

Abstract
Background
Newborn screening (NBS) algorithms for cystic fibrosis (CF) vary in the United State of America and include different cystic fibrosis transmembrane conductance regulator (CFTR) variants. CFTR variant distribution varies among racial and ethnic groups.
Objective
Our objectives were to identify differences in detection rate by race and ethnicity for CFTR variant panels, identify each US state detection rate for CFTR variant panels, and describe the rate of false‐negative NBS and delayed diagnoses by race and ethnicity.
Methods
This is a cross‐sectional analysis of the detection rate of at least 1 CFTR variant for seven panels by race and ethnicity in genotyped people with CF (PwCF) or CFTR‐related metabolic syndrome (CRMS)/CFTR‐related disorders in CF Foundation Patient Registry (CFFPR) in 2020. We estimated the case detection rate of CFTR variant panels by applying the detection rate to Census data. Using data from CFFPR, we compared the rate of delayed diagnosis or false‐negative NBS by race and ethnicity.
Results
For all panels, detection of at least 1 CFTR variant was highest in non‐Hispanic White PwCF (87.5%−97.0%), and lowest in Black, Asian, and Hispanic PwCF (41.9%−93.1%). Detection of at least 1 CFTR variant was lowest in Black and Asian people with CRMS/CFTR‐related disorders (48.4%−64.8%). States with increased racial and ethnic diversity have lower detection rates for all panels. Overall, 3.8% PwCF had a false‐negative NBS and 11.8% had a delayed diagnosis; Black, Hispanic, and mixed‐race PwCF were overrepresented.
Conclusion
CFTR variant panels have lower detection rates in minoritized racial and ethnic groups leading to false‐negative NBS, delayed diagnosis, and likely health disparities.
Keywords: CFTR‐related disorder, CRMS/CFSPID, cystic fibrosis, health disparities, newborn screening
1. INTRODUCTION
Since late 2009, all 50 states in the United State and the District of Columbia (DC) screen newborns for cystic fibrosis (CF). Early diagnosis and early treatment through newborn screening (NBS) reduces severe symptoms, such as failure to thrive or salt depletion, and are associated with improved nutrition, pulmonary outcomes, and survival. 1 , 2 , 3 , 4 , 5 , 6 NBS is performed using an initial biomarker test, immunoreactive trypsinogen (IRT); when elevated, a second‐tier, DNA‐based test has been used by all states and DC since 2020 to detect either the most common c.1521_1523del (p.Phe508del, legacy: F508del), allele or a panel of variants in the CF transmembrane conductance regulator (CFTR) gene. 7 , 8 NBS algorithms vary greatly among NBS programs, 7 and all require a sweat chloride test for confirmation or exclusion of diagnosis. 8 During the past decade, next‐generation sequencing (NGS) of CFTR has led to evolving, expanded variant panels. 9 , 10 These NGS‐generated panels have developed in association with the CFTR2 project 11 (cftr2.org) and evolved to include 139−372 variants. 12 , 13 For example, the Wisconsin‐expanded panel (WI‐expanded herein) evolved from 129 to 285 variants and currently tests for 372 pathogenic variants listed in the CFTR2 database plus c.350G>A (p.Arg117His, legacy R117H) is assessed in relationship to the poly‐T tract. 13 In most states, a screen is out‐of‐range, or positive, only when at least one CF‐causing CFTR variant is noted on the test. Some infants with a positive screen have an intermediate sweat test value with fewer than two CF‐causing variants, referred to as CFTR‐related metabolic syndrome (CRMS) or CF screen positive inconclusive diagnosis (CFSPID). 14 , 15 Follow‐up of these infants is recommended as a diagnosis of CF is sometimes made after an initial diagnosis of CRMS. 15
CF occurs in people of all ethnicities and races across the United State. 16 A study of 6359 new patients diagnosed through NBS and registered in the CF Foundation Patient Registry (CFFPR) during 2010−2018 revealed that about 20% were from racial and ethnic minority groups. 17 Although race and ethnicity are social constructs, CFTR genetic variant distribution varies between racial and ethnic groups and is dependent on ancestry admixture. 18 , 19 Minoritized racial and ethnic groups are more likely to have rare or de novo variants not represented on CFTR variant panels. 20 Similar to CF, variant panels screening for other diseases were developed based on a non‐Hispanic White population. 21
Most CF NBS algorithms usually require the detection of at least 1 CFTR variant for a test to be considered positive and would be falsely reported as negative if the infant had CF but no variants are detected. Given that CFTR variant frequency varies between racial and ethnic groups, there is a concern that CF NBS genetic panels can be associated with a disparity in diagnosis. This is an important equity issue that has ethical implications. 22 The first objective of our report is to identify differences in case detection rates for each racial and ethnic group for common CF genetic panels. Our next objective is to identify each US state, Washington DC, and Puerto Rico case detection rate for frequently used CFTR variant panels based on state population. Our third objective is to describe the predicted rate of false‐negative NBS for demographic racial and ethnic groups.
2. METHODS
2.1. Overall design
This is a cross‐sectional analysis of CFTR variants in all (N = 46,729) genotyped people in CFFPR in 2020 to estimate the detection of at least 1 CFTR variant or 2 CFTR variants using seven CFTR genetic panels, currently used by NBS Laboratories, by race and ethnicity. Detection of at least 1 CFTR variant includes those with 1 or 2 CFTR variants identified, which would trigger a positive NBS. Two groups were analyzed: people with CF (PwCF) and people with either CFTR‐related disorders or CRMS/CFSPID (PwCRMS). 23 We also performed a cross‐sectional analysis of all genotyped people in the CFFPR in 2020 who were born since 2010, when there was universal NBS for CF in the United State, to determine the number of missed newborn screen or delayed diagnoses. The CFFPR is a retrospective observational study of patients from accredited CF centers which includes 93.7% of (PwCF) who receive care in accredited CF centers in the United State. 24 Informed consent if given by either the PwCF or their guardian to have their data included in the CFFPR.
2.2. Measurements
2.2.1. Case detection estimation by race and ethnicity
We determined the detection rate of at least 1 or 2 CFTR variants for seven CFTR genetic panels for racial and ethnic groups in all genotyped PwCF in CFFPR in 2020. We determined the detection rate of at least 1 or 2 CFTR variants for seven CFTR genetic panels for racial and ethnic groups in all in all genotyped PwCRMS in CFFPR in 2020.
