

PURPOSE
Acquired resistance to anti–epidermal growth factor receptor (EGFR) inhibitor (EGFRi) therapy in colorectal cancer (CRC) has previously been explained by the model of acquiring new mutations in KRAS/NRAS/EGFR, among other MAPK-pathway members. However, this was primarily on the basis of single-agent EGFRi trials and little is known about the resistance mechanisms of EGFRi combined with effective cytotoxic chemotherapy in previously untreated patients.
METHODS
We analyzed paired plasma samples from patients with RAS/BRAF/EGFR wild-type metastatic CRC enrolled in three large randomized trials evaluating EGFRi in the first line in combination with chemotherapy and as a single agent in third line. The mutational signature of the alterations acquired with therapy was evaluated. CRC cell lines with resistance to cetuximab, infusional fluorouracil, leucovorin, and oxaliplatin, and SN38 were developed, and transcriptional changes profiled.
RESULTS
Patients whose tumors were treated with and responded to EGFRi alone were more likely to develop acquired mutations (46%) compared with those treated in combination with cytotoxic chemotherapy (9%). Furthermore, contrary to the generally accepted hypothesis of the clonal evolution of acquired resistance, we demonstrate that baseline resistant subclonal mutations rarely expanded to become clonal at progression, and most remained subclonal or disappeared. Consistent with this clinical finding, preclinical models with acquired resistance to either cetuximab or chemotherapy were cross-resistant to the alternate agents, with transcriptomic profiles consistent with epithelial-to-mesenchymal transition. By contrast, commonly acquired resistance alterations in the MAPK pathway do not affect sensitivity to cytotoxic chemotherapy.
CONCLUSION
These findings support a model of resistance whereby transcriptomic mechanisms of resistance predominate in the presence of active cytotoxic chemotherapy combined with EGFRi, with a greater predominance of acquired MAPK mutations after single-agent EGFRi. The proposed model has implications for prospective studies evaluating EGFRi rechallenge strategies guided by acquired MAPK mutations, and highlights the need to address transcriptional mechanisms of resistance.
INTRODUCTION
Despite advances in screening strategies and available therapies, colorectal cancer (CRC) remains the second leading cause of cancer-related death in the United States.1 Among patients who are diagnosed with CRC, 50% ultimately develop metastatic disease with a median survival of 2-3 years.2 In patients with metastatic disease, a combination of cytotoxic chemotherapy and targeted therapy remains the mainstay of treatment, but inevitably, duration of response is affected by acquired drug resistance.3-7 The mortality associated with CRC is related to development of secondary resistance after an initial response to therapy, particularly to targeted therapies such as anti–epidermal growth factor receptor (anti-EGFR) monoclonal antibodies used in the second- and third-line (3L) settings in patients with KRAS/NRAS (RAS) wild-type metastatic colorectal cancer (mCRC).
CONTEXT
Key Objective
How does chemotherapy and line of therapy affect acquired resistance to anti–epidermal growth factor receptor (EGFR) in RAS/RAF wild-type colorectal cancer?
Knowledge Generated
Rather than an outgrowth of preexisting clones, a transcriptional mechanism of acquired resistance appears to predominate in patients treated with a combination of an anti-EGFR and cytotoxic chemotherapy. This observation has implications for prospective studies evaluating anti-EGFR rechallenge strategies guided by acquired MAPK mutations, and highlights the need to address transcriptional mechanisms of resistance.
Relevance (A. Ko)
-
Mechanisms of resistance to anti-EGFR antibody therapy may differ depending on whether it is administered in first versus later lines of therapy, and as monotherapy versus in combination with chemotherapy. It is reasonable to consider repeat molecular interrogation at the time of progression in patients with metastatic colorectal cancer, via either circulating tumor DNA analysis or repeat tumor biopsy; however, further work is required to understand if and how this information can help guide subsequent treatment decisions.*
*Relevance section written by JCO Associate Editor Andrew Ko, MD.
Secondary mutations in the mitogen-activated protein (MAPK) signaling pathway have been implicated in resistance to anti-EGFR; however, the process by which resistant clones develop during therapy has not been well elucidated and there is a great deal of intrapatient and interpatient heterogeneity.8,9 Circulating tumor DNA (ctDNA) testing has allowed for the noninvasive detection of heterogeneous molecular alterations such as KRAS, NRAS, BRAF V600E, MAP2K1, and EGFR ectodomain (KRAS/NRAS/BRAF/MAP2K1/EGFR ECD), among others, which underlie the evolution of resistance to anti-EGFR.9-17 The conventional theory for the development of treatment resistance has been predominantly based on the model that therapy-resistant subclonal mutations proliferate and ultimately outnumber treatment-sensitive cells, leading to treatment resistance and disease progression.8 Our group, among others, has shown that during subsequent treatment without anti-EGFR, sensitive clones may be at least partially restored, explaining the clinical success of anti-EGFR rechallenge.12,15,18,19
However, clinical results from anti-EGFR rechallenge have been mixed, with response rates approximately 30% in various trials.15,19 Furthermore, acquired resistance mutations have been identified in only 35%-40% of patients, suggesting that there may be additional mechanisms of acquired resistance or novel pathways leading to escape from anti-EGFR therapy.9 Several novel pathways leading to escape from anti-EGFR therapy have been reported in recent years including transient deficiency in mismatch repair (adaptive mutability model)20,21 and adaptive changes in differentiation status and cell fate (epithelial-to-mesenchymal transition [EMT])—a complex biological process wherein epithelial cell procedurally lose their original morphology and simultaneously acquire mesenchymal characteristics potentially contributing to acquired anti-EGFR resistance.22
Despite this background, little is understood about how combination cytotoxic chemotherapy modulates the mechanisms of resistance to anti-EGFR and the dynamics of treatment-resistant subclones. To understand such processes, we analyzed the dynamics of ctDNA-detected subclones from three clinical trials and demonstrate clinically relevant differences in mechanisms of acquired resistance and subclonal dynamics. We used in vitro modeling to demonstrate cross-resistance to cytotoxic chemotherapy and anti-EGFR and describe the clinical relevance of the work to ongoing anti-EGFR rechallenge studies.
METHODS
Patients
We analyzed paired plasma samples from patients with ctDNA-confirmed RAS/BRAF/EGFR wild-type mCRC enrolled in three large randomized trials who had been treated with anti-EGFR and in whom paired baseline and time-of-progression plasma samples had been collected for sequencing of ctDNA on a platform optimized for very low allele frequencies (PlasmaSelect-R and Resolution Bio), as previously described.23,24 Of the 1,183 accrued patients in the first-line (1L) 203 study comparing infusional fluorouracil, leucovorin, and oxaliplatin to infusional fluorouracil, leucovorin, and oxaliplatin plus panitumumab, 147 had paired plasma samples available to establish the prevalence of mutations at baseline and progression. Of the 377 patients enrolled in the 3L 007 study comparing panitumumab plus best supportive care (BSC) versus BSC alone, 91 patients had paired samples available and were analyzed. In the 3L 763 study comparing panitumumab versus cetuximab, 331/999 enrolled patients from the panitumumab arm had paired plasma samples, which were analyzed (Fig 1). Patients receiving cetuximab in the 763 study were excluded from the analysis as plasma samples were not analyzed for these patients.
