

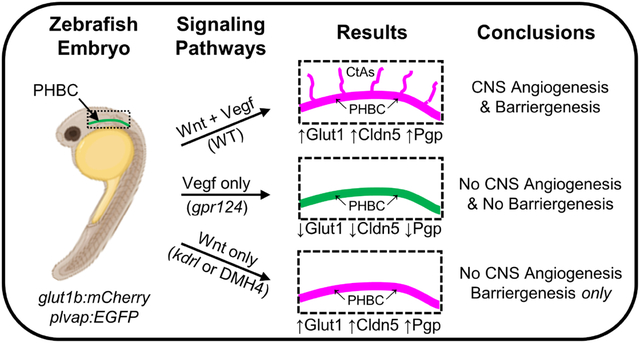
Abstract
During neurovascular development, brain endothelial cells (BECs) respond to secreted signals from the neuroectoderm that regulate CNS angiogenesis, the formation of new blood vessels in the brain, and barriergenesis, the acquisition of blood-brain barrier (BBB) properties. Wnt/β-catenin signaling and Vegf signaling are both required for CNS angiogenesis; however, the relationship between these pathways is not understood. Furthermore, while Wnt/β-catenin signaling is essential for barriergenesis, the role of Vegf signaling in this vital process remains unknown. Here, we provide the first direct evidence, to our knowledge, that Vegf signaling is not required for barriergenesis and that activation of Wnt/β-catenin in BECs is independent of Vegf signaling during neurovascular development. Using double transgenic glut1b:mCherry and plvap:EGFP zebrafish (Danio rerio) to visualize the developing brain vasculature, we performed a forward genetic screen and identified a new mutant allele of kdrl, an ortholog of mammalian Vegfr2. The kdrl mutant lacks CNS angiogenesis but, unlike the Wnt/β-catenin pathway mutant gpr124, acquires BBB properties in BECs. To examine Wnt/β-catenin pathway activation in BECs, we chemically inhibited Vegf signaling and found robust expression of the Wnt/β-catenin transcriptional reporter line 7xtcf-Xla.Siam:EGFP. Taken together, our results establish that Vegf signaling is essential for CNS angiogenesis but is not required for Wnt/β-catenin-dependent barriergenesis. Given the clinical significance of either inhibiting pathological angiogenesis or stimulating neovascularization, our study provides valuable new insights that are critical for the development of effective therapies that target the vasculature in neurological disorders.
Keywords: Angiogenesis, Barriergenesis, Blood-brain barrier, CNS, Vegf, Wnt/β-catenin
Graphical Abstract
Introduction
The blood-brain barrier (BBB) plays a vital role in the function of the central nervous system (CNS) by controlling the entry and exclusion of essential and harmful substances into the brain while maintaining a suitable microenvironment for normal neuronal function [1]. Physiologically, the BBB consists of differentiated brain endothelial cells (BECs) along with pericytes and glial cells that collectively form the neurovascular unit during BBB maturation [2–5]. During neurovascular development, BECs respond to extracellular cues that reduce expression of the structural component of fenestrae Plasmalemma vesicle-associated protein (PLVAP) [6], suppress transcytosis by expression of MFSD2a [7], and form tight junctions to regulate the passage of molecules between endothelial cells [8, 9]. Differentiated BECs also help to supply the brain with key nutrients through the expression of specialized transporters, such as the glucose transporter GLUT1 and amino acid transporter LAT1 [1, 10, 11]. Functionally, the BBB is critical for health and medicine for two main reasons. First, it makes delivering drugs to the brain a major challenge as many pharmaceuticals cannot effectively cross the BBB [12]. In addition, lipid-soluble compounds often have less penetrance than expected due to multi-drug resistance transporters that prohibit the transport of compounds into the brain [1, 12–14]. Second, the BBB is disrupted in many disease states, which may contribute to their neuropathologies [1, 14, 15]. To develop effective treatments and therapies for neurological conditions, a more thorough understanding of the BBB, including the signaling pathways involved in its development, maintenance, and function, is required.
Development of the BBB is a complex process, requiring multiple signal transduction pathways and cellular interactions. One crucial aspect of BBB development is CNS angiogenesis, the sprouting and migration of vascular endothelial cells into the brain parenchyma from preexisting blood vessels. As CNS angiogenesis occurs, BECs acquire BBB properties in a process called barriergenesis [11, 15, 16]. Importantly, this process does not suggest a fully mature and functional BBB. Instead, barriergenesis is the response of endothelial cells to extracellular cues within the brain microenvironment that initiate the BBB phenotype. To visualize this process, GLUT1 is often used as a functional indicator of BBB development in mice and zebrafish (Danio rerio) [2, 3, 17–23] because GLUT1 is the earliest known marker of BEC differentiation [24, 25]. Therefore, for our studies, we use the transgenic zebrafish line glut1b:mCherry as a reporter for barriergenesis [16].
Recent research has begun to elucidate the molecular mechanisms regulating neurovascular development, although our understanding is far from complete. Over the past decade, seminal studies discovered that Wnt/β-catenin signaling, while not required for peripheral angiogenesis, is required for both CNS angiogenesis and barriergenesis [17, 21, 26, 27]. For example, targeted disruption of embryonic β-catenin in mice is lethal and causes severe CNS angiogenesis defects and loss of GLUT1 expression in BECs [17, 21], and postnatal knockout of endothelial-specific β-catenin results in loss of BBB characteristics [26]. Subsequent studies in both mice and zebrafish discovered that the orphan G-protein coupled receptor GPR124 [16, 18, 28, 29] and the GPI-anchored membrane protein RECK [30–32] are also required for normal CNS angiogenesis and barriergenesis via their functions as ligand-specific Wnt coactivators [19, 20, 23, 33, 34]. Most recently, the endothelial Unc5B receptor was shown to promote BBB development and maintenance by facilitating Wnt/β-catenin signaling [35].
While current models of CNS angiogenesis and barriergenesis propose an absolute requirement for Wnt/β-catenin signaling [19, 34], the role of VEGF (vascular endothelial growth factor) signaling in these models remains poorly defined. Importantly, VEGF Receptor 2 (VEGFR2, alternately KDR/FLK-1), when activated by VEGF-A, is a key mediator of angiogenesis in development and disease [36–41]. Furthermore, studies have shown that Vegf signaling, like Wnt/β-catenin signaling, is absolutely required for angiogenesis to occur in the CNS [42–44]. For example, Raab et al. demonstrated that angiogenesis is severely impaired to all regions of the brain in mice lacking functional VEGF [44]. Additionally, whereas the specific requirement for VEGFR2 in CNS vascularization has been difficult to examine due to early lethality of the mouse knockout [45, 46], zebrafish Kdrl, a functional ortholog of mammalian VEGFR2, is required for normal CNS angiogenesis [47]. However, while both Wnt/β-catenin and Vegf signaling are required for CNS angiogenesis and Wnt/β-catenin signaling is required for barriergenesis, no prior studies, to our knowledge, have examined the role of Vegf signaling during barriergenesis. The relationship between Wnt/β-catenin and Vegf signaling in the regulation of CNS angiogenesis is also undetermined, with the possibilities including Vegf regulation of Wnt/β-catenin, Wnt/β-catenin regulation of Vegf signaling, or cooperation between the pathways.
In this study, we used zebrafish to genetically dissect the molecular mechanisms required for CNS angiogenesis and barriergenesis. Zebrafish provide a valuable animal model for studying vascular development due to the relative ease of visualizing blood vessels in vivo, generating transgenic reporter lines, and performing forward and reverse genetic analyses [48–55]. Here, we performed a forward genetic screen to identify genes that regulate neurovascular development. We previously established the glut1b:mCherry transgenic line to visualize barriergenesis and utilized it in a screen that identified a gpr124 mutant, which lacks both CNS angiogenesis and glut1b:mCherry expression [16]. In our current screen, we identified a new mutant that lacks CNS angiogenesis but expresses glut1b:mCherry, indicating defects in CNS angiogenesis but not barriergenesis. With this mutant, we demonstrate that Vegf signaling, while required for CNS angiogenesis, is not required for barriergenesis. Additionally, we discuss the implications this has on the interaction between the Vegf and Wnt/β-catenin signaling pathways in the context of neurovascular development.
Materials and methods
Zebrafish husbandry and experimental lines
Zebrafish were maintained and bred using standard practices [56]. Embryos and larvae were maintained at 28.5°C in egg water (0.03% Instant Ocean in reverse osmosis water). For imaging, 0.003% phenylthiourea (PTU) was used to inhibit melanin production. The transgenic zebrafish lines Tg(glut1b:mCherry)sj1 (hereafter glut1b:mCherry) and Tg(plvap:EGFP)sj3 (hereafter plvap:EGFP) and the gpr124 mutant line were previously generated in our lab [16]. The kdrlum19 mutant [57], provided in the Tg(fli1a:EGFP)y1 background [48], was a gift from Dr. Nathan Lawson (University of Massachusetts Medical School), Tg(7xtcf-Xla.Siam:EGFP)ia4 [58] (hereafter tcf:EGFP) was a gift from Dr. Junsu Kang (UW-Madison), and Tg(kdrl:HRAS-mCherry)s896 [59] (hereafter kdrl:mCherry) was a gift from Dr. Jan Huisken (Morgridge Institute for Research and UW-Madison). All experiments were performed in accordance with the University of Wisconsin-Madison Institutional Animal Care and Use Committee (protocol number M005020).
N-ethyl-N-nitrosourea (ENU) mutagenesis screen
Twelve adult male zebrafish homozygous for both glut1b:mCherry and plvap:EGFP transgenes were mutagenized with ENU as previously described [50]. After one month of recovery, F1 pairwise crosses between mutagenized males and glut1b:mCherry and plvap:EGFP transgenic females produced 96 F2 families. Pairwise crosses from F2 families were performed at least six times to identify F3 offspring with homozygous recessive mutations by screening embryos at 2–3 days post-fertilization (dpf) for brain vascular defects that disrupt transgene expression using a Nikon SMZ18 epifluorescence stereomicroscope. A total of twenty-four F2 families were screened. Unfortunately, this screen was terminated prematurely due to COVID-19 related research restrictions. A mutant line was confirmed if approximately 25% of the total offspring displayed a brain vasculature phenotype. The uw112 mutant line was identified as lacking CNS angiogenesis by observing glut1b:mCherry-positive primordial hindbrain channels (PHBCs) and the absence of plvap:EGFP-positive vessels within the brain parenchyma.
Confocal laser scanning microscopy
For live imaging, zebrafish embryos were anesthetized in 0.02% Tricaine and imbedded in 1.2% low melting point agarose (Invitrogen) in egg water with 0.003% PTU in a 35 mm glass-bottom dish, number 1.5 (MatTek). For fixed samples, larvae were imbedded in 1.2% low melting point agarose in 1× phosphate buffered saline (PBS) using the same dishes. Confocal microscopy was performed using a Nikon Eclipse Ti microscope equipped with a Nikon A1R. For time-lapse imaging, resonant scanning was used to acquire z-stacks at 10 min intervals for 3 h. All confocal images are 2D maximum intensity projections of 3D z-stacks generated using NIS-Elements software.
Complementation test
To determine if uw112 was allelic to kdrl, we performed a complementation test. Zebrafish heterozygous for the kdrlum19 mutation [57] were bred to uw112 heterozygotes expressing the glut1b:mCherry and plvap:EGFP transgenes, and the resulting offspring were evaluated for complementation at 3 dpf by visualizing brain vasculature using a Nikon SMZ18 epifluorescence stereomicroscope.
DNA sequencing and genotyping
WT and uw112 mutants (n=25 each) at 3 dpf were anesthetized in 0.02% Tricaine, transferred into RNase/DNase-free 1.5 ml microcentrifuge tubes with fitted pestle (Kontes), homogenized in TRIzol, and total RNA was extracted according to the manufacturer’s protocol (Ambion). cDNA was synthesized by reverse transcription from the RNA using the SuperScript IV First-Strand Synthesis System using Oligo(dT) primers according to the manufacturer’s protocol (Invitrogen). WT and uw112 mutant cDNA were amplified by PCR using AccuPrime Pfx DNA Polymerase (Invitrogen), 3’ A overhangs were added to PCR product using Go Taq DNA Polymerase (Promega), and the inserts were cloned into the pCRII-TOPO TA vector using the TOPO TA Cloning Dual Promoter kit (Invitrogen). TOP10 chemically competent E. coli cells were transformed, colonies were selected by blue/white screening, and plasmids were purified from E. coli cultures using QIAprep Spin Minipreps (Qiagen). Plasmids were sequenced by the UW-Madison Biotechnology Center using Sanger sequencing with forward and reverse primers covering the entire cDNA (primer sequences are available upon request). The sequencing data was analyzed using A plasmid Editor (ApE) software [60].