2.2.2. State case detection estimation
To determine the case detection rate by various CFTR genetic panels for each state, we first identified CFTR variant distribution for racial and ethnic groups in all genotyped PwCF in CFFPR in 2020. We then determined by racial and ethnic groups the percentage of cases that had at least 1 CFTR variant identified by different CFTR variant panels. Next, we applied the case detection rate for each racial and ethnic group overall in the United State and in each state plus DC and Puerto Rico to the 2020 US Census population data to determine the estimated case detection.
Racial and ethnic groups were defined as in the 2010 US Census in CFFPR. Individuals were categorized as: American Indian or Alaska Native, Asian, Black or African American, Hispanic, Native Hawaiian or Other Pacific Islander, or White. Mixed‐race included “other race” or more than one race. State data was based on the state of birth for each patient as recorded in the CFFPR. The 2020 US Census population data obtained from data.census.gov was used to estimate the minimum number of missed cases for each state with the assumption the distribution of CFTR variants by racial and ethnic group is the same as that in the diagnosed population with CF.
2.3. CFTR variant panels
Based on communications with NBS Laboratories, the following CFTR variant panels were analyzed: ACMG‐23 (23 variants), 25 Agena74 (74 variants), 26 Luminex39 (39 variants), 27 Luminex60 (60 variants), 28 Illumina139 (139 variants), 29 WI‐expanded (372 variants), 30 “CF‐Causing Variants & VVCCs panel” which is 280 pathogenic variants from the CFTR2 database plus the 49 variants with varying clinical consequences (VVCCs) (329 variants), and all 401 pathogenic variants in CFTR2 as of April 29, 2022. VVCCs are variants that, when paired with a CF disease causing variant, may cause CF in some people but not others. 31 In addition, the analysis was performed with F508del only since two states are including only this variant in the second tier of their CF NBS algorithms.
2.3.1. False‐negative NBS analysis
We did a cross‐sectional analysis of all patients in CFFPR from January 1, 2011 to December 31, 2020 diagnosed with CF after a false‐negative NBS. A yes/no question about diagnosis after a false‐negative NBS was first implemented in the CFFPR in 2013 and is not mandatory. We compared the race and ethnicity of the subjects with false‐negative NBS and then those with a false‐negative NBS and delayed diagnosis (>180 days of life).
Results with numbers 5 or fewer were reported as ≤5 rather than the exact number to avoid reidentification per CFF guidelines.
3. RESULTS
Detection of at least 1 CFTR variant ranged from 83.9% to 96.1% overall and detection of 2 CFTR variants ranged from 43.4% to 83.6% overall for each CFTR variant panel, but detection rates were lower in minoritized racial and ethnic groups (Tables 1 and 2). There was a higher detection rate of variants for all race and ethnicity groups with CFTR variant panels that included more variants compared to fewer variants. The Luminex139, WI‐expanded, and 280 pathogenic variant panel with 49 VVCCs, and CFTR2 list had higher case detection rates, while ACMG‐23 had the lowest case detection rate. Interestingly, and reflecting the rarity of most pathogenic CFTR variants, adding in 262 variants beyond the 139 of the Illumina139 panel to reach the current 401 in CFTR2 leads to only a few percentage points of increase in detection. In addition, the addition of VVCCs led to a larger increase in detection rates of 2 CFTR variants compared to at least 1 CFTR variant.
Table 1.
Detection of 1 variant in fully genotyped people with CF
| CFTR variant panel | Total population | African American/Black non‐Hispanic | American Indian, Alaskan Native, non‐Hispanic | Asian, non‐Hispanic | Hispanic | Mixed races, non‐Hispanic | Native Hawaiian Pacific Islander, non‐Hispanic | White, non‐Hispanic |
|---|---|---|---|---|---|---|---|---|
| Total N | 46,729 | 1699 | 144 | 217 | 4006 | 1128 | 7 | 39,528 |
| F508del | 39,192 (83.9%) | 913 (53.7%) | 92 (63.9%) | 91 (41.9%) | 2647 (66.1%) | 867 (76.9%) | 7 (100%) | 34,575 (87.5%) |
| ACMG‐23 | 43,376 (92.8%) | 1247 (73.4%) | 122 (84.7%) | 122 (56.2%) | 3243 (81.0%) | 979 (86.8%) | 7 (100.0%) | 37,656 (95.3%) |
| Luminex39 | 43,891 (93.9%) | 1332 (78.4%) | 123 (85.4%) | 135 (62.2%) | 3386 (84.5%) | 1005 (89.1%) | 7 (100.0%) | 37,903 (95.9%) |
| Luminex60 | 44,150 (94.5%) | 1365 (80.3%) | 125 (86.8%) | 145 (66.8%) | 3540 (88.4%) | 1022 (90.6%) | 7 (100.0%) | 38,046 (96.3%) |
| Agena | 44,176 (94.5%) | 1354 (79.7%) | 126 (87.5%) | 142 (65.4%) | 3534 (88.2%) | 1018 (90.2%) | 7 (100.0%) | 37,995 (96.1%) |
| Illumina139 | 44,589 (95.4%) | 1417 (83.4%) | 131 (91.0%) | 157 (72.4%) | 3601 (89.9%) | 1041 (92.3%) | 7 (100.0%) | 38,235 (96.7%) |
| WI_expanded | 44,683 (95.6%) | 1431 (84.2%) | 131 (91.0%) | 164 (75.6%) | 3699 (92.3%) | 1053 (93.4%) | 7 (100.0%) | 38,198 (96.6%) |
| CFTR2 database | 44,529 (95.3%) | 1429 (84.1%) | 131 (91.0%) | 167 (77.0%) | 3688 (92.1%) | 1050 (93.1%) | 7 (100.0%) | 38,057 (96.3%) |
| CF‐causing variants & VVCCs | 44,902 (96.1%) | 1462 (86.1%) | 131 (91.0%) | 168 (77.4%) | 3729 (93.1%) | 1062 (94.1%) | 7 (100.0%) | 38,343 (97.0%) |
Abbreviations: CF, cystic fibrosis; CFTR, cystic fibrosis transmembrane conductance regulator; VVCC,variants with varying clinical consequences.
