. 12 also suppressed the Warburg effect during the treatment of lenvatinib resistant thyroid cancers, which led to a decreased dependence on glycolysis.49
Figure 7.
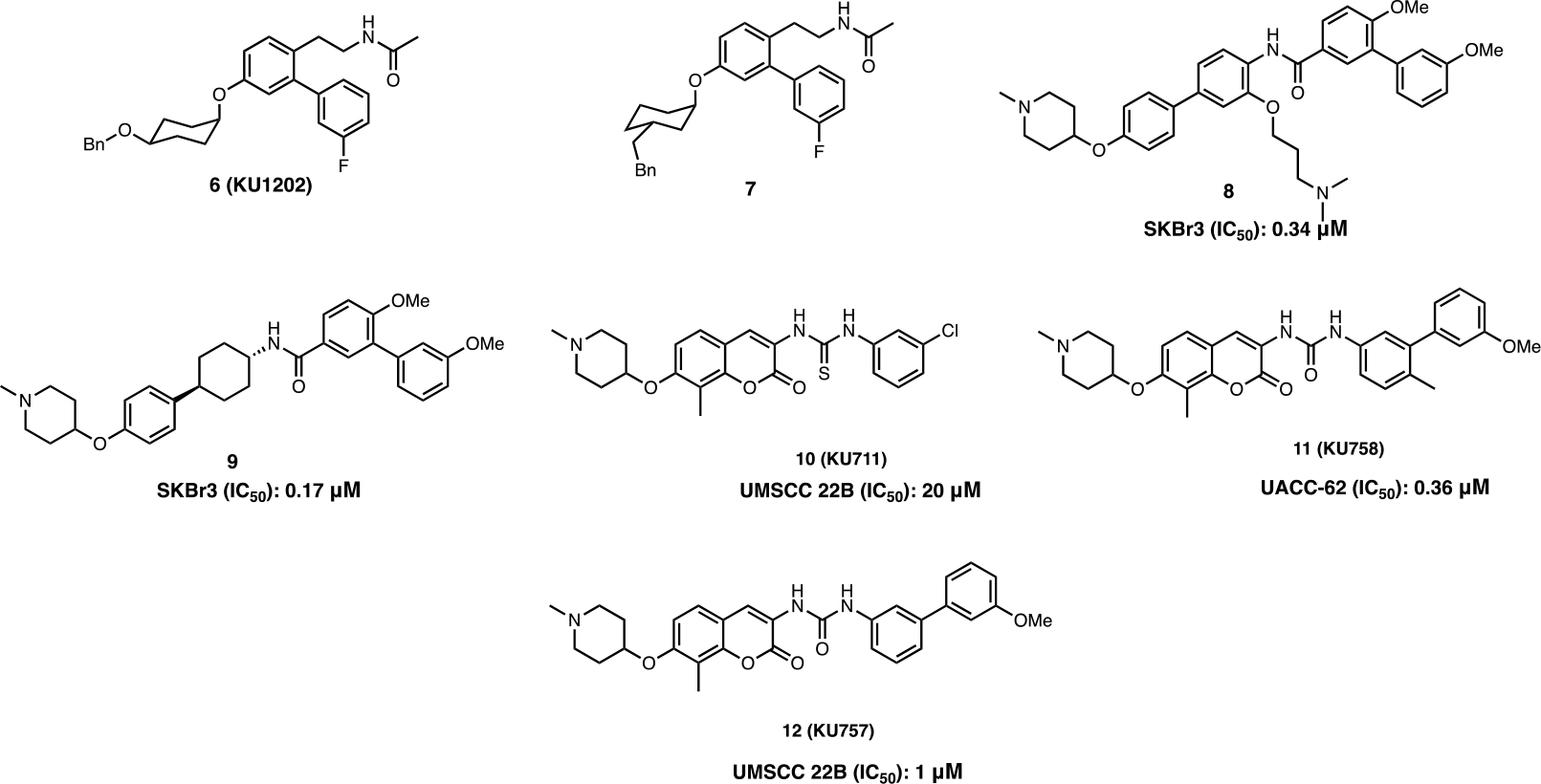
Novobiocin based CTIs.
Heat shock proteins (Hsp90 and Hsp70) are known to play a critical role in the folding and conformational maintenance of a diverse set of client proteins involved in cancer stem cell (CSC) developmental pathways and include kinases, transcription factors, and other proteins.50 Since these inhibitors target CSC’s, KU758’s (11) co-administration with standard chemotherapy drugs (vemurafenib and cobimetinib) led to synergistic activity against BRAF-mutant tumors and overcame several resistance pathways in metastatic melanoma. KU758 also induced apoptosis through poly(ADP-ribose) polymerase (PARP) cleavage, cell cycle arrest at G0/G1, cell migration, and mitigated drug-resistance pathways. Therefore, a combination drug strategy with CTIs could provide a new method to overcome metastatic melanoma and other resistant cancers.51
The combination of appendages from previously identified scaffolds into a single molecule can provide additional insights into the three-dimensional space within the Hsp90 C-terminal binding pocket. Therefore, a stilbene- and novobiocin chimera (13) which contains an N-methyl piperidine and a biaryl amide side chain was synthesized. Compounds that positioned the sugar and amide moieties between 7.7 to 12.1 Å apart at an angle of 180° led to enhanced anti-proliferative activity and Hsp90 inhibitory activity against multiple cancer cell lines (Figure 8).52 Optimization of 13 was pursued to minimize the entropic penalty paid upon binding and to study the local binding environment. Replacement of the biaryl amide side chain with a triazole-containing moiety (14) improved anti-proliferative activity and led to an optimal length of ~24 Å; which maximized Hsp90 CTD inhibition.53
Figure 8.
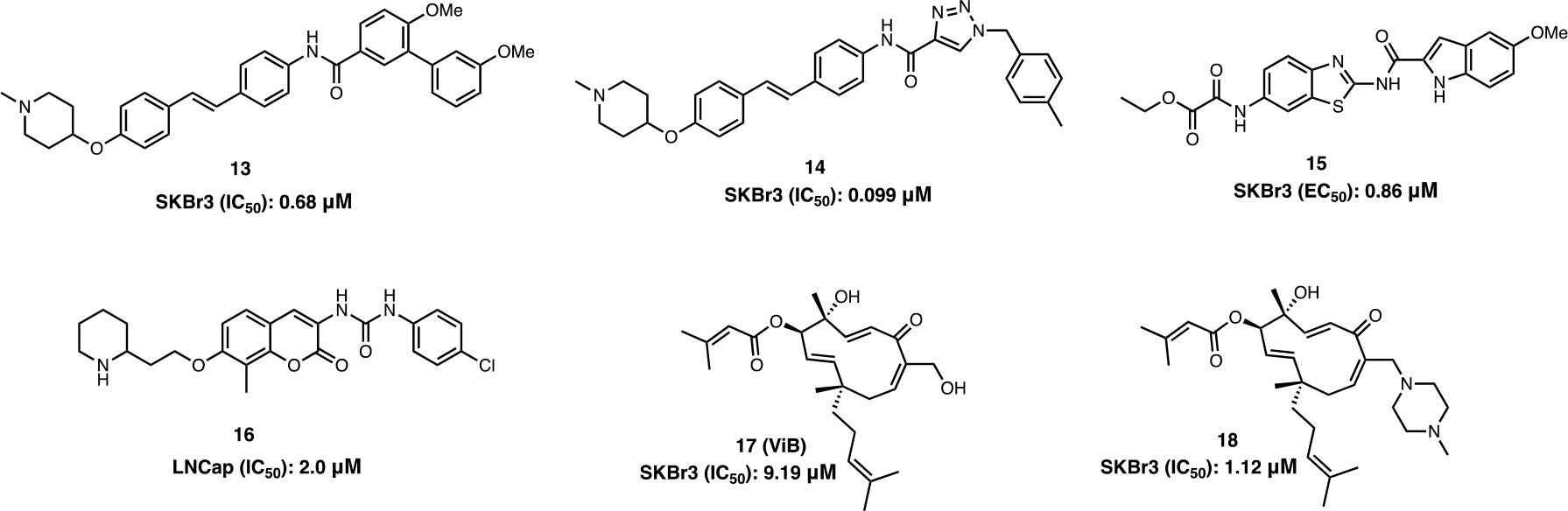
Stilbene and novobiocin based CTIs; Benzothiozole based CTIs; Isoform-selective CTIs.
