

Abstract
Lysine acetyltransferase (KAT) enzymes including the p300, MYST, and GCN5 families play major roles in modulating the structure of chromatin and regulating transcription. Because of their dysregulation in various disease states including cancer, efforts to develop inhibitors of KATs have steadily gained momentum. Here we provide an overview of recent progress on the development of high quality chemical probes of the p300 and MYST family of KATs and how they are emerging as useful tools for basic and translational investigation.
The reversible acetylation of Lys residues on histones and other proteins has emerged over the past 25 years as a major mechanism of gene and cellular regulation.1 Lysine acetyltransferases (KATs), originally known as histone acetyltransferases (HATs), catalyze the transfer of acetyl groups from acetyl-CoA to Lys side chains. This action is opposed by Lys deacetylases (KDACS, HDACs) which include Zn metallohydrolase HDACS and NAD-dependent sirtuins.1 Acetylation of a Lys sidechain neutralizes its positive charge and can create a binding surface for bromodomain containing proteins and proteins with YEATS domains.1–3 Where this serves an epigenetic function these three activities have been described as “writing” (acetylation), “erasing” (deacetylation), and “reading” (binding) the epigenetic code. More than 38,000 protein Lys acetylation sites have been mapped in mass spectrometry-based proteomics studies, making Lys acetylation among the most prevalent post-translational modifications (PTMs) in biology (https://www.phosphosite.org/staticSiteStatistics).4 Lys acetylation is mutually exclusive with other common PTMs, including Lys ubiquitination and methylation, contributing to a broad spectrum of chemical diversity. Despite its abundance, functional understanding of Lys acetylation is incomplete. With several notable exceptions, such as Lys acetylation on histone H3 and histone H4, defining the biological roles of protein Lys acetylation has been elusive.1
For more than a decade, small molecule inhibitors of HDACs and bromodomains have been extensively use as pharmacology tools for linking the general importance of Lys acetylation to regulation of gene expression and cell proliferation.5 There are at least six FDA-approved HDAC inhibitors used to treat malignancies, and numerous bromodomain antagonists have entered clinical trials for a range of conditions.1 In contrast, development of high potency, specific KAT inhibitors with desirable pharmacological properties has lagged.
Much of the effort in KAT inhibitor discovery has targeted three small families in humans. These include the p300 family which is comprised of paralogs p300 (KAT3B) and CBP (KAT3A), the MYST family which contains MOF (KAT8), MOZ (KAT6A), MORF (KAT6B), HBO1 (KAT7), and Tip60 (KAT5), and the GCN5 family that includes the paralogs GCN5 (KAT2A) and PCAF (KAT2B).6–8 Each of these KATs employs acetyl-CoA cofactor to directly transfer the acetyl group from the cofactor to protein Lys sidechains. The p300/CBP, MYST, and GCN5/PCAF family members are all predominantly localized to the nucleus. Each of these KATs are known to target Lys sites on histone N-terminal tails, but p300/CBP have long been shown to acetylate many Lys residues in non-histone proteins.6 The p300 and CBP enzymes have been implicated in a wide range of physiological and disease processes involving the heart, immune system, hematopoiesis, and endocrine function among others.6 They are widely known as transcriptional coactivators and prominent in chromatin regions associated with transcriptional enhancer activity. Loss of function p300/CBP mutations are commonly observed in B cell lymphomas. The MYST enzymes appear to play important roles in hematologic malignancies and GCN5 and PCAF are key in developmental biology and suggested to play roles in cancer.9, 10
Efforts to target p300/CBP and GCN5/PCAF KATs with small molecules date back more than two decades, shortly after they were discovered. Potent bisubstrate peptide-CoA conjugates were found to selectively block p300/CBP over GCN5/PCAF. These proved effective as structural tools, but were not cell penetrant due to their large size and charge.11 X-ray crystal structures of KATs in complex with CoA analogs revealed that the active sites of these enzymes interact most intimately with the phosphate and pantetheine groups of CoA, and minimally with the adenine moiety.12–14 Such molecular recognition contrasts the highly druggable protein kinase family which engulf the adenosine moiety of ATP.15 Beyond bisubstrate analogs, a range of hydrophobic natural products were reported as low potency KAT inhibitors between 2000 and 2010.6, 16 While cell permeable, most show quite promiscuous pharmacology.17 In silico screening led to development of the moderately potent and selective p300/CBP inhibitor C646 in 2010 (figure 1).18 It has shown some value as a tool compound for p300/CBP inhibition, but has since been supplanted by the more recently developed compounds.19
Figure 1. Small molecule KAT inhibitors.
