

Abstract
Giant Cell Arteritis (GCA) is an autoimmune disease of medium and large arteries, characterized by granulomatous inflammation of the three-layered vessel wall that results in vaso-occlusion, wall dissection and aneurysm formation. The immunopathogenesis of GCA is an accumulative process in which a prolonged asymptomatic period is followed by uncontrolled innate immunity, a breakdown in self-tolerance, the transition of autoimmunity from the periphery into the vessel wall and, eventually, the progressive evolution of vessel wall inflammation. Each of the steps in pathogenesis corresponds to specific immuno-phenotypes that provide mechanistic insights into how the immune system attacks and damages blood vessels.
Clinically evident disease begins with inappropriate activation of myeloid cells triggering the release of hepatic acute phase proteins and inducing extravascular manifestations, such as muscle pains and stiffness diagnosed as polymyalgia rheumatica (PMR). Loss of self-tolerance in the adaptive immune system is linked to aberrant signaling in the NOTCH pathway, leading to expansion of NOTCH1+CD4+ T cells and the functional decline of NOTCH4+ T regulatory cells (Checkpoint 1). A defect in the endothelial cell (EC) barrier of adventitial vasa vasorum networks marks Checkpoint 2; the invasion of monocytes, macrophages and T cells into the arterial wall. Due to the failure of the immuno-inhibitory PD-1/PD-L1 pathway, wall-infiltrating immune cells arrive in a permissive tissues microenvironment, where multiple T cell effector lineages thrive, shift towards high glycolytic activity and support the development of tissue-damaging macrophages, including multinucleated giant cells (Checkpoint 3). Eventually, the vascular lesions are occupied by self-renewing T cells that provide autonomy to the disease process and limit the therapeutic effectiveness of currently used immunosuppressants. The multi-step process deviating protective to pathogenic immunity offers an array of interception points that provide opportunities for the prevention and therapeutic management of this devastating autoimmune disease.
Keywords: arteritis, vasculitis, aortitis, macrophage, T cell, T regulatory cell, NOTCH signaling, PD-1/PD-L1 pathway
Giant Cell Arteritis
GCA is the most prevalent inflammatory vasculopathy and stands out amongst the arteritides due to its destructive potential. The unequivocal autoimmune character of GCA distinguishes it from atherosclerotic disease, which has elements of smoldering inflammation in the vessel wall. In GCA, the target of the autoimmune attack are medium and large arteries, specifically, the aorta, the extracranial, and the upper extremity aortic branch vessels. Clinical complications are ischemic in nature; best known is the arteritic ischemic optic neuropathy leading to sudden and painless vision loss. Patients with vasculitis-induced blindness in one eye are at high risk of ischemia in the second eye, emphasizing the systemic character of GCA. Patients with GCA in the vertebral and basilar arteries present with posterior circulation ischemic strokes. Vasculitic stenosis/occlusion of the subclavian arteries leads to aortic arch syndrome and upper extremity pulseless disease. Aortic aneurysms are typically localized in the ascending aorta, wall dissections occur along the entire thoracic aorta.
While the vascular manifestations of GCA are linked to vasculitis-induced vascular remodeling, including complete luminal occlusion, the pathogenic events underlying the extravascular manifestations remain enigmatic. Intense systemic inflammation accompanied by a vigorous hepatic acute-phase response causes constitutional symptoms (weight loss, fever, night sweats, headaches etc) and can be the only disease manifestation. Typically, patients complain about proximal muscle pain and stiffness, clinically diagnosed as polymyalgia rheumatica (PMR). PMR can occur as an isolated syndrome, can precede clinically detectable vasculitis, or can be combined with vasculitis. During the later stages of GCA, vascular and extravascular GCA are often asynchronous.
Formerly considered a transient condition, GCA is now recognized as a chronic disease and patients live for 10–20 years post diagnosis. Such patients are at high risk for passing through all stages of the disease process, including the progression of vessel wall inflammation to the aorta. Aortitis is a dangerous manifestation of GCA and becomes the major challenge of disease management during late disease.
Progress made in understanding the immunopathogenesis of GCA over the last decade has fundamentally changed the pathogenic concepts applied. After a prolonged period of asymptomatic autoimmunity, patients loose tissue tolerance and immune cells enter the blood vessel wall, where they establish granulomatous lesions that eventually become self-sustained (Fig.1). We will review the data supporting the distinct pathogenic steps leading to GCA.
Fig.1. Evolution of Autoimmunity in Giant Cell Arteritis.

At-risk individuals are born with genetic risk determinants, mostly in the HLA region. Metabolic phenotypes (low glucose, low cholesterol, low triglycerides) become evident during the period of asymptomatic pre-disease. Checkpoint 1: Distinguishing immune phenotypes (expansion of NOTCH1+ CD4+ T cells) are present in the naïve T cell compartment and indicate loss of self-tolerance. Protective T regulatory cells fail due to hyperactive NOTCH signaling. Late during pre-GCA, the bone marrow releases activated myeloid cells (neutrophils, monocytes), triggering a hepatic acute-phase response and clinical symptoms of polymyalgia rheumatica (PMR). Checkpoint 2: GCA becomes clinically evident with the breakdown of tissue tolerance. Monocytes and neutrophils break through the protective endothelial barrier and T cells follow to invade the vessel wall. Checkpoint 3: T cells and macrophages build granulomatous lesions. The defect in the PD1/PD-L1 immune checkpoint creates a “permissive” tissue environment, with multiple T cell and macrophage lineages prospering. Tissue-resident memory T cells render the lesion self-sustained and autonomous. The vessel wall responds to the immune attack with a maladaptive response-to-injury, including intrawall angiogenesis and intimal hyperplasia.
The vascular lesion.
GCA is a granulomatous vasculitis. T cells, mostly CD4+ T cells, and macrophages (Mø) form granulomatous infiltrates in the adventitial and medial layer of the affected artery. About 50% of patients have typical multinucleated giant cells. The cellular composition of the vessel wall infiltrate distinguishes GCA from Takayasu arteritis,1 indicative of fundamental differences in pathogenesis between the two large vessel vasculitides that map to an overlapping vascular territory but manifest in fundamentally different at-risk populations. In the aorta, but not in the peripheral arteries, B cells participate in forming perivascular tertiary lymphoid structures during late-stage GCA. Vessel wall infiltrates persist in about half of the patients after one year of treatment with high-dose glucocorticoids.2
Risk factors.
Risk factors for the development of GCA provide important clues towards immune system abnormalities that ultimately foster uncontrolled arterial wall inflammation. Patients are consistently older than 50 years of age, the majority is female. The aging process remains the strongest risk factor for clinically evident vasculitis, with considerable overlap between the immune abnormalities in GCA patients and in immune aging (Table 1). Descending from Northern European people adds significant risk for the development of PMR and GCA. Epidemiological studies have described lower body mass index and low fasting blood glucose levels many years before disease onset.3,4 The strongest genetic risk has been mapped to the HLA region, with particular HLA-DRB1*04 alleles conferring risk to develop PMR as well as GCA;5, 6, 7 compatible with a key role of T cells in disease pathogenesis. Other disease-associated polymorphisms (cytokines, adhesion molecules, signaling molecules) point towards abnormalities in innate and adaptive immunity.8 In Northern European populations and in Minnesota, yearly incidence rates reach about 20 per 100,000 people >50 years of age. In Southern European populations, 10 per 100,000 people >50 years of age are annually affected, and the incidence drops to about 1 per 100,000 people >50 years of age among American populations of Asian or African descent.9, 10, 11 The incidence and prevalence of PMR is about three-fold higher,12 with identical geographic distribution of at-risk populations.
Table 1.
Age as a risk factor for Giant Cell Arteritis
| Immune aging-associated phenotypes | GCA-associated phenotypes |
|---|---|
| Inflammaging (= age-dependent, subtle inflammatory activity resulting in tissue damage) |
Elevated acute-phase proteins; C-reactive protein, erythrocyte sedimentation rate, SAA, etc. |
| Hematopoiesis biased towards myelopoiesis | Emergency hematopoiesis |
| Clonal hematopoiesis giving rise to myeloid cells with inflammatory potential | Excess myelopoiesis |
| Loss of naïve T cells | Loss of CD8 T cells in GCA patients and their relatives |
| CD8 T cells age faster than CD4 T cells | Scarcity of CD8 T cells in GCA lesions (old patient) versus Takayasu lesions (young patient) |
| Decline of T regulatory (Treg) cell function | Loss of exosome-releasing NOTCH4+ CD8+ Treg cells |
| Accumulation of end-differentiated effector T cells | Accelerated maturation of NOTCH1+ CD4 T cells Vasculitic lesions occupied by multiple lineages of cytokine-releasing effector T cells |
| Accumulation of cytotoxic CD4 T cells with reactivity against endothelial cells | Vessel wall-destructive effector T cells |
| T cell exhaustion | Failed T cell exhaustion due to hypoactivity of the PD-1/PD-L1 pathway |
Although the mortality risk associated with inflammatory disease of medium and large arteries should be high, life expectancy of GCA patients is maintained or even better than in the general population,13, 14 compatible with the concept that immune abnormalities causing GCA include pro-survival factors. In support of this hypothesis, parents of GCA patients are enriched for nonagenarians.15
Inappropriate myeloid cell activation in GCA.
