

Abstract
Natural products have long served as rich sources of drugs possessing a wide range of pharmacological activities. The discovery and development of natural product drug candidates is often hampered by the inability to efficiently scale and produce a molecule of interest, due to inherent qualities of the native producer. Heterologous biosynthesis in an engineering and process-friendly host emerged as an option to produce complex natural products. Escherichia coli has previously been utilized to produce complex precursors to two popular natural product drugs, erythromycin and paclitaxel. These two molecules represent two of the largest classes of natural products, polyketides and isoprenoids, respectively. In this study, we have developed a platform E. coli strain capable of simultaneous production of both product precursors at titers greater than 15 mg l−1. The utilization of a two-phase batch bioreactor allowed for very strong in situ separation (having a partitioning coefficient of greater than 5,000), which would facilitate downstream purification processes. The system developed here could also be used in metagenomic studies to screen environmental DNA for natural product discovery and preliminary production experiments.
Keywords: Heterologous host, Escherichia coli, Polyketide, Isoprenoid, Two-phase bioreactor
Introduction
Natural products are a class of chemical compounds that originate from the secondary (non-growth-associated) metabolic processes of living organisms. They possess a range of useful pharmacological activities and of all small-molecule new chemical entities (NCEs) between 1981 and 2006, 34% were natural products or semi-synthetic derivatives of such molecules [44]. In fact, of the 109 antibacterial NCEs and the 83 anticancer NCEs, 74 and 45, respectively, were derived from natural products [44]. Therefore, even with the “blockbuster model” adopted by current pharmaceutical companies, natural products are still providing successful drugs and drug leads [35].
Unfortunately, most natural products have evolved independently of their useful therapeutic applications, and consequently, their native hosts tend to produce them in relatively small quantities [30]. As a result, production of natural products from their native hosts is oftentimes an expensive and slow process. To address this situation, researchers have explored the use of heterologous host organisms to produce such compounds, using the same recombinant strategies that spurred biopharmaceutical protein production. Through metabolic and genetic engineering of heterologous hosts, the complex natural product biosynthetic machinery can be redesigned with the goal of optimizing final product formation. Furthermore, bioprocess engineering options with heterologous hosts can also lead to higher production titers or productivities that may be unachievable with native producers. Typical heterologous hosts for secondary metabolite production range from various actinomycetes (particularly, Streptomyces coelicolor, S. lividans, and S. albus) [5], Escherichia coli [9], and Saccharomyces cerevisiae [25, 56]. The ideal heterologous host should have high specific growth rates, minimal nutrient requirements, established genetic engineering protocols, a fully sequenced and well-annotated genome, and easily scalable bioprocesses.
Polyketides and isoprenoids represent two of the largest classes of natural products. While derived from different molecular backbones, polyketides and isoprenoids have similar logic towards their biosyntheses. The most studied polyketide synthases (PKSs) are modular in nature and catalyze successive rounds of decarboxylative Claisen condensation reactions between an acyl thioester and thioesterified malonate derivatives [22]. Though many of the acyl-CoA starter units for priming PKSs (such as acetyl-CoA) are fairly common among prokaryotic and eukaryotic systems, many of the thioesterified malonate derivatives used as extender units are less common (such as (2S)-methylmalonyl-CoA, (2S)-ethylmalonyl-CoA, and chloroethylmalonyl-CoA) [10, 21, 42]. Once a polyketide has reached the end of the modular biosynthetic process, a terminal thioesterase domain is often responsible for polyketide cyclization and release from the PKS. Typical post-PKS tailoring includes glycosylation [7, 58], acylation, oxidation [63], and halogenation [43]. As an example, the polyketide 6-deoxyerythronolide B (6-dEB) is the 14-membered aglycone macrocyclic core of the wide-spectrum antibiotic erythromycin and is constructed by the deoxyerythronolide B synthase (DEBS) from one propionyl-CoA starter and six (2S)-methylmalonyl-CoA extender molecules (Fig. 1a). Natively produced from the soil-dwelling Saccharopolyspora erythraea [14, 18], the erythromycin system has been the subject of many seminal studies focused on modular polyketide biosynthesis [23, 28, 41, 51, 52, 61]. Although S. erythraea has been the host of choice for erythromycin production [60], and has spawned commercially successful semi-synthetic macrolides such as azithromycin, its 5–10 g l−1 production titer falls roughly an order of magnitude short of other popular antibiotic fermentation processes [1]. Recent efforts have been dedicated towards improving the genetic technologies [65], understanding the regulation of erythromycin production [13], and improving the erythromycin production titer [55] and purity [11] of S. erythraea-based erythromycin fermentation. However, to truly harness the combinatorial potential of the erythromycin modular biosynthetic process for the purpose of generating next-generation derivatives [40, 41, 46, 66], a heterologous production host will likely be necessary.
Fig. 1.
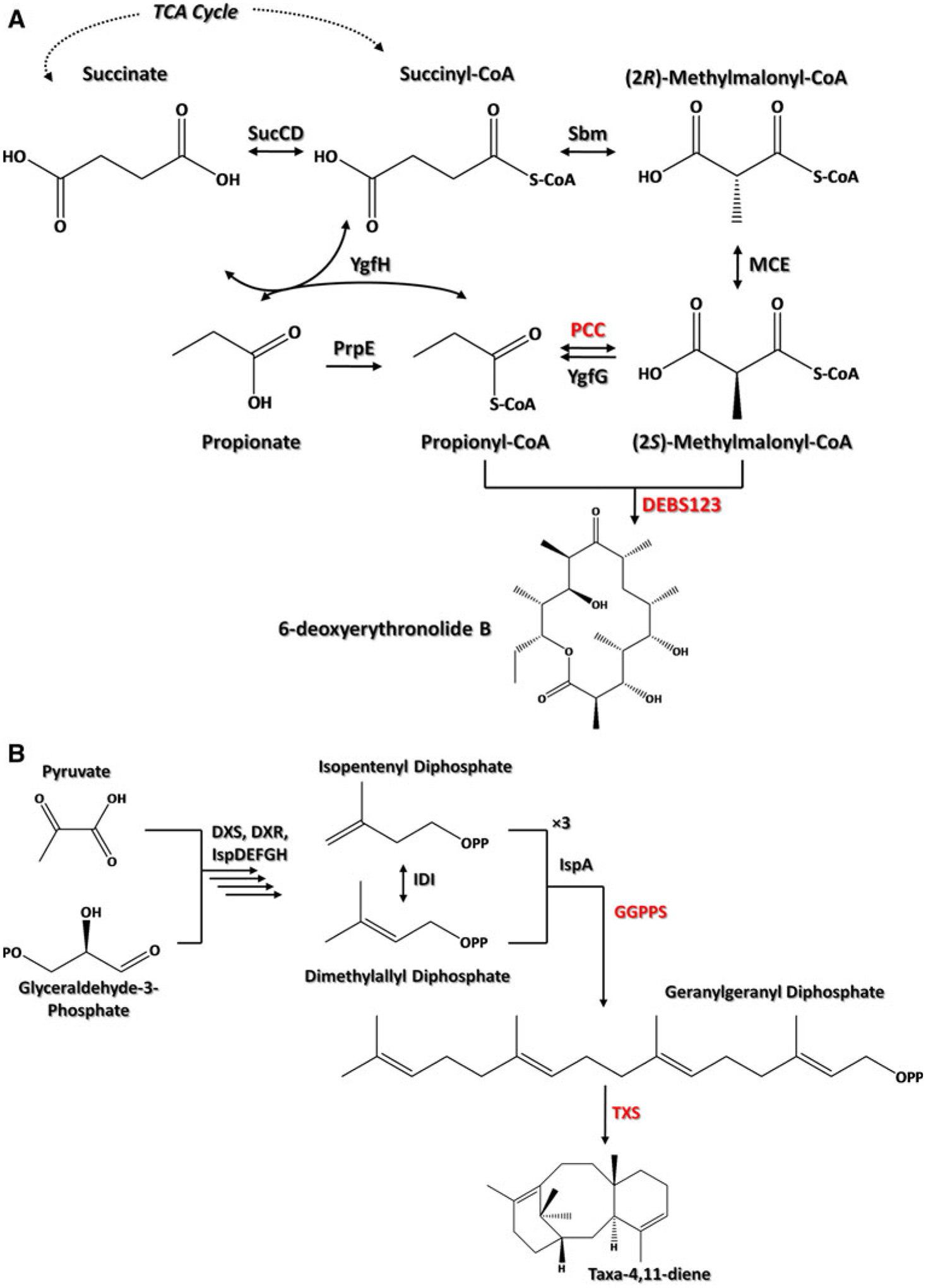
The metabolic pathways involved in a 6-dEB and b taxadiene biosynthesis. SucCD succinyl-CoA synthetase, Sbm “Sleeping beauty mutase”, MCE methylmalonyl-CoA epimerase, PCC propionyl-CoA carboxylase, YgfG methylmalonyl-CoA decarboxylase, DEBS123 deoxyerythronlide B synthase, PrpE propionyl-CoA synthetase, YgfH propionyl-CoA:succinate CoA transferase, DXS 1-deoxy-D-xylulose-5-phosphate synthase, DXR 1-deoxy-D-xylulose-5-phosphate reductoisomerase, IspD 4-diphosphocytidyl-2C-methyl-D-erythritol synthase, IspE 4-diphosphocytidyl-2-C-methylerythritol kinase, IspF 2C-methyl-D-erythritol 2,4-cyclodiphosphate synthase, IspG 1-hydroxy-2-methyl-2-(E)-butenyl 4-diphosphate synthase, IspH 1-hydroxy-2-methyl-2-(E)-butenyl 4-diphosphate reductase, IPP isopentenyl diphosphate isomerase, IspA geranyl diphosphate synthase/farnesyl diphosphate synthase, GGPPS geranylgeranyl diphosphate synthase, TXS taxadiene synthase
Isoprenoids are generated through a similar upstream and downstream pathway logic. Isoprenoids can be acyclic, monocyclic, or polycyclic molecules derived from successive head-to-tail condensation reactions from universal dimethylallyl diphosphate (DMAPP) and isopentenyl diphosphate (IPP) monomers resulting in C10, C15, C20, or higher carbon chain lengths. DMAPP and IPP derive from either the mevalonate [38, 53] or the 1-deoxy-D-xylulose 5-phosphate (DXP) pathways [19]. The mevalonate pathway exists in eukaryotes (mammals, plants, and fungi) while the DXP pathway exists in bacteria and plant plastids [2]. The mevalonate pathway begins with acetyl-CoA, while the DXP pathway begins with the condensation of pyruvate and glyceraldehyde 3-phosphate (Fig. 1b). Cyclization of C10, C15, and C20 chains through terpene synthases generate monoterpenes, sesquiterpenes, and diterpenes, respectively. Post-cyclization C–H functionalization leads to final products in these pathways.
