

Background and Aims:
Alagille syndrome (ALGS) is a multisystem disorder, characterized by cholestasis. Existing outcome data are largely derived from tertiary centers, and real‐world data are lacking. This study aimed to elucidate the natural history of liver disease in a contemporary, international cohort of children with ALGS.
Approach and Results:
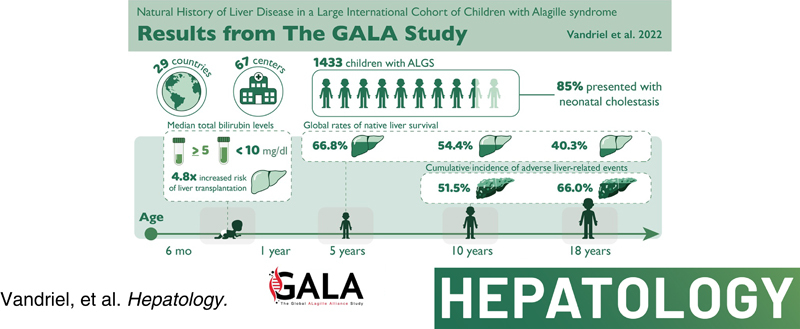
This was a multicenter retrospective study of children with a clinically and/or genetically confirmed ALGS diagnosis, born between January 1997 and August 2019. Native liver survival (NLS) and event‐free survival rates were assessed. Cox models were constructed to identify early biochemical predictors of clinically evident portal hypertension (CEPH) and NLS. In total, 1433 children (57% male) from 67 centers in 29 countries were included. The 10 and 18‐year NLS rates were 54.4% and 40.3%. By 10 and 18 years, 51.5% and 66.0% of children with ALGS experienced ≥1 adverse liver‐related event (CEPH, transplant, or death). Children (>6 and ≤12 months) with median total bilirubin (TB) levels between ≥5.0 and <10.0 mg/dl had a 4.1‐fold (95% confidence interval [CI], 1.6–10.8), and those ≥10.0 mg/dl had an 8.0‐fold (95% CI, 3.4–18.4) increased risk of developing CEPH compared with those <5.0 mg/dl. Median TB levels between ≥5.0 and <10.0 mg/dl and >10.0 mg/dl were associated with a 4.8 (95% CI, 2.4–9.7) and 15.6 (95% CI, 8.7–28.2) increased risk of transplantation relative to <5.0 mg/dl. Median TB <5.0 mg/dl were associated with higher NLS rates relative to ≥5.0 mg/dl, with 79% reaching adulthood with native liver (p < 0.001).
Conclusions:
In this large international cohort of ALGS, only 40.3% of children reach adulthood with their native liver. A TB <5.0 mg/dl between 6 and 12 months of age is associated with better hepatic outcomes. These thresholds provide clinicians with an objective tool to assist with clinical decision‐making and in the evaluation of therapies.
INTRODUCTION
Alagille syndrome (ALGS) is an autosomal dominant disorder, primarily characterized by hepatic involvement manifesting as high γ‐glutamyl transferase (GGT) cholestasis with variable extrahepatic involvement.1–3 Two causative genes encoding components of the Notch signaling pathway have been identified in ALGS: JAGGED1 (JAG1) and NOTCH2. Genetic testing yields a pathogenic JAG1 variant or deletion in 94.3% of patients with ALGS meeting phenotypic criteria, whereas pathogenic variants in NOTCH2 account for 2.5% of clinical cases.4 The prevalence of ALGS has historically been estimated to be approximately 1 in 70,000 live births; however, based on genetic analyses, the true disease burden is likely closer to 1 in 30,000.5
There is considerable variation in the clinical course of ALGS, and there are no known genotypic predictors of liver disease progression. To date, the phenotype and natural history of ALGS have been reported from single centers, often without molecular characterization of patients, and these data are somewhat outdated. A single recent multicenter study of ALGS comes from tertiary referral liver centers in North America and represents patients with cholestasis who are most severely affected.6 As a result, the full spectrum of ALGS‐related liver involvement remains unknown, and detailed analyses of real‐world natural history data are lacking. This is a crucial unmet need in an era when novel therapeutic strategies are being developed to target cholestasis‐induced pruritus, such as the apical sodium‐dependent bile acid transporter (ASBT) inhibitors.7
The Global ALagille Alliance (GALA) Study Group was formed to elucidate the natural history of liver disease in a contemporary international cohort of children with ALGS. Specifically, we sought to investigate rates of native liver survival (NLS) in children with ALGS and a history of neonatal cholestasis and to identify early laboratory predictors of long‐term hepatic outcomes. Furthermore, we aimed to evaluate global patient and graft survival following liver transplantation (LT) in children with ALGS.
PATIENTS AND METHODS
The GALA Study was established in 2018 and presently consists of 67 pediatric centers from 29 countries. Children with a clinically and/or genetically confirmed ALGS diagnosis born between January 1997 and August 2019 were eligible for inclusion. A diagnosis of ALGS was made in accordance with standard clinical criteria (Table S1). Children with ALGS born before January 1997 and who had a pathogenic or likely pathogenic variant in JAG1 or NOTCH2 were also included. The study was approved by the ethics committee at each participating center and a waiver of informed consent was granted by each institutional review committee, if applicable.