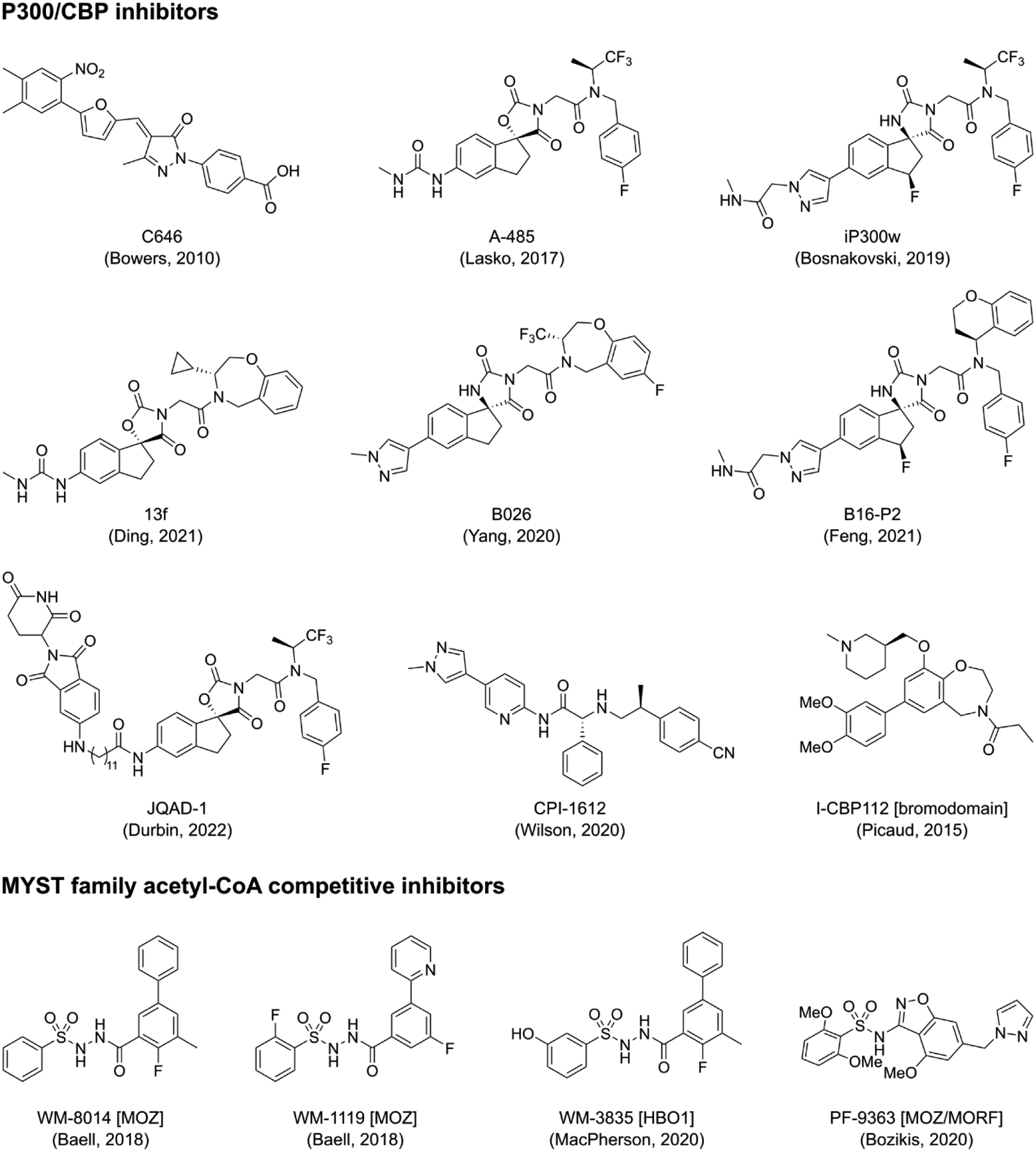
Representative small molecule inhibitors of p300/CBP (top) and MYST (bottom) family acetyltransferases that have been linked to pharmacological studies described here. All the depicted p300/CBP inhibitors are active site competitive, except where parenthetically noted. MYST family member isoform selectivity is noted parenthetically.
P300/CBP inhibitor A-485
The discovery of A-485, a spiro-oxazolidinedione, stemmed from virtual ligand screening and extensive medicinal chemistry (figure 1).20 A-485 showed low nanomolar potencies against both 300 and CBP acetyltransferase activities and an X-ray structure of A-485 in complex with the p300 HAT domain revealed that it occupies an overlapping binding site with the CoA phosphopantetheine moiety (figure 2).20 Kinetic studies confirm that A-485 is competitive with acetyl-CoA in p300 inhibition. Compared to prior p300/CBP small molecule inhibitors, A-485 is much more potent in cellular studies with IC50 for histone acetylation in the sub-micromolar range versus >10 μM observed with earlier agents. Notably, A-485 treatment shows clean blockade of histone H3 K18 and K27 acetylation, but not H3 K9 acetylation, mimicking p300/CBP gene knockdowns.20 Validation of the specificity of p300/CBP intracellular inhibition was also achieved using a closely related structural analog of A-485, A-486, in which the methyl-urea substituent is shifted by one carbon from its position in A-485. A-486 shows >100-fold reduced p300 affinity and, as expected, minimal effects on cellular acetylation.20 A-485 also displays practical pharmacokinetic properties permitting its applications in animal studies.20
Figure 2. P300 catalytic domain substrate interactions and drug bound state.
