

Abstract
ClpXP is an archetypical AAA+ protease, consisting of ClpX and ClpP. ClpX is an ATP-dependent protein unfoldase and polypeptide translocase, whereas ClpP is a self-compartmentalized peptidase. ClpXP is currently the only AAA+ protease for which high-resolution structures exist, the molecular basis of recognition for a protein substrate is understood, extensive biochemical and genetic analysis have been performed, and single-molecule optical trapping has allowed direct visualization of the kinetics of substrate unfolding and translocation. In this review, we discuss our current understanding of ClpXP structure and function, evaluate competing sequential and probabilistic mechanisms of ATP hydrolysis, and highlight open questions for future exploration.
Introduction
Molecular machines of the AAA+ superfamily (ATPases Associated with diverse cellular Activities) power important cellular functions that include protein degradation, remodeling of protein and nucleic-acid complexes, transport of cargo along microtubule tracks, recycling of vesicles, packaging of viral DNA, loading of the sliding clamp and/or replicative helicases onto DNA, cytokinesis, activation of innate-immune responses, translocation of chromosomes, and activation of transcription (Neuwald et al., 1999; Ogura and Wilkinson, 2001; Hanson and Whiteheart, 2005; Sauer and Baker, 2011). These AAA+ machines commonly function as hexameric rings with an axial channel or pore.
Active AAA+ proteases consist of a AAA+ hexameric ring that acts to engage protein substrates, to unfold any native structure present in this substrate, and to translocate the resulting polypeptide through the axial channel of the ring and into the degradation chamber of an associated self-compartmentalized peptidase (Figure 1A; Striebel et al., 2009; Sauer and Baker, 2011; Bard et al., 2018; Puchades et al., 2020; Zhang and Mao, 2020). Energy released by ATP binding, hydrolysis, and/or ADP/Pi release powers the substrate unfolding and translocation reactions needed for degradation. ClpXP is an archetypical and well-studied AAA+ protease present in most eubacteria and the mitochondria of eukaryotic cells (Baker and Sauer, 2012; Olivares et al., 2018). It consists of ClpX, a hexameric AAA+ unfoldase/translocase, and ClpP, a barrel-like peptidase assembled from two heptameric rings. Ring hexamers of ClpX can stack coaxially with one heptameric ring of ClpP to form singly capped complexes or with both heptamers to generate doubly capped complexes (Figure 1B). Both types of ClpXP complexes are active in protein degradation, whereas ClpP, by itself, is only able to cleave very small peptides that diffuse into its degradation chamber (Thompson and Maurizi, 1994; Lee et al., 2010a). Thus, intrinsically disordered proteins or nascent polypeptides in the cell are protected from degradation by free ClpP tetradecamers.
Figure 1.
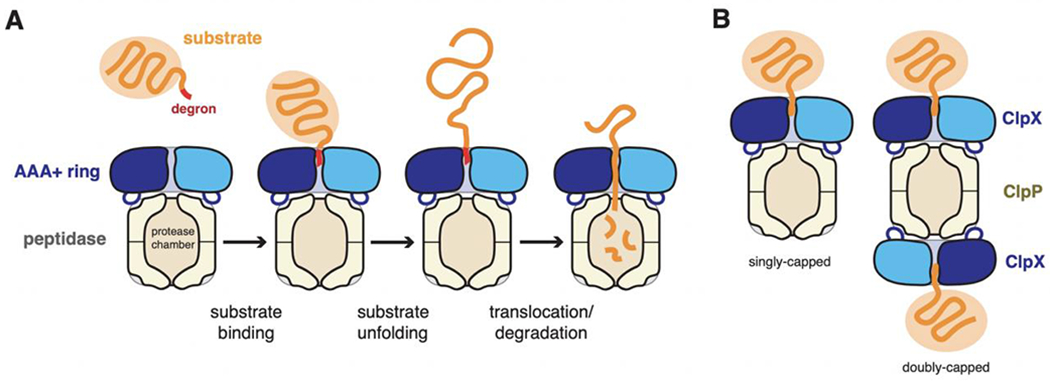
(A) AAA+ proteolytic mechanism. The AAA+ ring hexamer binds a peptide degron in a protein substrate, unfolds any native structure present, and then translocates the denatured polypeptide into a self-compartmentalized peptidase for degradation. (B). For the ClpXP protease, ClpX hexamers can bind to either heptameric ring of the ClpP peptidase.
Native proteins or large polypeptides are only degraded by ClpXP if they contain an unstructured peptide degron, typically at the N- or C-terminus, that is recognized by ClpX to allow unfolding and/or the initiation of translocation (Fig. 1A). In eubacteria, ClpXP helps ensure protein quality control by degrading ssrA-tagged protein fragments generated by premature termination of ribosomal synthesis (Moore and Sauer, 2007; Keiler, 2015). Bacterial ClpXP also affects cell fate by degrading transcription factors, cell-cycle regulators, and stress inhibitors (Jenal and Fuchs, 1998; Flynn et al., 2003; Baker and Sauer, 2012; Konovalova et al., 2014). In the absence of ClpP, ClpX alone can unfold proteins, disassemble macromolecular complexes, and catalyze incorporation of cofactors into metabolic enzymes (Burton and Baker, 2005; Kardon et al., 2015). Most studies of ClpXP have focused on the Escherichia coli enzymes and questions of fundamental mechanism, but ClpX and ClpP are also medically relevant. For example, mutations in mammalian ClpP or ClpX cause disease and developmental defects (Gispert et al., 2013; Yien et al., 2017; Bhandari et al., 2018; Luo et al., 2021), and antibiotics that hyperactivate ClpP and prevent its binding to ClpX kill Mycobacterium tuberculosis and eradicate Staphylococcus aureus persisters from biofilms (Brotz-Oesterhelt et al., 2005; Conlon et al., 2013; Carney et al., 2014).
In this review, we focus on current understanding of the structure and function of ClpXP, discuss how the predictions of competing sequential and probabilistic mechanisms of ATP hydrolysis concur or fail to accord with experiments, and highlight important unresolved mechanistic questions. For a discussion of the discovery of ClpX, ClpP, and biological substrates, we refer readers to previous reviews (Baker and Sauer, 2006; 2012; Mahmoud and Chien, 2018).
ClpX domains and sequence motifs
Each ClpX subunit comprises an N-terminal domain (~60 residues), a large AAA+ domain (~260 residues), and a small AAA+ domain (~100 residues) (Fig. 2A). The large and small AAA+ domains, together called the AAA+ module, have folds found in many AAA+ enzymes, whereas the N-terminal domain, which is a zinc-stabilized dimer in NMR and crystal structures, is only found in the ClpX family (Singh et al., 2001; Wojtyra et al., 2003; Donaldson et al., 2003; Park et al., 2007). The N-terminal domain, which is connected via a highly flexible linker to the large AAA+ domain, binds the auxiliary tags of some protein substrates and adaptor proteins but is not required for ClpXP degradation of ssrA-tagged substrates (Singh et al., 2001; Flynn et al., 2003; Neher et al., 2003; Wojtyra et al., 2003). Indeed, variants of E. coli ClpX lacking the N-terminal domain (ClpXΔN) can be genetically linked with short tethers to form pseudohexamers that combine with ClpP to degrade ssrA-tagged proteins with KM and Vmax kinetic parameters like those of wild-type ClpXP (Martin et al., 2005; Glynn et al., 2012). Thus, the AAA+ module executes the ATP-fueled protein-unfolding and polypeptide-translocation functions of ClpX.
Figure 2.
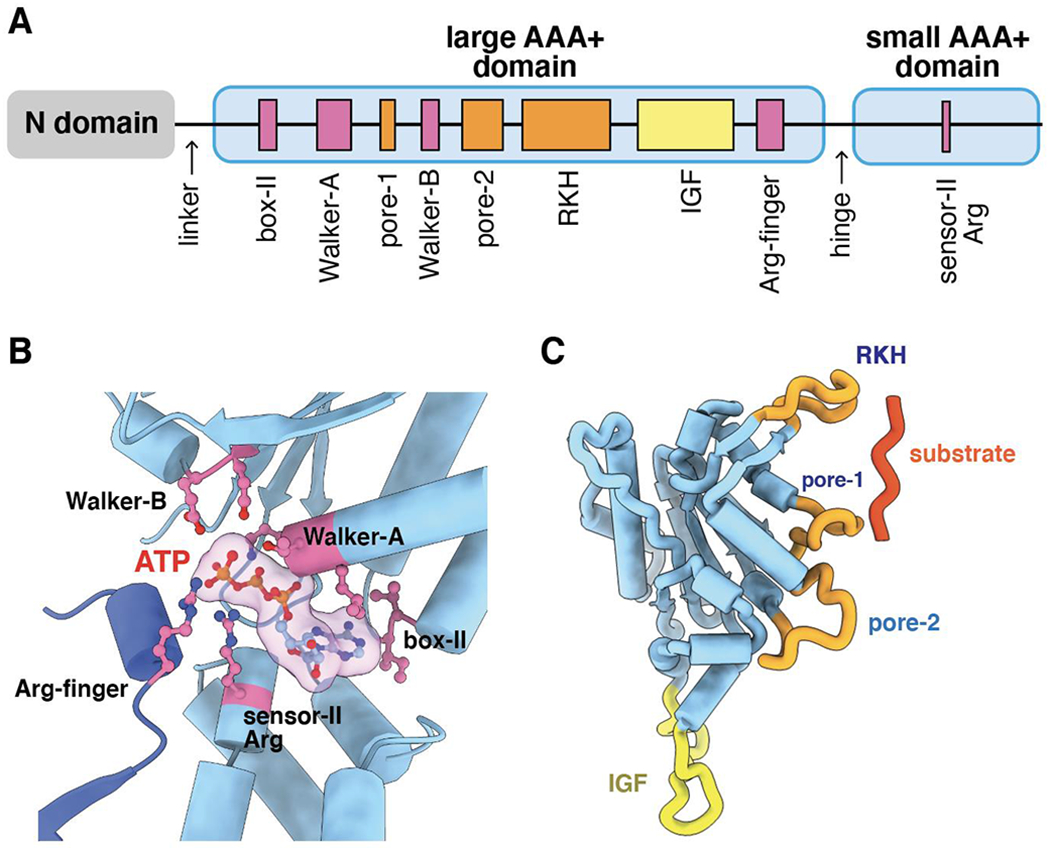
(A) Cartoon depiction of the domain structure and important sequence motifs in a ClpX subunit. (B) ATP (transparent surface) is contacted by box-II, Walker-A, Walker-B, and sensor-II residues from one subunit (light blue) and by the arginine finger of the neighboring subunit (darker blue). (C) Substrate and ClpP binding loops in a large AAA+ domain of a ClpX subunit.
