

ABSTRACT
Background
Elevated serum urea levels are common in moderate-to-advanced chronic kidney disease (CKD). Several studies have shown that urea is a direct and indirect uraemic toxin, especially with regard to cardiovascular disease. We sought to determine whether serum urea levels are associated with adverse cardiovascular events and death before renal replacement therapy (RRT) in patients with CKD.
Methods
CKD-REIN is a prospective cohort of CKD nephrology outpatients not receiving maintenance dialysis. The 2507 patients included in the analysis were divided into three groups according to the baseline serum urea level (T1 <10.5, T2 10.5–15.1 and T3 ≥15.1 mmol/L). Cox proportional hazard models were used to estimate hazard ratios (HRs) for first atheromatous or non-atheromatous cardiovascular (CV) events and all-cause mortality before RRT. The models were adjusted for baseline comorbidities, laboratory data and medications.
Findings
Of the 2507 included patients {median [interquartile range (IQR)] age: 69 [61–77]; mean (standard deviation) estimated glomerular filtration rate (eGFR) 33.5 (11.6) mL/min/1.73 m²}, 54% had a history of cardiovascular disease. After multiple adjustments for CV risk factors (including eGFR), patients in T3 had a higher risk of atheromatous and non-atheromatous CV events than patient in T1 (n events = 451, HR [95% CI]: 1.93 [1.39; 2.69]). The adjusted HRs for death before RRT (n events = 407) were 1.31 [0.97; 1.76] and 1.73 [1.22; 2.45] for patients T2 and those in T3, respectively.
Interpretation
Our data suggested that urea is a predictor of CV outcomes beyond CV risk factors including eGFR.
Keywords: cardiovascular disease, chronic kidney disease, urea, uraemic toxin
Graphical Abstract
Graphical Abstract.

KEY LEARNING POINTS.
What is already known about this subject?
Chronic kidney disease (CKD) is now considered as a major worldwide public health problem and the death rate due to cardiovascular disease (CVD) is significantly higher in patients with CKD than in the general population. However, conventional cardiovascular (CV) risk factors do not appear to fully account for this elevated risk of CVD.
For many years, urea was considered to be a relatively inert, nontoxic molecule. But several studies have shown that urea is a direct and indirect uraemic toxin. Although the mechanisms of urea's direct toxicity still require further investigation, in vitro and in vivo studies have shown that uraemia modulates the smooth muscle cell's phenotype, induces the expression of pro-apoptotic family genes. Other studies have shown that urea has an indirect toxic effect via protein carbamylation, which interferes with the proteins’ molecular and cellular functions and is associated with CKD progression after adjustment for conventional risk factors. However, it is still not clear whether serum urea levels are related to CV outcomes in patients with moderate-to-advanced CKD.
What this study adds?
Among 2507 patients with CKD not receiving maintenance dialysis attending nephrology outpatient facilities, we showed that patients with moderately elevated serum urea were at high risk of developing adverse CV outcomes. The association was independent of CV risk factors including estimated glomerular filtration rate.
What impact this may have on practice or policy?
Considering the toxic effect of elevated serum urea levels in the management of patients with moderate to advanced chronic renal failure is important, given the actual body of evidence. Given the dietary origin of a proportion of the urea in the circulation, nutritional therapy could be used to counter an elevation in urea levels. Interventional studies of various diets and their impacts on CV outcomes (and especially non-atheromatous events) in non-dialysed patients with CKD are warranted.
INTRODUCTION
Chronic kidney disease (CKD) is now considered as a major worldwide public health problem [1]. It directly affects the overall burden of morbidity and mortality and its increasing prevalence is being driven primarily by population ageing and increases in the prevalence of diabetes, hypertension and obesity [2]. Although the death rate due to cardiovascular disease (CVD) is significantly higher in patients with CKD than in the general population [3, 4], the underlying pathophysiological mechanisms are not fully understood. Indeed, conventional cardiovascular (CV) risk factors do not appear to fully account for this elevated risk of CVD.
Urea is a product of protein metabolism that is often used as a proxy for CKD severity and dialysis adequacy in clinical settings. For many years, urea was considered to be a relatively inert, nontoxic molecule. However, several studies have shown that urea is a direct and indirect uraemic toxin [5]. Although the mechanisms of urea's direct toxicity still require further investigation, in vitro and in vivo studies have shown that uraemia modulates the smooth muscle cell's phenotype and induces the expression of pro-apoptotic BCL-2 family genes; this might explain the elevated apoptosis rate observed in the arterial wall in uraemic patients [6, 7]. Furthermore, a high urea concentration in endothelial cell progenitor cultures has been associated with increased senescence and free radical formation [8]. In a study of nephrectomized mice, inhibition of the urea transporter was associated with reductions in hypertension and cardiac fibrosis and an improvement in cardiac function [9].
Other studies have shown that urea has an indirect toxic effect via protein carbamylation [10], which interferes with the proteins’ molecular and cellular functions. Protein carbamylation is associated with CKD progression after adjustment for conventional risk factors [11]. Furthermore, the exacerbation of vascular calcification by carbamylation [12], or atherosclerosis with lipoprotein carbamylation might explain the high incidence of CVD and mortality in CKD [13].
Thus, urea might contribute to the elevated CV risk in patients with CKD. However, no clinical studies we are aware of have reported an association between the occurrence of CV events (whether fatal or nonfatal) and serum urea levels in pre-dialysis patients with CKD. Thus, the objective of the present study was to determine whether serum urea levels are associated with adverse outcomes such as CV events (both atheromatous and non-atheromatous) and death before renal replacement therapy (RRT) in patients with CKD after adjustment for the estimated glomerular filtration rate (eGFR).
MATERIALS AND METHODS
Study design and participants
CKD-REIN is a prospective cohort study conducted by 40 nationally representative nephrology outpatient facilities in France. Details of the study protocol have been published [14]. Briefly, the main inclusion criteria are ≥18 years, a confirmed diagnosis of moderate or advanced CKD, an eGFR <60 mL/min/1.73 m² and the absence of dialysis or transplantation. Between July 2013 and April 2016, 3033 patients were enrolled during a routine nephrology outpatient appointment and are actively followed up for 5 years at most. The recruitment of outpatients ensured that the study participants did not present with acute illness (such as acute kidney injury or digestive tract haemorrhage) on inclusion. Furthermore, all patients were receiving stable background therapy. The study protocol was approved by the institutional review board at the French National Institute of Health and Medical Research (INSERM; reference: IRB00003888) and was registered at ClinicalTrials.gov (NCT03381950).