The extravascular component of GCA presents clinically with a strong systemic inflammatory response which is driven by myeloid cells.16 The myeloid compartment of the immune system consists of granulocytes, monocytes, dendritic cells, and tissue macrophages. Progress made in the understanding of the cellular ontogeny, differentiation pathways, activation mechanisms and tissue-specific functions of myeloid cells has fundamentally changed the concepts how myeloid cells contribute to protective and pathogenic immune responses.17 It is now recognized that tissue macrophages derive from the yolk sac and are seeded into tissue sites early in life to critically participate in maintaining healthy tissues. Bone-marrow derived myeloid cells are recruited in response to danger signals, combine with tissue macrophages to form tissue-residing populations. The bone marrow itself is highly responsive to inflammatory signals.18 Monocytes and macrophages respond to activating stimuli with a multidimensional pattern, creating intermediated states that do not fit to simplified polarization models. And, signals delivered by the tissue environment, be it the bone marrow or any peripheral tissue site, shape the functionality of myeloid cells. Ultimately, by presenting antigens to T cells, myeloid cells are critically involved in the breakdown of self-tolerance defining autoimmune disease and thus contribute to all stages of the disease process leading to GCA.
In response to “danger signals”, hematopoiesis in the bone marrow switches from steady-state mode to emergency hematopoiesis, utilizing a unique hematopoietic response program that dramatically increases myeloid cell turnover and output (Fig.2). Consequently, numbers of granulocytes and monocytes in the blood rise and shift towards less differentiated subtypes. This hematopoietic response pattern is present in patients with GCA, characterized by elevated numbers of monocytes and neutrophils19 (Fig.2). Interestingly, this “emergency hematopoiesis” is maintained in patients treated with glucocorticoids and appears to be a stable phenomenon throughout the disease course.
Figure 2. Myeloid cell activation in Giant Cell Arteritis.
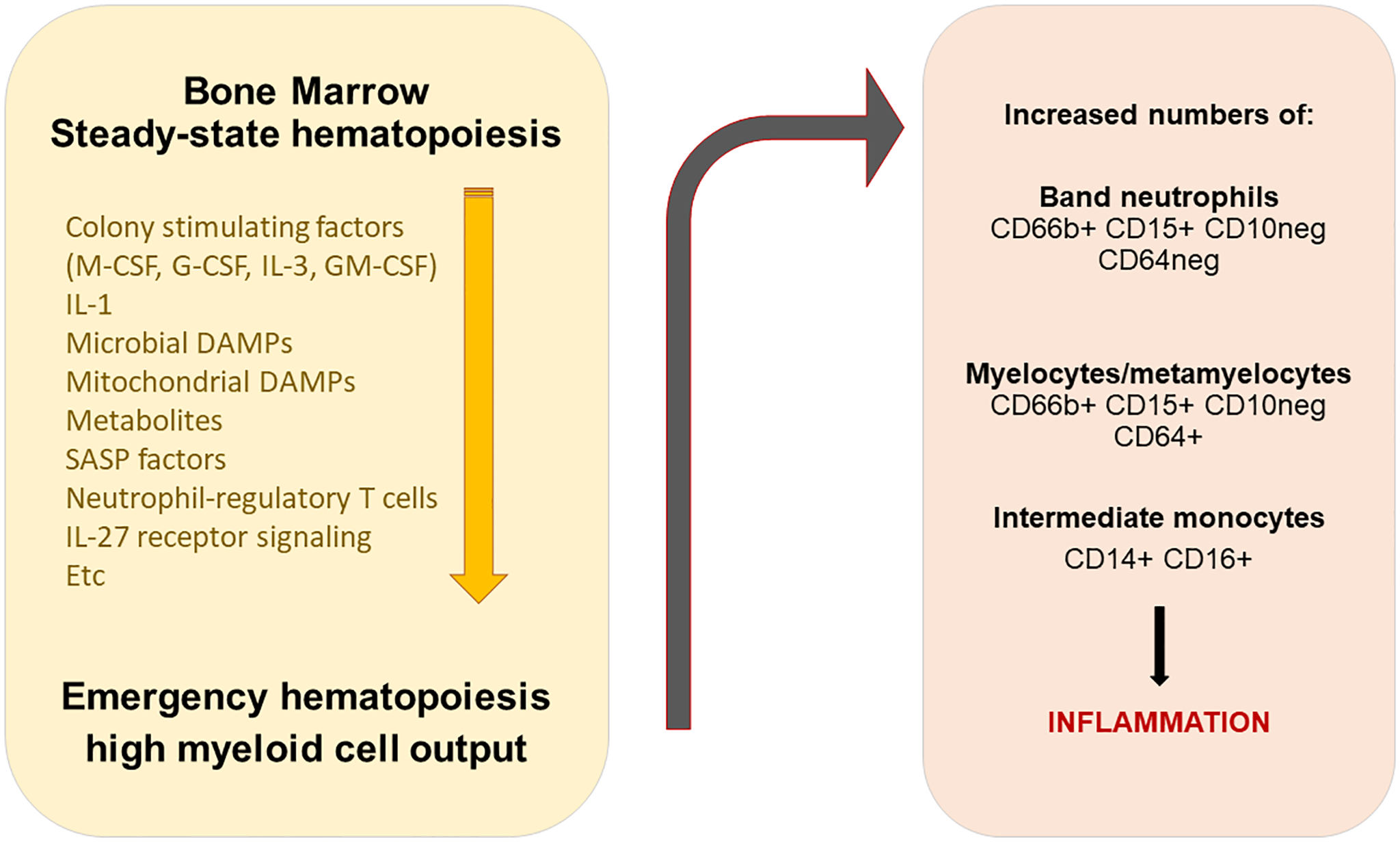
The bone marrow responds to an arsenal of danger-associated molecular patterns with a switch from steady-state hematopoiesis to emergency hematopoiesis, resulting in the release of high numbers of highly activated neutrophils and monocytes. The original triggers of bone marrow stimulation in patients with giant cell arteritis remain undefined. Circulating myeloid cells induce a hepatic acute phase response, giving rise to typical laboratory findings (elevated C-reactive protein and erythrocyte sedimentation rate) and in muscle pain/stiffness, diagnosed as polymyalgia rheumatica. Activated hematopoiesis is susceptible to corticosteroid suppression and to IL-6 blockade.
DAMP; danger-associated molecular pattern
Precise triggers of the myelopoietic bone marrow response in GCA are unknown. It is now clear that an arsenal of stimuli are sensed as danger signals and facilitate the switch of the bone marrow to emergency hematopoiesis (Fig. 2). DAMPs of microbial as well as mitochondrial origin are highly effective as inflammatory danger signals.20,21,22 The cytokine IL-1 appears to be more effective than other cytokines in mobilizing myeloid cell output.23,24 A growing body of work has implicated bone marrow sensing of metabolites in the regulation of myeloid cell production, release and activation status in atherosclerotic disease,25 and it will be interesting to see whether and which metabolic signals may play a role in GCA patients, given that low glucose and lipids and low body mass index are the relevant risk factors in this vasculitis. Recent work has implicated IL-27 receptor signaling in myelopoiesis regulation in abdominal aneurysm,26 indicating feedback regulation of bone marrow function through the adaptive immune system. In this context, neutrophil-regulatory T cells functioning through IL-23 and IL-17 may have a pathogenic role.27
Despite episodic claims that infectious microorganisms could set off exuberant activation of myeloid cells in GCA, no disease-specific triggers have been identified and unequivocally associated with disease onset.28 The advanced age of individuals at-risk for GCA complicates a clear link of infectious organisms with autoimmune vasculitis as the aging host notoriously harbors chronic infections, such as varicella zoster virus, cytomegalovirus, etc.
Increased output of granulocytes and monocytes, particularly more immature forms, is shared by patients with different vasculitides,29 emphasizing the non-specific character of this bone marrow response pattern. Immature granulocytes have a tendency to produce reactive oxygen species, which might affect endothelial cell function and release lytic enzymes and neutrophil extracellular traps (NETs), acting as an inflammatory nidus.30 NETs are typically encountered in neutrophilic autoimmune vasculitides,31 and have been reported as perivascular structures close to adventitial microvessels in GCA-affected arteries.32
Bone marrow-derived monocytes mobilized in GCA patients are increased in numbers and shifted towards the CD14++CD16+ intermediate phenotype.19 Accumulation of intermediate monocytes is typical for patients with coronary artery disease,33 and may well be a shared feature of patients along the spectrum of vascular diseases.34,35 Classical (CD14+CD16−) monocytes are considered critical drivers of inflammation, especially as they can differentiate into tissue macrophages and contribute to the tissue lesion. Non-classical CD14−CD16+ monocytes have been considered to be anti-inflammatory, maintaining vascular integrity by slowly patrolling on the endothelium, monitoring for danger signals and removing cell debris. Recent data call this distinction into question as non-classical monocytes appear to contribute to disease chronicity and burden.35 During the pre-vasculitic phase of disease, patients with PMR have elevated production of a number of monocyte-derived cytokines, including IL-6, IL-8, TNF, and IL-1b.36 Circulating monocytes from patients with GCA are prone to produce the tissue-digestive enzymes MMP-2 and MMP-937 and differentiate into chemokine-producing macrophages that are functionally and metabolically distinct from monocytes derived from patients with coronary artery disease.38
As humans age, they become more and more likely to show smoldering systemic inflammation, often called inflammaging39 and their risk to develop inflammatory disease progressively increases.40 A typical age-related abnormality is clonal hematopoiesis of indeterminate potential (CHIP), in which somatic mutations in hematopoietic stem cells can give rise to mutated immune effector cells, that have the potential to induce inflammatory disease.41 A pathogenic link between vasculitis, inappropriate hematopoiesis and intense inflammatory activity is exemplified by the recent identification of mutations in hematopoietic stem cells that cause to small vessel vasculitis.42 Acquired mutations in the UBA1 gene of blood cells lead to aggressive tissue inflammation and myelodysplasia/bone marrow failure. The syndrome has been named VEXAS, an acronym capturing clinical and pathogenic hallmarks: vacuoles, E1 ubiquitin activating enzyme mutations, X-chromosome location, auto- inflammation and somatic acquisition later in life. VEXAS patients and GCA patients share advanced age as a major risk factor.43 VEXAS patients can be misdiagnosed as biopsy-negative GCA. Both patient populations share intense acute phase responses, which give rise to an array of effector molecules, including C-reactive protein, fibrinogen, etc. These are important biomarkers in measuring disease activity in VEXAS and in extravascular GCA. Although sensitive indicators of inflammation, acute-phase proteins and cytokines lack the specificity to identify the offending cause and careful histologic and genetic examination is needed for proper patient assignment.