To genotype the kdrluw112 mutants, genomic DNA was extracted from individual embryos and PCR amplified to produce a 597 bp product using the following primers: Forward primer 5’-ACGTCACCGAAGAACCATCT-3’ and Reverse primer 5’-TGATCCCAAATTGCACTTCA-3’. Restriction fragment length polymorphism using EarI (New England Biolabs; R0528S) was used to distinguish WT and mutant sequences.
VEGFR inhibitor treatment
The VEGFR inhibitor DMH4 (Tocris Bioscience; CAS 515880-75-8) [61] was prepared as a 10 mM stock in 100% DMSO (dimethyl sulfoxide), and VEGFR inhibitor AV-951 (Tivozanib) (Selleckchem; CAS 475108-18-0) [62] was prepared as a 10 mM stock in 100% DMSO. To inhibit CNS angiogenesis, DMH4 (5 μM or 1 μM) or AV-951 (1 μM) was freshly prepared in egg water with 0.003% PTU and applied to embryos at 24 hours post-fertilization (hpf); then, treated embryos were imaged at 54 hpf using a Nikon Eclipse Ti confocal microscope equipped with a Nikon A1R.
Immunohistochemistry (IHC)
WT, kdrluw112, and gpr124 larvae at 5 dpf were fixed in 4% paraformaldehyde (Electron Microscopy Sciences) and were dehydrated with a 1× PBS/methanol series. Samples were stored in 100% methanol at −20°C for up to a month before use. To begin IHC, dehydrated larvae were rehydrated with a methanol/PBSTx [0.6% Triton X-100 (Sigma) in 1x PBS] series. Larvae were bleached [0.05 mg KOH (Sigma) and 150 μL H2O2 (Fisher) in 4.85 mL reverse osmosis water] for 40 min, placed in cold acetone for 8 min, and incubated overnight in blocking buffer [4% bovine serum albumin (OmniPur) in PBSTx] with PBSTx washes before each step. Primary antibodies were added the next day and incubated at 4°C overnight followed by secondary antibody incubation the next night. Samples were washed in PBSTx four times for 15 min after primary and secondary antibody incubations. Primary antibodies included rabbit anti-glut1 (1:200; Novus Biologicals NB300–666SS), mouse anti-Claudin 5 (1:100; Life Technologies 187364), and mouse anti-CD243/ABCB4 (P-glycoprotein 1) (1:200; BioLegend 903701). Secondary antibodies included Alexa Fluor goat anti-rabbit 568 (1:200; Invitrogen A11011) and Alexa Fluor goat anti-mouse 647 (1:200; Invitrogen A21235). Antibody dilutions were prepared in blocking buffer. For imaging, samples were imbedded and imaged using confocal microscopy as described above.
Morpholino injections
We used the previously published gpr124 morpholino sequence to knockdown Gpr124 function and phenocopy the zebrafish gpr124 mutant phenotype [60]. The gpr124 morpholino 5’-ACTGATATTGATTTAACTCACCACA-3’ was obtained from GeneTools and resuspended as a stock solution of 1 mM in dH2O. For microinjection, morpholinos were diluted to 0.2 mM in dH2O containing phenol red (0.05%) as an injection tracer. Microinjection needles were fabricated from 1.2 mm thin wall glass capillaries (WPI; TW120F-4) using a Sutter Instrument Flaming/Brown Micropipette Puller (Model P-97). Approximately 2 nL (~2 ng) were microinjected into the yolk of single-celled embryos. No obvious off-target effects were observed with the gpr124 morpholino.
Quantification of vasculature
Brain vasculature and fluorescence were quantified using NIS-Elements (Nikon) and FIJI (ImageJ) software. To quantify CNS angiogenesis in embryos at 2 dpf, the number of central artery loops (CtAs) were counted using the 3D rendering in NIS-Elements. For larvae at 3 dpf, the 3D renderings (showing either glut1b:mCherry or plvap:EGFP) were cropped in NIS-Elements to eliminate blood vessels outside of the brain parenchyma. In FIJI, background was removed and the commands Smooth (3D) > Skeletonize (2D/3D) > Analyze Skeleton (2D/3D) were used to find the total length of skeletonized vessels. Relative fluorescence intensities were calculated using the Measure feature of FIJI with the region of interest restricted to the hindbrain vasculature [63]. For quantification of IHC and tcf:EGFP signal, 2D maximum intensity projections were generated using NIS-Elements. In FIJI, the blood vessels in the hindbrain were traced by hand to calculate the length of the hindbrain vasculature in the image. Then the length from an image showing only the signal of interest was divided by the total length (from an image showing all channels) to calculate the fraction of positive vasculature in the hindbrain. Statistics (p-values) for all graphs were determined by ANOVA with Tukey HSD post hoc test (*p < 0.05; **p < 0.01; ***p < 0.001; ns = not significant).
Results
Zebrafish uw112, but not gpr124, mutants express the barriergenesis marker glut1b:mCherry in the absence of CNS angiogenesis
To identify genes involved in the regulation of CNS angiogenesis and barriergenesis, we performed a forward genetic screen utilizing N-ethyl-N-nitrosourea (ENU) mutagenesis to introduce random mutations in zebrafish expressing glut1b:mCherry as a marker of barriergenesis and plvap:EGFP as a marker of immature blood vessels [16]. Plvap is initially expressed in BECs, but this expression subsides during development and is absent from adult brain endothelium [16, 64]. Using a similar screening strategy, we previously identified a gpr124 mutant which has defects in CNS angiogenesis and no glut1b:mCherry expression [16]. For our new screen, the primary goal was to identify two independent classes of mutants: 1) mutants with reduced glut1b:mCherry expression but normal CNS angiogenesis, indicating defects in barriergenesis, and 2) mutants with reduced CNS angiogenesis but normal glut1b:mCherry expression, indicating defects in CNS angiogenesis but not barriergenesis. While no mutants with the former phenotype were found, one mutant line, uw112, distinctly showed the latter. Normally, CNS angiogenesis in zebrafish begins between 24–36 hpf with endothelial tip cells sprouting and migrating from the primordial hindbrain channels (PHBCs), resulting in the formation of central arteries (CtAs) by 2 dpf [49, 52, 54]. Both uw112 and gpr124 mutants fail to develop CtAs (Fig. 1A). The phenotypes are even more pronounced at 3 dpf, with a distinct lack of blood vessels in the brains of both mutants showing a severe CNS angiogenesis defect (Fig. 1B). However, unlike the gpr124 mutants, the uw112 mutants express glut1b:mCherry in the PHBCs at both 2 and 3 dpf (Fig. 1A, B).
Fig. 1. Zebrafish uw112 mutants express glut1b:mCherry in the absence of CNS angiogenesis.
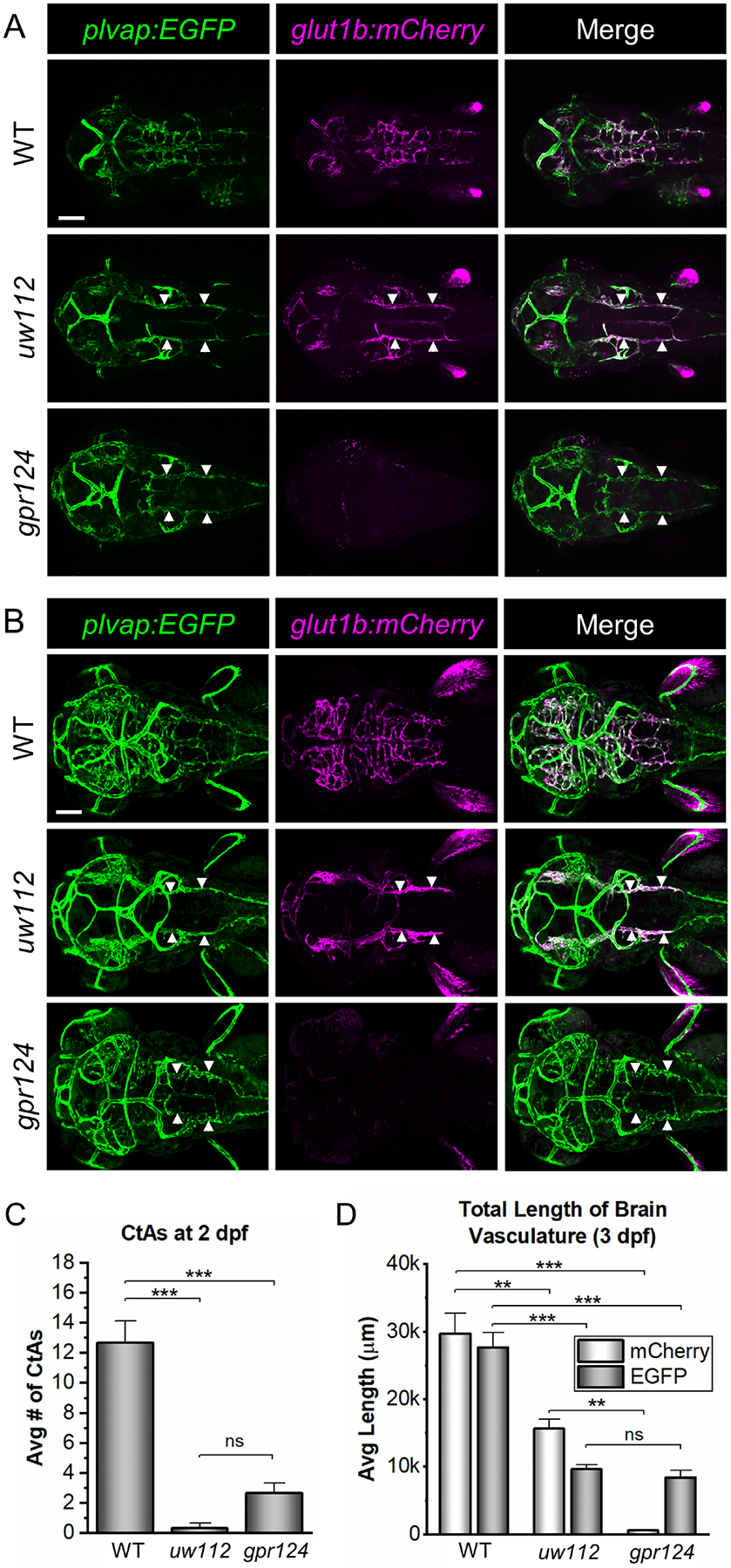
(A, B) Representative confocal microscopy images showing the glut1b:mCherry and plvap:EGFP labeled blood vessels in uw112 mutants in comparison to WT embryos and gpr124 mutants. Images are dorsal views of the head (anterior left) in live embryos at 2 and 3 dpf (A and B, respectively). At 2 dpf, uw112 and gpr124 mutants show a significant lack of central arteries (CtAs) as compared to WT (A). At 3 dpf, a distinct lack of blood vessels within the brain persists in both mutants (B). At both time points, uw112 mutants express glut1b:mCherry in the primordial hindbrain channels (PHBCs) (white arrows) while gpr124 mutants do not. Scale bars are 100 μm. (C) Quantification of the number of CtAs at 2 dpf (n=3). (D) Quantification of the vasculature within the brain parenchyma at 3 dpf. For each group (n=3), the blood vessels labeled with mCherry and those labeled with EGFP were quantified separately from the same fish. Data in both bar graphs (C, D) are presented as means ± standard error of the mean (SEM) (**p < 0.01; ***p < 0.001; ns = not significant).