Tablee 2.
Detection of 2 variants in fully genotyped people with CF
| CFTR variant panel | Total population | African American/Black non‐Hispanic | American Indian, Alaskan Native, non‐Hispanic | Asian, non‐Hispanic | Hispanic | Mixed races, non‐Hispanic | Native Hawaiian Pacific Islander, non‐Hispanic | White, non‐Hispanic |
|---|---|---|---|---|---|---|---|---|
| Total N | 46,729 | 1699 | 144 | 217 | 4006 | 1128 | 7 | 39,528 |
| F508del | 20,259 (43.4%) | 242 (14.2%) | 49 (34.0%) | 37 (17.1%) | 977 (24.4%) | 362 (32.1%) | ≤5 (≤71.4%) | 18,587 (47.0%) |
| ACMG‐23 | 31,446 (67.3%) | 537 (31.6%) | 94 (65.3%) | 55 (25.3%) | 1752 (43.7%) | 573 (50.8%) | ≤5 (≤71.4%) | 28,430 (71.9%) |
| Luminex39 | 33,020 (70.7%) | 692 (40.7%) | 96 (66.7%) | 70 (32.3%) | 1983 (49.5%) | 639 (56.6%) | ≤5 (≤71.4%) | 29,535 (74.7%) |
| Luminex60 | 34,630 (74.1%) | 752 (44.3%) | 102 (70.8%) | 80 (36.9%) | 2351 (58.7%) | 699 (62.0%) | ≤5 (≤71.4%) | 30,641 (77.5%) |
| Agena | 34,642 (74.1%) | 746 (43.9%) | 103 (71.5%) | 77 (35.5%) | 2326 (58.1%) | 684 (60.6%) | ≤5 (≤71.4%) | 30,701 (77.7%) |
| Illumina139 | 37,048 (79.3%) | 857 (50.4%) | 112 (77.8%) | 94 (43.3%) | 2499 (62.4%) | 773 (68.5%) | ≤5 (≤71.4%) | 32,708 (82.7%) |
| WI_expanded | 37,946 (81.2%) | 973 (57.3%) | 111 (77.1%) | 108 (49.8%) | 2796 (69.8%) | 809 (71.7%) | 6 (85.7%) | 33,143 (83.8%) |
| CFTR2 database | 36,560 (78.2%) | 969 (57.0%) | 108 (75.0%) | 112 (51.8%) | 2750 (68.6%) | 785 (69.6%) | 6 (85.7%) | 31,830 (80.5%) |
| CF‐causing variants & VVCCs | 39,078 (83.6%) | 1033 (60.8%) | 113 (78.5%) | 117 (53.9%) | 2952 (73.7%) | 857 (76.0%) | ≤5 (≤71.4%) | 34,001 (86.0%) |
Abbreviations: CF, cystic fibrosis; CFTR, cystic fibrosis transmembrane conductance regulator; VVCC,variants with varying clinical consequences.
Case detection, which is the identification of at least 1 CFTR variant, varied greatly between each race and ethnicity for all CFTR variant panels in PwCF (Table 1). Detection of at least 1 CFTR variant was 84.7%−91.0% in American Indian & Alaskan Native PwCF, 56.2%−77.4% in Asian PwCF, 73.4%−86.1% in Black/African American PwCF, 81.0%−94.1% in Hispanic PwCF, and 86.8%−94.1% in mixed‐race PwCF. There was little improvement in detection rate with an increased variant number in non‐Hispanic White PwCF. Hawaiian & Pacific Islander PwCF had 100% detection rate for all panels. There was very low detection of at least 1 copy of F508del in Black/African American, American Indian & Alaskan Native, Asian, and Hispanic PwCF.
Detection of 2 CFTR variants was lower than detection of at least 1 CFTR variant in all panels for each race and ethnicity group in PwCF. For all CFTR variant panels, detection of 2 CFTR variants was highest in non‐Hispanic White PwCF, ranging from 71.9% to 86.0% (Table 2). Detection of 2 CFTR variants was lower in all other races and ethnicities: 65.3%−78.5% in American Indian & Alaskan Native PwCF, 25.3%−53.9% in Asian PwCF, 31.6%−60.8% in Black/African American PwCF, 49.5%−76.0% in Hispanic PwCF, 36.7%−73.2% in mixed‐race PwCF, and ≤71.4%−85.7% in Hawaiian & Pacific Islander PwCF.
As expected, based on the definition of CRMS/CFSPID, PwCRMS had lower detection of both at least 1 CFTR variant with all variant panels compared to in PwCF. There was very low case detection of 2 CFTR variants for all races and ethnicities except for non‐Hispanic White PwCRMS (Table 3). Detection of at least 1 CFTR variant in PwCRMS varied greatly between race and ethnicity groups (Table 4). Detection of at least 1 CFTR variant was high in Hispanic, mixed‐race, and non‐Hispanic White PwCRMS.
Table 3.
Detection of 1 variant in fully genotyped people with CRMS and CFTR‐related disorder
| CFTR variant panel | Total population | African American/Black non‐Hispanic | American Indian, Alaskan Native, non‐Hispanic | Asian, non‐Hispanic | Hispanic | Mixed races, non‐Hispanic | Native Hawaiian Pacific Islander, non‐Hispanic | White, non‐Hispanic |
|---|---|---|---|---|---|---|---|---|
| Total N | 2604 | 122 | ≤5 | 21 | 392 | 112 | ≤5 | 1951 |
| ACMG‐23 | 1895 (72.8%) | 59 (48.4%) | ≤5 | 11 (52.4%) | 252 (64.3%) | 73 (65.2%) | ≤5 | 1490 (76.4%) |
| Luminex39 | 2015 (77.4%) | 69 (56.6%) | ≤5 | 12 (57.1%) | 279 (71.2%) | 81 (72.3%) | ≤5 | 1564 (80.2%) |
| Luminex60 | 2066 (79.3%) | 70 (57.4%) | ≤5 | 12 (57.1%) | 313 (79.8%) | 82 (73.2%) | ≤5 | 1579 (80.9%) |
| Agena | 1989 (76.4%) | 65 (53.3%) | ≤5 | 11 (52.4%) | 304 (77.6%) | 75 (67.0%) | ≤5 | 1524 (78.1%) |
| Illumina139 | 2079 (79.8%) | 70 (57.4%) | ≤5 | 12 (57.1%) | 309 (78.8%) | 83 (74.1%) | ≤5 | 1595 (81.8%) |
| WI_expanded | 2025 (77.8%) | 67 (54.9%) | ≤5 | 12 (57.1%) | 319 (81.4%) | 77 (68.8%) | ≤5 | 1540 (78.9%) |
| CFTR2 database | 1900 (73.0%) | 64 (52.5%) | ≤5 | 12 (57.1%) | 313 (79.8%) | 76 (67.9%) | ≤5 | 1425 (73.0%) |
| CF‐causing variants & VVCCs | 2139 (82.1%) | 79 (64.8%) | ≤5 | 12 (57.1%) | 332 (84.7%) | 85 (75.9%) | ≤5 | 1621 (83.1%) |
Abbreviations: CF, cystic fibrosis; CFTR, cystic fibrosis transmembrane conductance regulator; CRMS, CFTR‐related metabolic syndrome; VVCC,variants with varying clinical consequences.