Checkpoint 1 – the loss of immune tolerance in GCA.
As a hallmark of autoimmunity, GCA patients have loss of self-tolerance in the adaptive immune system (Fig.1). In contrast to other autoimmune diseases, production of autoantibodies is atypical for GCA.44 Instead, GCA patients have circulating and tissue-infiltrating CD4+ T cells that inappropriately express the NOTCH1 receptor and have a low threshold setting to respond to antigenic stimuli45 (Fig.3). In GCA patients, peripheral blood CD4+ T cells include a distinct population of NOTCH1+ cells, both in the naïve and memory population.46 NOTCH1 is typically expressed by malignant T cells in acute lymphoblastic leukemia (T-ALL).47 65% of T-ALL patients carry activating mutations in the NOTCH1 gene and the receptor drives uncontrolled activation, anabolism and cell cycle progression of lymphoblasts. While receptor activation in T-ALL patients is ligand-independent, NOTCH1+ CD4+ T cells require combined NOTCH ligand/antigenic stimulation.45 Several disease-relevant effector functions have been associated with NOTCH pathway signaling in GCA; including, T cell expansion, transmigration of T cells from the circulation to the vascular wall and metabolic reprogramming.46 It is currently unknown at which stage of PMR and GCA the aberrant expansion of NOTCH1+ CD4+ T cells begins, but like in other autoimmune disease, the loss of self-tolerance is likely occurring years before clinically evident disease. Similarly, how abnormalities in NOTCH signaling could be related to the emerging risk factors for GCA (low blood glucose, low body mass index) is not understood.
Figure 3. Hyperactivity of the NOTCH signaling pathway in Giant Cell Arteritis.

NOTCH signaling is critically involved in T cell fate decisions and NOTCH1 gain-of-function mutations lead to acute lymphocytic leukemia. Patients with GCA have expansion of a NOTCH1-high expressing population of CD4 T cells that continuously upregulate NOTCH target genes. The NOTCH ligand Jagged 1 is aberrantly expressed on microvascular endothelial cells of the vasa vasora, enabling NOTCH1+ T cells to transmigrate into the adventitial space and differentiate into pro-inflammatory effector cells. Also, the NOTCH4 variant appears on circulating CD8 T cells of GCA patients, where the NOTCH signaling pathway regulates the trafficking of intracellular vesicles. Intracellular packaging and exosomal release of NOX2-containing vesicles is a core process in regulatory T cells, which suppress neighboring T cells through exosomal communication. NOTCH4+ CD8 T regulatory cells no longer produce exosomes, thus failing in their suppressive function. CD8 T regulatory cells reside in secondary lymphoid organs and their failure leaves the patient with a hyper-reactive immune system. (Illustration credit: Sceyence Studios)
Evidence for a central role for the NOTCH signaling pathways in the GCA-specific breakdown of self-tolerance has been strengthened by recent reports that NOTCH4 is aberrantly expressed on a subset of regulatory T cells and leads to functional paralysis of such anti-inflammatory T cells48 (Fig.3). CD8+ FOXP3+ regulatory T cells reside in secondary lymphoid tissues and suppress activation of nearby CD4+ T cells through the release of exosomes that contain the enzyme NADPH oxidase 2 (NOX2).49 GCA patients have low numbers and low function of such NOX2-releasing CD8 Treg cells, thus disrupting control of CD4 T cell immunity. Notably, CD8 Treg cells mediate broad immune-controlling function by regulating the size of the CD4 T cell compartment and evidence has been provided that this pathway has disease relevance in vasculitis.48 Molecular mechanisms underlying this tolerance defect have been unraveled. Aberrant signaling of the NOTCH4 receptor leads to rerouting of the vesicular traffic in CD8 Treg cells.48 Precisely, NOX2 is packed into exosomes by recycling the oxidase from the cell surface and maturing RAB7+ late endosomes into exosomes.50 NOTCH4 signaling suppresses the RAB7 gene and instead activates the RAB5 and RAB11 gene, stopping the maturation of early endosomes into multivesicular bodies and trapping NOX2 in a RAB5+, RAB11+ intracellular compartment. NOTCH4+ CD8 Treg cells from GCA patients no longer release NOX2-containing exosomes and fail to restrain adaptive immunity.
So far, there is no evidence that the exosomal communication between Treg and target cells extends to CD4+ Treg cells. Like in other vasculitides and in other autoimmune diseases, GCA patient are well known to have numeric and qualitative deficiencies in CD4+ Treg cells.42 To which extent these shifts are upstream or downstream of inflammation is unknown and underlying molecular mechanisms are insufficiently understood.
The NOTCH4-imposed tolerance defect in GCA is a good example of the progressive accumulation of abnormalities that eventually become pathogenic.51 Loss-of-function of CD8+ Treg cells is strictly age-dependent in healthy individuals and occurs prematurely in GCA patients.49 Here, the process of immune aging is superimposed on GCA-specific risk factors51 amplifying age-related decline of immunosuppression that has relevance in establishing and upholding vasculitis.
In invertebrates, NOTCH signaling is a pleiotropic mediator of cell fate. It has the unique feature that receptors and ligands are membrane embedded, enforcing close physical contact of signal-sending and signal-receiving cells. It is now recognized as a facilitator of cell-cell communication of innate and adaptive immune cells. Most importantly, NOTCH signaling is essential to guide T cell identify during thymic T cell development and to modulate the intensity of T cell activation signals in peripheral T cells.52 This has nurtured hopes that targeting NOTCH signaling could be developed into a strategy for immunomodulatory therapy in disease conditions associated with chronic T cell activation.53 However, this evolutionary conserved signaling mechanism is engaged in the control of cell fate in a broad variety of cell types, which calls for cell-type specific interventions to avoid off target toxicity.54 The aberrant expression of NOTCH ligands on vasa vasorum microvessels provides a mechanism for NOTCH-expressing T cells to receive instructive signals in an unusual tissue microenvironment; the adventitial space of the arterial wall, a territory typically protected by immune privilege. Here, NOTCH signaling becomes an essential mechanism of breaking tissue tolerance.
Checkpoint 2 – the loss of tissue tolerance in GCA.
Arteries are non-reductant and life-sustaining organs and as such are protected from inappropriate inflammation. This protection has been named the immune privilege of the blood vessel. Shielding of the artery from immune attack is a multi-pronged process and relies on the intactness of the endothelial barrier that separates the blood flow from the vessel wall tissue site. Original concepts assumed that vessel wall-infiltrating cells come from the artery’s main lumen but studies completed in the 90’s provided evidence that the access route to the three-layered wall of medium and large arteries depended on the adventitia. Specifically, interferon-gamma-producing T cells were placed into the adventitia and antigen-presenting dendritic cells were localized at the adventitia-media border.55,56 It is now recognized that the T cells and monocytes/macrophages that form the granulomatous infiltrates in GCA-affected arteries invade the adventitia from vasa vasora and then move towards the media and intima. Thus, the loss of tissue tolerance requires a breakdown of the endothelial cell barrier of adventitial microvessels. Available data have implicated distinct mechanisms in damaging the protective endothelial cell barrier (Fig.1):
Immature, reactive oxygen-producing neutrophils from GCA patients can oxidize proteins and increase permeability of endothelial cell layers in an in vitro co-culture system.29 This process appears to be operational in all types of systemic vasculitides, including small vessel disease.
Circulating monocytes of GCA patients are biased to produce MMP-2 and MMP-9 and can digest the endothelial cell basement membrane in a MMP-9 dependent manner.37 This process is highly specific for GCA and not shared with other vasculitides or autoimmune diseases.57 Antibodies blocking MMP-9 enzyme activity have strong anti-vasculitogenic activity in vivo.37
Adventitial capillaries of GCA-affected arteries produce IL-23p19 protein associated with gp130, but lack the IL-12p40 chain, indicative of functional transformation of these microvessels.58
Vasa vasora EC express the NOTCH ligand Jagged, interact with NOTCH1+ CD4+ T cells, facilitate differentiation of such T cells into pro-inflammatory effector cells and enable transmigration of vasculitogenic T cells.46
Endothelial cells lining the vasa vasora networks are stimulated by circulating vascular endothelial cell growth factors (VEGF), one of the abundant inflammatory proteins in GCA sera59,60
While there is agreement that the EC barrier needs to be overcome by the innate and adaptive cells that ultimately build the granulomatous infiltrates, much needs to be learned on the precise role of ECs and other structural elements that lie between the circulating blood and the site of vasculitis. ECs are remarkably heterogeneous in structure and function, change over time and have tissue-specific imprinting. Recent single-cell studies have broadened the functional spectrum of EC to include subtypes with phagocytic, scavenging, antigen-presenting and immune cell recruiting capabilities.61 Each of these functional domains could make a disease-relevant contribution in GCA and EC may have a more important role than previously appreciated. Appropriately designed studies could also provide insights into the vascular tropism of this vasculitis which exclusively attacks blood vessels with a vasa vasorum tree.
