

Abstract
Background and Aims:
Intrahepatic cholangiocarcinoma (ICC) is a deadly but poorly understood disease, and its treatment options are very limited. The aim of this study was to identify the molecular drivers of ICC and search for therapeutic targets.
Approach and Results:
We performed a Sleeping Beauty transposon‐based in vivo insertional mutagenesis screen in liver‐specific Pten‐deficient mice and identified TNF receptor–related factor 3 (Traf3) as the most significantly mutated gene in murine ICCs in a loss‐of‐function manner. Liver‐specific Traf3 deletion caused marked cholangiocyte overgrowth and spontaneous development of ICC in Pten knockout and Kras G12D mutant mice. Hepatocyte‐specific, but not cholangiocyte‐specific, Traf3‐deficient and Pten‐deficient mice recapitulated these phenotypes. Lineage tracing and single‐cell RNA sequencing suggested that these ICCs were derived from hepatocytes through transdifferentiation. TRAF3 and PTEN inhibition induced a transdifferentiation‐like phenotype of hepatocyte‐lineage cells into proliferative cholangiocytes through NF‐κB‐inducing kinase (NIK) up‐regulation in vitro. Intrahepatic NIK levels were elevated in liver‐specific Traf3‐deficient and Pten‐deficient mice, and NIK inhibition alleviated cholangiocyte overgrowth. In human ICCs, we identified an inverse correlation between TRAF3 and NIK expression, with low TRAF3 or high NIK expression associated with poor prognosis. Finally, we showed that NIK inhibition by a small molecule inhibitor or gene silencing suppressed the growth of multiple human ICC cells in vitro and ICC xenografts in vivo.
Conclusions:
TRAF3 inactivation promotes ICC development through NIK‐mediated hepatocyte transdifferentiation. The oncogenic TRAF3–NIK axis may be a potential therapeutic target for ICC.
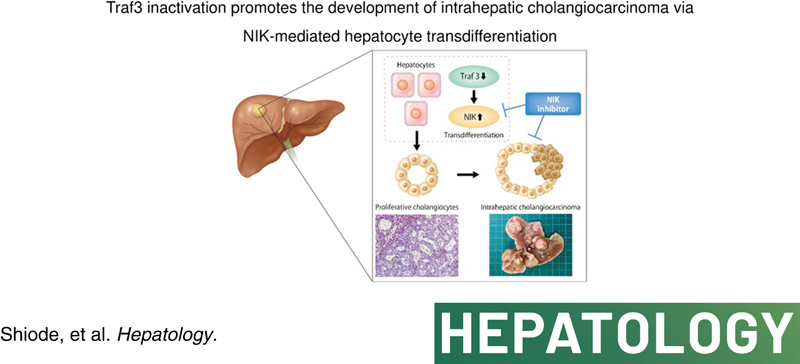
INTRODUCTION
Cholangiocarcinoma (CCA) is a biliary epithelial tumor that develops in the intrahepatic, perihilar, and distal biliary trees. CCA accounts for 15% of all primary hepatobiliary tumors and is the second most common primary liver malignancy after HCC.1 Intrahepatic CCA (ICC) arises above the second‐order bile ducts in the liver and accounts for 10%–20% of CCAs. Its incidence has increased by up to 10‐fold during the past three decades.1 Because approximately half of ICCs arise de novo, without identifiable risk factors, and there is no established screening program for this cancer type,2 ICC is often found in advanced stages and has a poor prognosis.3 To better understand the molecular mechanisms driving this deadly cancer, researchers involved in recent international projects, including The Cancer Genome Atlas project, sequenced hundreds of tumors from patients with ICC and identified several recurrent genomic and epigenomic abnormalities, including mutations in tumor protein P53, KRAS, PTEN, and AT‐rich interaction domain 1A.4–6 However, no therapies currently exist that address most of the potential driver genes. Although an FGF receptor (FGFR) inhibitor has recently been approved as the first molecularly targeted agent for patients with ICC and FGFR2 fusions or rearrangements, these treatments can be used only in up to 15% of patients.7 Thus, further understanding of its molecular pathogenesis and discovery of therapeutic targets are desired.
Transposon‐based insertional mutagenesis screens provide a versatile tool with which to identify cancer genes in a high‐throughput manner.8 We have previously performed insertional mutagenesis screens in a variety of cancer types using mice carrying high copies of the mutagenic Sleeping Beauty (SB) transposon.8–10 One of our recent screens aimed to identify cancer genes in the liver of mice carrying a liver‐specific Pten deletion, which is a known tumor suppressor gene in HCC and ICC.11,12 In this screen, transposon mutagenesis was found to significantly accelerate liver tumorigenesis, and pathological analysis revealed that HCC, ICC, and combined HCC–ICC (cHCC‐ICC) developed in these mice. Through the sequencing of histologically confirmed HCCs, we identified hundreds of candidate cancer genes (CCGs) involved in HCC development and progression.12
In the present study, we aimed to identify cancer genes involved in ICC. For this purpose, we focused on histologically confirmed ICC and cHCC‐ICC that developed in the screen.12 Through sequence analysis of ICCs and cHCC‐ICCs, we identified 405 ICC CCGs, including high‐frequency loss‐of‐function mutations in TNF receptor–related factor 3 (Traf3). We then further investigated the role of TRAF3 in the development of ICC and its clinical relevance in human ICCs. We also searched for therapeutic ICC targets that function downstream of TRAF3.
MATERIALS AND METHODS
Mice
All mice we used were male. We used Alb‐Cre/+; T2Onc2/+; reverse orientation splice acceptor βgeo line 26 (Rosa26)‐leptin, serum levels of (lsl)‐SB11/+; Pten fl/fl mice for the transposon mutagenesis screens; Alb‐Cre/+; Pten fl/fl; Traf3 fl/fl mice, Alb‐Cre/+; KrasG12D Tg ;Traf3 fl/fl mice, Alb‐Cre estrogen receptor targeting 2 (ERT2)/+; Pten fl/fl; Traf3 fl/fl mice, K19‐CreERT2/+; Pten fl/fl; Traf3 fl/fl mice, Cre recombinase under the control of the albumin promoter (Alb‐Cre)ERT2/+; Pten fl/fl; Traf3 fl/fl; ROSA26R/+ mice, K19‐CreERT2/+; Pten fl/fl; Traf3 fl/fl; ROSA26R/+ mice and Alb‐Cre/+; Pten fl/fl; Traf3 fl/fl; mixed lineage kinase domain like pseudokinase (Mlkl) fl/fl mice for the validation study; and nonobese diabetic/Shi severe combined immunodeficient Il2rγnull (NOG) mice for the xenograft study. All mice were housed in a specific pathogen‐free facility in microisolators in ventilated racks and fed a regular chow diet. Alb‐Cre,13 KrasG12D,14 K19‐Cre ERT2,15 Pten fl/fl,16 and ROSA26R17 mice were obtained from the Jackson Laboratory. The generation of Alb‐Cre ERT2 mice was described previously.18 Traf3 fl/fl mice19 were obtained from Prof. Robert Brink (Garvan Institute of Medical Research); Mlkl fl/fl mice20 were obtained from Prof. James Murphy (Walter and Eliza Hall Institute of Medical Research); T2Onc2 and Rosa26 lsl SB11 mice were obtained from the National Cancer Institute; and NOG mice were obtained from the Central Institute for Experimental Animals. All experiments were performed in accordance with relevant guidelines and regulations.
Human specimens
Formalin‐fixed and paraffin‐embedded human ICC tissues were collected from 46 patients with ICC who underwent surgical resection at the Department of Gastrointestinal Surgery in the Osaka University Graduate School of Medicine between 2011 and 2018. These samples were obtained with approval of the institutional research board of Osaka University Hospital (no. 15267). Informed consent was also obtained in writing from each patient. All experiments were performed in accordance with relevant guidelines and regulations.
Sequence, read processing, and common insertion site analysis
Transposon insertion sites were mapped using a previously reported splink HiSeq approach.12 CCGs were identified by applying the gene‐centric common insertion site (gCIS)‐calling method,8 and trunk driver analysis was performed as described.12 All 369,549 identified nonredundant transposon insertion sites are available upon request.