To quantify the early stages of CNS angiogenesis, we counted the number of CtAs at 2 dpf. As shown in Figure 1C, CtAs were essentially absent in the uw112 mutants and dramatically reduced in the gpr124 mutants. This was further shown at 3 dpf by quantifying the length of brain vasculature expressing plvap:EGFP (Fig. 1D). The uw112 and gpr124 mutants showed a similar reduction in the amount of brain vasculature (Fig. 1D) but no obvious difference in the level of plvap:EGFP expression within the vessels as compared to wildtype (WT) (Fig. 1B). As an indicator of barriergenesis, we also quantified brain vessels expressing glut1b:mCherry at 3 dpf as well as the relative fluorescence intensity (RFI) of glut1b:mCherry in the hindbrain vasculature at both 2 and 3 dpf (Fig. 1D and S1). Essentially none of the brain vessels in the gpr124 mutants expressed glut1b:mCherry at a significant level. In contrast, the uw112 mutants expressed glut1b:mCherry in the PHBCs at levels comparable to WT brain vessels (Fig. 1D and S1), suggesting that the induction of barriergenesis is intact in the uw112 mutants. Importantly, these results indicate that barriergenesis can occur in the absence of CNS angiogenesis.
Zebrafish uw112 is a new mutant allele of kdrl
We next identified the defective gene in the uw112 mutant. We determined that the uw112 mutants showed similar vascular phenotypes to previously published kdrl mutants (kdrlum19 and kdrlt20257) with confirmed null mutations [47, 65]. For example, uw112 and both null mutants have no CtAs, partial intersegmental vessels (ISVs) in the trunk, normal dorsal arteries and cardinal veins, and normal circulation. In contrast, kdrl mutants with missense mutations (kdrly17 and kdrlum6) show additional, more severe phenotypes, including loss of circulation (y17 and um6), pericardial edema (um6), discontinuous dorsal aorta (y17), and a single major trunk vessel (um6) [47, 65, 66]. Therefore, we performed a complementation test by breeding uw112 heterozygous fish to kdrlum19 heterozygous fish [57]. We found that uw112 and kdrlum19 failed to complement as one-quarter of the offspring exhibited the same brain vascular phenotype as uw112 and kdrlum19 homozygous mutants (Fig. 2A). Additionally, uw112, kdrlum19, and uw112/kdrlum19 mutant embryos showed similar ISV defects in the trunk, normal circulation, and normal dorsal arteries and cardinal veins (Fig. S2). These data strongly indicate that uw112 is a new kdrl null allele (i.e. kdrluw112). We therefore used the new kdrluw112 mutant for experimentation throughout the remainder of this study.
Fig. 2. Identification of zebrafish uw112 as a new mutant allele of kdrl.
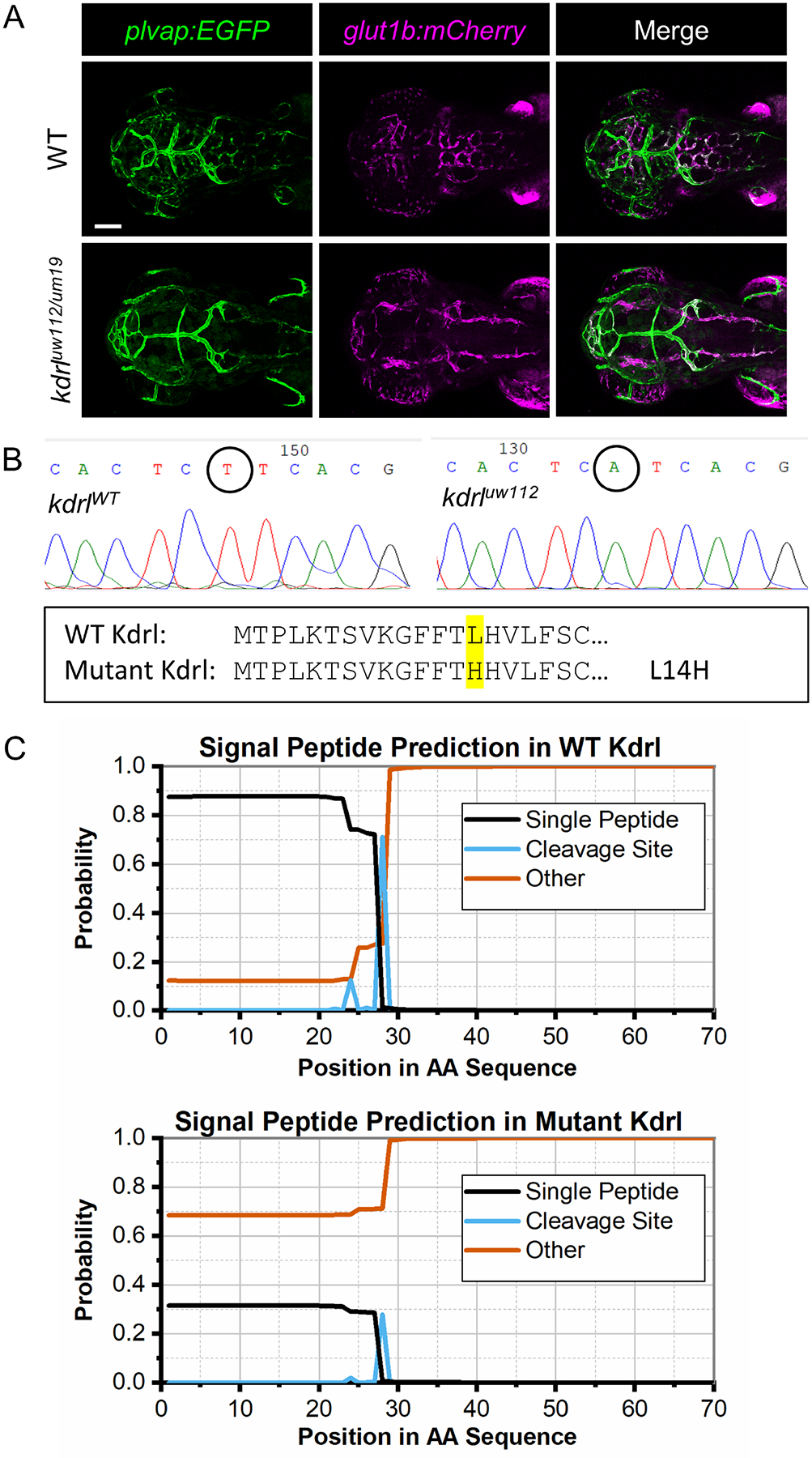
(A) Representative confocal images (dorsal views; anterior left) of the complementation test between uw112 and kdrlum19 mutants showing the same mutant phenotype as the uw112 mutants at 3 dpf. Scale bar is 100 μm. (B) DNA sequence for segments of kdrlWT and mutant kdrluw112 showing the T41A mutation (circled). The N-terminal protein sequence is also provided, showing the L14H protein mutation (highlighted). (C) SignalP-5.0 graphs predicting the presence of a signal peptide in the WT Kdrl protein verses the mutant Kdrl.
To identify the causal mutation in kdrluw112, full-length kdrl was sequenced from WT and mutant cDNA. Using this strategy, we found a T41A transversion in the open reading frame of kdrl (Fig. 2B). This results in a L14H missense mutation, which is located within the presumptive signal peptide [67]. To determine the functional consequences of this mutation, we used the SignalP-5.0 Server maintained by the Technical University of Denmark [68]. As shown in Figure 2C, the Kdrluw112 protein sequence is not predicted to include a signal peptide, likely indicating that the mutant protein is not correctly synthesized and expressed as a transmembrane protein. Together, the vascular phenotypes, complementation test, and DNA sequencing data strongly support that kdrluw112 is a null allele and demonstrate that Kdrl is required for CNS angiogenesis but not glut1b:mCherry expression in the PHBCs.
Vegf signaling is not required for glut1b:mCherry expression in BECs
Our finding that Kdrl is not required for the expression of the BBB marker glut1b:mCherry suggests that Vegf signaling is not required for barriergenesis. However, it is important to note that zebrafish have another Vegfr2 ortholog, Kdr (formerly called Kdrb or Vegfr2b) [66]. Previous studies demonstrated that kdrly17 mutants have partial ISVs, whereas kdrly17 mutants injected with kdr morpholinos or the combination of kdrl and kdr morpholinos completely lack ISVs in a manner similar to Vegfr inhibitors [66, 69]. In contrast, CtAs do not develop in kdrluw112 (Fig. 1) or in the other kdrl null mutants [47, 65], indicating that unlike the ISVs, kdr is not required for CtA formation. Indeed, a recent study by Vogrin et al generated a kdr null mutant (kdruq38bh) using CRISPR-genome editing and found normal brain vasculature in kdruq38bh mutants, further indicating that kdr in not required for CNS angiogenesis [70].
Therefore, to ensure that Vegf signaling was completely blocked, we treated embryos with potent Vegfr tyrosine kinase inhibitors that block angiogenesis in developing zebrafish. For these experiments, transgenic glut1b:mCherry, plvap:EGFP embryos were treated with Vegfr inhibitors at 24 hpf, 6 hours prior to the onset of CNS angiogenesis [16], then imaged at 2 dpf by confocal microscopy. We first treated embryos with 5 μM DMH4, a selective inhibitor of Vegf signaling that blocks angiogenesis in zebrafish [61], inhibits phosphorylation of VEGFR2 in human cell lines [71], and inhibits Vegfr-dependent phosphorylation of Erk (pErK), a key downstream component of Vegf signaling [72], in zebrafish endothelial cells. Treatment with 5 μM DMH4 completely blocked CNS angiogenesis as demonstrated by the absence of all CtAs (Fig. 3A, B). We found that, as in the kdrl mutants, 5 μM DMH4-treated embryos expressed the barriergenesis marker glut1b:mCherry in the PHBCs (Fig. 3A). To further demonstrate the effectiveness of DMH4, we treated embryos with a 5-fold lower concentration (1 μM) and observed very similar neurovascular phenotypes. These effects were also found in embryos treated with another Vegf inhibitor, AV-951 [62]. For all inhibitor-treated groups, CtAs were completely absent and showed a complete loss of CNS angiogenesis in comparison to the untreated and vehicle-only controls (Fig. 3B). Unlike the effect on angiogenesis, the fluorescence intensity of glut1b:mCherry in the hindbrain vasculature was not significantly different between the inhibitor-treated embryos and the controls (Fig. 3C).
Fig. 3. Vegfr tyrosine kinase inhibitors, DMH4 and AV-951, block CNS angiogenesis but not barriergenesis.