FIG 1.

A schematic of the three randomized trials used in the 1L and 3L settings. 1L, first-line; 3L, third-line; BSC, best supportive care; FOLFOX, infusional fluorouracil, leucovorin, and oxaliplatin.
Data Collection
KRAS/NRAS/BRAF/MAP2K1/EGFR mutational status of the tumor specimen, and baseline and at-progression plasma samples were collected per patient. Our analysis included all known mechanisms of resistance to anti-EGFR, including all KRAS/NRAS/BRAFV600E/MAP2K1 and EGFR ECD mutations. All patients with clonal mutations in these genes of interest at baseline were excluded. Subclonal mutations were those defined as having a relative mutational allelic frequency (rMAF) < 25%, consistent with prior literature.9,17,18,25 The rMAF approximates the percentage of tumor cells shedding the mutation of interest into the circulation and was defined as the mutant allele frequency (AF) of a mutation divided by the maximum mutant AF for any gene detected in that sample. Passenger mutations were defined as any mutation outside of RAS/BRAF/EGFR/MAP2K1 for purposes of this analysis. Only patients who stopped prior anti-EGFR because of progression were included in the analysis. Acquired mutations were defined as any mutation in KRAS/NRAS/BRAF/MAP2K1/EGFR in postprogression samples that were not present, at any frequency, at baseline. The 25/50/75 quantiles of time from the start of the study to the end of the study for 203 study are 127, 198, and 300 days (range, 1-1,310 days). The quantiles for the 007 study are 42, 98, and 155 days (range, 0-1,036 days). For the 763 study, they are 56, 140, and 196 days (range, 10-546 days).
Radiographic responses were assessed per RECISTv1.1.26 Overall survival was calculated as the time from the start of treatment to the date of death. Survival was censored at last follow-up if death was not observed.
See the Data Supplement (online only) for all statistical, mutational signature, and preclinical methods.
RESULTS
Acquired Anti-EGFR Resistance Mutations Are Rare in the 1L Setting
We found that acquisition of at least one KRAS/NRAS/EGFR ECD/BRAF/MAP2K1 mutation was significantly less common in postprogression samples in the EGFR-containing arms of the 1L 203 study compared with the two 3L 763 and 007 studies combined (9.1% [7/77] v 46% [181/395]; P < .001; Figs 2A-2C). The 1L 203 study evaluated the rates of these acquired mutations among all the patients without regards to response or duration of treatment. Here, we found that the rate of mutations was 9.1% (7/77) in the cytotoxic chemotherapy plus anti-EGFR arm and 5.7% (4/70) in the chemotherapy alone arm (Fig 2D). These rates were significantly lower compared with acquired mutations after 3L therapy with EGFR inhibitor alone. In the randomized 3L 007 study, acquired anti-EGFR resistance mutations were seen in 39% (25/64) and 0% (0/27) of patients in the panitumumab- and BSC-treated arms, respectively (P < .001; Fig 2E). Similarly, in the randomized 3L 763 study, 47% (156/331) of patients developed these resistance mutations. It has previously been demonstrated that codon 61 mutations are enriched in acquired resistance to EGFR inhibition. In the 1L 203 study, four KRAS acquired mutations were seen in the panitumumab-containing arm, with one being a codon 61 mutation. By contrast, in the 3L studies, there were 86 acquired KRAS mutations in the anti-EGFR arms, with 63% of them being in codon 61 (54/86). Notably, because responses in the 1L setting may be due to cytotoxic chemotherapy and EGFR inhibition, our analysis focused on comparisons of responses within a line of therapy rather than between first line and third line.
FIG 2.

(A) Rate of acquired resistance mutations in patients receiving FOLFOX plus panitumumab in the 1L setting (203 study). Resistance mutations (green) are defined as any mutations in KRAS, NRAS, BRAF V600E, MAP2K1, or EGFR ectodomain. Other mutations are in purple. (B) Rate of acquired resistance mutations in patients receiving panitumumab in the 3L setting (763 study). (C) Rate of acquired resistance mutations in patients receiving panitumumab in the 3L setting (007 study). (D) In the 1L study, the rate of anti-EGFR resistance mutation acquisition in the anti-EGFR arm (9.1% = 7/77) was no different than in the non–anti-EGFR–containing arm (5.7% = 4/70). (E) In the randomized 3L study, acquired anti-EGFR resistance mutations were seen in 39% (25/64) and 0% (0/27) of patients in the panitumumab- and BSC-treated arms, respectively (P < .001). *A statistically significant difference (P < .001) between subgroups. 1L, first-line; 3L, third-line; BSC, best supportive care; EGFR, epidermal growth factor receptor; FOLFOX, infusional fluorouracil, leucovorin, and oxaliplatin; MT, mutation.
Association of Tumor Response and Acquired Resistance Mutations
We also hypothesized that acquired mutations would occur at different rates when comparing responders and nonresponders to anti-EGFR, and that these would occur more commonly in responders in the 3L setting. There was no significant difference in the rate of acquired resistance mutations between patients in the 1L study who had a treatment response versus those with progressive disease (PD; 9% [6/67] v 12.5% [1/8]; P = .56; Fig 3A). There was no significant difference in the rate of acquired resistance mutations between patients in the 1L study who had a partial response (PR) versus non-PR (10.5% [4/38] v 8.1% [3/37]; P = 1). Similarly, in the 3L studies, there was only a modestly increased prevalence of acquired resistance mutations in patients receiving anti-EGFR who had complete response, PR, or stable disease versus those who had PD (49% [147/299] v 35% [34/96]; P = .02; Figs 3B and 3C). When restricted to those who had a complete response or PR only, there was no discernable difference in the rate of acquired resistance mutations (49.5% [55/111] v 44.4% [126/284] for stable disease or PD; P = .37).
FIG 3.

(A) Association with RECIST responses and acquired resistance mutations in the first-line 203 study. (B) Association with RECIST responses and acquired resistance mutations in the 3L 763 study. (C) Association with RECIST responses and acquired resistance mutations in the 3L 007 study. 3L, third-line; CR, complete response; MT, mutation; PD, progressive disease; PR, partial response; SD, stable disease.