Immunohistochemistry
Paraffin blocks were deparaffinized using xylene and ethanol. Hematoxylin and eosin staining was performed. For immunostaining, antigen activation was performed by heating with Target Retrieval Solution (Dako), followed by blocking with bovine serum albumin and overnight reaction at 4°C using primary antibody. Frozen sections were dried at room temperature, blocked with bovine serum albumin (Nakarai, Japan), and reacted with primary antibody included in the VECTASTAIN ABC Rabbit IgG Kit (Vector Laboratories). A DAB Substrate Kit (Vector Laboratories) was used for the staining reaction. The following antibodies were used for immunostaining: keratin 19 (KRT19; 1:500; Abcam; ab52625), TRAF3 (1:100; Abcam; ab217033), NF‐κB‐inducing kinase (NIK; 1:400; Novus Biologicals; NBP2‐23603), phosphorylated receptor interacting serine‐threonine protein kinase 3 (1:100; Genentech21) and KI67 (1:1000; Cell Signaling; 12202S). For β‐galactosidase (LacZ) staining, frozen sections were dried at room temperature and then fixed in 0.2% glutaraldehyde (Nakarai) for 10 min. The sections were then washed three times with PBS and incubated overnight in LacZ substrate including X‐gal (TaKaRa Bio) at room temperature. The cells were then washed three times with PBS, and nuclei were stained with nuclear stain red (ScyTek). The number of Ki67‐positive cells in the tumors was counted using ImageJ (Fiji). The immunostaining score for TRAF3 or NIK was calculated by multiplying the staining intensity (0–3) by the stained area (percentage). Patients with ICC were split into two groups based on the median immunostaining score of either TRAF3 or NIK, and their overall survival (OS) or disease‐free survival (DFS) was evaluated.
Single‐cell RNA sequencing
Portal vein puncture was performed in mice, and the liver was refluxed using a two‐step collagenase method. Twenty‐five milliliters of preperfusion solution was injected at 6 ml/min, followed by 3 ml/min of perfusion solution in which collagenase (Merck, Japan) was dissolved, and hepatocytes were collected by centrifugation at 50g for 1 min. The remaining tissues were then cut into small pieces and agitated with a solution that dissolved collagenase (Merck) and hyaluronidase (Sigma‐Aldrich) at 37°C for 40 min, followed by centrifugation at 50g for 1 min. After the pellet was removed, the supernatant was centrifuged at 350g for 4 min, and the nonparenchymal cell (NPC) fraction was collected. The hepatocytes and NPC fraction were mixed in a 1:1 ratio, dissolved in sorting buffer, and then processed with a BD Rhapsody Cartridge Reagent Kit (Becton, Dickinson and Company) according to the manufacturer’s instructions. Rhapsody libraries were converted to libraries for DNBSEQ using the MGIEasy Universal Library Conversion Kit (App‐A; MGI Tech, China). Sequencing was performed on a DNBSEQ‐G400RS (MGI Tech) platform in 2 × 100‐bp paired‐end mode. Reads were obtained from 14,393 cells from wild‐type (WT) mice and 10,083 cells from double knockout (DKO) mice and analyzed using Seqgeq (Becton, Dickinson and Company). After the data from both mice were merged, 10,364 cells remained after excluding the cells in poor condition with a low number of reads. These cells were analyzed using Seurat v3.2, with the percentage of genes set to 5%, the percentage of cells set to 10%, and perplexity set to 30 in t‐distributed stochastic neighbor embedding (t‐SNE) for all genes, and divided into 15 clusters.
Xenograft experiments
For the generation of xenograft tumors, 5 × 106 HuCCT1 cells were mixed with Matrigel at a 1:1 ratio and injected s.c. into NOG mice. The mice were randomly assigned to the control group or the treatment group with NIK small molecule inhibitor (NIKSMI; Genentech) when the tumor size reached 100–300 mm3. The mice were treated with NIKSMI orally twice a day at a dose of 200 mg/kg for 14 days. Tumor diameter was then measured every 3 days, and tumor size was calculated using the following formula22: tumor volume = [(minor diameter)2 × (major diameter)]/2. Furthermore, HuCCT1 cells were infected with negative control short hairpin RNA (shRNA) or NIK shRNA using lentivirus, and infected cells were selected with puromycin. A total of 5 × 106 cells were mixed with Matrigel at a 1:1 ratio and injected s.c. into NOG mice. Tumor diameters were measured starting at 3 weeks after implantation, and tumor volumes were measured in the same manner.
Statistical analysis
The data are presented as the mean ± SD. All data were subjected to appropriate statistical tests. Comparisons between two groups were performed by an unpaired two‐tailed t test for normally distributed variables and Mann‐Whitney’s U test for nonnormally distributed variables. Comparisons between three groups were carried out by the Tukey method. The Kaplan‐Meier method and log‐rank test were used to analyze differences in OS and DFS. The chi‐squared test was performed to examine the relationship between clinical data and immunostaining intensity for TRAF3 or NIK. p < 0.05 was considered to indicate a significant difference. Prism v.8.4.2 for Mac (GraphPad, San Diego, CA; research resource identifier [RRID] SCR_002798) and JMP 13 (SAS Institute, Inc., Cary, NC; RRID SCR_014242) were used for the analyses.
All other information regarding the Materials and Methods is provided in the Supporting Information.
RESULTS
SB mutagenesis identifies Traf3 as an ICC tumor suppressor gene in liver‐specific Pten‐deficient mice
Previously, we showed that SB transposon mutagenesis significantly accelerates the development of HCC and ICC in Pten KO mice.11,12,23 To comprehensively identify genes mutated by SB that drive ICC development, we sequenced the transposon insertion sites from 20 liver tumors with a CCA component (ICC or cHCC‐ICC) confirmed by tubular morphology and KRT19 staining (Figure 1A). This identified 369,549 nonredundant transposon insertion sites (average of 18,477 insertion sites per tumor). Using gCIS,12 which looks for a higher density of transposon insertions within the coding regions of all RefSeq genes than predicted by chance, we identified 405 statistically significant ICC CCGs (p < 0.05, calculated by the chi‐squared test followed by the Bonferroni correction) (Table S1). Trunk drivers that function early in tumor development can be identified by focusing on those with the highest sequence reads.12 We identified 10 potential trunk drivers, with Traf3 being the most significant (Figure 1B). Most of the transposon insertions in Traf3 were located throughout the coding region in the antisense orientation (Figure 1C). Because our mutagenic transposon is designed to inactivate genes when inserted in the reverse orientation, these data strongly suggest that transposon‐mediated loss‐of‐function Traf3 mutations contribute to ICC development in Pten KO mice.
FIGURE 1.