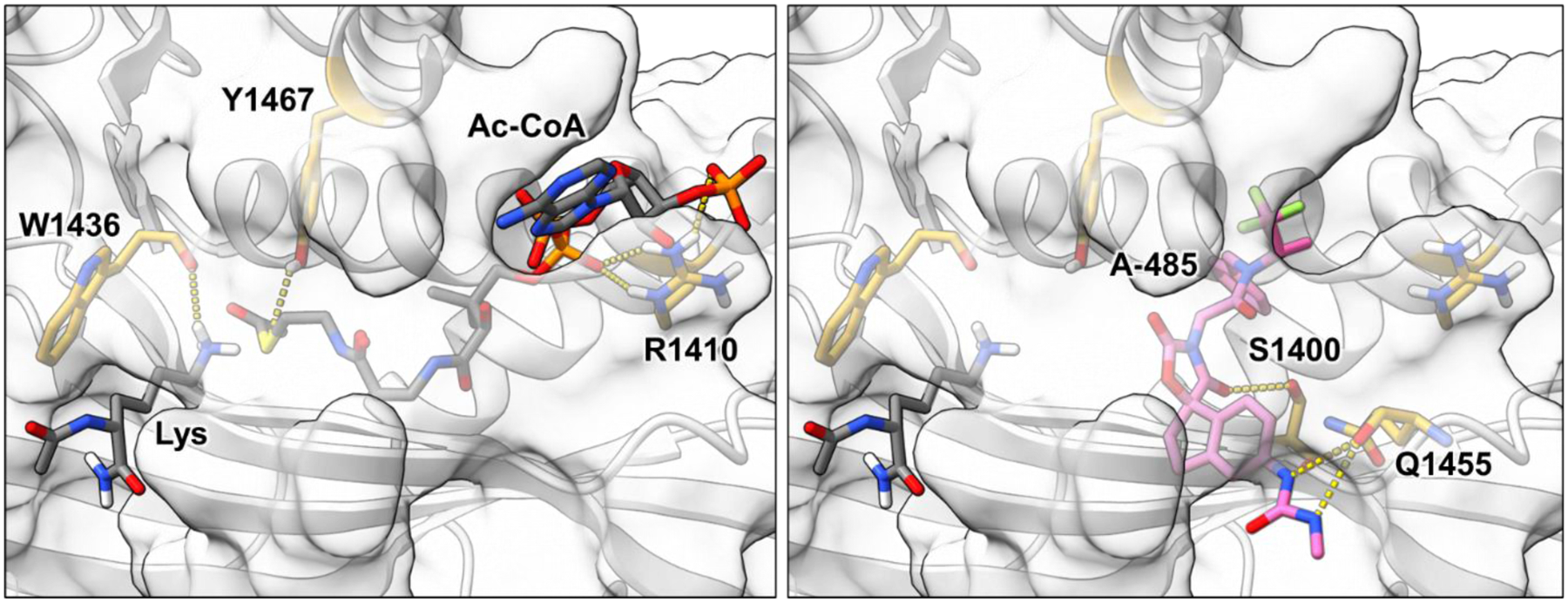
Left. Catalytic interactions of the p300 HAT domain (white) including electrostatic interactions between Arg1410 (yellow) and the phosphodiester and phosphate of CoA, stabilizing proton donation from Tyr1467 (yellow) to the CoA thioester, and hydrogen bonding between the carbonyl of Trp1436 (yellow) and the substrate lysine Nε (PDBID 3BIY, 4PZS).14, 54, 55 Composite substrate-bound structure was energy minimized using Rosetta 3.13 fast relax.50 Right. Occupancy of the acetyl-CoA binding site by competitive ligand A-485 (pink) with hydrogen bonding interactions between the N-methyl urea and carbonyl of Gln1455 (yellow), and hydrogen bonding between Ser1400 (yellow) and one carbonyl of the spiroxazolidinedione illustrated. This binding is anchored by numerous additional hydrophobic interactions that are not depicted here (PDBID 5KJ2).20
A-485 and global effects on Lys acetylation
Tens of thousands of cellular protein Lys acetylation sites have been mapped across multiple studies. In general, it has been difficult to assign an acetyltransferase to a specific Lys acetylation event using genetic approaches because of the potential for indirect effects of such knockdowns. A-485 and related analogs proved very useful in linking p300/CBP catalytic activity to about 1000 Lys acetylations, ~10% of the total acetyl-Lys population.21 Notably, the vast majority of the acetyl-Lys levels reduced by small molecule inhibition of p300/CBP overlapped with p300/CBP genetic knockdowns but were largely unaffected by p300/CBP bromodomain antagonism.21 Most of the A-485 modulated Lys acetylation sites were nuclear, as expected for the well-established cellular localization of p300/CBP.21 Many of the presumed p300/CBP acetyl-Lys sites were mapped to transcriptional machinery including RNA polymerase subunits, transcription factors, and epigenetic enzymes. Often these acetyl-Lys sites were found in basic clusters on the same polypeptide, giving rise to the description of p300/CBP activity as an “acetylspray.”21 Moreover, A-485 proved particularly useful in defining site-specific acetylation half-lives, revealing rapidly and slowly turned over acetyl-Lys sites, likely related to the accessibility of these positions to deacetylase enzymes.21 There were several hundred Lys acetylations with cellular half-lives of less than 1 hour following A-485 treatment. Several of these highly dynamic acetyl-Lys sites were found on the N-terminal tail of histone H2B.21 Relative to the intensively studied tails of histone H3 and H4, histone H2B tail PTMs have received little attention but these acetylomic studies suggest that H2B Lys acetylation might play a significant role in biology.22 Interestingly, using in vitro acetylated nucleosomes and HDAC complexes, it was found that several of these H2B acetyl-Lys sites are efficiently removed by the MIDAC and CoREST HDAC complexes.23 Of note, careful analysis of transcriptional rates using A-485 showed a tight correlation with mRNA synthesis and histone acetylation dynamics.21, 24 These results are consistent with the model that rapidly reversible Lys acetylation may drive transcription initiation and/or elongation as postulated.