We asked a similar question regarding acquisition of resistance mutations when comparing short versus long progression-free survival (PFS) in the 1L and 3L settings. Here too, there was a very low rate of acquired resistance mutations in patients receiving anti-EGFR in the 1L setting. When comparing acquired resistance mutations in patients with short versus long PFS (defined as < 10 months or ≥ 10 months in the 1L setting, and < 4 months or ≥ 4 months in the 3L setting), the rates were, respectively, 3/17 (18%) and 4/60 (7%) in the 1L setting (P = .18; Appendix Fig A1A, online only) and 67/178 (38%) and 114/217 (53%) in the 3L setting (P = .003; Appendix Fig A1B).
Increase in Rate of Passenger Mutations With Known Acquired Resistance Mutations on Anti-EGFR
The model of adaptive mutability as a mechanism of acquired resistance suggests that acquired resistance mutations may be accompanied by passenger mutations in the setting of globally reduced replication fidelity under targeted treatment. We thus sought to evaluate if other alterations in addition to KRAS/NRAS/EGFR/BRAF/MAP2K1 were enriched at the time of progression on anti-EGFR. We found a net percentage gain in the number of passenger mutations in patients treated with anti-EGFR versus not. Patients treated with anti-EGFR had an average of 1.4 passenger mutations compared with an average of 0.4 passenger mutations in patients not treated with anti-EGFR (P < .001; Fig 4A). We also found that among patients treated with anti-EGFR, those who acquired RAS/EGFR/BRAF/MAP2K1 mutations were more likely to acquire additional passenger mutations than those who did not (average 2.3 alternate mutations v 0.9, respectively; P < .001; Fig 4B).
FIG 4.

(A) Patients treated with anti-EGFR had an average of 1.4 passenger mutations (non-RAS/EGFR/BRAF/MAP2K1) compared with an average of 0.4 passenger mutations in patients not treated with anti-EGFR (P < .001). (B) Among patients treated with anti-EGFR, those who acquired RAS/EGFR/BRAF/MAP2K1 mutations were more likely to acquire additional passenger mutations than those who did not (average 2.1 alternate mutations v 0.9, respectively; P < .001). (C) Originally subclonal resistance mutations rarely expanded to become clonal at the time of progression. At baseline, 27% of patients treated with anti-EGFR had a baseline subclonal resistance alteration. There were a total of 171 subclonal resistance mutations among these patients. 8% of the 171 became clonal, 47% stayed subclonal, and 44% were absent at time of progression. EGFR, epidermal growth factor receptor; MT, mutation.
Preexisting Resistant Subclones Rarely Expand at the Time of Progression on Anti-EGFR
We then inquired about the impact of anti-EGFR on baseline subclonal resistance mutations. Subclonal mutations in KRAS/NRAS/BRAF/MAP2K1/EGFR were present at baseline in 27% of patients treated with anti-EGFR across all studies (n = 129). There were a total of 171 subclonal resistance mutations among these patients, with median rMAF and absolute AF of 0.6% and 0.21%, respectively. One model for the development of treatment resistance has been that preexisting therapy-resistant subclonal mutations proliferate and ultimately outnumber treatment-sensitive cells, leading to treatment resistance and disease progression. However, in contrast to expectations, these subclones rarely expanded to become clonal at the time of progression. Only 8% of the 171 baseline subclonal mutations in known resistance genes became clonal at progression, compared with 44% that remained subclonal, and 49% that were absent at the time of progression (Fig 4C). In the 1L study, there were six total resistance mutations (spread among five subjects) at baseline with median rMAF and absolute AF of 6.51% and 1.86%, respectively. Of the six resistance mutations, one became clonal, one became subclonal, and four were absent at time of progression. In the 3L studies, there were 165 total subclonal resistance mutations (spread among 124 subjects) at baseline, with median rMAF and absolute AF of 0.6% and 0.19%, respectively. Of the 165, 7% became clonal compared with 45% that remained subclonal and 48% that were absent at time of progression.
Mutational Signatures of Acquired Mutations
Given this higher rate of passenger alterations, we explored the mutational process driving such findings. We extracted mutational signatures from the somatic ctDNA data in patients treated with anti-EGFR in the 3L setting, analyzing 2,013 acquired single-nucleotide variant mutations on a per-patient basis, and aligning them to COSMIC-defined signatures. We demonstrated that patients treated in the 3L studies were characterized by the development of SBS17b with a cosine similarity score of 0.764. In addition, the mutation signatures were associated with SBS10b (attributed in part to defects in polymerase epsilon exonuclease repair; Fig 5). We validated this method by performing a second analysis to create a single composite profile. We then calculated the cosine similarity between this single composite profile and all COSMIC-defined signatures. This confirmed the findings of SBS17b and SBS10b, and identified an additional SBS89 mutational signature, with a modest cosine similarity (0.61). There were a cluster of mutational signatures that aligned with this mutation signature, including SBS25 (postchemotherapy signature), and most notably, SBS3 (strongly associated with homologous recombination [HR] deficiency, similar to BRCA).
FIG 5.

The mutational activities of corresponding extracted de novo SNV mutational signatures in patients treated with EGFR inhibitor in the third line, after exposure to anti-EGFR therapy. Post-EGFR exposure signatures are characterized by a chemotherapy exposure signature and a polymerase epsilon exonuclease domain mutation signature, with the highest cosine similarity, labeled per COSMIC version 2.0 SNV signature database. The x-axes correspond to the trinucleotide base mutation types and y-axes indicate the percentage of mutations in the signature attributed to each mutation type. EGFR, epidermal growth factor receptor; FU, fluorouracil; SNV, single-nucleotide variant.
Shared Mechanisms of Resistance Between Anti-EGFR and Cytotoxic Chemotherapy
To investigate the finding of lower acquired resistance when combined with active 1L chemotherapy, we first generated cell lines with and without acquired cetuximab resistance (where resistance was defined as a 10-fold increase or greater in the IC50; Figs 6A and 6B). Our analysis revealed increased expression of genes known to be involved in EMT, confirming prior findings that EMT is associated with resistance after long-term exposure to anti-EGFR.27 Additionally, these models confirmed the upregulation of growth factors and TGF-β as previously reported (Appendix Fig A2, online only).27 These changes were confirmed at a protein level for E-cadherin, slug, snail, and vimentin. We then evaluated the potential cross-resistance of these lines to cytotoxic chemotherapy agents fluorouracil, oxaliplatin, and SN38 (the active metabolite of irinotecan), and demonstrated that cetuximab-resistant cell lines were insensitive or less sensitive to the cytotoxic agents than the parent cell lines (Fig 6C).
FIG 6.