Data collection
From May 2018 to July 2021, the medical records of patients with ALGS were retrospectively reviewed at each participating center and each site abstracted data directly into REDCap (Research Electronic Data Capture), a remote web‐based database.8 To ensure the collection of rigorous, high‐quality data, the GALA Data Coordinating Center (DCC) at The Hospital for Sick Children, Toronto, Ontario, Canada, implemented several quality assurances measures and procedures as described in the Supporting Material. This study followed the Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) reporting guideline for cohort studies.9
Study definitions and variables
The pathogenicity of reported variants was classified according to the American College of Medical Genetics and Genomics guidelines.10 Neonatal cholestasis was defined as having at least one of the following features during the first 3 months of life: (1) direct/conjugated bilirubin (CB) greater than >2.0 mg/dl (34.0 μmol/L); (2) serum bile acids or GGT greater than 3 times the upper limit of normal; or (3) fat‐soluble vitamin deficiency, otherwise unexplainable.6 Liver involvement at any age was defined as a history of neonatal cholestasis, elevated liver aminotransferases, histological abnormalities, history of pruritus and/or xanthomas, or having underwent hepatobiliary surgery. Native liver biopsy reports were reviewed to characterize histopathological findings. Hepatic fibrosis was reported as a dichotomous variable (absent or present) based on review of histopathology reports. Pruritus and xanthomas were reported at each follow‐up visit as a categorical variable (present, absent, or unknown). Biochemical parameters including serum bile acids, total bilirubin (TB), CB, alanine aminotransferase (ALT), aspartate aminotransferase (AST), GGT, total cholesterol, and triglyceride, platelet count (PLT) were collected during the first year of life in those who presented with neonatal cholestasis. The AST to PLT Ratio Index (APRI) was calculated utilizing laboratory data from the first year of life.11 Cardiac involvement was determined by review of echocardiogram reports. Magnetic resonance imaging and/or computerized tomography scan reports were reviewed to determine the presence of cerebral and/or systemic vascular anomalies. Clinically evident portal hypertension (CEPH) was defined as the combination of splenomegaly, as noted on ultrasound and thrombocytopenia (PLT count below 150 × 109/L) and/or ascites requiring treatment with diuretics or esophageal or gastric varices requiring intervention at endoscopy.12 NLS was calculated from the date of birth until LT, death, or date of last clinical follow‐up, whichever event occurred first. To evaluate geographic differences, the cohort was stratified into one of seven geographic regions, (1) Africa, (2) Asia, (3) Europe, (4) the Middle East, (5) North America, (6) Oceania (Australia and New Zealand), and (7) South America, and compared. The time to the first adverse liver‐related event was calculated from the date of birth until one of the following events: CEPH, LT, death, or date of last known clinical follow‐up. Patient and graft survival following LT were calculated from the date of LT until retransplantation, death, or date of last known clinical follow‐up. Overall survival (all‐cause mortality, including post‐transplant) was calculated from the date of birth until death or date of last known clinical follow‐up, whichever event occurred first. In an analysis of the association between biochemistry data from the first year of life and long‐term events (manifestations of portal hypertension [PHT], CEPH, or NLS), time was calculated from 12 months of age until the event of interest or date of last known clinical follow‐up, whichever occurred first. Causes of death were classified into one of the following categories: liver or LT‐related complications, cardiac‐related complications, noncardiac vascular complications, multiorgan failure, sepsis, bleeding, other, or unknown. All follow‐up data were censored on August 31, 2019. A summary of the data elements is provided in Table S2.
Statistical analysis
Descriptive statistics were summarized with medians and interquartile ranges (IQR) and categorical variables are reported as counts and percentages. Clinical characteristics between groups were compared using Chi‐square test or Fisher's exact test, as appropriate. Laboratory values were log‐transformed to normalize distributions and adjusted for regional differences by site‐specific upper and lower limits of normal. For children who underwent Kasai portoenterostomy (KPE) during the first year of life, laboratory data were only reported up until the surgical procedure. All analyses were conducted in children with ALGS, and a history of neonatal cholestasis and a secondary analysis were performed in the entire cohort to determine the rate of all‐cause mortality. To assess the risk of NLS and LT in the presence of the competing risk of death, the Fine and Gray approach was applied.13 Time‐dependent Cox proportional‐hazards models were constructed to determine NLS in those who underwent a KPE or surgical biliary diversion (BD). Event‐free survival, patient and graft survival following LT, and overall patient survival rates were determined using the Kaplan–Meier method, and between‐group comparisons were made with log‐rank test. To determine the utility of liver biochemistry as a prognostic marker of developing manifestations of PHT (ascites or varices), CEPH, and NLS, median serial laboratory measurements from the first year of life were calculated and potential associations were assessed using log‐linear modeling. If a relationship was identified, thresholds were visually derived from the laboratory data distributions and the Akaike information criterion and C‐statistic values were compared. Cox proportional‐hazards models were then used to assess the effect of the threshold on the event(s). Hazard ratios (HRs) and 95% confidence intervals (CIs) were estimated using univariate and multivariate Cox regression analysis and adjusted for sex, year of birth, and geographical region by including this variable as a stratum. This allows each region to have its own background hazard rate, thereby allowing and repairing for heterogeneity. Those who had an event of interest (manifestations of PHT, CEPH, or LT/death) in the first year of life or follow‐up that ended before 1 year of age were excluded. A p value < 0.05 was considered statistically significant and the analysis was performed using Statistical Analysis System (SAS) version 9.4 and Statistical Package for the Social Sciences (SPSS, Chicago, IL) version 25.
RESULTS
Study cohort
At the time of data extraction, a total of 1543 children with ALGS were reported in the GALA database. Of these subjects, 110 (7%) did not meet the study eligibility criteria and were excluded from further analysis. Ascertainment of the cohort is summarized in Figure 1. The study cohort consisted of 1433 (57% male) clinically and/or genetically confirmed children with ALGS, with a median follow‐up of 6.0 years (IQR, 2.5–12.2). The largest number of patients were ascertained from Europe, 34% (n = 487), followed by North America, 29% (n = 420), and Asia, 23% (n = 333). Evaluable results of molecular genetic testing were available in 62% (n = 892/1433) of patients with ALGS and a pathogenic variant in JAG1 or NOTCH2 was identified in 98% (n = 878/892). Among genotyped patients, 2% (n = 14) were negative for a pathogenic variant in JAG1 or NOTCH2, despite meeting clinical criteria for ALGS (≥3 clinical characteristics). The majority of ALGS cases were de novo (56%).
FIGURE 1.
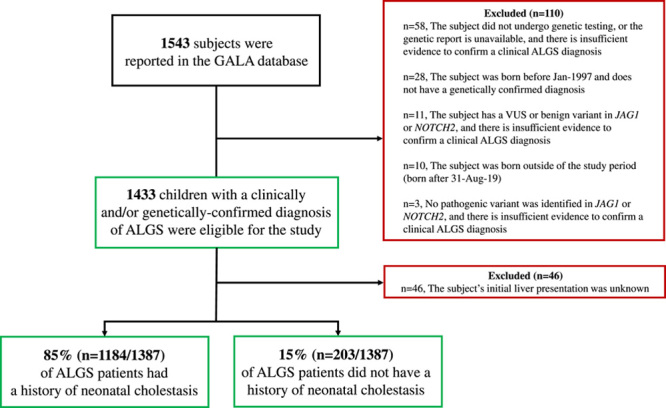
Ascertainment of the GALA Study cohort stratified by patients with Alagille syndrome (ALGS) with and without a history of neonatal cholestasis. Abbreviations: GALA, The Global ALagille Alliance (GALA) Study; JAG1, JAGGED1; VUS, variant of uncertain significance.
Table 1 summarizes the clinical characteristics of the entire study cohort and those with a history of neonatal cholestasis. Liver involvement at any age was reported in 95% (n = 1321/1387) of children with ALGS, and 85% (n = 1184/1387) presented with neonatal cholestasis. During the study period, pruritus was reported in 74% (n = 761/1028) of children with ALGS and first manifested at a median age of 12 months (IQR, 6.2–25.9). Nearly one in four children with ALGS reported xanthomas (24%, n = 243/980), first manifesting at a median age of 25 months (IQR, 15–43.2). At the first presentation of the xanthomas, patients with ALGS had a median serum cholesterol of 645.8 mg/dl (IQR, 398.3–1020.9; n = 198).