As observed in most AAA+ enzymes (Neuwald et al., 1999), ATP binds in a crevice between the large and small domains of each ClpX subunit, with important nucleotide contacts mediated by box-II, Walker-A, Walker-B, and sensor-II residues of that subunit and by the arginine finger of the adjacent subunit (Figure 2B). As expected, mutations in these sequence motifs eliminate or dramatically slow ATP hydrolysis by ClpX with concomitant deleterious effects on ClpXP degradation (Hersch et al., 2005; Stinson et al., 2013; Fei et al., 2020a). The large AAA+ domain also contains pore-1, pore-2, and RKH loops that line the axial channel of the hexameric ring (Figures 2A, 2C). Mutation of these pore sequences prevents or weakens substrate binding and can alter substrate specificity (Siddiqui et al., 2004; Farrell et al., 2007; Martin et al., 2007; 2008a; Iosefson et al., 2015a). The RKH loop is unique to the ClpX family, whereas pore-1 and pore-2 elements are common among AAA+ proteases and protein-remodeling machines. The large AAA+ domain of ClpX also harbors an IGF loop (Figures 2A, 2C) that mediates docking with ClpP (Kim et al., 2001), as discussed below. Deletion of this loop prevents ClpP binding without altering ClpX’s ability to unfold protein substrates (Joshi et al., 2004). In many bacteria, ClpP also partners with the double-ring ClpA or ClpC AAA+ enzymes, which have IGF-like loops, to allow ClpAP or ClpCP degradation of a diverse set of target proteins (Sauer and Baker, 2011).
Insights from crystal and cryo-EM structures
Crystal structures of ClpXΔN in monomeric and hexameric forms provided information about the folds of the large and small AAA+ domains and revealed that the large AAA+ domain of one subunit packs against the neighboring small domain in a rigid-body fashion (Kim and Kim, 2003; Glynn et al., 2009; Stinson et al., 2013). The pore-1, pore-2, RKH, and IGF loops were typically disordered in these structures, however, and two subunits of the ClpX hexamer did not bind nucleotide, as a consequence of a large rotation between the large and small AAA+ domains. Although these structures must represent low-energy conformations, they appear to be functionally irrelevant. For example, as discussed below, all six subunits of a ClpX hexamer are capable of binding nucleotide. In addition, crystal structures of single-chain ClpX hexamers (Glynn et al. 2009; Stinson et al., 2013) appear incapable of binding ClpP well, as their IGF loops would be inappropriately spaced to bind multiple docking clefts on the surface of a ClpP heptamer (see below).
Eight cryo-EM structures of E. coli or Neisseria meningitidis ClpXΔN bound to substrate and ClpP have recently been solved at near atomic resolution (Fei et al., 2020a; 2020b; Ripstein et al., 2020b). A lower-resolution cryo-EM structure of full-length Listeria monocytogenes ClpX bound to ClpP is also available (Gatsogiannis et al., 2019). In the high-resolution structures, the six subunits of the ClpXΔN hexamer form a shallow spiral (Figures 3A–3C). We label these subunits ABCDEF from the top to the bottom of the spiral (Figure 3C). Strict spiral or helical symmetry would generate an open lock-washer structure, whereas closure of the ClpX ring is mediated by modest changes in packing interactions between F/A or A/B subunits depending on the structure.
Figure 3.
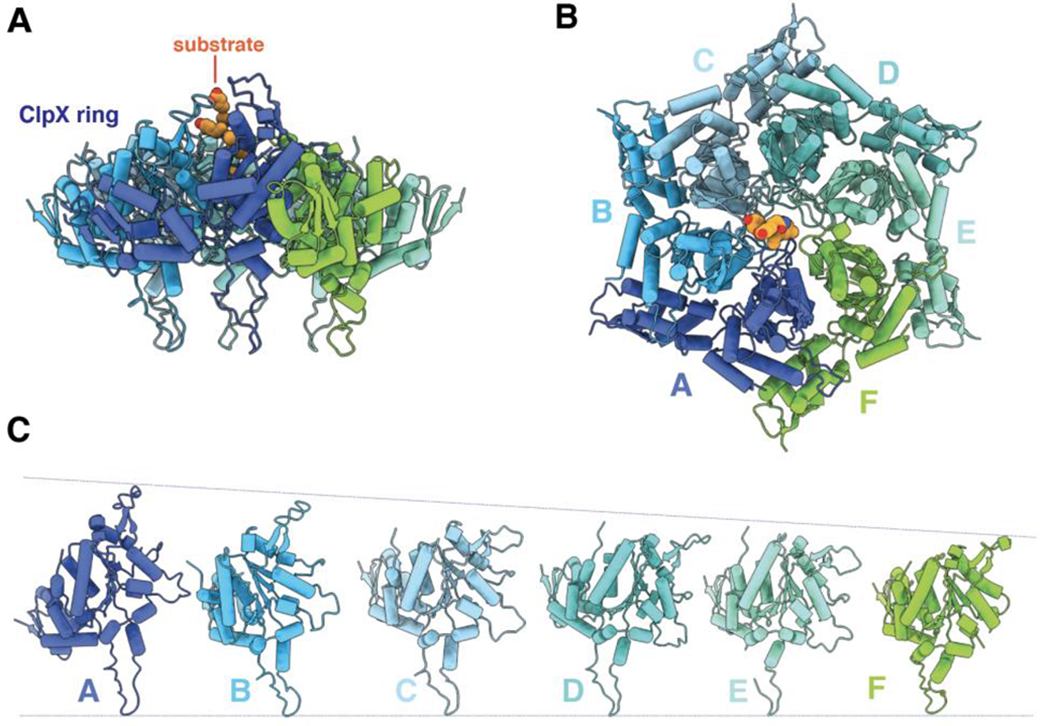
(A) Side view of ClpX hexamer (pdb code 6WRF) in cartoon representation. Substrate is colored orange and shown in sphere representation. (B) Top view of 6WRF ClpX hexamer. (C) Diagram showing the relative vertical positions of subunits in the 6WRF ClpX spiral.
In high-resolution cryo-EM structures, ClpX subunits B/C/D always bind ATP or ATPγS, whereas subunits A/E/F sometimes bind ADP or lack nucleotide (Figure 4A). Some high-resolution studies used hydrolytically impaired variants of ClpX and ATPγS (a slowly hydrolyzed ATP analog). The ADP observed in these structures likely originated from direct binding of ADP, an impurity in solutions of ATPγS, rather than hydrolysis (Fei et al., 2020a). These observations indicate that the hexamer is most stable with ADP bound at only certain spiral positions. Although ATP hydrolysis might only occur in subunits A/E/F, it is also possible that hydrolysis in subunits B/C/D rapidly moves them into an A/E/F position, where ADP is more stably accommodated.
Figure 4.
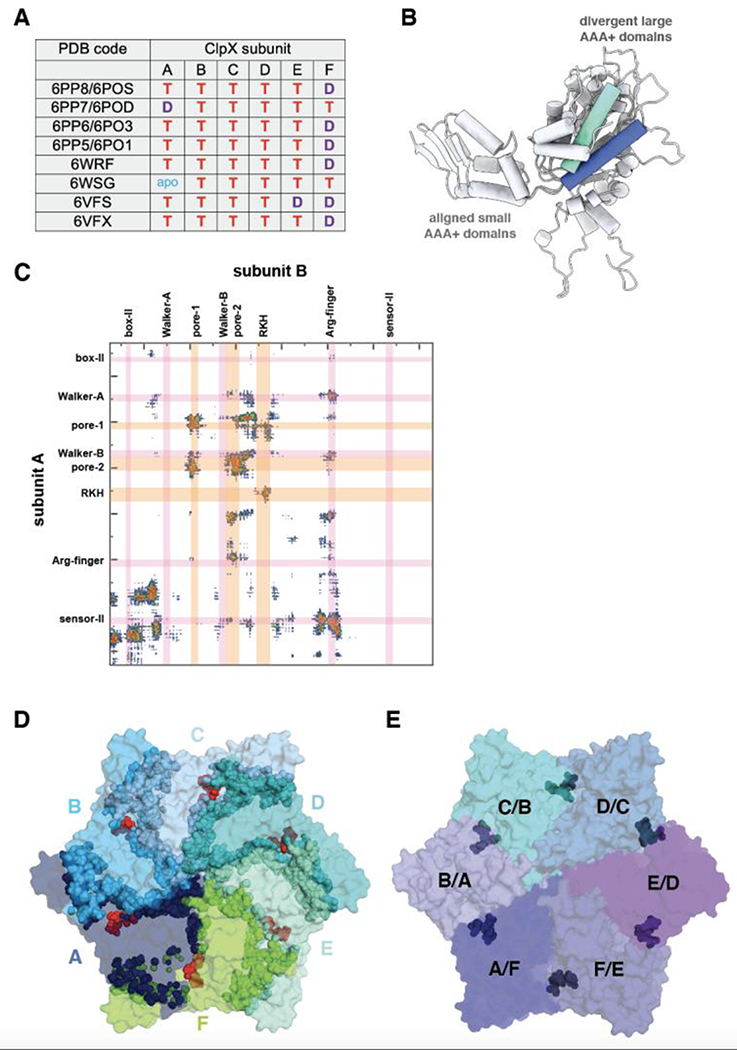
(A) Nucleotides bound in different subunits of ClpX hexamers (T = ATP/ATPγS; D = ADP; apo = nucleotide free). Subunit names in structure 6VFS were changed to be consistent with the other structures. (B) Cartoon representation of ClpX subunits A and E from structure 6WRF after aligning their small AAA+ domains. A helix consisting of residues 83-100 is colored blue in subunit A and aquamarine in subunit E. (C) Contact map prepared using the https://www.molnac.unisa.it/BioTools/cocomaps/ website for ClpX subunits A & B (pdb code 6WRF). The positions of sequence motifs involved in nucleotide binding/hydrolysis and substrate binding are shown. (D) Top view of ClpX hexamer (pdb code 6WRF) in surface representation with positions of subunit-subunit contacts indicated by spheres in the same color as the subunit surface and positions of nucleotide shown as red spheres. (E) Same view as in panel D, except the surface is colored by rigid-body units (residues 62-314 of one subunit and residues 318-412 of the counter-clockwise subunit) and the hinges (residues 315-318) between the large and small AAA+ subunits of individual subunits are shown as dark spheres.