For the purposes of the present analysis, we excluded patients with missing data for the serum urea level at baseline or who had an aberrant serum urea value (i.e. <2.5 or >100 mmol/L). To limit the effects of nonrenal factors on serum urea levels, patients who were on corticosteroids or had a history of metastatic cancer were excluded. Patients who had an eGFR <15 mL/min/1.73 m² at baseline were also excluded, considering that they had reached the stage of kidney failure (Figure 1). A total of 2507 patients were analysed here.
FIGURE 1:
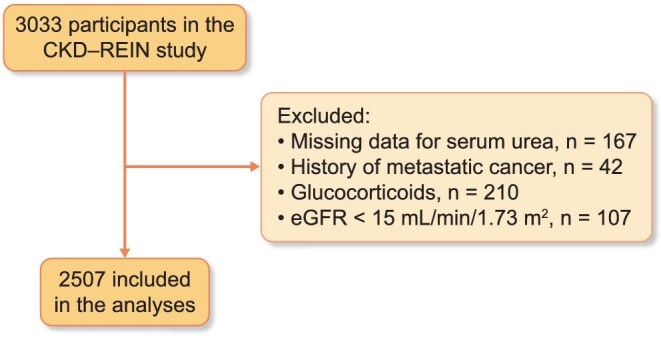
Study flowchart. eGFR, estimated glomerular filtration rate.
Study data
Trained clinical research associates (CRAs) collected data from patient interviews and medical records at baseline and then annually. The patients’ characteristics (age, sex and smoking status) were recorded and the patients were screened for a history of hypertension, diabetes, CVD or acute kidney injury (definitions in Supplementary data, Table S1). Blood pressure was measured, as were height and weight to calculate body mass index (BMI). All patients were prescribed a set of standard blood and urine tests (recommended by French health authorities for routine CKD care), with sample to be taken at their usual medical laboratory. Data on the serum creatinine, serum urea level, blood haemoglobin, serum albumin and albumin- or protein-to-creatinine ratio (Supplementary data, Table S1) were recorded. The GFR was estimated using the Chronic Kidney Disease Epidemiology Collaboration (CKD-EPI) creatinine equation and the isotope dilution mass spectrometry-traceable creatinine concentration determined in a Jaffe assay (in 38% of participants) or an enzymatic assay (in 57%; the assay type was unknown for 5%) [15].
Patients were asked to bring all their drug prescriptions for the preceding 3 months to the enrolment visit. The CRAs used an electronic case report form [linked to the international Anatomical Therapeutic and Chemical (ATC) thesaurus] to enter standardized ATC codes.
Longitudinal clinical data (nephrology consultations, hospital admissions, laboratory test results, medications and any transition to dialysis or transplantation) were collected every year from medical records and patient interviews.
Study outcomes
CV events were assessed carefully according to the Cardiovascular and Stroke Endpoint Definitions for Clinical Trials [16].
The primary endpoint was the occurrence of a first fatal or nonfatal atheromatous or non-atheromatous CVD. Fatal or nonfatal atheromatous CVD was defined as a composite of (i) death (fatal myocardial infarction or stroke), (ii) hospitalization for myocardial infarction, silent ischaemia, unstable angina, intrastent thrombosis, stroke or transient ischaemic attack, peripheral artery disease, percutaneous coronary intervention or coronary artery bypass graft, vascular surgery, or amputation and, (iii) revascularization for coronary or peripheral artery disease. Fatal or nonfatal non-atheromatous CVD was defined as a composite of (i) sudden cardiac death or death from heart failure, haemorrhagic stroke, or other CV causes, (ii) death associated with a CV procedure, such as noncoronary or peripheral vascular procedure, (iii) hospitalization for haemorrhagic stroke, heart failure, cardiac rhythm/conduction disorders, pulmonary embolism or deep vein thrombosis, valvular heart disease and, (iv) cardiac procedures such as valve prosthesis, cardioversion, implantation of a pacemaker and implantable cardioverter defibrillator.
The secondary outcomes included first major CV events (any CV death, myocardial infarction, stroke and hospitalization for heart failure) and all-cause death. Deaths before RRT were identified from medical records or reported by family members at the annual follow-up visit.
All the considered events occurred before RRT. RRT was defined in the present study as kidney dysfunction requiring initiation of chronic dialysis or preemptive transplantation (as identified from medical records or by linking to the French National Kidney Failure Registry) [17].
CV events were available for the first 3 years of follow-up at the time of analysis and so data were censored on the date of the patient's third annual follow-up visit, the date of the last follow-up before the third annual visit, the date of RRT or the date of death from a non-CV cause. Each death occurring during the 5-year follow-up period was used in the all-cause mortality model and the data were censored at the date of RRT or the date of the last follow-up.
Statistical analysis
The participants’ serum urea levels were divided into tertiles (T1 <10.5, T2 10.5–15.1, T3 ≥15.1 mmol/L). The baseline characteristics were described for the overall population (n = 2507) and the subgroups (serum urea tertiles) and reported as the median [interquartile range (IQR)] or the mean [standard deviation (SD)] for continuous data (depending on the distribution) and as the frequency (percentage) for qualitative variables. An analysis of variance (ANOVA) or the Kruskal–Wallis test was used to compare continuous variables, depending on the data distribution and homoscedasticity. Incidence rates and 95% confidence intervals (CIs) for each event were computed as a function of the serum urea levels. For each outcome studied, we used cause-specific Cox models to estimate crude and adjusted hazard ratios (HRs) [95% CI] associated with the serum urea level (in tertiles). HRs were adjusted for relevant demographic variables and baseline comorbidities (preselected in a review of the literature for each outcome). Risks associated with serum urea were adjusted for age at baseline, sex, smoking status, baseline eGFR, urine albumin- or protein-to-creatinine ratio, BMI, diabetes, systolic blood pressure, history of CVD, anaemia, serum albumin, high-sensitivity C-reactive protein and prescriptions of diuretics, statins and antiplatelet agents at baseline. The risk of death before RRT was further adjusted for a history of acute kidney injury and prescription of a proton pump inhibitor at baseline. Each model's validity (according to the proportional hazard assumption) was checked by testing the Schoenfeld residuals.