The diterpenoid natural product Taxol® (having the generic name of paclitaxel) possesses impressive anticancer properties and has shown efficacy against carcinomas of the ovary, breast, lung, head and neck, bladder and cervix, melanomas, and AIDS-related Kaposi’s sarcoma [59]. Extracted from the bark of Taxus brevifolia (the Pacific Yew tree), early stage production of paclitaxel resulted in extremely low yields; the sacrifice of a 100-year-old tree generated approximately 3 kg of bark, yielding approximately 300 mg of purified paclitaxel or the equivalent of approximately a single dose [26]. In 1988, a semi-synthetic method for producing paclitaxel from a common precursor (10-deacetylbaccatin III) was developed. The semi-synthetic route offered two advantages. First, relatively higher quantities of 10-deacetylbaccatin III could be isolated from the needles of other more prevalent Taxus species (specifically Taxus baccata L. (the common Yew)) [17]. Second, isolation from the renewable Yew needle presented an environmentally friendly alternative to the destructive production processes dependent upon Yew bark harvest. A fully synthetic route to paclitaxel was accomplished in 1994; however, the compound’s complex molecular architecture containing 11 chiral centers and a notably rare oxetane ring complicated efforts to establish an efficient and economic synthetic production process [47]. Paclitaxel has been extracted from T. brevifolia bark at a roughly 1,000 μg g biomass−1 [26] and produced in Taxus canadensis suspension cell culture at roughly 120 μg g biomass−1 [29]. Meanwhile, heterologous production of the first committed step to paclitaxel biosynthesis, taxa-4(5),11(12)-diene (henceforth referred to as taxadiene) has only reached 0.6 μg g biomass−1 for Arabidopsis thaliana [6], 471 μg g biomass−1 for transgenic tomato fruit [34], and 402 μg g biomass−1 for S. cerevisiae [20]. However, like the case for erythromycin, heterologous production of paclitaxel is viewed with great interest due to the potential to rationally control or alter the biosynthetic process towards improved bioactivity.
Heterologous production of erythromycin and paclitaxel intermediates has now been established using E. coli. Specifically, in separate applications, E. coli has been designed to support the production of 6-dEB and taxadiene. In this report, we explore the potential of producing both compounds simultaneously. Doing so presents a new form of consolidated bioprocessing in which two metabolically distinct natural products are produced through a heterologous platform, offering the potential to reduce the space, time, and cost associated with separate processing. E. coli was first genetically modified based upon the known requirements for polyketide and isoprenoid biosynthesis. A co-production process was then assessed, aided greatly by the ability to readily partition the two nascently formed products. The implications of this study range from future consolidated bioprocesses for producing additional therapeutic natural compounds to an E. coli host now capable of supporting continual drug discovery and development opportunities.
Materials and methods
Background strains and plasmids
E. coli, strain BAP1 has been developed previously for the heterologous production of polyketide and nonribosomal peptide natural products [51]. The Bacillus subtilis surf-actin phosphopantetheinyl transferase gene (sfp) [54] was inserted into the prpRBCD location of the BL21(DE3) chromosome, under the control of an inducible T7 promoter [51]. The sfp gene is required to post-translationally activate PKSs by transferring the 4′-phosphopantetheinyl moiety from coenzyme A to a conserved, reactive serine residue within each acyl carrier protein (ACP) domain of polyketide synthases (or the peptidyl carrier protein (PCP) domains of nonribosomal peptide synthetases) [54]. E. coli’s native holo-acyl carrier protein synthase (ACPS, required for activation of fatty acid synthases) does not recognize the apo-forms of many ACP and PCP domains [54]. Therefore, without sfp, 6-dEB cannot be produced through E. coli. During this genetic insertion, a T7 promoter was also inserted before the native prpE gene to increase metabolic flux towards propionyl-CoA, a direct precursor of 6-dEB. This strain was termed BAP1 [51]. Strain JW1 was derived from BAP1 [67], and contains a Flippase Recognition Target (FRT)-[12] flanked cat gene (coding for chloramphenicol acyl transferase, derived from pKD3 [16]) in the chromosome before the sfp gene (seen pictorially in Fig. 2), replacing the native prpR gene. Strain YW23 was derived from BL21(DE3), and contains an additional copy of the native dxs, idi, ispD, and ispF genes under the control of a single T7 promoter (also seen pictorially in Fig. 2) in the araA location of the chromosome (Table 1).
Fig. 2.
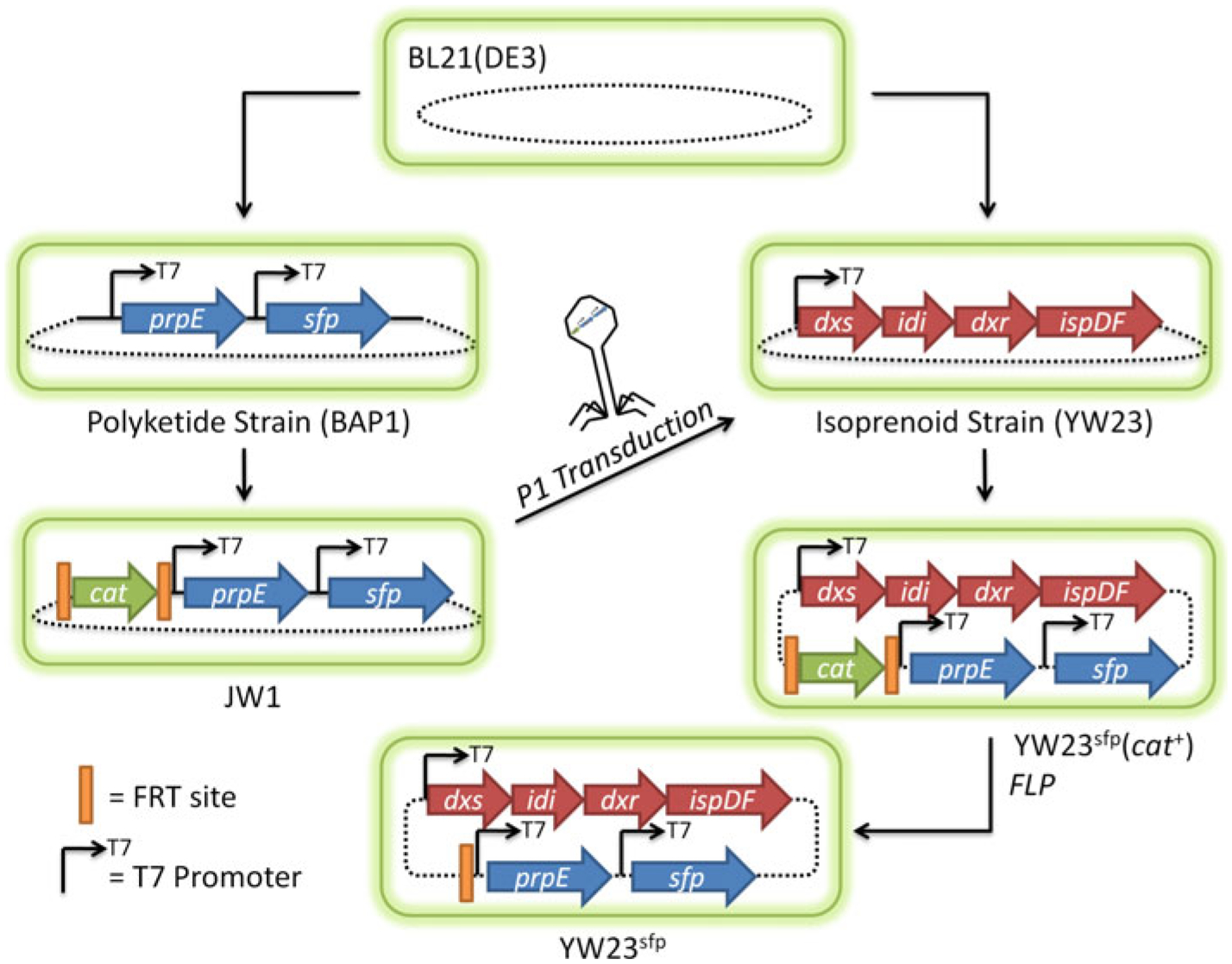
A pictorial representation of the strain construction workflow relevant to this study. The chromosome and chromosomal insertions are presented for clarification purposes and do not accurately reflect scale, exact locations, and distance between integrations
Table 1.
Plasmids and strains used in this study
| Name | Description | Source |
|---|---|---|
| pBP130 | bla; T7prom-eryA2-eryA3-T7term | [51] |
| pBP144 | kan; T7prom-pccB-accA1-T7prom-eryA1-T7term | [51] |
| pACYCDuet-TXS-GGPPS | cat; T7prom-txssyn-crtEsyn-T7term | (in preparation) |
| pKD3 | bla, cat; template for chloramphenicol cassette amplification | [16] |
| pKD46 | bla; encodes γ, β, and exo under the control of a pBAD promoter | [16] |
The genes required for the production of 6-dEB from propionate were previously inserted into plasmids pBP130 and pBP144 [51]. Briefly, pBP130 (approximately 26 kb and derived from pET21c (Novagen)) contains the eryA2 and eryA3 genes (coding for the DEBS2 and DEBS3 enzymes) under a single T7 promoter. Plasmid pBP144 (approximately 19 kb and derived from pET28 (Novagen)) contains eryA1 (coding for the DEBS1 enzyme) under a T7 promoter and genes coding for the two subunits of the S. coelicolor propionyl-CoA carboxylase enzyme (accA1 and pccB) [57] under another T7 promoter. All three eryA genes are from the native erythromycin producer, S. erythraea [14, 18].