Mechanical unfolding and translocation of substrate proteins requires major structural changes in the ClpX ring, which appear to arise as a consequence of reorientation of the large and small AAA+ domains of individual subunits. The domain-domain orientation within AAA+ modules is generally similar for subunits B/C/D/E/F but can be quite distinct for subunit A. For example, Figure 4B shows two of the most divergent orientations of the large AAA+ domain of subunit A, after aligning its small AAA+ domains. The ClpX ring, which functions as an integrated unit, is stabilized by interactions between neighboring large AAA+ domains and by packing of the small AAA+ domain against the adjacent large domain (Figure 4C, 4D). These subunit-subunit interaction interfaces include parts of the substrate-binding loops and the nucleotide-binding motifs (Figure 4C), providing a structural basis for coordinating ATP hydrolysis with substrate unfolding and translocation. Each small AAA+ domain in the ClpX ring interacts with the neighboring large domain in a very similar manner, although a small variation occurs in the subunits that connect the top and bottom of the spiral as a consequence of flexing of a residue 65-114 subdomain. Thus, the functional ring can be viewed as six rigid-body units, comprising neighboring large and small AAA+ domains, connected by short hinges that link the two domains of each AAA+ module (Fig. 4E). Engineered disulfide bonds across the rigid-body interfaces ensure a topologically closed ClpX ring and do not impair unfolding or degradation (Glynn et al., 2012). Thus, open-ring or lock-washer conformations are not required for ClpX function.
The topological closure of the ClpX ring ensures that altering the conformation of any single hinge would change the conformations of hinges in flanking subunits, which in turn could generate a concerted power stroke involving all six subunits of the hexamer. Moreover, as residues from both AAA+ domains of each ClpX subunit contact nucleotide, ATP binding, hydrolysis, or product release could change the local hinge conformation, providing a mechanism to initiate and then propagate the large structural changes needed for coordinated machine function. The importance of the hinges is highlighted by studies showing that small changes in their length or deletion of a single hinge reduces ClpX function to nearly undetectable levels (Glynn et al., 2012; Bell et al., 2018). As discussed later, differences between ClpX rings observed in individual cryo-EM structures also suggest conformational changes that underlie the molecular mechanisms of protein unfolding and translocation.
ClpP structure and interaction with ClpX
Although often viewed as a passive partner of ClpX, ClpP plays an essential role in energy-dependent ClpXP degradation and is an attractive antibacterial drug target. The two axially stacked heptameric rings of ClpP enclose a degradation chamber with Ser-His-Asp catalytic triads, which can be inactivated by classical serine-protease inhibitors or newly developed small-molecule drugs (Maurizi et al., 1990; Kessel et al., 1995; Wang et al., 1997; Zeiler et al., 2012; Bhandari et al., 2018; Moreno-Cinos et al., 2019).
As anticipated from biochemical and mutational experiments, cryo-EM structures show that docking of flexible IGF loops of ClpX into clefts or pockets between subunits on the ClpP ring accommodate the geometric mismatches between a spiral ClpX hexamer and a flat ClpP heptameric ring (Figures 5A–5C; Kim et al., 2001; Singh et al., 2001; Amor et al., 2019; Gatsogiannis et al., 2019; Fei et al., 2020a; Ripstein et al., 2020b). An Ile-Gly-Phe (IGF) tripeptide at the tip of each loop extends deeply into a ClpP cleft (Figure 5B), and other residues flanking the IGF tripeptide make additional important hydrophobic and polar contacts with ClpP. These interactions are very similar for all docked IGF loops, but the remaining portions of the loops adopt a wide variety of structures in different subunits of the ClpX ring (Figure 5C). This IGF-loop flexibility allows ClpP to remain tightly bound as ClpX adopts conformations that can vary by as much as 15 Å during its ATP-fueled mechanical cycle. Although the mutlivalent interactions between IGF-loops and ClpP clefts mediate high-affinity binding, individual loop-cleft interactions are highly dynamic and can break and reform on a time scale of seconds or faster (Amor et al., 2016).
Figure 5.
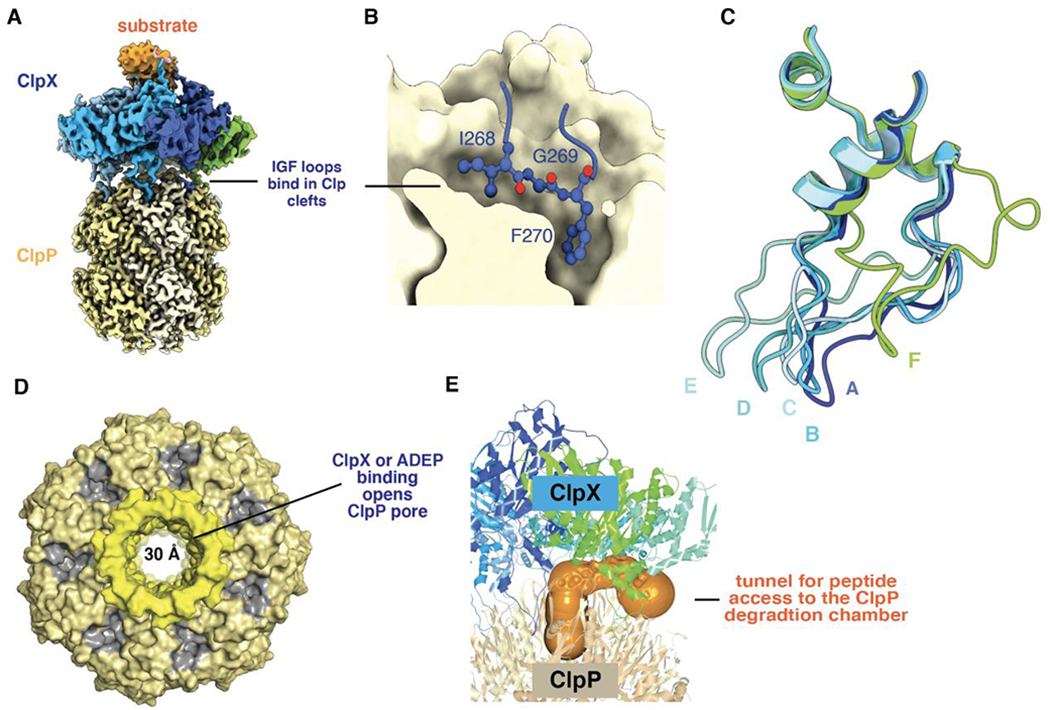
(A) Composite cryo-EM structure of ClpX bound to ClpP and protein substrate (pdb codes 6PPE and 6PP6). (B) An IGF sequence of ClpX binds deeply in a ClpP cleft. (C) IGF loops from aligned ClpX subunits (pdb code 6WRF) adopt a wide variety of conformations with respect to ClpP. This loop flexibility allows ClpX and ClpP to remain stably bound as ClpX adopts different conformations during its ATP-fueled mechanical cycle. (D) Axial view of ClpP (pdb code 6PPE) bound to ClpX (not shown) showing an open axial pore, the clefts that serve as docking sites for the IGF loops of ClpX or ADEPs (colored gray), and the collar of β hairpins that surround the axial pore (colored lighter yellow). (E) The orange surface shows a tunnel, calculated using CAVER (Pavelka et al., 2016), which may allow peptides to enter and leave the ClpP degradation chamber by passing between neighboring IGF loops of ClpX.
In most high-resolution cryo-EM structures, six ClpX IGF loops are docked into six ClpP clefts, leaving one cleft empty (Fei et al., 2020a; 2020b; Ripstein et al., 2020b). In one structure, however, there was good density for only five IGF loops, leaving two adjacent empty clefts. This finding, together with a strictly sequential model of function et al. (discussed below) led to the proposal that the ClpX ring rotates with respect to the ClpP ring during proteolysis (Ripstein et al., 2020b). An analogous rotation mechanism has been proposed for the ClpA and ClpP rings in ClpAP (Lopez et al., 2020). However, when rotation of ClpX or ClpA with respect to ClpP is prevented by crosslinking, both ClpXP and ClpAP remain active in degradation, albeit with reduced proteolytic velocities (Bell, 2020; Kim et al., 2020). Although these crosslinking results do not strictly exclude the possibility of rotation, direct single-molecule experiments demonstrating rotation will be needed to substantiate this intriguing model.