Table 4.
Detection of 2 variants in fully genotyped people with CRMS and CFTR‐related disorder
| CFTR variant panel | Total population | African American/Black non‐Hispanic | American Indian, Alaskan Native, non‐Hispanic | Asian, non‐Hispanic | Hispanic | Mixed races, non‐Hispanic | Native Hawaiian Pacific Islander, non‐Hispanic | White, non‐Hispanic |
|---|---|---|---|---|---|---|---|---|
| Total N | 2608 | 122 | ≤5 | 21 | 392 | 112 | ≤5 | 1951 |
| ACMG‐23 | 587 (22.5%) | 6 (4.9%) | ≤5 | 0 (0%) | 11 (2.8%) | 15 (13.4%) | 0 (0%) | 550 (28.2%) |
| Luminex39 | 879 (33.7%) | 14 (11.5%) | ≤5 | ≤5 (≤23.8%) | 52 (13.3%) | 28 (25.0%) | ≤5 | 770 (39.5%) |
| Luminex60 | 1001 (38.4%) | 15 (12.3%) | ≤5 | ≤5 (≤23.8%) | 90 (23.0%) | 32 (28.6%) | ≤5 | 849 (43.5%) |
| Agena | 755 (28.9%) | 8 (6.6%) | ≤5 | 0 (0%) | 54 (13.8%) | 21 (18.8%) | 0 (0%) | 667 (34.2%) |
| Illumina139 | 939 (36.0%) | 14 (11.5%) | ≤5 | ≤5 (≤23.8%) | 63 (16.1%) | 28 (25.0%) | ≤5 | 819 (42.0%) |
| WI_expanded | 667 (25.6%) | 7 (5.7%) | ≤5 | 0 (0%) | 24 (6.1%) | 17 (15.2%) | 0 (0%) | 614 (31.5%) |
| CFTR2 database | 65 (2.5%) | ≤5 | 0 (0%) | 0 (0%) | 12 (3.1%) | 0 (0%) | 0 (0%) | 48 (2.5%) |
| CF‐causing variants & VVCCs | 1205 (46.2%) | 35 (28.7%) | ≤5 | ≤5 (≤23.8%) | 148 (37.8%) | 40 (35.7%) | ≤5 | 967 (49.6%) |
Abbreviations: CF, cystic fibrosis; CFTR, cystic fibrosis transmembrane conductance regulator; VVCC,variants with varying clinical consequences.
States with increased racial and ethnic diversity had lower case detection rates for each CFTR variant panel. Case detection varied between states for all CFTR variant panels (Supporting Information: Tables 1–2). Detection rate of at least 1 CFTR variant was lowest in Hawaii and highest in Maine, while detection of 2 CFTR variants was lowest in Puerto Rico and highest in Maine (Figures 1 and 2).
Figure 1.
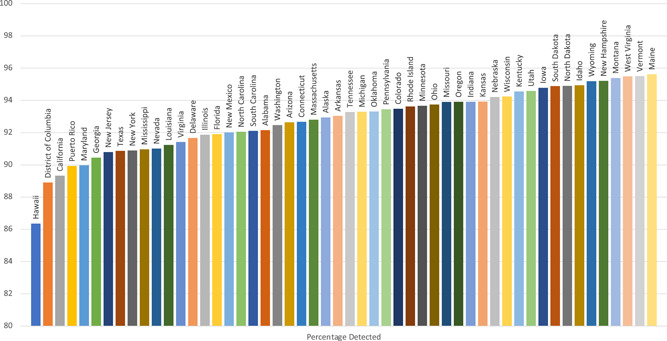
Estimated case detection of ≥1 CFTR variant by Illumina139. CFTR, cystic fibrosis transmembrane conductance regulator. [Color figure can be viewed at wileyonlinelibrary.com]
Figure 2.
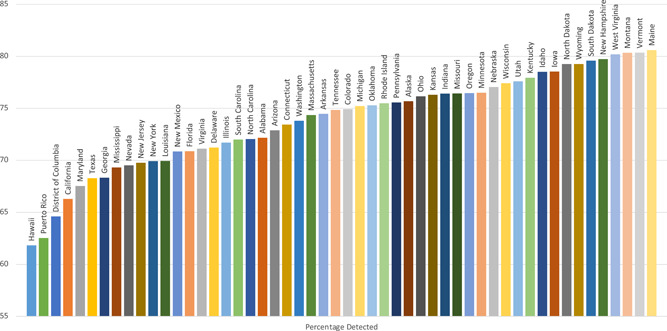
Estimated case detection of 2 CFTR variants by Illumina139. CFTR, cystic fibrosis transmembrane conductance regulator. [Color figure can be viewed at wileyonlinelibrary.com]
3.1. False‐negative NBS
From 2011 to 2020, 276 (3.8%) PwCF were reported to have a false‐negative NBS in CFFPR. This question was not answered in 2456 (33.5%) patients as it was not mandatory. From 2011 to 2020, 378 (11.8%) people were reported to have a delayed diagnosis (>180 days old), which is presumed to be a missed NBS. Adding together the 276 and 378 to estimate the minimal number of screening missed cases among a presumed total of 6354 new diagnoses reported by Martiniano et al. 17 suggests that the US only achieved 90% sensitivity with CF NBS algorithms used during the past decade and is consistent with previous estimates. 32 Black/African American, Hispanic, and mixed‐race people are overrepresented in both false‐negative NBS and delayed diagnosis compared to the entire CF population (Table 5). Non‐Hispanic White people are underrepresented in both false‐negative NBS and delayed diagnosis.