Checkpoint 3 – Autoimmunity presenting as granulomatous and self-renewing inflammation.
The typical presentation of GCA are non-necrotizing granulomatous infiltrates penetrating the arterial adventitia and media. Aggregates of epithelioid histiocytes are mixed with lymphocytes; necrotic centers are unusual and should trigger the search for alternative disease processes, e.g a different entity of vasculitis. Macrophages may coalesce to form pathognomic multinucleated giant cells, mostly lying around the fragmented lamina elastic interna. Cell populations have been mostly analyzed by immunohistochemical staining. Molecular approaches have been rare but are needed to lift studies beyond phenotypic descriptions. Ideally, cells could be isolated out of vasculitic arteries and tested for functional competency (Fig.1).
Lymphocytes and antigen-presenting cells.
CD4+ T cells dominate the infiltrates. The majority is activated, a subset is actively dividing and they express phenotypic markers of tissue-resident memory T cells.62 They are highly dependent on CD28-dependent co-stimulation and are programmed to rely on glucose metabolism to propagate and differentiate into effector T cells.62 A broad spectrum of effector T cell cytokines (IFNg, IL-2, IL-17, IL-21, IL-22, IL-9 etc) are produced in the granulomatous lesions, compatible with a generalized defect in T cell reactivity and differentiation44 (Fig.4). The co-existence of multiple T cell lineages questions the central importance of a small antigen-specific T cell population, is compatible with recruitment and local activation of many T cell specificities. While some of the effector cytokines may simply be an indicator of T cell activation, IFNγ-producing Th1 cells have been associated with the granulomatous pattern of vasculitis and have proven to be resistant to glucocorticoid therapy.63. IFNγ-producing Th1 cells are encountered almost exclusively in the adventitia,55 indicative of select antigens or select antigen-presenting cells relevant in their induction. IL-17-produicng T cells are consistently represented in the vasculitic infiltrates63,64 and essentially disappear with corticosteroid therapy.63 IL-21 is best known as the marker cytokine of follicular helper T cells (Tfh), T cells highly effective in providing help to B cells.65 The classical structure where Tfh cells interact with B cells are germinal centers. Such sophisticated lymphoid architectures are not part of the wall infiltrates in inflamed temporal arteries but tertiary lymphoid structures may occur with chronic-persistent disease. IL-21 may have costimulatory function in GCA, serving as a survival factor for TH1 and Th17 cells.66 It is also possible that T cells recruited to the vessel wall differentiate into a broad spectrum of effector T cells, because the stimulation condition in the tissue site are highly permissive. In that context it is interesting that transcriptomic and immunostaining experiments have indicated the presence of IL-22.67 IL-22 is now recognized as a critical regulator of epithelial homeostasis, serving as the connector between cells sensing microbes and dietary metabolites and an array for adaptive responses by epithelial cells.68,69 How this portfolio of effector functions could fit into the pathogenic cascades of GCA is unknown. Similarly, the search for T cell cytokines in inflamed temporal arteries has yielded evidence that IL-9 is produced within the infiltrates70 and that arteries with abundance of IL-17 and IL-9 display distinct distribution patterns of the inflammatory infiltrates. IL-9-producing T cells emerge through a unique developmental program, express distinct transcription factors (e.g. PU.1) and are considered powerful effector cells in allergic disease and in anticancer immunity,71 most compatible with a key role in Type 2 immunity.72 Originally associated with allergic disease and parasitic infection, type 2 immunity is now also recognized as a component of tissue regeneration and healing. Little is known about the immune pathways that regulate vascular remodeling and repair in GCA-affected arteries, but the outcome of disease is often connected to exuberant regeneration, such as lumen-occlusive intimal hyperplasia.
Fig.4. A diverse array of T cell effector lineages contributes to granulomatous vasculitis.

The population of T cells attracted to and retained in the inflamed vessel wall is assembled from many different T cell lineages. A series of different lineage-defining effector cytokines are produced in the lesions (A). T cells producing IFN-gamma, IL-21 or GM-CSF are mostly encountered in the adventitial layer. Effector T cell populations interact with multiple target cell populations (B). T cells attract, activate and sustain tissue-resident and tissue-infiltrating macrophage populations. Effector T cells interact with and receive stimulatory signals from endothelial cells. Effector T cell populations regulate pathways that involve stromal cell populations and lead to vascular remodeling, such as microvascular angiogenesis and intimal hyperplasia. Arrows indicate interactions that have been demonstrated or are suspected. The heterogeneity of effector T cells speaks against a single antigen driving GCA. One factor allowing the expansion of multiple effector lineages in the vascular lesions is the deficiency of the inhibitory PD-1/PD-L1 immune checkpoint. (Illustration credit: Sceyence Studios)
In transcriptomic analysis of temporal artery tissues STAT1, STAT2 and STAT4 target genes were upregulated, best compatible with dominance of interferon-dependent and IL-12-induced signaling.73 These data identified interferon-producing T cells as key drivers in GCA and encouraged treatment trials with JAK/STAT inhibitors.74 Notably, STAT3-dependent genes were not higher in inflamed than in non-inflamed arteries,73 indicating a minimal role for IL-6 in the vasculitic lesions.
A critical feature of the vasculitic lesions is the lack of expression of the immuno-inhibitory ligand PD-L1.75 (Fig. 5) Expressed on the surface of antigen-presenting cells, PD-L1 binds to the PD-1 receptor and provides a “stop signal” to T cells. Hyperactivity of the PD1-PD-L1 immune checkpoint is a hallmark of cancer cells evading T cell killing and has become an important therapeutic target to enhance anti-tumor immunity. Hypoactivity of the PD1/PD-L1 checkpoint in GCA removes one of the major countermechanisms to control adaptive immunity and allows T cells to respond to stimuli that would otherwise be insufficient to drive the T cell into differentiation. Lack of PD-L1 expression is shared between dendritic cells and macrophages, encompassing all relevant antigen-presenting cell populations.38 PD-L2 expression is intact on GCA macrophages and DC, but cannot substitute for the lacking PD-L1-dependent signals.61 While there is experimental evidence that lesional T cells acquire a tissue-resident phenotype and replenish the infiltrates through self-renewal,62 it is not entirely clear whether the deficiency of the PD-1/PD-L1 checkpoint is mechanistically involved in turning an inflammatory lesion into a self-sustained autonomous structure. The concept that the GCA lesions reach autonomy and renew from within without requiring external cellular supply has strong clinical support. A study harvesting a second biopsy in patients treated for 12 months with aggressive glucocorticoid therapy has reported persistent vasculitis in almost 50% of cases, enforcing reclassification of GCA from a transient into a permanent autoimmune disorder.2
Figure 5. Hypoactivity of the PD-1 (programmed cell death protein-1)/ PD-L1 (programmed cell death ligand-1) checkpoint in giant cell arteritis (GCA).

A functioning PD-1/PD-L1 checkpoint provides negative signals to end the T cell activation and expansion program. In GCA, antigen-presenting cells fail to express the inhibitory ligand PD-L1, preventing the transmission of inhibitory signals. At the same time, GCA T cells are highly responsive to co-stimulatory signals, favoring T cell activation, clonal expansion and effector differentiation. HLA indicates human leukocyte antigen; and TCR, T cell receptor.
Recent reports have indicated that inflamed arteries may contain organized lymphoid structures reminiscent of tertiary lymphoid tissues (TLT).76 In inflamed tissues and around tumors, resident fibroblasts can acquire lymphoid-stroma properties, organizing lymphocytes and antigen-presenting cells into efficient structures.77,78 These structures can give rise to ectopic germinal center and are typically a consequence of chronic, long-standing inflammation, attracting T cells, B cells and antigen presenting cells into the tissue. The formation of tertiary lymphoid tissue is part of the immune aging process, affecting most organ systems and should be expected to occur in the aged host under chronic inflammatory conditions.79 Whether TLT have a functional role in GCA is currently unknown.
Macrophages.
GCA is a granulomatous vasculitis, defining macrophages as key pathogenic cells. The histopathologic diagnosis of GCA requires the presence of highly activated macrophages (histiocytes), combined with multinucleated giant cells in about half of the cases. Macrophages are the most plastic cells of the hematopoietic system and offer enormous functional diversity. They are professional phagocytic cells specialized in the detection, phagocytosis and destruction of pathogens, apoptotic cells, and tissue debris. They also are professional antigen-presenting cells, critically important to trigger and maintain adaptive immunity.