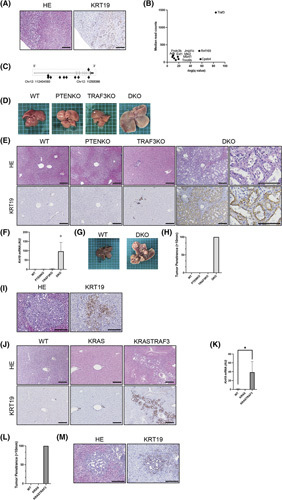
Liver‐specific inactivation of Traf3 induces cholangiocyte proliferation and promotes ICC development. (A) Hematoxylin and eosin and KRT19 staining of the liver of liver‐SB/Pten mice. Scale bars, 50 μm. (B) Trunk driver genes identified in ICC and cHCC‐ICC of liver‐SB/Pten mice. Transposon insertion sites with read counts ≥50 were selected, and 10 trunk drivers were identified. (C) Gene map showing the location of transposon insertions in the Traf3 locus. Each arrowhead indicates a single transposon insertion event, and the direction of the arrowhead denotes sense or antisense orientation. (D–I) Liver phenotypes were examined in WT mice, liver‐specific Pten knockout mice, liver‐specific Traf3 knockout mice, and liver‐specific Pten and Traf3 DKO mice. (D) Macro images of the liver at 6 weeks of age. (E) Hematoxylin and eosin staining and KRT19 staining of the liver at 6 weeks of age. Scale bars, 50 μm. (F) KRT19 liver expression levels at 6 weeks of age, n = 4–8 per group, *p < 0.05. (G) Macro images of the liver at 24 weeks of age. (H) Tumor penetrance (>10 mm) at 24 weeks of age. (I) Hematoxylin and eosin and KRT19 staining of the liver at 24 weeks of age. Scale bars, 50 μm. (J–M) Liver phenotypes were examined in WT mice, liver‐specific Kras‐mutant (KRAS) mice, and Kras‐mutant and Traf3 knockout (KRASTRAF3) mice. (J) Hematoxylin and eosin staining and KRT19 staining of the liver at 6 weeks of age. Scale bars, 50 μm. (K) KRT19 liver expression levels at 6 weeks of age, n = 3–4 per group, *p < 0.05. (L) Tumor penetrance (>10 mm) at 24 weeks of age. (M) Hematoxylin and eosin and KRT19 staining of the liver at 24 weeks of age. Scale bars, 50 μm. HE, hematoxylin and eosin; KO, knockout
Liver‐specific inactivation of Traf3 induces cholangiocyte proliferation and promotes ICC development
To validate the screening result, we generated liver‐specific Traf3/Pten DKO mice (L‐Traf3/PtenDKO) using mice expressing Alb‐Cre (Figure S1A,B). Macroscopically, these mice showed enlarged livers at 6 weeks of age (Figure 1D; Figure S1C) and histologically exhibited marked cholangiocyte overgrowth (Figure 1E), with a prominent increase in KRT19 expression (Figure 1F). Approximately 90% of the L‐Traf3/Pten DKO mice died early at approximately 8–12 weeks of age (Figure S1D), and all L‐Traf3/PtenDKO mice that survived longer exhibited multiple liver tumors at 24 weeks of age (Figure 1G, H). Most of the tumors were diagnosed as ICCs due to positive KRT19 staining and abnormalities in the bile duct structure, such as stratification and fusion of the bile ducts (Figure 1I). To further confirm the tumor suppressive role of TRAF3 in the development of ICC, we deleted Traf3 in mice that expressed liver‐specific constitutively active mutant KrasG12D , a known oncogene frequently mutated in human ICC.6 Liver‐specific Kras‐mutant and Traf3 knockout mice also showed a significant increase in KRT19‐positive cholangiocytes (Figure 1J,K; Figure S1E) and developed multiple liver tumors, including KRT19‐positive ICCs, at 24 weeks of age (Figure 1L,M). Taken together, these data suggested that TRAF3 inactivation induces cholangiocyte proliferation, contributing to ICC development.
ICCs originate from Traf3/Pten‐deficient hepatocytes
To further examine the role of TRAF3 in cholangiocarcinogenesis, we deleted Traf3/Pten specifically in hepatocytes or cholangiocytes of the adult liver using mice expressing a tamoxifen (TAM)–inducible Alb‐CreERT2 or keratin 19 (K19‐CreERT2) promoter, respectively. Hepatocyte‐specific Traf3/Pten DKO mice (HDKO) showed liver enlargement and intrahepatic cholangiocyte overgrowth, with elevated KRT19 levels 12 weeks after TAM administration (Figure 2A,B; Figure S2A,B). In contrast, cholangiocyte‐specific Traf3/Pten DKO mice (CDKO) did not show any of these phenotypes, even 24 weeks after TAM administration (Figure 2A). HDKO mice also developed multiple ICCs 12 weeks after TAM administration (Figure 2C–E), while CDKO mice did not develop any tumors. Collectively, hepatocyte‐specific deletion of Traf3/Pten recapitulated the phenotype of mice carrying liver‐specific deletions.
FIGURE 2.
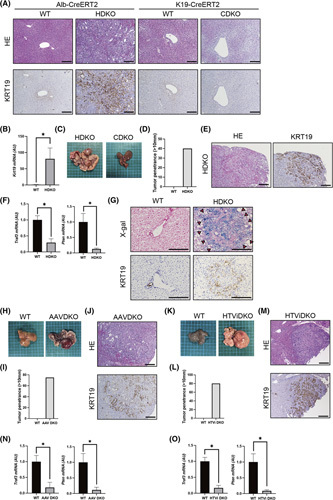
ICCs originate from Traf3/Pten‐deficient hepatocytes. Liver phenotypes were examined in WT mice, hepatocyte‐specific Pten and Traf3 DKO (HDKO) mice, cholangiocyte‐specific Pten and Traf3 DKO (CDKO) mice, and mice with hepatocyte‐specific Pten and Traf3 knockout following the administration of AAV8‐thyroxine binding globulin Cre (AAVDKO) or pEF‐iCre through HTVi (HTViDKO). (A) Hematoxylin and eosin and KRT19 staining of HDKO or CDKO and WT mice at 12 weeks of age. Scale bars, 50 μm. (B) Gene expression levels in HDKO and WT mice at 12 weeks of age. n = 3 per group, *p < 0.05. (C) Macro images of HDKO and CDKO mice at 24 weeks of age. (D) Tumor penetrance (>10 mm) of HDKO mice and WT mice at 24 weeks of age. (E) Hematoxylin and eosin staining and KRT19 staining of HDKO mice at 24 weeks of age. Scale bars, 50 μm. (F) Gene expression levels in tumors from HDKO mice and livers of WT mice. n = 3 per group, *p < 0.05. (G) X‐gal and KRT19 staining of HDKO and WT mice at 12 weeks of age. Scale bars, 50 μm. (H) Macro image of WT and AAV DKO mice. (I) Tumor penetrance (>10 mm) of AAV DKO mice and WT mice. (J) Hematoxylin and eosin and KRT19 staining of AAV DKO mice. Scale bars, 50 μm. (K) Macro image of WT and HTVi DKO mice. (L) Tumor penetrance (>10 mm) of HTVi DKO mice. (M) Hematoxylin and eosin and KRT19 staining of HTVi DKO mice. Scale bars, 50 μm. (N,O) Gene expression levels in AAV DKO tumors and WT mice (N) and HTVi DKO tumors and WT mice (O), n = 3 per group, *p < 0.05. HE, hematoxylin and eosin
We then investigated the cell of origin of ICCs in these mice and found that the expression levels of Traf3 and Pten in these tumors were significantly lower than those in WT liver tissues (Figure 2F), suggesting that ICCs may be derived from Traf3/Pten‐deficient hepatocytes. This prompted us to perform lineage tracing using Rosa26 Cre reporter mice expressing LacZ in Cre‐expressing tissues (Rosa26‐LacZ).17 For this purpose, we crossed CDKO or HDKO mice with Rosa26‐LacZ mice and generated CDKO/Rosa26‐LacZ or HDKO/Rosa26‐LacZ mice, respectively. The specificity of Cre recombinase expression in only cholangiocytes or hepatocytes was confirmed in the CDKO/Rosa26‐LacZ or HDKO/Rosa26‐LacZ mice, respectively (Figure S3A,B). Importantly, KRT19‐positive proliferating cholangiocytes and ICCs were positive for LacZ staining in HDKO/Rosa26‐LacZ mice (Figure 2G), strongly suggesting that proliferating cholangiocytes and ICCs originated from Traf3/Pten‐deficient hepatocytes.
To further confirm these results, we selectively deleted Traf3/Pten in adult hepatocytes by expressing Cre recombinase in adult hepatocytes of Traf3/Pten compound homozygous floxed (Traf3/Pten‐floxed) mice by hydrodynamic tail vein injection (HTVi) or an adenovirus‐associated vector (AAV8).24 These mice also developed ICCs 16 weeks after Cre delivery with high penetrance (Figure 2H–M), and the expression levels of Traf3/Pten in tumor tissue were significantly down‐regulated compared with those in surrounding nontumorous liver tissue (Figure 2N,O). Taken together, these results further confirmed that ICCs originate from Traf3/Pten‐deficient hepatocytes.
Single‐cell RNA sequencing confirms that cholangiocytes are produced from transdifferentiated Traf3/Pten‐deficient hepatocytes
To further investigate the process by which Traf3/Pten‐deficient hepatocytes produce ICCs, we performed single‐cell RNA sequencing of liver cells obtained from HDKO mice and control WT littermates 12 weeks after TAM administration. t‐SNE clustering of the RNA expression results was then performed using previously reported markers25 (Figure S4). While t‐SNE clustering revealed a variety of hepatic cell types common in both mice, including hepatocytes (Clusters 3, 6, 15), endothelial cells (Cluster 8), and immune cells (Clusters 10–14), large hepatocyte populations (Clusters 1, 2, 4, 5, 7, 9) were only identified in HDKO mice (Figure 3A,B). These unique populations expressed not only hepatocyte markers (Alb, apolipoprotein A1, argininosuccinate synthase 1) but also cholangiocyte markers (secreted phosphoprotein 1, SRY [sex‐determining region Y]‐box transcription factor 9 [Sox9]) (Figure 3C). Furthermore, single‐cell trajectory analysis using the Monocle algorithm suggested that these double‐positive hepatocytes were transdifferentiated from normal hepatocytes (Figure 3D).