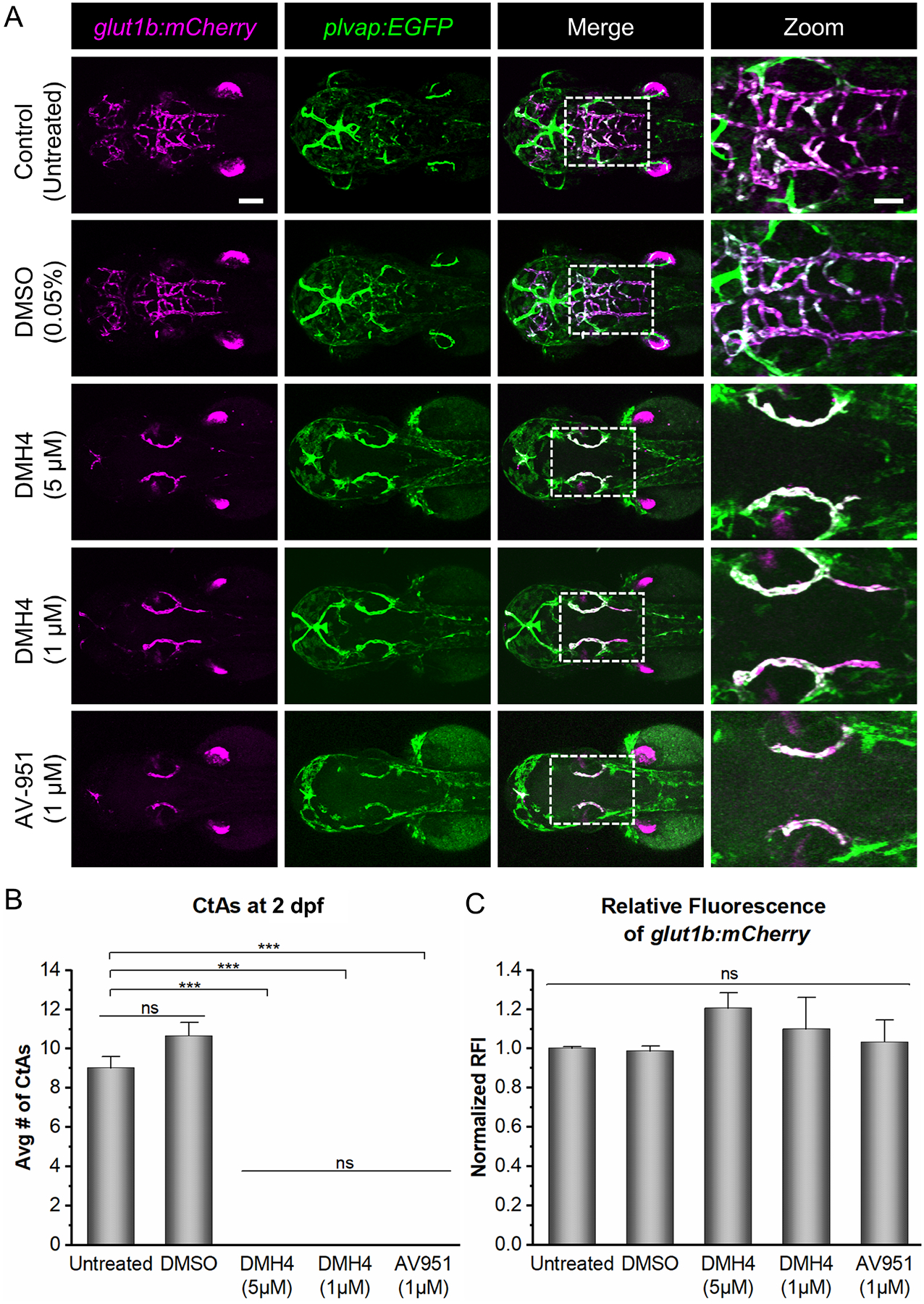
(A) Representative confocal images (dorsal views; anterior left) of untreated, DMSO-treated, or inhibitor-treated glut1b:mCherry, plvap:EGFP embryos. Embryos were treated at 24 hpf then imaged at 54 hpf. Note that the inhibitor-treated embryos lack CtAs but express glut1b:mCherry in the PHBCs indicating that barriergenesis occurs in the absence of CNS angiogenesis. In addition, 1 μM DMH4 was as effective as 5 μM DMH4 at blocking CNS angiogenesis. AV-951 (1 μM) showed similar effects as 5 μM DMH4. Scale bars are 100 μm (top left) and 50 μm (top right). (B) Quantification of the number of CtAs at 2 dpf for untreated, DMSO-treated, or inhibitor-treated embryos (n=3). (C) Quantification of the normalized RFI of glut1b:mCherry in untreated, DMSO-treated, or inhibitor-treated embryos at 2 dpf (n=3). Data for (B) and (C) are presented as means ± SEM (***p < 0.001; ns = not significant).
To further compare CNS angiogenesis between kdrluw112 mutants and DMH4-treated embryos, we performed time-lapse imaging. We found an indistinguishable absence of CNS angiogenesis using both genetic and chemical methods of disrupting Vegf signaling (Fig. S3). Additionally, there was no evidence of filopodia extensions from the PHBCs or other obvious indications of CNS angiogenesis in either the kdrl mutant or the DMH4-treated embryo (Movies S1–6). Finally, the fluorescence intensity of glut1b:mCherry in kdrl mutants was not significantly different from that of DMH4-treated WT embryos nor that of DMH4-treated kdrl mutants (Fig. S4). The lack of differences between these various experimental conditions demonstrates that inhibiting Vegf signaling has no effect on barriergenesis as indicated by glut1b:mCherry expression.
Zebrafish kdrl, but not gpr124, mutants express BBB-enriched proteins in BECs
Given that kdrl mutants lack CNS angiogenesis but maintain glut1b:mCherry expression as a marker of barriergenesis, we predicted that Vegf signaling plays a fundamental role in CNS angiogenesis but not barriergenesis. However, GLUT1 expression on its own is not necessarily indicative of a complete and functioning BBB. We therefore examined kdrl mutants for the expression of other commonly used BBB markers using whole-mount immunohistochemistry (IHC). We first performed IHC against the tight junction protein Claudin-5, which previous studies have used to label the BBB in zebrafish [73–77]. WT, kdrl, and gpr124 larvae at 5 dpf were stained with both an anti-Claudin-5 antibody (α-Claudin-5) and an anti-Glut1 antibody (α-Glut1). We performed these experiments at 5 dpf as this timepoint produced a consistently stronger signal due to more complete BBB development. Both the Glut1 staining and the transgenic plvap:EGFP expression were used as references showing the vasculature in the brain of each fish. In WT and kdrl larvae, Claudin-5 staining was detected in nearly all vessels in the hindbrain. However, in gpr124 larvae, serving as a negative control, only a very small fraction of the hindbrain vasculature had any discernable Claudin-5 labeling (Fig. 4A). The Claudin-5 staining was quantified as the fraction of total hindbrain vasculature labeled with α-Claudin-5 (Fig. 4B). This fraction was significantly decreased in gpr124 mutants, but not kdrl mutants, further suggesting that barriergenesis is not disrupted by the mutation in kdrl.
Fig. 4. Zebrafish kdrl, but not gpr124, mutants, express BBB markers.
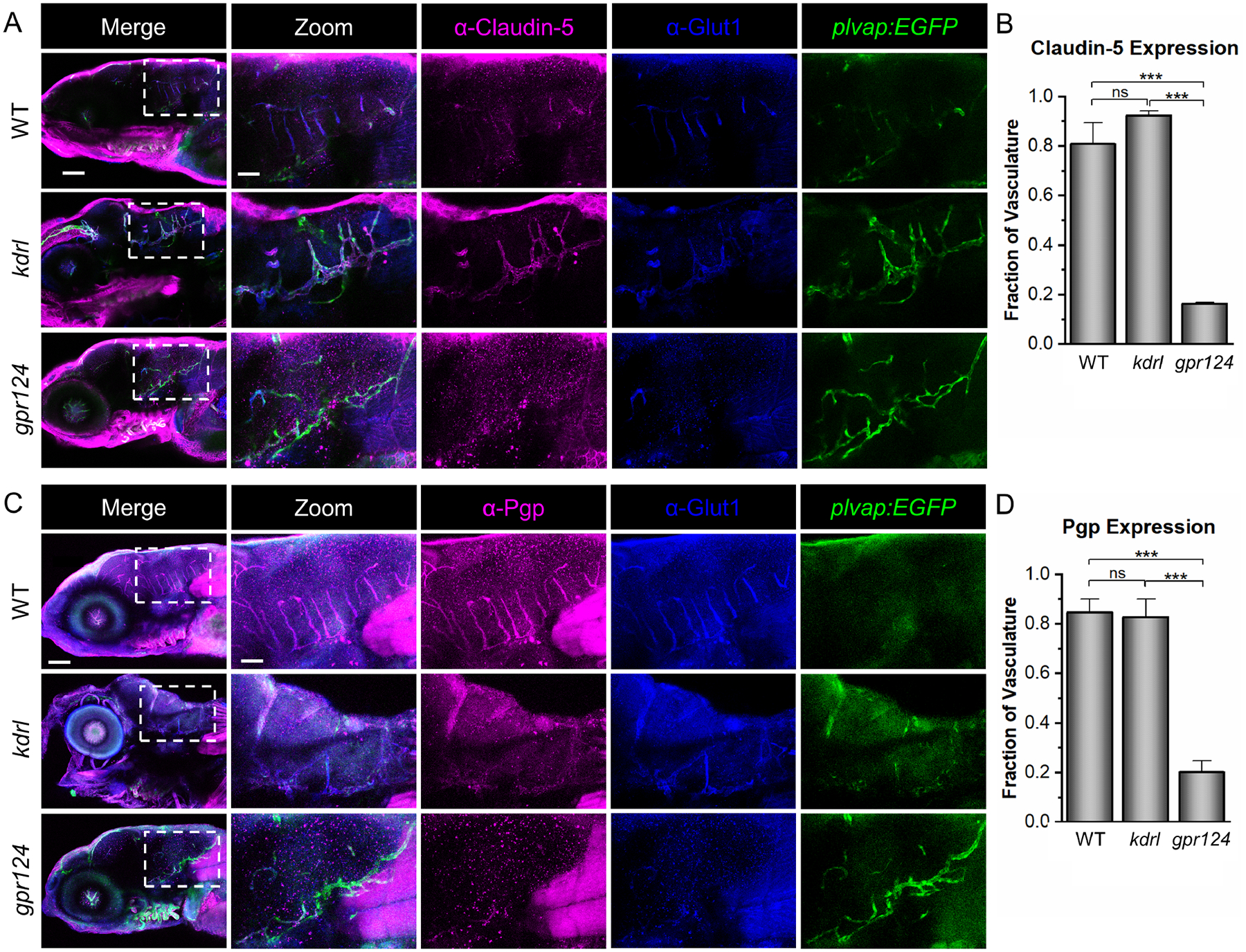
(A) Representative confocal microscopy images showing Claudin-5 staining. WT, kdrl, and gpr124 larvae were stained with α-Claudin-5 and α-Glut1. The plvap:EGFP transgene was used as a blood vessel marker. (B) Quantification was calculated from the fraction of the total blood vessels in the hindbrain labeled with α-Claudin-5. (C, D) Representative confocal images of α-Pgp staining at 5 dpf with the same controls as in (A) and quantification strategy as in (B). All images are lateral views (anterior left; dorsal top) of whole-mount stained larvae at 5 dpf. Scale bars are 100 μm for the merged images (left panels) and 40 μm for the zoomed images. Data in B and D are presented as means (n = 3) ± SEM (***p < 0.001; ns = not significant).
We also performed IHC against the multidrug resistance protein P-glycoprotein (Pgp) [78]. We previously demonstrated that Pgp is expressed at the zebrafish BBB during early development [79]. As with Claudin-5, WT, kdrl, and gpr124 larvae at 5 dpf were stained with an anti-Pgp antibody (α-Pgp). The α-Glut1 and plvap:EGFP were again used as references. These results showed the same pattern as the Claudin-5 staining, with Pgp expressed in the kdrl PHBCs but nearly absent in gpr124 (Fig. 4C). The Pgp staining was quantified as the fraction of total hindbrain vasculature labeled with α-Pgp (Fig. 4D). Our IHC results, together with the glut1b:mCherry expression in live embryos, demonstrate that BBB markers are not disrupted in the kdrl mutants but are essentially absent in the gpr124 mutants. These observations further support the conclusion that Wnt signaling (via Gpr124) is required for both CNS angiogenesis and barriergenesis while Vegf signaling is essential for CNS angiogenesis but not required for barriergenesis.
Vegf signaling is not required for the activation of Wnt/β-catenin-dependent transcription
Knowing that Vegf signaling and Wnt/β-catenin signaling both regulate CNS angiogenesis and that Vegf signaling is not required for barriergenesis, we next examined activation of Wnt/β-catenin signaling in the absence of Vegf signaling. Here, we utilized a transgenic Wnt/β-catenin transcriptional reporter line, tcf:EGFP, to visualize activation of the Wnt/β-catenin signaling pathway [58]. Previous studies used a similar strategy to demonstrate that gpr124 and reck mutants lack Wnt/β-catenin transcriptional activity [20]. To disrupt Vegf signaling, we treated double transgenic tcf:EGFP, kdrl:mCherry embryos with 5 μM DMH4. We reasoned that if VEGF is required for Wnt signaling, then DMH4 should block the activation of tcf:EGFP. We found, however, that tcf:EGFP co-localized with kdrl:mCherry-labeled PHBCs in DMH4-treated embryos, indicating that Wnt/β-catenin signaling is intact in the absence of Vegf signaling (Fig. 5A). Similarly, we performed the same experiment with kdrl mutants and again found tcf:EGFP expression in the PHBCs (Fig. S5), further confirming that Vegf signaling is not required for the activation of Wnt/β-catenin-dependent transcription.
Fig. 5. Vegf signaling is not required for Wnt/β-catenin transcriptional activity.