TABLE 1. Baseline clinical features for 1433 children with Alagille syndrome (ALGS) and those who presented with neonatal cholestasis (n = 1184).
| All | History of neonatal cholestasisa | ||
|---|---|---|---|
| n | 1433 | 1184 | |
| Male, % (n) | 57% (n = 823) | 59% (n = 694) | |
| Age at first clinical suspicion, % ( n ) | |||
| 0–1 years | 82% (n = 1148/1408) | 88% (n = 1033/1171) | |
| De novo, % (n) | 55% (n = 415/762) | 58% (n = 348/597) | |
| Genetically confirmed diagnoses, % (n) | 61% (n = 878) | 60% (n = 715) | |
| Diagnostic criteria, % ( n ) | |||
| Liver involvement, anyb | 95% (n = 1321/1387) | 100% | |
| Echo‐confirmed cardiac anomaly, any | 91% (n = 1231/1347) | 91% (n = 1017/1114) | |
| Characteristic facies | 90% (n = 1193/1325) | 90% (n = 984/1091) | |
| Posterior embryotoxon | 51% (n = 605/1179) | 51% (n = 503/992) | |
| Butterfly vertebrae | 44% (n = 549/1262) | 44% (n = 472/1063) | |
| Renal anomaly, any | 39% (n = 500/1275) | 39% (n = 422/1071) | |
| Vascular involvement, any | 36% (n = 189/532) | 34% (n = 152/448) | |
| Median laboratory values in the first year of life in children with ALGS who presented with neonatal cholestasis c | ≤6 months | >6 and ≤12 months | 12 months |
| Bile acids, μmol/L | 139.2 (IQR 98.6–205.7) (n = 192) | 181.8 (IQR 76.4–285.8) (n = 100) | 139.9 (IQR 95.8–221.0) (n = 247) |
| Total bilirubin, mg/dl | 8.0 (IQR 5.8–10.4) (n = 497) | 6.3 (IQR 1.2–11.8) (n = 419) | 7.9 (IQR 4.1–11.0) (n = 600) |
| Conjugated bilirubin, mg/dl | 5.5 (IQR 3.5–7.5) (n = 440) | 4.4 (IQR 0.8–8.2) (n = 338) | 5.5 (IQR 2.7–7.9) (n = 529) |
| ALT, IU/L | 154.0 (IQR 96.0–228.0) (n = 503) | 153.0 (IQR 96.0–224.0) (n = 439) | 153.0 (IQR 99.0–222.8) (n = 605) |
| AST, IU/L | 178.0 (IQR 116.0–263.5) (n = 479) | 154.0 (IQR 106.0–223.0) (n = 407) | 165.0 (IQR 118.0–246.0) (n = 577) |
| GGT, IU/L | 510.5 (IQR 258.3–830.5) (n = 490) | 427.5 (IQR 222.0–782.0) (n = 409) | 463.0 (IQR 250.5–826.5) (n = 599) |
| Cholesterol, mg/dl | 228.0 (IQR 170.0–309.0) (n = 275) | 340.0 (IQR 224.0–619.0) (n = 151) | 251.0 (IQR 186.0–387.0) (n = 355) |
| Triglycerides, mg/dl | 381.0 (IQR 274.0–531.0) (n = 226) | 522.0 (IQR 381.0–726.0) (n = 123) | 416.0 (IQR 301.0 –584.0) (n = 298) |
| Platelet count, 109/L | 504.0 (IQR 389.0–605.0) (n = 451) | 386.0 (IQR 312.7–482.5) (n = 377) | 436.5 (IQR 349.8–550.3) (n = 562) |
Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase; Echo, echocardiogram; GGT, gamma‐glutamyl transferase.
Neonatal cholestasis was defined as having at least one of the following features during the first three months of life: (1) direct/conjugated bilirubin greater than >2.0 mg/dl (34.0 μmol/L); (2) serum bile acids or GGT >3 times the upper limit of normal (ULN); (3) fat‐soluble vitamin deficiency, otherwise unexplainable.
Liver involvement at any age was defined as a history of neonatal cholestasis, elevated liver aminotransferase, histological abnormalities, history of pruritus and/or xanthomas or underwent hepatobiliary surgery.
In children who underwent Kasai portoenterostomy during the first year of life, laboratory data was only reported up until the procedure. Those who underwent LT or died in the first year of life or their follow‐up ended before 1 year of age were excluded from the laboratory analysis.
Outcomes of liver disease
Among the 1184 patients with a history of neonatal cholestasis, 345 underwent an isolated LT and 4 underwent combined liver‐kidney transplantation during the study period. Of note, an additional 14 patients with ALGS who did not present with neonatal cholestasis, or their initial liver presentation was unknown, underwent LT.
To determine the rate of NLS, LT, or risk of death without LT in those with a history of neonatal cholestasis (n = 1184), a competing risk analysis was performed to evaluate the risk of each of these three independent outcomes (Figure 2). At 5, 10, and 18 years, the rate of NLS was 66.8%, 54.4%, and 40.3%. The cumulative incidence of LT at 5, 10, and 18 years was 27.1% (95% CI, 24.3–30.1), 37.8% (95% CI, 34.2–41.3), and 50.4% (95% CI, 45.4–55.2). The risk of death without transplantation was 6.1% (95% CI, 4.7–7.7), 7.8% (95% CI, 6.1–9.8), and 9.3% (95% CI, 7.1–11.8). There were no significant differences between male and female patients in terms of NLS rates (log‐rank, p = 0.35). Rates of NLS were significantly different across the seven geographic regions (log‐rank, p < 0.001; Figure S2A and Table S3).
FIGURE 2.
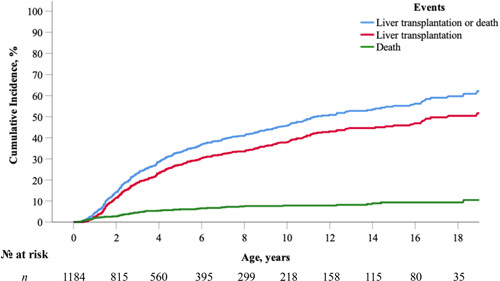
Cumulative incidence of native liver survival (NLS) in the presence of competing events (liver transplantation [LT] or risk of death without LT; n = 1184) in children with Alagille syndrome (ALGS) who presented with neonatal cholestasis. At 5, 10, and 18 years, the rate of NLS was 66.8%, 54.4%, and 40.3%.
Among children with ALGS, 9% (n = 102/1184) underwent a KPE at a median of 61 days (IQR, 40.0–75.0; n = 99). All patients with KPE met the classic clinical criteria for ALGS, and the mutation detection rate was 88% (n = 66/75). Infants who underwent KPE (n = 99) were found to have a 3.2‐fold increased risk of LT/death (95% CI, 2.5–4.3, p < 0.001). To account for the increased risk of LT/death among children who underwent a KPE, a sensitivity competing risk analysis was conducted to determine whether these patients were influencing the overall rate of NLS. In this analysis, children who underwent KPE were truncated at the time of their surgical procedure, and the cumulative risk for LT/death was found to be comparable with the primary analysis: at 5, 10, and 18 years, the rate of NLS was 70.7% (95% CI, 26.2–32.5), 58.3% (95% CI, 37.8–45.5), and 43.0% (95% CI, 51.5–62.2) (Figure S1).