P300/CBP inhibition effects in physiology and disease
The discovery of A-485 and next generation p300/CBP inhibitor analogs has enabled a number of studies to characterize the roles of p300/CBP acetyltransferase activity in development and disease mechanisms. In the context of endocrinology, p300/CBP has been implicated in glucose homeostasis. The beta cell of pancreatic islets is responsible for insulin production. It has been shown that A-485 treatment impairs glucose-stimulated insulin secretion from beta cells.25 Molecular studies revealed that this occurred through a transcriptional network in which A-485 prevented the expression of glucose sensing genes including the transcription factors Hnf1α and Foxo1.25 As a result, beta cell maturation was blocked. However, A-485 could also block lipogenesis in white adipose tissue and liver, which would tend to have therapeutic value in metabolic disease.26 Moreover, A-485 was also shown to limit inflammation in acute liver injury.27
Osteoporosis involves a decrease in bone density that can increase fractures and is a common affliction of the elderly, especially post-menopausal women. Bone density is regulated in part by the reciprocal action of osteoclasts and osteoblasts, cells that resorb and fortify bone tissue, respectively. It was found in a mouse model of osteoporosis that A-485 can block osteoclast differentiation by antagonizing the RANKL molecular program.28 In this fashion, A-485 was found to prevent bone loss in this ovariectomized mouse model of post-menopausal osteoporosis.28
A-485 has been shown to possess antineoplastic activity in a wide range of cancer types. Among several melanoma cell lines, the strength of the anti-proliferative effects of p300/CBP inhibitor A-485 was shown to correlate with high levels of expression of the transcription factor MITF.29, 30 MITF is an established melanocyte and melanoma transcriptional activator and has been linked to driving the growth a subset of melanomas. A-485 treatment was shown to reduce MITF levels by blocking its transcription and this in turn impeded the production of the growth-related forkhead box M1 (FOXM1) transcription factor.30
Multiple myeloma proliferation has been shown to be especially sensitive to p300/CBP inhibition.31 Interestingly, a determinant of multiple myeloma sensitivity was observed to be the histone deacetylase complex HDAC3/NCOR1/SMRT. Genetic depletion of the NCOR1 complex promoted resistance to A-485 mediated p300/CBP inhibition and the pharmacological effects of HDAC3-selective inhibitor RGFP966 antagonized the hypoacetylation induced by A-485. As H3K27 acetylation is an important acetylation sites associated with p300/CBP, A-485 was shown to enhance H3K27me3 at the expense of H3K27ac. Based on this H3K27 acetyl to methyl swapping, Hogg et al hypothesized that A-485 treated multiple myeloma might be especially vulnerable to antagonism of the H3K27me3 demethylase KDM6A. Using genetic or pharmacological manipulation of KDM6A with GSK-J4, they validated this potential.31
Prostate cancer has also been shown to be sensitive to p300/CBP inhibition.20 This was most clearly observed with androgen receptor-dependent prostate cancer. Unlike the anti-androgen effects of androgen receptor splice variant prostate cancer which becomes anti-androgen-resistant, A-485 maintains its potency in such splice variant prostate cancer.20 Interestingly, combining A-485 with the p300/CBP bromodomain antagonist I-CBP112 was shown to be synergistic in slowing prostate cancer cell growth. This appeared to be connected to the depletion of p300 from chromatin conferred by the combination of A-485 and I-CBP112, presumably because of the diminished histone acetylation conferred by A-485.32
Recent studies with A-485 and even newer potent and selective p300/CBP HAT inhibitors have shown promise in blocking the growth of prostate cancer (B026 & B16-P2),33, 34 breast cancer,35 glioblastoma (CPI-1612),36, 37 mantle cell lymphoma,38 Myb-dependent acute leukemia, NOTCH-dependent acute leukemia,39 NUT midline carcinoma,40 CIC-DUX4 sarcoma (iP300w),41 ovarian cancer (13f),42 and pituitary adenoma (figure 1).43 On the other hand, A-485 enhanced the proliferative capacity of Tet2 mutant leukemia.44 It remains to be seen whether the therapeutic window of even the high quality p300/CBP inhibitors is sufficiently large to be effective for treating cancer in people, given the “on-target” toxicity of such compounds. In addition, recent studies suggest that A-485 inhibition of cancer cell proliferation may engender resistance by upregulating acetyl-CoA biosynthesis.45