Two heterologous enzymes are needed to produce taxadiene through E. coli: (1) a geranylgeranyl-diphosphate synthase (to catalyze: farnesyl-diphosphate + isopentenyl-diphosphate → geranylgeranyl-diphosphate + diphosphate) and (2) a cyclizing taxadiene synthase (to catalyze: geranylgeranyl-diphosphate → taxa-4,11-diene + diphosphate). The original template sequences were crtE (for the geranylgeranyl-diphosphate synthase) from Erwinia herbicola (also known as Enterobacter agglomerans) [15] and txs (for the taxadiene synthase) from Taxus baccata [6]. The sequences were codon optimized for E. coli, constructed synthetically [31], and cloned into the MCS1 of pACYCDuet-1 (Novagen) between NcoI and SalI sites (under the control of a single T7 promoter). This plasmid was named pACYCDuet-TXS-GGPPS (approximately 7.5 kb).
Strain construction
Figure 2 shows a pictorial representation of the workflow of strain construction relevant to this study. YW23sfp was generated through P1 bacteriophage transduction [39] in which JW1 was used as the donor strain and YW23 was the recipient. The infected YW23 was plated on LB agar containing 20 mg l−1 chloramphenicol to select for integrants containing the expected genomic insertion, incubated overnight at 37°C, and successful integrants were verified by PCR, as described previously [67]. This new strain was stored as a glycerol stock, prepared electrocompetent, and transformed with pCP20 [12], which contains the gene coding for the Saccharomyces cerevisiae Flp recombinase [12], used to excise the cat gene between the integrated FRT sites. The ultimate result was a chloramphenicol sensitive strain, YW23sfp. This strain was stored as a glycerol stock, prepared electrocompetent, and transformed with (1) pBP130 and pBP144, (2) pACYCDuet-TXS-GGPPS, or (3) all three plasmids.
Shake-flask production cultures
Glycerol stocks were used to inoculate 2-ml Luria–Bertani (LB) medium cultures with appropriate antibiotics for overnight incubation at 37°C and 250 rpm. Production cultures were conducted in 125-ml non-baffled Erlenmeyer flasks in a rich medium containing 5 g l−1 yeast extract, 10 g l−1 tryptone, 10 g l−1 sodium chloride, 15 g l−1 glycerol, 3 ml l−1 50% (v v−1) Antifoam B, 100 mM HEPES, and pH 7.60. Production cultures (15 ml) were inoculated with the precultures to an OD600nm = 0.1 and supplemented with 20 mM appropriate precursors, appropriate antibiotics, and 100 μM isopropyl β-D-1-thioga-lactopyranoside (IPTG). When pBP130 and pBP144 were present in the cell (when 6-dEB was being generated), 20 mM propionate was added to the medium. When n-dodecane was used as the organic phase, 12 ml of culture medium was supplemented with 3 ml n-dodecane (for 20% v v−1). Cultures were incubated for 120 h at 22°C and 250 rpm. At the end of the culture period, cell-density was measured spectrophotometrically at 600 nm, and 1-ml aliquots were stored at −20°C for subsequent analyses. Cell-density in gram dry cell weight per liter (gDCW l−1) was calculated using an experimentally determined correlation of 1 OD600nm = 0.52 gDCW l−1 (data not shown). When needed, antibiotics were supplemented at concentrations of 100 mg l−1 for carbenicillin, 50 mg l−1 for kanamycin, and 34 mg l−1 for chloramphenicol.
Two-phase batch bioprocess
Strain YW23sfp(pBP130/pBP144/pACYCDuet-TXS-GGPPS) was first inoculated in 2 ml of selective LB medium and grown for approximately 8 h at 37°C and 250 rpm. This culture was then used to inoculate a larger culture containing 100 ml of LB medium with appropriate antibiotics. After an overnight incubation at 37°C and 250 rpm, 1.2 l of medium (as described above) was inoculated with this starter culture in a 3-l bioreactor (New Brunswick Scientific BioFlo 110) to an OD600nm = 0.1. The system was then charged with 300 ml of n-dodecane, bringing the final volume to 1.5 l. Air was supplied at 0.5 vessel volumes per minute (VVM), temperature was controlled with a water bath at 22°C, and pH was controlled at 7.60 with the addition of 5 M NH4OH. The bioprocess was run for 5 days at 22°C and 500 rpm. The medium also contained 100 mg l−1 carbenicillin, 50 mg l−1 kanamycin, and 34 mg l−1 chloramphenicol to maintain plasmid selection and 100 μM IPTG to induce gene-expression. Sample aliquots (1 ml) were taken from the bioreactor every 6–12 h over the course of the culture period and stored at −20°C until subsequent analysis.
6-dEB quantification by RP-HPLC-ELSD
The HPLC method for 6-dEB separation and quantification has been described previously [64]. Briefly, quantification of 6-dEB was carried out on an Agilent 1100 series HPLC coupled with an Alltech 800 series evaporative light-scattering detector (ELSD). The guard column used was an Inertsil ODS3 C18 5 μm, 4.6 × 10 mm while the analytical column used was an Inertsil ODS3 C18 5 μm, 4.6 × 150 mm (GL Sciences). Column temperature was maintained at 25°C while the autosampler was maintained at 4°C. Ultra-high-purity-grade nitrogen gas (AirGas East) was used as the mobile phase for the ELSD at a pressure of3.00 ± 0.05 bar, while the ELSD drift tube temperature was maintained at 55°C and the gain setting was set at 16.
Culture samples were first centrifuged for 10 min at 10,000 × g to remove insolubles. A 20.0-μl supernatant injection volume was then used with a solvent system of 100% distilled, deionized water (ddH2O) from 0–2.0 min as the mobile phase passed through the guard column and to a waste collection. A six-port switching valve then directed the mobile phase to the analytical column as a gradient to 100% HPLC-grade acetonitrile (Sigma–Aldrich) began from 2.0–5.0 min followed by 100% acetonitrile from 5.0–8.0 min, a quick gradient back to 100% ddH2O from 8.0–8.1 min, and finally 100% ddH2O maintained from 8.1–9.5 min. The mobile-phase flow-rate was 1 ml min−1. Under these conditions, 6-dEB eluted at7.92 ± 0.05 min. Quantification was carried out against a five-point calibration curve of purified 6-dEB (kindly provided by Kosan Biosciences).
Taxadiene quantification by GC-MS
For taxadiene quantification from the aqueous phase, samples were first centrifuged for 10 min at 10,000 × g to remove insolubles. Aqueous supernatant (750 μl) was supplemented with (−)-trans-caryophyllene (TC) at a final concentration of 1 μg l−1 to serve as an internal standard for quantification. The samples were then extracted with an equal volume of hexane, followed by 20 s of vortexing and centrifugation for 10 min at 10,000 × g. The hexane layer (150 μl) was removed and stored in glass vials at −20°C until analysis with gas chromatography-mass spectroscopy (GC–MS) could be conducted. For quantification of taxadiene in the organic phase, an aliquot of the n-dodecane was diluted 100-fold in hexane containing 1 μg l−1 TC. Samples were placed in glass vials stored at −20°C until GC–MS analysis was conducted.
Samples were analyzed on a Shimadzu QP5050A GC–MS using splitless injection. Gas chromatography was run on a non-polar Rxi®-XLB column (30 m × 0.25 mm ID, 0.25 μm). The inlet pressure for the column was set at 120 kPa and column flow velocity was 1.6 ml min−1. The flowrate of the ultra-high-purity helium carrier gas was 20 ml min−1. The temperature of the column was initially set and maintained at 100°C for 2 min and was then increased to 235°C at a rate of 15.0°C min−1. The column was then maintained at this temperature for 1 min. Mass spectrometry was performed in single ion monitoring (SIM) mode scanning for mass to charge ratios of 107 m z−1, 122 m z−1, and 272 m z−1, corresponding to principle daughter ions and parent ion of taxadiene, respectively [32]. Under these conditions, TC and taxadiene eluted at approximately 6.8 and 11.4 min, respectively. Quantification of taxadiene was accomplished based on a calibration curve of taxadiene (kindly provided by Dr. Ajikumar Parayil and Prof. Gregory Stephanopoulos) concentration versus the peak area ratio of TC to taxadiene.
Metabolite quantification by CEX-HPLC-RID
The clarified aqueous phase was also analyzed for quantities of precursor and byproduct metabolites. This analysis was conducted with HPLC (Agilent 1100 Series) coupled with a refractive index detector (RID). Clarified culture supernatant (20 μl) was applied to a Bio-Rad Aminex® HPX-87H Ion Exchange (300 mm × 7.8 mm, 9 μm) column, preceded by a 30-mm guard column of the same resin. The isocratic analysis used a 9.5 mM H2SO4 solvent held at a flow rate of 0.3 ml min−1. Under these conditions, the elution order of the analyzed compounds was pyruvate (16.7 min), glycerol (25.1 min), acetate (29.1 min), and propionate (36.6 min). Quantification of these compounds was conducted against a five-point calibration curve of purchased standards (Sigma–Aldrich).
Results
Production of 6-dEB and taxadiene separately and together
The production of 6-dEB and taxadiene, both separately and together, was first analyzed in the newly constructed YW23sfp. Both 6-dEB and taxadiene were quantified in the culture medium after 120 h of shake-flask cultivation at 22°C and 250 rpm. As can be seen in Fig. 3a, YW23sfp(pBP130/pBP144) produced 48.8 ± 7.1 mg l−1 6-dEB while the titer decreased to 17.9 ± 7.3 mg l−1 (p < 0.001) with the addition of the pACYCDuet-TXS-GGPPS plasmid. The taxadiene titers were approximately an order of magnitude lower than that of 6-dEB (Fig. 3b); however, the titers are similar to those previously reported in the parent to YW23sfp (in preparation). The YW23sfp(pACYCDuet-TXS-GGPPS) strain produced taxadiene at 1.53 ± 0.27 mg l−1, and there was no statistically significant difference when pBP130 and pBP144 were added to the system (p = 0.772).
Fig. 3.