A detailed profiling of lesions macrophages in GCA is not available, but a large array of macrophages products have been placed into the vasculitic infiltrates (Fig.6). With few exceptions, the functional relevance of such macrophage products has been implied and loss-of-function and gain-of-function experiments are not available to provide proof for the pathogenic relevance of macrophage-associated functions. Like in other autoimmune diseases, it is expected that macrophages in the vasculitic lesions fall into multiple functional subsets. For example, single cell RNA sequencing approaches have allowed to distinguish 6 different macrophage populations in the synovial tissue of patients with rheumatoid arthritis80 and these 6 macrophage subsets differ in transcriptomics, positioning and function. All macrophage subpopulation in rheumatoid synovitis shared the high expression of HLA-DR molecules, were engaged in antigen-presentation and had excellent adaptability to the glucose deplete conditions in the tissue niche.80 Based on data available, GCA macrophages excel in a number of functional properties:81
Figure 6. Macrophage heterogeneity in granulomatous vasculitis.
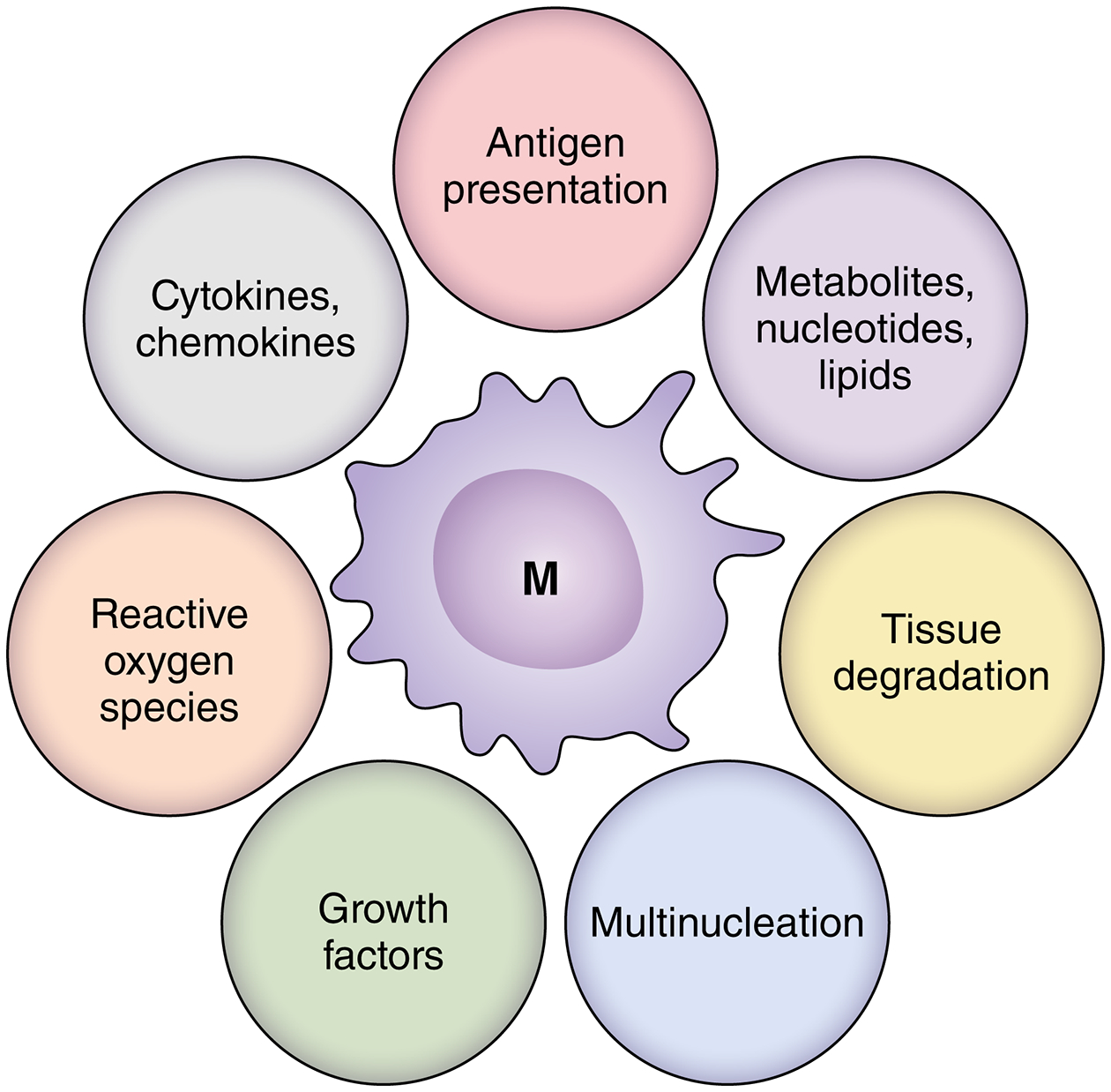
Given the granulomatous character of the vasculitic lesions, macrophages are key effector cells in giant cell arteritis. Some macrophages differentiate into multinucleated giant cells. An array of macrophage products have been localized in the inflamed vessel wall, supporting the concept of extensive heterogeneity of lesional macrophages. The product portfolio implicates macrophages in a multitude of functional domains, including inflammatory responses but also tissue degradative and reparative processes, such as vascular remodeling. Little is known about antigen-presenting functions, but most lesion macrophages are high expressors for HLA-DR. Regional diversity of macrophages suggest that the immediate tissue microenvironment may instruct macrophage activation and differentiation. (Illustration credit: Sceyence Studios).
chemokine expression relevant for recruitment of inflammatory cell38
anti-oxidant defense by upregulating aldose reductase.82 This functional property is particularly important as macrophages accumulating in the inflamed medial layer are characterized by strong mitochondrial activity83
production of growth and angiogenesis factors, driving the vascular remodeling process84,85,86
production of the metalloproteinase MMP-9. MMP-9 is highly expressed in the patient’s macrophages and blocking of MMP-9 enzyme activity is highly efficient in suppressing vasculitis. The bias towards exuberant MMP-9 is shared by monocytes, peripheral macrophages, lesional macrophages and multinucleated giant cells,37 suggestive for a regulatory abnormality controlling this potent tissue destructive enzyme in GCA.
co-stimulating in a CD28-dependent manner62
adopting a permissive state for T cell co-stimulation by failing to express the co-inhibitory ligand PD-L1.75
In aggregate, unique features of lesional macrophages in GCA include their propensity to differentiate into multinucleated giant cells, participate in tissue destruction and co-ordinate the wall remodeling process. Molecularly, their most intriguing abnormality lies in the defective expression of PD-L1, which must have profound impact on their antigen presenting function87,75 and on the breakdown of immune tolerance in GCA.
Conclusions and Perspective
The autoimmune vasculitis GCA manifests in elastin-rich arteries and produces a distinguishing histopathologic pattern: granulomatous infiltrates in the adventitia and media which elicit a fast and concentric remodeling process, including neoangiogenesis in the media and intima and lumen-stenosing intimal hyperplasia. The granulomatous nature of the vessel wall inflammation identifies T cells and macrophages as key pathogenic players in the disease process.
Like in other autoimmune diseases, the loss of self-tolerance (Checkpoint 1) occurs long before clinically evident disease and additional pathogenic events are needed before the asymptomatic host can transition to vascular inflammation. Loss of tissue tolerance (Checkpoint 2) involves tissue-invasive monocytes and T cells entering the vessel wall through a vasa vasorum access port. Several defects in peripheral immune tolerance support the transition of granulomatous arteritis into a self-sustained, tissue-destructive lesion (Checkpoint 3).
Newly identified risk factors for GCA include low body mass, low fasting blood glucose, low triglycerides and low cholesterol, preceding disease onset by years. Thus, metabolic signaling, possibly inverse to that in atherosclerotic disease, may shape early steps of tolerance breakdown. Metabolic signaling also moves early disease steps outside of the blood vessel.
Work over the last decade has identified the following critical T cell defects:
aberrant NOTCH1 signaling on peripheral and lesion CD4 T cells, promoting T cell effector differentiation
aberrant NOTCH4 signaling on peripheral regulatory T cells, destroying their anti-inflammatory capabilities by redirecting intracellular vesicle trafficking
retention of multiple lineages of effector T cells in the granulomatous lesions, producing an array of effector cytokines
The following macrophage defects have been implicated in tissue invasion, antigen presentation and chemokine/cytokine production:
systemic inflammatory response “priming” neutrophils and macrophages and inducing a strong hepatic acute phase response
excessive MMP-9 production, digesting the basement membrane of vasa vasora
defective expression of PD-L1, breaking the protective PD-1/PD-L1 immune checkpoint
pluripotent macrophages, some of them differentiating into multinucleated giant cells
pluripotent macrophages supporting multiple lineages of effector T cells in the vasculitic lesions
The emerging disease paradigm offers multiple interception points to diagnose and manage asymptomatic pre-disease and fully developed vasculitis beyond cytokine blockade:
define metabolic signals that jeopardize immune tolerance in at-risk individuals
define upstream abnormalities that cause hyperactivity of the NOTCH signaling pathway
define functional outcomes of immune checkpoint signaling
identify upstream signals leading to “emergency hematopoiesis”
define the molecular elements of the vasa vasorum-tissue junction to reestablish tissue tolerance
define disease-relevant abnormalities in the innate and adaptive immune system of at-risk individuals before they lose tolerance
The community awaits publication of single cell omics studies which might provide insights into the pathogenic events that lead to GCA.