MYST family inhibitors
Recent advances have also been made in the development of MYST acetyltransferase inhibitors. The human MYST enzymes Tip60 (KAT5), MOZ (KAT6A), MORF (KAT6B), HBO1 (KAT7), and MOF (KAT8) have been shown to be embedded in specific and large multi-protein complexes. Tip60 is part of the NuA4 complex, and KAT6A/B are centerpieces of the MOZ/MORF complex, and KAT7 is a member of the HBO1 complex. Each of these complexes acts are mostly in the nucleus and have multiple histone reader domain containing proteins that helps recruit them to specific sites in chromatin. Loss of function mutations in these enzymatic complexes have been linked to developmental disorders. Recent genetic and biochemical studies have indicated that the NuA4 complex is responsible for H4K16 acetylation,46 the MOZ/MORF complex is associated with H3K9 and K23 acetylation,47 the HBO1 complex primarily acetylates H3K1410, while the KAT8/MOF complex appears to be responsible for acetylating H4K16.48
Based on a combination of high throughput screening, medicinal chemistry, and structure-based design, the acyl-sulfonylhydrazides were shown to be broad spectrum MYST family inhibitors.9 The initial successful compounds including WM-8014 and WM-1119 were especially potent against KAT6A/B with WM-1119 having in vivo bioavailability.9 These MYST family inhibitors compete against acetyl-CoA and X-ray structures have revealed their detailed molecular recognition with the acetyltransferase domain active sites (figure 3). Various follow-up analogs of these compounds have been reported with heightened selectivity for either KAT6A/B or KAT7 including PF-9363 which appears to be specific for KAT6A/KAT6B.49, 50 These MYST small molecule inhibitors have helped establish the histone Lys targeting of these enzyme complexes. These compounds have also shown promise in the treatment in model systems of various hematologic malignancies including leukemias, lymphomas, as well as hepatocellular carcinoma. In particular, KAT7 appears to be critical for the maintenance of leukemic stem cells and KAT6A/KAT6B in ER+ breast cancer.10, 50 KAT6A is often involved in chromosomal translocations that lead to KAT6A fusion proteins that can drive blood cancers rendering them sensitive to agents like WM-1119.51 It has recently been shown that WM-1119 can upregulate cyclin dependent kinase inhibitor 2A (p16) thereby blocking cyclin dependent kinases Cdk4 and Cdk6 in acute myeloid leukemia and synergizing with chemotherapy to slow the growth of this malignancy.52
Figure 3. KAT6 catalytic domain substrate interactions and drug bound state.
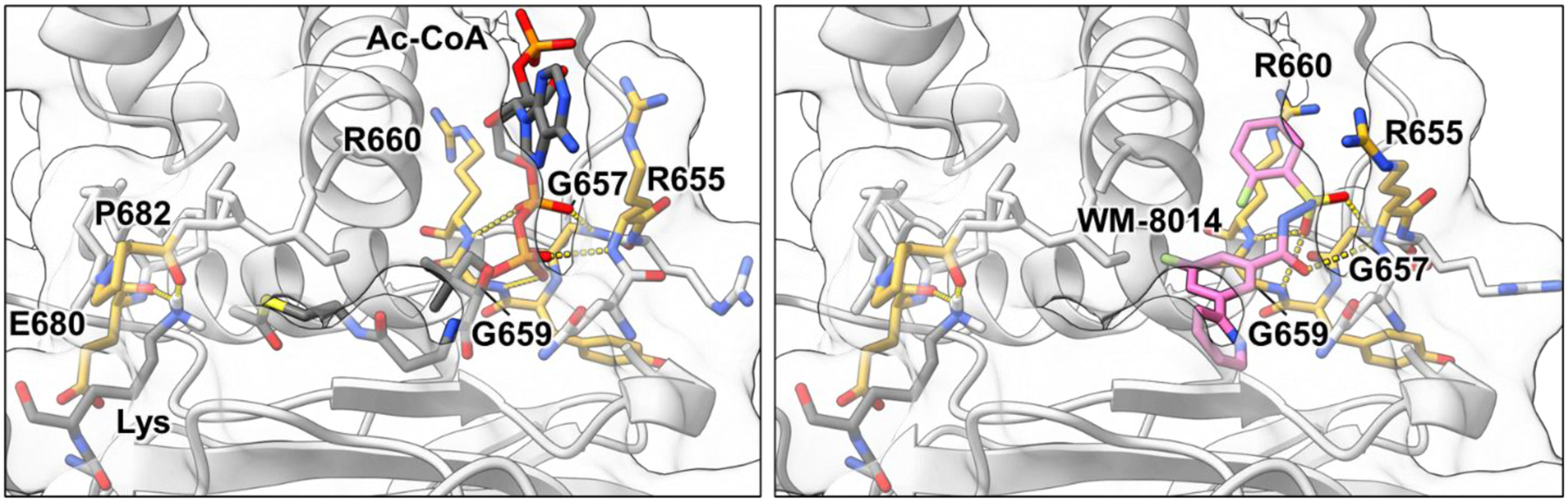
Left. Catalytic interactions of the MYST1 HAT domain (white) including hydrogen bonding between the phosphodiester and phosphate of CoA and the backbone amides of Arg655, Gly657, Gly659 and R660 (yellow). Hydrogen bonding between the carbonyl of Glu680 Pro682 (yellow) direct the substrate lysine Nε (PDBID 6BA4, 6VO5).9, 56 Composite substrate-bound structure was energy minimized using Rosetta 3.13 fast relax.50 Right. Occupancy of the acetyl-CoA binding site by competitive ligand WM-8014 (pink) including hydrogen bonding interactions between the acylsulfonylhydrazide core and the backbone amides of Arg655, Gly657, Gly659 and R660 (yellow). This binding is anchored by additional hydrophobic interactions that are not depicted here (PDBID 6CT2).9
Summary and outlook
The past 5 years have been a fertile time in the development of potent and selective pharmacological inhibitors of several lysine acetyltransferase enzymes in the p300/CBP and MYST family. These compounds have become powerful mechanistic probes to study the basic functions of these enzymes and their potential as therapeutic targets. Next generation p300/CBP inhibitors may offer even more exquisite specificity such as reported for the p300 degrader compound based on A-485 (JQAD-1).53 However, high quality pharmacological probes are still lacking for PCAF/GCN5 acetyltransferase enzymes. Current challenges include how to achieve strong in vivo inhibition of the requisite acetyltransferase for treating cancer and other disorders, understanding mechanisms of acquired resistance, and identifying relevant biomarkers. It is likely that specific combinations of acetyltransferase inhibitors and other epigenetic and non-epigenetic agents will be key elements of therapeutic strategies for the treatment of cancers and other diseases.