We used penalized splines in fully adjusted Cox models to represent the functional relationship between serum urea and the CVD risk. As a sensitivity analysis, we built a cause-specific Cox model for each type of CVD outcome, i.e. an analysis of (i) fatal and nonfatal atheromatous CVD events and (ii) fatal and nonfatal non-atheromatous CVD events.
Missing covariate data were managed by multivariate imputation by chained equations (MICE) [18]. The assumption whereby data were missing at random was plausible. By using the MICE package in R statistical software (version 4.0.3) [19], we created 50 datasets (20 iterations). All covariates present in the Cox models and baseline serum bicarbonate were included in the imputation model. Fitted Cox models were generated for each dataset and pooled regression coefficients were obtained using Rubin's rules. All statistical analyses were performed with R software.
RESULTS
Characteristics of the patients at baseline
Of the 3033 patients, 2507 patients were analysed (Table 1). The median age was 69 years [61–77] and the mean (SD) eGFR was 33.5 (11.6) mL/min/1.73 m²; 54% of the patients had a history of CVD, 45% had diabetes and 38% had anaemia. The mean (SD) serum urea was 8.3 (1.5) for T1, 12.6 (1.3) for T2 and 20.3 (5.1) mmol/L for T3. We observed significant difference in eGFR levels and the albumin- or protein-to-creatinine ratio at baseline between the three groups. A strong negative correlation was found between serum urea and eGFR (r = −0.64). The BMI was significantly higher in patients in T3. Compared with patients in T1, patients in the other tertiles were more likely to have a history of CVD, diabetes, anaemia, diuretic use and/or acute kidney injury. Lower serum bicarbonates levels were found in patients in T3. The proportion of patients with heart failure at baseline was significantly higher in T3 (19%) than in T1 (9%) and T2 (16%) (P < 0.001, not shown).
Table 1.
The baseline characteristics of the study population
| Baseline serum urea (mmol/L) | ||||||
|---|---|---|---|---|---|---|
| Total (N = 2507) | T1 <10.5 (N = 829) | T2 10.5–15.1 (N = 835) | T3 ≥ 15.1 (N = 843) | P- value | Imputed data (N = 2507) | |
| Serum urea (mmol/L) | 13.8 (5.9) | 8.3 (1.5) | 12.6 (1.3) | 20.3 (5.1) | 0% | |
| Age at baseline (years) | 69 [61–77] | 68 [60–76] | 69 [61–77] | 69 [61–77] | 0.13 | 0% |
| Men, n (%) | 66 | 65 | 66 | 67 | 0.71 | 0 |
| Smoking, n (%) | 0.03 | 0.8 | ||||
| Never-smoker, n (%) | 40.6 | 44.5 | 39.6 | 37.7 | ||
| Current smoker, n (%) | 12.6 | 11.7 | 11.8 | 14.4 | ||
| Former smoker, n (%) | 46.8 | 43.8 | 48.5 | 47.9 | ||
| eGFR at baseline (mL/min/1.73 m²) | 33.5 (11.6) | 43.5 (9.9) | 32.6 (8.9) | 24.5 (7.0) | <0.001 | 0% |
| Albumin- or protein-to-creatinine ratio | <0.001 | 8.0 | ||||
| A1 (normal to mildly increased), n (%) | 28.6 | 42.1 | 27.0 | 16.9 | ||
| A2 (moderately increased), n (%) | 31.8 | 31.8 | 33.7 | 29.7 | ||
| A3 (severely increased), n (%) | 39.6 | 26.1 | 39.2 | 53.4 | ||
| Body mass index (kg/m²) | 28.8 (5.8) | 28.3 (5.2) | 28.7 (5.9) | 29.5 (6.3) | <0.001 | 2.0% |
| Diabetes, n (%) | 44.8 | 36.8 | 43.9 | 53.6 | <0.001 | 0.2 |
| Systolic blood pressure (mmHg) | 142 (20) | 142 (20) | 142 (21) | 143 (20) | 0.32 | 2.3% |
| History of cardiovascular disease, n (%) | 53.9 | 47.3 | 54.6 | 59.6 | <0.001 | 1.3 |
| Anaemia, n (%) | 38.3 | 21.1 | 35.8 | 57.8 | <0.001 | 0.3 |
| Serum bicarbonate (mmol/L) | 25.0 (3.4) | 25.8 (3.1) | 24.9 (3.3) | 24.1 (3.6) | <0.001 | 6.9% |
| Serum albumin (g/L) | 40.4 (4.5) | 40.6 (4.4) | 40.5 (4.2) | 39.9 (4.9) | 0.009 | 15.2% |
| High-sensitivity C-reactive protein (mg/L) | 2.5 [1.1–5.9] | 2.2 [1.1–5.0] | 2.5 [1.1–5.4] | 2.9 [1.2–7.1] | <0.001 | 17.6% |
| History of acute kidney injury, n (%) | 22.5 | 20.0 | 20.9 | 26.6 | 0.004 | 7.7 |
| Diuretic prescription at baseline, n (%) | 54.0 | 39.9 | 51.1 | 70.7 | <0.001 | 0.3 |
| PPI prescription at baseline, n (%) | 31.4 | 27.9 | 33.9 | 32.4 | 0.02 | 0.3 |
| RASi prescription at baseline, n (%) | 76.9 | 72.3 | 80.2 | 78.0 | <0.001 | 0.3 |
| Statin prescription at baseline, n (%) | 58.9 | 51.4 | 61.0 | 64.1 | <0.001 | 0.% |
| Antiplatelet prescription at baseline, n (%) | 42.1 | 37.6 | 43.1 | 45.6 | 0.003 | 0.3 |
eGFR, estimated glomerular filtration rate, based on the CKD-EPI equation; RASi, renin-angiotensin system inhibitor; PPI, proton pump inhibitor; T, tertile.