Acknowledgments
This work was supported in part by the National Institutes of Health (R01AR042527, R01AI108906, R01HL142068, and P01HL129941 to CMW and R01AI108891, R01AG045779, U19AI057266, R01AI129191 to JJG) and the Encrantz Family Discovery Fund.
Non-standard Abbreviations and Acronyms
- GCA
giant cell arteritis
- PMR
polymyalgia rheumatica
- Treg
regulatory T cell
- PD-1
Programmed cell death protein 1
- PD-L1
Programmed Cell Death Ligand 1
- EC
endothelial cell
- NET
neutrophil extracellular traps
- NOX2
NADPH oxidase 2
- DAMP
danger-associated molecular pattern
Footnotes
Disclosures
The authors declare that no conflict of interest exists.
References
- 1.Watanabe R, Berry GJ, Liang DH, Goronzy JJ, Weyand CM. Pathogenesis of Giant Cell Arteritis and Takayasu Arteritis-Similarities and Differences. Curr Rheumatol Rep. 2020;22:68. doi: 10.1007/s11926-020-00948-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maleszewski JJ, Younge BR, Fritzlen JT, Hunder GG, Goronzy JJ, Warrington KJ, Weyand CM. Clinical and pathological evolution of giant cell arteritis: a prospective study of follow-up temporal artery biopsies in 40 treated patients. Mod Pathol. 2017;30:788–796. doi: 10.1038/modpathol.2017.10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wadstrom K, Jacobsson L, Mohammad AJ, Warrington KJ, Matteson EL, Turesson C. Negative associations for fasting blood glucose, cholesterol and triglyceride levels with the development of giant cell arteritis. Rheumatology (Oxford). 2020;59:3229–3236. doi: 10.1093/rheumatology/keaa080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jakobsson K, Jacobsson L, Warrington K, Matteson EL, Liang K, Melander O, Turesson C. Body mass index and the risk of giant cell arteritis: results from a prospective study. Rheumatology (Oxford). 2015;54:433–440. doi: 10.1093/rheumatology/keu331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weyand CM, Hicok KC, Hunder GG, Goronzy JJ. The HLA-DRB1 locus as a genetic component in giant cell arteritis. Mapping of a disease-linked sequence motif to the antigen binding site of the HLA-DR molecule. J Clin Invest. 1992;90:2355–2361. doi: 10.1172/JCI116125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weyand CM, Hunder NN, Hicok KC, Hunder GG, Goronzy JJ. HLA-DRB1 alleles in polymyalgia rheumatica, giant cell arteritis, and rheumatoid arthritis. Arthritis Rheum. 1994;37:514–520. doi: 10.1002/art.1780370411 [DOI] [PubMed] [Google Scholar]
- 7.Carmona FD, Mackie SL, Martin JE, Taylor JC, Vaglio A, Eyre S, Bossini-Castillo L, Castaneda S, Cid MC, Hernandez-Rodriguez J, et al. A large-scale genetic analysis reveals a strong contribution of the HLA class II region to giant cell arteritis susceptibility. Am J Hum Genet. 2015;96:565–580. doi: 10.1016/j.ajhg.2015.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carmona FD, Gonzalez-Gay MA, Martin J. Genetic component of giant cell arteritis. Rheumatology (Oxford). 2014;53:6–18. doi: 10.1093/rheumatology/ket231 [DOI] [PubMed] [Google Scholar]
- 9.Piram M, Maldini C, Mahr A. Effect of race/ethnicity on risk, presentation and course of connective tissue diseases and primary systemic vasculitides. Curr Opin Rheumatol. 2012;24:193–200. doi: 10.1097/BOR.0b013e32835059e5 [DOI] [PubMed] [Google Scholar]
- 10.Saadoun D, Vautier M, Cacoub P. Medium- and Large-Vessel Vasculitis. Circulation. 2021;143:267–282. doi: 10.1161/CIRCULATIONAHA.120.046657 [DOI] [PubMed] [Google Scholar]
- 11.Sharma A, Mohammad AJ, Turesson C. Incidence and prevalence of giant cell arteritis and polymyalgia rheumatica: A systematic literature review. Semin Arthritis Rheum. 2020;50:1040–1048. doi: 10.1016/j.semarthrit.2020.07.005 [DOI] [PubMed] [Google Scholar]
- 12.Crowson CS, Matteson EL. Contemporary prevalence estimates for giant cell arteritis and polymyalgia rheumatica, 2015. Semin Arthritis Rheum. 2017;47:253–256. doi: 10.1016/j.semarthrit.2017.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hill CL, Black RJ, Nossent JC, Ruediger C, Nguyen L, Ninan JV, Lester S. Risk of mortality in patients with giant cell arteritis: A systematic review and meta-analysis. Semin Arthritis Rheum. 2017;46:513–519. doi: 10.1016/j.semarthrit.2016.08.015 [DOI] [PubMed] [Google Scholar]
- 14.Garen T, Lerang K, Hoffmann-Vold AM, Andersson H, Midtvedt O, Brunborg C, Kilian K, Gudbrandsson B, Gunnarsson R, Norby G, et al. Mortality and causes of death across the systemic connective tissue diseases and the primary systemic vasculitides. Rheumatology (Oxford). 2019;58:313–320. doi: 10.1093/rheumatology/key285 [DOI] [PubMed] [Google Scholar]
- 15.Milchert M, Brzosko M. Familial aggregation of longevity in giant cell arteritis and polymyalgia rheumatica. Rheumatol Int. 2020;40:2071–2075. doi: 10.1007/s00296-020-04649-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weyand CM, Goronzy JJ. Immune mechanisms in medium and large-vessel vasculitis. Nat Rev Rheumatol. 2013;9:731–740. doi: 10.1038/nrrheum.2013.161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bassler K, Schulte-Schrepping J, Warnat-Herresthal S, Aschenbrenner AC, Schultze JL. The Myeloid Cell Compartment-Cell by Cell. Annu Rev Immunol. 2019;37:269–293. doi: 10.1146/annurev-immunol-042718-041728 [DOI] [PubMed] [Google Scholar]
- 18.Boettcher S, Manz MG. Regulation of Inflammation- and Infection-Driven Hematopoiesis. Trends Immunol. 2017;38:345–357. doi: 10.1016/j.it.2017.01.004 [DOI] [PubMed] [Google Scholar]
- 19.van Sleen Y, Graver JC, Abdulahad WH, van der Geest KSM, Boots AMH, Sandovici M, Brouwer E. Leukocyte Dynamics Reveal a Persistent Myeloid Dominance in Giant Cell Arteritis and Polymyalgia Rheumatica. Front Immunol. 2019;10:1981. doi: 10.3389/fimmu.2019.01981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mitroulis I, Ruppova K, Wang B, Chen LS, Grzybek M, Grinenko T, Eugster A, Troullinaki M, Palladini A, Kourtzelis I, et al. Modulation of Myelopoiesis Progenitors Is an Integral Component of Trained Immunity. Cell. 2018;172:147–161.e112. doi: 10.1016/j.cell.2017.11.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McDonald B, Pittman K, Menezes GB, Hirota SA, Slaba I, Waterhouse CC, Beck PL, Muruve DA, Kubes P. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science. 2010;330:362–366. doi: 10.1126/science.1195491 [DOI] [PubMed] [Google Scholar]
- 22.Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, Brohi K, Itagaki K, Hauser CJ. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464:104–107. doi: 10.1038/nature08780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pietras EM, Mirantes-Barbeito C, Fong S, Loeffler D, Kovtonyuk LV, Zhang S, Lakshminarasimhan R, Chin CP, Techner JM, Will B, et al. Chronic interleukin-1 exposure drives haematopoietic stem cells towards precocious myeloid differentiation at the expense of self-renewal. Nat Cell Biol. 2016;18:607–618. doi: 10.1038/ncb3346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kovtonyuk LV, Caiado F, Garcia-Martin S, Manz EM, Helbling P, Takizawa H, Boettcher S, Al-Shahrour F, Nombela-Arrieta C, Slack E, et al. IL-1 mediates microbiome-induced inflammaging of hematopoietic stem cells in mice. Blood. 2022;139:44–58. doi: 10.1182/blood.2021011570 [DOI] [PubMed] [Google Scholar]
- 25.Flynn MC, Kraakman MJ, Tikellis C, Lee MKS, Hanssen NMJ, Kammoun HL, Pickering RJ, Dragoljevic D, Al-Sharea A, Barrett TJ, et al. Transient Intermittent Hyperglycemia Accelerates Atherosclerosis by Promoting Myelopoiesis. Circ Res. 2020;127:877–892. doi: 10.1161/circresaha.120.316653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Peshkova IO, Aghayev T, Fatkhullina AR, Makhov P, Titerina EK, Eguchi S, Tan YF, Kossenkov AV, Khoreva MV, Gankovskaya LV, et al. IL-27 receptor-regulated stress myelopoiesis drives abdominal aortic aneurysm development. Nat Commun. 2019;10:5046. doi: 10.1038/s41467-019-13017-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stark MA, Huo Y, Burcin TL, Morris MA, Olson TS, Ley K. Phagocytosis of apoptotic neutrophils regulates granulopoiesis via IL-23 and IL-17. Immunity. 2005;22:285–294. doi: 10.1016/j.immuni.2005.01.011 [DOI] [PubMed] [Google Scholar]
