

Abstract
The clinical effectiveness of adrenocorticotropin in inducing remission of steroid-resistant nephrotic syndrome points to a steroidogenic-independent anti-proteinuric activity of melanocortins. However, which melanocortin receptors (MCR) convey this beneficial effect and if systemic or podocyte-specific mechanisms are involved remain uncertain. In vivo, wild-type (WT) mice developed heavy proteinuria and kidney dysfunction following Adriamycin insult, concomitant with focal segmental glomerulosclerosis (FSGS) and podocytopathy, marked by loss of podocin and synaptopodin, podocytopenia and extensive foot process effacement on electron microscopy. All these pathologic findings were prominently attenuated by NDP-MSH, a potent non-steroidogenic pan-MCR agonist. Surprisingly, MC1R deficiency in MC1R-null mice barely affected the severity of Adriamycin-elicited injury. Moreover, the beneficial effect of NDP-MSH was completely preserved in MC1R-null mice, suggesting that MC1R is likely non-essential for the protective action. A direct podocyte effect seems to contribute to the beneficial effect of NDP-MSH, because Adriamycin-inflicted cytopathic signs in primary podocytes prepared from WT mice were all mitigated by NDP-MSH, including apoptosis, loss of podocyte markers, de novo expression of the podocyte injury marker desmin, actin cytoskeleton derangement and podocyte hypermotility. Consistent with in vivo findings, the podoprotective activity of NDP-MSH was fully preserved in MC1R-null podocytes. Mechanistically, MC1R expression was predominantly distributed to glomerular endothelial cells in glomeruli but negligibly noted in podocytes in vivo and in vitro, suggesting that MC1R signaling is unlikely involved in direct podocyte protection. Ergo, melanocortin therapy protects against podocyte injury and ameliorates proteinuria and glomerulopathy in experimental FSGS, at least in part, via a podocyte-specific non-MC1R-mediated melanocortinergic signaling.
Keywords: glomerular disease, adrenocorticotropic hormone, podocytes, apoptosis, cytoskeleton
Introduction
Focal segmental glomerulosclerosis (FSGS) is a common cause of nephrotic syndrome in adults and ranks among the most frequently encountered primary glomerular disease causing end-stage renal disease (ESRD) [1, 2]. FSGS is a histologic lesion rather than a specific disease. According to the etiology, it can be classified into primary, secondary and genetic forms of FSGS. In the past several decades, the incidence and prevalence of idiopathic FSGS have been progressively increasing. It is estimated that FSGS affects approximately 40,000 people in the United States with a similar prevalence in Europe, and more than 5400 patients in the U.S. are diagnosed with FSGS every year with new cases on the rise [3, 4]. While the cause of primary FSGS still remains largely unknown, the pathogenesis of renal lesions of FSGS may be multifactorial involving both immune and non-immune mediated mechanisms as well as genetic predisposition, epigenetic modifiers and environmental cues[5]. Nevertheless, it has been widely accepted that the pathologic basis of FSGS lesion is injury and dysfunction of the glomerular podocytes, a key structural constituent of the glomerular filtration barrier that ensures retention of essential plasma proteins in blood while the filtrate is passed on as urine [1, 6]. Because the exact molecular mechanism responsible for FSGS is unclear, clinical treatments are currently not pathogenesis-targeted but mostly empirical, confined largely to the use of angiotensin blockades and immunosuppressants like corticosteroids, alkylating agents, calcineurin inhibitors and others [7]. Such regimens are, however, of limited utility with unsatisfying responsiveness and severe adverse effects. There is a pressing need to develop novel therapeutic approaches that are able to both improve the remission rate of FSGS and reduce its relapse. A growing body of evidence recently suggests that melanocortins may fulfill this unmet need [8].
Melanocortins are a group of peptide hormones released upon stress by the anterior pituitary gland, including adrenocorticotropic hormone (ACTH) and α,β,γ-MSH, and exert biological functions via their cognate receptors (MC1~5R) expressed in many peripheral organ systems [9–13]. As the only melanocortin peptide with steroidogenic activity via agonizing MC2R [10, 12], ACTH has been one of the very few FDA-approved first-line treatments for nephrotic glomerular disease since the 1950s and has a demonstrable anti-edematous, proteinuria-reducing and renoprotective effect [14, 15]. However, these beneficial effects of ACTH have long been believed to be mediated indirectly by glucocorticoids secondary to its steroidogenic activity[16], though the FDA-approved ACTH for treating nephrosis is a propriety mixture (Acthar gel) of authentic, native ACTH1-39 and a variety of degradation products of proopiomelanocortin, such as melanocortins. As such, ACTH has been replaced by synthetic corticosteroids for clinical use. Recently, more and more studies revealed that ACTH is still effective in patients with steroid resistant glomerular diseases like FSGS [17–21]. entailing a steroidogenic-independent anti-proteinuric activity of the melanocortinergic pathway. However, which melanocortin receptor (MCR) conveys this beneficial effect is controversial. Moreover, it remains uncertain if a systemic or podocyte-specific mechanism is involved. It is critical to address these issues for translating melanocortin therapy into clinical practice, because existing melanocortinergic drugs, like ACTH and α-MSH, are pan MCR agonists, and despite a likely beneficial effect in glomerulopathies, may cause considerable side effects due to non-selective activation of all MCRs, including Cushing’s syndrome, hyperglycemia, aldosteronism, hypertension, increased risk for melanoma, anorexia, mood and sleep disorders and others [8]. In addition, existing formulation of ACTH are potentially antigenic and may trigger the generation of neutralizing antibodies that may undermine the therapeutic efficacy [22]. Identification of the MCR conveying the protective effect in glomerular diseases like FSGS will pave the way for developing highly selective small molecule agonists that are able to avoid the off-target adverse activities and minimize the antigenic issues.
Recently there is evidence suggesting that MC1R is expressed on podocytes and its activation mediates the anti-proteinuric and renoprotective effect of melanocortin therapy [23]. However, this is in sharp conflict with the latest clinical evidence indicating that MC1R is dispensable for the beneficial effect of melanocortins in glomerular disease [24]. Indeed, MC1R-mutant red-haired patients with steroid-resistant FSGS responded still very well to ACTH therapy and were able to achieve complete remission of proteinuria [24]. Therefore, whether MC1R is exerting a podocyte protective and anti-proteinuric effect is highly controversial. Here, by using NDP-MSH, a potent non-steroidogenic pan-MCR agonist, the present study tested in a model of FSGS induced by Adriamycin (ADR) whether the steroidogenic-independent melanocortinergic pathway is protective in experimental FSGS. To avoid the confounding effect of steroidogenesis, no parallel experiments with the FDA-approved ACTH (e.g. Acthar gel) or recombinant human ACTH were performed. Furthermore, by harnessing the naturally occurring MC1R-null mice, the present study also attempted to validate if MC1R is responsible for the anti-proteinuric and podocyte protective effect.
Materials and Methods
Animal experiment design
Animal studies were carried out at the Rhode Island Hospital Central Animal Facility and were approved by the Rhode Island Hospital Institutional Animal Care and Use Committee. All animal experiments conformed to the United States Department of Agriculture regulations and the National Institutes of Health guidelines for humane care and use of laboratory animals. Mc1rE/e mice on a C57BL/6 genetic background were originally purchased from the Jackson Laboratory (Bar Harbor, ME, U.S.A) and maintained as previously described[24]. They were bred to generate the recessive yellow Mc1re/e mice that are null for MC1R as well as the wild-type (WT) control littermates. The mouse model of ADR-induced FSGS was established by a single tail-vein injection of ADR (25 mg/kg body wt, Sigma-Aldrich, St. Louis, MO) [25]. From day 0 to day 5, mice were given two daily intraperitoneal injections of 1 ml of a glucose-electrolyte solution to prevent weight loss due to low appetite. MC1R-null and WT male mice were randomly assigned to the following groups to receive different treatments: (1)Control groups: MC1R-null or WT mice received saline (0.1 ml) treatment as a single tail-vein injection; (2) ADR groups: MC1R-null or WT mice received ADR (25 mg/kg)via a single tail-vein injection and co-treatment with vehicle; (3) ADR+NDP-MSH groups: MC1R-null or WT mice received subcutaneous injection of NDP-MSH (0.6 μmol/kg wt, GL Biochem Ltd., Boston, MA, U.S.A) [26] 6 h before and alternative day (on day 2, 4, 6) after the single tail vein injection of ADR (25 mg/kg). On day 0, 3, 5 and 7 after diverse treatments, spot urine was collected. Throughout the animal experiments, no anesthetics were required and used. All animals were killed on day 7, and kidneys resected and serum collected for further examination.
Urinary protein analysis and creatinine measurements
To discern the composition of urine proteins, urine samples were processed for sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) followed by staining with Coomassie Brilliant Blue (Sigma-Aldrich). The concentrations of albumin in urine samples were measured by using mouse albumin ELISA quantitation kit (Bethyl Laboratories Inc., Montgomery, TX, U.S.A). Urine and serum creatinine levels were measured using a creatinine assay kit (Biovision, Mountain View, CA, U.S.A) according to the manufacturer’s instructions. Urine albumin excretion was expressed as urine albumin-to-creatinine ratios (mg/mg).