Table 5.
Racial and ethnic differences in missed NBS and delayed diagnosis
| Race and ethnicity | CF population in CFFPR in 2020 (N = 46,729) | False negative NBS (N = 278) | Diagnosis >180 days old (N = 368) | False negative NBS & diagnosis >180 days old (N = 103) |
|---|---|---|---|---|
| American Indian/Alaskan Native, non‐Hispanic | 144 (0.3%) | ≤5 (≤1.8%) | ≤5 (≤1.4%) | ≤5 (≤4.9%) |
| Asian, non‐Hispanic | 217 (0.5%) | ≤5 (≤1.8%) | ≤5 (≤1.4%) | ≤5 (≤4.9%) |
| Black/African American, non‐Hispanic | 1699 (3.6%) | 18 (6.5%) | 24 (6.5%) | 6 (5.8%) |
| Hawaiian, Pacific Islander, non‐Hispanic | 7 (0.01%) | ≤5 (≤1.8%) | 0 (0%) | 0 (0%) |
| Hispanic | 4006 (8.6%) | 43 (15.5%) | 85 (23.1%) | 17 (16.5%) |
| Mixed races, non‐Hispanic | 1128 (2.4%) | 14 (5.0%) | 21 (5.7%) | 6 (5.8%) |
| White, non‐Hispanic | 39,538 (84.6%) | 195 (70.1%) | 230 (62.5%) | 70 (68.0%) |
Abbreviations: CF, cystic fibrosis; CFFPR, CF Foundation Patient Registry; NBS, newborn screening.
4. DISCUSSION
In this cross‐sectional analysis, we found that CFTR variant panels did not perform uniformly across racial and ethnic groups but showed wide variation in the detection rate of at least 1 or 2 CFTR variants in PwCF of different races and ethnicities. There were larger differences between races and ethnicities in detecting 2 CFTR variants compared to detecting at least 1 CFTR variant for all variant panels. Detection by CFTR variant panels was highest in the non‐Hispanic White PwCF while the detection rate was lowest in Black/African American and Asian PwCF for all variant panels. For many of the commonly used variant panels, detection of at least 1 CFTR variant was also very low in Hispanic and mixed‐race PwCF. The findings of very low detection rates of infants with at least 1 CFTR variant in many races and ethnicities is quite troubling, as CF NBS protocols for many states' NBS for CF only report positive cases if at least 1 CFTR variant is detected and if no variants are detected, it would be falsely reported as negative. Using these variant panels in many races and ethnicities will lead to missed cases by NBS, that is, delays in diagnoses and potentially irreversible malnutrition and/or lung disease.
In PwCF in all racial and ethnic groups, there was a greater increase in detection rate of 2 CFTR variants compared to at least 1 CFTR variant with an increased number of variants on panels. This is important because detecting 2 pathogenic variants on a dried blood spot specimen establishes a presumptive genetic diagnosis of CF that can lead to expedited diagnosis and rapid implementation of therapies. With the addition of more CFTR variants to screening panels, there was a large improvement in case detection for PwCF in many races and ethnicities, especially in Asian and Black/African American PwCF. However, there was very little or no improvement in detection rate of at least 1 CFTR variant with increased variants on panels in non‐Hispanic White and Hawaiian or Pacific Islander PwCF. These differences highlight the need to choose CFTR variant panels that reflect the racial and ethnic diversity of a population.
There was very low case detection of 2 CFTR variants in PwCRMS using all variant panels in all races and ethnicities. Panels detected at least 1 CFTR variant often in non‐Hispanic White and mixed‐race PwCRMS, but not in all other races and ethnicities. Many PwCRMS have variants of variable or unknown clinical significance. Identifying CRMS is not a goal of CF NBS, despite the risk that some PwCRMS may evolve a CF phenotype. Nevertheless, these data are important for understanding the impact of variant panels on identifying PwCRMS.
We estimate that about 10% of PwCF were either diagnosed after a false‐negative NBS or had a delayed diagnosis. Importantly, Hispanic and Black/African American people represented a higher percentage of the missed or delayed diagnoses than expected, while non‐Hispanic White people were a smaller percentage than expected. Our findings of false‐negative NBS are the minimum estimates of missed cases and represent failures of NBS. The 90% sensitivity for CF NBS is undoubtedly the lowest for genetic disorders on the Recommended Uniform Screening Panel. The actual number of missed cases is undoubtedly higher due to children who had false‐negative NBS and are not yet diagnosed with CF, or children who died before being diagnosed with CF. 33 This is consistent with our findings of low detection rates of at least 1 CFTR variant on commonly used screening panels for many races and ethnicities. Delayed diagnoses or diagnoses after false‐negative NBS in many races and ethnicities are concerning as the goal is to start treatments and therapies before the onset of malnutrition and pulmonary disease. The US urgently needs nationwide quality improvement in CF NBS algorithms and follow‐up activities.
For NBS to be equitable, algorithms must be modified to have high case detection for all diverse subpopulations. In 2012, the birthrate of non‐Hispanic White infants was first lower than the combined birthrate of infants of other demographically defined racial and ethnic backgrounds. 34 Since in the United States, the majority of PwCF are non‐Hispanic White, it may appear overall that there is no benefit with adding variants to NBS panels. However, this creates stark disparities that vary by race and ethnicity. State NBS Laboratories should review their CFTR variant panels for potential disparities, and those who currently have a racially and ethnically diverse population should act swiftly to improve the identification of variants and increase detection rates. As the US demographics continue to evolve, missed cases due to variant panels in NBS will become an issue even in states that currently have homogenous populations. Public health measures, such as NBS, should be created to benefit all groups in the population equally.