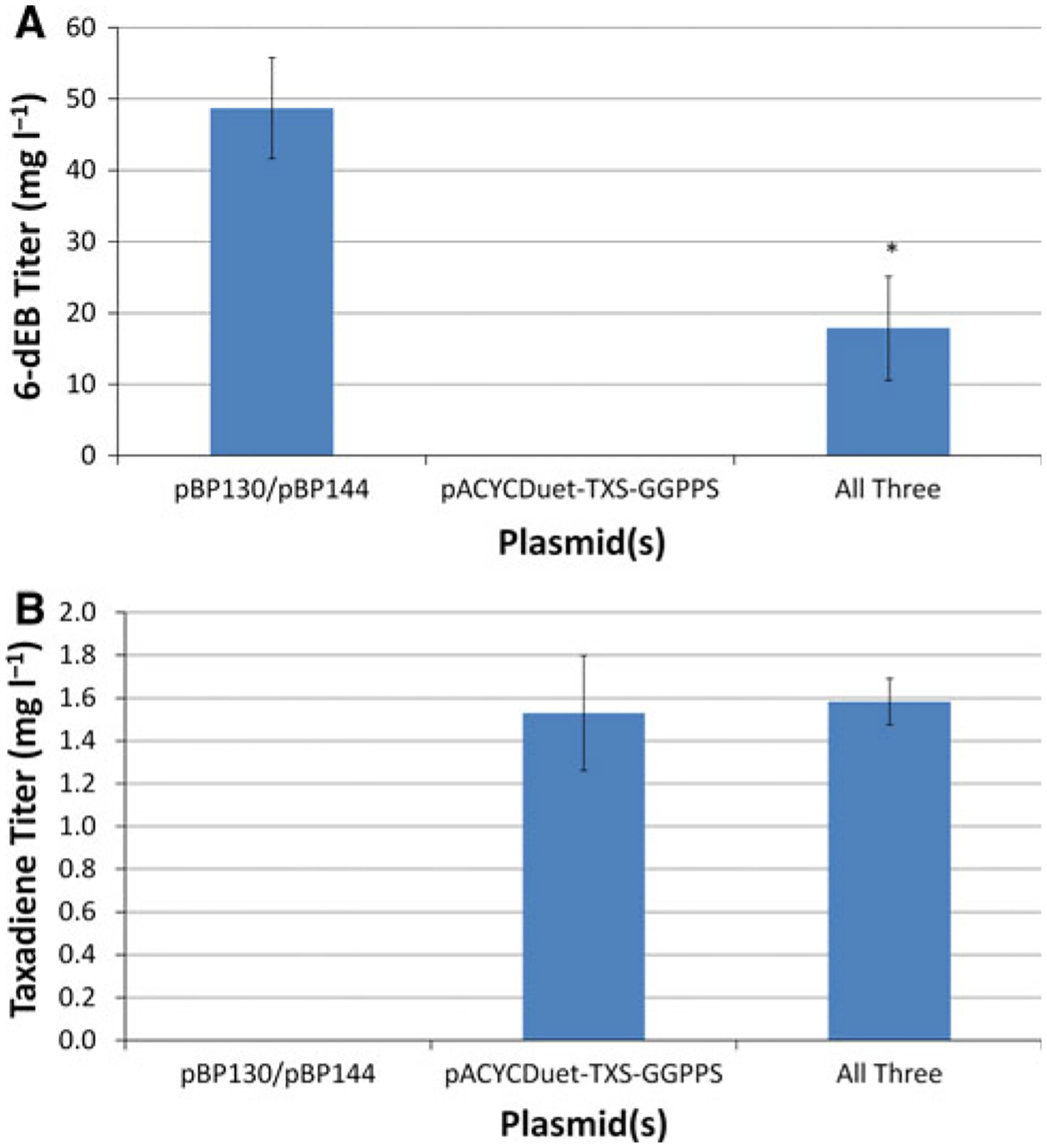
Production of a 6-dEB and b taxadiene in YW23sfp containing either the plasmids required for 6-dEB biosynthesis (pBP130/pBP144), the plasmid required for taxadiene biosynthesis (pACYCD-uet-TXS-GGPPS), or all three plasmids together. Propionate (20 mM) was supplemented in the medium for experiments in columns one and three. Error bars represented ± one standard deviation from three independent replicates. *Indicates statistically significant results at p < 0.05
Co-production of 6-dEB and taxadiene in a two-phase system
It was previously reported that taxadiene has a strong preference for partitioning into organic environments in situ, improving titer dramatically [3]. In these studies, n-dodecane was chosen as an organic phase based on its successful use in sequestering E. coli-produced amorpha-4,11-diene (a sesquiterpene) [45]. A concentration of 20% n-dodecane (v v−1) was previously determined to be the optimal concentration for improving specific titer (data not shown). As a result, the two-phase systems reported here contain an aqueous medium for cell growth and 20% n-dodecane (v v−1) to capture secreted taxadiene.
Figure 4a presents 6-dEB titer from YW23sfp(pBP130/pBP144/pACYCDuet-TXS-GGPPS) with the inclusion of n-dodecane in the production medium. Of note, 6-dEB was not found in the n-dodecane phase (<1 mg l−1) though this compound also exhibits non-polar properties and is readily extracted by ethyl acetate. The two-phase 6-dEB titers for YW23sfp(pBP130/pBP144) and YW23sfp(pBP130/pBP144/pACYCDuet-TXS-GGPPS) were similar to single-phase production, indicating that the addition of n-dodecane had no effect on polyketide levels. As in the single-phase system, 6-dEB production decreased approximately two-fold with the addition of the pACYCDuet-TXS-GGPPS plasmid (p = 0.023).
Fig. 4.
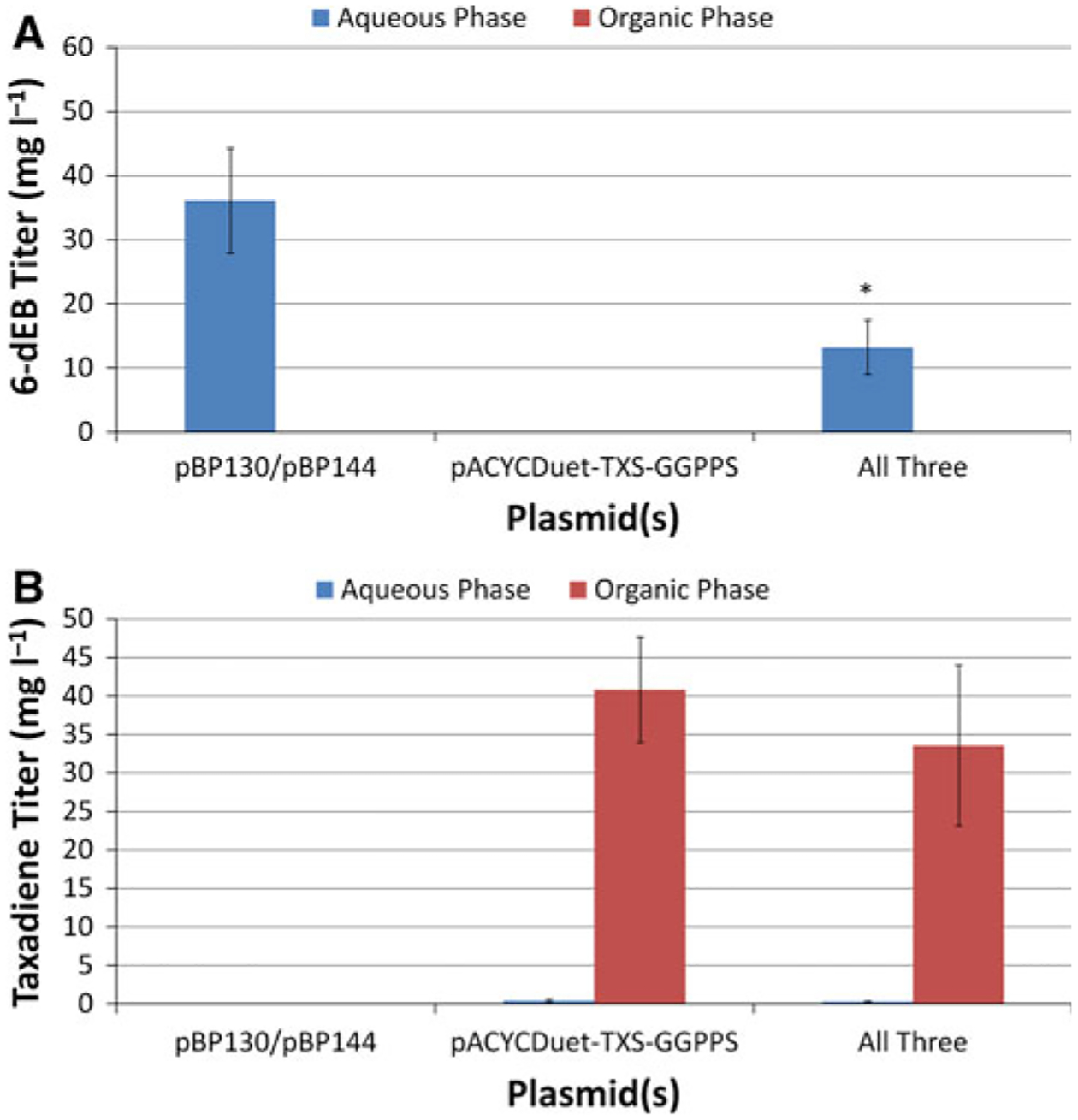
Production of a 6-dEB and b taxadiene in YW23sfp in the two-phase shake flask cultures under the same plasmid combinations as before. The concentration in both the aqueous and the organic phases are shown. Propionate (20 mM) was supplemented in the medium for experiments in columns one and three. Error bars represented ± one standard deviation from three independent replicates. *Indicates statistically significant results at p < 0.05
Figure 4b presents taxadiene titer in the same three scenarios. As expected, the addition of n-dodecane to the shake-flask cultures increased the titer of taxadiene significantly to 40.8 ± 6.9 mg l−1. There was residual taxadiene found in the aqueous phase (0.40 ± 0.21 mg l−1). As before, there was no statistically significant difference (p = 0.588 for the dodecane phase and p = 0.374 in the aqueous phase) in taxadiene titer when the 6-dEB plasmids were included. The organic:aqueous partitioning coefficient for taxadiene () is roughly 102, while the same partitioning coefficient for 6-dEB () is at most 10−1.65. This separation factor () of over 5,000 is a conservative estimate due to the roughly 1 mg l−1 detection limit for 6-dEB in the HPLC-ELSD system used here.