Histologic studies
Formalin fixed kidneys were embedded in paraffin and prepared into sections (3μm thick). For general histologic analysis, sections were processed for periodic acid-Schiff (PAS) staining. A semiquantitative morphometric glomerular damage score was used to evaluate the degree of glomerular injury, which was featured by one or more of the following histological signs: podocytic swelling and vacuolization, glomerular synechiae, glomerular capillary congestion, collapse and/or obliteration accompanied by hyaline material and/or mesangial matrix expansion. Ten glomeruli per animal were counted and graded as 0, 1, 2, 3, or 4 respectively, according to 0, 75% injury changes across a kidney section. The scores were the averagedto generate a composite glomerular damage score [27]. Under low magnification, the number of protein casts in 10 randomly selected non-overlapping fields from the tubulointerstitia was counted and averaged as the protein cast score [28]. The morphologic features of all sections were assessed by a single observer in a blinded manner.
Electron microscopy
The kidney cortical specimens were cut into small pieces (1mm3), fixed with 2.5% glutaraldehyde in phosphate buffered saline, pH 7.4, and embedded in Epon 812 (Polysciences, Warrington, PA, U.S.A). Then ultrathin sections were cut and mounted on a copper grid. Conventional electron micrographs were obtained using an EM-10 microscope (Zeiss) operated at 80 kV with absolute magnifications of 8000 or 20,000. For counting the number of podocyte foot processes, 6 random electron microscopic fields of glomeruli per animal were examined. The morphologic features were assessed by a single observer in a blinded manner.
Western immunoblot analysis
Cultured cells were lysed and kidney tissues were homogenized in radioimmunoprecipitation assay buffer containing protease inhibitors. Total cell lysates or subcellular protein fractions were prepared and processed for immunoblot analysis as described previously [29]. Antibodies against the following molecules were used as primary antibodies to probe blots: synaptopodin (1:1000, PROGEN Biotechnik GmbH, Heidelberg, Germany), desmin (1:1000, Santa Cruz Biotechnology, CA), cleaved caspase-3 (1:2000, Cell Signaling Technology, Danvers, MA, U.S.A) and GAPDH (1:5000, Santa Cruz Biotechnology, Santa Cruz, CA). For immunoblot analysis, the integrated pixel density of bands was determined using the Image J software.
Glomerular isolation and primary culture of glomerular podocytes
Isolation of glomeruli from the WT and MC1R-null mice was performed as described previously [24], with minor modifications. For isolation of primary podocyte, all procedures were performed in a ventilated dissection hood under clean-contaminated condition. Mice were euthanized and the kidney was perfused with 5 ml of phosphate-buffered saline containing 8×107 Dynabeads M-450 (Dynal Biotech ASA, Oslo, Norway). The kidneys were then cut into 1mm3 pieces and then digested in a digestion buffer containing 1 mg/ml of collagenase A and 100 U/ml of DNase I for 30 min at 37°C. The tissue was then pressed gently through a 100μm cell strainer (Falcon, Bedford, MA, U.S.A) and washed with ice-cold sterilized phosphate-buffered saline (PBS). Washed cells were centrifuged for 5 min at 200g, and supernatants were aspirated and discarded. Cell pellets were resuspended in ice-cold PBS and glomeruli-containing Dynabeads were gathered using a magnetic particle concentrator. An aliquot (1:1500) of the glomerular isolate was visualized under a microscope to ensure that the sample contained <5 tubular fragments per ×200 field. The majority of the isolated glomeruli (80%) were decapsulated, which was similar to what had been reported previously [30]. During the procedure, kidney tissues were kept at 4°C except for the collagenase digestion at 37°C.
The enriched glomeruli were plated on collagen type I-coated dishes at 37°C in RPMI 1640 medium (Life Technologies, Grand Island, NY, USA) with 10% fetal bovine serum (Life Technologies), 0.075% sodium pyruvate (Sigma), 100U/ml penicillin, 100μg/ml streptomycin (Life Technologies) in a humidified incubator with 5% CO2. Subculture of primary podocytes was performed by detaching the glomerular cells with 0.25% trypsin-EDTA (Invitrogen, Carlsbad, CA, USA), followed by sieving through a 40 μm cell strainer (Falcon), and culture on collagen type I-coated dishes as reported before [31]. Podocytes of passages 1 or 2 were characterized by the expression of multiple podocyte specific markers and used in all experiments. Confluent primary podocytes were pretreated with 10−7M NDP-MSH or vehicle for 30 min and then stimulated with with ADR (0.25μg/ml; Sigma-Aldrich) in PBS or vehicle for 24 h.
Cell migration assay
Confluent monolayers of primary podocytes were scraped with a 10μl pipette after different treatments and visualized at 24h using an inverted microscope. Phase contrast micrographs were obtained at 0h and 24h after scratching. Data were calculated and expressed as the percent area closure, which represents the percent difference in wound area after 24 h and were analyzed using Image J version 32 (NIH, Bethesda, MD) image processing program [32].
Albumin influx assay
A paracellular permeability influx assay was used to evaluate the filtration barrier function of podocyte monolayers [33, 34]. Primarily cultured podocytes were seeded into the collagen coated transwell filters (3μm pore, Corning, NY, U.S.A) in the top chamber and grew to confluence. After different treatments for 24h, podocytes were washed twice with PBS supplemented with 1mmol/l MgCl2 and CaCl2. The top chambers were then refilled with 0.15ml RPMI 1640 and the bottom chambers were refilled with 1ml RPMI 1640 supplemented with 40mg/ml of Bovine Serum Albumin (BSA) and then incubated at 37°C. At 1h and 3h, 20μl medium in the top chambers were collected and prepared for albumin concentration assay by using a bicinchoninic acid protein assay kit.
Cellular viability assay
Podocyte viability was estimated by the tetrazolium (MTT) assay. The assay is based on the principle that active dehydrogenases of living cells can convert the tetrazolium ring, which is water soluble and gives yellow color, into insoluble formazan crystals, which can be dissolved using acidified isopropanol and are purple in color, whereas dead cells cannot make the conversion. Thus, the concentration of the dye is proportional to the percentage of viable cells. Briefly, following ADR and/or NDP-MSH treatment for 24 h, primary podocytes were treated with tetrazolium for 1 h. After incubation, formazan crystals were dissolved by adding equal volumes of acidified isopropanol. After the crystals were completely dissolved, absorbance was measured spectrophotometrically at 570 nm. Absorbance values obtained were employed to calculate the relative cell viability.
Cell apoptosis analysis
Cultured cells or frozen tissue sections were fixed with 4% paraformaldehyde in phosphate-buffered saline and processed for staining with a TdT-mediated dUTP nick end labeling (TUNEL) kit (Promega, Madison, WI, U.S.A) [35]. TUNEL positive cells were visualized by using the fluorescence microscope (BX43, Olympus, Tokyo, Japan).
RNA interference
B16F10 melanoma cells were cultured as previously described [22] and maintained in Dulbecco’s Modified Eagle’s Medium supplemented with 10% fetal bovine serum. Cells were plated at 50% confluence in the medium containing 5% fetal bovine serum for 24 h and then underwent RNA interference (RNAi). Predesigned siRNA duplex was designed by the BLOCK-iT™ RNAi Designer (Invitrogen). MC1R-specific siRNA sequence (5′-UGCUGGCACUCAUGGCGAUUCUGUAtt-3′) was designed according to the complete coding sequence of murine MC1R gene (GenBank accession no. AB973126.1). In addition, a scrambled siRNA sequence (5′-GCGAGUAGCGCUAGGAAGUtt-3′) without sequence similarity to any known gene sequences from mouse, rat, or human was designed as control for RNAi. RNAi was performed by using the lipofectamine (Invitrogen)-mediated transient transfection with siRNA. Cells were processed for immunblot analysis or immunostaining 24 h after transfection [36].
Immunofluorescent staining
Cultured cells or frozen kidney cryostat sections were fixed and processed for fluorescent staining. Samples were stained with primary antibodies against synaptopodin (1:100, PROGEN Biotechnik GmbH, Heidelberg, Germany), desmin (1:100, Santa Cruz Biotechnology, CA), Wilms’ tumor 1 (WT-1; 1:100, Santa Cruz Biotechnology, CA), MC1R (Alomone Labs, Jerusalem, Israel) , or CD31 (Cell Signaling Technology) overnight at 4°C, followed by Alexa Fluor-conjugated secondary antibodies (Invitrogen). As a negative control, primary antibodies were replaced by preimmune serum and no specific staining was noted. F-actin in podocytes was stained by rhodamine phalloidin (Molecular Probes, OR, U.S.A). Finally, some samples were counterstained with propidium iodide or 4’,6-diamidino-2-phenylindole, mounted with Vectashield mounting medium (Vector Laboratories, Burlingame, CA, U.S.A) and visualized by fluorescence microscope (BX43, Olympus, Tokyo,Japan). ImageJ software was used for post processing of the images (scaling, merging, and colocalization analysis).
Statistical analyses
All results are expressed as the means±SD. Statistical analyses were performed by SPSS 22 (IBM Corporation, Armonk, New York, U.S.A). All data are expressed as mean ± SD. Unless otherwise indicated, all experimental observations were repeated six times. Statistical analysis of the data from multiple groups was performed by repeated measures ANOVA followed by Fisher’s Least Significant Difference (LSD) tests. Data from two groups were compared by Student’s t-test. P<0.05 was considered significant.