Acknowledgments
We thank the NIH (GM62437), the Leukemia and Lymphoma Society, and the American Cancer Society (PF-20-105-01-DMC to S.D.W) for financial support. P.A.C. declares that he is a cofounder and equity holder of Acylin Therapeutics and has been a paid consultant for Abbvie Inc. which have developed p300/CBP acetyltransferase inhibitors.
Declaration of interests
Philip Cole reports financial support was provided by National Institute of General Medical Sciences.
Philip Cole reports was provided by AbbVie Inc. Philip Cole reports a relationship with Acylin Therapeutics and Abbvie Inc that includes: consulting or advisory and equity or stocks. Philip Cole has patent #U.S. patent# 9,005,670 issued April 14, 2015 with royalties paid to Acylin Therapeutics. Co-editor for COCB
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- (1).Wang ZA; Cole PA The Chemical Biology of Reversible Lysine Post-translational Modifications. Cell Chem Biol. 2020, 27, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Filippakopoulos P; Picaud S; Mangos M; et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell. 2012, 149 (1), 214–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Li X; Liu S; Li X; Li XD YEATS Domains as Novel Epigenetic Readers: Structures, Functions, and Inhibitor Development. ACS Chem Biol. 2021. [DOI] [PubMed] [Google Scholar]
- (4).Hornbeck PV; Zhang B; Murray B; Kornhauser JM; Latham V; Skrzypek E PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res. 2015, 43 (D1), D512–D520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Shorstova T; Foulkes WD; Witcher M Achieving clinical success with BET inhibitors as anti-cancer agents. Br J Cancer. 2021, 124 (9), 1478–1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Dancy BM; Cole PA Protein Lysine Acetylation by p300/CBP. Chem Rev. 2015, 115 (6), 2419–2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Sapountzi V; Côté J MYST-family histone acetyltransferases: Beyond chromatin. Cell Mol Life Sci. 2011, 68 (7), 1147–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Koutelou E; Farria AT; Dent SYR Complex functions of Gcn5 and Pcaf in development and disease. Biochim Biophys Acta - Gene Regul Mech. 2021, 1864 (2), 194609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9)*.Baell JB; Leaver DJ; Hermans SJ; et al. Inhibitors of histone acetyltransferases KAT6A/B induce senescence and arrest tumour growth. Nature. 2018, 560 (7717), 253–257. [DOI] [PubMed] [Google Scholar]; Described the first MYST family competitive inhibitors with robust in vivo activity and selectivity.
- (10).MacPherson L; Anokye J; Yeung MM; et al. HBO1 is required for the maintenance of leukaemia stem cells. Nature. 2020, 577 (7789), 266–270. [DOI] [PubMed] [Google Scholar]
- (11).Lau OD; Kundu TK; Soccio RE; et al. HATs off: Selective synthetic inhibitors of the histone acetyltransferases p300 and PCAF. Mol Cell. 2000, 5 (3), 589–595. [DOI] [PubMed] [Google Scholar]
- (12).Yan Y; Barlev NA; Haley RH; Berger SL; Marmorstein R Crystal structure of yeast Esa1 suggests a unified mechanism for catalysis and substrate binding by histone acetyltransferases. Mol Cell. 2000, 6 (5), 1195–1205. [DOI] [PubMed] [Google Scholar]
- (13).Poux AN; Cebrat M; Kim CM; Cole PA; Marmorstein R Structure of the GCN5 histone acetyltransferase bound to a bisubstrate inhibitor. Proc Natl Acad Sci U S A. 2002, 99 (22), 14065–14070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Liu X; Wang L; Zhao K; et al. The structural basis of protein acetylation by the p300/CBP transcriptional coactivator. Nature. 2008, 451 (7180), 846–850. [DOI] [PubMed] [Google Scholar]
- (15).Wang Z; Cole PA Catalytic Mechanisms and Regulation of Protein Kinases. In: Shokat KM, ed. Protein Kinase Inhib. Res. Med Vol. 548. Academic Press; 2014:1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Arif M; Pradhan SK; Thanuja GR; et al. Mechanism of p300 specific histone acetyltransferase inhibition by small molecules. J Med Chem. 2009, 52 (2), 267–277. [DOI] [PubMed] [Google Scholar]
- (17).Dahlin JL; Nelson KM; Strasser JM; et al. Assay interference and off-target liabilities of reported histone acetyltransferase inhibitors. Nat Commun 2017 81. 2017, 8 (1), 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Bowers EM; Yan G; Mukherjee C; et al. Virtual ligand screening of the p300/CBP histone acetyltransferase: Identification of a selective small molecule inhibitor. Chem Biol. 2010, 17 (5), 471–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19)*.Ogiwara H; Sasaki M; Mitachi T; et al. Targeting p300 addiction in CBP-deficient cancers causes synthetic lethality by apoptotic cell death due to abrogation of MYC expression. Cancer Discov. 2016, 6 (4), 430–445. [DOI] [PubMed] [Google Scholar]; Described a p300/CBP competitive inhibitor with robust in vivo activity and selectivity.