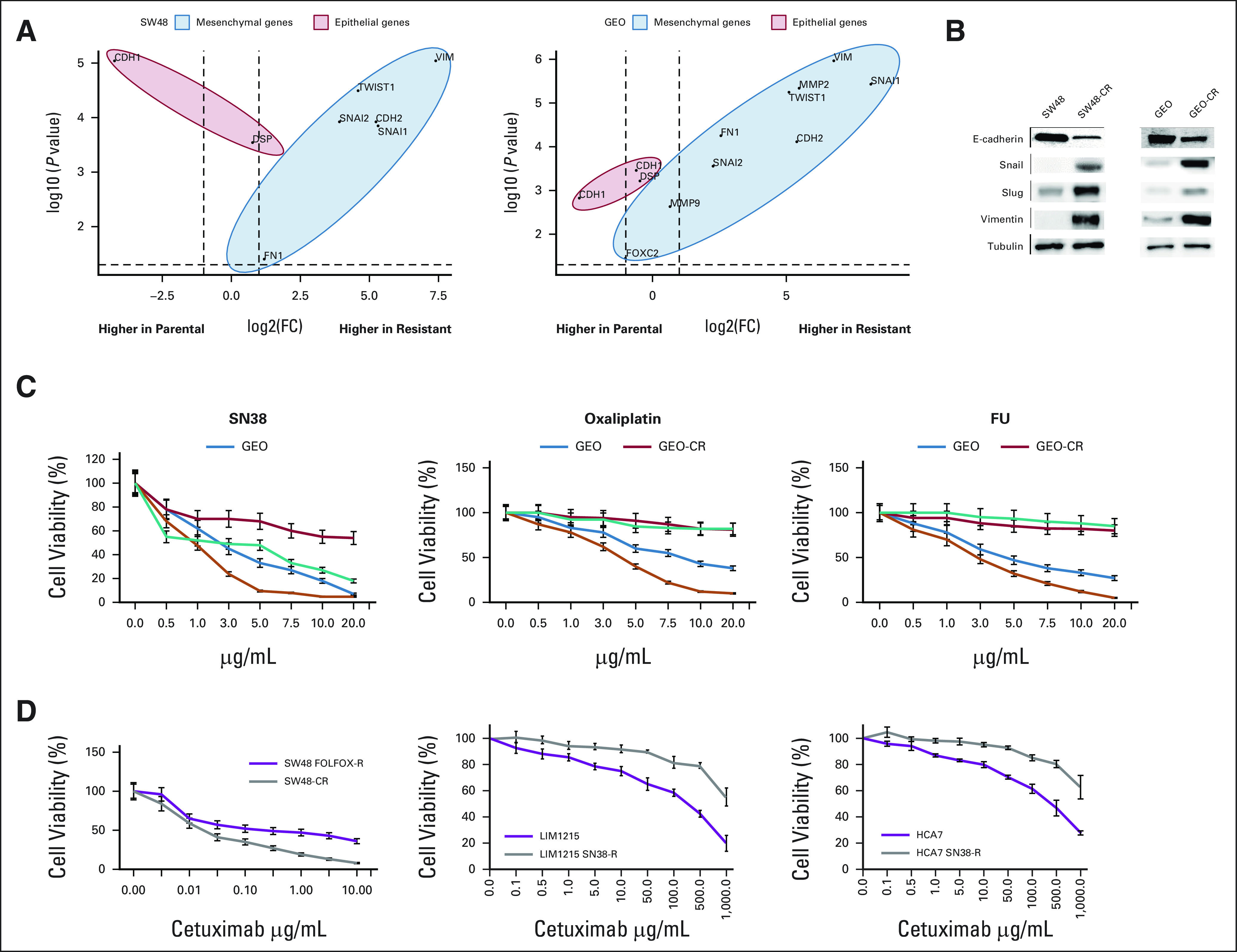
(A) Agilent microarray analyses were performed to assess baseline gene expression profile for SW48-CR versus SW48 and for GEO-CR versus GEO colorectal cancer cell lines, as described in the Materials and Methods section. Principle component analysis plot representing differentially expressed genes for GEO-CR versus GEO cells and for SW48-CR versus SW48 cells. The –log10 (P values) plotted against log2 fold change values for all the significantly differentially expressed genes at 5% false discovery rate with at least a two-fold change. The dots on the negative and positive values of x-axis represent downregulated and upregulated genes, respectively. (B) To determine whether these phenotypic changes were associated with epithelial-to-mesenchymal transition, we examined the expression of epithelial and mesenchymal marker proteins. Consistent with gene expression profile changes, cetuximab-resistant cells showed downregulation of the epithelial marker E-cadherin and upregulation of the mesenchymal markers. (C) Sensitivity of GEO, GEO-CR, SW48, and SW48-CR cells to the increasing concentrations of FU, oxaliplatin, and SN38 at concentrations ranging from 0.05 to 20 μg/mL after 72 hours of treatment and evaluated for proliferation by MTT staining. The results are the average ± standard deviation of three independent experiments each done in triplicate. (D) Sensitivity of SW48 FOLFOX-R cells, LIM1215 SN38-R cells, and HCA7 SN38-R cells to increasing concentrations of cetuximab at concentrations ranging from 0.1 to 100 μg/mL after 72 hours of treatment and evaluated for proliferation by MTT staining. The results are the average ± standard deviation of three independent experiments each done in triplicate. FC, fold change; FOLFOX, infusional fluorouracil, leucovorin, and oxaliplatin; FU, fluorouracil.
To evaluate the converse, we treated cetuximab-sensitive lines with cytotoxic chemotherapy until acquired resistance and assessed changes in cetuximab sensitivity. Cetuximab was less effective against the fluorouracil, oxaliplatin, and SN38-resistant cell lines than the parental line (Fig 6D). As RAS mutations are well established as a genomic mechanism of acquired resistance to anti-EGFR, we evaluated the impact of isogenic lines with engineered KRASG12D and did not identify any impact of the mutation on sensitivity to cytotoxic chemotherapy (Appendix Figs A3A-A3C, online only).
DISCUSSION
The growing utilization of ctDNA has allowed for a noninvasive dynamic evaluation of the complex molecular heterogeneity of mCRC. This has led to the discovery of the molecular underpinnings of acquired resistance to anti-EGFR therapy; however, little is known regarding the molecular profile of patients after 1L therapy with anti-EGFR, as most experience with anti-EGFR has been beyond the 1L setting. Acquired resistance mutations have been identified in only 35%-40% of patients,9 and other transcriptomic mechanisms of acquired resistance have been identified but have not been robustly quantified in clinical trial cohorts.27
To our knowledge, we are the first to demonstrate that patients whose tumors were treated with and responded to anti-EGFR alone in the 3L were more likely to develop acquired mutations compared with those treated in combination with chemotherapy in the 1L (46% v 9%; P < .0001). Our preclinical work is limited to a demonstration of a high level of cross-resistance between cetuximab and chemotherapy resistant models, driven by transcriptional changes that have been previously described by Woolston et al, among others.27,28 These changes include increased growth factors and EMT, a multistep morphogenetic process in which cells switch from an epithelial to a mesenchymal phenotype, and has been accepted as a mechanism of resistance to various targeted and cytotoxic therapies.29-33 Taken together, our results lead to the hypothesis that tumors may favor a single transcriptomic mechanism of resistance sufficient for both anti-EGFR and cytotoxic components of the treatment regimen in the 1L setting, in contrast to the genomic mechanisms that predominate in later lines, where cytotoxic agents are not used in combination or where the cytotoxic regimens provide less selective pressure (Appendix Fig A4, online only). Our limited in vitro data cannot exclude the possibility that there may be differences in the resistance profiles between irinotecan and oxaliplatin combinations, and should be complemented with more extensive preclinical modeling to explore this interaction of transcriptional and genetic resistance mechanisms.