In the absence of ClpX, each ClpP ring has a narrow axial pore that restricts entry of large peptides into the degradation chamber (Wang et al., 1997). Notably, ClpX binding restructures the N-terminal β hairpins that form the ClpP pore, opening this aperture to a width of ~30 Å (Figure 5D; Fei et al., 2020a; Ripstein et al., 2020b). This pore opening allows polypeptides translocated though the ClpX channel to enter the degradation chamber. When ClpX binds only one ring of ClpP, the axial pore of the unbound ClpP ring remains closed, demonstrating that the two rings are not allosterically coupled with respect to pore opening (Fei et al., 2020b). Early EM studies of doubly capped ClpXP complexes showed that simultaneous translocation from both ClpX rings is rare (Ortega et al., 2002). This observation was interpreted to mean that substrate unfolding and not translocation is rate limiting but also raised the possibility of negatively cooperative interactions between the two ClpP rings. If such negative cooperativity is operative, its structural basis is unknown.
ClpP alone degrades peptides with 10 or more amino acids very slowly, but ClpXP degrades these peptides ~10-fold faster without requiring ATP hydrolysis (Lee et al., 2010a). The ability of ClpX to stimulate peptide degradation by ClpP depends on its ability to open the ClpP pore. It is often assumed that such peptides pass through the ClpX channel and then through the ClpP pore, but, as we discuss below, the channel is likely to be closed in the absence of a translocating protein substrate. Moreover, the rate of decapeptide degradation by ClpXP barely slows when saturating concentrations of an ssrA-tagged protein substrate are also present, suggesting that these peptides do not traverse the ClpX channel (T. Bell and R. Sauer, unpublished results). Openings between the upper parts of the IGF loops of ClpX, especially those spanning the empty ClpP cleft, provide a likely path for such peptides to directly enter the open pore of ClpP without passing through the ClpX channel (Figure 5E), which is likely to be closed. This route into the degradation chamber would be even more accessible when only five IGF loops were stably docked. Microscopic reversibility also makes it feasible that the peptide products of degradation, which are typically 5-20 residues in length (Tremblay et al., 2020; Mawla et al., 2021), exit the ClpP chamber by the same route. Indeed, the 30-Å diameter of the open ClpP pore is wide enough to accommodate an incoming translocating polypeptide from ClpX at the same time that outgoing peptide products exit. An alternative model posits that structural fluctuations at the equatorial ring-ring interface of ClpP create transient windows that allow release of peptide products (Sprangers et al., 2005).
In a fascinating illustration of biological mimicry, acyldepsipeptides (ADEPs), which are synthesized by some bacteria, bind in the same clefts on the ClpP ring that are the docking sites for the ClpX IGF loops, open the axial pores into the degradation chamber, and thereby enhance ClpP proteolysis of unstructured polypeptides (Brötz-Oesterhelt et al., 2005; Kirstein et al., 2009; Lee et al., 2010b; Li et al., 2010). In addition to competitively blocking binding of ClpX or other partner enzymes, ADEPs cause rapid dissociation of ClpXP complexes (Amor et al., 2016). The ability of ADEPs to kill many bacteria appears to depend on hyperactivated rogue degradation of cellular proteins (Brötz-Oesterhelt et al., 2005; Kirstein et al., 2009), but killing of M. tuberculosis results from inhibition of ClpXP or ClpCP function (Famulla et al., 2016).
Some structures of ClpP show an axially compressed and inactive conformation, but whether these structures are populated during ClpXP degradation is unknown (Kimber et al., 2010; Zhang et al., 2011; Liu et al., 2014; Ripstein et al., 2020a). A compressed ClpP structure has yet to be observed in complex with ClpX or ADEPs. For human ClpP in the absence of ClpX, individual heptameric rings are stable (Kang et al., 2005). These heptamers have an exposed degradation chamber but are inactive as a consequence of conformational changes in their catalytic triads, thereby preventing uncontrolled degradation of cellular proteins prior to assembly of active tetradecamers. Most structures of ClpP tetradecamers from other species resemble the E. coli enzyme. However, some bacteria encode multiple ClpP genes, allowing for tetradecamers with two distinct heptameric ClpP rings (Kahne and Darwin, 2021). For example, M. tuberculosis ClpP consists of ClpP1 and ClpP2 heptameric rings, only one of which binds ADEPs (Schmitz et al., 2014; Li et al., 2016). Determining the biological significance of these mixed-ring ClpP enzymes is an area of active interest.
Degron recognition
To be degraded by ClpXP, a protein must contain an unstructured peptide, called a degron or degradation tag, that is initially engaged in the axial channel or pore of the ClpX ring. For example, E. coli ClpXP degrades proteins containing C-terminal tags ending in Ala-Ala, as well as additional classes of C- and N-terminal degrons (Gottesman et al., 1998; Flynn et al., 2003). The E. coli ssrA tag (-Ala-Ala-Asn-Asp-Glu-Asn-Tyr-Ala-Leu-Ala-Ala-COO−), which is added to partially synthesized proteins during tmRNA-mediated ribosome rescue (Keiler et al., 1995; Roche and Sauer, 1999), is the best studied ClpX degron. Appending the ssrA tag to proteins, like green fluorescent protein (GFP), which are not natural substrates, renders them susceptible to ClpXP degradation (Gottesman et al., 1998; Kim et al., 2000; Singh et al., 2000; Burton et al., 2001; Lee et al., 2001; Kenniston et al., 2003; 2004).
Biochemical and genetic experiments show that the terminal -Tyr-Ala-Leu-Ala-Ala-COO− residues of the ssrA tag are the critical determinants of ClpX recognition (Flynn et al., 2001). A cryo-EM structure reveals how ClpX initially binds this sequence (Fei et al., 2020b). In this recognition complex, the C-terminal portion of the ssrA tag is bound in the upper part of the axial channel of the ClpX ring, with the pore-2 loop of subunit A blocking access to the lower channel (Figure 6A). Contacts with the -Ala-Ala-COO− and nearby tag residues are made by Thr199 and Val202 from this ClpX pore-2 loop, by Tyr153 and Val154 from the pore-1 loops of subunits A and B, and by Arg228 and His230 from multiple RKH loops. These results provide a gratifying structural explanation for the importance of the pore-1, pore-2, and RKH loops of ClpX in ssrA-tag recognition (Siddiqui et al., 2004; Farrell et al., 2007; Martin et al., 2007; 2008a; Iosefson et al., 2015a; Fei et al., 2020b). For example, human ClpX has leucines at positions corresponding to Thr199 and His230 in E. coli ClpX and fails to recognize ssrA-tagged substrates, but a human variant containing pore-2 and RKH loops from the E. coli enzyme acquires this recognition activity (Martin et al., 2008a). Although it has yet to be shown that the axial channel of ClpX is also closed prior to degron binding, this model seems likely as it provides a mechanism to prevent promiscuous binding of many different peptide sequences in an open channel, thereby accounting for the high degree of specificity that ClpXP exhibits with respect to substrate choice.
Figure 6.
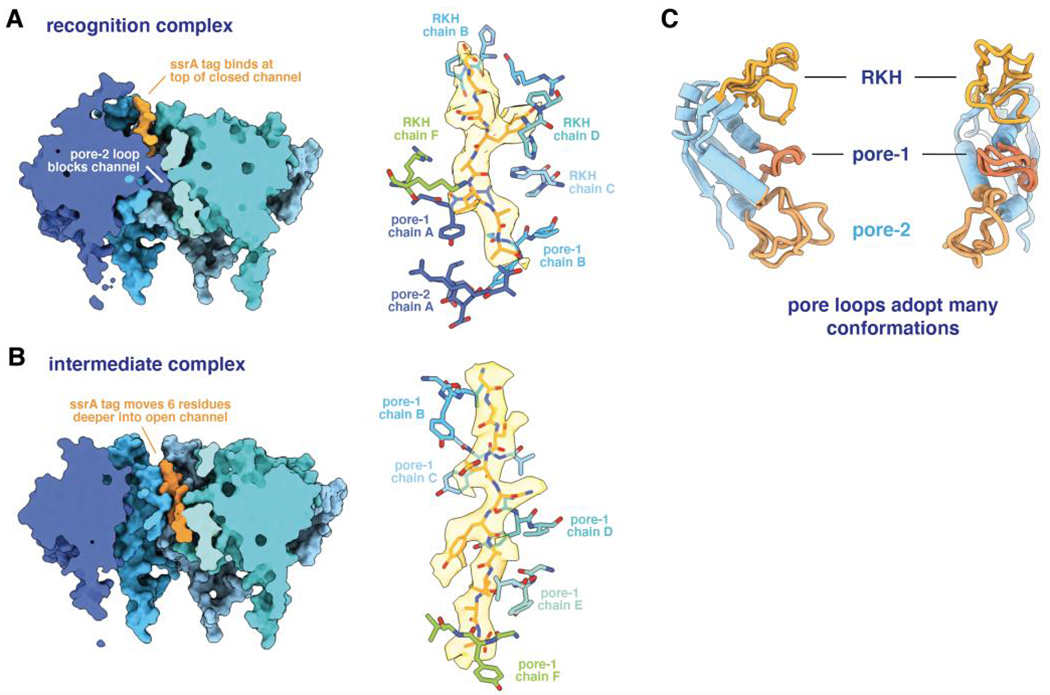
(A) The left panel shows a cutaway view of the ssrA tag in the recognition complex (pdb code 6WRF), emphasizing the blocked axial channel. The right panel shows the ssrA tag in this complex in stick representation with semi-transparent density, and emphasizes key contacts with specific pore-1, pore-2, or RKH loops of ClpX. (B) The left panel shows a cutaway view of the ssrA tag in the intermediate complex (pdb code 6WSG), highlighting the open ClpX channel and movement of the degron deeper into the axial channel. The right panel shows that five pore-1 loops of ClpX interact with the tag with a two-residue periodicity. (C) The pore loops of ClpX can adopt multiple conformations, facilitating flexible interactions with substrate peptides.