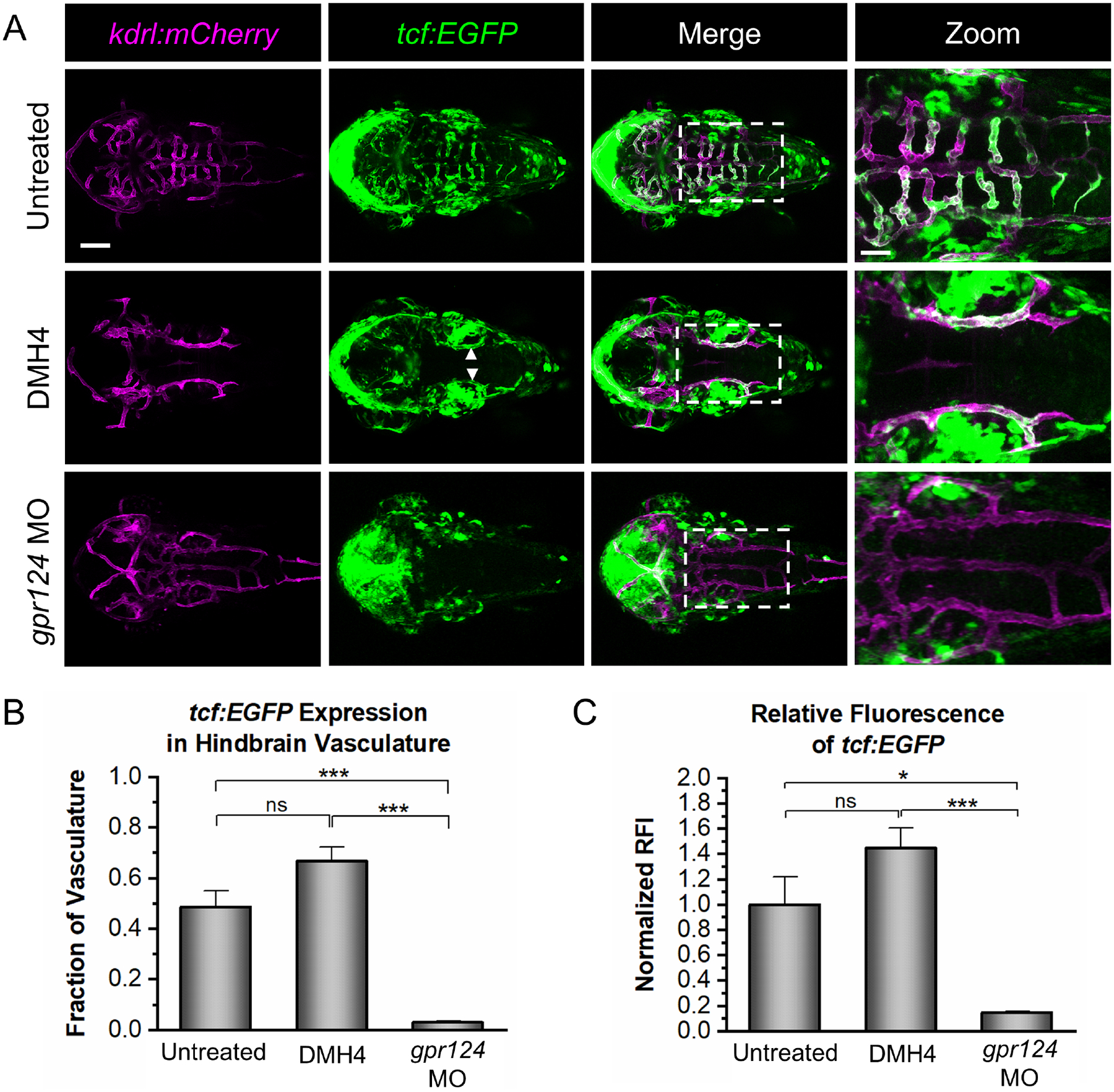
(A) Representative confocal images of tcf:EGFP, kdrl:mCherry embryos at 2 dpf that were untreated, DMH4-treated (DMH4), or injected with a gpr124 morpholino (gpr124 MO). tcf:EGFP signal is present in the WT brain vasculature (top panels) and in the PHBCs of the DMH4-treated embryos (middle panels; white arrows), but not the gpr124 MO-injected embryos (bottom panels). Scale bar is 100 μm for the first three columns and 40 μm for the zoomed images (right panels). (B) Quantification of the fraction of kdrl:mCherry-labeled hindbrain vasculature expressing tcf:EGFP. (C) Quantification of the normalized relative fluorescence intensity (RFI) of tcf:EGFP in the hindbrain vasculature. Data in B and C are presented as means (n = 4) ± SEM (*p < 0.05; ***p < 0.001; ns = not significant).
As a negative control, we also examined tcf:EGFP expression in the PHBCs when Wnt/β-catenin signaling was inhibited through loss of Gpr124. For this experiment, gpr124 morpholinos were used to obtain the phenotype of the gpr124 mutants in tcf:EGFP, kdrl:mCherry embryos. As others have previously demonstrated, we found that the gpr124 morpholino phenocopies the gpr124 mutants with no obvious off-target effects [20]. As expected, the gpr124 morphants did not express tcf:EGFP in the PHBCs (Fig. 5A; bottom panels), showing a clear difference in comparison to the DMH4-treated embryos. Activation of Wnt signaling in each group was quantified using the fraction of the kdrl:mCherry-labeled hindbrain vasculature expressing tcf:EGFP (Fig. 5B) and the relative fluorescence intensity of the tcf:EFGP within the hindbrain vasculature (Fig. 5C). In DMH4-treated embryos, both the fraction of vasculature expressing tcf:EFGP and the intensity of that signal are comparable to the untreated embryos while gpr124 morphants show minimal tcf:EGFP expression in vessels. The presence of tcf:EGFP signal in the PHBCs of DMH4-treated embryos but not the gpr124 morphants supports the conclusion that Vegf signaling is not required for Wnt/β-catenin activation during CNS angiogenesis.
Discussion
Our findings highlight the role of Vegf signaling in neurovascular development as BECs acquire BBB properties. Here, we performed an unbiased genetic screen in zebrafish that utilized the glut1b:mCherry and plvap:EGFP transgenic lines as markers of BEC differentiation during developmental CNS angiogenesis. Using this strategy, we identified a zebrafish mutant that maintained expression of the BBB marker glut1b:mCherry but completely lacked CNS angiogenesis in a manner similar to gpr124 mutants. Genetic analyses revealed that this mutant represents a new allele of kdrl, an ortholog of mammalian Vegfr2. Unlike gpr124 mutants, which lack normal CNS angiogenesis and barriergenesis due to defective Wnt/β-catenin signaling, the kdrl mutants acquire barrier properties in BECs as demonstrated by the expression of BBB markers Glut1, Claudin-5, and Pgp. Furthermore, we show that activation of Wnt/β-catenin signaling does not require Vegf signaling during neurovascular development. Taken together, our results establish that Vegf signaling is essential for CNS angiogenesis but is not required for Wnt/β-catenin-dependent barriergenesis.
Previous studies regarding the induction of BBB properties in BECs have primarily focused on the Wnt/β-catenin pathway, where several key genes have been discovered. For example, endothelial-specific expression of β-catenin [17, 21, 26], Gpr124 [16, 18–20, 28, 29, 33, 34], and Reck [20, 23, 30–32, 34] and non-endothelial expression of Wnt7a/7b [17, 19–21, 33, 34] in the neuroepithelium are required for proper BBB formation. Disruption of these genes in mice and zebrafish revealed similar phenotypes that include impaired developmental CNS angiogenesis, vascular malformations, defective angiogenic sprouting, and loss of Glut1 expression in the endothelium. Most recently, endothelial Unc5B was also found to control BBB integrity by functioning as a Wnt co-receptor that activates Wnt/β-catenin signaling [35]. In addition to Wnt/β-catenin, genes essential for Vegf signaling, including Vegfa [43, 44] and Nrp1 [80, 81], are also required for normal CNS angiogenesis. Yet, to our knowledge, we are the first to report that Vegf signaling is not required for barriergenesis.
Given that both Wnt/β-catenin and Vegf signaling are required for normal developmental CNS angiogenesis, crosstalk between these pathways seems likely. Although direct evidence for interaction is lacking, recent studies have proposed some intriguing possibilities and identified additional complications. For example, Tam et al identified death receptors, DR6 and TROY, as potential regulators of CNS angiogenesis in mice and zebrafish [22]. This study suggested that these death receptors are targets of Wnt/β-catenin signaling and are required for Vegf-mediated JNK activation. However, loss of DR6 and TROY exhibited only modest effects on CNS angiogenesis and no reduction of Glut1 expression. In addition, studies by Ulrich et al found that zebrafish reck mutants displayed weak kdrl transcriptional reporter expression in PHBCs, suggesting that inactivation of Wnt/β-catenin signaling might impair Vegf activity [23]. Conversely, Vanhollebeke et al reasoned that reduced Vegf signaling is an unlikely explanation for defective CNS angiogenesis in both the gpr124 mutants and reck morphants because basilar artery formation is unaffected and no significant differences in the transcriptional level of vegf ligands were found in zebrafish gpr124 mutants [20]. Our results, similar to Vanhollebeke et al, did not reveal any obvious reduction of kdrl:mCherry signal in gpr124 morphants. We propose that the phenotypic discrepancies between the gpr124 and reck mutants may be due to functional differences of these gene products as the effects of inactivating these genes are not fully equivalent as previously described [23]. Moreover, reck encodes a GPI-anchored glycoprotein that inhibits the activity of several matrix metalloproteinases (MMPs) [82] and functions as a key regulator of angiogenesis [32, 83]. Thus, reck mutants may exhibit additional phenotypes that could be independent of Wnt/β-catenin signaling.
Additional mechanisms may involve crosstalk of Wnt/β-catenin and Vegf signaling with other developmental pathways, such as Notch [84, 85]. During angiogenic sprouting, Vegf signaling functions upstream of Notch signaling to control tip/stalk cell specification [86–88]. To examine tip cell function in Wnt/β-catenin mutants, Vanhollebeke et al used transplantation experiments to demonstrate a tip cell-specific requirement for Gpr124 and Reck during CNS angiogenesis. However, these experiments did not discriminate whether the Wnt/β-catenin signaling pathway provides either a permissive role during CNS angiogenesis or a selective role during tip cell specification. Under Vegf stimulation, endothelial tip cells upregulate expression of the Notch receptor ligand Dll4, which activates Notch signaling in neighboring stalk cells and suppresses the tip cell phenotype [38]. To assess dll4 in Wnt/β-catenin mutants, the zebrafish gpr124 and reck mutants were examined by in situ hybridization and both showed dll4 transcript expression in the PHBCs [20, 23]. While these results potentially indicate that Vegf signaling is intact in the absence of Wnt/β-catenin, endothelial-specific β-catenin gain-of-function studies have also shown upregulation of Dll4, complicating this analysis [89].
Paradoxically, previous studies have also demonstrated that Vegf signaling upregulates Plvap expression [64, 90] while Wnt/β-catenin signaling downregulates Plvap expression [91, 92]. During neurovascular development, Plvap, a structural component of fenestrae, is initially expressed in BECs, but this expression subsides during blood vessel maturation and is absent from adult brain endothelium except for the vasculature of circumventricular organs and in certain disease states [6, 93, 94]. Interestingly, a recent study by Parab et al used our transgenic plvap:EGFP reporter line to demonstrate that specific combinations of Vegf ligands are required to selectively drive expression of plvap:EGFP during vessel formation in the zebrafish myelencephalic choroid plexus [95]. In addition, our previous work showed that plvap:EGFP is initially expressed in the early brain vasculature, decreases during BBB maturation, and is absent from the adult zebrafish brain [16]. These results demonstrate that the zebrafish plvap:EGFP transgene recapitulates the developmental expression of mammalian PLVAP [6]. In our current study, we find plvap:EGFP expression in the PHBCs of both the gpr124 and kdrl mutants as well as embryos treated with Vegfr inhibitors, indicating that loss of either pathway does not disrupt plvap:EGFP expression. Thus, while both Wnt and Vegf pathways are activated during CNS angiogenesis, it remains unclear how Plvap expression is regulated by these or other pathways during neurovascular development.