A surgical BD was reported in 5% (n = 56/1184) of children with ALGS, at a median age of 2.4 years (IQR, 1.9–4.4; n = 53). NLS rates in patients with ALGS who underwent a surgical BD, revealed a 1.9‐fold greater risk of LT/death (95% CI, 1.3–3.0; p < 0.001).
The median age of LT was 2.8 years (IQR, 1.6–5.4) and 72% (n = 247) of transplants were performed during the first 5 years of life. The primary indications for LT were complications of persistent cholestasis (intractable pruritus, growth failure, xanthomas, metabolic bone disease, and/or fat‐soluble vitamin deficiency) in 72% (n = 235/328) and manifestations of PHT (ascites or varices) in 30% (n = 97/328; Table S4). Not surprisingly, 53% (n = 174/328) of transplant recipients with ALGS had more than one indication for LT. Of the 97 patients who underwent LT because of manifestations of PHT, 39% (n = 38/97) also reported pruritus as an indication for LT. PHT as the indication for transplantation was more prevalent among older transplant recipients with ALGS (median 4.2 years vs. 2.5 years, p = 0.001).
The majority of children with ALGS underwent a deceased donor transplant (68%), and the median follow‐up duration after LT was 4.0 years (IQR 1.3–8.5). Figure 3 illustrates patient and graft survival rates following isolated LT. Patient survival rates following LT at 5, 10, and 20 years were 92.0%, 91.0%, and 88.0%, respectively (Figure 3A). Graft survival rates in children with ALGS at 5, 10, and 20 years after LT were 88.0%, 86.3%, and 83.4% (Figure 3B).
FIGURE 3.
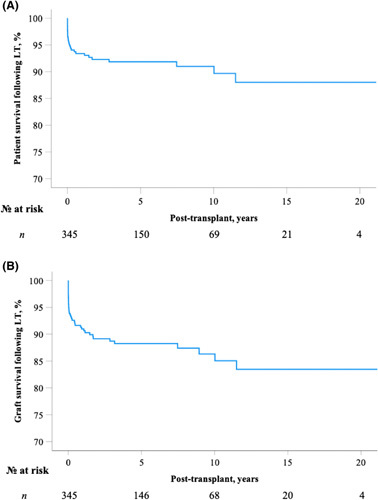
Patient and graft survival following isolated liver transplantation (LT) in 345 children with Alagille syndrome (ALGS). Patient and graft survival following LT is calculated from the date of LT until retransplantation, death or the date of last known clinical follow‐up. (A) After LT, 10‐ and 20‐year patient survival rates were 91.0% and 88.0%, respectively. (B) After LT, 10‐ and 20‐year graft survival rates were 86.3% and 83.4%, respectively.
Analysis of adverse liver‐related events
Six hundred and fifty (650) adverse liver‐related events were reported in 471 children with ALGS and a history of neonatal cholestasis. By 5, 10, and 18 years, 36.3%, 51.5%, and 66.0% of children with ALGS experienced at least one adverse liver‐related event (Figure 4). At 10 and 18 years, there were no significant differences between male and female patients in terms of rates of event‐free survival (53.6% vs. 47.2% and 69.3% vs. 63.4%, respectively; log‐rank, p = 0.22).
FIGURE 4.
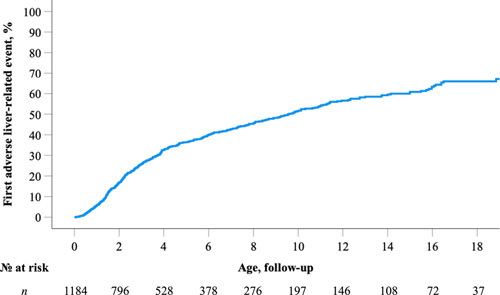
Time to first adverse liver‐related event in 1184 children with Alagille syndrome (ALGS). A Kaplan–Meier analysis revealed by 5, 10, and 18 years, 36.3%, 51.5%, and 66.0% of children with ALGS will experience at least one adverse liver‐related event, respectively.
To further explore the progression of liver disease in this ALGS cohort, the development of CEPH was investigated (Figure 5). The cumulative incidence of developing a combination of ultrasound‐confirmed splenomegaly and PLT <150 × 109/L was 39.2% at 10 years and 65.8% at 18 years. By 18 years of age, 22.3% of children with ALGS developed ascites requiring treatment with diuretics and 12.7% developed varices requiring intervention at endoscopy. By 18 years of age, 68.9% of patients developed CEPH.
FIGURE 5.
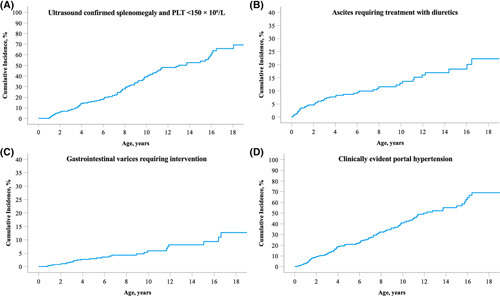
Cumulative incidence of clinically evident portal hypertension in an international cohort of children with Alagille syndrome (ALGS). (A) Ultrasound‐confirmed splenomegaly and PLT <150 × 109/L; (B) ascites requiring treatment with diuretics; (C) gastrointestinal varices requiring intervention; (D) clinically evident portal hypertension. Abbreviation: PLT, platelet count.
Predictors of NLS
Among the 1184 patients with ALGS, serial laboratory measurements were available in 605 with 3777 follow‐up visits during the first year of life. The median laboratory values were evaluated at the following three time points: (1) ≤6 months, (2) between >6 and ≤12 months, and (3) 12 months.
TB, CB, ALT, AST, and APRI showed a significant log‐linear relationship with LT or death (Table S5). Based on the distribution of TB values, three thresholds were applied: (1) <5.0 mg/dl, (2) ≥5.0 and <10.0 mg/dl, and (3) ≥10.0 mg/dl. As shown in Figure 6A, a univariate Cox regression analysis showed children with ALGS (>6 and ≤12 months) with median TB levels <5.0 mg/dl had significantly higher rates of NLS compared with those ≥5.0 mg/dl, with 79.0% reaching adulthood with their native liver compared with 31.6% and 18.2% (log‐rank, p < 0.001). Median TB levels between ≥5.0 and <10.0 mg/dl were associated with a 4.8‐fold (95% CI, 2.4–9.7) increased risk of LT. When utilizing a median serum TB cutoff of ≥10.0 mg/dl, the risk of LT increased to HR 15.6 (95% CI, 8.7–28.2) relative to those <5.0 mg/dl.