For some substrates, degron recognition is enhanced and KM for degradation is lowered by secondary peptide signals that bind the N-domain or other non-pore regions of ClpX directly or via adaptor proteins (Levchenko et al., 2000; Baker and Sauer, 2012; Mahmoud and Chien, 2018). These auxiliary peptide interactions tether the substrate to ClpX and increase the effective concentration of the primary degron with respect to its binding site in the axial channel (McGinness et al., 2007). Determining structures that illuminate how ClpX recognizes different classes of degrons and how adaptors improve substrate recognition is a future challenge.
Structures of translocation complexes
The ssrA tag binds in a closed ClpX channel in the recognition complex, whereas processive translocation and subsequent ClpXP degradation requires an open channel. Insight into the first step in this transition is provided by an ‘intermediate complex’ in which the ssrA tag moves six residues deeper into an open ClpX channel (Figure 6B; Fei et al., 2020b). In this structure, the tag interacts with a two amino-acid periodicity with five ClpX pore-1 loops (Figure 6B), as well as with additional pore-2 and RKH loops. Similar ClpX-substrate interactions are observed in multiple structures (Fei et al., 2020a; Ripstein et al., 2020b), although the peptide side chains in these structures were not modeled, presumably because the density represents multiple peptide species or a single sequence bound in multiple overlapping registers. A two-residue interaction periodicity is also frequently observed in structures of different AAA+ proteases and protein-remodeling machines with substrates (Puchades et al., 2020, and references therein). These open-channel complexes appear to represent snapshots of translocation complexes.
Contacts between ClpX and the ssrA tag in the intermediate complex are largely independent of the peptide sequence. This structural finding was foreshadowed by biochemical studies showing that ClpXP translocation has little sequence specificity (Barkow et al., 2009). For example, ClpXP can translocate peptides with D-amino acids, peptides with as many as nine additional methylene groups between successive peptide bonds, and peptides with polymeric Arg, Lys, Glu, Gln, Gly, or Pro sequences. Thus, degron recognition is highly sequence specific, whereas subsequent translocation is largely sequence independent. The ability of the RKH, pore-1, and pore-2 loops to adopt many different conformations (Figure 6C) allows ClpX to accommodate changes in side-chain size and polarity in substrate sequences and thus efficiently translocate any unfolded protein into ClpP for degradation.
Impressively, ClpXP degrades disulfide bonded, branched, and knotted substrates, a feat that requires simultaneous translocation of two or three polypeptides through the axial channel (Burton et al., 2001; Bolon et al., 2004; Kenniston et al., 2004; San Martín et al., 2017; Sivertsson et al., 2019; Sriramoju et al., 2020). How this action is accomplished structurally is unclear, but a model in which the ClpX ring cracks open to accommodate multiple chains is unlikely (Glynn et al., 2012). A narrow constriction point in the channel in cryo-EM structures would need to open to accommodate one or more additional peptide strands (Bell et al., 2019; Fei et al., 2020a). Channel expansion might happen by local unfolding of sequences adjacent to one or more of the hinges, as inserting a Gly12 sequence into one hinge in single-chain ClpX allows substantial degradation of some protein substrates (Bell et al., 2018).
Unfolding models and degron length
Degron length dictates whether ClpXP can unfold a protein directly from an initial recognition complex or requires some number of translocation steps to pull the native portion of the substrate against the ClpX ring and thereby generate a mechanical unfolding force. For example, the structure of the recognition complex suggested that degrons as short as five residues would suffice for ClpXP unfolding and degradation (Fei et al., 2020b). When GFP substrates with the C-terminal three, five, seven, nine, or eleven amino acids of the ssrA tag were tested, ClpXP degraded GFP-yalaa and variants with longer tags but did not degrade GFP-laa, whose degron is too short to form the recognition complex (Figure 7A). GFP is a very stable protein, which exhibits spontaneous denaturation on a timescale of years (Kim et al., 2000). Thus, these results indicate that ClpX has a powerful unfolding power stroke, which can initiate from the closed-channel recognition complex of short-degron substrates.
Figure 7.
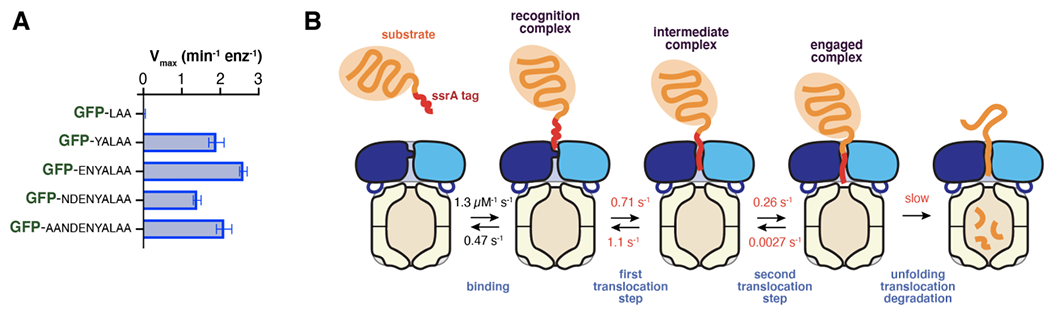
(A) Maximal rates of ClpXP degradation of GFP variants with degradation tags derived from the C-terminal 3, 5, 7, 9, or 11 amino acids of the ssrA tag (Fei et al., 2020b). (B) Multistep model for degradation of substrates with degrons of ~20 residues. The rate constants shown were determined for a slowly degraded titinI27 substrate (Saunders et al., 2020). Rate constants colored red become substantially smaller when ATPγS is substituted for ATP.
Unlike GFP-yalaa, a longer-degron substrate such as GFP-g3yg9senyalaa (the substrate in the recognition and intermediate complexes) would require extra translocation steps following recognition to pull the native GFP β-barrel against the ClpX ring as a prelude to unfolding from an open-channel structure (Figure 7B). The cryo-EM structures of the recognition and intermediate complexes of ClpXP with GFP-g3yg9senyalaa were determined from images collected in a single experiment in which the enzyme and substrate were rapidly mixed and vitrified (Fei et al., 2020b). A low-resolution structure of a fully engaged complex in which the native portion of GFP is pulled against the top of the ClpX ring was recently identified within this dataset, supporting progression from the recognition to the intermediate to the engaged complex as a prelude to unfolding GFP-g3yg9senyalaa (X. Fei, E. Zhong, R. Sauer, and J. Davis, in preparation).
Biophysical studies confirm that ClpXP binding and engagement of a substrate with a longer degron is a multistep reaction. Specifically, the polyphasic kinetics of association and dissociation between ClpXP and a titinI27 domain bearing a 20-residue ssrA degron provide strong support for the model of Figure 7B (Saunders et al., 2020). Indeed, the rate constants determined by global fitting for individual steps in this model generate kinetic trajectories that match the experimental data extremely well over a wide range of substrate concentrations. Several findings emerge from these studies. First, all steps are reversible. Following initial binding, direct progress to the engaged complex is only about 20% as probable as dissociation. Prior studies using multidomain substrates show that ClpXP often dissociates after unfolding and degrading only one or two native domains, directly demonstrating that ClpXP can dissociate following substrate engagement (Lee et al., 2001; Kenniston et al., 2005). In the kinetic studies, the rates of the binding and dissociation steps are independent of the rate of ATP hydrolysis, whereas subsequent forward and reverse kinetic steps slow as the rate of ATP hydrolysis slows (Saunders et al., 2020). The hydrolysis dependence of the latter steps is expected if these steps are linked to power strokes. An intriguing possibility raised by these studies is that degradation of proteins with terminal peptide sequences that bind poorly in the ClpX channel is further minimized by kinetic proof reading.
The importance of substrate grip during unfolding
Although polypeptide translocation by ClpXP has little sequence dependence, unfolding can fail in a fashion that depends on sequence. For example, sequences rich in glycine immediately adjacent to a native domain prevent or greatly reduce the rate of ClpXP unfolding and degradation (Kraut, 2013; Too et al., 2013; Vass and Chen, 2013). In fact, GFP-g13senyalaa (Figure 8A) with a continuous stretch of thirteen glycines is not degraded by ClpXP, but introducing single Tyr→Gly substitutions at certain positions of the poly-glycine region rescues degradation, with a substitution at position 4 (GFP-G3YG9SENYALAA) having the largest effect (Figure 8B; Bell et al., 2019).
Figure 8.
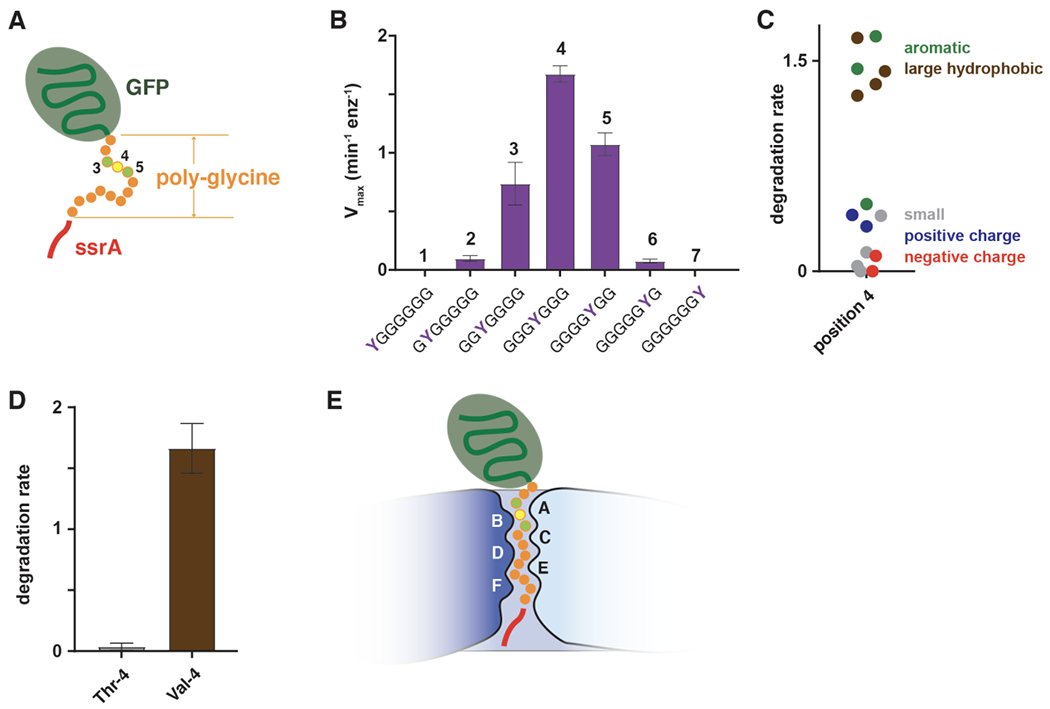
(A) GFP-g13senyalaa contains 13 glycines between the native barrel of GFP and a partial ssrA degron and is not degraded by ClpXP. (B) Effects of tyrosine substitutions at the N-terminal seven positions of the poly-glycine region of GFP-g13senyalaa on the maximal rate of ClpXP degradation (Bell et al., 2019). (C) Effects of position-4 side chain properties on ClpXP degradation (Bell et al., 2019). (D) Degradation of variants with threonine or valine at position 4 of the polyglycine region (Bell et al., 2019). (E) During unfolding, degron side chains 3-5 amino acids from the native domain of a substrate are the most important determinants of grip and are positioned to interact with the pore-1 loops of subunits A and B, near the top of the ClpX channel.