Results
WT and MC1R-null mice are equally protected by NDP-MSH therapy against the ADR elicited albuminuria and glomerular injury.
The ADR nephropathy in mice is a standard model for human nephrotic syndrome and recapitulates key features of podocytopathy and FSGS in humans with high fidelity, including podocyte foot process effacement, massive proteinuria, progressive glomerulosclerosis and other renal lesions of FSGS [37]. Admittedly, because of the etiologic complexity of human FSGS, this model may not exactly reproduce the same pathogenic pathways that are acting in primary or other forms of FSGS lesions. In our study, WT and MC1R-null mice were injured with a single tail vein injection of ADR 6 h after subcutaneous NDP-MSH or vehicle treatment and were subsequently treated daily with NDP-MSH or vehicle. Shown in Figure 1A, urine protein was barely detected in non-ADR-injured WT or MC1R-null mice by PAGE of urine followed by staining with Coomassie Brilliant Blue, but started to appear as early as 3 days after ADR injury and was substantially evident on day 7. The molecular size of the major component of urine protein is line with that of the standard albumin, suggesting the presence of massive albuminuria. NDP-MSH treatment markedly mitigated the ADR induced albumin excretion in urine in both WT and MC1R-null mice. Quantification of albumin levels in urine with creatinine adjustment revealed that ADR injury caused heavy albuminuria that peaked on days 5 to 7 and was comparable in WT and MC1R-null mice (Figure 1B), implying that loss of constitutive MC1R function did not worsen the injurious effect of ADR. NDP-MSH treatment substantially attenuated albuminuria in WT and MC1R-null mice to a similar extent and resulted in approximately 40% reduction in urine albumin to creatinine ratios (on day 5, the ACR reduction in WT and MC1R-null mice is 41.55% and 36.75%, and on day 7, the ACR reduction in WT and MC1R-null mice is 37.16% and 37.20%), denoting that NDP-MSH has a potent anti-proteinuric effect, and that MC1R is unlikely essential for this beneficial effect. Massive proteinuria is indicative of glomerulopathy. Indeed, as shown by periodic acid-Schiff staining in Figure 1C, following ADR injury, WT and MC1R-null mice both developed prominent glomerular injury, featured by glomerular synechiae, glomerular capillary congestion, collapse and/or obliteration accompanied by hyaline material, mesangial matrix expansion, and podocytic swelling and vacuolization. This was associated with prominent focal protein casts in the tubulointerstitia in both WT and MC1R-null mice, in consistency with the manifestation of heavy proteinuria. NDP-MSH treatment markedly ameliorated the glomerular and tubulointerstitial lesions induced by ADR. Semi-quantitative morphometric analysis demonstrated that WT and MC1R-null mice developed comparable glomerular and interstitial injury after ADR injury and responded equally to NDP-MSH treatment, as reflected by a similar glomerular injury score (Figure 1D) and protein cast score (Figure 1E). Moreover, WT and MC1R-null mice developed comparable kidney dysfunction after ADR injury, and responded equally to NDP-MSH treatment, as reflected by a similar change in serum creatinine levels (Figure 1F). Taken together, these data suggest that endogenous MC1R is unlikely a modifying factor of ADR-elicited proteinuria or glomerular injury and that MC1R is seemingly dispensable for the beneficial effect of NDP-MSH in mice with ADR nephropathy.
Figure 1. MC1R is not involved in ADR nephropathy and is non-essential for the protective effect of NDP-MSH.
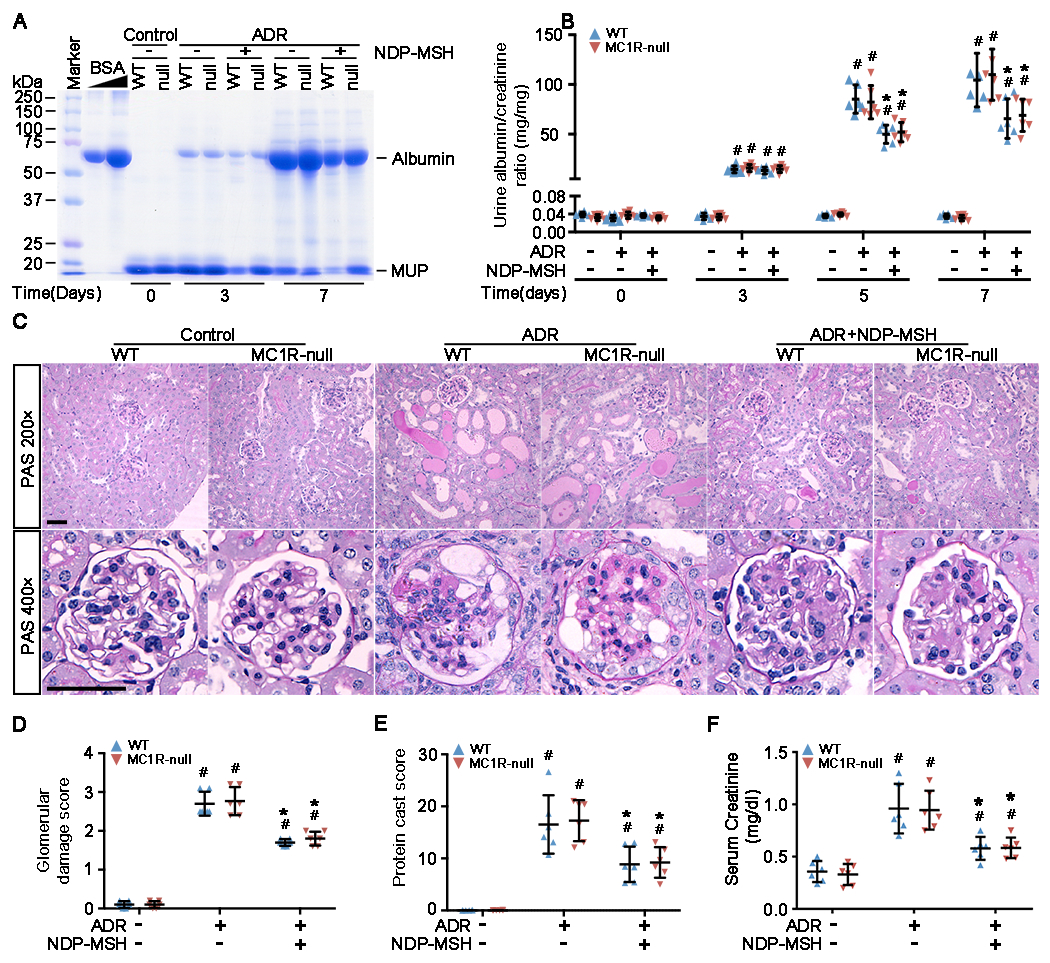
Male WT and MC1R-null (null) mice were treated with vehicle or Adriamycin (ADR) in the presence or absence of NDP-MSH co-treatment. Spot urine was collected on indicated time points after ADR or vehicle treatment. All animals were killed on Day 7, and kidneys and serum harvested and examined. (A) Urine samples (1.5 μl) were subjected to SDS-PAGE and staining with Coomassie Brilliant Blue. Bovine serum albumin (BSA, 20, 40, μg) served as standard controls; (B) Quantification of urine albumin levels adjusted with urine creatinine concentrations. #P<0.05 versus non-ADR-injured animals with the same MC1R mutation status at each time point; *P<0.05 versus ADR alone-treated animals with the same MC1R mutation status at each time point (n=6); (C) Representative micrographs demonstrate periodic acid–Schiff (PAS) staining (scale bar=50μm) of mouse kidneys. ADR-induced injury is featured by podocytic swelling and vacuolization, glomerular synechiae, glomerular capillary congestion, collapse and/or obliteration accompanied by hyaline material and/or mesangial matrix expansion as well as protein casts in tubulointerstitium; (D) Semi-quantitative morphometric analysis of PAS-stained kidney sections for glomerular damage scores. #P<0.05 versus non-ADR-injured animals with the same MC1R mutation status; *P<0.05 versus ADR alone-treated animals with the same MC1R mutation status (n=6); (E) Semi-quantitative morphometric analysis of PAS-stained kidney sections for protein cast scores. #P<0.05 versus non-ADR-injured animals with the same MC1R mutation status; *P<0.05 versus ADR alone-treated animals with the same MC1R mutation status (n=6); (F) Serum samples were processed for creatinine assay. #P<0.05 versus non-ADR-injured animals with the same MC1R mutation status; *P<0.05 versus ADR alone-treated animals with the same MC1R mutation status (n=6).