Effect of precursor supplementation on 6-dEB and taxadiene production
The exogenous feeding of propionate is required for >1 mg l−1 production of 6-dEB. We reasoned that this was one of the reasons why the 6-dEB titer was much higher than that of taxadiene in the single-phase system (Fig. 3). As a result, we tested the effect of the addition of 20 mM propionate, 20 mM pyruvate, or 20 mM propionate and pyruvate on 6-dEB and taxadiene titers. Pyruvate, as illustrated in Fig. 1b, can be considered a precursor of the DXP-based isoprenoid biosynthetic pathway, though it also serves as a highly connected metabolite in primary metabolism. The dual producer YW23sfp(pBP130/pBP144/pACYCDuet-TXS-GGPPS) was solely used in this study. As shown in Fig. 5a, the addition of pyruvate had no effect on 6-dEB titer, while the sole feeding of pyruvate was not able to produce 6-dEB > 1 mg l−1. However, the supplementation of precursors in the medium in any combination had no effect on taxadiene titer (ANOVA p = 0.782).
Fig. 5.
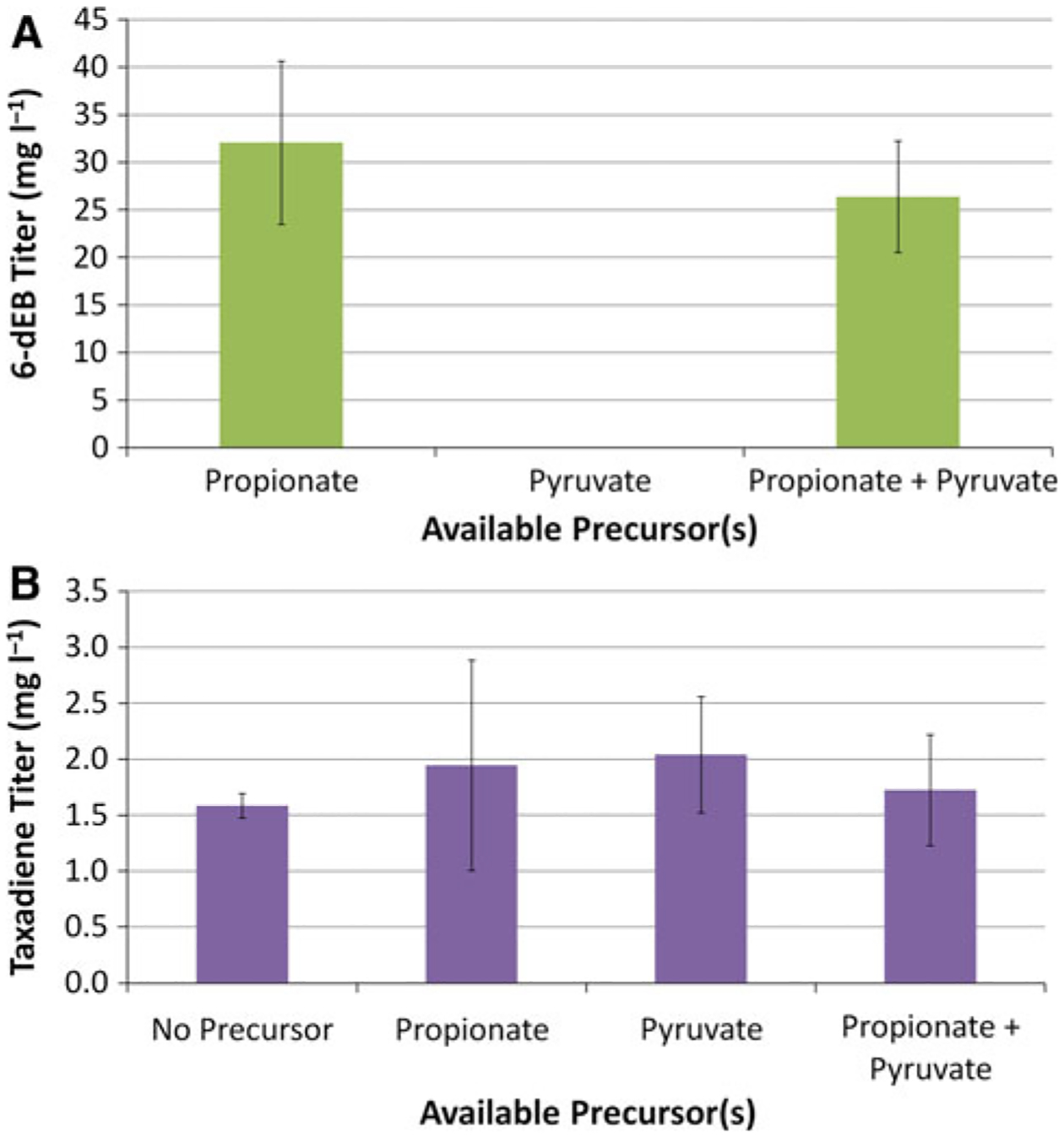
a Production of 6-dEB with the supplementation of 20 mM propionate, 20 mM pyruvate, or 20 mM propionate and 20 mM pyruvate. b Production of taxadiene with the supplementation of nothing, 20 mM propionate, 20 mM pyruvate, or 20 mM propionate and 20 mM pyruvate. Error bars represented ± one standard deviation from three independent replicates
Propionate consumption was next analyzed between the single- and two-phase production systems across the three different plasmid combinations (Fig. 6a). Interestingly, regardless of whether the 6-dEB production plasmids were used, propionate uptake was unaffected. This indicates that the production of 6-dEB has no influence on propionate uptake, at least in the production scenarios here. Across all three single-phase systems, propionate consumption did not vary (ANOVA p = 0.139). This same result was observed in the dual phase system as well (ANOVA p = 0.688). Somewhat unexpectedly, pyruvate was completely consumed at the end of the culture period (Fig. 6b). Neither propionate nor pyruvate consumption was affected by the presence of the other compound in the culture medium.
Fig. 6.
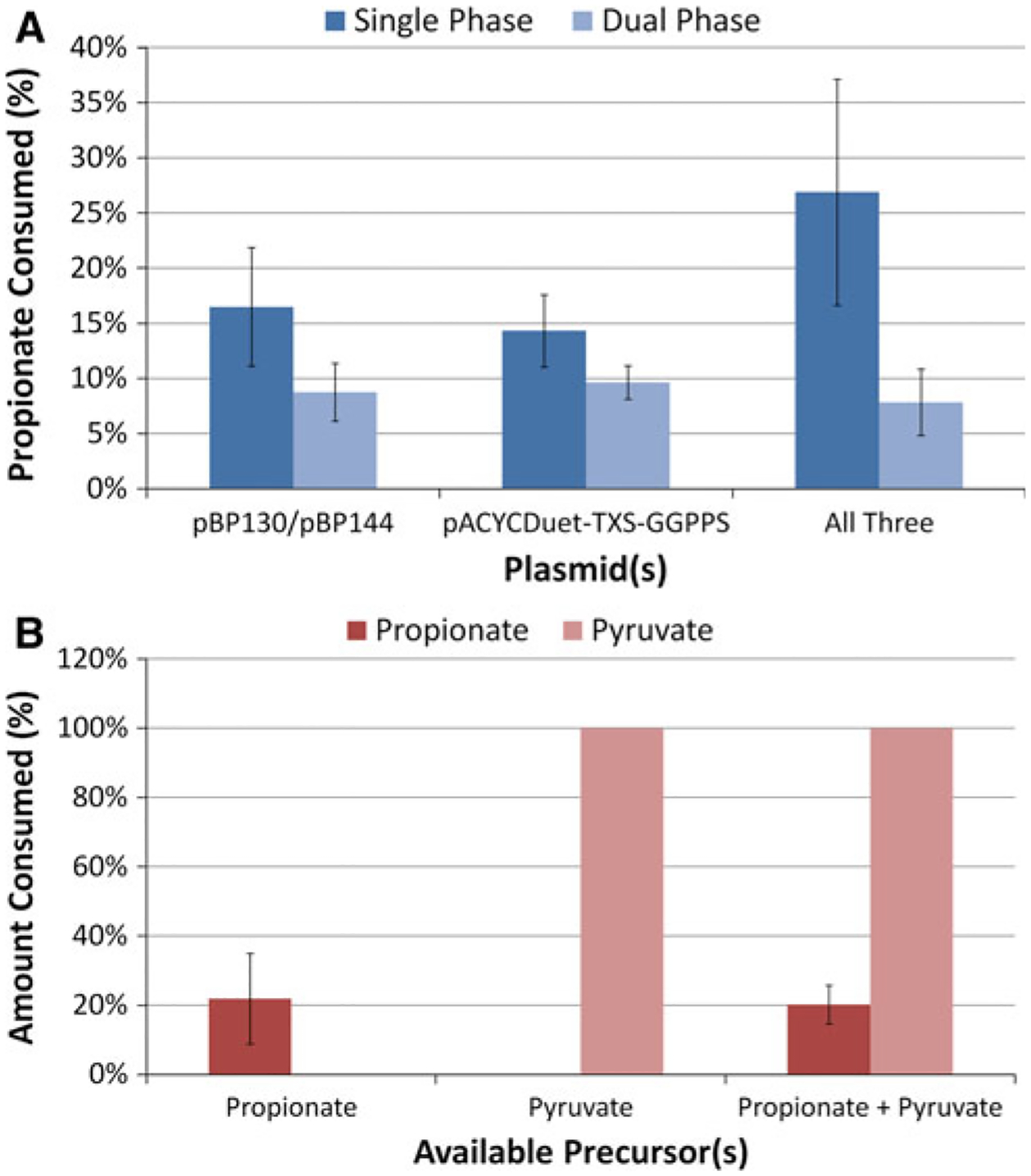
a Consumption of propionate in YW23sfp across the three plasmid systems described previously and in the single- vs. dual-phase culture systems. b Consumption of propionate and pyruvate by themselves or together in YW23sfp(pBP130/pBP144/pACYCDuet-TXS-GGPPS)
Co-production of 6-dEB and taxadiene in a two-phase bioreactor
To further test our polyketide and isoprenoid co-production platform, we scaled the culture to a 3-l bioreactor system. In this setting, aeration, temperature, and pH were all controlled, and 1.2 l of culture medium was again charged with 20% n-dodecane. A batch run was conducted over the course of 120 h and sampled every 6–12 h.