In contrast to our study, Ulrich et al used in situ hybridization to examine plvap transcript expression and found relatively higher levels of plvap transcript in the PHBCs of reck mutants and in embryos with ubiquitous, heat shock-induced Axin-1 in comparison to the level of plvap transcript in the PHBCs of WT and kdrlum19 mutants [23]. These results are somewhat surprising given that this study indicated that plvap transcript was present in WT CtAs but absent from WT and kdrlum19 mutant PHBCs. Based upon these observations, the authors suggested that Vegf activity is dispensable for cerebrovascular canonical Wnt signaling. Presumably, this suggestion is based upon the concept that reduced Wnt/β-catenin signaling in reck mutants may result in the upregulation of the plvap transcript, whereas WT and kdrlum19 mutants maintain normal Wnt/β-catenin signaling. Conversely, as described above, we did not observe upregulation of the plvap:EGFP transgenic reporter in gpr124 morphants nor did we detect reduced plvap:EGFP expression in the PHBCs of WT or kdrl mutants. Thus, we conclude that the inconsistencies between Ulrich et al and our study may be due to differences between plvap expression by in situ hybridization versus plvap:EGFP transgene expression and/or functional differences between reck and gpr124.
Despite the essential role for Wnt/β-catenin and Vegf signaling during neurovascular development, no studies to date have definitively determined the specific interactions between these pathways. Based upon our findings, we propose that Wnt/β-catenin and Vegf signaling act either sequentially or cooperatively (Fig. 6). For sequential activation, one pathway is required for the other pathway to be functional. Based upon the expression of barriergenesis markers and activation of TCF/LEF transcription, we found that Wnt/β-catenin signaling is intact when Vegf signaling is disrupted, suggesting that Vegf signaling is not upstream of Wnt/β-catenin signaling or barriergenesis (Fig. 6, red arrows). Therefore, if the pathways are sequential, then Wnt/β-catenin signaling must be required for Vegf signaling (Fig. 6, dashed arrow 1). This model is consistent with previous studies that demonstrated GPR124 regulation of Vegf-induced tumor angiogenesis [96] and endothelial-specific β-catenin regulation of VEGFR2 and VEGFR3 during postnatal brain and retinal angiogenesis [97]. These interactions are likely transcription-dependent, where Wnt/β-catenin signaling activates the transcription of one or more genes required for Vegf signaling. However, other studies showed endothelial-specific expression of dll4 transcript in the absence of Wnt signaling [20, 23]. As Vegf is known to regulate Dll4 during tip cell specification [38], these results potentially indicate that Vegf signaling can function independently of Wnt/β-catenin signaling in BECs. These findings support a cooperative activation model as an alternative mechanism, in which Wnt/β-catenin and Vegf pathways converge in the regulation of CNS angiogenesis (Fig. 6, dashed arrow 2). This could occur through co-regulation of transcriptional targets, in which one or more genes essential for angiogenesis requires transcription factors from both pathways to activate transcription.
Fig. 6. Schematic model showing the potential interactions between Wnt/β-catenin signaling and Vegf signaling.
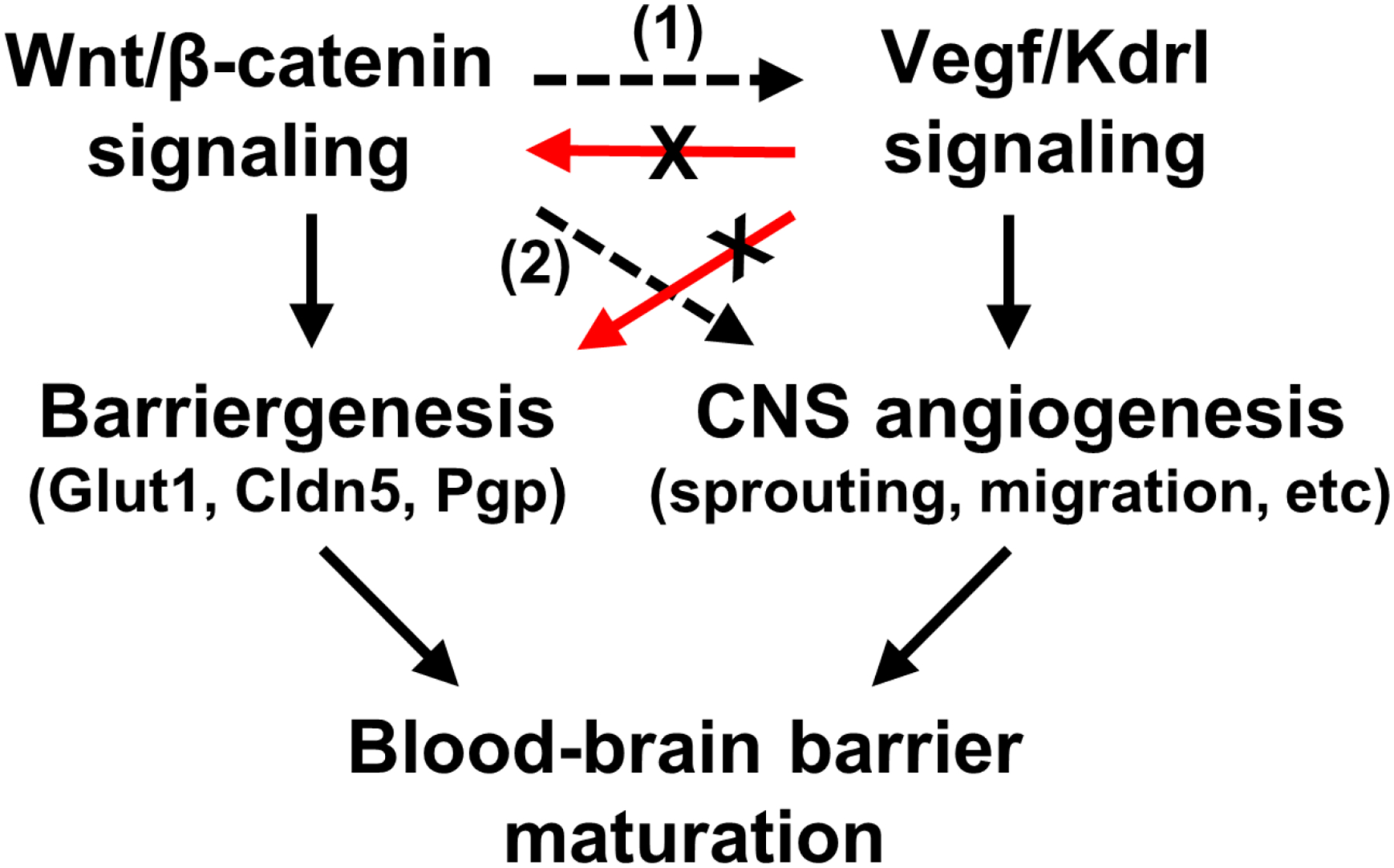
Our data indicate that Wnt/β-catenin signaling and barriergenesis do not require Vegf signaling (red arrows with X), whereas CNS angiogenesis requires either (1) Wnt/β-catenin regulation of Vegf signaling or (2) Wnt/β-catenin and Vegf co-regulation (dashed black arrows).
In conclusion, our findings provide the first direct evidence, to our knowledge, that Vegf signaling is not required for Wnt/β-catenin-dependent barriergenesis. We also show that activation of Wnt/β-catenin signaling occurs independently of Vegf signaling in brain endothelial cells. Our results provide important new insights for unraveling the complexities of neurovascular development and discovering effective therapies that target the brain vasculature. Future studies examining the molecular and cellular connections between CNS angiogenesis and barriergenesis, the interdependent relationships between multiple signal transduction pathways, and the individual contributions of both Wnt/β-catenin and Vegf signaling will be essential to fully understand the fundamental biology of the BBB.
Supplementary Material
Highlights.
Vegf signaling is required for CNS angiogenesis, but not barriergenesis
Zebrafish kdrl, but not gpr124, mutants acquire blood-brain barrier properties
Wnt/β-catenin-dependent barriergenesis does not require Vegf signaling
Acknowledgments
We thank Dr. Nathan Lawson (University of Massachusetts Medical School) for providing the kdrlum19 mutant line, Dr. Junsu Kang (UW-Madison) for providing the Tg(7xtcf-Xla.Siam:EGFP)ia4 transgenic line, and Dr. Jan Huisken (Morgridge Institute for Research and UW-Madison) for providing the Tg(kdrl:HRAS-mCherry)s896 transgenic line. We also wish to thank Randall Kopielski and Jessica Fairbanks for assistance with zebrafish husbandry and animal facility care and maintenance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, Begley DJ. Structure and function of the blood-brain barrier. Neurobiol Dis. 2010;37(1):13–25. Epub 2009/08/12. doi: S0969–9961(09)00208–3 [pii] 10.1016/j.nbd.2009.07.030. [DOI] [PubMed] [Google Scholar]
- 2.Armulik A, Genove G, Mae M, Nisancioglu MH, Wallgard E, Niaudet C, et al. Pericytes regulate the blood-brain barrier. Nature. 2010;468(7323):557–61. Epub 2010/10/15. doi: nature09522 [pii] 10.1038/nature09522. [DOI] [PubMed] [Google Scholar]
- 3.Daneman R, Zhou L, Kebede AA, Barres BA. Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature. 2010;468(7323):562–6. Epub 2010/10/15. doi: nature09513 [pii] 10.1038/nature09513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stewart PA, Wiley MJ. Developing nervous tissue induces formation of blood-brain barrier characteristics in invading endothelial cells: a study using quail--chick transplantation chimeras. Dev Biol. 1981;84(1):183–92. Epub 1981/05/01. doi: 0012–1606(81)90382–1 [pii]. [DOI] [PubMed] [Google Scholar]
- 5.Abbott NJ, Ronnback L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci. 2006;7(1):41–53. Epub 2005/12/24. doi: nrn1824 [pii] 10.1038/nrn1824. [DOI] [PubMed] [Google Scholar]
- 6.Hallmann R, Mayer DN, Berg EL, Broermann R, Butcher EC. Novel mouse endothelial cell surface marker is suppressed during differentiation of the blood brain barrier. Dev Dyn. 1995;202(4):325–32. doi: 10.1002/aja.1002020402. [DOI] [PubMed] [Google Scholar]
- 7.Ben-Zvi A, Lacoste B, Kur E, Andreone BJ, Mayshar Y, Yan H, et al. Mfsd2a is critical for the formation and function of the blood-brain barrier. Nature. 2014;509(7501):507–11. Epub 20140514. doi: 10.1038/nature13324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reese TS, Karnovsky MJ. Fine structural localization of a blood-brain barrier to exogenous peroxidase. J Cell Biol. 1967;34(1):207–17. Epub 1967/07/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Saunders NR, Liddelow SA, Dziegielewska KM. Barrier mechanisms in the developing brain. Front Pharmacol. 2012;3:46. doi: 10.3389/fphar.2012.00046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chow BW, Gu C. The molecular constituents of the blood-brain barrier. Trends Neurosci. 2015;38(10):598–608. doi: 10.1016/j.tins.2015.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hagan N, Ben-Zvi A. The molecular, cellular, and morphological components of blood-brain barrier development during embryogenesis. Semin Cell Dev Biol. 2015;38:7–15. Epub 20141227. doi: 10.1016/j.semcdb.2014.12.006. [DOI] [PubMed] [Google Scholar]
- 12.Pardridge WM. Blood-brain barrier delivery. Drug Discov Today. 2007;12(1–2):54–61. Epub 2006/11/13. doi: 10.1016/j.drudis.2006.10.013. [DOI] [PubMed] [Google Scholar]
- 13.Saunders NR, Ek CJ, Habgood MD, Dziegielewska KM. Barriers in the brain: a renaissance? Trends Neurosci. 2008;31(6):279–86. Epub 2008/05/13. doi: S0166–2236(08)00118–5 [pii] 10.1016/j.tins.2008.03.003. [DOI] [PubMed] [Google Scholar]
- 14.Zlokovic BV. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron. 2008;57(2):178–201. Epub 2008/01/25. doi: S0896–6273(08)00034–2 [pii] 10.1016/j.neuron.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 15.Obermeier B, Daneman R, Ransohoff RM. Development, maintenance and disruption of the blood-brain barrier. Nat Med. 2013;19(12):1584–96. Epub 20131205. doi: 10.1038/nm.3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Umans RA, Henson HE, Mu F, Parupalli C, Ju B, Peters JL, et al. CNS angiogenesis and barriergenesis occur simultaneously. Dev Biol. 2017;425(2):101–8. doi: 10.1016/j.ydbio.2017.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Daneman R, Agalliu D, Zhou L, Kuhnert F, Kuo CJ, Barres BA. Wnt/beta-catenin signaling is required for CNS, but not non-CNS, angiogenesis. Proc Natl Acad Sci U S A. 2009;106(2):641–6. Epub 2009/01/09. doi: 0805165106 [pii] 10.1073/pnas.0805165106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Anderson KD, Pan L, Yang XM, Hughes VC, Walls JR, Dominguez MG, et al. Angiogenic sprouting into neural tissue requires Gpr124, an orphan G protein-coupled receptor. Proc Natl Acad Sci U S A. 2011;108(7):2807–12. doi: 10.1073/pnas.1019761108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou Y, Nathans J. Gpr124 controls CNS angiogenesis and blood-brain barrier integrity by promoting ligand-specific canonical wnt signaling. Dev Cell. 2014;31(2):248–56. Epub 2014/10/16. doi: 10.1016/j.devcel.2014.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vanhollebeke B, Stone OA, Bostaille N, Cho C, Zhou Y, Maquet E, et al. Tip cell-specific requirement for an atypical Gpr124- and Reck-dependent Wnt/beta-catenin pathway during brain angiogenesis. Elife. 2015;4. doi: 10.7554/eLife.06489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stenman JM, Rajagopal J, Carroll TJ, Ishibashi M, McMahon J, McMahon AP. Canonical Wnt signaling regulates organ-specific assembly and differentiation of CNS vasculature. Science. 2008;322(5905):1247–50. Epub 2008/11/22. doi: 322/5905/1247 [pii] 10.1126/science.1164594. [DOI] [PubMed] [Google Scholar]