In addition to Hsp90 CTD inhibition, novobiocin is a clinically approved DNA Gyrase B inhibitor that is used to treat bacterial infections. Originally, Neckers and co-workers postulated that novobiocin may also inhibit Hsp90 because both Hsp90 and DNA gyrase both contain a structurally similar Bergerat fold.19,35 This hypothesis led to the investigation of whether other DNA Gyrase B inhibitors can inhibit Hsp90 also. 15 is a DNA gyrase B inhibitor that was demonstrated to induce Hsp90-dependent client protein degradation without induction of the HSR in SKBr3 breast cancer cells.54 Therefore, it is possible that other DNA Gyrase B inhibitors can be used as a starting point for the development of new Hsp90 C-terminal inhibitors (Figure 8).55
The Hsp90 family of chaperone includes four isoforms (Hsp90α, Hsp90β, GRP94 and TRAP-1) that exhibit >85% sequence identity within the N-terminal ATP binding pocket. Simultaneous inhibition of all four isoforms can lead to detrimental side effects such as cardio and ocular toxicities.56 Therefore, the development of isoform-selective inhibitors is important to overcome these detriments. KU675 (16) (an analog of novobiocin) binds directly to the Hsp90 CTD and suppresses the proliferation of androgen-dependent and -independent prostate cancer cell lines (Figure 8). 16 displayed improved antiproliferative activity against the LNCap-LN3 and PC3MM2 cells lines as compared to 1. However, treatment of these prostate cancer cell lines with 16 led to selective degradation of the Hsp90α-specific client proteins, B-Raf and survivin, whereas the Hsp90 -specific client protein, CXCR4, was marginally affected. The molecule β was also found to exhibit differential binding affinities against Hsp90α and Hsp90β, respectively (Kd’s = of 191 μM (Hsp90α) and 726 μM (Hsp90β)).57 On the other hand, vibsanin B (ViB) (17) exhibited higher affinity for Hsp90β when compared to Hsp90α, and also inhibited leukocyte chemotactic migration and improved experimental autoimmune encephalomyelitis (EAE) in-vitro and in-vivo.58 18, a ViB based derivative displayed an IC50 = 1.12 μM against SKBr3 cells, while retaining selectivity for Hsp90β.59 This data suggests there is potential to develop CTIs that preferentially inhibit a single isoform.
Deguelin based CTIs:
Another class of CTIs is based on the natural product deguelin (19), which is a biosynthetic rotenoid that binds the nucleotide-binding site within the Hsp90 CTD and appears to form key interactions with Ser677 and Lys615.60 The binding of 19 to the CTD results in the degradation of Hsp90’s client protein, hypoxia-inducible factor (HIF-1α). Deguelin destabilizes HIF-1α interactions and leads to inhibition of cell proliferation and metastasis in several cancer cell lines both in vitro and in vivo.62,63,64 However, at higher doses, 19 is neurotoxic. After extensive SAR studies by the Lee group, a novel deguelin derivative, L80 (20a) was developed (Figure 9). 20a did not produce neurotoxic activity, but did manifest potent antiproliferative and apoptotic activity against multiple cancer cell lines. In vivo, 20a displayed antitumor and antiangiogenic activities against H1299 xenografts.60 Eventually, studies led to 20b, which induced HIF-1α degradation in a dose-dependent manner and improved anti-proliferative activity as compared to 20a. 20b also inhibited hypoxia-mediated angiogenic processes in human retinal microvascular endothelial cells (HRMEC) in vitro.63
Figure 9.
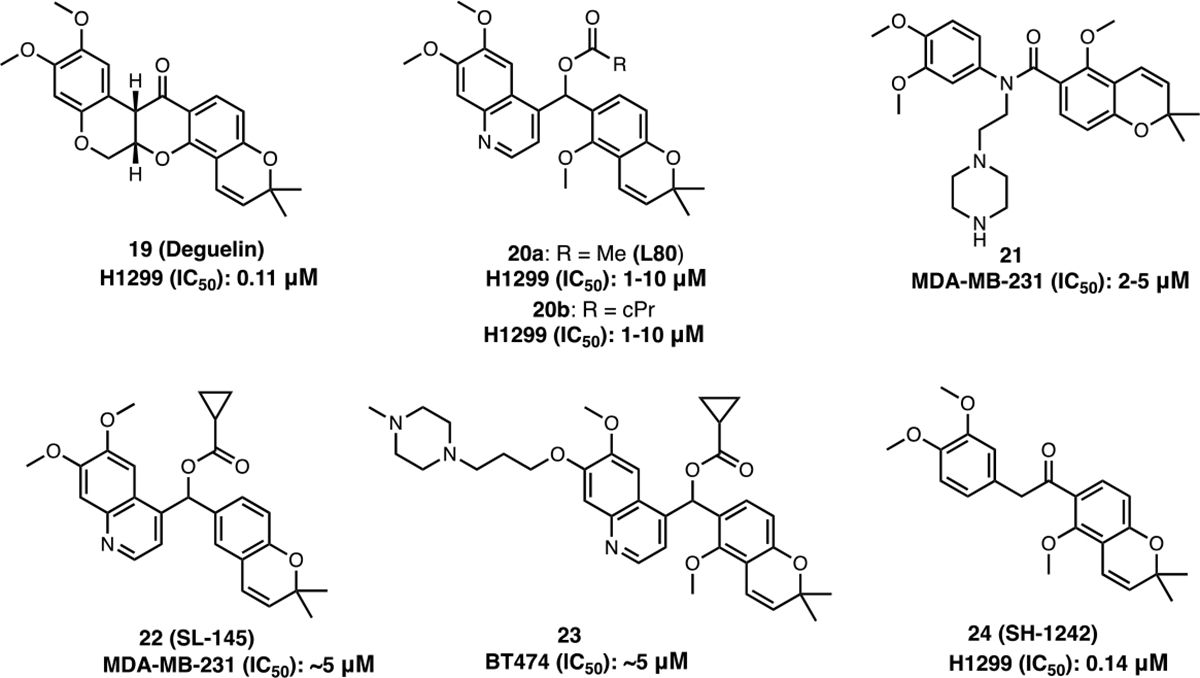
Deguelin based CTIs.