Data are quoted as the frequency (%), mean (standard deviation) or the median [interquartile range]. An ANOVA or a Kruskal–Wallis or Chi-squared test was used to compare groups.
Fatal and nonfatal atheromatous or non-atheromatous cardiovascular events
Of the 2507 patients, 451 experienced a first atheromatous or non-atheromatous CV event (fatal or nonfatal) over a median [IQR] follow-up period of 3.0 [2.2–3.1] years, leading to a crude incidence rate [95% CI] of 7.1 [6.4; 7.7] per 100 person-years (PY). The incidence rate was highest in patients in the third urea tertile (Supplementary data, Table S2, Figure 2). After multiple adjustments, the risk of fatal and nonfatal atheromatous or non-atheromatous CV events was found to be significantly higher for patients in T3 than in patients in T1 (HR [95% CI]: 1.93 [1.39; 2.69]; Figure 2, Supplementary data, Table S3). A nonsignificant trend towards a higher risk was also noted for patients in T2 (HR [95% CI]: 1.24 [0.93; 1.66]). Penalized spline regression showed that serum urea was linearly related to the CVD risk (Figure 3). The interaction between serum urea and heart failure was tested and turned out not to be significant (P = .15), as well as the interaction between serum urea and eGFR (P = .69).
FIGURE 2:
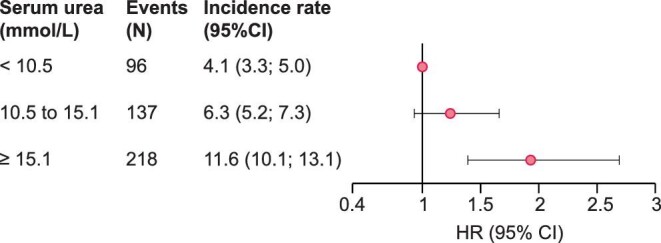
Adjusted HRs for fatal and nonfatal atheromatous or non-atheromatous cardiovascular events, according to the baseline serum urea level. HR, hazard ratio; CI, confidence interval. Adjusted for age at baseline, sex, smoking status, baseline estimated glomerular filtration rate, urine albumin- or protein-to-creatinine ratio, body mass index, diabetes, systolic blood pressure, history of cardiovascular disease, anaemia, serum albumin, high-sensitivity C-reactive protein and prescriptions of diuretics, statins and antiplatelet agents at baseline.
FIGURE 3:
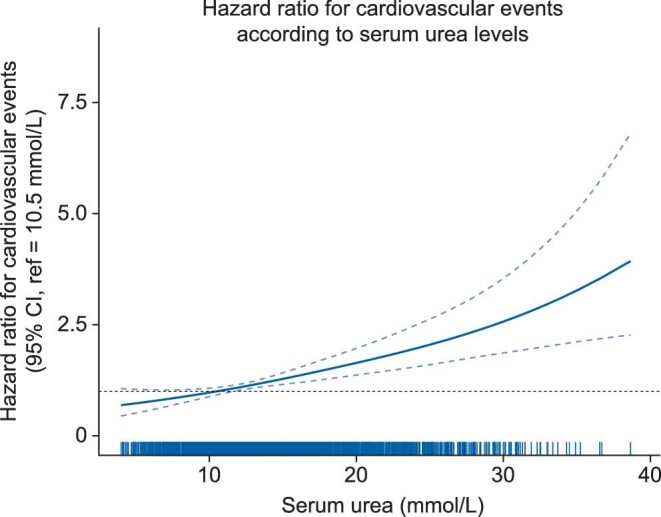
Hazard ratio for fatal and nonfatal atheromatous or non-atheromatous cardiovascular events, according to the baseline serum urea level (mmol/L). The continuous line represents predictions with penalized splines in Cox models (95% confidence intervals). Ticks on the x-axis represent the distribution of the baseline serum urea level. Hazard ratios are adjusted for age at baseline, sex, smoking status, baseline estimated glomerular filtration rate, urine albumin- or protein-to-creatinine ratio, body mass index, diabetes, systolic blood pressure, history of cardiovascular disease, anaemia, serum albumin, high-sensitivity C-reactive protein and prescriptions of diuretics, statins and antiplatelet agents at baseline.
A sensitivity analysis failed to show an association between the baseline serum urea level and atheromatous CV events (fatal or nonfatal) (Supplementary data, Table S4). In contrast, a significantly higher risk of non-atheromatous CV events (fatal or nonfatal) was observed in patients in T3 (HR [95% CI]: 2.13 [1.40; 3.24]), and a trend towards a greater risk was observed in patients in T2 (HR [95% CI], 1.28 [0.88; 1.88]); Supplementary data, Table S5).
Major CV events
Over the first 3 years of follow-up, 275 patients first experienced a major CV event. The crude incidence rate was 4.1 [3.6; 4.6] per 100 PY. The incidence rate was 3.5 times higher in patients in T3 than in patients in T1 (Supplementary data, Table S2, Figure 4). The adjusted HR [95% CI] for major CV events associated with the baseline serum urea level was 1.13 [0.77; 1.67] for patients in T2 and 1.94 [1.27; 2.98] for patients in T3 (Figure 4, Supplementary data, Table S6).
FIGURE 4:
Adjusted HRs for major cardiovascular events, according to the baseline serum urea level. HR, hazard ratio; CI, confidence interval. Adjusted for age at baseline, sex, smoking status, baseline estimated glomerular filtration rate, urine albumin- or protein-to-creatinine ratio, body mass index, diabetes, systolic blood pressure, history of cardiovascular disease, anaemia, serum albumin, high-sensitivity C-reactive protein and prescriptions of diuretics, statins and antiplatelet agents at baseline of diuretics, prescription of statins and prescription of antiplatelet agents at baseline.
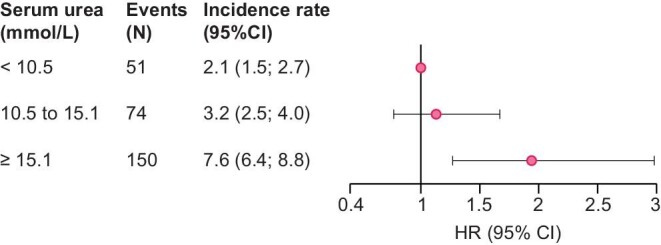
Death before RRT
Over a median follow-up period of 4.8 [3.3; 5.1] years in the CKD-REIN cohort, 407 patients died before RRT, leading to an incidence rate of 4.0 [3.6; 4.3] per 100 person-years (Supplementary data, Table S2, Figure 5). The adjusted HRs [95% CI] for death before RRT were 1.31 [0.97; 1.76] and 1.73 [1.22; 2.45] for patients in T2 and T3, respectively (Figure 5, Supplementary data, Table S7).