- 28.Ostrowski RA, Metgud S, Tehrani R, Jay WM. Varicella Zoster Virus in Giant Cell Arteritis: A Review of Current Medical Literature. Neuroophthalmology. 2019;43:159–170. doi: 10.1080/01658107.2019.1604763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang L, Ai Z, Khoyratty T, Zec K, Eames HL, van Grinsven E, Hudak A, Morris S, Ahern D, Monaco C, et al. ROS-producing immature neutrophils in giant cell arteritis are linked to vascular pathologies. JCI Insight. 2020;5. doi: 10.1172/jci.insight.139163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Margraf A, Lowell CA, Zarbock A. Neutrophils in acute inflammation: current concepts and translational implications. Blood. 2022;139:2130–2144. doi: 10.1182/blood.2021012295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee KH, Kronbichler A, Park DD, Park Y, Moon H, Kim H, Choi JH, Choi Y, Shim S, Lyu IS, et al. Neutrophil extracellular traps (NETs) in autoimmune diseases: A comprehensive review. Autoimmun Rev. 2017;16:1160–1173. doi: 10.1016/j.autrev.2017.09.012 [DOI] [PubMed] [Google Scholar]
- 32.Palamidas DA, Argyropoulou OD, Georgantzoglou N, Karatza E, Xingi E, Kapsogeorgou EK, Anagnostopoulos CD, Lazaris AC, Ritis K, Goules AV, et al. Neutrophil extracellular traps in giant cell arteritis biopsies: presentation, localization and co-expression with inflammatory cytokines. Rheumatology (Oxford). 2022;61:1639–1644. doi: 10.1093/rheumatology/keab505 [DOI] [PubMed] [Google Scholar]
- 33.Shirai T, Nazarewicz RR, Wallis BB, Yanes RE, Watanabe R, Hilhorst M, Tian L, Harrison DG, Giacomini JC, Assimes TL, et al. The glycolytic enzyme PKM2 bridges metabolic and inflammatory dysfunction in coronary artery disease. J Exp Med. 2016;213:337–354. doi: 10.1084/jem.20150900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tahir S, Steffens S. Nonclassical monocytes in cardiovascular physiology and disease. Am J Physiol Cell Physiol. 2021;320:C761–C770. doi: 10.1152/ajpcell.00326.2020 [DOI] [PubMed] [Google Scholar]
- 35.Narasimhan PB, Marcovecchio P, Hamers AAJ, Hedrick CC. Nonclassical Monocytes in Health and Disease. Annu Rev Immunol. 2019;37:439–456. doi: 10.1146/annurev-immunol-042617-053119 [DOI] [PubMed] [Google Scholar]
- 36.Galbo H, Kall L. Circadian variations in clinical symptoms and concentrations of inflammatory cytokines, melatonin, and cortisol in polymyalgia rheumatica before and during prednisolone treatment: a controlled, observational, clinical experimental study. Arthritis Res Ther. 2016;18:174. doi: 10.1186/s13075-016-1072-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Watanabe R, Maeda T, Zhang H, Berry GJ, Zeisbrich M, Brockett R, Greenstein AE, Tian L, Goronzy JJ, Weyand CM. MMP (Matrix Metalloprotease)-9-Producing Monocytes Enable T Cells to Invade the Vessel Wall and Cause Vasculitis. Circ Res. 2018;123:700–715. doi: 10.1161/CIRCRESAHA.118.313206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Watanabe R, Hilhorst M, Zhang H, Zeisbrich M, Berry GJ, Wallis BB, Harrison DG, Giacomini JC, Goronzy JJ, Weyand CM. Glucose metabolism controls disease-specific signatures of macrophage effector functions. JCI Insight. 2018;3. doi: 10.1172/jci.insight.123047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhao TV, Sato Y, Goronzy JJ, Weyand CM. T-Cell Aging-Associated Phenotypes in Autoimmune Disease. Front Aging. 2022;3:867950. doi: 10.3389/fragi.2022.867950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.De Maeyer RPH, Chambers ES. The impact of ageing on monocytes and macrophages. Immunol Lett. 2021;230:1–10. doi: 10.1016/j.imlet.2020.12.003 [DOI] [PubMed] [Google Scholar]
- 41.Evans MA, Sano S, Walsh K. Cardiovascular Disease, Aging, and Clonal Hematopoiesis. Annu Rev Pathol. 2020;15:419–438. doi: 10.1146/annurev-pathmechdis-012419-032544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Beck DB, Ferrada MA, Sikora KA, Ombrello AK, Collins JC, Pei W, Balanda N, Ross DL, Ospina Cardona D, Wu Z, et al. Somatic Mutations in UBA1 and Severe Adult-Onset Autoinflammatory Disease. N Engl J Med. 2020;383:2628–2638. doi: 10.1056/NEJMoa2026834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gloor AD, Berry GJ, Goronzy JJ, Weyand CM. Age as a risk factor in vasculitis. Semin Immunopathol. 2022;44:281–301. doi: 10.1007/s00281-022-00911-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Akiyama M, Ohtsuki S, Berry GJ, Liang DH, Goronzy JJ, Weyand CM. Innate and Adaptive Immunity in Giant Cell Arteritis. Front Immunol. 2020;11:621098. doi: 10.3389/fimmu.2020.621098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Piggott K, Deng J, Warrington K, Younge B, Kubo JT, Desai M, Goronzy JJ, Weyand CM. Blocking the NOTCH pathway inhibits vascular inflammation in large-vessel vasculitis. Circulation. 2011;123:309–318. doi: 10.1161/CIRCULATIONAHA.110.936203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wen Z, Shen Y, Berry G, Shahram F, Li Y, Watanabe R, Liao YJ, Goronzy JJ, Weyand CM. The microvascular niche instructs T cells in large vessel vasculitis via the VEGF-Jagged1-Notch pathway. Sci Transl Med. 2017;9. doi: 10.1126/scitranslmed.aal3322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sanchez-Martin M, Ferrando A. The NOTCH1-MYC highway toward T-cell acute lymphoblastic leukemia. Blood. 2017;129:1124–1133. doi: 10.1182/blood-2016-09-692582 [DOI] [PubMed] [Google Scholar]
- 48.Jin K, Wen Z, Wu B, Zhang H, Qiu J, Wang Y, Warrington KJ, Berry GJ, Goronzy JJ, Weyand CM. NOTCH-induced rerouting of endosomal trafficking disables regulatory T cells in vasculitis. J Clin Invest. 2021;131. doi: 10.1172/JCI136042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wen Z, Shimojima Y, Shirai T, Li Y, Ju J, Yang Z, Tian L, Goronzy JJ, Weyand CM. NADPH oxidase deficiency underlies dysfunction of aged CD8+ Tregs. J Clin Invest. 2016;126:1953–1967. doi: 10.1172/JCI84181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Huotari J, Helenius A. Endosome maturation. EMBO J. 2011;30:3481–3500. doi: 10.1038/emboj.2011.286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jin K, Parreau S, Warrington KJ, Koster MJ, Berry GJ, Goronzy JJ, Weyand CM. Regulatory T Cells in Autoimmune Vasculitis. Front Immunol. 2022;13:844300. doi: 10.3389/fimmu.2022.844300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Radtke F, MacDonald HR, Tacchini-Cottier F. Regulation of innate and adaptive immunity by Notch. Nat Rev Immunol. 2013;13:427–437. doi: 10.1038/nri3445 [DOI] [PubMed] [Google Scholar]
- 53.Christopoulos PF, Gjølberg TT, Krüger S, Haraldsen G, Andersen JT, Sundlisæter E. Targeting the Notch Signaling Pathway in Chronic Inflammatory Diseases. Front Immunol. 2021;12:668207. doi: 10.3389/fimmu.2021.668207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Siebel C, Lendahl U. Notch Signaling in Development, Tissue Homeostasis, and Disease. Physiol Rev. 2017;97:1235–1294. doi: 10.1152/physrev.00005.2017 [DOI] [PubMed] [Google Scholar]
- 55.Wagner AD, Bjornsson J, Bartley GB, Goronzy JJ, Weyand CM. Interferon-gamma-producing T cells in giant cell vasculitis represent a minority of tissue-infiltrating cells and are located distant from the site of pathology. Am J Pathol. 1996;148:1925–1933. [PMC free article] [PubMed] [Google Scholar]
- 56.Pryshchep O, Ma-Krupa W, Younge BR, Goronzy JJ, Weyand CM. Vessel-specific Toll-like receptor profiles in human medium and large arteries. Circulation. 2008;118:1276–1284. doi: 10.1161/CIRCULATIONAHA.108.789172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Watanabe R, Berry GJ, Liang DH, Goronzy JJ, Weyand CM. Cellular Signaling Pathways in Medium and Large Vessel Vasculitis. Front Immunol. 2020;11:587089. doi: 10.3389/fimmu.2020.587089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Espigol-Frigole G, Planas-Rigol E, Ohnuki H, Salvucci O, Kwak H, Ravichandran S, Luke B, Cid MC, Tosato G. Identification of IL-23p19 as an endothelial proinflammatory peptide that promotes gp130-STAT3 signaling. Sci Signal. 2016;9:ra28. doi: 10.1126/scisignal.aad2357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Baldini M, Maugeri N, Ramirez GA, Giacomassi C, Castiglioni A, Prieto-Gonzalez S, Corbera-Bellalta M, Di Comite G, Papa I, Dell’antonio G, et al. Selective up-regulation of the soluble pattern-recognition receptor pentraxin 3 and of vascular endothelial growth factor in giant cell arteritis: relevance for recent optic nerve ischemia. Arthritis Rheum. 2012;64:854–865. doi: 10.1002/art.33411 [DOI] [PubMed] [Google Scholar]