Other postion-4 residues with aromatic or large hydrophobic side chains also support robust ClpXP degradation, whereas those with small, polar, and charged side chains result in lower degradation rates (Figure 8C). The deleterious effect of polar side chains is illustrated dramatically by the isosteric Thr/Val pair, in which substituting the hydroxyl group of threonine with the methyl group of valine results in a 17-fold increase in the degradation rate (Figure 8D; Bell et al., 2019). Single tyrosines at tag positions 3 and 5 also support substantial ClpXP degradation but the effect falls off sharply at neighboring positions (Figure 8B), indicating that the distance of the tyrosine from the native domain is an important determinant of grip. Based on structural considerations, degron positions 3-5 in a fully engaged complex would interact with the pore-1 loops of chains A and B in the ClpX spiral (Figure 8E). Notably, these same pore-1 loops form the narrowest point in the ClpX axial channel and also interact with ssrA tag residues in the recognition complex (Fei et al., 2020a; 2020b). Thus, ClpX contacts with a substrate near the top of the channel are critical for the unfolding of both short- and long-degron substrates.
During open-channel unfolding in optical-trap experiments, multiple pore-1 loops synergistically maintain grip on substrates when challenged by opposing forces (Iosefson et al., 2015a; 2015b; Rodriguez-Aliaga et al., 2016). Grip is likely to be force dependent and may also come into play during translocation. For example, ClpXP can unfold and translocate substrates containing long proline tracts in the absence of opposing force but stalls at these sequences at forces of 10-20 pN in optical trapping experiments (O. Iosefson and R. Sauer, in preparation).
Effects of protein stability on unfolding and degradation
In biochemical studies, decreasing the stability of ssrA-tagged titinI27 variants increases the rate at which they are degraded by ClpXP and reduces the ATP cost (Kenniston et al., 2003). The kinetics of ClpXP degradation of single molecules containing multiple titinI27, filamin-A, GFP, and/or Halo domains have also been studied by optical trapping, which allows direct observation of unfolding and translocation events (Aubin-Tam et al., 2011; Maillard et al., 2011; Sen et al., 2013; Cordova et al., 2014; Iosefson et al., 2015b; Rodriguez-Aliaga et al., 2016; Olivares et al., 2017). Major conclusions from these studies include: (i) most power strokes fail to unfold stable substrates, and hundreds of ATPs can be hydrolyzed before unfolding occurs; (ii) the average time required for ClpXP to unfold a domain decreases as the mechanical stability of the substrate decreases; (iii) successful unfolding typically results from a single power stroke and is complete in less than 1 millisecond; (iv) resistance to unfolding can change markedly depending on degron location and whether ClpXP pulls from the N-terminus or C-terminus of a native domain; (v) ClpXP translocates a wide variety of different peptide sequences at comparable rates, with only occasional pauses or stalling events; (vi) a single power stroke translocates 5-8 amino acids of the substrate through the axial channel, often in kinetic bursts involving multiple steps; and (vii) each power stroke can perform ~5 kT of mechanical work, corresponding to ~50% recovery of the free energy of ATP hydrolysis.
The effect of the pulling direction on the resistance of a protein to ClpXP unfolding can be dramatic. For example, unfolding is the slow step in ClpXP degradation of titinI27 with a C-terminal degron but is ~60-fold faster for titinI27 with an N-terminal degron, resulting in translocation becoming the rate-determining step in degradation (Olivares et al., 2017). It is not known if effects of this magnitude would be observed for other protein substrates, but the titinI27 experiments suggest that evolution selects for degradation tags at the terminus of a target protein from which ClpXP unfolding is fastest and degradation therefore requires less energy in the form of ATP consumption.
ClpX can function using probabilistic ATP hydrolysis
The hexameric motors of a large number of AAA+ proteases and protein-remodeling machines share similar spiral architectures with pore-1 loops in different subunits interacting with adjacent two-residue segments of substrate peptides in their axial channels (Puchades et al., 2017; 2020; de la Peña et al., 2018; Dong et al. 2019, and references therein). These structural features led to a widely accepted proposal that substrate translocation occurs in two-residue steps, by a mechanism in which each subunit sequentially cycles through each position in the spiral as a consequence of ATP hydrolysis in a special subunit at one position (usually corresponding to subunit E in ClpX). In this model, each ATP-hydrolysis event advances the five highest subunits and substrate down one position in the spiral, hence generating a two-residue translocation step, and shifts the bottom subunit to the top. Below, we review experimental evidence that ClpXP does not operate by this sequential mechanism and discuss an alternative probabilistic model.
A major prediction of the prevailing model is a translocation step of two residues, but optical-trapping experiments indicate that ClpXP translocates substrates in minimum segments of 5 to 7 residues (Aubin-Tam et al., 2011; Maillard et al., 2011; Sen et al., 2013; Cordova et al., 2014). Importantly, this larger step size is strongly supported by two cryo-EM structures of ClpXP bound to GFP-g3yg9senyalaa, in which the degron in the channel is observed at offset positions differing by 6 amino acids (Figure 6; Fei et al., 2020b). As these two structures accounted for most images in this dataset, additional structures with the substrate offset by 2 residues are unlikely to be significantly populated. Proponents of the two-residue translocation model might argue that the six-residue offset of substrate observed in ClpXP structures represents an unfolding step rather than a translocation step, but parsimony supports the idea that the same basic mechanical power stroke drives unfolding and translocation. Moreover, if unfolding and translocation power strokes did differ in length, it would be logical to have a lower-gear and shorter step to maximize force application during unfolding and a longer translocation step to minimize the energetic cost of overall degradation. How might ClpX take a fundamental translocation step of ~6 residues? Differences between the cryo-EM structures of the recognition and translocation complexes suggest that a step of this approximate size could occur if a power stroke moved the top subunit and bound substrate to the bottom of the spiral with concomitant movement of each additional subunit up one position in the spiral (see Video 4 in Fei et al., 2020b; https://elifesciences.org/articles/61496#video4). Notably, this mechanism simply reverses the direction of subunit movement in the 2-residue translocation model and thus ensures that highly populated structural intermediates will share the canonical spiral architecture.
In optical-trapping experiments, some translocation events move ~12, ~18, or ~24 amino acids through the channel, with these longer steps and the fundamental ~6-residue steps occurring randomly rather than in any repeating pattern (Aubin-Tam et al., 2011; Maillard et al., 2011; Sen et al., 2013; Cordova et al., 2014). The longer steps in these experiments are thought to represent kinetic bursts of two, three, or four power strokes. In principle, a six-residue translocation step might also represent a kinetic burst of three very rapid 2-residue steps. However, the kinetics of ATP binding and hydrolysis are substantially too slow for the longest observed ClpXP translocation steps to represent kinetic bursts of twelve power strokes (see Fei et al., 2020a for a more detailed discussion).
Another prediction of the mainstream model is that eliminating ATP hydrolysis in one or more subunits of the ClpX hexamer should prevent machine function when a ‘dead’ subunit moves into the single hydrolytically active position in the spiral. Notably, however, a single-chain ClpX hexamer with just two ATPase-active subunits supports unfolding, translocation, and degradation (Martin et al., 2005; Cordova et al., 2014). Although this hobbled ClpX motor unfolds, translocates, and degrades a destabilized titinI27 variant at one-third the rate of the parental enzyme, it performs these tasks with wild-type thermodynamic efficiency. The ability of the ClpX motor to function with only a subset of hydrolytically active subunits indicates that a power stroke can initiate from ATP hydrolysis in subunits at different positions in the spiral, which implies that the chance of hydrolysis in any given subunit is probabilistic. This probability could in turn be influenced by the detailed interactions of each subunit with substrate. Two additional observations support this idea. First, in cryo-EM structures, the ATP-binding sites in many ClpX subunits appear to be catalytically competent, in the sense that Walker-B, Arg-finger, and sensor-II side chains critical for hydrolysis are properly positioned with respect to bound nucleotide. Second, the closed topology of the ClpX ring would allow hydrolysis in any competent subunit to alter the conformation of the entire ring and drive a power stroke.