- (20).Lasko LM; Jakob CG; Edalji RP; et al. Discovery of a selective catalytic p300/CBP inhibitor that targets lineage-specific tumours. Nature. 2017, 550 (7674), 128–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21)*.Weinert BT; Narita T; Satpathy S; et al. Time-Resolved Analysis Reveals Rapid Dynamics and Broad Scope of the CBP/p300 Acetylome. Cell. 2018, 174 (1), 231–244.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]; Identified unique cellular targets of p300/CBP through acetylation-focused proteomic analysis following catalytic inhibition. Site-specific acetylation half-lives were measured and found to range from minutes to hours. Data set is searchable online through a graphic user interface at http://p300db.choudharylab.org.
- (22)*.Narita T; Higashijima Y; Kilic S; Liebner T; Walter J; Choudhary C A unique H2B acetylation signature marks active enhancers and predicts their target genes. bioRxiv. 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]; Characterizes gene enhancers by H2B acetylation, suggesting that these short half-life sites provide a more accurate picture of recent enhancer activity than the canonical H3K27ac.
- (23).Wang ZA; Whedon SD; Wu M; et al. Histone H2B Deacylation Selectivity: Exploring Chromatin’s Dark Matter with an Engineered Sortase. J Am Chem Soc. 2022, 144 (8), 3360–3364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Hsu E; Zemke NR; Berk AJ Promoter-specific changes in initiation, elongation, and homeostasis of histone H3 acetylation during CBP/p300 inhibition. Elife. 2021, 10,. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Zhang L; Sheng C; Zhou F; et al. CBP/p300 HAT maintains the gene network critical for β cell identity and functional maturity. Cell Death Dis. 2021, 12 (5), 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Zhou F; Liu Q; Zhang L; et al. Selective inhibition of CBP/p300 HAT by A-485 results in suppression of lipogenesis and hepatic gluconeogenesis. Cell Death Dis. 2020, 11 (9), 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Peng J; Li J; Huang J; et al. P300/CBP inhibitor A-485 alleviates acute liver injury by regulating macrophage activation and polarization. Theranostics. 2019, 9 (26), 8344–8361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Huo S; Liu X; Zhang S; et al. p300/CBP inhibitor A-485 inhibits the differentiation of osteoclasts and protects against osteoporotic bone loss. Int Immunopharmacol. 2021, 94, 1567–5769. [DOI] [PubMed] [Google Scholar]
- (29).Wang R; He Y; Robinson V; et al. Targeting Lineage-specific MITF Pathway in Human Melanoma Cell Lines by A-485, the Selective Small-molecule Inhibitor of p300/CBP. Mol Cancer Ther. 2018, 17 (12), 2543–2550. [DOI] [PubMed] [Google Scholar]
- (30).Kim E; Zucconi BE; Wu M; et al. MITF expression predicts therapeutic vulnerability to p300 inhibition in human melanoma. Cancer Res. 2019, 79 (10), 2649–2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Hogg SJ; Motorna O; Cluse LA; et al. Targeting histone acetylation dynamics and oncogenic transcription by catalytic P300/CBP inhibition. Mol Cell. 2021, 81 (10), 2183–2200.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Zucconi BE; Makofske JL; Meyers DJ; et al. Combination Targeting of the Bromodomain and Acetyltransferase Active Site of p300/CBP. Biochemistry. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Yang Y; Zhang R; Li Z; et al. Discovery of Highly Potent, Selective, and Orally Efficacious p300/CBP Histone Acetyltransferases Inhibitors. J Med Chem. 2020, 63 (3), 1337–1360. [DOI] [PubMed] [Google Scholar]
- (34).Feng L; Yu S; Wang H; et al. Synthesis and biological evaluation of spirocyclic chromane derivatives as a potential treatment of prostate cancer. Molecules. 2021, 26 (11), 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Waddell A; Mahmud I; Ding H; Huo Z; Liao D Pharmacological Inhibition of CBP/p300 Blocks Estrogen Receptor Alpha (ERα) Function through Suppressing Enhancer H3K27 Acetylation in Luminal Breast Cancer. Cancers (Basel). 2021, 13 (11), 2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Wilson JE; Patel G; Patel C; et al. Discovery of CPI-1612: A Potent, Selective, and Orally Bioavailable EP300/CBP Histone Acetyltransferase Inhibitor. ACS Med Chem Lett. 2020, 11 (6), 1324–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Mladek AC; Yan H; Tian S; et al. Neuro-Oncology RBBP4-p300 axis modulates expression of genes essential for cell survival and is a potential target for therapy in glioblastoma. Neuro Oncol. 