FIGURE 5:
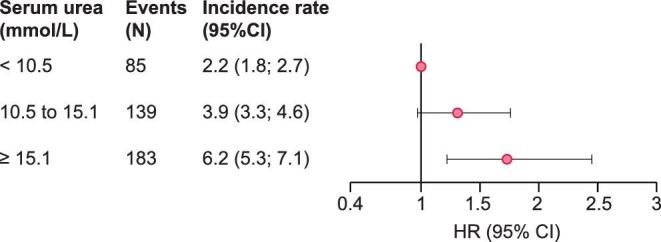
Adjusted HRs for death before renal replacement therapy according to the baseline serum urea level. HR, hazard ratio; CI, confidence interval. Adjusted for age at baseline, sex, smoking status, baseline estimated glomerular filtration rate, urine albumin- or protein-to-creatinine ratio, body mass index, diabetes, systolic blood pressure, history of cardiovascular disease, anaemia, serum albumin, high-sensitivity C-reactive protein, history of acute kidney injury, diuretic prescription, proton pump inhibitor prescription and prescriptions of statins and antiplatelet agents at baseline.
DISCUSSION
Our analysis of a large prospective cohort of CKD patients not receiving maintenance dialysis attending nephrology outpatient facilities showed that higher serum urea levels were associated with a higher incidence of adverse CV outcomes and a higher all-cause mortality rate. Our findings are in line with the current body of epidemiological evidence on kidney function biomarkers and the risk of CVD, and suggest that urea should be taken into account when seeking to predict and prevent CV disease in patients with CKD.
Several serum biomarkers [such as serum creatinine or urea, or blood urea nitrogen (BUN), which reflects only the nitrogen content of urea], are routinely used in clinical settings to evaluate kidney function. Urea is the main metabolite derived from dietary proteins and tissue protein turnover. The compound is almost exclusively excreted by the kidneys in the urine, after filtration in the glomerulus and a certain degree of reabsorption from the filtrate. Although several nonrenal factors affect the serum urea concentration [20], reduced urinary elimination of urea (due to CKD) is the main factor that increases serum urea levels. Volume depletion by diuretics or a decrease in the effective circulating volume induced by heart failure might contribute to the elevation of urea levels in our CKD patients. However, the fact that bicarbonate levels were significantly lower in T3 than in T1 (Table 1) tends to rule out volume depletion by diuretics. In contrast, the difference between the tertiles in the prevalence of heart failure suggests that the elevated urea levels could be due (at least in part) to a decrease in the effective circulating volume caused by heart failure.
Under normal conditions, the serum urea level ranges from 2.2 to 7.2 mmol/L (or 13–43 mg/dL) [21]. Patients in the first urea tertile in our study can be considered as having a normal level, those in the second tertile have a slightly elevated level and those in the third tertile have a moderately elevated level. During the course of the CKD, serum urea levels can easily reach or exceed 10 times the upper normal limit—especially when kidney failure occurs [21]. Until recently, urea was considered to be a biologically inert marker. However, studies of animal models of CKD have shown that the accumulation of urea is toxic [21].
CKD is a common disease and its prevalence will continue to increase in the coming years [2]. CVD is one of the leading contributors to morbidity and mortality in patients with CKD [22]. The limited effectiveness of most conventional risk factor modification strategies in patients with CKD may suggest that various metabolic pathways underlie the development of CVD in this population [23]. The results of animal studies suggest that several direct and indirect pathophysiological mechanisms underlie the relationship between urea levels and CV adverse events in CKD [5]. Uraemic conditions lead to the overexpression of proapoptotic genes in animal tissues and in cultured human vascular smooth muscle cells [6]. Moreover, the urea levels found in the serum of CKD patients directly increase levels of reactive oxygen species (ROS) and oxidative stress in several types of cells [24–26]. Interestingly, endothelial progenitor cell number and function decrease with advancing CKD [27], which might be due to the acceleration of senescence in endothelial progenitor cells by urea-induced ROS [8]. In fact, these endothelial progenitor cells have a key role in the repair and maintenance of the vascular system [28]. Furthermore, there is a growing body of evidence on indirect toxic effects of urea via post-translational modifications of proteins; the resulting biochemical alterations might have an impact on CV outcomes [5, 21]. Indeed, urea's dissociation products (e.g. cyanate) irreversibly carbamylate proteins [29] and so change the latter's physical properties and molecular and cellular functions. CKD is associated with elevated protein carbamylation [10]. Low-density lipoproteins are sensitive to carbamylation; this damage can cause endothelial cell death and smooth muscle cell proliferation in vitro [30] and other molecular alterations leading to vascular damage in vivo [5]. In the 4D study, protein carbamylation was associated with heart failure and death in diabetic patients with end-stage renal disease [31]. Several comparative studies in patients with CKD have suggested that BUN is a strong predictor of all-cause mortality in patients with heart failure and in those with acute coronary syndrome [32–34]. Although the mechanisms underlying these associations have yet to be characterized, these data suggest that carbamylation has an important role in CV outcomes in patients with CKD.
Interestingly, in our study, the highest serum urea level was associated with both CV events and death before RRT in the CKD-REIN cohort and these associations persisted after adjustment for the current eGFR—suggesting that more attention should be paid to the prevention of CVD in patients with high serum urea levels. We found that the risk of atheromatous CVD was not significant but that the association between high serum urea levels and non-atheromatous CVD was strong and significant. Our hypothesis is that urea might have limited effects (directly, or indirectly via carbamylation) on atheromatous CVD, relative to traditional risk factors. Urea is the most potent uraemic toxin for inducing the expression of the pro-apoptotic BCL2 family protein BAD in human smooth muscle cells. However, the induction of BAD by urea alone did not induce apoptosis but sensitized cells to the pro-apoptotic effect of oxidized cholesterol—a physiologically relevant inducer of this form of programmed death [6]. Moreover, urea might have a significant impact on nonatherosclerotic events, such as cardiac fibrosis. Indeed, it has been shown that the inhibition of urea transporters reduces cardiac fibrosis and improves heart function [9].