FIGURE 3.
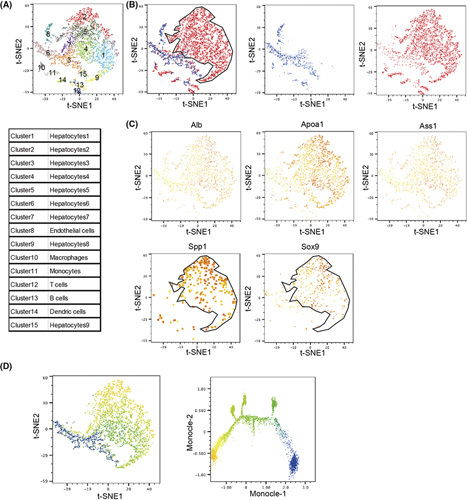
Single‐cell RNA sequencing reveals the transdifferentiation of Traf3/Pten‐deficient hepatocytes into cholangiocytes. (A–C) t‐SNE plot of hepatocyte‐specific Pten and Traf3 DKO mice and WT mice. (A) Each cluster of the t‐SNE plot. (B) WT‐derived cells (blue dots) and DKO‐derived cells (red dots). (C) General markers based on principal component analysis clustering. Low expression intensity in yellow and high in red. (D) t‐SNE plots and monocle pseudotime analysis. Dots are pseudotime, with blue indicating less time elapsed and yellow indicating more time elapsed. Apoa1, apolipoprotein A1; Ass1, argininosuccinate synthase 1; Spp1, secreted phosphoprotein 1
TRAF3/PTEN inhibition promotes the transdifferentiation of hepatocytes into cholangiocytes in vitro
Next, we sought to determine whether we could recapitulate this transdifferentiation phenotype in vitro. For this purpose, we first inhibited TRAF3 and PTEN in HepG2 cells (Figure S5A) and performed RNA sequencing. TRAF3/PTEN inhibition resulted in the down‐regulation of a variety of hepatocyte markers as well as the up‐regulation of multiple cholangiocyte markers (Figure 4A),25,26 suggesting activation of the transdifferentiation program from hepatocytes to cholangiocytes. Up‐regulation of KRT19 mRNA and protein levels upon TRAF3/PTEN inhibition was also confirmed by quantitative PCR and western blotting in HepG2 cells (Figure 4B,C). Moreover, TRAF3/PTEN inhibition significantly increased the proliferation of HepG2 cells (Figure 4D). To validate this transdifferentiation phenotype in noncancerous cells, we established Traf3/Pten‐proficient or Traf3/Pten‐deficient murine liver organoids (Figure 4E; Figure S5B,C) and induced hepatocyte differentiation (Figure S5D). Hepatocytes derived from Traf3/Pten‐deficient organoids (CRE Hep) showed significantly higher Krt19 expression levels and lower Alb levels than hepatocytes derived from WT organoids (green fluorescent protein [GFP] Hep) (Figure 4F). Furthermore, we examined cholangiocyte marker expression in primary human hepatocytes (PHHs) (Figure S5E). Similarly, TRAF3/PTEN inhibition resulted in the up‐regulation of KRT19 mRNA and protein levels (Figure 4G,H). Taken together, these data suggested that TRAF3/PTEN inactivation was the driving force for the transdifferentiation of hepatocytes into cholangiocytes.
FIGURE 4.
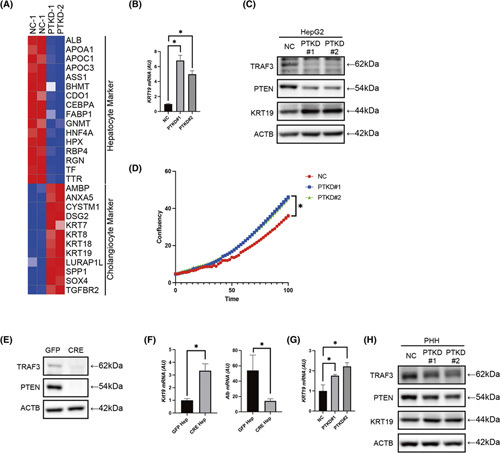
TRAF3/PTEN inhibition promotes the transdifferentiation of hepatocytes into cholangiocytes in vitro. Cell phenotypes were examined in HepG2 cells or PHHs with knockdown of PTEN and TRAF3 or control siRNA, liver organoids established from the liver of Traf3/Pten‐floxed mice transfected with GFP‐expressing lentivirus or CRE‐expressing lentivirus, hepatocytes derived from GFP organoids (GFP Hep) and CRE organoids (CRE Hep). (A) Heatmap of hepatocyte markers and cholangiocyte markers in HepG2 cells. (B) Gene expression levels in HepG2 cells with control and phenotypes examined in HepG2 cells or PHHs with knockdown of PTEN and TRAF3. (C) Western blot in HepG2 with control and phenotypes examined in HepG2 or PHHs with knockdown of PTEN and TRAF3. (D) Cell growth curve of HepG2 with control or phenotypes examined in HepG2 or PHHs with knockdown of PTEN and TRAF3 analyzed by IncuCyte. n = 8 per group, *p < 0.05. (E) Western blot in organoids. (F) Gene expression levels in GFP Hep and CRE Hep. n = 3 per group, *p < 0.05. (G) Gene expression levels in PHHs with control or phenotypes examined in HepG2 or PHHs with knockdown of PTEN and TRAF3. n = 4 per group, *p < 0.05. (H) Western blot in PHHs with control or phenotypes examined in HepG2 or PHHs with knockdown of PTEN and TRAF3. ACTB, actin beta; ALB, Albumin; AMBP, Alpha‐1‐microglobulin/bikunin precursor; ANXA5, Annexin A5; APOA1, Apolipoprotein A1; APOC1, Apolipoprotein C1; APOC3, Apolipoprotein C3; ASS1, Argininosuccinate synthase 1; BHMT, Betaine–homocysteine S‐methyltransferase; CDO1, Cysteine dioxygenase type 1; CEBPA, CCAAT enhancer binding protein alpha; CYSTM1, Cysteine rich transmembrane module containing 1; DSG2, Desmoglein 2; FABP1, Fatty acid binding protein 1; GNMT, Glycine N‐methyltransferase; HNF4A, Hepatocyte nuclear factor 4 alpha; HPX, Hemopexin; KRT7, Keratin 7; KRT8, Keratin 8; KRT18, Keratin 18; KRT19, Keratin 19; LURAP1L, Leucine rich adaptor protein 1 like; NC, negative control; PTKD#1 and PTKD#2, phenotypes examined in HepG2 or PHHs with knockdown of PTEN and TRAF3, respectively; RBP4, Retinol binding protein 4; RGN, Regucalcin; SOX4, SRY‐box transcription factor 4; SPP1, Secreted phosphoprotein 1; TF, Transferrin; TGFBR2, Transforming growth factor beta receptor 2; TTR, Transthyretin
Inactivation of TRAF3/PTEN induces the transdifferentiation of hepatocytes into proliferative cholangiocytes through NIK activation
To identify the mechanisms driving the transdifferentiation of hepatocytes into cholangiocytes, we collected liver tissue from HDKO and WT control littermates 2 weeks after TAM administration, when a significant reduction in Traf3/Pten levels was observed without apparent cholangiocyte overgrowth (Figure S6A,B). Subsequently, we examined the activities of pathways known to be involved in bile duct proliferation, such as the Hippo signaling pathway, the NOTCH signaling pathway, the c‐Jun N‐terminal kinase (JNK) signaling pathway, the Hedgehog signaling pathway,27–29 and the necroptosis pathway, which has recently been shown to be involved in hepatocyte‐derived ICC development.30 While we did not find apparent activation of JNK and Hedgehog signaling, we observed up‐regulation of some NOTCH and Yes‐associated protein (YAP) target gene expression (Figure S6C,D) and phosphorylation of receptor interacting serine/threonine kinase 3, an important mediator of necroptosis (Figure S6E), in the livers of HDKO mice (Figure S6F,G). To explore the involvement of necroptosis, we genetically deleted MLKL (executer of necroptosis) in the liver of L‐Traf3/Pten‐DKO mice, but this did not rescue the phenotype (Figure S6H,I). This result suggested that necroptosis is not important for transdifferentiation in our mice. Next, to address the involvement of the Hippo and NOTCH signaling pathways in the transdifferentiation process, we further examined their pathway activation in TRAF3/PTEN‐inhibited HepG2 cells in vitro and found up‐regulation of some of the NOTCH and YAP target gene expression levels (Figure S6J). We thus evaluated the effect of inhibiting Hippo or NOTCH signaling on the transdifferentiation process in vitro. Small interfering RNA (siRNA)–mediated silencing of recombination signal binding protein for immunoglobulin kappa J region, the major transcriptional effector of NOTCH signaling, significantly suppressed the up‐regulation of KRT19 (Figure S6K), suggesting the involvement of the NOTCH signaling pathway in the transdifferentiation process. Meanwhile, inhibition of the HIPPO pathway by siRNA‐mediated silencing of YAP or TAZ (WW domain‐containing transcription regulator protein 1) did not suppress this process (Figure S6L,M).