The protective effect of NDP-MSH on ADR-elicited podocytopathy is completely retained in MC1R-null mice
Podocytes are one of the cell types directly and primarily targeted by ADR toxicity, resulting in disruption of the glomerular filtration barrier. In WT mice, ADR injury caused prominent podocytopathy, marked by ultrastructural lesions, like podocyte loss, microvillous transformation and massive foot process effacement, as shown by electron microscopy of glomeruli (Figure 2A) and quantified by the drastic reduction of the number of podocyte foot processes per micrometer glomerular basement membrane (Figure 2B). In addition, glomerular expressions of typical podocyte marker proteins, including synaptopodin and WT-1, were substantially diminished in WT mice after ADR injury, as revealed by fluorescent immunohistochemistry staining (Figure 2A) and by immunoblot analysis of isolated glomeruli (Figure 2D) and densitometric analysis (Figure 2E), inferring podocyte loss or dedifferentiation. In agreement, increased expression of desmin, a muscle specific intermediate filament and also a podocyte injury marker, was noted in ADR injured WT kidneys (Figure 2A,F) and mostly located to the cytoplasm of WT-1 positive glomerular cells (Figure 2A), suggesting transdifferentiation of podocytes. In addition, dual color immunofluorescent staining demonstrated that ADR injury resulted in appearance of cells positive for both WT-1 and TUNEL staining in WT mice, indicative of podocyte apoptosis (Figure 2A,C). This was corroborated by immunoblot analysis (Figure 2D) of isolated glomeruli for caspase 3 cleavage, an essential signaling event involved in apoptosis, followed by densitometric quantification (Figure 2G). All these signs of podocyte injury, including ultrastructural lesions, loss of podocyte markers, de novo expression of desmin in podocytes and podocyte apoptosis, were also observed, to a similar extent, in MC1R-null mice after ADR injury, entailing that endogenous MC1R signaling is unlikely involved in ADR-elicited podocytopathy. Following NDP-MSH treatment, all of the signs of podocytopathy were considerably attenuated in both WT and MC1R-null mice to the same magnitude, denoting that melanocortin therapy protects against podocytopathy and that MC1R signaling is seemingly unrequired for this beneficial activity.
Figure 2. MC1R is not required for the NDP-MSH-improved podocytopathy in ADR injured mice.

Male WT and ADR mice were treated as elaborated in Figure 1. (A) Kidney specimens were procured from mice on day 7 after ADR or saline treatment and processed for electron microscopy (scale bar=1μm) and fluorescent immunohistochemistry staining (scale bar=20μm) for indicated molecules, including synaptopodin (SYNPO), desmin and WT-1, or in combination with TUNEL staining for apoptotic cells. Black arrowheads highlight injured podocytes with evident foot process effacement. White arrowheads indicate WT-1+ podocytes with positive staining for desmin or TUNEL; (B) Absolute counting of the number of apoptotic podocytes in glomeruli, expressed as the number of cells positive for both TUNEL and WT-1 in each glomerulus. #P<0.05 versus non-ADR-injured animals with the same MC1R mutation status; *P<0.05 versus ADR alone-treated animals with the same MC1R mutation status(n=6); (C) Absolute counting of the number of foot processes per unit length of glomerular basement membrane (GBM) on electron micrographs#P<0.05 versus non-ADR-injured animals with the same MC1R mutation status; *P<0.05 versus ADR alone-treated animals with the same MC1R mutation status(n=6); (D) Glomeruli were isolated from kidneys by the magnetic beads-based approach and processed for immunoblot analysis for indicated molecules. (E-G) Immunoblots were subjected to densitometric analysis. Data were presented as arbitrary units of densitometric ratios of indicated proteins to GAPDH as folds of the non-ADR-injured WT mice. #P<0.05 versus non-ADR-injured animals with the same MC1R mutation status; *P<0.05 versus ADR alone-treated animals with the same MC1R mutation status (n=6).
NDP-MSH restores the homeostatic motility of podocytes and preserves podocyte filtration barrier function in an MC1R independent manner
Human and experimental podocytopathy may involve both podocyte autonomous injury and podocyte non-autonomous or extra-renal pathogenic components. To determine if the beneficial effect of NDP-MSH observed above in ADR nephropathy is attributable to a possible direct protection of podocytes, in vitro studies were performed using primary cultures of podocytes isolated from WT and MC1R-null mice by the magnetic bead-based approach. Primary podocytes were cultured on collagen-coated Transwell filters to confluence and formed an intact monolayer. A paracellular permeability influx assay was applied to evaluate the filtration barrier function of podocyte monolayers (Figure 3A). After 24 h injury with ADR, the filtration barrier function of podocytes derived from WT mice was drastically disrupted, as evidenced by the time course measurement of the amount of albumin fluxed across the podocyte monolayers from the bottom to the top chambers. In contrast, in cells co-treated with NDP-MSH, the albumin fluxed across the podocyte monolayers was significantly mitigated, suggestive of a protective effect of NDP-MSH on the filtration barrier function. In podocytes derived from MC1R-null mice, a similar impairment of the filtration barrier function was observed upon ADR insult and protection by NDP-MSH was noted to the same extent, entailing that MC1R is non-essential for the podocyte protective activity of NDP-MSH.
Figure 3. NDP-MSH directly preserves podocyte filtration barrier function and rectifies podocyte hypermotility in an MC1R independent manner.
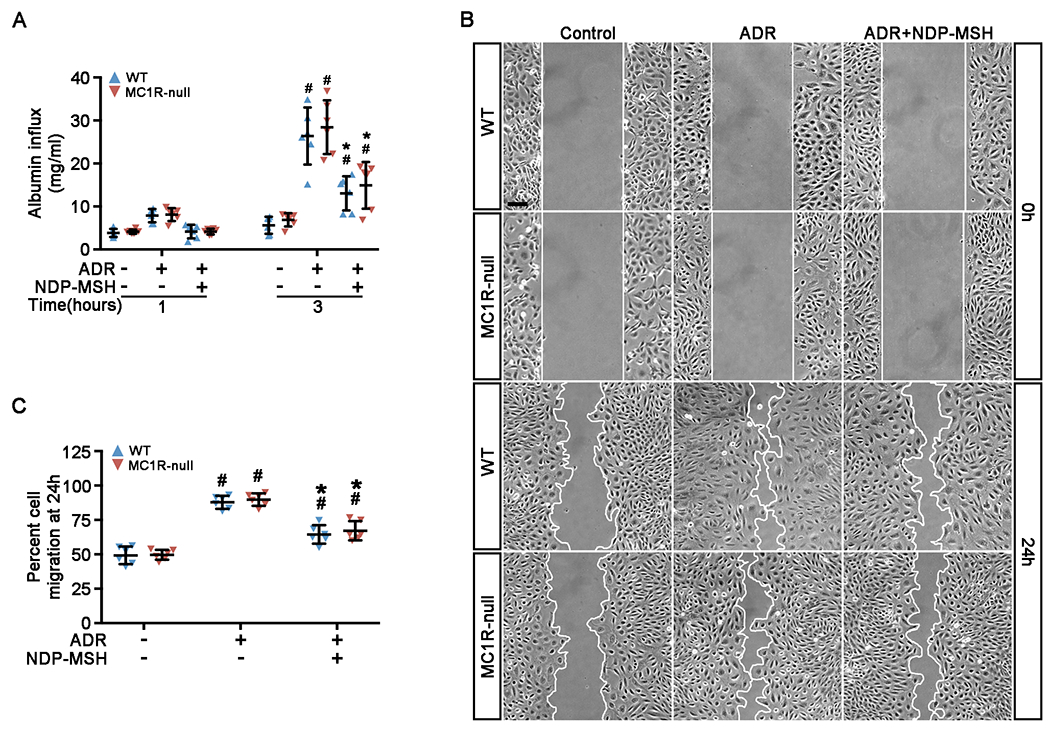
Primary podocytes derived from WT and MC1R-null mice were grown to confluence and treated with ADR or vehicle in the presence or absence of NDP-MSH (10−7M). (A) Cells were grown on the collagen coated transwell filters and after indicated treatments for 24h paracellular permeability assay was carried out to determine the filtration barrier function of podocytes monolayers. Culture media in the top chamber were collected at 1 or 3h during the paracellular permeability assay and subjected to quantification of the albumin influx across podocyte monolayers. Duration of albumin incubation is shown next to the x-axis. #P < 0.05 vs non-ADR-injured podocytes with the same MC1R mutation status (n = 6); *P < 0.05 vs ADR alone-treated podocytes with the same MC1R mutation status. (n = 6). (B) Primary podocytes were treated and subsequently scratch was processed using a 10μL pipette. Phase-contrast micrographs were taken immediately after wounding (0 h) and after migration for 24 h. Scale bar = 100 μm. (C) Quantification by computerized morphometric analysis of the cell migration area following the indicated treatments. #P < 0.05 vs non-ADR-injured podocytes with the same MC1R mutation status (n = 6); *P < 0.05 vs ADR alone-treated podocytes with the same MC1R mutation status. (n = 6).
Evidence indicates that podocytes are motile cells and that podocyte foot process motility is vital for maintaining the structural and functional homeostasis of the glomerular filtration barrier. As shown by a traditional cell migration assay for assessing cellular motility (Figure 3B), podocytes derived from both WT and MC1R-null mice possessed a comparable migratory capacity under basal conditions that lessened the distances between the leading edges of the migrating podocyte sheets. ADR injury accelerated the closure of the gap between the invading fronts of both cells to a comparable magnitude (Figure 3C), suggesting an enhanced podocyte motility. This effect was mitigated by NDP-MSH co-treatment in both types of cells to the same extent.