Further research is needed to understand how additional variants should be added to panels to increase case detection. CFTR variant type and frequency vary across races and ethnicities dependent on ancestry admixture and migration patterns, but race and ethnicity are not reliable proxies for ancestry. For example, Hispanic populations have different CFTR variant type and frequency, even when closely geographically located. 35 Furthermore, CFTR variants are not well mapped in the African diaspora, which has the most genetic heterogeneity globally. Many CFTR variant panels were originally developed to target CFTR variants for non‐Hispanic White populations and do not include CFTR variants that occur relatively frequently in other populations. Guidelines for developing NBS programs for CF suggest to use “the DNA‐mutation frequencies identified in the CF Foundation's patient registry and then expanding the data to reflect the population subgroups in that state.” 36 Including variants that are rare overall but occur more frequently in population subgroups will improve the detection rates of NBS programs. This is not only a problem in CF; in other diseases, variant screening panels were developed based on a non‐Hispanic White population and, when applied to other populations, led to misdiagnoses. 21
While the addition of VVCCs only increased the detection rate of at least 1 CFTR variant by 0%−2% for each race and ethnicity group, there was a greater increase in the detection rate for 2 CFTR variants (2.1%−6.4%). This is likely due to VVCCS being infrequent in the CF population as most people with 1 copy of a VVCCs has a second more common CFTR variant found on other panels. The addition of less frequent variants to panels will only have small increases in the detection of at least 1 CFTR variant.
To date, three states with diverse racial and ethnic populations have updated their NBS algorithms to the increase detection rate. 33 Their protocols include CFTR sequencing rather than reliance solely on variant panels. California and New York NBS protocols do IRT level, CFTR variant panel, and then CFTR sequencing in infants with only 1 variant identified. Wisconsin takes a different approach with a NGS determined panel that evolves and expands with additions to the CFTR2 list. Wisconsin's NBS protocol does IRT level and a current CFTR panel of 372 pathogenic variants, created by modifications of the Illumina reporting software. Then in those newborns with a sweat chloride level 30 mmol/L of higher and only 1 pathogenic CFTR variants identified, the variant calling file is assessed to analyze the full CFTR sequence in an effort to identify another variant. Novel variants are often found, interpreted, and reported. New York has an extra step that does NGS of CFTR in infants with very high (top 0.1%) IRT levels even if no variants are detected on the variant panel. 12 NGS allows for an evolving variant panel with the latest science to ensure accurate up to‐date diagnoses are being made by clinicians. These NBS programs with CFTR sequencing have improved detection rates for all race and ethnicity groups and should be considered by all NBS programs to increase the detection rates.
These CFTR sequencing protocols, unfortunately, will potentially miss CF cases with low IRT levels or with 2 rare or de novo variants that are not on the initial variant panel. The former can be improved by using a floating IRT cutoff of 4% to increase the detection rate of NBS. 17 Detection by NBS of infants with 2 rare or de novo variants can be improved by marking the NBS as positive in those with a very high IRT level even if zero variants are identified as panels. Unintended consequences of the approach of using CFTR sequencing include delays in diagnosis and increased detection of infants with inconclusive genetic and sweat chloride concentration resulting in categorization of CRMS/CFSPID. While there are real concerns about increased identification of CF carriers and CRMS/CFSPID patients and parental anxiety, these concerns should not outweigh disproportionately missing diagnoses in racial and ethnic minoritized people, leading to increased morbidity and mortality from CF.
NBS protocols for diseases other than CF alter cutoffs to maximize detecting cases and minimize the risk of false negatives with consideration of the lethality of the disease and urgency needed for treatment. CF has a higher rate of missed cases compared to other screened diseases. The false negative rate was about 2% for Congenital Adrenal Hypoplasia in NBS programs. 37 , 38 There were zero false negative cases for citrin deficiency 39 and NBS for lysosomal storage diseases were optimized with the goal of zero false negative cases. 40 , 41
Low case detection with CFTR variant panels leading to missed or delayed diagnosis is likely a major contributor to existing health disparities in racial and ethnic minoritized people. Early diagnosis through NBS is associated with improved nutritional outcomes and improved lung disease. 42 Diagnosis after a false‐negative NBS is often at an older age when the child presents with symptoms such as failure to thrive. PwCF who are Black/African American or Hispanic have worse outcomes, including lower lung function, 43 , 44 increased respiratory infections, 45 and higher mortality 46 ; false‐negative NBS in these groups may be contributing to these disparities.
4.1. Limitations
Race and ethnicity descriptions may vary between the US Census and CFFPR as data for US Census is directly reported by a household member, and data for CFFPR is reported by CF Center Staff. However, misclassification of race and ethnicity in the CFFPR is not likely to be a significant factor as less than 2% of race and ethnicity have been found to be inaccurate in the CFFPR. 47
One limitation of using people of all ages in the CFFPR is that this is not an accurate description of the variant frequency of newborns as race and ethnicity have shifted over time. The Census data only includes descriptions of race and ethnicity of those 18 years and older but not a description of infants born over the last year. Our results may be an overestimation of rate detections as there are likely PwCF who are undiagnosed since variant panels are also used for diagnosis. The overestimation is likely greater in minoritized race and ethnicity populations who have rare or de novo mutations. We only included people who had 2 CFTR variants identified. This disproportionately excluded more people of minoritized race and ethnicity as they are more likely not to have 2 CFTR variants identified. This ascertainment bias likely resulted in overestimation of case detection. The CFFPR includes a very small number of people who identify as Hawaiian or Pacific Islander, which limits the generalizability of our findings to all Hawaiian and Pacific Islander people.
A yes/no question regarding whether a participant in CFFPR had a false‐negative NBS was implemented in CFFPR in 2013. Before 2013, there was no data collected about false‐negative NBS; after 2013 it was not a mandatory reporting question leading to a significant amount of missing information. However, the findings of the racial and ethnic breakdown of false‐negative screening were consistent with delayed diagnosis.
5. CONCLUSIONS
From a comprehensive, unique assessment of CFTR variants in United State. PwCF who are registered in the CFFPR, we conclude that detection of at least 1 or 2 CFTR variants through CF NBS is different by demographic racial and ethnic group. This can contribute to missed or delayed diagnosis of CF, furthering CF health disparities. These results indicate that nationwide quality improvement is needed.