- 60.Pulsatelli L, Boiardi L, Assirelli E, Pazzola G, Muratore F, Addimanda O, Dolzani P, Versari A, Casali M, Bottazzi B, et al. Imbalance between angiogenic and anti-angiogenic factors in sera from patients with large-vessel vasculitis. Clin Exp Rheumatol. 2020;38 Suppl 124:23–30. [PubMed] [Google Scholar]
- 61.Amersfoort J, Eelen G, Carmeliet P. Immunomodulation by endothelial cells - partnering up with the immune system? Nat Rev Immunol. 2022;22:576–588. doi: 10.1038/s41577-022-00694-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang H, Watanabe R, Berry GJ, Nadler SG, Goronzy JJ, Weyand CM. CD28 Signaling Controls Metabolic Fitness of Pathogenic T Cells in Medium and Large Vessel Vasculitis. J Am Coll Cardiol. 2019;73:1811–1823. doi: 10.1016/j.jacc.2019.01.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Deng J, Younge BR, Olshen RA, Goronzy JJ, Weyand CM. Th17 and Th1 T-cell responses in giant cell arteritis. Circulation. 2010;121:906–915. doi: 10.1161/CIRCULATIONAHA.109.872903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Samson M, Audia S, Fraszczak J, Trad M, Ornetti P, Lakomy D, Ciudad M, Leguy V, Berthier S, Vinit J, et al. Th1 and Th17 lymphocytes expressing CD161 are implicated in giant cell arteritis and polymyalgia rheumatica pathogenesis. Arthritis Rheum. 2012;64:3788–3798. doi: 10.1002/art.34647 [DOI] [PubMed] [Google Scholar]
- 65.Crotty S Follicular helper CD4 T cells (TFH). Annu Rev Immunol. 2011;29:621–663. doi: 10.1146/annurev-immunol-031210-101400 [DOI] [PubMed] [Google Scholar]
- 66.Terrier B, Geri G, Chaara W, Allenbach Y, Rosenzwajg M, Costedoat-Chalumeau N, Fouret P, Musset L, Benveniste O, Six A, et al. Interleukin-21 modulates Th1 and Th17 responses in giant cell arteritis. Arthritis Rheum. 2012;64:2001–2011. doi: 10.1002/art.34327 [DOI] [PubMed] [Google Scholar]
- 67.Zerbini A, Muratore F, Boiardi L, Ciccia F, Bonacini M, Belloni L, Cavazza A, Cimino L, Moramarco A, Alessandro R, et al. Increased expression of interleukin-22 in patients with giant cell arteritis. Rheumatology (Oxford). 2018;57:64–72. doi: 10.1093/rheumatology/kex334 [DOI] [PubMed] [Google Scholar]
- 68.Keir M, Yi Y, Lu T, Ghilardi N. The role of IL-22 in intestinal health and disease. J Exp Med. 2020;217:e20192195. doi: 10.1084/jem.20192195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dudakov JA, Hanash AM, van den Brink MR. Interleukin-22: immunobiology and pathology. Annu Rev Immunol. 2015;33:747–785. doi: 10.1146/annurev-immunol-032414-112123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ciccia F, Rizzo A, Guggino G, Cavazza A, Alessandro R, Maugeri R, Cannizzaro A, Boiardi L, Iacopino DG, Salvarani C, et al. Difference in the expression of IL-9 and IL-17 correlates with different histological pattern of vascular wall injury in giant cell arteritis. Rheumatology (Oxford). 2015;54:1596–1604. doi: 10.1093/rheumatology/kev102 [DOI] [PubMed] [Google Scholar]
- 71.Angkasekwinai P, Dong C. IL-9-producing T cells: potential players in allergy and cancer. Nat Rev Immunol. 2021;21:37–48. doi: 10.1038/s41577-020-0396-0 [DOI] [PubMed] [Google Scholar]
- 72.Gieseck RL 3rd, Wilson MS, Wynn TA. Type 2 immunity in tissue repair and fibrosis. Nat Rev Immunol. 2018;18:62–76. doi: 10.1038/nri.2017.90 [DOI] [PubMed] [Google Scholar]
- 73.Zhang H, Watanabe R, Berry GJ, Tian L, Goronzy JJ, Weyand CM. Inhibition of JAK-STAT Signaling Suppresses Pathogenic Immune Responses in Medium and Large Vessel Vasculitis. Circulation. 2018;137:1934–1948. doi: 10.1161/circulationaha.117.030423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Koster MJ, Crowson CS, Giblon RE, Jaquith JM, Duarte-García A, Matteson EL, Weyand CM, Warrington KJ. Baricitinib for relapsing giant cell arteritis: a prospective open-label 52-week pilot study. Ann Rheum Dis. 2022;81:861–867. doi: 10.1136/annrheumdis-2021-221961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhang H, Watanabe R, Berry GJ, Vaglio A, Liao YJ, Warrington KJ, Goronzy JJ, Weyand CM. Immunoinhibitory checkpoint deficiency in medium and large vessel vasculitis. Proc Natl Acad Sci U S A. 2017;114:E970–E979. doi: 10.1073/pnas.1616848114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ciccia F, Rizzo A, Maugeri R, Alessandro R, Croci S, Guggino G, Cavazza A, Raimondo S, Cannizzaro A, Iacopino DG, et al. Ectopic expression of CXCL13, BAFF, APRIL and LT-beta is associated with artery tertiary lymphoid organs in giant cell arteritis. Ann Rheum Dis. 2017;76:235–243. doi: 10.1136/annrheumdis-2016-209217 [DOI] [PubMed] [Google Scholar]
- 77.Gago da Graca C, van Baarsen LGM, Mebius RE. Tertiary Lymphoid Structures: Diversity in Their Development, Composition, and Role. J Immunol. 2021;206:273–281. doi: 10.4049/jimmunol.2000873 [DOI] [PubMed] [Google Scholar]
- 78.Sautes-Fridman C, Petitprez F, Calderaro J, Fridman WH. Tertiary lymphoid structures in the era of cancer immunotherapy. Nat Rev Cancer. 2019;19:307–325. doi: 10.1038/s41568-019-0144-6 [DOI] [PubMed] [Google Scholar]
- 79.Sato Y, Oguchi A, Fukushima Y, Masuda K, Toriu N, Taniguchi K, Yoshikawa T, Cui X, Kondo M, Hosoi T, et al. CD153/CD30 signaling promotes age-dependent tertiary lymphoid tissue expansion and kidney injury. J Clin Invest. 2022;132. doi: 10.1172/JCI146071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hu Z, Zhao TV, Huang T, Ohtsuki S, Jin K, Goronzy IN, Wu B, Abdel MP, Bettencourt JW, Berry GJ, et al. The transcription factor RFX5 coordinates antigen-presenting function and resistance to nutrient stress in synovial macrophages. Nat Metab. 2022;4:759–774. doi: 10.1038/s42255-022-00585-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Weyand CM, Watanabe R, Zhang H, Akiyama M, Berry GJ, Goronzy JJ. Cytokines, growth factors and proteases in medium and large vessel vasculitis. Clin Immunol. 2019;206:33–41. doi: 10.1016/j.clim.2019.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rittner HL, Hafner V, Klimiuk PA, Szweda LI, Goronzy JJ, Weyand CM. Aldose reductase functions as a detoxification system for lipid peroxidation products in vasculitis. J Clin Invest. 1999;103:1007–1013. doi: 10.1172/JCI4711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rittner HL, Kaiser M, Brack A, Szweda LI, Goronzy JJ, Weyand CM. Tissue-destructive macrophages in giant cell arteritis. Circ Res. 1999;84:1050–1058. doi: 10.1161/01.res.84.9.1050 [DOI] [PubMed] [Google Scholar]
- 84.Kaiser M, Weyand CM, Bjornsson J, Goronzy JJ. Platelet-derived growth factor, intimal hyperplasia, and ischemic complications in giant cell arteritis. Arthritis Rheum. 1998;41:623–633. doi: 10.1002/1529-0131(199804)41:4<623::AID-ART9>3.0.CO;2-6 [DOI] [PubMed] [Google Scholar]
- 85.Kaiser M, Younge B, Bjornsson J, Goronzy JJ, Weyand CM. Formation of new vasa vasorum in vasculitis. Production of angiogenic cytokines by multinucleated giant cells. Am J Pathol. 1999;155:765–774. doi: 10.1016/S0002-9440(10)65175-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.van Sleen Y, Jiemy WF, Pringle S, van der Geest KSM, Abdulahad WH, Sandovici M, Brouwer E, Heeringa P, Boots AMH. A Distinct Macrophage Subset Mediating Tissue Destruction and Neovascularization in Giant Cell Arteritis: Implication of the YKL-40/Interleukin-13 Receptor alpha2 Axis. Arthritis Rheumatol. 2021;73:2327–2337. doi: 10.1002/art.41887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Watanabe R, Shirai T, Namkoong H, Zhang H, Berry GJ, Wallis BB, Schaefgen B, Harrison DG, Tremmel JA, Giacomini JC, et al. Pyruvate controls the checkpoint inhibitor PD-L1 and suppresses T cell immunity. J Clin Invest. 2017;127:2725–2738. doi: 10.1172/JCI92167 [DOI] [PMC free article] [PubMed] [Google Scholar]