Subsequent studies on 20a, led to the development of 21 and SL-145 (22), which induced the degradation of Hsp90-dependent client proteins (Akt, ERK, STAT3) and exhibited cytotoxic activity against triple-negative breast cancer (TNBC) cell lines (MDA-MB-231 and 4T1) in a concentration-dependent manner.64,65 Molecular docking with 21 suggested 21 to bind the CTD nucleotide-binding pocket in an open conformation, while maintaining a T-shape configuration through hydrogen bonding and π-cation interactions.64 23 was prepared to exhibit increased solubility as compared to 20a. 23 inhibited the viability of trastuzumab-sensitive (BT474 and SKBr3) and trastuzumab-resistant (JIMT-1 and MDA-MB-453) cells in a concentration-dependent manner, while improving the drug-like properties of the scaffold.66 Another deguelin analog, SH-1242 (24) was developed and exhibits similar activity as 20a. The binding of 24 to Hsp90 disrupts interactions between Hsp90 and the co-chaperone, Hsp90-Hsp70 organizing protein (HOP), and manifests good anti-cancer activity against three different lung cancers (NSCLC, KRAS driven and PDX tumors) in vivo.67
Given the success of deguelin and novobiocin derived CTIs, the potential to combine scaffolds into a single chimera was pursued. The deguelin and novobiocin derived series (NCT) of CTIs were designed and synthesized via the hybridization of novobiocin and deguelin (Figure 10). NCT-50 (25) displayed higher inhibitory activity and lower toxicity than both novobiocin and deguelin. 25 exhibited inhibitory activity against both chemo-resistant and chemo-naïve NSCLC cells and inhibited the function of Hsp90 by stabilizing the open conformation of the homodimer.68 The related analog, NCT-80 (26) inhibited STAT3 activation and disrupted interactions between Hsp90 and STAT3 as determined by immunoprecipitation and pull-down assays. Since activation of Wnt/β-catenin signaling plays an important role in CSCs and anticancer drug resistance, treatment of CSCs and non-CSCs in NSCLC with 26 resulted in antitumor activity both in vitro and in vivo without activation of the STAT3 or Wnt/β-catenin signaling pathways.69,70
Figure 10.
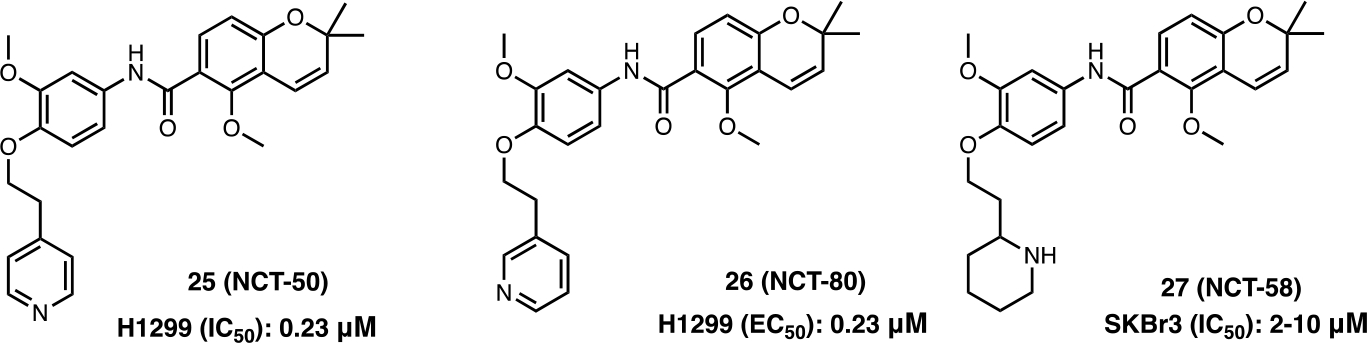
Deguelin and novobiocin based CTIs.
Higher levels of HSF-1 exist in Her2-positive breast cancers and correlate directly with tumor volume and reduced survival.71 Treatment of JIMT-1 xenograft tumors with NCT-58 (27) reduced the expression of CD44/ALDH1, a progenitor marker of breast cancer stem cells (BCSC). In addition, 27 led to the induction of apoptosis via activation of caspase-3/caspase-7 and the downregulation of HSF-1, Hsp70 and Hsp90 expression.72 Moreover, 27 exhibited the ability to eradicate trastuzumab-resistant cells and induce the degradation of Her2, Her3, EGFR, truncated p95Her2 and Akt.73 As a result, 27 represents a novel scaffold for the optimization of small molecules to treat trastuzumab-resistant Her2-positive breast cancers.
Peptide based CTIs:
The use of peptides as therapeutic agents can improve target affinity, selectivity and specificity as peptides can enter a cell by taking advantage of transporters.74 Peptides that directly block TPR-containing cochaperones from binding to the Hsp90 C-terminus can modulate multiple cell growth pathways simultaneously and offer an insight into SAR. An example of such is peptide LB51 (28), which bound the MEEVD region of the CTD and disrupted interactions with PPID with an IC50 value of 4μM.75 A structurally similar cyclic pentapeptide, LB76 (29) (Figure 11) was identified as the first de novo protein–protein disruptor that used an amino acid sequence complementary to the MEEVD sequence of the CTD. 29 blocked the interactions between Hsp90 and TPR-containing co-chaperones (eg. FKBP) as determined by a pull-down assay, but was observed to be a less effective disruptor of interactions between Hsp90 and its co-chaperone, FKBP.76
Figure 11.