- 22.Tam SJ, Richmond DL, Kaminker JS, Modrusan Z, Martin-McNulty B, Cao TC, et al. Death receptors DR6 and TROY regulate brain vascular development. Developmental cell. 2012;22(2):403–17. Epub 2012/02/22. doi: 10.1016/j.devcel.2011.11.018. [DOI] [PubMed] [Google Scholar]
- 23.Ulrich F, Carretero-Ortega J, Menéndez J, Narvaez C, Sun B, Lancaster E, et al. Reck enables cerebrovascular development by promoting canonical Wnt signaling. Development. 2016;143(1):147–59. Epub 20151210. doi: 10.1242/dev.123059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bauer H, Sonnleitner U, Lametschwandtner A, Steiner M, Adam H, Bauer HC. Ontogenic expression of the erythroid-type glucose transporter (Glut 1) in the telencephalon of the mouse: correlation to the tightening of the blood-brain barrier. Brain Res Dev Brain Res. 1995;86(1–2):317–25. [DOI] [PubMed] [Google Scholar]
- 25.Pardridge WM, Boado RJ, Farrell CR. Brain-type glucose transporter (GLUT-1) is selectively localized to the blood-brain barrier. Studies with quantitative western blotting and in situ hybridization. J Biol Chem. 1990;265(29):18035–40. [PubMed] [Google Scholar]
- 26.Liebner S, Corada M, Bangsow T, Babbage J, Taddei A, Czupalla CJ, et al. Wnt/beta-catenin signaling controls development of the blood-brain barrier. J Cell Biol. 2008;183(3):409–17. Epub 2008/10/29. doi: jcb.200806024 [pii] 10.1083/jcb.200806024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou Y, Wang Y, Tischfield M, Williams J, Smallwood PM, Rattner A, et al. Canonical WNT signaling components in vascular development and barrier formation. J Clin Invest. 2014;124(9):3825–46. Epub 2014/08/01. doi: 10.1172/jci76431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cullen M, Elzarrad MK, Seaman S, Zudaire E, Stevens J, Yang MY, et al. GPR124, an orphan G protein-coupled receptor, is required for CNS-specific vascularization and establishment of the blood-brain barrier. Proc Natl Acad Sci U S A. 2011;108(14):5759–64. Epub 2011/03/18. doi: 10.1073/pnas.1017192108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kuhnert F, Mancuso MR, Shamloo A, Wang HT, Choksi V, Florek M, et al. Essential regulation of CNS angiogenesis by the orphan G protein-coupled receptor GPR124. Science. 2010;330(6006):985–9. doi: 10.1126/science.1196554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miki T, Shamma A, Kitajima S, Takegami Y, Noda M, Nakashima Y, et al. The ß1-integrin-dependent function of RECK in physiologic and tumor angiogenesis. Mol Cancer Res. 2010;8(5):665–76. Epub 20100420. doi: 10.1158/1541-7786.MCR-09-0351. [DOI] [PubMed] [Google Scholar]
- 31.Chandana EP, Maeda Y, Ueda A, Kiyonari H, Oshima N, Yamamoto M, et al. Involvement of the Reck tumor suppressor protein in maternal and embryonic vascular remodeling in mice. BMC Dev Biol. 2010;10:84. Epub 20100806. doi: 10.1186/1471-213X-10-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oh J, Takahashi R, Kondo S, Mizoguchi A, Adachi E, Sasahara RM, et al. The membrane-anchored MMP inhibitor RECK is a key regulator of extracellular matrix integrity and angiogenesis. Cell. 2001;107(6):789–800. doi: 10.1016/s0092-8674(01)00597-9. [DOI] [PubMed] [Google Scholar]
- 33.Posokhova E, Shukla A, Seaman S, Volate S, Hilton MB, Wu B, et al. GPR124 functions as a WNT7-specific coactivator of canonical beta-catenin signaling. Cell Rep. 2015;10(2):123–30. doi: 10.1016/j.celrep.2014.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cho C, Smallwood PM, Nathans J. Reck and Gpr124 Are Essential Receptor Cofactors for Wnt7a/Wnt7b-Specific Signaling in Mammalian CNS Angiogenesis and Blood-Brain Barrier Regulation. Neuron. 2017;95(5):1056–+. doi: 10.1016/j.neuron.2017.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boyé K, Geraldo LH, Furtado J, Pibouin-Fragner L, Poulet M, Kim D, et al. Endothelial Unc5B controls blood-brain barrier integrity. Nat Commun. 2022;13(1):1169. Epub 20220304. doi: 10.1038/s41467-022-28785-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ferrara N Vascular Endothelial Growth Factor: Basic Science and Clinical Progress. Endocrine Reviews. 2004;25(4):581–611. doi: 10.1210/er.2003-0027. [DOI] [PubMed] [Google Scholar]
- 37.Shibuya M. Vascular Endothelial Growth Factor (VEGF) and Its Receptor (VEGFR) Signaling in Angiogenesis: A Crucial Target for Anti- and Pro-Angiogenic Therapies. Genes Cancer. 2011;2(12):1097–105. doi: 10.1177/1947601911423031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gerhardt H, Golding M, Fruttiger M, Ruhrberg C, Lundkvist A, Abramsson A, et al. VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J Cell Biol. 2003;161(6):1163–77. Epub 2003/06/16. doi: 10.1083/jcb.200302047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gille H, Kowalski J, Li B, LeCouter J, Moffat B, Zioncheck TF, et al. Analysis of biological effects and signaling properties of Flt-1 (VEGFR-1) and KDR (VEGFR-2). A reassessment using novel receptor-specific vascular endothelial growth factor mutants. J Biol Chem. 2001;276(5):3222–30. Epub 20001031. doi: 10.1074/jbc.M002016200. [DOI] [PubMed] [Google Scholar]
- 40.Carmeliet P, Ferreira V, Breier G, Pollefeyt S, Kieckens L, Gertsenstein M, et al. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature. 1996;380(6573):435–9. doi: 10.1038/380435a0. [DOI] [PubMed] [Google Scholar]
- 41.Ferrara N, Carver-Moore K, Chen H, Dowd M, Lu L, O’Shea KS, et al. Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature. 1996;380(6573):439–42. doi: 10.1038/380439a0. [DOI] [PubMed] [Google Scholar]
- 42.Engelhardt B Development of the blood-brain barrier. Cell Tissue Res. 2003;314(1):119–29. Epub 2003/09/05. doi: 10.1007/s00441-003-0751-z. [DOI] [PubMed] [Google Scholar]
- 43.Haigh JJ, Morelli PI, Gerhardt H, Haigh K, Tsien J, Damert A, et al. Cortical and retinal defects caused by dosage-dependent reductions in VEGF-A paracrine signaling. Dev Biol. 2003;262(2):225–41. doi: 10.1016/s0012-1606(03)00356-7. [DOI] [PubMed] [Google Scholar]
- 44.Raab S, Beck H, Gaumann A, Yüce A, Gerber HP, Plate K, et al. Impaired brain angiogenesis and neuronal apoptosis induced by conditional homozygous inactivation of vascular endothelial growth factor. Thromb Haemost. 2004;91(3):595–605. doi: 10.1160/TH03-09-0582. [DOI] [PubMed] [Google Scholar]
- 45.Rosenstein JM, Krum JM, Ruhrberg C. VEGF in the nervous system. Organogenesis. 2010;6(2):107–14. doi: 10.4161/org.6.2.11687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shalaby F, Rossant J, Yamaguchi TP, Gertsenstein M, Wu XF, Breitman ML, et al. Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature. 1995;376(6535):62–6. doi: 10.1038/376062a0. [DOI] [PubMed] [Google Scholar]
- 47.Habeck H, Odenthal J, Walderich B, Maischein H, Schulte-Merker S, consortium Ts. Analysis of a zebrafish VEGF receptor mutant reveals specific disruption of angiogenesis. Curr Biol. 2002;12(16):1405–12. doi: 10.1016/s0960-9822(02)01044-8. [DOI] [PubMed] [Google Scholar]
- 48.Lawson ND, Weinstein BM. In vivo imaging of embryonic vascular development using transgenic zebrafish. Dev Biol. 2002;248(2):307–18. Epub 2002/08/09. doi: S0012160602907116 [pii]. [DOI] [PubMed] [Google Scholar]
- 49.Ulrich F, Ma LH, Baker RG, Torres-Vazquez J. Neurovascular development in the embryonic zebrafish hindbrain. Dev Biol. 2011;357(1):134–51. doi: 10.1016/j.ydbio.2011.06.037. [DOI] [PubMed] [Google Scholar]
- 50.Driever W, Solnica-Krezel L, Schier AF, Neuhauss SC, Malicki J, Stemple DL, et al. A genetic screen for mutations affecting embryogenesis in zebrafish. Development. 1996;123:37–46. [DOI] [PubMed] [Google Scholar]
- 51.Haffter P, Granato M, Brand M, Mullins MC, Hammerschmidt M, Kane DA, et al. The identification of genes with unique and essential functions in the development of the zebrafish, Danio rerio. Development. 1996;123:1–36. [DOI] [PubMed] [Google Scholar]
- 52.Fujita M, Cha YR, Pham VN, Sakurai A, Roman BL, Gutkind JS, et al. Assembly and patterning of the vascular network of the vertebrate hindbrain. Development. 2011;138(9):1705–15. Epub 2011/03/25. doi: 10.1242/dev.058776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rafferty SA, Quinn TA. A beginner’s guide to understanding and implementing the genetic modification of zebrafish. Prog Biophys Mol Biol. 2018;138:3–19. Epub 2018/07/19. doi: 10.1016/j.pbiomolbio.2018.07.005. [DOI] [PubMed] [Google Scholar]
- 54.Bussmann J, Wolfe SA, Siekmann AF. Arterial-venous network formation during brain vascularization involves hemodynamic regulation of chemokine signaling. Development. 2011;138(9):1717–26. Epub 20110323. doi: 10.1242/dev.059881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bill BR, Petzold AM, Clark KJ, Schimmenti LA, Ekker SC. A primer for morpholino use in zebrafish. Zebrafish. 2009;6(1):69–77. Epub 2009/04/21. doi: 10.1089/zeb.2008.0555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Westerfield M. The zebrafish book: A guide for the laboratory use of zebrafish (Danio rerio). 4th Edition ed. Eugene, OR: University of Oregon Press; 2000. [Google Scholar]
- 57.Meng X, Noyes MB, Zhu LJ, Lawson ND, Wolfe SA. Targeted gene inactivation in zebrafish using engineered zinc-finger nucleases. Nat Biotechnol. 2008;26(6):695–701. Epub 20080525. doi: 10.1038/nbt1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Moro E, Ozhan-Kizil G, Mongera A, Beis D, Wierzbicki C, Young RM, et al. In vivo Wnt signaling tracing through a transgenic biosensor fish reveals novel activity domains. Dev Biol. 2012;366(2):327–40. Epub 20120421. doi: 10.1016/j.ydbio.2012.03.023. [DOI] [PubMed] [Google Scholar]