AUTHOR CONTRIBUTIONS
Meghan E. McGarry: writing−original draft; writing−review & editing; visualization; investigation; formal analysis; conceptualization. Clement L. Ren: investigation; writing−review & editing; formal analysis; conceptualization. Runyu Wu: visualization; formal analysis; methodology; data curation; writing−review & editing; conceptualization; investigation; software. Philip M. Farrell: conceptualization; investigation; writing−review & editing; visualization; formal analysis. Susanna A. McColley: conceptualization; investigation; writing − review & editing; visualization; formal analysis.
Supporting information
Supplementary information.
ACKNOWLEDGMENTS
The authors would like to thank the CF Foundation for the use of the CFFPR data to conduct this study. Additionally, we would like to thank the people with CF, care providers, and care coordinators at CF centers throughout the US for their contributions to the CFFPR. This study was supported by Cystic Fibrosis Foundation (MCCOLL19QI0, NHLBI K23HL133437) and Cystic Fibrosis Foundation (MCGARR20A0‐KB).
McGarry ME, Ren CL, Wu R, Farrell PM, McColley SA. Detection of disease‐causing CFTR variants in state newborn screening programs. Pediatric Pulmonology. 2023;58:465‐474. 10.1002/ppul.26209
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from Cystic Fibrosis Foundation Patient Registry. Restrictions apply to the availability of these data, which were used under license for this study. Data are available from https://www.cff.org/researchers/patient-registry-data-requests with the permission of Cystic Fibrosis Foundation Patient Registry.
REFERENCES
- 1. Farrell PM, Li Z, Kosorok MR, et al. Bronchopulmonary disease in children with cystic fibrosis after early or delayed diagnosis. Am J Respir Crit Care Med. 2003;168(9):1100‐1108. [DOI] [PubMed] [Google Scholar]
- 2. Farrell PM, Lai HJ, Li Z, et al. Evidence on improved outcomes with early diagnosis of cystic fibrosis through neonatal screening: enough is enough! J Pediatr. 2005;147(3):S30‐S36. [DOI] [PubMed] [Google Scholar]
- 3. Lai HJ, Cheng Y, Farrell PM. The survival advantage of patients with cystic fibrosis diagnosed through neonatal screening: evidence from the United States Cystic Fibrosis Foundation registry data. J Pediatr. 2005;147(3):S57‐S63. [DOI] [PubMed] [Google Scholar]
- 4. Coffey MJ, Whitaker V, Gentin N, et al. Differences in outcomes between early and late diagnosis of cystic fibrosis in the newborn screening era. J Pediatr. 2017;181:137‐145. [DOI] [PubMed] [Google Scholar]
- 5. Waters DL, Wilcken B, Irwing L, et al. Clinical outcomes of newborn screening for cystic fibrosis. Archiv Dis Childhood‐Fetal Neonatal Ed. 1999;80(1):F1‐F7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Grosse SD, Boyle CA, Botkin JR, et al. Newborn screening for cystic fibrosis: evaluation of benefits and risks and recommendations for state newborn screening programs. MMWR Recomm Rep. 2004;53:1‐36. [PubMed] [Google Scholar]
- 7. Bender LM, Cotten SW, Willis MS. Kids in America: newborn screening for cystic fibrosis. Lab Med. 2011;42(10):595‐601. [Google Scholar]
- 8. Farrell PM, White TB, Ren CL, et al. Diagnosis of cystic fibrosis: consensus guidelines from the cystic fibrosis foundation. J Pediatr. 2017;181:S4. [DOI] [PubMed] [Google Scholar]
- 9. Baker MW, Atkins AE, Cordovado SK, Hendrix M, Earley MC, Farrell PM. Improving newborn screening for cystic fibrosis using next‐generation sequencing technology: a technical feasibility study. Genet Med. 2016;18(3):231‐238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hughes EE, Stevens CF, Saavedra‐Matiz CA, et al. Clinical sensitivity of cystic fibrosis mutation panels in a diverse population. Hum Mutat. 2016;37(2):201‐208. [DOI] [PubMed] [Google Scholar]
- 11. Sosnay PR, Salinas DB, White TB, et al. Applying cystic fibrosis transmembrane conductance regulator genetics and CFTR2 data to facilitate diagnoses. J Pediatr. 2017;181:S27‐S32. [DOI] [PubMed] [Google Scholar]
- 12. Sicko RJ, Stevens CF, Hughes EE, et al. Validation of a custom next‐generation sequencing assay for cystic fibrosis newborn screening. Int J Neonatal Screening. 2021;7(4):73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Furnier SM, Durkin MS, Baker MW. Translating molecular technologies into routine newborn screening practice. Int J Neonatal Screening. 2020;6(4):80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ren CL, Borowitz DS, Gonska T, et al. Cystic fibrosis transmembrane conductance regulator‐related metabolic syndrome and cystic fibrosis screen positive, inconclusive diagnosis. J Pediatr. 2017;181:S45‐S51. [DOI] [PubMed] [Google Scholar]
- 15. Cystic Fibrosis F, Borowitz D, Parad RB, et al. Cystic Fibrosis Foundation practice guidelines for the management of infants with cystic fibrosis transmembrane conductance regulator‐related metabolic syndrome during the first two years of life and beyond. J Pediatr. 2009;155(6):S106‐S116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Andersen DH. Cystic fibrosis of the pancreas and its relation to celiac disease: a clinical and pathologic study. Am J Dis Child. 1938;56(2):344‐399. [Google Scholar]
- 17. Martiniano SL, Croak K, Bonn G, Sontag MK, Sagel SD. Improving outcomes for Colorado's IRT‐IRT‐DNA cystic fibrosis newborn screening algorithm by implementing floating cutoffs. Mol Gen Metab. 2021;134(1‐2):65‐67. [DOI] [PubMed] [Google Scholar]
- 18. Schrijver I, Pique L, Graham S, Pearl M, Cherry A, Kharrazi M. The spectrum of CFTR variants in nonwhite cystic fibrosis patients: implications for molecular diagnostic testing. J Mol Diagn. 2016;18(1):39‐50. [DOI] [PubMed] [Google Scholar]
- 19. Sugarman EA, Rohlfs EM, Silverman LM, Allitto BA. CFTR mutation distribution among US Hispanic and African American individuals: evaluation in cystic fibrosis patient and carrier screening populations. Genet Med. 2004;6(5):392‐399. [DOI] [PubMed] [Google Scholar]
- 20. McGarry ME, McColley SA. Cystic fibrosis patients of minority race and ethnicity less likely eligible for CFTR modulators based on CFTR genotype. Pediatr Pulmonol. 2021;56(6):1496‐1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Manrai AK, Funke BH, Rehm HL, et al. Genetic misdiagnoses and the potential for health disparities. N Engl J Med. 2016;375(7):655‐665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ross LF. Newborn screening for cystic fibrosis: a lesson in public health disparities. J Pediatr. 2008;153(3):308‐313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Barben J, Castellani C, Munck A, et al. Updated guidance on the management of children with cystic fibrosis transmembrane conductance regulator‐related metabolic syndrome/cystic fibrosis screen positive, inconclusive diagnosis (CRMS/CFSPID). Journal of Cystic Fibrosis. 2021;20(5):810‐819. [DOI] [PubMed] [Google Scholar]
- 24.Cystic Fibrosis Foundation Patient Registry. Annual data report. 2020.