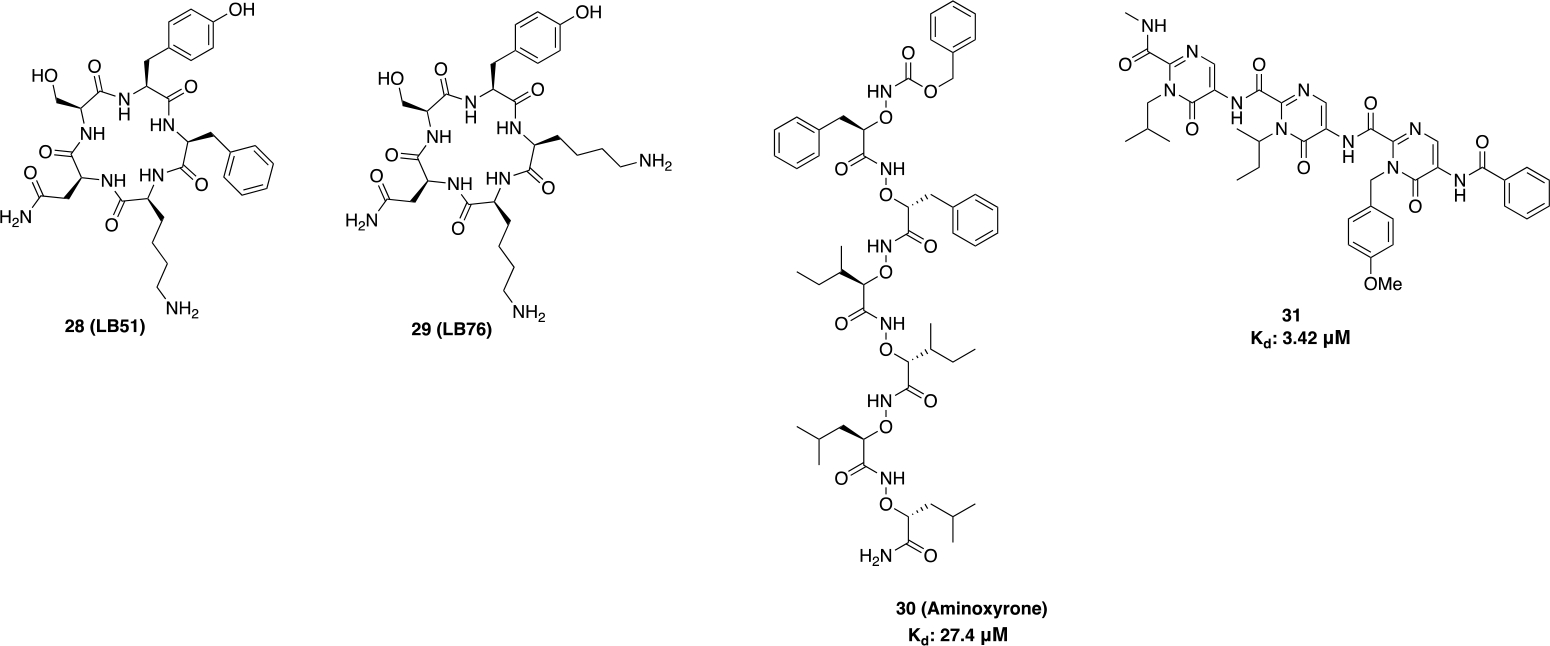
Peptide based CTIs.
A different peptide-based CTI, aminoxyrone (AX) (30), an α-aminoxy hexapeptide, binds the dimerization interface of Hsp90 CTD with a Kd value of 27.4 μM.31,77 This affinity led to the disruption of Hsp90’s interaction with its co-chaperone, p53. 30 displayed antiproliferative activity against BCR-ABL1+ TKI resistant leukemia cells and manifested activity without induction of the HSR.77 Further studies suggest that 30 operates via disruption of Hsp90 dimerization or destabilization of the already formed homodimer. Based on 30 and molecular dynamics (MD) simulations with the putative binding residues (I688, Y689, I692, and L696) located within the α-helix (H5) in the CTD dimerization interface, compound 31 was designed. 31, which consists of a tripyrimidonamide ring system demonstrated affinity towards the Hsp90α CTD (Kd = 3.42 ± 1.0 μM) and displayed good antiproliferative activity (IC50= 1.3 ± 0.3 μM) against a BCR-ABL1+ tested leukemia cell line, K562.29
Discovery of New Inhibitory Scaffolds:
Small molecule Hsp90 CTI’s with novel scaffolds (Figure 12) have been developed and validated using multiple approaches (Table 1).78 Dihydropyridine derivative, LA1011(32), binds the Hsp90 CTD as determined by isothermal titration calorimetry. In Alzheimer’s disease (AD), there are decreased levels of Hsp27 and Hsp70. Hence, treatment of the APPxPS1 Alzheimer’s disease mouse model with 32 led to induction of the HSR and manifested neuroprotective activity as evidenced by protection of dendritic spines, viable neurons in the hippocampus, and a decrease in tau accumulation.79 32 bound human Hsp90α with Kd = 3.8 ± 0.7 μM and human Hsp90β with Kd = 9.7 ± 0.7 μM, while simultaneously activating their ATPase activity. Therefore, this new scaffold exhibits allosteric modulation of the Hsp90 chaperone-mediated process and prevented the aggregation of citrate synthase and the refolding of luciferase.80 The discovery of interactions and a sensitive steric environment within the CTD was used to design 3,4-dihydropyrimidin-2(1H)-one (DHPM). Optimization of the aromatic ring at C-4 of DHPM led to a second generation lead compound (33). 33 represents one of the few unnatural compounds that was synthesized to target the Hsp90 CTD, and displayed an IC50 value of 15.2 μM against human T lymphocyte Jurkat cell lines.81
Figure 12.
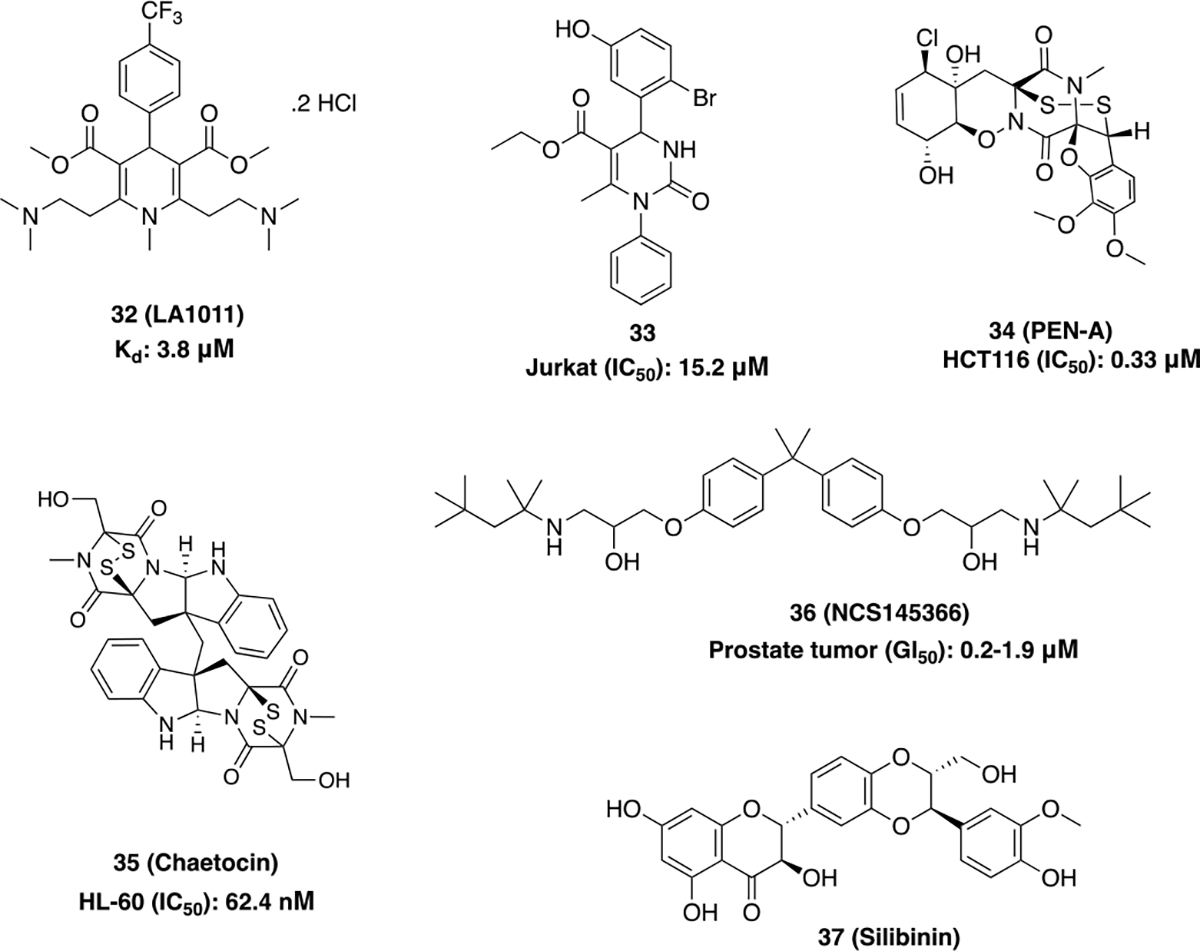
New Scaffolds for CTIs.