To further globally search for genes that may be important for transdifferentiation, we sequenced the RNAs expressed in these liver tissues and looked for genes that were up‐regulated in DKO livers compared with WT livers, which revealed 197 up‐regulated genes (Table S2). Among them, we focused on mitogen‐activated protein kinase kinase kinase 14 (Map3k14), also known as NIK (Figure 5A), because NIK functions downstream of TRAF3 and is important in noncanonical NF‐κB signaling.31 Subsequently, we confirmed that Map3k14 expression was significantly up‐regulated in the livers of HDKO mice 12 weeks after TAM administration and in mouse liver‐derived organoids, human hepatocytes, and hepatoma cells upon knockdown of TRAF3/PTEN expression (Figure 5B–E).
FIGURE 5.
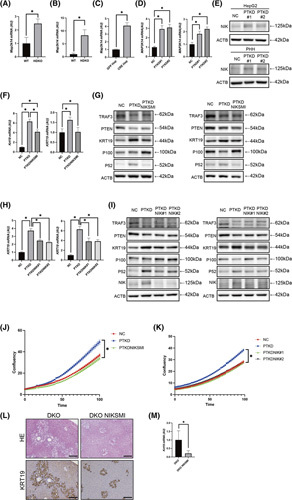
Inactivation of TRAF3/PTEN induces the transdifferentiation of hepatocytes into proliferative cholangiocytes through NIK activation. Liver phenotypes were examined in WT mice, HDKO mice, mice with liver‐specific Pten and Traf3 double knockouts (DKO), and mice with liver‐specific Pten and Traf3 double knockout following NIKSMI treatment twice a day at a dose of 200 mg/kg. Cell phenotypes were examined in hepatocytes derived from GFP‐expressing lentivirus‐infected organoids (GFP Hep); CRE‐expressing lentivirus‐infected organoids (CRE Hep); HepG2 cells or PHHs with knockdown of PTEN and TRAF3 or of PTEN, TRAF3, and MAP3K14; or control siRNA, HepG2 cells, or PHHs with knockdown of PTEN and TRAF3 treated with NIKSMI at a concentration of 9 μM. (A,B) Gene expression levels in HDKO mice at 2 weeks (n = 3) (A) and at 12 weeks (n = 4) (B) after TAM administration, *p < 0.05. (C) Gene expression levels in GFP Hep and CRE Hep. n = 3 per group, *p < 0.05. (D) Gene expression levels in HepG2 (left) or PHHs (right) with control or with knockdown of PTEN and TRAF3. n = 4 per group, *p < 0.05. (E) Western blot in HepG2 cells or PHHs with control or with knockdown of PTEN and TRAF3. (F) Gene expression levels in HepG2 (left) or PHHs (right) with control, with knockdown of PTEN and TRAF3, and with knockdown of PTEN and TRAF3 treated with NIKSMI at a concentration of 9 μM. n = 4 per group, *p < 0.05. (G) Western blot in HepG2 cells (left) or PHHs (right) with control, with knockdown of PTEN and TRAF3, and with knockdown of PTEN and TRAF3 treated with NIKSMI at a concentration of 9 μM. (H) Gene expression levels in HepG2 (left) or PHHs (right) with control or with knockdown of PTEN and TRAF3 or of PTEN, TRAF3, and MAP3K14. n = 4 per group, *p < 0.05. (I) Western blot in HepG2 (left) or PHHs (right) with control or with knockdown of PTEN and TRAF3 or of PTEN, TRAF3, and MAP3K14. (J,K) Cell growth curve of HepG2 with control or with knockdown of PTEN and TRAF3 or of PTEN, TRAF3, and MAP3K14 (J) or with knockdown of PTEN and TRAF3 or of PTEN, TRAF3, and MAP3K14 (K) analyzed by IncuCyte, n = 8 per group, *p < 0.05. (L) Hematoxylin and eosin and KRT19 staining of DKO mice and DKO NIKSMI mice. Scale bars, 50 μm. (M) Gene expression levels in DKO mice and DKO NIKSMI mice. n = 5 per group, *p < 0.05. ACTB, actin beta; HE, hematoxylin and eosin; NC, negative control; PTKD#1 and PTKD#2, phenotypes examined in HepG2 or PHHs with knockdown of PTEN and TRAF3, respectively
To determine whether NIK is important for transdifferentiation, we blocked its kinase activity using a small molecule inhibitor, NIKSMI,32 or inhibited NIK expression using NIK siRNA. While TRAF3/PTEN inhibition in PHH cells and HepG2 cells activated noncanonical NF‐κB signaling, as demonstrated by the increase in p52 expression, both NIKSMI treatment and NIK siRNA suppressed noncanonical NF‐κB signaling and the up‐regulation of KRT19 expression (Figure 5F–I). Both NIKSMI and siRNA also blocked the increase in cell proliferation resulting from TRAF3/PTEN inhibition in HepG2 cells (Figure 5J,K). These inhibitory effects of NIKSMI were dose‐dependent (Figure S7A,B) and were not caused by apoptosis (Figure S7C). Treatment of L‐Traf3/PtenDKO mice with NIKSMI in vivo also significantly suppressed the cholangiocyte overgrowth observed in L‐Traf3/PtenDKO mice (Figure 5L,M). Collectively, these data indicated that inactivation of TRAF3/PTEN induces the transdifferentiation of hepatocytes into proliferative cholangiocytes through NIK activation.
Dysregulation of the TRAF3–NIK axis is associated with an advanced stage and poor prognosis of human ICC
We then evaluated the clinical relevance of the TRAF3–NIK axis in 46 surgically resected patients with ICC. Clinical information on these patients is provided in Table S3. We first performed immunostaining for TRAF3 and NIK in the normal liver and found that TRAF3 expression levels in normal hepatocytes were higher than those in normal cholangiocytes, whereas NIK expression levels in normal hepatocytes were lower than those in normal cholangiocytes (Figure S8). We then quantified the protein levels in ICC tissues (Figure 6A) and observed a significant negative correlation between TRAF3 and NIK protein expression in tumor tissues (Figure 6B). We then evaluated the association of TRAF3 and NIK expression levels with clinicopathological factors and patient prognosis. Interestingly, low TRAF3 or high NIK expression was significantly associated with advanced tumor stage (Tables S4 and S5). Importantly, patients with low TRAF3 expression or high NIK expression showed significantly shorter DFS and OS than those with high TRAF3 levels or low NIK levels, respectively, after surgical resection (Figure 6C–F). These findings suggested that human ICCs with dysregulation of the TRAF3–NIK axis may have higher malignant potential.
FIGURE 6.