MC1R is non-essential for the protective effect of NDP-MSH on ADR-induced podocyte injury
To determine if the molecular events accounting for the ADR induced changes in podocyte motility and filtration barrier function were also modified by NDP-MSH, primary podocytes were subjected to further molecular testing. Primary podocytes in culture exhibited typical arborized morphologic features with rich phalloidin-labeled filamentous actin stress fibers and were characterized as expressing typical podocyte markers like synaptopodin. ADR-injured podocytes demonstrated cellular shrinkage and a spindle-like or asterlike cell shape, associated with diminished ventral stress fibers (Figure 4A). ADR also evidently caused loss of podocyte markers like synaptopodin, concomitant with de novo expression of the mesenchymal marker desmin, as shown by fluorescent immunocytochemistry staining (Figure 4A) and immunoblot analysis combined with densitometry (Figure 4B–D). This is indicative of podocyte dedifferentiation and transdifferentiation and is comparably detected in both WT and MC1R-null podocytes, consistent with the findings in vivo. NDP-MSH co-treatment attenuated the ADR-elicited changes in cell shape and F-actin stress fibers, mitigated the loss of synaptopodin and diminished the expression of desmin in podocytes derived from WT and MC1R-null mice to the same extent (Figure 4A,B), signifying that NDP-MSH preserves the integrity of actin cytoskeleton and overrides dedifferentiation and transdifferentiation, and that MC1R is dispensable for this protective effect.
Figure 4. The protective effect of NDP-MSH against the ADR-elicited cell shape changes, cytoskeleton disarrangement and transdifferentiation is fully preserved in MC1R-null podocytes.
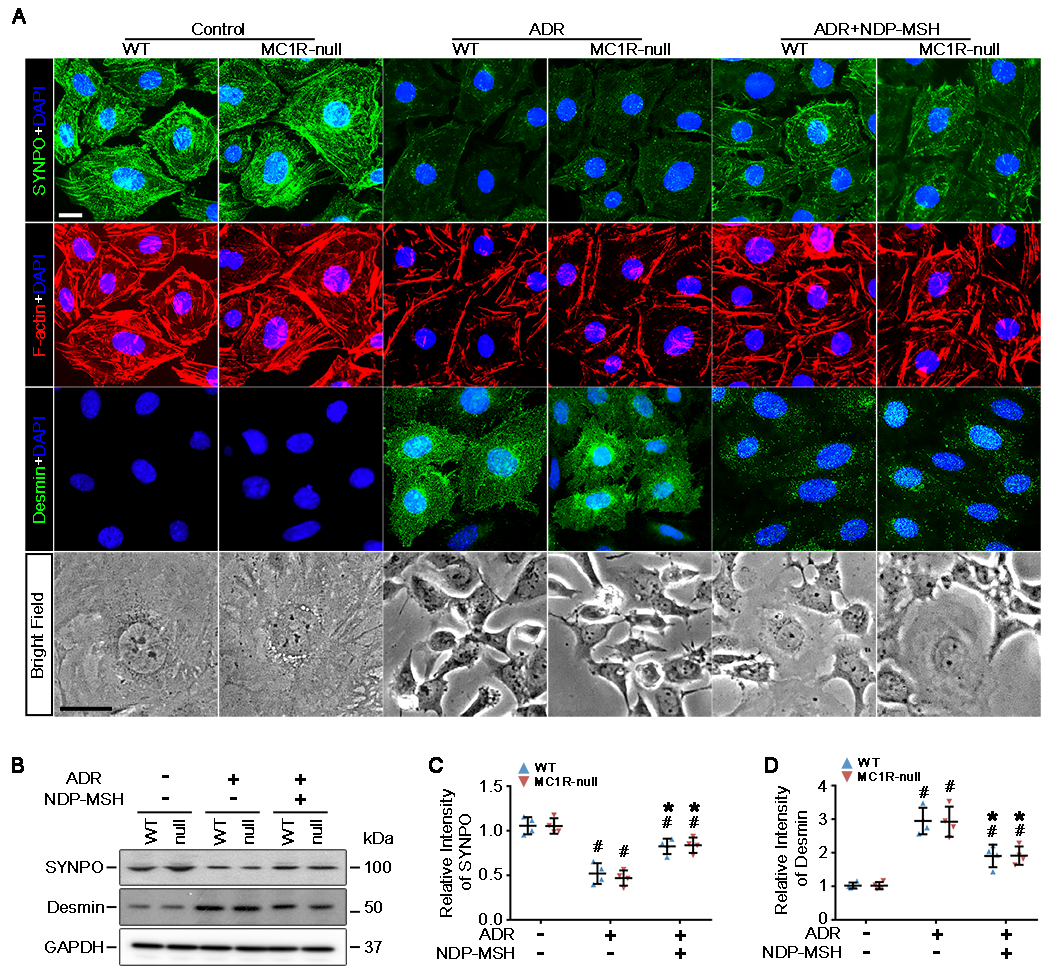
Primary podocytes derived from WT and MC1R-null mice were treated with ADR or vehicle in the presence or absence of NDP-MSH (10−7M) for 24h. (A) Cells were fixed and subjected to staining for cytoskeletal F-actin with rhodamine phalloidin and to fluorescent immunocytochemistry staining for synaptopodin (SYNPO) and desmin. Cells were counterstained with 4′,6-diamidino-2-phenylindole (DAPI). Representative micrographs of phase contrast microscopy and florescent microscopy are shown (Scale bar=20μm). (B) Cell lysates were subjected to immunoblot analysis for indicated molecules. (C and D) Immunoblots were subjected to densitometric analysis. Data were presented as arbitrary units of densitometric ratios of indicated proteins to GAPDH as folds of the non-ADR-injured WT cells. #P<0.05 versus non-ADR-injured cells with the same MC1R mutation status; *P<0.05 versus ADR alone treated cells with the same MC1R mutation status (n=4).
NDP-MSH improves cell viability and protects against apoptosis equally in WT and MC1R-null podocytes
In agreement with the in vivo finding that ADR induces podocyte death and loss, exposure of primary WT podocytes to ADR resulted in a marked impairment of cellular viability (Figure 5A,B), which was dramatically obliterated by NDP-MSH co-treatment, as assessed by the MTT assay (Figure 5B). Cellular apoptosis is, at least in part, responsible for this impaired viability, because apoptotic cells, marked by TUNEL staining (Figure 5A,C), were prominently amplified in podocytes exposed to ADR. This was further confirmed by immunoblot analysis of cell lysates for caspase 3 cleavage (Figure 5D) followed by densitometry (Figure 5E). NDP-MSH co-treatment considerably counteracted the proapoptotic effect of ADR in primary WT podocytes. The injurious effect of ADR and the protective effect of NDP-MSH on cell viability were equally observed in primary MC1R-null podocytes, signifying that NDP-MSH improves viability and protects against apoptosis in ADR-injured podocytes and that MC1R is not required for this beneficial activity.
Figure 5. NDP-MSH directly protects podocytes against the ADR-elicited apoptosis and improves cellular viability via an MC1R-independent mechanism.
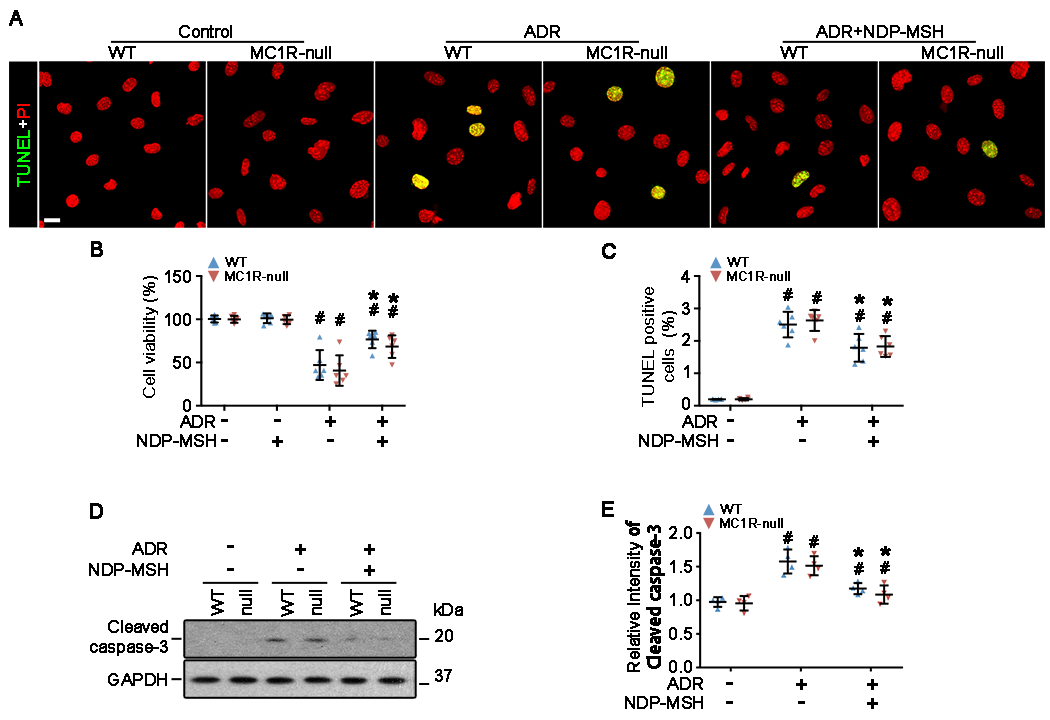
Primary podocytes derived from WT and MC1R-null mice were treated with ADR or vehicle in the presence or absence of NDP-MSH (10−7M) for 24 h. (A) Cells were fixed and subjected to TUNEL staining for apoptotic cells. Cells were counterstained with propidium iodide (PI) (Scale bar=20μm). (B) Absolute counting of the numbers of TUNEL positive apoptotic podocytes expressed as percentage of the total number of podocyte nuclei per high-power field. #P < 0.05 vs non-ADR-injured podocytes with the same MC1R mutation status (n = 6); *P < 0.05 vs ADR alone-treated podocytes with the same MC1R mutation status. (n = 6). (C) Cellular viability was assessed by the MTT assay. #P < 0.05 vs non-ADR-injured podocytes with the same MC1R mutation status (n = 6); *P < 0.05 vs ADR alone-treated podocytes with the same MC1R mutation status. (n = 6). (D) Cell lysates were subjected to immunoblot analysis for indicated molecules. (E) Immunoblots were subjected to densitometric analysis. Data were presented as arbitrary units of densitometric ratios of cleaved caspase-3 to GAPDH as folds of the non-ADR-injured WT cells. #P<0.05 versus non-ADR-injured cells with the same MC1R mutation status; *P<0.05 versus ADR alone treated cells with the same MC1R mutation status (n=4).