Table 1.
Methods employed to discover and validate newly discovered CTIs.
| Approaches for drug discovery | Experimental methods employed |
|---|---|
| medicinal chemistry-based approaches used to develop Hsp90 C-terminal modulators | • homology modeling • molecular docking • virtual screening • pharmacophore development • molecular dynamics (MD) simulation • computational modelling and the application of computer-aided drug design (CADD)90 • quantitative structure-activity relationship (QSAR) • nuclear magnetic resonance (NMR) |
| Methods used to validate target engagement | • photo-cross linking and mass spectrometry to identify proximal residues91,92 • alpha (amplified luminescence proximity homogeneous assay)-based assay that employs biotin-labelled Hsp90 CTD and glutathione S-transferase (GST)-tagged PPID to disrupt interactions between the Hsp90 CTD and TPR-containing peptidylprolyl isomerase D (PPID) • surface plasmon resonance (SPR) studies87,93 • proteolytic fingerprinting92 • isothermal titration calorimetry (ITC)80 • luciferase-refolding assay76 • molecular dynamics receptor exploration88 • fluorescence resonance energy transfer (TR-FRET) assay85 • fluorescence polarization (FP) assay with FITC labeled Hsp90 N-terminal inhibitors to confirm a lack of binding to the NTD94 |
Penisuloxazin A (PEN-A) (34) strongly binds the Hsp90α C-terminus at a site distinct from the ATP binding domain that was utilized via an ATP-agarose pulldown assay. The disulfide bond present in this molecule can oxidize cysteine residues (C572, C597 and C598) within the CTD and consequently, disrupt Hsp90 dimerization and the interactions between Hsp90 and its co-chaperones (CDC37, p23, FKBP5 and PP5). 34 inhibited the proliferation of cancer cells, enhanced apoptosis and suppressed tumor growth in an HCT116 colon cancer xenograft model.30,82
Chaetocin (35) is another novel Hsp90α C-terminal inhibitor that was confirmed through surface plasmon resonance (SPR) and proteolytic fingerprinting assays. 35 exhibited antiproliferative activity against K562 and HL-60 cells with an IC50 value of 125 nM and 62.4 nM, respectively. 35 induced the degradation of various Hsp90-dependent client proteins (AMl1-ETO and BCL-ABL) and disrupted interactions between Hsp90 and cochaperones HOP and pp5 in a dose-dependent manner. 35 led to the degradation of SUV39H1, an Hsp90 client protein that interacts with Hsp90 via Hsp90-Hsp70 Organizing protein (HOP). SUV39H1 is a transcriptional repressor that serves an important role in the regulation of proliferation and self-renewal of hematopoietic stem cells involved in acute myeloid leukemia (AML).83
NSC145366 (36) which shares the symmetrical scaffold present in bisphenol A (BPA), binds to a novel site within the C-terminal dimerization interface, independent of the CTD nucleotide-binding site. Upon binding the CTD, 36 induces allosteric inhibition of ATPase activity at the NTD (IC50= 119 μM) by increasing Hsp90 oligomerization. NSC145366 displayed antiproliferative activity against prostate tumor cells (GI50= 0.2–1.9 μM), leading to the downregulation of Hsp90-dependent client proteins (AR and BRCA1) without induction of the HSR.84
Silibinin (37), a natural product derived from milk thistle seeds, interacts with Hsp90α and β and induces conformational changes that lead to disruption of Hsp90 interactions with other co-chaperones. 37 circumvented the hepatotoxic side effects observed with N-terminal inhibitors and led to the downregulation of signal transducers and the activator of transcription (STAT3) signaling, ultimately displaying efficacious activity in in vivo metastasis models of non-small cell lung cancer (NSCLC).85
In silico Design of CTIs:
Rapid Overlay of Chemical Structures (ROCS) is an extensively used screening tool for ligand based drug design. After decades of failed efforts to obtain co-crystal structures of small molecules bound to the CTD, this method of C-terminal inhibitor development appears promising. ROCS modeling with novobiocin as a reference generated compound 38 (Figure 13). A homology modeling study performed with yeast Hsp90 (PDB code: 2CG9) predicted 38 to bind the Hsp90 C-terminal ATP binding pocket. SAR studies on 38 led to the development of compound 39, which manifested potent anti-proliferative activities against various breast cancer cells both in vitro and in vivo and induced the degradation of Hsp90 clients.86 Using an orthogonal screening approach, two new C-terminal inhibitors (compounds 40 and 41) were discovered. These compounds induced apoptosis and downregulated oncogenic client proteins without induction of the HSR in Jurkat (human T-lymphocyte) and U937 (human monocyte form histiocytic lymphoma) cell lines.87 Compound 42 was identified via molecular dynamics (MD) simulations using novobiocin in a structure-based pharmacophore model. This compound displayed antiproliferative activity against different cancer cell lines while inhibiting the Hsp90 dependent refolding of luciferase.88 Compounds 43 and 44 are the first examples of small molecules shown to directly disrupt Hsp90 CTD-TPR-domain co-chaperone interactions, which were discovered by virtual screening and intrinsic protein fluorescence quenching binding assays. These compounds bind the dimerization pocket within the CTD across both monomers as predicted by docking studies. Using an ALPHALISA assay, the IC50 values of 43 and 44 were determined to be 35 μM and 220 μM, respectively.25 45 represents a unique scaffold that was discovered via a structure-based virtual screening against the Hsp90β CTD. 45 displayed antiproliferative activity against two different cancer cells lines (IC50= 1.4 ± 0.4 μM against MCF-7 and IC50= 2.8 ± 0.4 μM against SK-N-MC), wherein the hydrophobic interactions between the biphenyl group and Ala608A were found essential for activity.89
Figure 13.