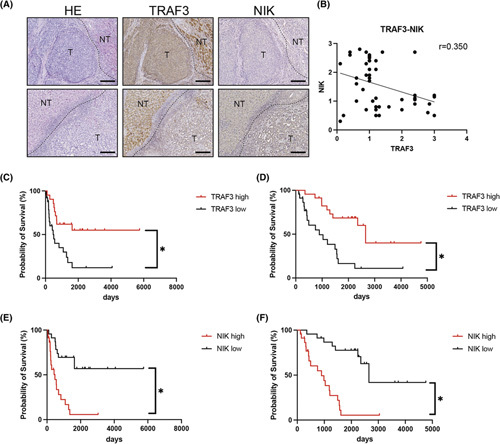
Dysregulation of the TRAF3–NIK axis is associated with advanced stage and poor prognosis of human ICC. (A) Representative images of ICCs with low TRAF3 expression and high NIK expression (upper panels) and ICCs with high TRAF3 expression and low NIK expression (lower panels). (B) Correlation between TRAF3 and NIK staining intensity in tumor tissue of patients with ICC. Scale bars, 50 μm. DFS (C) and OS (D) based on TRAF3 staining intensity in patients with ICC. DFS (E) and OS (F) based on NIK staining intensity in patients with ICC. HE, hematoxylin and eosin; NT, nontumor; T, tumor
NIK inhibition could represent an ICC therapeutic
Finally, we sought to determine whether dysregulation of the TRAF3–NIK axis may represent an ICC therapeutic. We first tested this possibility in vitro using NIK siRNA or NIKSMI. Interestingly, both treatments significantly suppressed the proliferation of multiple human ICC cell lines in vitro (Figure 7A–I). This effect was not mediated by apoptosis (Figure S9A). We then assessed the therapeutic potential of NIK shRNA (Figure S9B,C) or NIKSMI in vivo. NIKSMI treatment did not cause any weight loss or change in blood test results in the mice, suggesting good tolerance of this drug in vivo (Figure S9D,E). We also confirmed that NIKSMI treatment significantly suppressed P52 expression (Figure S9F,G). Both NIK shRNA and NIKSMI treatments significantly suppressed the growth of xenografts derived from HuCCT1 cells (Figure 7J,K). The number of Ki67‐positive cells was significantly lower in tumors treated with NIKSMI or NIK shRNA than in tumors treated with vehicle or negative control shRNA (Figure 7L,M). NIKSMI treatment also reduced the number of proliferating cell nuclear antigen–positive cells (Figure S9H,I). Meanwhile, no induction of apoptosis was observed (Figure S9J–M). Taken together, these results suggest that NIK inhibition could represent a potential therapeutic target for ICC.
FIGURE 7.
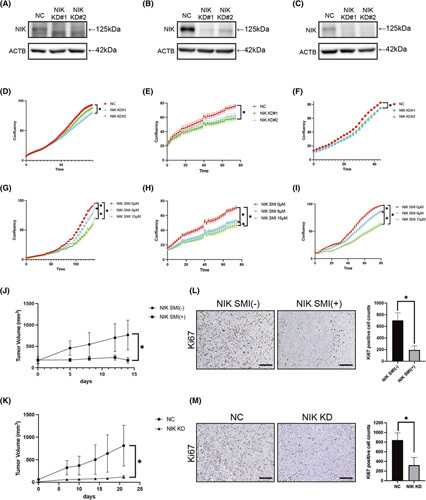
NIK inhibition could represent a potential therapeutic for ICC. Cell phenotypes were examined in HuCCT1, HuCCA1, and KKU232 cells with knockdown of MAP3K14, MAP3K14 shRNA, or control. (A–C) Western blot in HuCCT1 (A), HuCCA1 (B), and KKU213 (C) with control or cells with knockdown of MAP3K14. (D–F) Cell growth curve of HuCCT1 (D), HuCCA1 (E), and KKU213 (F) cells treated with knockdown of NIK analyzed by IncuCyte. n = 8 per group, *p < 0.05. (G–I) Cell growth curves of HuCCT1 (G), HuCCA1 (H), and KKU213 (I) cells with NIKSMI analyzed by IncuCyte. n = 8 per group, *p < 0.05. (J,L) HuCCT‐1 cells were injected s.c. into NOG mice. Mice were orally treated with or without NIKSMI twice a day at a dose of 200 mg/kg. Tumor volumes were measured at various times after treatment, n = 7 per group, *p < 0.05 (J). Ki67‐positive cell counts of HuCCT1 xenograft tumor tissues, n = 7 per group, *p < 0.05. Scale bars, 50 μm (L). (K,M) HuCCT‐1 cells lentivirally transfected with NIK shRNA or negative control shRNA wereinjected s.c. into NOG mice. Tumor volumes were measured at various times, n = 7 per group, *p < 0.05 (K). Ki67‐positive cell counts of HuCCT1 xenograft tumor tissues, n = 7 per group, *p < 0.05. Scale bars, 50 μm (M). ACTB, actin beta; NC, negative control; NIK KD, MAP3K14 shRNA; NIK KD#1 and NIK KD#2, cells with knockdown of MAP3K14
DISCUSSION
In this study, we used SB transposon mutagenesis in Pten‐deficient mice to identify 405 statistically significant ICC CCGs,9 including frequent loss‐of‐function mutations in the Traf3 tumor suppressor gene. TRAF3 is one of seven members of the TRAF adapter protein family. The involvement of TRAF3 in cancer has previously been studied in hematological cancers, where inactivating mutations in TRAF3 have been found in ~10% and 20% of patients with diffuse large B‐cell lymphoma and multiple myeloma, respectively.33,34 Experimentally, TRAF3 acts as a key inhibitor of homeostatic B‐cell survival, and its deletion leads to B‐cell lymphoma.35 More recently, several tumor suppressive roles for TRAF3 have also been reported in solid tumors, including breast, lung, and head and neck cancers and osteosarcoma.36 However, the role of TRAF3 in ICC carcinogenesis has not been reported.
ICC has been considered to develop exclusively from intrahepatic cholangiocytes because it expresses a variety of cholangiocyte markers, including KRT19 and SOX9, and has a luminal structure. However, several recent lineage tracing studies have strongly suggested that ICCs can originate from hepatocytes through transdifferentiation.37–41 These reports showed that ICC could develop from mature hepatocytes in mice following the activation of one or more oncogenes, including KRAS, AKT, YAP, or NOTCH, specifically in hepatocytes through AAV8‐mediated or HTVi–mediated delivery or by using Alb‐CreERT2.38–41 Recent phylogenetic tree analysis of mutational profiles in cHCC‐ICC also suggested the existence of oncogenic reprogramming.42
In this study, we report experimental evidence for hepatocyte‐derived ICC development through TRAF3 inactivation. In our studies, we ensured hepatocyte‐specific gene editing by Cre recombinase delivery through HTVi and AAV8, as well as by using Alb‐CreERT2, and confirmed hepatocyte specificity by lineage tracing using ROSA26 mice. All knockout mice generated in these studies showed a similar cholangiocarcinogenesis phenotype, which strongly supports our claim that ICC originated from hepatocytes through transdifferentiation. In addition, we showed the process of transdifferentiation from mature hepatocytes into cholangiocyte‐like cells by pseudotime analysis using single‐cell RNA sequencing.
TRAF3 is known to have multifaceted roles in the cell. It forms homomultimers or heteromultimers through the coiled‐coil/TRAF‐N domain and has a C‐terminal TRAF‐C domain that is used to mediate many of its protein–receptor and protein–protein interactions.43 One of the important but complex physiological roles of TRAF3 is to mediate Toll‐like receptor (TLR) signaling in immune cells. Whereas TRAF3 positively controls the TLR‐mediated and retinoic acid–inducible gene I–like receptor–mediated type I interferon response, it negatively regulates myeloid differentiation factor 88–dependent mitogen‐activated protein kinase activation and the production of inflammatory cytokines such as TNF, IL‑6, and IL‑12.44 Another important role of TRAF3 is to mediate the alternative NF‑κB pathway. TRAF3 negatively regulates the intracellular levels of NIK by binding with the TRAF2‐cellular inhibitor of apoptosis (cIAP) complex, which leads to cIAP‐mediated K48‑linked polyubiquitylation of NIK and its subsequent proteasomal degradation.43 Upon cluster of differentiation 40 or B‐cell activating factor receptor engagement, TRAF3 is degraded by the proteasome, and NIK protein accumulation leads to activation of the alternative NF‑κB signaling pathway.44 The importance of TRAF3/NIK‐mediated alternative NF‑κB signaling in maintaining tissue homeostasis has been demonstrated by the early postnatal lethal phenotypes caused by systemic TRAF3 deletion, which was rescued by the additional deletion of Nfkb2, a transcription factor of the noncanonical NF‑κB signaling pathway.45 In the current study, we found that TRAF3 inactivation up‐regulated NIK and activated noncanonical NF‑κB signaling (Figure 5G,I). Subsequently, we showed that NIK inhibition suppressed hepatocyte transdifferentiation into proliferative cholangiocytes. Our studies shed light on the important role of TRAF3/NIK‐mediated activation of the noncanonical NF‐κB pathway in hepatocyte‐derived cholangiocarcinogenesis.