FIGURE 6.
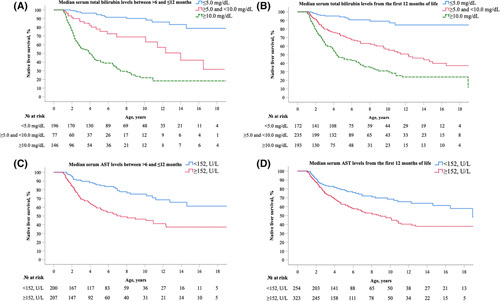
Early biochemical predictors for native liver survival in children with Alagille syndrome (ALGS). (A) Median serum total bilirubin levels between >6 and ≤12 months; (B) Median serum total bilirubin levels from the first 12 months of life; (C) Median serum aspartate aminotransferase (AST) levels between >6 and ≤12 months; (D) Median serum AST levels from the first 12 months of life.
To further explore the predictive value of TB, the applied thresholds were used to investigate the risk for developing CEPH. This analysis revealed that children with ALGS (>6 and ≤12 months) with median TB levels between ≥5.0 and <10.0 mg/dl had a 4.1‐fold (95% CI, 1.6–10.8) increased risk of developing CEPH. For a median serum TB threshold of ≥10.0 mg/dl, the risk was HR 8.0‐fold (95% CI, 3.4–18.4) versus <5.0 mg/dl (Figure S3).
The prognostic significance of the serum TB thresholds was also assessed for predicting manifestations of PHT (ascites or varices). Children (>6 and ≤12 months) with median TB levels between ≥5.0 and <10.0 mg/dl and those with ≥10.0 mg/dl had a significant increase in the likelihood of developing manifestations of PHT: 4.2‐fold (95% CI, 0.9–19.0) and 4.5‐fold (95% CI, 1.1–18.0), respectively.
Based on the distribution of AST, a median AST value ≥152 U/L between >6 and ≤12 months of life was identified and associated with an increased risk of LT (HR, 1.9; 95% CI, 1.4–2.6: Figure 6C). No other specific cutoffs for serum liver biochemistries, including serum bile acids, from the first year of life, were significantly associated with LT or death.
To explore potential bias, a sensitivity analysis was performed to assess rates of NLS between children with (n = 600) and without (n = 406) TB data in the first year of life using the Kaplan‐Meir method and a log‐rank. In this analysis, there were no significant differences in rates of NLS between those with and without available laboratory data (log‐rank, p = 0.78).
Hepatocellular carcinoma
Histologically proven hepatocellular carcinoma (HCC) was reported in <1.0% (n = 9/1433) of the study cohort, and the median age of diagnosis was 4.1 years (IQR, 1.5–8.3). Hepatic fibrosis was reported in 55% (n = 5/9) of native liver biopsies, and 75% (n = 3/4) had CEPH. Among these patients, six underwent LT, two died before LT, and one patient underwent surgical resection and remains alive at last clinical follow‐up. The majority of patients were diagnosed with HCC prior to LT (n = 4/6).
Histopathology
Seven hundred and fifty‐four native liver biopsy reports were retrospectively reviewed in 604 patients with ALGS and a history of neonatal cholestasis. Bile duct paucity was reported in only 65% (n = 202/31) of liver biopsies performed during the first 3 months of life. Features of biliary obstruction, including bile duct proliferation and/or ductular bile plugs, were reported in 22% (n = 69/311) of patients aged ≤3 months. Among these 69 patients with ALGS and histologic features of obstruction, 44 underwent genetic testing, and a pathogenic variant in JAG1 or NOTCH2 was identified in 31 patients (70%). In a subgroup analysis, patients were stratified according to the presence or absence of baseline bile duct paucity (Table S6). Children with ALGS without baseline bile duct paucity were significantly more likely to have giant cell transformation (p < 0.001) and features of biliary obstruction (p < 0.001).
Histological features were compared based on age at first native liver biopsy: (1) ≤6 months, (2) between >6 and ≤24 months, and (3) >24 months. As shown in Table S7, the presence of bile duct paucity and fibrosis increased significantly with advancing age (p < 0.001; p < 0.001). As expected, the frequency of giant cell transformation significantly decreased with age (p < 0.001).
Of the 604 children with ALGS, 14% (n = 85/604) underwent two or more native liver biopsies at least 12 months apart during the study period (Table S8). The patient's first and second biopsies were compared, and the median interval between biopsies was 31.0 months (IQR 20.5–61.0). Second biopsies were significantly more likely than baseline biopsies to show liver fibrosis (28% vs. 62%, p = 0.01) but not bile duct paucity (66% vs. 86%, p = 0.53). Bile duct paucity was not reported on either baseline or follow‐up biopsy in 6% (n = 5/86) of children with ALGS who underwent a second biopsy. Among these five patients without bile duct paucity on either biopsy, a pathogenic variant in JAG1 or NOTCH2 was identified in 80% (n = 4/5).
Mortality
During the study period, 108 deaths were reported in all 1433 children with ALGS. At 5, 10, and 18 years, the rate of overall patient survival was 92.8%, 91.2%, and 88.1%, respectively (Figure S4). Significant differences in overall survival rates across geographic regions were observed (log‐rank, p = 0.002; Figure S2B). The median age of death was 2.6 years (1.2–4.7). Liver‐related complications, including LT complications, were the leading cause of death (22%; median age 2.8 years, IQR, 1.6–6.7), followed by cardiac‐related complications in 18% (median age 1.1 years, IQR, 0.5–4.5), and multiorgan failure (median age 3.9 years, IQR, 3.0–7.0) and noncardiac vascular complications (median age 2.2 years, IQR, 1.5–3.2) accounted for 15% each.
A survival analysis was performed to compare overall survival in ALGS with (n = 1184) and without (n = 203) a history of neonatal cholestasis. A Kaplan–Meier analysis showed the cumulative rate of survival at 10 and 18 years was significantly lower in children with ALGS who presented with neonatal cholestasis in comparison with those who did not (log‐rank p < 0.001; Figure S5). The 10 and 18‐year patient survival rates in patients with ALGS presenting with cholestasis were 89% and 86% compared with 100% and 97%, respectively.