MC1R expression is negligible, if not absent, in glomerular podocytes in vitro, and in vivo
The above findings suggest that MC1R is unlikely responsible for the protective effect of melanocortin therapy in podocytes. As such, there is reason to doubt whether podocytes actually express MC1R. To address this issue, the expression profile of MC1R was explored in podocytes in vitro and in vivo. Primary podocytes derived from WT and MC1R-null mice were subjected to fluorescent immunocytochemistry staining for MC1R. In parallel, B16F10 murine melanoma cells known to express MC1R [38] served as a positive control and those with MC1R silencing served as a negative control. As expected, B16F10 cells demonstrated strong positive signal of MC1R expression, marked by intense punctate staining that is consistent with a typical staining pattern of cytoplasmic membrane receptors (Figure 6A). In stark contrast, B16F10 cells with MC1R silencing barely showed any staining signal, denoting a successful RNA interference and a high specificity of MC1R staining. Subsequently, in primary podocytes derived from WT and MC1R-null mice, the staining of MC1R was found to be negligible, if not absent, suggesting that MC1R is unlikely expressed in podocytes (Figure 6A). Immunoblot analysis of cell lysates (Figure 6B) combined with densitometry (Figure 6C) corroborated the morphologic findings. Despite some non-specific bands equally shown by all cells, the MC1R specific band was found in the lane of B16F10 cells but drastically diminished in the lane of B16F10 cells with MC1R silencing. This MC1R specific band was clearly absent from the lanes of WT or MC1R-null podocytes, thus negating the possibility of MC1R expression in glomerular podocytes. Apart from the podocytes, renal glomeruli are composed of multiple other cell types, including glomerular parietal epithelial cells, glomerular mesangial cells and vascular endothelial cells. Consistent with previous observations [35], fluorescent immunohistochemistry staining of mouse kidneys for MC1R revealed strong staining in renal tubules (Figure 6D). However, very weak staining was noted in glomeruli, where MC1R staining was largely colocalized with the standing for the endothelial marker CD31, entailing that MC1R is predominantly expressed by capillary endothelial cells in glomeruli (Figure 6D), in agreement with previous reports that vascular endothelial cells express MC1R [39, 40]. Collectively, these data are consistent with the above findings in animal models that MC1R is unlikely involved in the experimental FSGS.
Figure 6. MC1R expression is negligibly noted in podocytes in vitro and in vivo.
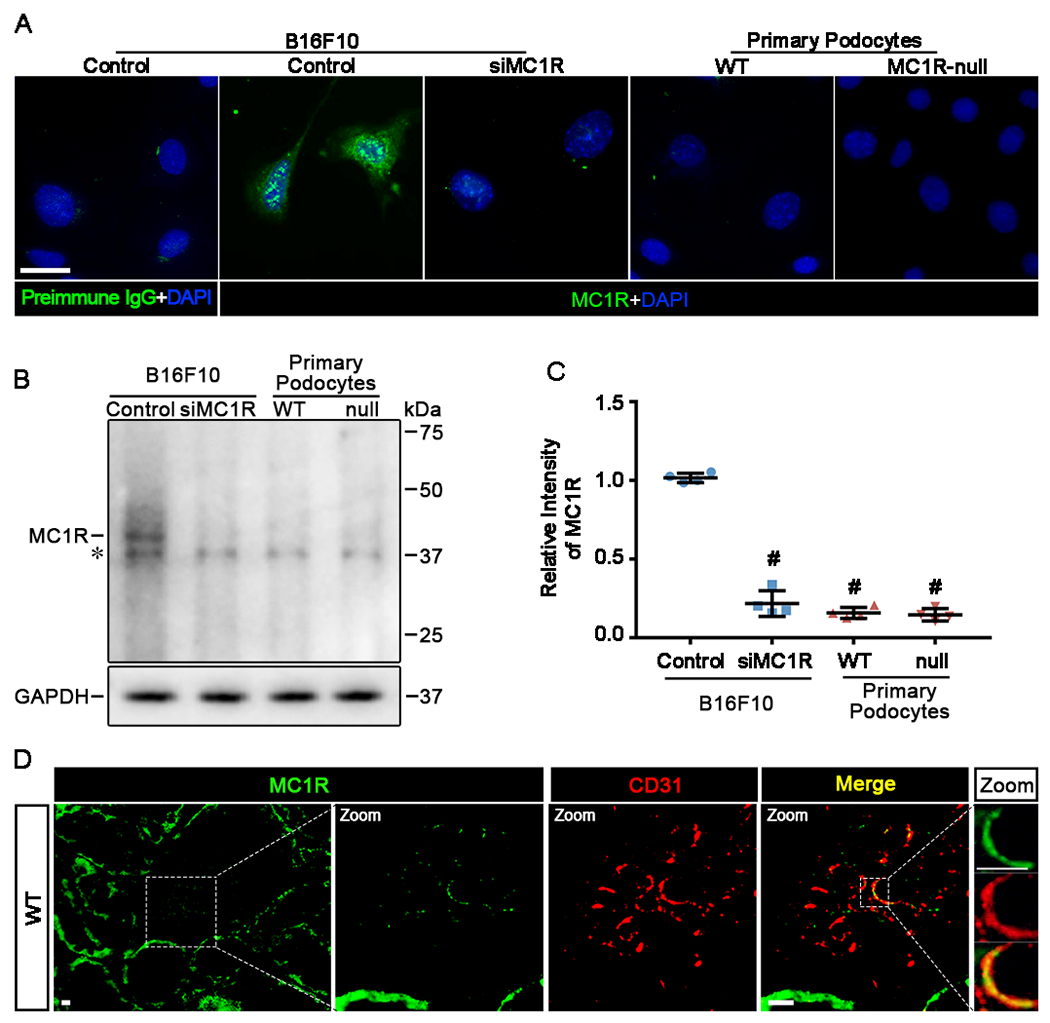
(A) Primary podocytes derived from WT and MC1R-null mice were cultured under physiologic conditions and processed for fluorescent immunocytochemistry staining for MC1R with 4′,6-diamidino-2-phenylindole (DAPI) counterstaining. B16F10 cells served as a positive control and preimmune IgG control for MC1R staining. B16F10 cells with MC1R silencing (siMC1R) served as a negative control. (Scale bar=30μm). (B) Cells lysate were subjected to immunoblot analysis for MC1R. The MC1R specific band is indicated. The asterisk indicates nonspecific bands probed by the antibody. (C) Immunoblots were subjected to densitometric analysis. Data were presented as arbitrary units of densitometric ratios of MC1R to GAPDH as folds of the control group. #P<0.05 versus control group (n=4). (D) Cryosections of WT kidneys were processed for fluorescent immunohistochemistry staining for MC1R (green). Representative micrographs are shown. Enlarged views show colocalization of MC1R staining (green) with CD31 staining (red) in glomeruli (Scale bars=10μm).
Discussion
Intractable nephrotic syndrome associated with FSGS represents a paramount challenge for clinical practice. The present study by using a murine model of FSGS, demonstrated that treatment with NDP-MSH, a potent non-steroidogenic pan-MCR agonist, is capable of protecting against podocytopathy and ameliorating proteinuria and glomerular injury. To the best of our knowledge, this is the first report describing the beneficial effect of melanocortin therapy in experimental FSGS. Remarkably, this beneficial effect seems independent of MC1R signaling, because WT and MC1R-null mice are equally protected by NDP-MSH against ADR-elicited proteinuria and FSGS.