Aberrant NIK expression and its oncogenic roles have been reported in a variety of cancer types, including hematological and solid cancers (e.g., melanoma, glioma, breast cancer, and pancreatic cancer).46,47 However, its involvement in the pathogenesis or therapeutics of ICC has not been exploited. Herein, we showed a clinical association of the TRAF3–NIK axis with poor prognosis in human patients with ICC and provided experimental evidence indicating that NIK is a promising ICC therapeutic target. In addition, we identified a differentiation abnormality as the mode of action of NIK‐mediated cholangiocarcinogenesis. Interestingly, activation of NIK and the alternative NF‐κB pathway has also been shown to inhibit the differentiation of hematopoietic stem cells, which is important for leukemogenesis, and to promote cancer stemness in breast cancer.46,47 Taken together, these data suggest an important role for NIK and the alternative NF‐κB signaling pathway in cellular differentiation and show that targeting the differentiation program could be a therapeutic option for cancer types with aberrant NIK expression, including ICC.
In conclusion, our studies have revealed a mechanism of intrahepatic cholangiocarcinogenesis resulting from hepatocyte transdifferentiation induced by TRAF3/NIK‐mediated noncanonical NF‐κB signaling. We have also demonstrated the clinical relevance of the TRAF3–NIK axis in human ICC and identified NIK as a promising therapeutic target for treating ICC.
Supplementary Material
Acknowledgments
AUTHOR CONTRIBUTIONS
Conceptualization: Yuto Shiode, Takahiro Kodama, and Tetsuo Takehara. Methodology: Yuto Shiode, Takahiro Kodama, Justin Y. Newberg, Jumpei Kondo, Nancy A. Jenkins, Neal G. Copeland, and Tetsuo Takehara. Experimentation: Yuto Shiode, Satoshi Shigeno, Kazuhiro Murai, and Satoshi Tanaka. Writing: Yuto Shiode and Takahiro Kodama. Funding acquisition: Takahiro Kodama, Tomohide Tatsumi, and Tetsuo Takehara. Resources: Yuto Shiode, Justin Y. Newberg, Jumpei Kondo, Shogo Kobayashi, Ryoko Yamada, Hayato Hikita, Ryotaro Sakamori, Hiroshi Suemizu, and Hidetoshi Eguchi. Supervision: Tetsuo Takehara.
ACKNOWLEDGMENT
Alb‐Cre ERT2 mice were provided by Daniel Metzger and Pierre Chambon (Institute for Genetics and Cellular and Molecular Biology). KKU‐213 cells were provided by Dr. Banchob Spira (Khon Kaen University).
CONFLICT OF INTEREST
Nothing to report.
DATA DEPOSITION
The RNA‐sequencing and single‐cell RNA‐sequencing data reported in this study have been deposited in the Gene Expression Omnibus database under the following accession numbers: GSE178418, GSE184276 (RNA‐sequencing data), and GSE178814 (single‐cell RNA‐sequencing data).
Footnotes
Abbreviations: AAV, adenovirus‐associated vector; Alb‐Cre, Cre recombinase under the control of the albumin promoter; CCA, cholangiocarcinoma; CCG, candidate cancer gene; CDKO, cholangiocyte‐specific Pten and Traf3 DKO; cHCC‐ICC, combined HCC and ICC; DFS, disease‐free survival; DKO, double knockout; ERT2, estrogen receptor targeting 2; GFP, green fluorescent protein; HDKO, hepatocyte‐specific Pten and Traf3 DKO; HTVi, hydrodynamic tail vein injection; ICC, intrahepatic CCA; KRT19, keratin 19; LacZ, β‐galactosidase; Map3k14, mitogen‐activated protein kinase kinase kinase 14; MLKL, mixed lineage kinase domain like pseudokinase; NIK, NF‐κB‐inducing kinase; NIKSMI, NIK small molecule inhibitor; NOG, nonobese diabetic/Shi severe combined immunodeficient Il2rγnull; OS, overall survival; PHH, primary human hepatocyte; ROSA26, reverse orientation splice acceptor βgeo line 26; SB, Sleeping Beauty; shRNA, short hairpin RNA; siRNA, small interfering RNA; Sox9, SRY (sex‐determining region Y)‐box transcription factor 9; TAM, tamoxifen; TAZ, WW domain‐containing transcription regulator protein 1; Traf3, TNF receptor–related factor 3; t‐SNE, t‐distributed stochastic neighbor embedding; WT, wild‐type; YAP, Yes‐associated protein.
Yuto Shiode and Takahiro Kodama contributed equally to this work and share first authorship.
Funding information Supported by the Japan Agency for Medical Research and Development (P21fk0310108h0005, to T.Tak.; JP21fk0210074, to T.K.; JP21fk021055h, to T.K.; and JP21fk0210091s, to T.K.) and by a Grant-in- Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology, Japan (20H03661, to T.K.)
Supplemental Digital Content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's website, www.hepjournal.com.
REFERENCES
- 1.Rizvi S, Khan SA, Hallemeier CL, Kelley RK, Gores GJ. Cholangiocarcinoma—evolving concepts and therapeutic strategies. Nat Rev Clin Oncol. 2018;15(2):95–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Banales JM, Marin JJG, Lamarca A, Rodrigues PM, Khan SA, Roberts LR, et al. Cholangiocarcinoma 2020: the next horizon in mechanisms and management. Nat Rev Gastroenterol Hepatol. 2020;17(9):557–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Banales JM, Cardinale V, Carpino G, Marzioni M, Andersen JB, Invernizzi P, et al. Expert consensus document. Cholangiocarcinoma: current knowledge and future perspectives consensus statement from the European Network for the Study of Cholangiocarcinoma (ENS‐CCA). Nat Rev Gastroenterol Hepatol. 2016;13(5):261–80. [DOI] [PubMed] [Google Scholar]
- 4.Farshidfar F, Zheng S, Gingras M‐C, Newton Y, Shih J, Robertson AG, et al. Integrative genomic analysis of cholangiocarcinoma identifies distinct IDH‐mutant molecular profiles. Cell Rep. 2017;19(13):2878–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wardell CP, Fujita M, Yamada T, Simbolo M, Fassan M, Karlic R, et al. Genomic characterization of biliary tract cancers identifies driver genes and predisposing mutations. J Hepatol. 2018;68(5):959–69. [DOI] [PubMed] [Google Scholar]
- 6.Zou S, Li J, Zhou H, Frech C, Jiang X, Chu JSC, et al. Mutational landscape of intrahepatic cholangiocarcinoma. Nat Commun. 2014;5:5696. [DOI] [PubMed] [Google Scholar]
- 7.Goyal L, Kongpetch S, Crolley VE, Bridgewater J. Targeting FGFR inhibition in cholangiocarcinoma. Cancer Treat Rev. 2021;95:102170. [DOI] [PubMed] [Google Scholar]
- 8.Kodama T, Bard‐Chapeau EA, Newberg JY, Kodama M, Rangel R, Yoshihara K, et al. Two‐step forward genetic screen in mice identifies ral GTPase‐activating proteins as suppressors of hepatocellular carcinoma. Gastroenterology. 2016;151(2):324–37.e12. [DOI] [PubMed] [Google Scholar]
- 9.Copeland NG, Jenkins NA. Harnessing transposons for cancer gene discovery. Nat Rev Cancer. 2010;10(10):696–706. [DOI] [PubMed] [Google Scholar]
- 10.Kodama T, Newberg JY, Kodama M, Rangel R, Yoshihara K, Tien JC, et al. Transposon mutagenesis identifies genes and cellular processes driving epithelial–mesenchymal transition in hepatocellular carcinoma. Proc Natl Acad Sci U S A. 2016;113(24):E3384–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Horie Y, Suzuki A, Kataoka EI, Sasaki T, Hamada K, Sasaki J, et al. Hepatocyte‐specific Pten deficiency results in steatohepatitis and hepatocellular carcinomas. J Clin Invest. 2004;113(12):1774–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kodama T, Yi J, Newberg JY, Tien JC, Wu H, Finegold MJ, et al. Molecular profiling of nonalcoholic fatty liver disease‐associated hepatocellular carcinoma using SB transposon mutagenesis. Proc Natl Acad Sci U S A. 2018;115(44):E10417–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Postic C, Shiota M, Niswender KD, Jetton TL, Chen Y, Moates JM, et al. Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic beta cell‐specific gene knock‐outs using Cre recombinase. J Biol Chem. 1999;274(1):305–15. [DOI] [PubMed] [Google Scholar]