DISCUSSION
To the best of our knowledge, the GALA database is the largest cohort of children with ALGS ever ascertained and encompasses a broad spectrum of sites in terms of center size and geography, with 29 countries represented. This analysis of the GALA data set provides insights into the natural history of liver disease and outcomes of this complex disorder. In this real‐world global view, 40% of children with ALGS‐cholestasis reached adulthood with their native liver. A competing risk analysis revealed that this rate of NLS is largely driven by LT and, perhaps surprisingly, not death, despite the multisystem nature of this disorder with substantial cardiac involvement. As expected, heterogeneity in rates of NLS was observed across geographic regions and likely represents differences in allocation policies and clinical resources. The burden of liver disease is greatest among young children, with the median age of LT being only 2.8 years and almost three‐quarters of all LTs occurring in the first 5 years of life. The majority of early LTs occur because of complications associated with cholestasis, including pruritus, with a smaller number occurring later in childhood due to the onset of PHT. The NLS of 40% at 18 years is higher than the 24% rate recently reported by the Childhood Liver Disease Research Network (ChiLDReN), which is composed of North American tertiary referral centers and therefore may select a more severely affected group of patients.6 In addition to LT, a time to first adverse liver‐related event analysis demonstrates the extent of clinically significant liver comorbidities occurring during childhood, with more than 60% of children experiencing an adverse liver event during the study period. These data highlight the consequences of profound cholestasis arising from developmental defects of the biliary tree and reveal the unmet need to develop targeted therapies for ALGS‐related cholestasis for the youngest children. Because the majority of transplants in ALGS have pruritus as a leading indication, antipruritic agents that target cholestasis (rather than fibrosis) may also offer hope to change disease biology and extend NLS. The natural history data and outcomes presented here are essential to evaluate therapies already in clinical trials and to identify rational therapies.
Serum TB level in children with ALGS aged 6 and ≤12 months was identified as a prognostic marker for long‐term hepatic outcomes. Median TB levels <5.0 mg/dl during this early stage of life were associated with significantly higher rates of NLS, with 79% reaching adulthood with their native liver. Children with ALGS (>6 and ≤12 months) and median TB levels between ≥5.0 and <10.0 mg/dl had an almost 5‐fold increased risk of LT, and >10 was 15.6‐fold as compared with <5.0 mg/dl. It is important to note that these TB thresholds are not only associated with LT but also appear to be associated with progressive liver disease, as shown by their association with the development of CEPH. Children with ALGS (>6 and ≤12 months) with median TB levels between ≥5.0 and <10.0 mg/dl had a 4.1‐fold increased risk of developing CEPH, and those with TB >10.0 mg/dl had a 8.0‐fold increased risk. It is intriguing that a median AST >152 in the first year of life is also associated with NLS, which may portend the onset of PHT.13
These data provide clinically valuable information to the clinician managing infants with ALGS liver disease. One of the challenges associated with the management of ALGS liver disease is the decision‐making surrounding timing of LT. Unlike biliary atresia (BA), in which cholestasis is often progressive even after KPE, in ALGS, a patient's cholestasis can resolve or stabilize.14 Therefore, not all children with cholestasis and ALGS inevitably require LT, and there is a strong need for early predictors of NLS. In a previous study, Mouzaki et al. identified a serum TB cutoff of 3.8 mg/dl between 12 and 24 months of life as a threshold for poor hepatic outcome (surgical biliary diversion or LT); however, this threshold was applied at an older age and is therefore less clinically useful early on. Furthermore, the sample size was limited in this analysis, and patients were stratified into outcome groups at the age of 10 years, thereby not accounting for events beyond this age.14
It may be considered surprising that serum bile acids in the first year of life were not predictive of NLS in ALGS. Observations from real‐world practice at GALA sites clearly show that serum bile acid levels are not routinely sent on a clinical basis in ALGS. Only 247 patients had serum bile acid levels available in the first year of life for this analysis. This is in stark contrast to other laboratory variables evaluated as predictors for which levels were available for >600 patients. Therefore, the lack of association of serum bile acids and liver disease outcomes in ALGS warrants further prospective investigation, especially in an era when ASBT inhibitors that target serum bile acid levels are now approved therapies.15
This series represents the largest review of liver biopsy reports in ALGS. Examination of liver tissue histopathology in this extensive cohort also provides insights that will aid diagnosis. It is noteworthy that bile duct paucity was reported in only 65% of liver biopsies performed during the first 3 months of life, the period during which there are diagnostic challenges with distinguishing ALGS from syndromic BA. It is crucial for clinicians to appreciate that more than one‐third of infants with ALGS will not have bile duct paucity and that, further, almost one‐quarter have histologic features of biliary obstruction. These observations suggest that ductular proliferation and/or bile duct plugs are not pathognomonic of BA and are present in a sizeable subset of infants with ALGS, potentially leading to misdiagnosis. Clearly, a future histopathologic review of early ALGS liver biopsies is warranted, though even the current data alone can inform clinical practice. These data highlight the limitations of relying on histopathology for diagnostic purposes in infants with cholestasis and support the incorporation of rapid genetic testing into diagnostic algorithms.16 Consistent with earlier reports, though in a substantially larger cohort, this study also found that the frequency at which bile duct paucity is identified on liver biopsy increases with advancing age.3 However, in individual patients who have undergone repeat liver biopsies, the progression of bile duct paucity over time did not reach statistical significance.
The GALA Study demonstrates that global patient and graft survival rates in children with ALGS are very good, exceeding 83% at 18 years of age. These observations are comparable with two prior North American analyses of ALGS.17,18 This reflects the global clinical practice of carefully selecting recipients with ALGS. Some children with ALGS may be deemed ineligible for LT because of a severe cardiac phenotype, though, with expert input from cardiologists, this number of patients should diminish over time.19 It is also notable that there is substantial early mortality 52% (<30 days post‐LT) in ALGS. This concerning observation was also reported by the prior SPLIT study in which early post‐LT mortality rates were significantly higher in ALGS in comparison with patients transplanted for BA.18
In this contemporary cohort of children with ALGS, the all‐cause mortality rate was 8.5%. This rate is considerably lower than earlier reports and likely reflects an increased availability of LT, earlier interventions for cardiac defects, and subspecialty clinical teams working synergistically to manage the multisystem disease burden.1–3,20,21 Furthermore, although previous studies have estimated long‐term outcomes in ALGS, most of them were conducted without considering competing risks. Among reported deaths, liver or LT‐related complications were the most common, followed closely by cardiac‐related complications, together accounting for almost half of all deaths. Notably, most deaths occurred during the first 5 years of life. Patient survival data in ALGS underscore the need for adult clinicians to be aware of ALGS and the medical needs of these individuals as they transition from pediatric to adult care. There is currently a paucity of research in this age bracket, and specific recommendations for the clinical management of adults with ALGS are lacking.
Although our study provides insight into the natural history and outcomes of ALGS, there are certain limitations that require comment. This is a retrospective study, and therefore it was not feasible to assess the severity and evolution of pruritus and xanthomas from medical records. This reflects the variability with which clinicians assess these two clinically important features of ALGS and speaks to the need for standardized methods to assess and record these on a clinical basis. In addition, the histopathology data reported were extracted from local liver biopsy reports rather than central review. Histologic staging systems clearly vary by site, and to overcome this to some extent, fibrosis was reported as a binary variable for this analysis, though this does limit its value. Finally, the prevalence and spectrum of hepatic involvement may still be underestimated as individuals who present with partial or subclinical disease expression may go undetected and are not represented in this analysis. Overall, despite the retrospective nature of the GALA, the data were rigorously collected and queried by the DCC to account for incompleteness.