- 25. Watson MS, Cutting GR, Desnick RJ, et al. Cystic fibrosis population carrier screening: 2004 revision of American College of Medical Genetics mutation panel. Genet Med. 2004;6(5):387‐391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Agena 74 CFTR panel. https://www.agenabio.com/products/panel/cftr-panel/
- 27.Luminex CFTR Panel. https://www.luminexcorp.com/cystic-fibrosis/#overview
- 28.Illumina CFTR Panel. https://www.illumina.com/products/by-type/ivd-products/miseqdx-cystic-fibrosis-139-variant-assay.html
- 29.Wisconsin Newborn Screening Program. http://www.slh.wisc.edu/clinical/newborn/health-care-professionals-guide/nbs-test-panel-of-diseases/
- 30.CFTR2 Database. https://cftr2.org/ accessed 04/29/2022
- 31.The Clinical and Functional TRanslation of CFTR (CFTR2). Accessed December 10, 2021. http://cftr2.org
- 32. Fritz A, Farrell P. Estimating the annual number of false negative cystic fibrosis newborn screening tests. Pediatr Pulmonol. 2012;47(2):207‐208. [DOI] [PubMed] [Google Scholar]
- 33. Kharrazi M, Yang J, Bishop T, et al. Newborn screening for cystic fibrosis in California. Pediatrics. 2015;136(6):1062‐1072. [DOI] [PubMed] [Google Scholar]
- 34.Most children younger than age 1 are minorities, Census Bureau Reports. 2012. https://www.census.gov/newsroom/releases/archives/population/cb12-90.html
- 35. Zeiger AM, McGarry ME, Mak A, et al. Identification of CFTR variants in Latino patients with cystic fibrosis from the Dominican Republic and Puerto Rico. Pediatr Pulmonol. 2020;55(2):533‐540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Comeau AM, Accurso FJ, White TB, et al. Guidelines for implementation of cystic fibrosis newborn screening programs: cystic Fibrosis Foundation workshop report. Pediatrics. 2007;119(2):e495‐e518. [DOI] [PubMed] [Google Scholar]
- 37. Chan CL, McFann K, Taylor L, Wright D, Zeitler PS, Barker JM. Congenital adrenal hyperplasia and the second newborn screen. J Pediatr. 2013;163(1):109‐113. [DOI] [PubMed] [Google Scholar]
- 38. Held PK, Shapira SK, Hinton CF, Jones E, Hannon WH, Ojodu J. Congenital adrenal hyperplasia cases identified by newborn screening in one‐and two‐screen states. Mol Gen Metab. 2015;116(3):133‐138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chen H‐A, Hsu R‐H, Chen Y‐H, et al. Improved diagnosis of citrin deficiency by newborn screening using a molecular second‐tier test. Mol Gen Metab. 2022;136:330‐336. [DOI] [PubMed] [Google Scholar]
- 40. Sanders KA, Gavrilov DK, Oglesbee D, et al. A comparative effectiveness study of newborn screening methods for four lysosomal storage disorders. Int J Neonatal Screening. 2020;6(2):44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Burton BK, Charrow J, Hoganson GE, et al. Newborn screening for lysosomal storage disorders in Illinois: the initial 15‐month experience. J Pediatr. 2017;190:130‐135. [DOI] [PubMed] [Google Scholar]
- 42. Farrell PM, Kosorok MR, Laxova A, et al. Nutritional benefits of neonatal screening for cystic fibrosis. N Engl J Med. 1997;337(14):963‐969. [DOI] [PubMed] [Google Scholar]
- 43. McGarry ME, Neuhaus J, Nielson DW, Burchard EG, Ly NP. Pulmonary function disparities exist and persist in Hispanic patients with cystic fibrosis: a longitudinal analysis. Pediatr Pulmonol. 2017;52(12):1550‐1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. McGarry ME, Neuhaus JM, Nielson DW, Ly NP. Regional variations in longitudinal pulmonary function: a comparison of Hispanic and non‐Hispanic subjects with cystic fibrosis in the United States. Pediatr Pulmonol. 2019;54:1382‐1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. McGarry ME, Huang C‐Y, Nielson DW, Ly NP. Early acquisition and conversion of Pseudomonas aeruginosa in Hispanic youth with cystic fibrosis in the United States. J Cys Fibrosis. 2021;20(3):424‐431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Buu MC, Sanders LM, Mayo JA, Milla CE, Wise PH. Assessing differences in mortality rates and risk factors between Hispanic and non‐Hispanic patients with cystic fibrosis in California. CHEST J. 2016;149(2):380‐389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Knapp EA, Fink AK, Goss CH, et al. The Cystic Fibrosis Foundation Patient Registry . Design and methods of a national observational disease registry. Ann Am Thorac Soc. 2016;13(7):1173‐1179. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information.
Data Availability Statement
The data that support the findings of this study are available from Cystic Fibrosis Foundation Patient Registry. Restrictions apply to the availability of these data, which were used under license for this study. Data are available from https://www.cff.org/researchers/patient-registry-data-requests with the permission of Cystic Fibrosis Foundation Patient Registry.