2022, 24 (8), 1261–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Zhou X. ru.; Li X; Liao L. ping.; et al. P300/CBP inhibition sensitizes mantle cell lymphoma to PI3Kδ inhibitor idelalisib. Acta Pharmacol Sin. 2021, 43 (2), 457–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Tottone L; Zhdanovskaya N; Pestaña ÁC; et al. Histone modifications drive aberrant notch3 expression/activity and growth in T-ALL. Front Oncol. 2019, 9 (APR), 198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Zhang X; Zegar T; Lucas A; et al. Therapeutic targeting of p300/CBP HAT domain for the treatment of NUT midline carcinoma. Oncogene. 2020, 39 (24), 4770–4779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Bosnakovski D; Ener ET; Cooper MS; et al. Inactivation of the CIC-DUX4 oncogene through P300/CBP inhibition, a therapeutic approach for CIC-DUX4 sarcoma. Oncogenesis. 2021, 10 (10), 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Ding H; Pei Y; Li Y; et al. Design, synthesis and biological evaluation of a novel spiro oxazolidinedione as potent p300/CBP HAT inhibitor for the treatment of ovarian cancer. Bioorganic Med Chem. 2021, 52 (October), 116512. [DOI] [PubMed] [Google Scholar]
- (43).Ji C; Xu W; Ding H; et al. The p300 Inhibitor A-485 Exerts Antitumor Activity in Growth Hormone Pituitary Adenoma. J Clin Endocrinol Metab. 2022, 107 (6), e2291–e2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Man N; Mas G; Karl DL; et al. p300 suppresses the transition of myelodysplastic syndromes to acute myeloid leukemia. JCI Insight. 2021, 6 (19),. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Bishop TR; Subramanian C; Bilotta EM; et al. Metabolic adaptations underpin resistance to histone acetyltransferase inhibition. bioRxiv. 2022, 2022.08.12.503669. [Google Scholar]
- (46).Qu K; Chen K; Wang H; Li X; Chen Z Structure of the NuA4 acetyltransferase complex bound to the nucleosome. Nature. 2022, 610, 569. [DOI] [PubMed] [Google Scholar]
- (47)*.Klein BJ; Jang SM; Lachance C; et al. Histone H3K23-specific acetylation by MORF is coupled to H3K14 acylation. Nat Commun. 2019, 10 (1),. [DOI] [PMC free article] [PubMed] [Google Scholar]; Provides structural, biochemical, and cellular evidence for recognition of H3K14 acylation by KAT6B, which directs its K23 acylation activity.
- (48).Proietti G; Wang Y; Punzo C; Mecinović J Substrate Scope for Human Histone Lysine Acetyltransferase KAT8. Int J Mol Sci. 2021, 22 (2), 846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Bozikis YE; Brodsky O; Camerino MA; et al. Benzisoxazole sulfonamide derivatives. WO 2020/254946 A1. 2020. [Google Scholar]
- (50).Sharma S; Chung J; Uryu S; et al. Abstract 1130: First-in-class KAT6A/KAT6B inhibitor CTx-648 (PF-9363) demonstrates potent anti-tumor activity in ER+ breast cancer with KAT6A dysregulation. Cancer Res. 2021, 81 (13_Supplement), 1130. [Google Scholar]
- (51).Su J; Wang X; Bai Y; Sun M; Yao Y; Duan Y The role of MOZ/KAT6A in hematological malignancies and advances in MOZ/KAT6A inhibitors. Pharmacol Res. 2021, 174, 105930. [DOI] [PubMed] [Google Scholar]
- (52).Ling VY; Straube J; Godfrey W; et al. Targeting cell cycle and apoptosis to overcome chemotherapy resistance in acute myeloid leukemia. Leuk 2022. 2022, 1–11. [DOI] [PubMed] [Google Scholar]
- (53)*.Durbin AD; Wang T; Wimalasena VK; et al. EP300 Selectively Controls the Enhancer Landscape of MYCN-Amplified Neuroblastoma. Cancer Discov. 2022, 12 (3), 730–751. [DOI] [PMC free article] [PubMed] [Google Scholar]; Describes a p300-selective degrader using A-485
- (54).Maksimoska J; Segura-Peña D; Cole PA; Marmorstein R Structure of the p300 histone acetyltransferase bound to acetyl-coenzyme a and its analogues. Biochemistry. 2014, 53 (21), 3415–3422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Leaver-Fay A; Tyka M; Lewis SM; et al. Rosetta3: An Object-Oriented Software Suite for the Simulation and Design of Macromolecules. Methods Enzymol. 2011, 487 (C), 545–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Halabelian L; Zeng H; Dong A; et al. Crystal structure of Human histone acetytransferase 1 (HAT1) in complex with isobutyryl-COA and K12A mutant variant of histone H4. 2020.