Given the dietary origin of a proportion of the urea in the circulation, nutritional therapy could be used to counter an elevation in urea levels. Indeed, Di Iorio et al. have shown that serum urea levels were lower in patients on a Mediterranean diet and on a very low protein diet supplemented with ketoanalogues, relative to patients with an unrestricted diet [35]. Interventional studies of various diets and their impacts on cardiovascular outcomes (and especially non-atheromatous events) in non-dialysed patients with CKD are warranted.
The present study had several strengths. Firstly, and to the best of our knowledge, the present prospective study is the first to have investigated the association between urea levels and CVD in non-dialysed patients with CKD. Secondly, we studied a large number of patients, which increased the study's statistical power and enabled extensive adjustment for confounders (including the eGFR). Thirdly, all the CV events were carefully adjudicated according to standardized definitions [16]. Our study also had limitations. Firstly, the observational design means that we cannot rule out the possibility of residual confounding and we did not explore time-varying risk associations between serum urea and outcomes. However, our study could be considered to be a hypothesis-generating study. Secondly, we did not assess the protein intake at the time of the blood test, nor the time at which this blood test was taken; these variables might have affected the baseline serum urea levels. Thirdly, elevated urea could be a marker for occult heart failure. Lastly, urea is one of many uraemic toxins and so these other toxins (not assayed here) might have influenced the study outcomes. However, urea is a useful biomarker because it is measured in routine clinical practice—in contrast to various other uraemic toxins, which require specific assays.
CONCLUSION
The present prospective, epidemiological study is the first to our knowledge to show that serum urea levels are associated with CVD and mortality in non-dialysed patients with CKD, independently of renal function. Indeed, our analysis of a large cohort of patients with CKD demonstrated that individuals with elevated serum urea levels had a higher risk of CV outcomes and death.
Supplementary Material
ACKNOWLEDGEMENTS
We thank the CKD-REIN study coordination staff for their efforts in setting up the cohort: Elodie Speyer, Céline Lange, Reine Ketchemin, Natalia Alencar de Pinho and all the clinical research associates. The authors thank Thao Nguyen Khoa for dosing high-sensitivity C-reactive protein.
Appendix
The CKD-REIN study collaborators:
The CKD-REIN Study Group steering committee and coordinators include: Carole Ayav, Serge Briançon, Dorothée Cannet, Christian Combe, Denis Fouque, Luc Frimat, Yves-Edouard Herpe, Christian Jacquelinet, Maurice Laville, Ziad A. Massy, Christophe Pascal, Bruce M. Robinson, Bénédicte Stengel, Céline Lange, Karine Legrand, Sophie Liabeuf, Marie Metzger and Elodie Speyer.
The CKD-REIN investigators/collaborators include: Thierry Hannedouche, Bruno Moulin, Sébastien Mailliez, Gaétan Lebrun, Eric Magnant, Gabriel Choukroun, Benjamin Deroure, Adeline Lacraz, Guy Lambrey, Jean-Philippe Bourdenx, Marie Essig, Thierry Lobbedez, Raymond Azar, Hacène Sekhri, Mustafa Smati, Mohamed Jamali, Alexandre Klein, Michel Delahousse, Christian Combe, Séverine Martin, Isabelle Landru, Eric Thervet, Ziad A. Massy, Philippe Lang, Xavier Belenfant, Pablo Urena, Carlos Vela, Luc Frimat, Dominique Chauveau, Viktor Panescu, Christian Noel, François Glowacki, Maxime Hoffmann, Maryvonne Hourmant, Dominique Besnier, Angelo Testa, François Kuentz, Philippe Zaoui, Charles Chazot, Laurent Juillard, Stéphane Burtey, Adrien Keller, Nassim Kamar, Denis Fouque and Maurice Laville.
Contributor Information
Solène M Laville, Department of Clinical Pharmacology, Amiens University Hospital, Amiens, France; MP3CV Laboratory, EA7517, University of Picardie Jules Verne, Amiens, France.
Aymeric Couturier, Department of Nephrology, Ambroise Paré University Hospital, APHP, Boulogne-Billancourt, Paris, France.
Oriane Lambert, Centre for Research in Epidemiology and Population Health (CESP), Paris-Saclay University, Versailles Saint Quentin University, INSERM UMRS, 1018 Villejuif, France.
Marie Metzger, Centre for Research in Epidemiology and Population Health (CESP), Paris-Saclay University, Versailles Saint Quentin University, INSERM UMRS, 1018 Villejuif, France.
Nicolas Mansencal, Centre for Research in Epidemiology and Population Health (CESP), Paris-Saclay University, Versailles Saint Quentin University, INSERM UMRS, 1018 Villejuif, France; Department of Cardiology, Ambroise Paré University Hospital, APHP, Boulogne-Billancourt, Paris, France.
Christian Jacquelinet, Biomedecine Agency, Saint Denis La Plaine, France.
Maurice Laville, Université de Lyon, CarMeN INSERM, Lyon, France.
Luc Frimat, Nephrology Department, CHRU de Nancy, Vandoeuvre-lès-Nancy, France; Lorraine University, APEMAC, Vandoeuvre-lès-Nancy, France.
Denis Fouque, Université de Lyon, CarMeN INSERM, Lyon, France; Nephrology Department, Centre Hospitalier Lyon Sud, Pierre-Bénite, France.
Christian Combe, Service de Néphrologie Transplantation Dialyse Aphérèse, Centre Hospitalier Universitaire de Bordeaux, Bordeaux, France; INSERM, U1026, Univ Bordeaux Segalen, Bordeaux, France.
Bruce M Robinson, Arbor Research Collaborative for Health, Ann Arbor MI, USA.
Bénédicte Stengel, Centre for Research in Epidemiology and Population Health (CESP), Paris-Saclay University, Versailles Saint Quentin University, INSERM UMRS, 1018 Villejuif, France.