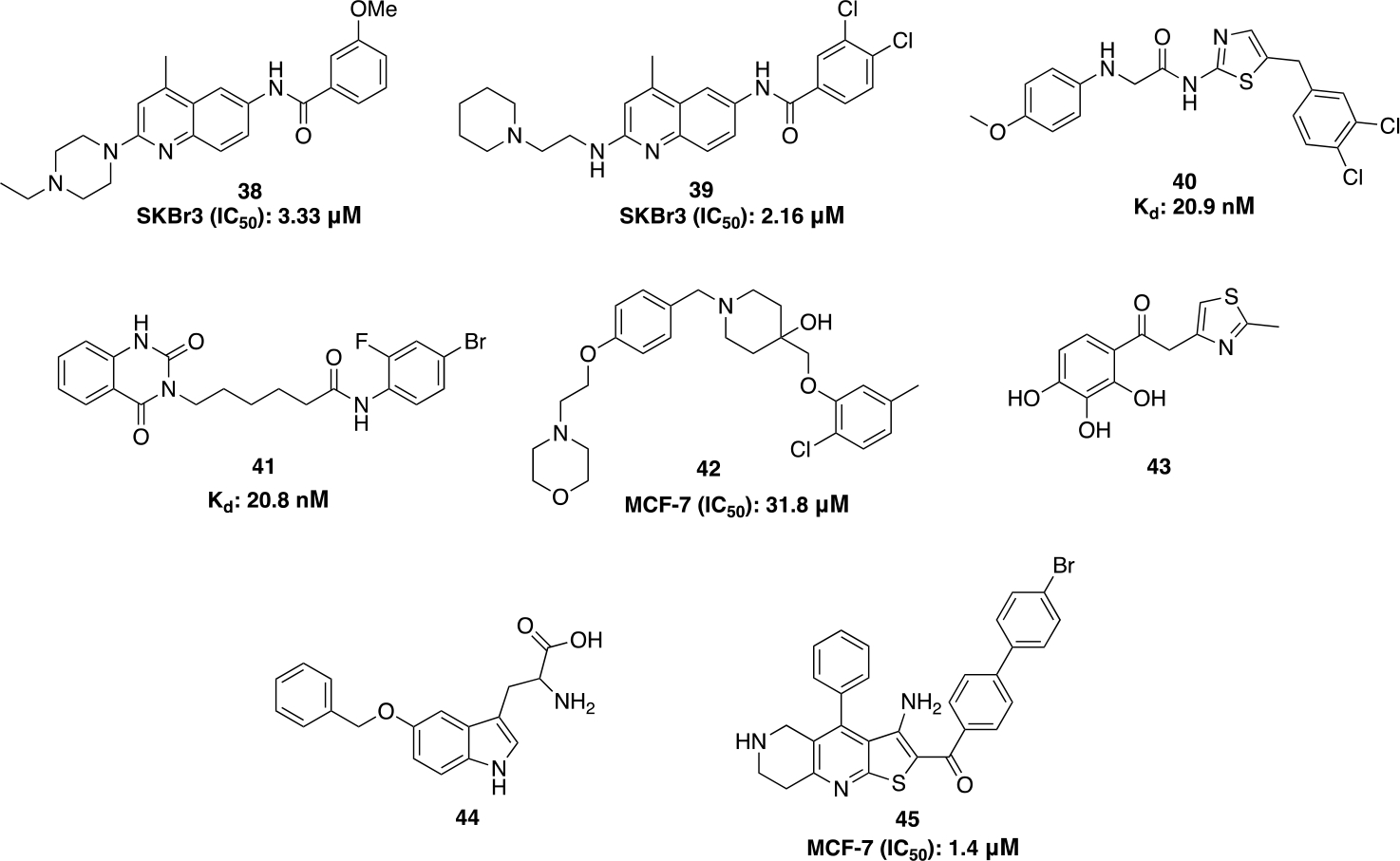
Computational based CTIs.
In conclusion, targeting the Hsp90 CTD offers an exciting opportunity to disrupt the maturation of ~400 client proteins associated with numerous pathologies, including cancer. Moreover, C-terminal modulators have demonstrated and displayed the ability to segregate neuroprotective activity from anticancer activity by segregating induction of the HSR from inhibition of the protein folding machinery. Therefore, the development of CTI’s provide an alternative strategy to overcome the dose-limiting toxicities that were observed with inhibition of the Hsp90 NTD. Despite progress made thus far, the mechanism of action for CTI’s remain underinvestigated and requires significant biochemical interrogation. Hsp90 C-terminal compounds that exhibit greater inhibitory activity are needed along with solution of the co-crystal structures in order to further advance this field. Such studies would allow for a more thorough SAR analysis between inhibitors and the C-terminal binding site that could lead to the advancement of additional clinical candidates. Despite these concerns, a number of CTI have been developed and this review provides an update on the current status of C-terminal modulators used to explore Hsp90 biology and their clinical applications.
Acknowledgments
This work is supported by grants from The National Institutes of Health to B.S.J.B [CA120458]. All biological images used in this review were created with Biorender.com
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Makhnevych T, Houry WA. Biochim Biophys Acta BBA - Mol Cell Res. 2012;1823:674–682. doi: 10.1016/j.bbamcr.2011.09.001 [DOI] [PubMed] [Google Scholar]
- 2.Ritossa F Experientia. 1962;18:571–573. doi: 10.1007/BF02172188 [DOI] [Google Scholar]
- 3.Serwetnyk MA, Blagg BSJ. Acta Pharm Sin B. 2021;11:1446–1468. doi: 10.1016/j.apsb.2020.11.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Donnelly A, Blagg BSJ. Curr Med Chem. 2008;15:2702–2717. doi: 10.2174/092986708786242895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Söti C, Rácz A, Csermely P. J Biol Chem. 2002;277(9):7066–7075. doi: 10.1074/jbc.M105568200 [DOI] [PubMed] [Google Scholar]
- 6.Prodromou C EMBO J. 1999;18:754–762. doi: 10.1093/emboj/18.3.754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Murphy PJM, Kanelakis KC, Galigniana MD, Morishima Y, Pratt WB. J Biol Chem. 2001;276:30092–30098. doi: 10.1074/jbc.M103773200 [DOI] [PubMed] [Google Scholar]
- 8.Caplan AJ, Mandal AK, Theodoraki MA. Trends Cell Biol. 2007;17:87–92. doi: 10.1016/j.tcb.2006.12.002 [DOI] [PubMed] [Google Scholar]
- 9.Kosano H, Stensgard B, Charlesworth MC, McMahon N, Toft D. J Biol Chem. 1998;273:32973–32979. doi: 10.1074/jbc.273.49.32973 [DOI] [PubMed] [Google Scholar]
- 10.Prodromou C EMBO J. 2000;19:4383–4392. doi: 10.1093/emboj/19.16.4383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ali MMU, Roe SM, Vaughan CK, et al. Nature. 2006;440:1013–1017. doi: 10.1038/nature04716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chaudhury S, Keegan BM, Blagg BSJ. Med Res Rev. 2021;41:202–222. doi: 10.1002/med.21729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hanahan D, Weinberg RA. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- 14.Taipale M, Krykbaeva I, Koeva M, et al. Cell. 2012;150:987–1001. doi: 10.1016/j.cell.2012.06.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peterson LB, Blagg BS. Future Med Chem. 2009;1:267–283. doi: 10.4155/fmc.09.17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang H, Burrows F. J Mol Med. 2004;82. doi: 10.1007/s00109-004-0549-9 [DOI] [PubMed] [Google Scholar]
- 17.Kamal A, Thao L, Sensintaffar J, et al. Nature. 2003;425:407–410. doi: 10.1038/nature01913 [DOI] [PubMed] [Google Scholar]
- 18.Whitesell L, Lindquist SL. Nat Rev Cancer. 2005;5:761–772. doi: 10.1038/nrc1716 [DOI] [PubMed] [Google Scholar]
- 19.Marcu MG, Schulte TW, Neckers L. JNCI J Natl Cancer Inst. 2000;92:242–248. doi: 10.1093/jnci/92.3.242 [DOI] [PubMed] [Google Scholar]
- 20.Morimoto RI. Science. 1993;259:1409–1410. doi: 10.1126/science.8451637 [DOI] [PubMed] [Google Scholar]
- 21.Gupta A, Bansal A, Hashimoto-Torii K. Neurosci Lett. 2020;716:134678. doi: 10.1016/j.neulet.2019.134678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lackie RE, Maciejewski A, Ostapchenko VG, et al. Front Neurosci. 2017;11:254. doi: 10.3389/fnins.2017.00254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Atalay M, Oksala N, Lappalainen J, Laaksonen D, Sen C, Roy S. Curr Protein Pept Sci. 2009;10:85–95. doi: 10.2174/138920309787315202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Urban MJ, Li C, Yu C, et al. ASN Neuro. 2010;2:AN20100015. doi: 10.1042/AN20100015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mak OW, Sharma N, Reynisson J, Leung IKH. Bioorg Med Chem Lett. 2021;38:127857. doi: 10.1016/j.bmcl.2021.127857 [DOI] [PubMed] [Google Scholar]