- 14.Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, Montoya R, et al. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K‐ras. Genes Dev. 2001;15(24):3243–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Means AL, Xu Y, Zhao A, Ray KC, Gu G. A CK19(CreERT) knockin mouse line allows for conditional DNA recombination in epithelial cells in multiple endodermal organs. Genesis. 2008;46(6):318–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lesche R, Groszer M, Gao J, Wang Y, Messing A, Sun H, et al. Cre/loxP‐mediated inactivation of the murine Pten tumor suppressor gene. Genesis. 2002;32(2):148–9. [DOI] [PubMed] [Google Scholar]
- 17.Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21(1):70–1. [DOI] [PubMed] [Google Scholar]
- 18.Schuler M, Dierich A, Chambon P, Metzger D. Efficient temporally controlled targeted somatic mutagenesis in hepatocytes of the mouse. Genesis. 2004;39(3):167–72. [DOI] [PubMed] [Google Scholar]
- 19.Gardam S, Sierro F, Basten A, Mackay F, Brink R. TRAF2 and TRAF3 signal adapters act cooperatively to control the maturation and survival signals delivered to B cells by the BAFF receptor. Immunity. 2008;28(3):391–401. [DOI] [PubMed] [Google Scholar]
- 20.Murphy J, Czabotar P, Hildebrand J, Lucet I, Zhang J‐G, Alvarez‐Diaz S, et al. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity. 2013;39(3):443–53. [DOI] [PubMed] [Google Scholar]
- 21.Newton K, Wickliffe KE, Maltzman A, Dugger DL, Strasser A, Pham VC, et al. RIPK1 inhibits ZBP1‐driven necroptosis during development. Nature. 2016;540(7631):129–33. [DOI] [PubMed] [Google Scholar]
- 22.Ding X, Zhang B, Pei Q, Pan J, Huang S, Yang Y, et al. Triptolide induces apoptotic cell death of human cholangiocarcinoma cells through inhibition of myeloid cell leukemia‐1. BMC Cancer. 2014;14:271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu X, Kobayashi S, Qiao W, Li C, Xiao C, Radaeva S, et al. Induction of intrahepatic cholangiocellular carcinoma by liver‐specific disruption of Smad4 and Pten in mice. J Clin Invest. 2006;116(7):1843–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yanger K, Knigin D, Zong Y, Maggs L, Gu G, Akiyama H, et al. Adult hepatocytes are generated by self‐duplication rather than stem cell differentiation. Cell Stem Cell. 2014;15(3):340–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xiong X, Kuang H, Ansari S, Liu T, Gong J, Wang S, et al. Landscape of intercellular crosstalk in healthy and NASH liver revealed by single‐cell secretome gene analysis. Mol Cell. 2019;75(3):644–60.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ramachandran P, Dobie R, Wilson‐Kanamori JR, Dora EF, Henderson BEP, Luu NT, et al. Resolving the fibrotic niche of human liver cirrhosis at single‐cell level. Nature. 2019;575(7783):512–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nishikawa Y, Sone M, Nagahama Y, Kumagai E, Doi Y, Omori Y, et al. Tumor necrosis factor‐alpha promotes bile ductular transdifferentiation of mature rat hepatocytes in vitro. J Cell Biochem. 2013;114(4):831–43. [DOI] [PubMed] [Google Scholar]
- 28.Nishio M, Sugimachi K, Goto H, Wang J, Morikawa T, Miyachi Y, et al. Dysregulated YAP1/TAZ and TGF‐beta signaling mediate hepatocarcinogenesis in Mob1a/1b‐deficient mice. Proc Natl Acad Sci U S A. 2016;113(1):E71–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tang L, Tan Y‐X, Jiang B‐G, Pan Y‐F, Li S‐X, Yang G‐Z, et al. The prognostic significance and therapeutic potential of hedgehog signaling in intrahepatic cholangiocellular carcinoma. Clin Cancer Res. 2013;19(8):2014–224. [DOI] [PubMed] [Google Scholar]
- 30.Seehawer M, Heinzmann F, D’Artista L, Harbig J, Roux P‐F, Hoenicke L, et al. Necroptosis microenvironment directs lineage commitment in liver cancer. Nature. 2018;562(7725):69–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Parvatiyar K, Pindado J, Dev A, Aliyari SR, Zaver SA, Gerami H, et al. A TRAF3‐NIK module differentially regulates DNA vs RNA pathways in innate immune signaling. Nat Commun. 2018;9(1):2770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brightbill HD, Suto E, Blaquiere N, Ramamoorthi N, Sujatha‐Bhaskar S, Gogol EB, et al. NF‐κB inducing kinase is a therapeutic target for systemic lupus erythematosus. Nat Commun. 2018;9(1):179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Annunziata CM, Davis RE, Demchenko Y, Bellamy W, Gabrea A, Zhan F, et al. Frequent engagement of the classical and alternative NF‐κB pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell. 2007;12(2):115–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bushell KR, Kim Y, Chan FC, Ben‐Neriah S, Jenks A, Alcaide M, et al. Genetic inactivation of TRAF3 in canine and human B‐cell lymphoma. Blood. 2015;125(6):999–1005. [DOI] [PubMed] [Google Scholar]
- 35.Moore CR, Liu Y, Shao C, Covey LR, Morse HC, 3rd, Xie P. Specific deletion of TRAF3 in B lymphocytes leads to B‐lymphoma development in mice. Leukemia. 2012;26(5):1122–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang B, Shetti D, Fan C, Wei K. miR‐29b‐3p promotes progression of MDA‐MB‐231 triple‐negative breast cancer cells through downregulating TRAF3. Biol Res. 2019;52(1):38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fan B, Malato Y, Calvisi DF, Naqvi S, Razumilava N, Ribback S, et al. Cholangiocarcinomas can originate from hepatocytes in mice. J Clin Invest. 2012;122(8):2911–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sekiya S, Suzuki A. Intrahepatic cholangiocarcinoma can arise from Notch‐mediated conversion of hepatocytes. J Clin Invest. 2012;122(11):3914–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang J, Dong M, Xu Z, Song X, Zhang S, Qiao YU, et al. Notch2 controls hepatocyte‐derived cholangiocarcinoma formation in mice. Oncogene. 2018;37(24):3229–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hill MA, Alexander WB, Guo B, Kato Y, Patra K, O'Dell MR, et al. Kras and Tp53 mutations cause cholangiocyte‐ and hepatocyte‐derived cholangiocarcinoma. Cancer Res. 2018;78(16):4445–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yimlamai D, Christodoulou C, Galli G, Yanger K, Pepe‐Mooney B, Gurung B, et al. Hippo pathway activity influences liver cell fate. Cell. 2014;157(6):1324–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xue R, Chen LU, Zhang C, Fujita M, Li R, Yan S‐M, et al. Genomic and transcriptomic profiling of combined hepatocellular and intrahepatic cholangiocarcinoma reveals distinct molecular subtypes. Cancer Cell. 2019;35(6):932–47.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bishop GA. TRAF3 as a powerful and multitalented regulator of lymphocyte functions. J Leukoc Biol. 2016;100(5):919–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hacker H, Tseng PH, Karin M. Expanding TRAF function: TRAF3 as a tri‐faced immune regulator. Nat Rev Immunol. 2011;11(7):457–68. [DOI] [PubMed] [Google Scholar]
- 45.He JQ, Zarnegar B, Oganesyan G, Saha SK, Yamazaki S, Doyle SE, et al. Rescue of TRAF3‐null mice by p100 NF‐kappa B deficiency. J Exp Med. 2006;203(11):2413–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pflug KM, Sitcheran R. Targeting NF‐κB‐inducing kinase (NIK) in immunity, inflammation, and cancer. Int J Mol Sci. 2020;21(22):8470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Maubach G, Feige MH, Lim MCC, Naumann M. NF‐kappaB‐inducing kinase in cancer. Biochim Biophys Acta Rev Cancer. 2019;1871(1):40–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The RNA‐sequencing and single‐cell RNA‐sequencing data reported in this study have been deposited in the Gene Expression Omnibus database under the following accession numbers: GSE178418, GSE184276 (RNA‐sequencing data), and GSE178814 (single‐cell RNA‐sequencing data).