The pattern of acquired alterations identified in these cohorts supports the adaptive mutability model, which was recently described by Russo and colleagues as a process by which drug resistance may emerge as a result of a transient ability to acquire therapy-resistant mutations.20,34 Russo and colleagues showed that CRC cells under therapeutic stress downregulate DNA-repair pathways, such as mismatch repair and HR, in a transient manner, allowing development of resistant clones such as RAS/EGFR ECD mutations in the case of EGFR inhibition. This altered protein expression was restored back to baseline when the therapeutic stress of the targeted therapy was removed, suggesting a transient process. In support of this work, we showed that the rate of passenger mutations increased in patients treated with anti-EGFR, implying higher rates of adaptive mutability in response to targeted therapy. Our finding that preexisting subclones rarely expanded to become clonal at the time of progression, and mostly remained subclonal or absent, also implies that there may be a transient mutational process driving resistance rather than expansion of preexisting clones. This mutational process may be a rapid adaptive process, as many patients without clinical evidence of radiographic response in the 3L studies or with short PFS still acquired detectable new resistance mutations, suggesting that acquired resistance mechanisms may be responsible for more treatment failures than previously appreciated.
We validated these clinical findings by analyzing mutational signatures after therapy with anti-EGFR. In recent years, the analysis of mutational signatures has emerged as a valuable tool with which to document the mutational processes operative in cells.35 By analyzing post-EGFR mutational activities of corresponding extracted de novo single-nucleotide variants in patients treated with EGFR in the 3L, we demonstrated that at a patient level, post-3L EGFR mutational signatures are characterized by a SBS17b signature, which has been previously reported.36-38 We also identified other signatures associated with HR deficiency and defects in polymerase epsilon exonuclease functions, consistent with some of the features of adaptive mutability previously shown preclinically with the downregulation of HR effectors such as BRCA2 and RAD51 and upregulation of error-prone polymerase repair enzymes with EGFR inhibition in CRC cell lines.20
Our work has several limitations. First, although our data set cannot address whether the variation in acquired mutations arises from line of therapy or by a combination of first exposure to chemotherapy, we believe we are justified in our conclusions as other studies with 3L chemotherapy plus anti-EGFR combinations are demonstrating acquired resistance mutations at rates similar to single-agent anti-EGFR in our analysis.8,10,12,14,39,40 Ultimately, a 1L single-agent anti-EGFR study would be required to further characterize these findings. Also, recognizing that not all resistant mutations have the same pattern of clonal evolution, we attempted to analyze absolute differences in rMAF in each gene of interest; however, we were unable to assess any differences because of small sample sizes, and preliminary data found that the line of therapy effect is larger than the between-gene effect. It is possible that additional genomic mechanisms of resistance may be present and undetected by ctDNA, because of the limits of detection with current methodologies. Also, ctDNA is still an emerging field, and standardization remains a challenge. In general, ctDNA is best positioned to assess DNA-based mechanisms of resistance, thus highlighting the need for comprehensive integrated DNA- and RNA-based mechanisms to assess a tumor's transcriptional profile and predict transcriptional activation. Finally, no gene amplifications were included in this analysis because of methodological limitations of the assays, and these too may contribute to EGFR resistance.
These findings have implications for ongoing and prospective studies of anti-EGFR rechallenge. Several recently published anti-EGFR rechallenge studies have used ctDNA to evaluate for acquired MAPK mutations before rechallenge.15,19 However, as demonstrated here, monitoring with ctDNA may not accurately detect the anti-EGFR resistance mechanisms with 1L chemotherapy. Therefore, if our findings are confirmed, ctDNA-guided rechallenge studies may not be applicable for anti-EGFR in early lines. Furthermore, unlike data on ctDNA-identified MAPK mutations, where regressions to wild-type status have been documented, it is unclear if transcriptomic resistance mechanisms will regress, and noninvasive monitoring of these processes is not currently feasible. One could hypothesize that such transcriptomic resistance mechanisms would be maintained through the use of continued cytotoxic therapy; however, this remains unknown, and further research is needed. Until these key biologic questions are resolved, the implications for optimal timing of EGFR inhibition into treatment regimens remain unclear. Comprehensive tumor characterization at progression on anti-EGFR therapies to evaluate transcriptomic and genomic changes may be required for prediction of anti-EGFR rechallenge response.
APPENDIX
FIG A1.

Acquired resistance mutations occur more frequently in the 3L setting, and in patients experiencing long PFS. (A) In the first-line setting, acquired resistance mutations occurred in 4/60 (7%) of patients with short PFS (< 10 months) and 3/17 (18%) with long PFS (≥ 10 months; P = .18). (B) In the two 3L studies combined, 59/182 patients (32%) with short PFS, and 104/181 (58%) with long PFS had acquired resistance mutations (P < .0001). *A statistically significant difference (P < .0001) between subgroups. 3L, third-line. MT, mutation; PFS, progression-free survival.
FIG A2.

Agilent microarray analyses were performed to assess baseline gene expression profile for SW48-CR versus SW48 and for GEO-CR versus GEO colorectal cancer cell lines, as described in the Materials and Methods section. Principle component analysis plot representing differentially expressed genes for GEO-CR versus GEO cells and for SW48-CR versus SW48 cells. The –log10 (P values) plotted against log2 fold change values for all the significantly differentially expressed genes at 5% false discovery rate with at least a two-fold change. The dots on the negative and positive values of x-axis represent downregulated and upregulated genes, respectively. The findings represent the upregulation of growth factors and transforming growth factor-β in resistant cells. FC, fold change.
FIG A3.

Sensitivity of SW48 and SW48 G12D cell lines to increasing concentrations of (A) FU, (B) oxaliplatin and (C) SN38 at concentrations ranging from 0.05 to 20 μg/mL after 72 hours of treatment and evaluated for proliferation by XTT staining. The results are the average ± standard deviation of three independent experiments each done in triplicate. FU, fluorouracil.