The GALA study provides a comprehensive real‐world analysis of ALGS liver disease in more than 1400 children with ALGS from 29 countries. A TB below 5.0 mg/dl between 6 and 12 months of age appears to be associated with better NLS and may help guide complex decisions regarding timing of LT, which is challenging in ALGS. These natural history data are important to evaluate novel therapies that are currently in clinical trials for ALGS. With 40% of children with ALGS surviving to adulthood with native liver, adult hepatologists also need to be aware of ALGS and its complex manifestations.
Supplementary Material
Acknowledgments
AUTHOR CONTRIBUTIONS
Shannon M. Vandriel: participated in the study concept and design, acquisition of data, analysis and interpretation of data, drafting of the manuscript, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Li‐Ting Li: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Huiyu She: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Jian‐She Wang: participated in the study concept and design, data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Melissa A. Gilbert: analysis and interpretation of data, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Irena Jankowska: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Piotr Czubkowski: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Dorota Gliwicz‐Miedzińska: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Emmanuel M. Gonzales: participated in the study concept and design, data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Emmanuel Jacquemin: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Jérôme Bouligand: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Nancy B. Spinner: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Kathleen M. Loomes: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; David A. Piccoli: participated in the study concept and design, data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Lorenzo D'Antiga: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Emanuele Nicastro: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Étienne Sokal: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Tanguy Demaret: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Noelle H. Ebel: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Jeffrey A. Feinstein: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Rima Fawaz: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Silvia Nastasio: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Florence Lacaille: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Dominique Debray: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Henrik Arnell: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Björn Fischler: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Susan Siew: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Michael Stormon: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Saul J. Karpen: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Rene Romero: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Kyung Mo Kim: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Woo Yim Baek: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Winita Hardikar: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Sahana Shankar: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Amin J. Roberts: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Helen M. Evans: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; M. Kyle Jensen: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Marianne Kavan: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Shikha S. Sundaram: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Alexander Chaidez: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Palaniswamy Karthikeyan: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Maria Camila Sanchez: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Maria Lorena Cavalieri: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Henkjan J. Verkade: participated in the study concept and design, data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Way Seah Lee: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; James E. Squires: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Christina Hajinicolaou: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Chatmanee Lertudomphonwanit: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Ryan T. Fischer: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Catherine Larson‐Nath: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Yael Mozer‐Glassberg: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Cigdem Arikan: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Henry C. Lin: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Jesus Quintero Bernabeu: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Seema Alam: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Deirdre A. Kelly: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Elisa Carvalho: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Cristina Targa Ferreira: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Giuseppe Indolfi: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Ruben E. Quiros‐Tejeira: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Pinar Bulut: data acquisition,critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Pier Luigi Calvo: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Zerrin Önal: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Pamela L. Valentino: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Dev M. Desai: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; John Eshun: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Maria Rogalidou, data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Antal Dezsőfi: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Sabina Wiecek: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Gabriella Nebbia: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Raquel Borges Pinto: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Victorien M. Wolters: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; María Legarda Tamara: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Andréanne N. Zizzo: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Jennifer Garcia: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Kathleen Schwarz: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Marisa Beretta: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Thomas Damgaard Sandahl: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Carolina Jimenez‐Rivera: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Nanda Kerkar: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Jernej Brecelj: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Quais Mujawar: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Nathalie Rock: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Cristina Molera Busoms: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Wikrom Karnsakul: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Eberhard Lurz: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Ermelinda Santos‐Silva: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Niviann Blondet: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Luis Bujanda: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Uzma Shah: data acquisition, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Richard J. Thompson: participated in the study concept and design, drafting of the manuscript, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Bettina E. Hansen: participated in the study concept and design, statistical analysis and interpretation of data, drafting of the manuscript, critical revision of the manuscript for important intellectual content, and final approval of the version to be published; Binita M. Kamath: participated in the study concept and design, analysis and interpretation of data, drafting of the manuscript, critical revision of the manuscript for important intellectual content, final approval of the version to be published, obtaining funding, and overall study supervision.
ACKNOWLEDGMENTS
We would like to thank the following agencies for their generous funding support: The Alagille Syndrome Alliance, Mirum Pharmaceuticals, Inc., and Albireo Pharma, Inc., who provided unrestricted educational grants to the Hospital for Sick Children (SickKids Foundation). The National Natural Science Foundation of China (81873543 and 81570468) provided funding to the Children's Hospital of Fudan University, The Center for Pediatric Liver Diseases, Shanghai, China. The authors would also like to acknowledge Mikayla Sonnenberg, Aly Fawzy, Mila Valcic, and Corey Forster from the GALA DCC at the Hospital for Sick Children. Finally, the authors would like to thank all local research teams who helped with data collection.
FUNDING INFORMATION
This study received funding support from the following agencies: The Alagille Syndrome Alliance, Mirum Pharmaceuticals, Inc., and Albireo Pharma, Inc., who provided unrestricted educational grants to the Hospital for Sick Children (SickKids Foundation). The study sponsors were not involved in the conduct of the research study or preparation of the manuscript.
CONFLICT OF INTEREST
Binita M. Kamath consults for and received grants from Mirum and Albireo. She consults for Audentes Therapeutics and Third Rock Ventures. Bettina E. Hansen consults for Mirum, Albireo, Chemomab, Calliditas, Intercept and Cyma Bay. She received grants from Mirum, Albireo, Intercept and Cyma Bay. Deirdre A. Kelly consults for, advises, and received grants from Mirum and Albireo. Emmanuel M. Gonzales consults for Albireo, CTRS, Mirum, and Vivet. Henrik Arnell consults for Albireo and Baxter. Henry C. Lin advises Albireo. Emmanuel Jacquemin consults for Laboratoire CTRS France and Vivet Therapeutics France. Kathleen Schwarz consults for and received grants from Gilead. She consults for Mirum. She received grants from Albireo. Kathleen M. Loomes consults for and received grants from Albireo and Mirum. She consults for Travere Therapeutics. Lorenzo D'Antiga consults for Albireo and Mirum. M. Kyle Jensen consults for Albireo and Guidepoint Global. Nanda Kerkar advises Albireo and Mirum. Emanuele Nicastro advises and received grants from Mirum Pharmaceuticals. Noelle H. Ebel consults for Mirum Pharmaceuticals. Rene Romero consults for Albireo and Mirum. He received grants from Gilead. Richard J. Thompson owns stock in, consults for, and advises Generation Bio and Rectify. He consults for, advises, and is on the speakers' bureau for Albireo and Mirum. Rima Fawaz consults for Mirum. She advises Albireo. Ryan T. Fischer is on the speakers' bureau for Albireo and Mirum. Saul J. Karpen consults for Albireo, Intercept, and Mirum. Nancy B. Spinner consults for Mirum and Travere. Wikrom Karnsakul consults for and received grants from Albireo and Gilead. He consults for Mirum.