- 59.Chi NC, Shaw RM, De Val S, Kang G, Jan LY, Black BL, et al. Foxn4 directly regulates tbx2b expression and atrioventricular canal formation. Genes Dev. 2008;22(6):734–9. doi: 10.1101/gad.1629408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Davis MW, Jorgensen EM. ApE, A Plasmid Editor: A Freely Available DNA Manipulation and Visualization Program. Frontiers in Bioinformatics. 2022;2. doi: 10.3389/fbinf.2022.818619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hao J, Ho JN, Lewis JA, Karim KA, Daniels RN, Gentry PR, et al. In vivo structure-activity relationship study of dorsomorphin analogues identifies selective VEGF and BMP inhibitors. ACS chemical biology. 2010;5(2):245–53. Epub 2009/12/22. doi: 10.1021/cb9002865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chimote G, Sreenivasan J, Pawar N, Subramanian J, Sivaramakrishnan H, Sharma S. Comparison of effects of anti-angiogenic agents in the zebrafish efficacy-toxicity model for translational anti-angiogenic drug discovery. Drug Des Devel Ther. 2014;8:1107–23. Epub 2014/08/19. doi: 10.2147/DDDT.S55621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shihan MH, Novo SG, Le Marchand SJ, Wang Y, Duncan MK. A simple method for quantitating confocal fluorescent images. Biochem Biophys Rep. 2021;25:100916. Epub 20210201. doi: 10.1016/j.bbrep.2021.100916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Strickland LA, Jubb AM, Hongo JA, Zhong F, Burwick J, Fu L, et al. Plasmalemmal vesicle-associated protein (PLVAP) is expressed by tumour endothelium and is upregulated by vascular endothelial growth factor-A (VEGF). J Pathol. 2005;206(4):466–75. doi: 10.1002/path.1805. [DOI] [PubMed] [Google Scholar]
- 65.Covassin LD, Siekmann AF, Kacergis MC, Laver E, Moore JC, Villefranc JA, et al. A genetic screen for vascular mutants in zebrafish reveals dynamic roles for Vegf/Plcg1 signaling during artery development. Dev Biol. 2009;329(2):212–26. Epub 2009/03/06. doi: 10.1016/j.ydbio.2009.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Covassin LD, Villefranc JA, Kacergis MC, Weinstein BM, Lawson ND. Distinct genetic interactions between multiple Vegf receptors are required for development of different blood vessel types in zebrafish. Proc Natl Acad Sci U S A. 2006;103(17):6554–9. Epub 2006/04/14. doi: 10.1073/pnas.0506886103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Roskoski R VEGF receptor protein-tyrosine kinases: structure and regulation. Biochem Biophys Res Commun. 2008;375(3):287–91. Epub 20080803. doi: 10.1016/j.bbrc.2008.07.121. [DOI] [PubMed] [Google Scholar]
- 68.Almagro Armenteros JJ, Tsirigos KD, Sønderby CK, Petersen TN, Winther O, Brunak S, et al. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat Biotechnol. 2019;37(4):420–3. Epub 20190218. doi: 10.1038/s41587-019-0036-z. [DOI] [PubMed] [Google Scholar]
- 69.Bahary N, Goishi K, Stuckenholz C, Weber G, Leblanc J, Schafer CA, et al. Duplicate VegfA genes and orthologues of the KDR receptor tyrosine kinase family mediate vascular development in the zebrafish. Blood. 2007;110(10):3627–36. Epub 20070814. doi: 10.1182/blood-2006-04-016378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Vogrin AJ, Bower NI, Gunzburg MJ, Roufail S, Okuda KS, Paterson S, et al. Evolutionary Differences in the Vegf/Vegfr Code Reveal Organotypic Roles for the Endothelial Cell Receptor Kdr in Developmental Lymphangiogenesis. Cell Rep. 2019;28(8):2023–36.e4. doi: 10.1016/j.celrep.2019.07.055. [DOI] [PubMed] [Google Scholar]
- 71.Li H, Ha HL, Ding X, Bae C, Gazy N, Hao J, et al. DMH4, a VEGFR2 inhibitor, effectively suppresses growth and invasion of lung cancer cells. J Appl Biomed. 2018;16:46–50. Epub 2017. doi: 10.1016/j.jab.2017.10.006. [DOI] [Google Scholar]
- 72.Costa G, Harrington KI, Lovegrove HE, Page DJ, Chakravartula S, Bentley K, et al. Asymmetric division coordinates collective cell migration in angiogenesis. Nat Cell Biol. 2016;18(12):1292–301. Epub 20161121. doi: 10.1038/ncb3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jeong JY, Kwon HB, Ahn JC, Kang D, Kwon SH, Park JA, et al. Functional and developmental analysis of the blood-brain barrier in zebrafish. Brain Res Bull. 2008;75(5):619–28. Epub 2008/03/22. doi: S0361–9230(07)00327–9 [pii] 10.1016/j.brainresbull.2007.10.043. [DOI] [PubMed] [Google Scholar]
- 74.Morita K, Sasaki H, Furuse M, Tsukita S. Endothelial claudin: claudin-5/TMVCF constitutes tight junction strands in endothelial cells. J Cell Biol. 1999;147(1):185–94. doi: 10.1083/jcb.147.1.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nitta T, Hata M, Gotoh S, Seo Y, Sasaki H, Hashimoto N, et al. Size-selective loosening of the blood-brain barrier in claudin-5-deficient mice. J Cell Biol. 2003;161(3):653–60. Epub 2003/05/14. doi: 10.1083/jcb.200302070 jcb.200302070 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Xie J, Farage E, Sugimoto M, Anand-Apte B. A novel transgenic zebrafish model for blood-brain and blood-retinal barrier development. BMC Dev Biol. 2010;10:76. Epub 2010/07/27. doi: 1471–213X-10–76 [pii] 10.1186/1471-213X-10-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zheng PP, Romme E, van der Spek PJ, Dirven CM, Willemsen R, Kros JM. Glut1/SLC2A1 is crucial for the development of the blood-brain barrier in vivo. Ann Neurol. 2010;68(6):835–44. doi: 10.1002/ana.22318. [DOI] [PubMed] [Google Scholar]
- 78.Schumacher U, Mollgård K. The multidrug-resistance P-glycoprotein (Pgp, MDR1) is an early marker of blood-brain barrier development in the microvessels of the developing human brain. Histochem Cell Biol. 1997;108(2):179–82. doi: 10.1007/s004180050159. [DOI] [PubMed] [Google Scholar]
- 79.Umans RA, Taylor MR. Zebrafish as a model to study drug transporters at the blood-brain barrier. Clin Pharmacol Ther. 2012;92(5):567–70. Epub 2012/10/10. doi: 10.1038/clpt.2012.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kawasaki T, Kitsukawa T, Bekku Y, Matsuda Y, Sanbo M, Yagi T, et al. A requirement for neuropilin-1 in embryonic vessel formation. Development. 1999;126(21):4895–902. [DOI] [PubMed] [Google Scholar]
- 81.Gu C, Rodriguez ER, Reimert DV, Shu T, Fritzsch B, Richards LJ, et al. Neuropilin-1 conveys semaphorin and VEGF signaling during neural and cardiovascular development. Dev Cell. 2003;5(1):45–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Meng N, Li Y, Zhang H, Sun XF. RECK, a novel matrix metalloproteinase regulator. Histol Histopathol. 2008;23(8):1003–10. doi: 10.14670/HH-23.1003. [DOI] [PubMed] [Google Scholar]
- 83.Noda M, Oh J, Takahashi R, Kondo S, Kitayama H, Takahashi C. RECK: a novel suppressor of malignancy linking oncogenic signaling to extracellular matrix remodeling. Cancer Metastasis Rev. 2003;22(2–3):167–75. doi: 10.1023/a:1023043315031. [DOI] [PubMed] [Google Scholar]
- 84.Collu GM, Hidalgo-Sastre A, Brennan K. Wnt-Notch signalling crosstalk in development and disease. Cell Mol Life Sci. 2014;71(18):3553–67. Epub 20140619. doi: 10.1007/s00018-014-1644-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Blanco R, Gerhardt H. VEGF and Notch in tip and stalk cell selection. Cold Spring Harb Perspect Med. 2013;3(1):a006569. Epub 20130101. doi: 10.1101/cshperspect.a006569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hellström M, Phng LK, Hofmann JJ, Wallgard E, Coultas L, Lindblom P, et al. Dll4 signalling through Notch1 regulates formation of tip cells during angiogenesis. Nature. 2007;445(7129):776–80. Epub 20070128. doi: 10.1038/nature05571. [DOI] [PubMed] [Google Scholar]
- 87.Leslie JD, Ariza-McNaughton L, Bermange AL, McAdow R, Johnson SL, Lewis J. Endothelial signalling by the Notch ligand Delta-like 4 restricts angiogenesis. Development. 2007;134(5):839–44. Epub 20070124. doi: 10.1242/dev.003244. [DOI] [PubMed] [Google Scholar]
- 88.Siekmann AF, Lawson ND. Notch signalling limits angiogenic cell behaviour in developing zebrafish arteries. Nature. 2007;445(7129):781–4. Epub 20070128. doi: 10.1038/nature05577. [DOI] [PubMed] [Google Scholar]
- 89.Corada M, Nyqvist D, Orsenigo F, Caprini A, Giampietro C, Taketo MM, et al. The Wnt/beta-catenin pathway modulates vascular remodeling and specification by upregulating Dll4/Notch signaling. Dev Cell. 2010;18(6):938–49. doi: 10.1016/j.devcel.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hamilton BJ, Tse D, Stan RV. Phorbol esters induce PLVAP expression via VEGF and additional secreted molecules in MEK1-dependent and p38, JNK and PI3K/Akt-independent manner. J Cell Mol Med. 2019;23(2):920–33. Epub 20181105. doi: 10.1111/jcmm.13993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wang Y, Sabbagh MF, Gu X, Rattner A, Williams J, Nathans J. Beta-catenin signaling regulates barrier-specific gene expression in circumventricular organ and ocular vasculatures. Elife. 2019;8. Epub 20190401. doi: 10.7554/eLife.43257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Benz F, Wichitnaowarat V, Lehmann M, Germano RF, Mihova D, Macas J, et al. Low wnt/β-catenin signaling determines leaky vessels in the subfornical organ and affects water homeostasis in mice. Elife. 2019;8. Epub 20190401. doi: 10.7554/eLife.43818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Phoenix TN, Patmore DM, Boop S, Boulos N, Jacus MO, Patel YT, et al. Medulloblastoma Genotype Dictates Blood Brain Barrier Phenotype. Cancer Cell. 2016;29(4):508–22. Epub 20160331. doi: 10.1016/j.ccell.2016.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bosma EK, van Noorden CJF, Schlingemann RO, Klaassen I. The role of plasmalemma vesicle-associated protein in pathological breakdown of blood-brain and blood-retinal barriers: potential novel therapeutic target for cerebral edema and diabetic macular edema. Fluids Barriers CNS. 2018;15(1):24. Epub 20180920. doi: 10.1186/s12987-018-0109-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Parab S, Quick RE, Matsuoka RL. Endothelial cell-type-specific molecular requirements for angiogenesis drive fenestrated vessel development in the brain. Elife. 2021;10. Epub 20210118. doi: 10.7554/eLife.64295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wang Y, Cho SG, Wu X, Siwko S, Liu M. G-protein coupled receptor 124 (GPR124) in endothelial cells regulates vascular endothelial growth factor (VEGF)-induced tumor angiogenesis. Curr Mol Med. 2014;14(4):543–54. doi: 10.2174/1566524014666140414205943. [DOI] [PubMed] [Google Scholar]
- 97.Martowicz A, Trusohamn M, Jensen N, Wisniewska-Kruk J, Corada M, Ning FC, et al. Endothelial β-Catenin Signaling Supports Postnatal Brain and Retinal Angiogenesis by Promoting Sprouting, Tip Cell Formation, and VEGFR (Vascular Endothelial Growth Factor Receptor) 2 Expression. Arterioscler Thromb Vasc Biol. 2019;39(11):2273–88. Epub 20190919. doi: 10.1161/ATVBAHA.119.312749. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.