FIG A4.

Schema of mutational changes in patients treated with anti-EGFR with and without chemotherapy. (A) Upon treatment with anti-EGFR alone, tumors experience a gain of new mutations (light green) and transcriptional changes (blue) while on treatment. At the time of progression, acquired resistance mutations predominate with few anti-EGFR sensitive clones remaining (light pink). (B) Upon treatment with anti-EGFR plus a backbone chemotherapy, tumors experience a gain of new mutations and transcriptional changes while on treatment, but transcriptional resistance mutations dominate at progression. EGFR, epidermal growth factor receptor.
Ryan Sun
Consulting or Advisory Role: Boehringer Ingelheim
Research Funding: Sanofi
Jason Willis
Honoraria: Cor2Ed
Kanwal P. Raghav
Consulting or Advisory Role: AstraZeneca, Bayer, Eisai, Daiichi Sankyo, Seattle Genetics
Speakers' Bureau: Bayer
Research Funding: Bayer (Inst), Roche/Genentech (Inst), Guardant Health (Inst), Daiichi Sankyo/Astra Zeneca (Inst), HiberCell (Inst), Merck Serono (Inst)
Van K. Morris
Consulting or Advisory Role: Incyte, Boehringer Ingelheim, Axiom Healthcare Strategies, BioMedical Insights, Bicara Therapeutics
Research Funding: Bristol Myers Squibb (Inst), EMD Serono (Inst), Immatics, Pfizer (Inst), BioNTech (Inst), Bicara Therapeutics (Inst)
John P. Shen
Stock and Other Ownership Interests: Agios, Syndax
Consulting or Advisory Role: Engine Biosciences
Research Funding: Celsius Therapeutics
Eduardo Vilar
Consulting or Advisory Role: Janssen Research & Development, Recursion Pharmaceuticals, Guardant Health
Research Funding: Janssen Research & Development
Patents, Royalties, Other Intellectual Property: The University of Texas MD Anderson Cancer Center, Vilar E, Wu W, Katayama H, Hanash S, Bommi P. Methods for Prognosing, Diagnosing, and Treating Colorectal Cancer, United States, 63/152,751, 2/23/2021, Filed (Provisional), The University of Texas MD Anderson Cancer Center, Vilar E, Chang K, Wu W, Bowen CM, Sinha K. Methods and Compositions Comprising MHC Class I Peptides, United States, 63/171,137, 4/6/2021, Filed (Provisional)
Travel, Accommodations, Expenses: Janssen Research & Development
Marko Rehn
Employment: Amgen
Stock and Other Ownership Interests: Amgen
Agnes Ang
Employment: Amgen
Stock and Other Ownership Interests: Amgen (Inst)
Research Funding: Amgen
Teresa Troiani
Consulting or Advisory Role: BMS Italy, Novartis, Servier, Pfizer
Scott Kopetz
Stock and Other Ownership Interests: MolecularMatch, Lutris, Iylon, Frontier Medicines
Consulting or Advisory Role: Genentech, EMD Serono, Merck, Holy Stone Healthcare, Novartis, Lilly, Boehringer Ingelheim, Boston Biomedical, AstraZeneca/MedImmune, Bayer Health, Pierre Fabre, EMD Serono, Redx Pharma, Ipsen, Daiichi Sankyo, Natera, HalioDx, Lutris, Jacobio, Pfizer, Repare Therapeutics, Inivata, GlaxoSmithKline, Jazz Pharmaceuticals, Iylon, Xilis, AbbVie, Amal Therapeutics, Gilead Sciences, Mirati Therapeutics, Flame Biosciences, Servier, Carina Biotech, Bicara Therapeutics, Endeavor BioMedicines, Numab, Johnson & Johnson/Janssen, Genomic Health, Frontier Medicines, Replimune, Taiho Pharmaceutical
Research Funding: Sanofi, Biocartis, Guardant Health, Array BioPharma, Genentech/Roche, EMD Serono, MedImmune, Novartis, Amgen, Lilly, Daiichi Sankyo
No other potential conflicts of interest were reported.
See accompanying editorial on page 436
PRIOR PRESENTATION
Presented in part at the 2021 ASCO annual meeting, Chicago, IL, June 4-8, 2021 and the 2022 ASCO annual meeting, Chicago, IL, June 3-7, 2022.
SUPPORT
Supported by the Andrew Sabin Family Fellowship (no grant number applies) and the NIH GI SPORE Grant (Grant No. P50 CA221707) (C.M.P.) and by NIH Grant (Grant No. R01 CA187238 and P50 CA221707) (S.K.).
C.M.P., R.S., M.W., and S.N. contributed equally to this work as co-first authors.
AUTHOR CONTRIBUTIONS
Conception and design: Christine M. Parseghian, Ryan Sun, Stefania Napolitano, Jumanah Alshenaifi, Agnes Ang, Scott Kopetz
Financial support: Christine M. Parseghian, Scott Kopetz
Administrative support: Christine M. Parseghian, Scott Kopetz
Provision of study materials or patients: Christine M. Parseghian, Stefania Napolitano
Collection and assembly of data: Christine M. Parseghian, Ryan Sun, Melanie Woods, Hey Min Lee, Jumanah Alshenaifi, Shakayla Nunez, Van K. Morris, Alexey Sorokin, Preeti Kanikarla, Agnes Ang, Scott Kopetz
Data analysis and interpretation: Christine M. Parseghian, Ryan Sun, Stefania Napolitano, Jumanah Alshenaifi, Jason Willis, Shakayla Nunez, Kanwal P. Raghav, Van K. Morris, John P. Shen, Madhulika Eluri, Eduardo Vilar, Marko Rehn, Agnes Ang, Teresa Troiani, Scott Kopetz
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Resistance Mechanisms to Anti–Epidermal Growth Factor Receptor Therapy in RAS/RAF Wild-Type Colorectal Cancer Vary by Regimen and Line of Therapy
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/authors/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Ryan Sun
Consulting or Advisory Role: Boehringer Ingelheim
Research Funding: Sanofi
Jason Willis
Honoraria: Cor2Ed
Kanwal P. Raghav
Consulting or Advisory Role: AstraZeneca, Bayer, Eisai, Daiichi Sankyo, Seattle Genetics
Speakers' Bureau: Bayer
Research Funding: Bayer (Inst), Roche/Genentech (Inst), Guardant Health (Inst), Daiichi Sankyo/Astra Zeneca (Inst), HiberCell (Inst), Merck Serono (Inst)
Van K. Morris
Consulting or Advisory Role: Incyte, Boehringer Ingelheim, Axiom Healthcare Strategies, BioMedical Insights, Bicara Therapeutics
Research Funding: Bristol Myers Squibb (Inst), EMD Serono (Inst), Immatics, Pfizer (Inst), BioNTech (Inst), Bicara Therapeutics (Inst)
John P. Shen
Stock and Other Ownership Interests: Agios, Syndax
Consulting or Advisory Role: Engine Biosciences
Research Funding: Celsius Therapeutics
Eduardo Vilar