The anti-proteinuric and glomerular protective effect of melanocortin therapy has also been described in several other models of proteinuric glomerulopathies, including rats with passive Heymann nephritis (PHN), a model of MN [41], and mice with LPS-elicited podocytopathy [24]. However, the type of MCR mediating this beneficial action remains uncertain and it is unclear if systemic or kidney-specific melanocortinergic signaling contributes. Notably, MCR expression in the kidney is not well defined. To date, conflicting data exist regarding the type of MCR expressed by glomerular podocytes and possibly mediating the proteinuria reducing effect. In a study by Lindskog et al [23], only MC1R was detected by RT-PCR assay as the major MCR in human and rat kidneys as well as in cultured human podocytes, glomerular endothelial cells, mesangial cells and tubular epithelial cells. However, because all MCRs are products of small intronless genes, genomic contamination may complicate the RT-PCR -based expression analysis [42–44]. In their subsequent confirmatory study, MC1R was detected virtually everywhere in the kidney by immunohistochemistry and partially colocalized with synaptopodin [23]. However, MC1R is a cellular membrane receptor but synaptopodin is a cytoplasmic protein associated with actin cytoskeleton. This unusual colocalization of cellular membrane receptor staining with cytoplasmic protein staining implies the possibility of artifacts or non-specific staining. This concern is further accentuated by a subsequent in-depth study, in which selective MC1R agonists barely induced any cAMP response in wild-type podocytes [45], again casting doubt on the claim that glomerular podocytes express MC1R. More recently, a post-hoc analysis of glomerulus-specific transcriptome data in a variety of nephrotic glomerulopathies revealed that MC1R was the only MCR that is augmented in diverse glomerular diseases [46], somewhat in support of the notion that MC1R may underlie the beneficial effect of ACTH in nephrotic patients. Nevertheless, this analysis is likely to have the risk of subjective bias because a number of samples in the examined datasets were arbitrarily designated as outliers and excluded from the analysis. Considering that MC1R is extensively expressed by vascular endothelia, including glomerular endothelia, it is tempting to speculate that injuries to glomerular capillary tufts [47] may contribute to altered glomerular expression of MC1R in FSGS.
Despite the uncertainty regarding if MC1R is expressed by glomerular podocytes and mediates the glomerular protective effect, almost all studies unequivocally demonstrated that melanocortin therapy is beneficial in experimental proteinuric glomerulopathies. For instance, in rats with PHN, a model of MN, pan MCR agonists, such as ACTH and α-MSH as well as selective MC1R agonist MS05 [23], improved proteinuria and podocyte injury. However, because PHN is a model of immune-mediated glomerulopathy and MC1R-mediated signaling is well known to exert a systemic immunemodulatory activity, thus the benefit in PHN may be likely ascribed to systemic immune regulation during the autologous phase of PHN, rather than an MC1R-mediated direct protection of podocytes. Indeed, in murine models of nephrotoxin (ADR)-induced nephropathy that involves less immunopathogenic mechanisms, highly selective MC1R agonists failed to attenuate proteinuria and podocyte injuries [41], suggesting that MC1R signaling is unlikely responsible for podocyte protection. This is in agreement with the aforementioned in vitro finding that MC1R agonists failed to trigger any cAMP response in wild-type podocytes [45]. Conversely, MC1R non-functional mutation barely affects the therapeutic efficacy of pan-MCR agonists in experimental podocytopathy. Indeed, in murine models of rapid podocytopathy elicited by LPS, WT and MC1R-null mice are equally protected by NDP-MSH against podocyte injury and proteinuria [24]. Likewise, the present study shows that in murine models of FSGS, the beneficial effect of NDP-MSH on proteinuria, podocyte injury and glomerular destruction are completely preserved in MC1R-null mice. The present study is not without limitations. For instance, the WT and MC1R-null mice used in the present study were in the C57BL/6 background, which is not sensitive to ADR injury and thus necessitates a very high dose (25 mg/kg). Nevertheless, the findings made in the present study are consistent with what was described recently in nephrotic FSGS patients bearing congenital red hair color and dominant-negative MC1R mutations, in whom ACTH monotherapy still induced complete remission of proteinuria and disease despite steroid resistance [24]. These data suggest that MC1R is unlikely responsible for the beneficial effect of melanocortin therapy. Furthermore, in combination with the in vitro finding that NDP-MSH is potently effective in protecting against injury in cultured podocytes and this effect is fully preserved in MC1R-null podocytes, there is ample reason to conceive that melanocortinergic signaling transmitted by MCR other than MC1R may exert a direct podocyte protective activity. The exact identity of the podocyte specific MCR warrants further investigation, so does the underlying signaling mechanism.
In summary, melanocortin therapy by using NDP-MSH is effective in protecting against podocyte injury, reducing proteinuria and ameliorating glomerular destruction in experimental FSGS. In vitro in primarily cultured podocytes, NDP-MSH demonstrates a direct podoprotective effect. This beneficial effect is comparably retained in MC1R-null mice and podocytes, and thus is likely mediated by a podocyte specific non-MC1R-mediated melanocortinergic signaling.
Clinical Perspectives.
Melanocortin peptides like ACTH and α-MSH, exert a proteinuria reducing and glomerular protective effect in clinical and experimental nephrotic glomerulopathies. However, the type of melanocortin receptor conveying this beneficial effect is controversial. Moreover, it remains uncertain if a systemic or podocyte-specific melanocortinergic signaling contributes.
In the present study, we demonstrated that melanocortin therapy by using NDP-MSH is effective in protecting against podocyte injury, reducing proteinuria and ameliorating glomerular destruction in experimental FSGS. This beneficial effect is likely mediated by a podocyte specific non-MC1R-mediated melanocortinergic signaling.
Our findings may pave the way for defining the podocyte-specific MCR and for developing targeted melanocortin therapy for the treatment of proteinuric glomerulopathies.
Funding
Part of this study was presented at the American Society of Nephrology Kidney Week 2018 in San Diego. The research work of the authors was supported in part by the Foundation for Health, the U.S. National Institutes of Health [grant number R01DK114006 (to R.G.)] and Research Incentive Fund from Brown University and University of Toledo. The funders had no role in the design and conduct of this study, collection and interpretation of the data, or preparation and approval of the manuscript.
Competing Interests
Dr. Gong and Dr.Dworkin report research funding from the Mallincrodt Pharmaceuticals, which is not related to this study. Dr. Gong served as a consultant to the Questcor Pharmaceuticals and the Mallincrodt Pharmaceuticals. All other authors declared no competing interests.
Abbreviations
- ACTH
adrenocorticotropic hormone
- ADR
Adriamycin
- DAPI
4′,6-diamidino-2-phenylindole
- ESRD
end-stage renal disease
- FSGS
focal segmental glomerulosclerosis
- MCR
melanocortin receptor
- α-MSH
α-melanocyte-stimulating hormone
- NDP-MSH
[Nle4,D-phe7]-alpha-melanocyte-stimulating hormone
- PAS
periodic acid–Schiff
- WT
wild-type
References
- 1.Rosenberg AZ and Kopp JB (2017) Focal Segmental Glomerulosclerosis. Clinical journal of the American Society of Nephrology : CJASN. 12, 502–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.D’Agati VD, Kaskel FJ and Falk RJ (2011) Focal segmental glomerulosclerosis. The New England journal of medicine. 365, 2398–2411 [DOI] [PubMed] [Google Scholar]
- 3.Reiser J, Nast CC and Alachkar N (2014) Permeability factors in focal and segmental glomerulosclerosis. Advances in chronic kidney disease. 21, 417–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hogg R, Middleton J and Vehaskari VM (2007) Focal segmental glomerulosclerosis--epidemiology aspects in children and adults. Pediatric nephrology (Berlin, Germany). 22, 183–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fogo AB (2015) Causes and pathogenesis of focal segmental glomerulosclerosis. Nature reviews. Nephrology. 11, 76–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jefferson JA and Shankland SJ (2014) The pathogenesis of focal segmental glomerulosclerosis. Advances in chronic kidney disease. 21, 408–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Korbet SM (2012) Treatment of primary FSGS in adults. Journal of the American Society of Nephrology : JASN. 23, 1769–1776 [DOI] [PubMed] [Google Scholar]
- 8.Gong R (2014) Leveraging melanocortin pathways to treat glomerular diseases. Advances in chronic kidney disease. 21, 134–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gantz I and Fong TM (2003) The melanocortin system. American journal of physiology. Endocrinology and metabolism. 284, E468–474 [DOI] [PubMed] [Google Scholar]