- 26.Yun BG, Huang W, Leach N, Hartson SD, Matts RL. Biochemistry. 2004;43:8217–8229. doi: 10.1021/bi0497998 [DOI] [PubMed] [Google Scholar]
- 27.Allan RK, Mok D, Ward BK, Ratajczak T. J Biol Chem. 2006;281:7161–7171. doi: 10.1074/jbc.M512406200 [DOI] [PubMed] [Google Scholar]
- 28.Roy SS, Kapoor M. J Mol Graph Model. 2016;70:253–274. doi: 10.1016/j.jmgm.2016.10.002 [DOI] [PubMed] [Google Scholar]
- 29.Bhatia S, Spanier L, Bickel D, et al. Chemistry. 2021. doi: 10.26434/chemrxiv-2021-qwxbh [DOI] [Google Scholar]
- 30.Dai J, Chen A, Zhu M, et al. Biochem Pharmacol. 2019;163:404–415. doi: 10.1016/j.bcp.2019.03.012 [DOI] [PubMed] [Google Scholar]
- 31.Bickel D, Gohlke H. Bioorg Med Chem. 2019;27:115080. doi: 10.1016/j.bmc.2019.115080 [DOI] [PubMed] [Google Scholar]
- 32.Hartson SD, Thulasiraman V, Huang W, Whitesell L, Matts RL. Biochemistry. 1999;38:3837–3849. doi: 10.1021/bi983027s [DOI] [PubMed] [Google Scholar]
- 33.Hall JA, Forsberg LK, Blagg BS. Future Med Chem. 2014;6:1587–1605. doi: 10.4155/fmc.14.89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marcu MG, Schulte TW, Neckers L. JNCI J Natl Cancer Inst. 2000;92:242–248. doi: 10.1093/jnci/92.3.242 [DOI] [PubMed] [Google Scholar]
- 35.Marcu MG, Chadli A, Bouhouche I, Catelli M, Neckers LM. J Biol Chem. 2000;275:37181–37186. doi: 10.1074/jbc.M003701200 [DOI] [PubMed] [Google Scholar]
- 36.Soti C, Vermes A, Haystead TAJ, Csermely P. Eur J Biochem. 2003;270:2421–2428. doi: 10.1046/j.1432-1033.2003.03610.x [DOI] [PubMed] [Google Scholar]
- 37.Peng S, Woodruff J, Pathak PK, Matts RL, Deng J. Acta Crystallogr Sect Struct Biol. 2022;78. doi: 10.1107/S2059798322002261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chatterjee BK, Jayaraj A, Kumar V, et al. J Biol Chem. 2019;294:6450–6467. doi: 10.1074/jbc.RA118.002502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kumar MV V, Ebna Noor R, Davis RE, et al. MedChemComm. 2018;9:1323–1331. doi: 10.1039/C8MD00151K [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Birar VC, Gelis I, Zuo A, Blagg BSJ. Bioorg Med Chem Lett. 2020;30:127303. doi: 10.1016/j.bmcl.2020.127303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yu XM, Shen G, Neckers L, et al. J Am Chem Soc. 2005;127:12778–12779. doi: 10.1021/ja0535864 [DOI] [PubMed] [Google Scholar]
- 42.Ghosh S, Liu Y, Garg G, et al. ACS Med Chem Lett. 2016;7:813–818. doi: 10.1021/acsmedchemlett.6b00224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ghosh S, Shinogle HE, Garg G, et al. ACS Chem Biol. 2015;10:577–590. doi: 10.1021/cb5008713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Anyika M, McMullen M, Forsberg LK, Dobrowsky RT, Blagg BSJ. ACS Med Chem Lett. 2016;7:67–71. doi: 10.1021/acsmedchemlett.5b00331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Narasimharao Meka P, Amatya E, Kaur S, et al. Bioorg Med Chem. 2022;70:116940. doi: 10.1016/j.bmc.2022.116940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Garg G, Zhao H, Blagg BSJ. Bioorg Med Chem. 2017;25:451–457. doi: 10.1016/j.bmc.2016.11.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Garg G, Forsberg LK, Zhao H, Blagg BSJ. Chem - Eur J. 2017;23:16574–16585. doi: 10.1002/chem.201703206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Subramanian C, Kovatch KJ, Sim MW, et al. Neoplasia. 2017;19:1003–1011. doi: 10.1016/j.neo.2017.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Subramanian C, Gorney R, Wang T, et al. Surgery. 2021;169:34–42. doi: 10.1016/j.surg.2020.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chiosis G, Neckers L. ACS Chem Biol. 2006;1:279–284. doi: 10.1021/cb600224w [DOI] [PubMed] [Google Scholar]
- 51.Sanchez JN, Subramanian C, Chanda M, et al. Melanoma Res. 2021;31:197–207. doi: 10.1097/CMR.0000000000000734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Byrd KM, Subramanian C, Sanchez J, et al. Chem - Eur J. 2016;22:6921–6931. doi: 10.1002/chem.201504955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Byrd KM, Kent CN, Blagg BSJ. ChemMedChem. 2017;12:2022–2029. doi: 10.1002/cmdc.201700630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pugh KW, Zhang Z, Wang J, et al. ACS Med Chem Lett. 2020;11:1535–1538. doi: 10.1021/acsmedchemlett.0c00100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dernovšek J, Zajec Ž, Durcik M, et al. Pharmaceutics. 2021;13:1283. doi: 10.3390/pharmaceutics13081283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hong DS, Banerji U, Tavana B, George GC, Aaron J, Kurzrock R. Cancer Treat Rev. 2013;39:375–387. doi: 10.1016/j.ctrv.2012.10.001 [DOI] [PubMed] [Google Scholar]
- 57.Liu W, Vielhauer GA, Holzbeierlein JM, et al. Mol Pharmacol. 2015;88:121–130. doi: 10.1124/mol.114.097303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ye BX, Deng X, Shao LD, et al. J Immunol. 2015;194:4489–4497. doi: 10.4049/jimmunol.1402798 [DOI] [PubMed] [Google Scholar]
- 59.Shao LD, Su J, Ye B, et al. J Med Chem. 2017;60:9053–9066. doi: 10.1021/acs.jmedchem.7b01395 [DOI] [PubMed] [Google Scholar]
- 60.Lee SC, Min HY, Choi H, et al. Mol Pharmacol. 2015;88:245–255. doi: 10.1124/mol.114.096883 [DOI] [PubMed] [Google Scholar]