Figure 7a presents cell density and specific product titers as a function of time. Not surprisingly, with better aeration (0.5 VVM) and a higher charge of principal carbon source (45 g l−1 glycerol), the cell-density increased from approximately 4 gDCW l−1 in the shake-flask (data not shown) to approximately 20 gDCW l−1 in stationary phase growth. The specific titers of 6-dEB and taxadiene reached2.73 and 4.90 mg gDCW−1, resulting in raw titers of 32.8 and 94.2 mg l−1, respectively. Figure 7b presents the consumption of glycerol and propionate, as well as the production of acetate, as a function of time. By 100 h, the glycerol had been completely consumed, resulting in an averaged specific uptake rate of 0.28 mmol gDCW−1 h−1. Interestingly, propionate consumption stalled around 50 h at approximately 5 mM. This corresponded to an accompanying plateau in 6-dEB production titer. Oddly, this occurred during the middle of the exponential growth phase and no changes were observed in glycerol consumption, taxadiene production, or acetate production. However, in general, the acetate by-product titers were relatively low, increasing to approximately 22 mM when glycerol was still present in the medium and then consumed by the culture after the glycerol was exhausted.
Fig. 7.
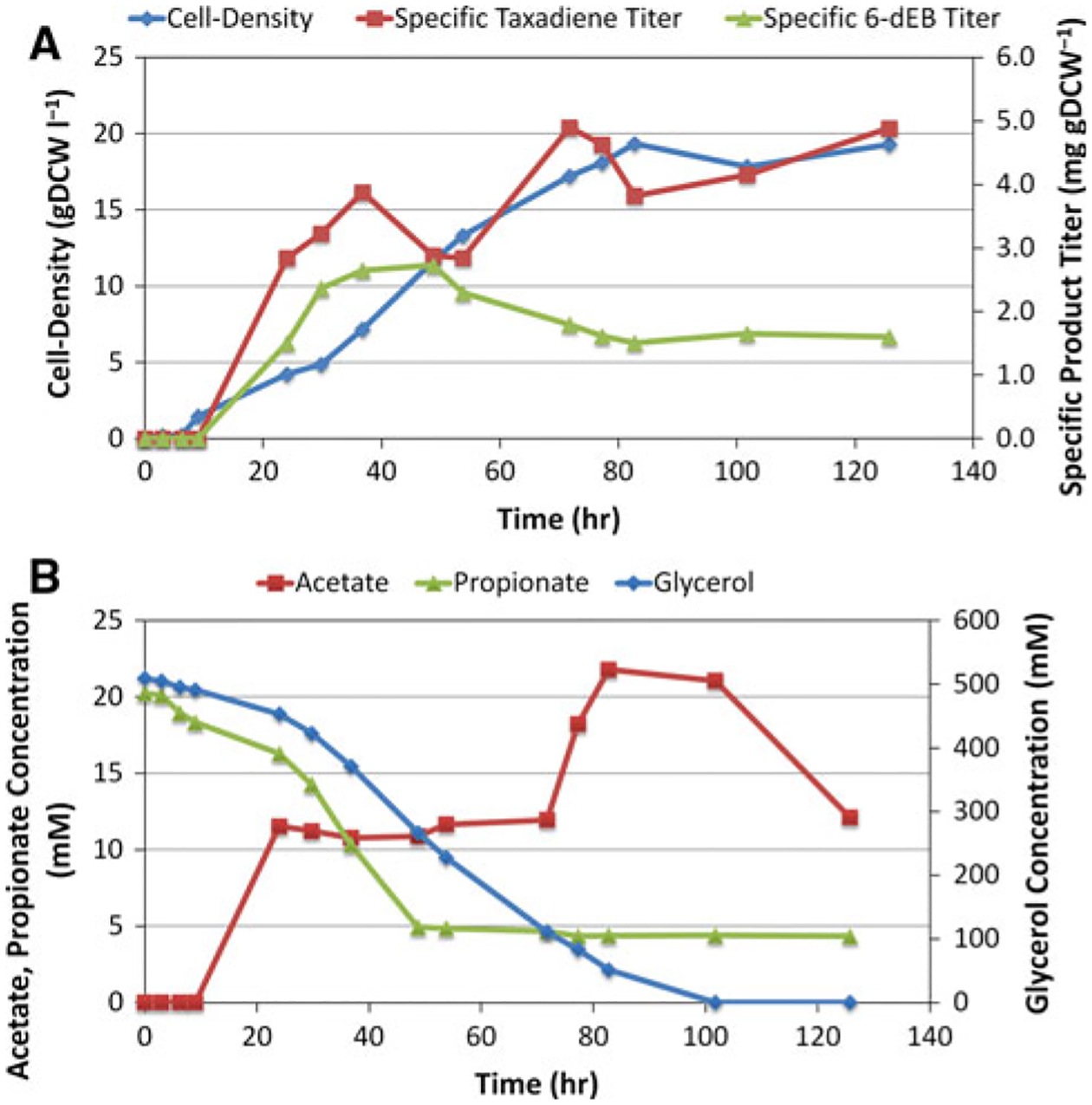
Two-phase bioreactor culture of YW23sfp(pBP130/pBP144/pACYCDuet-TXS-GGPPS) supplemented with 20 mM propionate. a Cell-density, specific taxadiene titer, and specific 6-dEB titer are shown as a function of time. b Propionate, acetate, and glycerol concentrations in the medium are shown as a function of time
Discussion
Due to the immense therapeutic potential of natural products, there has been a growing interest in developing efficient and scalable heterologous production processes. Here, we reported the co-production of a polyketide and a diterpenoid in mg l−1 quantities with a two-phase bioreactor. Doing so offers the future potential to simultaneously produce two valuable products with a reduction in the operating costs that would normally accompany individual production processes. First, we constructed a strain of E. coli with a chromosomally over-expressed isoprenoid biosynthetic pathway and a separate chromosomal modification to allow PKS (and NRPS) posttranslational modification and precursor supply. E. coli is considered relatively free of metabolism capable of supporting the over-production of compounds like polyketides, nonribosomal peptides, and isoprenoids. Therefore, it offers the opportunity to introduce and design metabolism capable of supporting diverse natural product classes without the concern of unwanted crosstalk between existing native pathways (a situation common to Streptomyces hosts which typically possess multiple secondary metabolite pathways) or overlap between the heterologously introduced pathways. The resulting strain in this study, YW23sfp, was tested for the production of a polyketide (6-dEB) and an isoprenoid (taxadiene) separately and together. While the 6-dEB titer decreased during co-production, the taxadiene titer was unchanged. We then tested the production capabilities of this strain in a two-phase culture system; here, we anticipated an in situ product separation capability resulting from the differences in our polyketide and isoprenoid compounds. It was previously reported that amorpha-4,11-diene titers decreased due to compound volatilization in a highly aerated bioreactor [45]. After confirming the same result for taxadiene, we adopted a similar two-phase culture strategy. The 6-dEB titer was unchanged with the addition of the second phase, while the taxadiene titer was greatly improved. The substantial difference in the organic:aqueous partitioning coefficients for taxadiene and 6-dEB resulted in a separation factor of greater than 5,000 and allowed the simultaneously production and separation of these two complex natural products. The co-production strategy should be extendable to other products that similarly match the characteristics and properties of the 6dEB and taxadiene compounds featured in this study.
Next, we determined the effects of exogenous precursors on the simultaneous production of 6-dEB and taxadiene in E. coli. Results showed that the addition of either pyruvate or propionate, alone or together, had no significant effect on either 6-dEB or taxadiene production. Results did, however, verify that an exogenous supply of propionate is essential for 6-dEB production. Equal amounts of propionate and pyruvate were consumed regardless of the product(s) produced or the quantities in which they were being generated. This phenomenon is particularly noteworthy because it suggests other metabolic pathways for propionate consumption. Scenarios characterized by less production of 6-dEB but equal consumption of propionate clearly suggest that the bacteria are utilizing the substrate for purposes other than 6-dEB production.
Finally, a scaled production scenario was conducted in a bioreactor with a working volume of 1.5 l and consisting of 20% n-dodecane. Results verified that production trends observed in the smaller-scale 15-ml shake flasks. Final specific product titers in the 15-ml shake flasks were nearly equivalent to those achieved in the bioreactor for 6-dEB and slightly higher for taxadiene. Drawing parallels to other dual-purpose bioprocesses, the polyketide/isoprenoid co-production system could be considered a type of consolidated production process. Similar terminology has been coined to describe the efficient conversion of biomass to useful biofuels where the goal is to simultaneously combine cellulosic biomass degradation with subsequent biofuel fermentation using S. cerevisiae as a host [36, 62]. In both settings, the design strives to minimize the operational steps and costs of the overall process.
The concept of co-production could also be extended to other heterologous systems. For example, a genome-minimized strain of S. avermitilis was recently constructed and reported for the heterologous production of streptomycin (from S. griseus) and cephamycin C (from S. clavuligerus) [33]. These titers reached nearly 200 mg l−1 and 120 mg l−1 for streptomycin and cephamycin C, respectively. The strain was then used to express a codon optimized amorpha-4,11-diene synthase gene (ads, from Artemisia annua) under the control of the rpsJ promoter to produce amorpha-4,11-diene at an estimated titer of 10–30 mg l−1 [33]. While this genome-minimized strain was able to produce approximately 101–102 mg l−1 of an aminoglycoside, a β-lactam, and a sesquiterpene, the co-production of these compounds was not explored, though presumably this would be feasible.