Sophie Liabeuf, Department of Clinical Pharmacology, Amiens University Hospital, Amiens, France; MP3CV Laboratory, EA7517, University of Picardie Jules Verne, Amiens, France.
Ziad A Massy, Department of Nephrology, Ambroise Paré University Hospital, APHP, Boulogne-Billancourt, Paris, France; Centre for Research in Epidemiology and Population Health (CESP), Paris-Saclay University, Versailles Saint Quentin University, INSERM UMRS, 1018 Villejuif, France.
the CKD-REIN study collaborators:
Carole Ayav, Serge Briançon, Dorothée Cannet, Christian Combe, Denis Fouque, Luc Frimat, Yves-Edouard Herpe, Christian Jacquelinet, Maurice Laville, Ziad A Massy, Christophe Pascal, Bruce M Robinson, Bénédicte Stengel, Céline Lange, Karine Legrand, Sophie Liabeuf, Marie Metzger, Elodie Speyer, Thierry Hannedouche, Bruno Moulin, Sébastien Mailliez, Gaétan Lebrun, Eric Magnant, Gabriel Choukroun, Benjamin Deroure, Adeline Lacraz, Guy Lambrey, Jean Philippe, Bourdenx, Marie Essig, Thierry Lobbedez, Raymond Azar, Hacène Sekhri, Mustafa Smati, Mohamed Jamali, Alexandre Klein, Michel Delahousse, Christian Combe, Séverine Martin, Isabelle Landru, Eric Thervet, Ziad A Massy, Philippe Lang, Xavier Belenfant, Pablo Urena, Carlos Vela, Luc Frimat, Dominique Chauveau, Viktor Panescu, Christian Noel, François Glowacki, Maxime Hoffmann, Maryvonne Hourmant, Dominique Besnier, Angelo Testa, François Kuentz, Philippe Zaoui, Charles Chazot, Laurent Juillard, Stéphane Burtey, Adrien Keller, Nassim Kamar, Denis Fouque, and Maurice Laville
AUTHORS’ CONTRIBUTIONS
S.M.L., S.L. and Z.A.M. designed this project. S.M.L., O.L., M.M., S.L. and Z.A.M. analysed and/or contributed to the interpretation of the data. S.M.L., S.L. and Z.A.M. wrote the first draft of the article. A.C., O.L., N.M., M.M., C.J., M.L., L.F., D.F., C.C., B.M.R. and B.S. provided critical revision for important intellectual content. All co-authors approved the final version of the article.
FUNDING
CKD-REIN is funded by the Agence Nationale de la Recherche through the 2010 ‘Cohortes-Investissements d'Avenir’ program (ANR) and by the 2010 national Programme Hospitalier de Recherche Clinique. CKD-REIN is also supported through a public-private partnership with Amgen, Fresenius Medical Care, and GlaxoSmithKline (GSK) since 2012, Lilly France since 2013, Otsuka Pharmaceutical since 2015, Baxter and Merck Sharp & Dohme-Chibret (MSD France) from 2012 to 2017, Sanofi-Genzyme from 2012 to 2015, and Vifor Fresenius and AstraZeneca, since 2018. INSERM Transfert set up and has managed this partnership since 2011. The funding source had no role in the study's design, conduct, and reporting.
CONFLICT OF INTEREST STATEMENT
S.M.L., S.L., O.L. and N.M. have nothing to declare. D.F. reports grants and lecture fees form Sanofi, Fresenius Kabi, Astra-Zeneca and Lilly. Z.A.M. reports grants for CKD-REIN and other research projects from Amgen, Baxter, Fresenius Medical Care, GlaxoSmithKline, Merck Sharp and Dohme-Chibret, Sanofi-Genzyme, Lilly, Otsuka and the French government, as well as fees and grants to charities from Amgen, Daichii, and Sanofi-Genzyme. These sources of funding are not necessarily related to the content of the present manuscript. B.S. reports grants for the CKD-REIN cohort study from Amgen, Baxter, Fresenius Medical Care, GlaxoSmithKline, Merck Sharp and Dohme-Chibret, Sanofi Genzyme, Lilly, Otsuka and Vifor Fresenius, as well as speaker from Lilly and MSD, unrelated to the content of this manuscript.
AUTHORIZATIONS
All legal authorizations were obtained, including those from the Comité consultatif sur le traitement de l'information en matière de recherche dans le domaine de la santé (CCTIRS N°12.360), the Commission nationale de l'informatique et des libertés (CNIL N°DR-2012-469) and the Kremlin-Bicêtre Comité de protection des personnes (CPP N°IDRCB 2012-A00902-41). The CKD-REIN sample collection is registered in the management application by the COnservation D'Eléments du COrps Humain biological resource center (CODECOCH N°-2012-1624). The Institut national de la santé et de la recherche médical (INSERM) institutional review board approved the study protocol (IRB00003888), which was registered at ClinicalTrials.gov (NCT03381950).