Consulting or Advisory Role: Janssen Research & Development, Recursion Pharmaceuticals, Guardant Health
Research Funding: Janssen Research & Development
Patents, Royalties, Other Intellectual Property: The University of Texas MD Anderson Cancer Center, Vilar E, Wu W, Katayama H, Hanash S, Bommi P. Methods for Prognosing, Diagnosing, and Treating Colorectal Cancer, United States, 63/152,751, 2/23/2021, Filed (Provisional), The University of Texas MD Anderson Cancer Center, Vilar E, Chang K, Wu W, Bowen CM, Sinha K. Methods and Compositions Comprising MHC Class I Peptides, United States, 63/171,137, 4/6/2021, Filed (Provisional)
Travel, Accommodations, Expenses: Janssen Research & Development
Marko Rehn
Employment: Amgen
Stock and Other Ownership Interests: Amgen
Agnes Ang
Employment: Amgen
Stock and Other Ownership Interests: Amgen (Inst)
Research Funding: Amgen
Teresa Troiani
Consulting or Advisory Role: BMS Italy, Novartis, Servier, Pfizer
Scott Kopetz
Stock and Other Ownership Interests: MolecularMatch, Lutris, Iylon, Frontier Medicines
Consulting or Advisory Role: Genentech, EMD Serono, Merck, Holy Stone Healthcare, Novartis, Lilly, Boehringer Ingelheim, Boston Biomedical, AstraZeneca/MedImmune, Bayer Health, Pierre Fabre, EMD Serono, Redx Pharma, Ipsen, Daiichi Sankyo, Natera, HalioDx, Lutris, Jacobio, Pfizer, Repare Therapeutics, Inivata, GlaxoSmithKline, Jazz Pharmaceuticals, Iylon, Xilis, AbbVie, Amal Therapeutics, Gilead Sciences, Mirati Therapeutics, Flame Biosciences, Servier, Carina Biotech, Bicara Therapeutics, Endeavor BioMedicines, Numab, Johnson & Johnson/Janssen, Genomic Health, Frontier Medicines, Replimune, Taiho Pharmaceutical
Research Funding: Sanofi, Biocartis, Guardant Health, Array BioPharma, Genentech/Roche, EMD Serono, MedImmune, Novartis, Amgen, Lilly, Daiichi Sankyo
No other potential conflicts of interest were reported.
REFERENCES
- 1. Siegel RL, Miller KD, Goding Sauer A, et al. Colorectal cancer statistics, 2020. CA Cancer J Clin. 2020;70:145–164. doi: 10.3322/caac.21601. [DOI] [PubMed] [Google Scholar]
- 2. Nielsen DL, Palshof JA, Larsen FO, et al. A systematic review of salvage therapy to patients with metastatic colorectal cancer previously treated with fluorouracil, oxaliplatin and irinotecan +/- targeted therapy. Cancer Treat Rev. 2014;40:701–715. doi: 10.1016/j.ctrv.2014.02.006. [DOI] [PubMed] [Google Scholar]
- 3. Cremolini C, Loupakis F, Antoniotti C, et al. FOLFOXIRI plus bevacizumab versus FOLFIRI plus bevacizumab as first-line treatment of patients with metastatic colorectal cancer: Updated overall survival and molecular subgroup analyses of the open-label, phase 3 TRIBE study. Lancet Oncol. 2015;16:1306–1315. doi: 10.1016/S1470-2045(15)00122-9. [DOI] [PubMed] [Google Scholar]
- 4. Douillard JY, Oliner KS, Siena S, et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med. 2013;369:1023–1034. doi: 10.1056/NEJMoa1305275. [DOI] [PubMed] [Google Scholar]
- 5. Grothey A, Van Cutsem E, Sobrero A, et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): An international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381:303–312. doi: 10.1016/S0140-6736(12)61900-X. [DOI] [PubMed] [Google Scholar]
- 6. Mayer RJ, Van Cutsem E, Falcone A, et al. Randomized trial of TAS-102 for refractory metastatic colorectal cancer. N Engl J Med. 2015;372:1909–1919. doi: 10.1056/NEJMoa1414325. [DOI] [PubMed] [Google Scholar]
- 7. Pessino A, Artale S, Sciallero S, et al. First-line single-agent cetuximab in patients with advanced colorectal cancer. Ann Oncol. 2008;19:711–716. doi: 10.1093/annonc/mdm516. [DOI] [PubMed] [Google Scholar]
- 8. Misale S, Yaeger R, Hobor S, et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature. 2012;486:532–536. doi: 10.1038/nature11156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Strickler JH, Loree JM, Ahronian LG, et al. Genomic landscape of cell-free DNA in patients with colorectal cancer. Cancer Discov. 2018;8:164–173. doi: 10.1158/2159-8290.CD-17-1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Arena S, Bellosillo B, Siravegna G, et al. Emergence of multiple EGFR extracellular mutations during cetuximab treatment in colorectal cancer. Clin Cancer Res. 2015;21:2157–2166. doi: 10.1158/1078-0432.CCR-14-2821. [DOI] [PubMed] [Google Scholar]
- 11. Pietrantonio F, Vernieri C, Siravegna G, et al. Heterogeneity of acquired resistance to anti-EGFR monoclonal antibodies in patients with metastatic colorectal cancer. Clin Cancer Res. 2017;23:2414–2422. doi: 10.1158/1078-0432.CCR-16-1863. [DOI] [PubMed] [Google Scholar]
- 12. Siravegna G, Mussolin B, Buscarino M, et al. Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat Med. 2015;21:827. doi: 10.1038/nm0715-827b. [DOI] [PubMed] [Google Scholar]
- 13. Van Emburgh BO, Arena S, Siravegna G, et al. Acquired RAS or EGFR mutations and duration of response to EGFR blockade in colorectal cancer. Nat Commun. 2016;7:13665. doi: 10.1038/ncomms13665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Siena S, Sartore-Bianchi A, Garcia-Carbonero R, et al. Dynamic molecular analysis and clinical correlates of tumor evolution within a phase II trial of panitumumab-based therapy in metastatic colorectal cancer. Ann Oncol. 2018;29:119–126. doi: 10.1093/annonc/mdx504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cremolini C, Rossini D, Dell'Aquila E, et al. Rechallenge for patients with RAS and BRAF wild-type metastatic colorectal cancer with acquired resistance to first-line cetuximab and irinotecan: A phase 2 single-arm clinical trial. JAMA Oncol. 2019;5:343–350. doi: 10.1001/jamaoncol.2018.5080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Francipane MG, Lagasse E. mTOR pathway in colorectal cancer: an update. Oncotarget. 2014;5:49–66. doi: 10.18632/oncotarget.1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Johnson B, Loree JM, Morris VK, et al. Activity of EGFR inhibition in atypical (non-V600E) BRAF-mutated metastatic colorectal cancer. J Clin Oncol. 2019;37(suppl 4) abstr 596. [Google Scholar]