In addition to a bioprocess production platform, a strain capable of supporting multiple classes of natural products could also be of great utility in drug discovery efforts. It has long been known that actinomycetes (particularly from the soil-dwelling Streptomyces genus) produce a vast number of secondary metabolites and that drug discovery through isolating and culturing actinomycetes has proven successful. Daptomycin (marketed as Cubicin® by Cubist Pharmaceuticals) [4] was isolated from an extended search of almost 107 actinomycetes. Furthermore, advances in annotation and bioinformatic approaches have aided the identification of secondary metabolite gene clusters from those organisms characterized by genome sequencing. For example, the native erythromycin producer, S. erythraea, contains sequence information for a predicted 25 polyketide, nonribosomal peptide, and terpene compounds [49], even though no products derived from these clusters (besides erythromycin) were identified on 50 different types of solid and liquid media [8]. Similar phenomena have been observed for S. avermitilis [27] and S. griseus [48]. Accessing this potential may involve innovative culturing techniques, which have been used as a means to stimulate expression of dormant secondary metabolite pathways [50], but this is not necessarily a high-throughput, process-friendly approach. In addition, it has been cited that 99% of all microorganisms cannot be cultured with conventional laboratory methods [35], therefore, severely limiting current drug screening approaches through these methods. To address these concerns, the concept of metagenomics was introduced [24]. This approach generally consists of the following steps: (1) collection of environmental samples, (2) isolation of genomic DNA from these samples, (3) cloning of the environmental DNA into bacterial artificial chromosome (BAC) vectors, (4) transformation into a heterologous host, and (5) screening for biological activity. This workflow, which can be designed in a high-throughput manner, has been successfully utilized to screen soil DNA for new antimicrobial natural products [37]. Even though E. coli is a primary choice for the heterologous host used in metagenomic efforts, a significant shortcoming is the lack of native metabolism needed to support many complex secondary metabolites. For example, even if an entire PKS cluster was captured in a single BAC, a host lacking a functional phosphopantetheinyl transferase (PPTase) would be unable to support subsequent biosynthesis. In addition, wild-type E. coli hosts will typically not provide the needed acyl-CoA precursors, such as the (2S)-ethylmalonyl-CoA and chloroethylmalonyl-CoA extender units described previously, to support complex natural product biosynthesis.
Although not the particular focus of this study, the strain developed here could be used for metagenomic studies, having the ability to now support the production of three classes of natural products (polyketides, nonribosomal peptides, and isoprenoids). The over-expression of a promiscuous PPTase gene and numerous genes to support PKS and isoprenoid precursor supply should facilitate the production of a wide range of natural products. From a development standpoint, a single strain capable of discovery and production has the potential to greatly decrease strain and process development time, highlighted in this study by the potential to simultaneously produce two meaningful products.
Acknowledgments
The authors recognize support from the National Institutes of Health (GM085323) and the Milheim Foundation (Grant for Cancer Research No. 2006-17). MM was supported through the Tufts University Summer Scholars program. JW was supported through the Chinese Scholarship Council as a visiting student from East China University of Science and Technology.
Contributor Information
Brett A. Boghigian, Department of Chemical and Biological Engineering, Science and Technology Center, Tufts University, 4 Colby Street, Medford, MA 02155, USA
Melissa Myint, Department of Chemical and Biological Engineering, Science and Technology Center, Tufts University, 4 Colby Street, Medford, MA 02155, USA.
Jiequn Wu, Department of Chemical and Biological Engineering, Science and Technology Center, Tufts University, 4 Colby Street, Medford, MA 02155, USA; State Key Laboratory of Bioreactor Engineering, National Engineering Research Center for Biotechnology, East China University of Science and Technology, 200237 Shanghai, People’s Republic of China.
Blaine A. Pfeifer, Department of Chemical and Biological Engineering, Science and Technology Center, Tufts University, 4 Colby Street, Medford, MA 02155, USA
References
- 1.Adrio JL, Demain AL (2006) Genetic improvement of processes yielding microbial products. FEMS Microbiol Rev 30:187–214 [DOI] [PubMed] [Google Scholar]
- 2.Ajikumar PK, Tyo K, Carlsen S, Mucha O, Phon TH, Stephanopoulos G (2008) Terpenoids: opportunities for biosynthesis of natural product drugs using engineered microorganisms. Mol Pharm 5:167–190 [DOI] [PubMed] [Google Scholar]
- 3.Ajikumar PK, Xiao WH, Tyo KE, Wang Y, Simeon F, Leonard E, Mucha O, Phon TH, Pfeifer B, Stephanopoulos G (2010) Isoprenoid pathway optimization for Taxol precursor overproduction in Escherichia coli. Science 330:70–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baltz RH (2008) Renaissance in antibacterial discovery from actinomycetes. Curr Opin Pharmacol 8:557–563 [DOI] [PubMed] [Google Scholar]
- 5.Baltz RH (2010) Streptomyces and Saccharopolyspora hosts for heterologous expression of secondary metabolite gene clusters. J Ind Microbiol Biotechnol 37:759–772 [DOI] [PubMed] [Google Scholar]
- 6.Besumbes O, Sauret-Gueto S, Phillips MA, Imperial S, Rodriguez-Concepcion M, Boronat A (2004) Metabolic engineering of isoprenoid biosynthesis in Arabidopsis for the production of taxadiene, the first committed precursor of Taxol. Biotechnol Bioeng 88:168–175 [DOI] [PubMed] [Google Scholar]
- 7.Blanchard S, Thorson JS (2006) Enzymatic tools for engineering natural product glycosylation. Curr Opin Chem Biol 10:263–271 [DOI] [PubMed] [Google Scholar]
- 8.Boakes S, Oliynyk M, Cortes J, Bohm I, Rudd BA, Revill WP, Staunton J, Leadlay PF (2004) A new modular polyketide synthase in the erythromycin producer Saccharopolyspora erythraea. J Mol Microbiol Biotechnol 8:73–80 [DOI] [PubMed] [Google Scholar]
- 9.Boghigian BA, Pfeifer BA (2008) Current status, strategies, and potential for the metabolic engineering of heterologous polyketides in Escherichia coli. Biotechnol Lett 30:1323–1330 [DOI] [PubMed] [Google Scholar]
- 10.Chan YA, Podevels AM, Kevany BM, Thomas MG (2009) Biosynthesis of polyketide synthase extender units. J Nat Prod 26:90–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen Y, Deng W, Wu J, Qian J, Chu J, Zhuang Y, Zhang S, Liu W (2008) Genetic modulation of the overexpression of tailoring genes eryK and eryG leading to the improvement of erythromycin A purity and production in Saccharopolyspora erythraea fermentation. Appl Environ Microbiol 74:1820–1828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cherepanov PP, Wackernagel W (1995) Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 158:9–14 [DOI] [PubMed] [Google Scholar]
- 13.Chng C, Lum AM, Vroom JA, Kao CM (2008) A key developmental regulator controls the synthesis of the antibiotic erythromycin in Saccharopolyspora erythraea. Proc Natl Acad Sci USA 105:11346–11351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cortes J, Haydock SF, Roberts GA, Bevitt DJ, Leadlay PF (1990) An unusually large multifunctional polypeptide in the erythromycin-producing polyketide synthase of Saccharopolyspora erythraea. Nature 348:176–178 [DOI] [PubMed] [Google Scholar]
- 15.Cunningham FX Jr, Sun Z, Chamovitz D, Hirschberg J, Gantt E (1994) Molecular structure and enzymatic function of lycopene cyclase from the cyanobacterium Synechococcus sp strain PCC7942. Plant Cell 6:1107–1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Datsenko KA, Wanner BL (2000) One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA 97:6640–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Denis JN, Greene AE, Guenard D, Gueritte-Voegelein F, Mangatal L, Potier P (1988) A highly efficient, practical approach to natural taxol. J Am Chem Soc 110:5917–5919 [Google Scholar]
- 18.Donadio S, Staver MJ, McAlpine JB, Swanson SJ, Katz L (1991) Modular organization of genes required for complex polyketide biosynthesis. Science 252:675–679 [DOI] [PubMed] [Google Scholar]
- 19.Eisenreich W, Bacher A, Arigoni D, Rohdich F (2004) Biosynthesis of isoprenoids via the non-mevalonate pathway. Cell Mol Life Sci 61:1401–1426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Engels B, Dahm P, Jennewein S (2008) Metabolic engineering of taxadiene biosynthesis in yeast as a first step towards Taxol (Paclitaxel) production. Metab Eng 10:201–206 [DOI] [PubMed] [Google Scholar]
- 21.Eustaquio AS, McGlinchey RP, Liu Y, Hazzard C, Beer LL, Florova G, Alhamadsheh MM, Lechner A, Kale AJ, Kobayashi Y, Reynolds KA, Moore BS (2009) Biosynthesis of the salinos-poramide A polyketide synthase substrate chloroethylmalonyl-coenzyme A from S-adenosyl-l-methionine. Proc Natl Acad Sci USA 106:12295–12300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fischbach MA, Walsh CT (2006) Assembly-line enzymology for polyketide and nonribosomal peptide antibiotics: logic, machinery, and mechanisms. Chem Rev 106:3468–3496 [DOI] [PubMed] [Google Scholar]