- 61.Chang DJ, An H, suk Kim K, et al. J Med Chem. 2012;55:10863–10884. doi: 10.1021/jm301488q [DOI] [PubMed] [Google Scholar]
- 62.Hadden MK, Galam L, Gestwicki JE, Matts RL, Blagg BSJ. J Nat Prod. 2007;70:2014–2018. doi: 10.1021/np070190s [DOI] [PubMed] [Google Scholar]
- 63.Kim HS, Hong M, Ann J, et al. Bioorg Med Chem. 2016;24:6082–6093. doi: 10.1016/j.bmc.2016.09.067 [DOI] [PubMed] [Google Scholar]
- 64.Kim HS, Hoang VH, Hong M, et al. Bioorg Med Chem. 2019;27:1370–1381. doi: 10.1016/j.bmc.2019.02.040 [DOI] [PubMed] [Google Scholar]
- 65.Kim JY, Cho TM, Park JM, et al. Oncogene. 2022;41:3289–3297. doi: 10.1038/s41388-022-02269-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nguyen CT, Thanh La M, Ann J, et al. Bioorg Med Chem Lett. 2021;45:128134. doi: 10.1016/j.bmcl.2021.128134 [DOI] [PubMed] [Google Scholar]
- 67.Lee SC, Min HY, Choi H, et al. Cancer Res. 2016;76:686–699. doi: 10.1158/0008-5472.CAN-15-1492 [DOI] [PubMed] [Google Scholar]
- 68.Hyun SY, Le HT, Nguyen CT, et al. Sci Rep. 2018;8:13924. doi: 10.1038/s41598-018-32196-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Katoh M Int J Oncol. 2017;51:1357–1369. doi: 10.3892/ijo.2017.4129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lee HJ, Min HY, Yong YS, et al. Theranostics. 2022;12:105–125. doi: 10.7150/thno.63788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Santagata S, Hu R, Lin NU, et al. Proc Natl Acad Sci. 2011;108:18378–18383. doi: 10.1073/pnas.1115031108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Park JM, Kim YJ, Park S, et al. Mol Cancer. 2020;19:161. doi: 10.1186/s12943-020-01283-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Park S, Kim YJ, Park JM, et al. Cell Death Discov. 2021;7:354. doi: 10.1038/s41420-021-00743-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lamers C Future Drug Discov. 2022;4:FDD75. doi: 10.4155/fdd-2022-0005 [DOI] [Google Scholar]
- 75.Rahimi MN, Buckton LK, Zaiter SS, et al. ACS Med Chem Lett. 2018;9:73–77. doi: 10.1021/acsmedchemlett.7b00310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rahimi MN, McAlpine SR. Chem Commun. 2019;55:846–849. doi: 10.1039/C8CC07576J [DOI] [PubMed] [Google Scholar]
- 77.Bhatia S, Diedrich D, Frieg B, et al. Blood. 2018;132:307–320. doi: 10.1182/blood-2017-10-810986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Banerjee M, Hatial I, Keegan BM, Blagg BSJ. Pharmacol Ther. 2021;221:107747. doi: 10.1016/j.pharmthera.2020.107747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Klettner A Drug News Perspect. 2004;17:299. doi: 10.1358/dnp.2004.17.5.829033 [DOI] [PubMed] [Google Scholar]
- 80.Roe MS, Wahab B, Török Z, Horváth I, Vigh L, Prodromou C. Front Mol Biosci. 2018;5:51. doi: 10.3389/fmolb.2018.00051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Terracciano S, Foglia A, Chini MG, et al. RSC Adv. 2016;6:82330–82340. doi: 10.1039/C6RA17235K [DOI] [Google Scholar]
- 82.Kurop MK, Huyen CM, Kelly JH, Blagg BSJ. Eur J Med Chem. 2021;226:113846. doi: 10.1016/j.ejmech.2021.113846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lian B, Lin Q, Tang W, Qi X, Li J. Biomol Ther. 2021;29:73–82. doi: 10.4062/biomolther.2020.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Goode KM, Petrov DP, Vickman RE, et al. Biochim Biophys Acta BBA - Gen Subj 2017;1861:1992–2006. doi: 10.1016/j.bbagen.2017.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cuyàs E, Verdura S, Micol V, et al. Food Chem Toxicol. 2019;132:110645. doi: 10.1016/j.fct.2019.110645 [DOI] [PubMed] [Google Scholar]
- 86.Jiang F, ping Guo A, cheng Xu J, et al. Eur J Med Chem. 2017;141:1–14. doi: 10.1016/j.ejmech.2017.07.080 [DOI] [PubMed] [Google Scholar]
- 87.Terracciano S, Russo A, Chini MG, et al. Sci Rep. 2018;8:1709. doi: 10.1038/s41598-017-14902-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tomašič T, Durcik M, Keegan BM, et al. Int J Mol Sci. 2020;21:6898. doi: 10.3390/ijms21186898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zajec Ž, Dernovšek J, Gobec M, Tomašič T. Biomolecules. 2022;12:884. doi: 10.3390/biom12070884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Magwenyane AM, Ugbaja SC, Amoako DG, Somboro AM, Khan RB, Kumalo HM. Comput Math Methods Med. 2022;2022:1–20. doi: 10.1155/2022/2147763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sgobba M, Forestiero R, Degliesposti G, Rastelli G. EJ Chem Inf Model. 2010;50:1522–1528. doi: 10.1021/ci1001857 [DOI] [PubMed] [Google Scholar]
- 92.Matts RL, Dixit A, Peterson LB, et al. ACS Chem Biol. 2011;6:800–807. doi: 10.1021/cb200052x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yasgar A, Jadhav A, Simeonov A, Coussens NP. Methods in Molecular Biology. Springer; New York; 2016:77–98. doi: 10.1007/978-1-4939-3673-1_5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Khandelwal A, Kent CN, Balch M, et al. Nat Commun. 2018;9:425. doi: 10.1038/s41467-017-02013-1 [DOI] [PMC free article] [PubMed] [Google Scholar]