Footnotes
Abbreviations: ALGS, Alagille syndrome; ALT, alanine aminotransferase; AST, aspartate aminotransferase; BA, biliary atresia; BD, biliary diversion; CB, conjugated bilirubin; CEPH, clinically evident portal hypertension; CI, confidence interval; DCC, Data Coordinating Center; GALA, Global ALagille Alliance; GGT, γ‐glutamyl transferase; HR, hazard ratio; IQR, interquartile range; JAG1, JAGGED1; KPE, Kasai portoenterostomy; LT, liver transplantation; NLS, native liver survival; PHT, portal hypertension; PLT, platelet count; TB, total bilirubin.
Supplemental Digital Content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal’s website, www.hepjournal.com.
Funding information Albireo Pharma, Inc, Grant/Award Number: unrestricted educational grants; Mirum Pharmaceuticals, Inc, Grant/Award Number: unrestricted educational grant; The Alagille Syndrome Alliance
REFERENCES
- 1.Alagille D, Odièvre M, Gautier M, Dommergues JP. Hepatic ductular hypoplasia associated with characteristic facies, vertebral malformations, retarded physical, mental, and sexual development, and cardiac murmur. J Pediatr. 1975;86:63–71. [DOI] [PubMed] [Google Scholar]
- 2.Alagille D, Estrada A, Hadchouel M, Gautier M, Odièvre M, Dommergues JP. Syndromic paucity of interlobular bile ducts (Alagille syndrome or arteriohepatic dysplasia): review of 80 cases. J Pediatr. 1987;110:195–200. [DOI] [PubMed] [Google Scholar]
- 3.Emerick KM, Rand EB, Goldmuntz E, Krantz ID, Spinner NB, Piccoli DA. Features of Alagille syndrome in 92 patients: frequency and relation to prognosis. Hepatology. 1999;29:822–9. [DOI] [PubMed] [Google Scholar]
- 4.Gilbert MA, Bauer RC, Rajagopalan R, Grochowski CM, Chao G, McEldrew D, et al. Alagille syndrome mutation update: comprehensive overview of JAG1 and NOTCH2 mutation frequencies and insight into missense variant classification. Hum Mutat. 2019;40:2197–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Leonard LD, Chao G, Baker A, Loomes K, Spinner NB. Clinical utility gene card for: Alagille Syndrome (ALGS). Eur J Hum Genet. 2014;22(3). 10.1038/ejhg.2013.140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kamath BM, Ye W, Goodrich NP, Loomes KM, Romero R, Heubi JE, et al. Outcomes of childhood cholestasis in Alagille syndrome: results of a multicenter observational study. Hepatol Commun. 2020;4:387–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shneider BL, Spino C, Kamath BM, Magee JC, Bass LM, Setchell KD, et al. Placebo‐controlled randomized trial of an intestinal bile salt transport inhibitor for pruritus in Alagille syndrome. Hepatol Commun. 2018;2:1184–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harris PA, Taylor R, Thielke R, Payne J, Gonzalez N, Conde JG. Research electronic data capture (REDCap)‐‐a metadata‐driven methodology and workflow process for providing translational research informatics support. J Biomed Inform. 2009;42:377–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aghazadeh‐Attari J, Mobaraki K, Ahmadzadeh J, Mansorian B, Mohebbi I. Quality of observational studies in prestigious journals of occupational medicine and health based on Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) Statement: a cross‐sectional study. BMC Res Notes. 2018;11:266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier‐Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wai CT, Greenson JK, Fontana RJ, Kalbfleisch JD, Marrero JA, Conjeevaram HS, et al. A simple noninvasive index can predict both significant fibrosis and cirrhosis in patients with chronic hepatitis C. Hepatology. 2003;38:518–26. [DOI] [PubMed] [Google Scholar]
- 12.Bass LM, Shneider BL, Henn L, Goodrich NP, Magee JC, Childhood Liver Disease Research Network. Clinically evident portal hypertension: an operational research definition for future investigations in the pediatric population. J Pediatr Gastroenterol Nutr. 2019;68:763–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shneider BL, Abel B, Haber B, Karpen SJ, Magee JC, Romero R, et al. Portal hypertension in children and young adults with biliary atresia. J Pediatr Gastroenterol Nutr. 2012;55:567–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mouzaki M, Bass LM, Sokol RJ, Piccoli DA, Quammie C, Loomes KM, et al. Early life predictive markers of liver disease outcome in an international, multicentre cohort of children with Alagille syndrome. Liver Int. 2016;36:755–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gonzales E, Hardikar W, Stormon M, Baker A, Hierro L, Gliwicz D, et al. Efficacy and safety of maralixibat treatment in patients with Alagille syndrome and cholestatic pruritus (ICONIC): a randomised phase 2 study. Lancet. 2021;398:1581–92. [DOI] [PubMed] [Google Scholar]
- 16.Karpen SJ, Kamath BM, Alexander JJ, Ichetovkin I, Rosenthal P, Sokol RJ, et al. Use of a comprehensive 66‐gene cholestasis sequencing panel in 2171 cholestatic infants, children, and young adults. J Pediatr Gastroenterol Nutr. 2021;72:654–60. [DOI] [PubMed] [Google Scholar]
- 17.Arnon R, Annunziato R, Schiano T, Miloh T, Baisley M, Sogawa H, et al. Orthotopic liver transplantation for adults with Alagille syndrome. Clin Transplant. 2012;26:E94–100. [DOI] [PubMed] [Google Scholar]
- 18.Kamath BM, Yin W, Miller H, Anand R, Rand EB, Alonso E, et al. Outcomes of liver transplantation for patients with Alagille syndrome: the studies of pediatric liver transplantation experience. Liver Transpl. 2012;18:940–8. [DOI] [PubMed] [Google Scholar]
- 19.Luong R, Feinstein JA, Ma M, Ebel NH, Wise‐Faberowski L, Zhang Y, et al. Outcomes in patients with Alagille syndrome and complex pulmonary artery disease. J Pediatr. 2021;229:86–94.e4. [DOI] [PubMed] [Google Scholar]
- 20.Hoffenberg EJ, Narkewicz MR, Sondheimer JM, Smith DJ, Silverman A, Sokol RJ. Outcome of syndromic paucity of interlobular bile ducts (Alagille syndrome) with onset of cholestasis in infancy. J Pediatr. 1995;127:220–4. [DOI] [PubMed] [Google Scholar]
- 21.Quiros‐Tejeira RE, Ament ME, Heyman MB, Martin MG, Rosenthal P, Hall TR, et al. Variable morbidity in Alagille syndrome: a review of 43 cases. J Pediatr Gastroenterol Nutr. 1999;29:431–7. [DOI] [PubMed] [Google Scholar]