Summary
The recent cryo-EM revolution has allowed determination of numerous high-resolution ClpXP structures (Fei et al., 2020a; 2020b; Ripstein et al., 2020b). These structures reveal that the ClpX ring shares a common spiral architecture with many other AAA+ unfoldases and protein-remodeling machines, which are proposed to take two-residue translocation steps and operate by a sequential ATP-hydrolysis mechanism. However, an extensive body of single-molecule and bulk biochemical experiments for ClpXP reveals major conflicts with this model. Allowing ATP hydrolysis to occur at different ClpX spiral positions, with probabilities potentially linked to differences in detailed substrate contacts, accounts for the ability of ClpX to function with only a subset of active subunits. Combining probabilistic hydrolysis with a model in which each power stroke moves substrate from the top to the bottom of the spiral also explains the large translocation step sizes observed in optical trapping experiments. Furthermore, a model of this type accounts for unfolding of GFP with a short-degron tail in a single power stroke and provides a satisfying mechanistic rationale for strong substrate grip by the uppermost pore-1 loops in the spiral. Testing this model for ClpXP and interrogating its generalizability to different AAA+ proteases and protein-remodeling machines through structural biology, biophysics, and biochemistry remains an important and exciting future challenge.
Funding
Work in our labs was supported by grants from the NIH (RO1-GM101988; RO1-AI016892; R35-GM141517; 5R01-DK115558; 5T32-GM007287) and by the Howard Hughes Medical Institute.
Footnotes
Disclosure statement
The authors declare no conflict of interest.
References
- Amor AJ, Schmitz KR, Baker TA, Sauer RT. 2019. Roles of the ClpX IGF loops in ClpP association, dissociation, and protein degradation. Protein Sci. 28(4):756–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amor AJ, Schmitz KR, Sello JK, Baker TA, Sauer RT. 2016. Highly dynamic interactions maintain kinetic stability of the ClpXP protease during the ATP-fueled mechanical cycle. ACS Chem Biol. 11(6): 1552–1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aubin-Tam ME, Olivares AO, Sauer RT, Baker TA, Lang MJ. 2011. Single-molecule protein unfolding and translocation by an ATP-fueled proteolytic machine. Cell. 145(2):257–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker TA, Sauer RT. 2006. ATP-dependent proteases of bacteria: recognition logic and operating principles. Trends Biochem Sci. 31(12):647–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker TA, Sauer RT. 2012. ClpXP, an ATP-powered unfolding and protein-degradation machine. Biochim Biophys Acta. 1823(1): 15–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bard JAM, Goodall EA, Greene ER, Jonsson E, Dong KC, Martin A. 2018. Structure and function of the 26S proteasome. Annu Rev Biochem. 87:697–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkow SR, Levchenko I, Baker TA, Sauer RT. 2009. Polypeptide translocation by the AAA+ ClpXP protease machine. Chem Biol. 16(6):605–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell TA, Baker TA, Sauer RT. 2018. Hinge-linker elements in the AAA+ protein unfoldase ClpX mediate intersubunit communication, assembly, and mechanical activity. Biochemistry. 57(49):6787–6796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell TA, Baker TA, Sauer RT. 2019. Interactions between a subset of substrate side chains and AAA+ motor pore loops determine grip during protein unfolding. Elife. 8:e46808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell TA. (2020). Intersubunit communication and coordinated mechanical activity in the AAA+ protease ClpXP. PhD thesis. Massachusetts Institute of Technology, Cambridge, MA. [Google Scholar]
- Bhandari V, Wong KS, Zhou JL, Mabanglo MF, Batey RA, Houry WA. 2018. The role of ClpP protease in bacterial pathogenesis and human diseases. ACS Chem Biol. 13(6): 1413–1425. [DOI] [PubMed] [Google Scholar]
- Bolon DN, Grant RA, Baker TA, Sauer RT. 2004. Nucleotide-dependent substrate handoff from the SspB adaptor to the AAA+ ClpXP protease. Mol Cell. 16(3):343–350. [DOI] [PubMed] [Google Scholar]
- Brötz-Oesterhelt H, Beyer D, Kroll HP, Endermann R, Lade IC, Schroeder W, Hinzen B, Raddatz S, Paulsen H, Henninger K, Bandow JE, Sahl HG, Labischinski H. 2005. Dysregulation of bacterial proteolytic machinery by a new class of antibiotics. Nat Med. 11:1082–1087. [DOI] [PubMed] [Google Scholar]
- Burton BM, Baker TA. 2005. Remodeling protein complexes: insights from the AAA+ unfoldase ClpX and Mu transposase. Protein Sci. 14(8): 1945–1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton RE, Siddiqui SM, Kim YI, Baker TA, Sauer RT. 2001. Effects of protein stability and structure on substrate processing by the ClpXP unfolding and degradation machine. EMBO J. 20(12):3092–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carney D, Schmitz KR, Truong J, Sauer RT, Sello JK. 2014. Restriction of the conformational dynamics of the cyclic acyldepsipeptide macrocycle improves antibacterial activity by enhancing both ClpP peptidase binding and activation. J Amer Chem Soc. 136:1922–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conlon BP, Nakayasu ES, Fleck LE, LaFleur MD, Isabella VM, Coleman K, Leonard SN, Smith RD, Adkins JN, Lewis K. 2013. Activated ClpP kills persisters and eradicates a chronic biofilm infection. Nature. 503:365–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordova JC, Olivares AO, Shin Y, Stinson BM, Calmat S, Schmitz KR, Aubin-Tam ME, Baker TA, Lang MJ, Sauer RT. 2014. Stochastic but highly coordinated protein unfolding and translocation by the ClpXP proteolytic machine. Cell. 158(3):647–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Peña AH, Goodall EA, Gates SN, Lander GC, Martin A. 2018. Substrate-engaged 26S proteasome structures reveal mechanisms for ATP-hydrolysis-driven translocation. Science. 362(6418): eaav0725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaldson LW, Wojtyra U, Houry WA. 2003. Solution structure of the dimeric zinc binding domain of the chaperone ClpX. J Biol Chem. 278(49):48991–48996. [DOI] [PubMed] [Google Scholar]
- Dong Y, Zhang S, Wu Z, Li X, Wang WL, Zhu Y, Stoilova-McPhie S, Lu Y, Finley D, Mao Y. 2019. Cryo-EM structures and dynamics of substrate-engaged human 26S proteasome. Nature. 565(7737):49–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Famulla K, Sass P, Malik I, Akopian T, Kandror O, Alber M, Flinzen B, Ruebsamen-Schaeff H, Kalscheuer R, Goldberg AL, Brötz-Oesterhelt H. 2016. Acyldepsipeptide antibiotics kill mycobacteria by preventing the physiological functions of the ClpP1P2 protease. Mol Microbiol. 101 (2): 194–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrell CM, Baker TA, Sauer RT. 2007. Altered specificity of a AAA+ protease. Mol Cell. 25(1): 161–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fei X, Bell TA, Barkow SR, Baker TA, Sauer RT. 2020b. Structural basis of ClpXP recognition and unfolding of ssrA-tagged substrates. Elife. 9:e61496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fei X, Bell TA, Jenni S, Stinson BM, Baker TA, Harrison SC, Sauer RT. 2020a. Structures of the ATP-fueled ClpXP proteolytic machine bound to protein substrate. Elife. 9:e52774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn JM, Levchenko I, Seide IM, Wickner SH, Sauer RT, Baker TA. 2001. Overlapping recognition determinants within the ssrA degradation tag allow modulation of proteolysis. Proc Natl Acad Sci USA. 98(19): 10584–10589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn JM, Neher SB, Kim YI, Sauer RT, Baker TA. 2003. Proteomic discovery of cellular substrates of the ClpXP protease reveals five classes of ClpX-recognition signals. Mol Cell. 11 (3):671–683. [DOI] [PubMed] [Google Scholar]
- Gatsogiannis C, Balogh D, Merino F, Sieber SA, Raunser S. 2019. Cryo-EM structure of the ClpXP protein degradation machinery. Nat Struct Mol Biol. 26(10):946–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gispert S, Parganlija D, Klinkenberg M, Dröse S, Wittig I, Mittelbronn M, Grzmi IP, Koob S, Hamann A, Walter M, Büche IF, Adler T, Hrabé de Angelis M, Busch DH, Zell A, Reichert AS, Brandt U, Osiewacz HD, Jendrach M, Auburger G. 2013. Loss of mitochondrial peptidase ClpP leads to infertility, hearing loss plus growth retardation via accumulation of CLPX, mtDNA and inflammatory factors. Hum Mol Genet. 22(24):4871–4887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glynn SE, Martin A, Nager AR, Baker TA, Sauer RT. 2009. Structures of asymmetric ClpX hexamers reveal nucleotide-dependent motions in a AAA+ protein-unfolding machine. Cell. 139(4): 744–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glynn SE, Nager AR, Baker TA, Sauer RT. 2012. Dynamic and static components power unfolding in topologically closed rings of a AAA+ proteolytic machine. Nat Struct Mol Biol. 19(6):616–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottesman S, Roche E, Zhou Y, Sauer RT. 1998. The ClpXP and ClpAP proteases degrade proteins with carboxy-terminal peptide tails added by the ssrA-tagging system. Genes Dev. 12(9): 1338–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson PI, Whiteheart SW. 2005. AAA+ proteins: have engine, will work. Nat Rev Mol Cell Biol. 6(7):519–529. [DOI] [PubMed] [Google Scholar]
- Hersch GL, Burton RE, Bolon DN, Baker TA, Sauer RT. 2005. Asymmetric interactions of ATP with the AAA+ ClpX6 unfoldase: allosteric control of a protein machine. Cell. 121 (7): 1017–1027. [DOI] [PubMed] [Google Scholar]