- 18. Parseghian CM, Loree JM, Morris VK, et al. Anti-EGFR resistant clones decay exponentially after progression: Implications for anti-EGFR re-challenge. Ann Oncol. 2019;30:243–249. doi: 10.1093/annonc/mdy509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sartore-Bianchi A, Pietrantonio F, Lonardi S, et al. Phase II study of anti-EGFR rechallenge therapy with panitumumab driven by circulating tumor DNA molecular selection in metastatic colorectal cancer: The CHRONOS trial. J Clin Oncol. 2021;39(suppl 15) abstr 3506. [Google Scholar]
- 20. Russo M, Crisafulli G, Sogari A, et al. Adaptive mutability of colorectal cancers in response to targeted therapies. Science. 2019;366:1473–1480. doi: 10.1126/science.aav4474. [DOI] [PubMed] [Google Scholar]
- 21. Hobor S, Van Emburgh BO, Crowley E, et al. TGFalpha and amphiregulin paracrine network promotes resistance to EGFR blockade in colorectal cancer cells. Clin Cancer Res. 2014;20:6429–6438. doi: 10.1158/1078-0432.CCR-14-0774. [DOI] [PubMed] [Google Scholar]
- 22. Buck E, Eyzaguirre A, Barr S, et al. Loss of homotypic cell adhesion by epithelial-mesenchymal transition or mutation limits sensitivity to epidermal growth factor receptor inhibition. Mol Cancer Ther. 2007;6:532–541. doi: 10.1158/1535-7163.MCT-06-0462. [DOI] [PubMed] [Google Scholar]
- 23. Leary RJ, Sausen M, Kinde I, et al. Detection of chromosomal alterations in the circulation of cancer patients with whole-genome sequencing. Sci Transl Med. 2012;4:162ra154. doi: 10.1126/scitranslmed.3004742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sabari JK, Offin M, Stephens D, et al. A prospective study of circulating tumor DNA to guide matched targeted therapy in lung cancers. J Natl Cancer Inst. 2019;111:575–583. doi: 10.1093/jnci/djy156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Thierry AR, Pastor B, Jiang ZQ, et al. Circulating DNA demonstrates convergent evolution and common resistance mechanisms during treatment of colorectal cancer. Clin Cancer Res. 2017;23:4578–4591. doi: 10.1158/1078-0432.CCR-17-0232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1) Eur J Cancer. 2009;45:228–247. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 27. Woolston A, Khan K, Spain G, et al. Genomic and transcriptomic determinants of therapy resistance and immune landscape evolution during anti-EGFR treatment in colorectal cancer. Cancer Cell. 2019;36:35–50.e9. doi: 10.1016/j.ccell.2019.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Garvey CM, Lau R, Sanchez A, et al. Anti-EGFR therapy induces EGF secretion by cancer-associated fibroblasts to confer colorectal cancer chemoresistance. Cancers (Basel) 2020;12:1393. doi: 10.3390/cancers12061393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2:442–454. doi: 10.1038/nrc822. [DOI] [PubMed] [Google Scholar]
- 30. Rosano L, Cianfrocca R, Spinella F, et al. Acquisition of chemoresistance and EMT phenotype is linked with activation of the endothelin A receptor pathway in ovarian carcinoma cells. Clin Cancer Res. 2011;17:2350–2360. doi: 10.1158/1078-0432.CCR-10-2325. [DOI] [PubMed] [Google Scholar]
- 31. Rho JK, Choi YJ, Lee JK, et al. Epithelial to mesenchymal transition derived from repeated exposure to gefitinib determines the sensitivity to EGFR inhibitors in A549, a non-small cell lung cancer cell line. Lung Cancer. 2009;63:219–226. doi: 10.1016/j.lungcan.2008.05.017. [DOI] [PubMed] [Google Scholar]
- 32. Kajiyama H, Shibata K, Terauchi M, et al. Chemoresistance to paclitaxel induces epithelial-mesenchymal transition and enhances metastatic potential for epithelial ovarian carcinoma cells. Int J Oncol. 2007;31:277–283. [PubMed] [Google Scholar]
- 33. Hiscox S, Jiang WG, Obermeier K, et al. Tamoxifen resistance in MCF7 cells promotes EMT-like behaviour and involves modulation of beta-catenin phosphorylation. Int J Cancer. 2006;118:290–301. doi: 10.1002/ijc.21355. [DOI] [PubMed] [Google Scholar]
- 34. Hata AN, Niederst MJ, Archibald HL, et al. Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat Med. 2016;22:262–269. doi: 10.1038/nm.4040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Petljak M, Alexandrov LB, Brammeld JS, et al. Characterizing mutational signatures in human cancer cell lines reveals episodic APOBEC mutagenesis. Cell. 2019;176:1282–1294.e20. doi: 10.1016/j.cell.2019.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Woolston A, Barber LJ, Griffiths B, et al. Mutational signatures impact the evolution of anti-EGFR antibody resistance in colorectal cancer. Nat Ecol Evol. 2021;5:1024–1032. doi: 10.1038/s41559-021-01470-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lee CA, Abd-Rabbo D, Reimand J. Functional and genetic determinants of mutation rate variability in regulatory elements of cancer genomes. Genome Biol. 2021;22:133. doi: 10.1186/s13059-021-02318-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pleasance E, Titmuss E, Williamson L, et al. Pan-cancer analysis of advanced patient tumors reveals interactions between therapy and genomic landscapes. Nat Cancer. 2020;1:452–468. doi: 10.1038/s43018-020-0050-6. [DOI] [PubMed] [Google Scholar]
- 39. Raghav KPS, Ou F-S, Venook AP, et al. Circulating tumor DNA dynamics on front-line chemotherapy with bevacizumab or cetuximab in metastatic colorectal cancer: A biomarker analysis for acquired genomic alterations in CALGB/SWOG 80405 (Alliance) randomized trial. J Clin Oncol. 2022;40(suppl 4) abstr 193. [Google Scholar]
- 40. Montagut C, Dalmases A, Bellosillo B, et al. Identification of a mutation in the extracellular domain of the epidermal growth factor receptor conferring cetuximab resistance in colorectal cancer. Nat Med. 2012;18:221–223. doi: 10.1038/nm.2609. [DOI] [PubMed] [Google Scholar]