- 23.Gokhale RS, Tsuji SY, Cane DE, Khosla C (1999) Dissecting and exploiting intermodular communication in polyketide synthases. Science 284:482–485 [DOI] [PubMed] [Google Scholar]
- 24.Handelsman J, Rondon MR, Brady SF, Clardy J, Goodman RM (1998) Molecular biological access to the chemistry of unknown soil microbes: a new frontier for natural products. Chem Biol 5:R245–R249 [DOI] [PubMed] [Google Scholar]
- 25.Hawkins KM, Smolke CD (2008) Production of benzylisoquinoline alkaloids in Saccharomyces cerevisiae. Nat Chem Biol 4:564–573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Horwitz SB (1994) How to make taxol from scratch. Nature 367:593–594 [DOI] [PubMed] [Google Scholar]
- 27.Ikeda H, Ishikawa J, Hanamoto A, Shinose M, Kikuchi H, Shiba T, Sakaki Y, Hattori M, Omura S (2003) Complete genome sequence and comparative analysis of the industrial microorganism Streptomyces avermitilis. Nat Biotechnol 21:526–531 [DOI] [PubMed] [Google Scholar]
- 28.Kao CM, Katz L, Khosla C (1994) Engineered biosynthesis of a complete macrolactone in a heterologous host. Science 265:509–512 [DOI] [PubMed] [Google Scholar]
- 29.Ketchum RE, Gibson DM, Croteau RB, Shuler ML (1999) The kinetics of taxoid accumulation in cell suspension cultures of Taxus following elicitation with methyl jasmonate. Biotechnol Bioeng 62:97–105 [DOI] [PubMed] [Google Scholar]
- 30.Khosla C, Keasling JD (2003) Metabolic engineering for drug discovery and development. Nat Rev Drug Discov 2:1019–1025 [DOI] [PubMed] [Google Scholar]
- 31.Kodumal SJ, Patel KG, Reid R, Menzella HG, Welch M, Santi DV (2004) Total synthesis of long DNA sequences: synthesis of a contiguous 32-kb polyketide synthase gene cluster. Proc Natl Acad Sci USA 101:15573–15578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Koepp AE, Hezari M, Zajicek J, Vogel BS, LaFever RE, Lewis NG, Croteau R (1995) Cyclization of geranylgeranyl diphosphate to taxa-4(5), 11(12)-diene is the committed step of taxol biosynthesis in Pacific yew. J Biol Chem 270:8686–8690 [DOI] [PubMed] [Google Scholar]
- 33.Komatsu M, Uchiyama T, Omura S, Cane DE, Ikeda H (2010) Genome-minimized Streptomyces host for the heterologous expression of secondary metabolism. Proc Natl Acad Sci USA 107:2646–2651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kovacs K, Zhang L, Linforth RS, Whittaker B, Hayes CJ, Fray RG (2007) Redirection of carotenoid metabolism for the efficient production of taxadiene [taxa-4(5), 11(12)-diene] in transgenic tomato fruit. Transgenic Res 16:121–126 [DOI] [PubMed] [Google Scholar]
- 35.Li JW, Vederas JC (2009) Drug discovery and natural products: end of an era or an endless frontier? Science 325:161–165 [DOI] [PubMed] [Google Scholar]
- 36.Lynd LR, van Zyl WH, McBride JE, Laser M (2005) Consolidated bioprocessing of cellulosic biomass: an update. Curr Opin Biotechnol 16:577–583 [DOI] [PubMed] [Google Scholar]
- 37.MacNeil IA, Tiong CL, Minor C, August PR, Grossman TH, Loiacono KA, Lynch BA, Phillips T, Narula S, Sundaramoorthi R, Tyler A, Aldredge T, Long H, Gilman M, Holt D, Osburne MS (2001) Expression and isolation of antimicrobial small molecules from soil DNA libraries. J Mol Microbiol Biotechnol 3:301–308 [PubMed] [Google Scholar]
- 38.Martin VJ, Pitera DJ, Withers ST, Newman JD, Keasling JD (2003) Engineering a mevalonate pathway in Escherichia coli for production of terpenoids. Nat Biotechnol 21:796–802 [DOI] [PubMed] [Google Scholar]
- 39.Masters M (1977) The frequency of P1 transduction of the genes of Escherichia coli as a function of chromosomal position: preferential transduction of the origin of replication. Mol Gen Genet 155:197–202 [DOI] [PubMed] [Google Scholar]
- 40.Menzella HG, Reeves CD (2007) Combinatorial biosynthesis for drug development. Curr Opin Microbiol 10:238–245 [DOI] [PubMed] [Google Scholar]
- 41.Menzella HG, Reid R, Carney JR, Chandran SS, Reisinger SJ, Patel KG, Hopwood DA, Santi DV (2005) Combinatorial polyketide biosynthesis by de novo design and rearrangement of modular polyketide synthase genes. Nat Biotechnol 23:1171–1176 [DOI] [PubMed] [Google Scholar]
- 42.Moore BS, Hertweck C (2002) Biosynthesis and attachment of novel bacterial polyketide synthase starter units. Nat Prod Rep 19:70–99 [DOI] [PubMed] [Google Scholar]
- 43.Neumann CS, Fujimori DG, Walsh CT (2008) Halogenation strategies in natural product biosynthesis. Chem Biol 15:99–109 [DOI] [PubMed] [Google Scholar]
- 44.Newman DJ, Cragg GM (2007) Natural products as sources of new drugs over the last 25 years. J Nat Prod 70:461–477 [DOI] [PubMed] [Google Scholar]
- 45.Newman JD, Marshall J, Chang M, Nowroozi F, Paradise E, Pitera D, Newman KL, Keasling JD (2006) High-level production of amorpha-4, 11-diene in a two-phase partitioning bioreactor of metabolically engineered Escherichia coli. Biotechnol Bioeng 95:684–691 [DOI] [PubMed] [Google Scholar]
- 46.Nguyen KT, Ritz D, Gu JQ, Alexander D, Chu M, Miao V, Brian P, Baltz RH (2006) Combinatorial biosynthesis of novel antibiotics related to daptomycin. Proc Natl Acad Sci USA 103:17462–17467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nicolaou KC, Yang Z, Liu JJ, Ueno H, Nantermet PG, Guy RK, Claiborne CF, Renaud J, Couladouros EA, Paulvannan K et al. (1994) Total synthesis of taxol. Nature 367:630–634 [DOI] [PubMed] [Google Scholar]
- 48.Ohnishi Y, Ishikawa J, Hara H, Suzuki H, Ikenoya M, Ikeda H, Yamashita A, Hattori M, Horinouchi S (2008) Genome sequence of the streptomycin-producing microorganism Streptomyces griseus IFO 13350. J Bacteriol 190:4050–4060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Oliynyk M, Samborskyy M, Lester JB, Mironenko T, Scott N, Dickens S, Haydock SF, Leadlay PF (2007) Complete genome sequence of the erythromycin-producing bacterium Saccharopolyspora erythraea NRRL23338. Nat Biotechnol 25:447–453 [DOI] [PubMed] [Google Scholar]
- 50.Pettit RK (2009) Mixed fermentation for natural product drug discovery. Appl Microbiol Biotechnol 83:19–25 [DOI] [PubMed] [Google Scholar]
- 51.Pfeifer BA, Admiraal SJ, Gramajo H, Cane DE, Khosla C (2001) Biosynthesis of complex polyketides in a metabolically engineered strain of E. coli. Science 291:1790–1792 [DOI] [PubMed] [Google Scholar]
- 52.Pieper R, Luo G, Cane DE, Khosla C (1995) Cell-free synthesis of polyketides by recombinant erythromycin polyketide synthases. Nature 378:263–266 [DOI] [PubMed] [Google Scholar]
- 53.Pitera DJ, Paddon CJ, Newman JD, Keasling JD (2007) Balancing a heterologous mevalonate pathway for improved isoprenoid production in Escherichia coli. Metab Eng 9:193–207 [DOI] [PubMed] [Google Scholar]
- 54.Quadri LE, Weinreb PH, Lei M, Nakano MM, Zuber P, Walsh CT (1998) Characterization of Sfp, a Bacillus subtilis phosphopantetheinyl transferase for peptidyl carrier protein domains in peptide synthetases. Biochemistry 37:1585–1595 [DOI] [PubMed] [Google Scholar]
- 55.Reeves AR, Brikun IA, Cernota WH, Leach BI, Gonzalez MC, Weber JM (2007) Engineering of the methylmalonyl-CoA metabolite node of Saccharopolyspora erythraea for increased erythromycin production. Metab Eng 9:293–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ro DK, Paradise EM, Ouellet M, Fisher KJ, Newman KL, Ndungu JM, Ho KA, Eachus RA, Ham TS, Kirby J, Chang MC, Withers ST, Shiba Y, Sarpong R, Keasling JD (2006) Production of the antimalarial drug precursor artemisinic acid in engineered yeast. Nature 440:940–943 [DOI] [PubMed] [Google Scholar]
- 57.Rodriguez E, Gramajo H (1999) Genetic and biochemical characterization of the α and β components of a propionyl-CoA carboxylase complex of Streptomyces coelicolor A3(2). Micro-biology 145(Pt 11):3109–3119 [DOI] [PubMed] [Google Scholar]
- 58.Salas JA, Mendez C (2007) Engineering the glycosylation of natural products in actinomycetes. Trends Microbiol 15:219–232 [DOI] [PubMed] [Google Scholar]
- 59.Skeel RT (1999) Handbook of cancer chemotherapy. Lippincott Williams & Wilkins, Philadelphia, PA, USA [Google Scholar]
- 60.Smith RL, Bungay HR, Pittenger RC (1962) Growth-biosynthesis relationships in erythromycin fermentation. Appl Microbiol 10: 293–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tang Y, Kim CY, Mathews II, Cane DE, Khosla C (2006) The2.7-Å crystal structure of a 194-kDa homodimeric fragment of the 6-deoxyerythronolide B synthase. Proc Natl Acad Sci USA 103:11124–11129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.van Zyl WH, Lynd LR, den Haan R, McBride JE (2007) Consolidated bioprocessing for bioethanol production using Saccharomyces cerevisiae. Adv Biochem Eng Biotechnol 108:205–235 [DOI] [PubMed] [Google Scholar]
- 63.Walsh CT (2008) The chemical versatility of natural-product assembly lines. Acc Chem Res 41:4–10 [DOI] [PubMed] [Google Scholar]
- 64.Wang Y, Boghigian BA, Pfeifer BA (2007) Improving heterologous polyketide production in Escherichia coli by overexpression of an S-adenosylmethionine synthetase gene. Appl Microbiol Biotechnol 77:367–373 [DOI] [PubMed] [Google Scholar]
- 65.Wang Y, Zhang S (2008) High-frequency transformation of the industrial erythromycin-producing bacterium Saccharopolyspora erythraea. Biotechnol Lett 30:357–361 [DOI] [PubMed] [Google Scholar]
- 66.Weissman KJ, Leadlay PF (2005) Combinatorial biosynthesis of reduced polyketides. Nat Rev Microbiol 3:925–936 [DOI] [PubMed] [Google Scholar]
- 67.Wu J, Boghigian BA, Myint M, Zhang H, Zhang S, Pfeifer BA (2010) Construction and performance of heterologous polyketide-producing K-12- and B-derived Escherichia coli. Lett Appl Microbiol 51:196–204 [DOI] [PubMed] [Google Scholar]