- 10.Cone RD (2006) Studies on the physiological functions of the melanocortin system. Endocr Rev. 27, 736–749 [DOI] [PubMed] [Google Scholar]
- 11.Hadley ME and Haskell-Luevano C (1999) The proopiomelanocortin system. Annals of the New York Academy of Sciences. 885, 1–21 [DOI] [PubMed] [Google Scholar]
- 12.Cone RD, Lu D, Koppula S, Vage DI, Klungland H, Boston B, Chen W, Orth DN, Pouton C and Kesterson RA (1996) The melanocortin receptors: agonists, antagonists, and the hormonal control of pigmentation. Recent progress in hormone research. 51, 287–317; discussion 318 [PubMed] [Google Scholar]
- 13.Abdel-Malek ZA (2001) Melanocortin receptors: their functions and regulation by physiological agonists and antagonists. Cellular and molecular life sciences : CMLS. 58, 434–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arneil GC and Wilson HE (1953) A.C.T.H. in nephrosis. Archives of disease in childhood. 28, 372–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Piel CF (1956) Management of nephrosis; the use of long continued hormone therapy. California medicine. 85, 152–156 [PMC free article] [PubMed] [Google Scholar]
- 16.Gong R (2011) The renaissance of corticotropin therapy in proteinuric nephropathies. Nature reviews. Nephrology. 8, 122–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Berg AL and Arnadottir M (2004) ACTH-induced improvement in the nephrotic syndrome in patients with a variety of diagnoses. Nephrol Dial Transplant. 19, 1305–1307 [DOI] [PubMed] [Google Scholar]
- 18.Bomback AS, Tumlin JA, Baranski J, Bourdeau JE, Besarab A, Appel AS, Radhakrishnan J and Appel GB (2011) Treatment of nephrotic syndrome with adrenocorticotropic hormone (ACTH) gel. Drug design, development and therapy. 5, 147–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bomback AS, Canetta PA, Beck LH Jr., Ayalon R, Radhakrishnan J and Appel GB (2012) Treatment of resistant glomerular diseases with adrenocorticotropic hormone gel: a prospective trial. American journal of nephrology. 36, 58–67 [DOI] [PubMed] [Google Scholar]
- 20.Hogan J, Bomback AS, Mehta K, Canetta PA, Rao MK, Appel GB, Radhakrishnan J and Lafayette RA (2013) Treatment of idiopathic FSGS with adrenocorticotropic hormone gel. Clin J Am Soc Nephrol. 8, 2072–2081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Anwar S, Larson DS, Naimi N, Ashraf M, Culiberk N, Liapis H, Wei C, Reiser J and Brennan DC (2015) A case report of adrenocorticotropic hormone to treat recurrent focal segmental glomerular sclerosis post-transplantation and biomarker monitoring. Frontiers in medicine. 2, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang P, Zhang Y, Wang Y, Brem AS, Liu Z and Gong R (2017) Acquired Resistance to Corticotropin Therapy in Nephrotic Syndrome: Role of De Novo Neutralizing Antibody. Pediatrics. 140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lindskog A, Ebefors K, Johansson ME, Stefansson B, Granqvist A, Arnadottir M, Berg AL, Nystrom J and Haraldsson B (2010) Melanocortin 1 receptor agonists reduce proteinuria. Journal of the American Society of Nephrology : JASN. 21, 1290–1298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qiao Y, Berg AL, Wang P, Ge Y, Quan S, Zhou S, Wang H, Liu Z and Gong R (2016) MC1R is dispensable for the proteinuria reducing and glomerular protective effect of melanocortin therapy. Scientific reports. 6, 27589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jeansson M, Bjorck K, Tenstad O and Haraldsson B (2009) Adriamycin alters glomerular endothelium to induce proteinuria. J Am Soc Nephrol. 20, 114–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Botte DA, Noronha IL, Malheiros DM, Peixoto TV and de Mello SB (2014) Alpha-melanocyte stimulating hormone ameliorates disease activity in an induced murine lupus-like model. Clin Exp Immunol. 177, 381–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li C, Ge Y, Dworkin L, Peng A and Gong R (2016) The beta isoform of GSK3 mediates podocyte autonomous injury in proteinuric glomerulopathy. The Journal of pathology. 239, 23–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Takiue K, Sugiyama H, Inoue T, Morinaga H, Kikumoto Y, Kitagawa M, Kitamura S, Maeshima Y, Wang DH, Masuoka N, Ogino K and Makino H (2012) Acatalasemic mice are mildly susceptible to adriamycin nephropathy and exhibit increased albuminuria and glomerulosclerosis. BMC nephrology. 13, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jiang Y, Bao H, Ge Y, Tang W, Cheng D, Luo K, Gong G and Gong R (2015) Therapeutic targeting of GSK3beta enhances the Nrf2 antioxidant response and confers hepatic cytoprotection in hepatitis C. Gut. 64, 168–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Takemoto M, Asker N, Gerhardt H, Lundkvist A, Johansson BR, Saito Y and Betsholtz C (2002) A new method for large scale isolation of kidney glomeruli from mice. Am J Pathol. 161, 799–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou S, Wang P, Qiao Y, Ge Y, Wang Y, Quan S, Yao R, Zhuang S, Wang LJ, Du Y, Liu Z and Gong R (2016) Genetic and Pharmacologic Targeting of Glycogen Synthase Kinase 3beta Reinforces the Nrf2 Antioxidant Defense against Podocytopathy. Journal of the American Society of Nephrology : JASN. 27, 2289–2308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu W, Ge Y, Liu Z and Gong R (2014) Glycogen synthase kinase 3beta dictates podocyte motility and focal adhesion turnover by modulating paxillin activity: implications for the protective effect of low-dose lithium in podocytopathy. The American journal of pathology. 184, 2742–2756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pegoraro AA, Singh AK, Arruda JA, Dunea G and Bakir AA (2000) A simple method to detect an albumin permeability factor in the idiopathic nephrotic syndrome. Kidney Int. 58, 1342–1345 [DOI] [PubMed] [Google Scholar]
- 34.Li Y, Kang YS, Dai C, Kiss LP, Wen X and Liu Y (2008) Epithelial-to-mesenchymal transition is a potential pathway leading to podocyte dysfunction and proteinuria. Am J Pathol. 172, 299–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Si J, Ge Y, Zhuang S, Wang LJ, Chen S and Gong R (2013) Adrenocorticotropic hormone ameliorates acute kidney injury by steroidogenic-dependent and -independent mechanisms. Kidney Int. 83, 635–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen S, Ge Y, Si J, Rifai A, Dworkin LD and Gong R (2008) Candesartan suppresses chronic renal inflammation by a novel antioxidant action independent of AT1R blockade. Kidney international. 74, 1128–1138 [DOI] [PubMed] [Google Scholar]
- 37.Lee VW and Harris DC (2011) Adriamycin nephropathy: a model of focal segmental glomerulosclerosis. Nephrology (Carlton, Vic.). 16, 30–38 [DOI] [PubMed] [Google Scholar]
- 38.Scott TL, Wakamatsu K, Ito S and D’Orazio JA (2009) Purification and growth of melanocortin 1 receptor (Mc1r)- defective primary murine melanocytes is dependent on stem cell factor (SFC) from keratinocyte-conditioned media. In vitro cellular & developmental biology. Animal. 45, 577–583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Saporiti F, Piacentini L, Alfieri V, Bono E, Ferrari F, Chiesa M and Colombo GI (2019) Melanocortin-1 Receptor Positively Regulates Human Artery Endothelial Cell Migration. Cellular physiology and biochemistry : international journal of experimental cellular physiology, biochemistry, and pharmacology. 52, 1339–1360 [DOI] [PubMed] [Google Scholar]
- 40.Rinne P, Ahola-Olli A, Nuutinen S, Koskinen E, Kaipio K, Eerola K, Juonala M, Kahonen M, Lehtimaki T, Raitakari OT and Savontaus E (2015) Deficiency in Melanocortin 1 Receptor Signaling Predisposes to Vascular Endothelial Dysfunction and Increased Arterial Stiffness in Mice and Humans. Arteriosclerosis, thrombosis, and vascular biology. 35, 1678–1686 [DOI] [PubMed] [Google Scholar]
- 41.Lindskog Jonsson A, Granqvist A, Elvin J, Johansson ME, Haraldsson B and Nystrom J (2014) Effects of melanocortin 1 receptor agonists in experimental nephropathies. PloS one. 9, e87816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sanchez MS, Celis ME and Schioth HB (2000) Evidence that alpha-MSH induced grooming is not primarily mediated by any of the cloned melanocortin receptors. Neuropeptides. 34, 77–82 [DOI] [PubMed] [Google Scholar]
- 43.Szardenings M, Muceniece R, Mutule I, Mutulis F and Wikberg JE (2000) New highly specific agonistic peptides for human melanocortin MC(1) receptor. Peptides. 21, 239–243 [DOI] [PubMed] [Google Scholar]
- 44.Khuu LA, Tayyari F, Sivak JM, Flanagan JG, Singer S, Brent MH, Huang D, Tan O and Hudson C (2017) Aqueous humor endothelin-1 and total retinal blood flow in patients with non-proliferative diabetic retinopathy. Eye (Lond). 31, 1443–1450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Elvin J, Buvall L, Lindskog Jonsson A, Granqvist A, Lassen E, Bergwall L, Nystrom J and Haraldsson B (2016) Melanocortin 1 receptor agonist protects podocytes through catalase and RhoA activation. American journal of physiology. Renal physiology. 310, F846–856 [DOI] [PubMed] [Google Scholar]
- 46.Bergwall L, Wallentin H, Elvin J, Liu P, Boi R, Sihlbom C, Hayes K, Wright D, Haraldsson B, Nystrom J and Buvall L (2018) Amplification of the Melanocortin-1 Receptor in Nephrotic Syndrome Identifies a Target for Podocyte Cytoskeleton Stabilization. Scientific reports. 8, 15731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.D’Agati V (2003) Pathologic classification of focal segmental glomerulosclerosis. Seminars in nephrology. 23, 117–134 [DOI] [PubMed] [Google Scholar]