REFERENCES
- 1. Levey AS, Atkins R, Coresh Jet al. Chronic kidney disease as a global public health problem: approaches and initiatives – a position statement from kidney disease improving global outcomes. Kidney Int 2007; 72: 247–259 [DOI] [PubMed] [Google Scholar]
- 2. Jha V, Garcia-Garcia G, Iseki Ket al. Chronic kidney disease: global dimension and perspectives. Lancet North Am Ed 2013; 382: 260–272 [DOI] [PubMed] [Google Scholar]
- 3. Go AS, Chertow GM, Fan Det al. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N Engl J Med 2004; 351: 1296–1305 [DOI] [PubMed] [Google Scholar]
- 4. Vanholder R, Massy Z, Argiles Aet al. Chronic kidney disease as cause of cardiovascular morbidity and mortality. Nephrol Dial Transplant 2005; 20: 1048–1056 [DOI] [PubMed] [Google Scholar]
- 5. Massy ZA, Pietrement C, Touré F. Reconsidering the lack of urea toxicity in dialysis patients. Semin Dial 2016; 29: 333–337 [DOI] [PubMed] [Google Scholar]
- 6. Trécherel E, Godin C, Louandre Cet al. Upregulation of BAD, a pro-apoptotic protein of the BCL2 family, in vascular smooth muscle cells exposed to uremic conditions. Biochem Biophys Res Commun 2012; 417: 479–483 [DOI] [PubMed] [Google Scholar]
- 7. Shroff RC, McNair R, Figg Net al. Dialysis accelerates medial vascular calcification in part by triggering smooth muscle cell apoptosis. Circulation 2008; 118: 1748–1757 [DOI] [PubMed] [Google Scholar]
- 8. D'Apolito M, Colia AL, Lasalvia Met al. Urea-induced ROS accelerate senescence in endothelial progenitor cells. Atherosclerosis 2017; 263: 127–136 [DOI] [PubMed] [Google Scholar]
- 9. Kuma A, Wang XH, Klein JDet al. Inhibition of urea transporter ameliorates uremic cardiomyopathy in chronic kidney disease. FASEB J 2020; 34: 8296–8309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kalim S, Karumanchi SA, Thadhani RIet al. Protein carbamylation in kidney disease: pathogenesis and clinical implications. Am J Kidney Dis 2014; 64: 793–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kalim S, Berg AH, Karumanchi SAet al. Protein carbamylation and chronic kidney disease progression in the chronic renal insufficiency cohort study. Nephrol Dial Transplant 2021; 37: 139–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mori D, Matsui I, Shimomura Aet al. Protein carbamylation exacerbates vascular calcification. Kidney Int 2018; 94: 72–90 [DOI] [PubMed] [Google Scholar]
- 13. Apostolov EO, Ray D, Savenka AVet al. Chronic uremia stimulates LDL carbamylation and atherosclerosis. J Am Soc Nephrol 2010; 21: 1852–1857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Stengel B, Metzger M, Combe Cet al. Risk profile, quality of life and care of patients with moderate and advanced CKD: The French CKD-REIN Cohort Study. Nephrol Dial Transplant 2019; 34: 277–286 [DOI] [PubMed] [Google Scholar]
- 15. Levey AS, Stevens LA, Schmid CHet al. A new equation to estimate glomerular filtration rate. Ann Intern Med 2009; 150: 604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hicks KA, Mahaffey KW, Mehran Ret al. 2017 Cardiovascular and stroke endpoint definitions for clinical trials. Circulation 2018; 137: 961–972 [DOI] [PubMed] [Google Scholar]
- 17. Couchoud C, Lassalle M. Rapport annuel 2017 - Réseau Epidémiologie, Information, Néphrologie.; 2017; https://www.agence-biomedecine.fr/IMG/pdf/rapportrein2017.pdf (29 March 2020, date last accessed) [Google Scholar]
- 18. van Buuren S, Groothuis-Oudshoorn K. mice: Multivariate Imputation by Chained Equations in R. J Stat Softw 2011; 45: 1–67 [Google Scholar]
- 19. R Core Team. R: A Language and Environment for Statistical Computing., Vienna, Austria. R Foundation for Statistical Computing; 2020; https://www.R-project.org/ [Google Scholar]
- 20. Luke RG. Uremia and the BUN. N Engl J Med 1981; 305: 1213–1215 [DOI] [PubMed] [Google Scholar]
- 21. Vanholder R, Gryp T, Glorieux G. Urea and chronic kidney disease: the comeback of the century? (in uraemia research). Nephrol Dial Transplant 2018; 33: 4–12 [DOI] [PubMed] [Google Scholar]
- 22. Thompson S, James M, Wiebe Net al. Cause of death in patients with reduced kidney function. J Am Soc Nephrol 2015; 26: 2504–2511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gregg LP, Hedayati SS. Management of traditional cardiovascular risk factors in CKD: what are the data? Am J Kidney Dis 2018; 72: 728–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. D'Apolito M, Du X, Zong Het al. Urea-induced ROS generation causes insulin resistance in mice with chronic renal failure. J Clin Invest 2010; 120: 203–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Koppe L, Nyam E, Vivot Ket al. Urea impairs β cell glycolysis and insulin secretion in chronic kidney disease. J Clin Invest 2016; 126: 3598–3612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. D'Apolito M, Du X, Pisanelli Det al. Urea-induced ROS cause endothelial dysfunction in chronic renal failure. Atherosclerosis 2015; 239: 393–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Krenning G, Dankers PYW, Drouven JWet al. Endothelial progenitor cell dysfunction in patients with progressive chronic kidney disease. Am J Physiol Renal Physiol 2009; 296: F1314–F1322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Werner N, Nickenig G. Endothelial progenitor cells in health and atherosclerotic disease. Ann Med 2007; 39: 82–90 [DOI] [PubMed] [Google Scholar]
- 29. Kraus LM, Kraus AP. Carbamoylation of amino acids and proteins in uremia. Kidney Int 2001; 78: S102–S107 [DOI] [PubMed] [Google Scholar]
- 30. Ok E, Basnakian AG, Apostolov EOet al. Carbamylated low-density lipoprotein induces death of endothelial cells: a link to atherosclerosis in patients with kidney disease. Kidney Int 2005; 68: 173–178 [DOI] [PubMed] [Google Scholar]
- 31. Drechsler C, Kalim S, Wenger JBet al. Protein carbamylation is associated with heart failure and mortality in diabetic patients with end-stage renal disease. Kidney Int 2015; 87: 1201–1208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Aronson D, Mittleman MA, Burger AJ. Elevated blood urea nitrogen level as a predictor of mortality in patients admitted for decompensated heart failure. Am J Med 2004; 116: 466–473 [DOI] [PubMed] [Google Scholar]
- 33. Kirtane AJ, Leder DM, Waikar SSet al. Serum blood urea nitrogen as an independent marker of subsequent mortality among patients with acute coronary syndromes and normal to mildly reduced glomerular filtration rates. J Am Coll Cardiol 2005; 45: 1781–1786 [DOI] [PubMed] [Google Scholar]
- 34. Cauthen CA, Lipinski MJ, Abbate Aet al. Relation of blood urea nitrogen to long-term mortality in patients with heart failure. Am J Cardiol 2008; 101: 1643–1647 [DOI] [PubMed] [Google Scholar]
- 35. Di Iorio BR, Marzocco S, Bellasi Aet al. Nutritional therapy reduces protein carbamylation through urea lowering in chronic kidney disease. Nephrol Dial Transplant 2018; 33: 804–813 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.