- Iosefson O, Nager AR, Baker TA, Sauer RT. 2015a. Coordinated gripping of substrate by subunits of a AAA+ proteolytic machine. Nat Chem Biol. 11(3):201–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iosefson O, Olivares AO, Baker TA, Sauer RT. 2015b. Dissection of axial-pore loop function during unfolding and translocation by a AAA+ proteolytic machine. Cell Rep. 12(6): 1032–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jena IU, Fuchs T 1998. An essential protease involved in bacterial cell-cycle control. EMBO J. 17(19):5658–5669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi SA, Hersch GL, Baker TA, Sauer RT. 2004. Communication between ClpX and ClpP during substrate processing and degradation. Nat Struct Mol Biol. 11(5):404–411. [DOI] [PubMed] [Google Scholar]
- Kahne SC, Darwin KH. 2021. Structural determinants of regulated proteolysis in pathogenic bacteria by ClpP and the proteasome. Curr Opin Struct Biol. 67:120–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang SG, Dimitrova MN, Ortega J, Ginsburg A, Maurizi MR. 2005. Human mitochondrial ClpP is a stable heptamer that assembles into a tetradecamer in the presence of ClpX. J Biol Chem. 280(42):35424–35432. [DOI] [PubMed] [Google Scholar]
- Kardon JR, Yien YY, Huston NC, Branco DS, Hildick-Smith GJ, Rhee KY, Paw BH, Baker TA. 2015. Mitochondrial ClpX activates a key enzyme for heme biosynthesis and erythropoiesis. Cell. 161(4):858–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keiler KC, Waller PR, Sauer RT. 1996. Role of a peptide tagging system in degradation of proteins synthesized from damaged messenger RNA. Science. 271 (5251 ):990–993. [DOI] [PubMed] [Google Scholar]
- Keiler KC. 2015. Mechanisms of ribosome rescue in bacteria. Nat Rev Microbiol. 13(5):285–297. [DOI] [PubMed] [Google Scholar]
- Kenniston JA, Baker TA, Fernandez JM, Sauer RT. 2003. Linkage between ATP consumption and mechanical unfolding during the protein processing reactions of an AAA+ degradation machine. Cell. 114(4):511–520. [DOI] [PubMed] [Google Scholar]
- Kenniston JA, Baker TA, Sauer RT. 2005. Partitioning between unfolding and release of native domains during ClpXP degradation determines substrate selectivity and partial processing. Proc Natl Acad Sci USA. 102(5): 1390–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenniston JA, Burton RE, Siddiqui SM, Baker TA, Sauer RT. 2004. Effects of local protein stability and the geometric position of the substrate degradation tag on the efficiency of ClpXP denaturation and degradation. J Struct Biol. 146(1–2): 130–140. [DOI] [PubMed] [Google Scholar]
- Kesse IM, Maurizi MR, Kim B, Kocsis E, Trus BL, Singh SK, Steven AC. 1995. Homology in structural organization between E. coli ClpAP protease and the eukaryotic 26 S proteasome. J Mol Biol. 250(5):587–594. [DOI] [PubMed] [Google Scholar]
- Kimber MS, Yu AY, Borg M, Leung E, Chan HS, Houry WA. 2010. Structural and theoretical studies indicate that the cylindrical protease ClpP samples extended and compact conformations. Structure. 18(7):798–808. [DOI] [PubMed] [Google Scholar]
- Kim DY, Kim KK. 2003. Crystal structure of ClpX molecular chaperone from Helicobacter pylori. J Biol Chem. 278(50):50664–50670. [DOI] [PubMed] [Google Scholar]
- Kim S, Zuromski KL, Bell TA, Sauer RT, Baker TA. 2020. ClpAP proteolysis does not require rotation of the ClpA unfoldase relative to ClpP. Elife. 9:e61451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YI, Burton RE, Burton BM, Sauer RT, Baker TA. 2000. Dynamics of substrate denaturation and translocation by the ClpXP degradation machine. Mol Cell. 5(4):639–648. [DOI] [PubMed] [Google Scholar]
- YI Kim, Levchenko I, Fraczkowska K, Woodruff RV, Sauer RT, Baker TA. 2001. Molecular determinants of complex formation between Clp/Hsp100 ATPases and the ClpP peptidase. Nat Struct Biol. 8(3):230–233. [DOI] [PubMed] [Google Scholar]
- Kirstein J, Hoffmann A, Lilie H, Schmidt R, Rübsamen-Waigmann H, Brötz-Oesterhelt H, Mogk A, Turgay K. 2009. The antibiotic ADEP reprogrammes ClpP, switching it from a regulated to an uncontrolled protease. EMBO Mol Med. 1(1 ):37–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konovalova A, Søgaard-Anderse L, Kroos L. 2014. Regulated proteolysis in bacterial development. FEMS Microbiol Rev. 38(3):493–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraut DA. 2013. Slippery substrates impair ATP-dependent protease function by slowing unfolding. J Biol Chem. 288(48):34729–34735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BG, Park EY, Lee KE, Jeon H, Sung KH, Paulsen H, Rübsamen-Schaeff H, Böotz-Oesterhelt H, Song HK. 2010b. Structures of ClpP in complex with acyldepsipeptide antibiotics reveal its activation mechanism. Nat Struct Mol Biol. 17(4):471–478. [DOI] [PubMed] [Google Scholar]
- Lee C, Schwartz MP, Prakash S, Iwakura M, Matouschek A. 2001. ATP-dependent proteases degrade their substrates by processively unraveling them from the degradation signal. Mol Cell. 7(3):627–637. [DOI] [PubMed] [Google Scholar]
- Lee ME, Baker TA, Sauer RT. 2010a. Control of substrate gating and translocation into ClpP by channel residues and ClpX binding. J Mol Biol. 399(5):707–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levchenko I, Seide IM, Sauer RT, Baker TA. 2000. A specificity-enhancing factor for the ClpXP degradation machine. Science. 289(5488):2354–2356. [DOI] [PubMed] [Google Scholar]
- Li DH, Chung YS, Gloyd M, Joseph E, Ghirlando R, Wright GD, Cheng YQ, Maurizi MR, Guarné A, Ortega J. 2010. Acyldepsipeptide antibiotics induce the formation of a structured axial channel in ClpP: A model for the ClpX/ClpA-bound state of ClpP. Chem Biol. 17(9):959–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Kandror O, Akopian T, Dharkar P, Wlodawer A, Maurizi MR, Goldberg AL. 2016. Structure and functional properties of the active form of the proteolytic complex, ClpP1P2, from Mycobacterium tuberculosis. J Biol Chem. 291(14):7465–7476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K, Ologbenla A, Houry WA. 2014. Dynamics of the ClpP serine protease: a model for self-compartmentalized proteases. Crit Rev Biochem Mol Biol. 49(5):400–412. [DOI] [PubMed] [Google Scholar]
- Lopez KE, Rizo AN, Tse E, Lin J, Scull NW, Thwin AC, Lucius AL, Shorter J, Southworth DR. 2020. Conformational plasticity of the ClpAP AAA+ protease couples protein unfolding and proteolysis. Nat Struct Mol Biol. 27(5):406–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo B, Ma Y, Zhou Y, Zhang N, Luo Y. 2021. Human ClpP protease, a promising therapy target for diseases of mitochondrial dysfunction. Drug Discov Today. 15:S1359-6446(21)00034-9. [DOI] [PubMed] [Google Scholar]
- Mahmoud SA, Chien P. 2018. Regulated proteolysis in bacteria. Annu Rev Biochem. 87:677–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maillard RA, Chistol G, Sen M, Righini M, Tan J, Kaiser CM, Hodges C, Martin A, Bustamante C. 2011. ClpX(P) generates mechanical force to unfold and translocate its protein substrates. Cell. 145(3):459–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin A, Baker TA, Sauer RT. 2007. Distinct static and dynamic interactions control ATPase-peptidase communication in a AAA+ protease. Mol Cell. 27(1 ):41–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin A, Baker TA, Sauer RT. 2008a. Diverse pore loops of the AAA+ ClpX machine mediate unassisted and adaptor-dependent recognition of ssrA-tagged substrates. Mol Cell. 29(4):441–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin A, Baker TA, Sauer RT. 2008b. Pore loops of the AAA+ ClpX machine grip substrates to drive translocation and unfolding. Nat Struct Mol Biol. 15(11): 1147–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurizi MR, Clark WP, Katayama Y, Rudikoff S, Pumphrey J, Bowers B, Gottesman S. 1990. Sequence and structure of Clp P, the proteolytic component of the ATP-dependent Clp protease of Escherichia coli. J Biol Chem. 265(21): 12536–12545. [PubMed] [Google Scholar]
- Maurizi MR, Clark WP, Kim SH, Gottesman S. 1990. Clp P represents a unique family of serine proteases. J Biol Chem. 265(21): 12546–12552. [PubMed] [Google Scholar]
- Mawla GD, Hall BM, Cárcamo-Oyarce G, Grant RA, Zhang JJ, Kardon JR, Ribbeck K, Sauer RT, Baker TA. 2021. ClpP1P2 peptidase activity promotes biofilm formation in Pseudomonas aeruginosa. Mol Microbiol. 115(6): 1094–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinness KE, Bolon DN, Kaganovich M, Baker TA, Sauer RT. 2007. Altered tethering of the SspB adaptor to the ClpXP protease causes changes in substrate delivery. J Biol Chem. 282(15): 11465–11473. [DOI] [PubMed] [Google Scholar]
- Moore SD, Sauer RT. 2007. The tmRNA system for translational surveillance and ribosome rescue. Annu Rev Biochem. 76:101–124. [DOI] [PubMed] [Google Scholar]
- Moreno-Cinos C, Goossens K, Saladol G, Van Der Veken P, De Winter H, Augustyns K. 2019. ClpP Protease, a Promising Antimicrobial Target. Int J Mol Sci. 20(9):2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nager AR, Baker TA, Sauer RT. 2011. Stepwise unfolding of a β barrel protein by the AAA+ ClpXP protease. J Mol Biol. 413(1):4–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher SB, Sauer RT, Baker TA. 2003. Distinct peptide signals in the UmuD and Umu D’ subunits of UmuD/D’ mediate tethering and substrate processing by the ClpXP protease. Proc Natl Acad Sci USA. 100(23): 13219–13224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuwald A, Aravind L, Spouge JL, Koonin EV. 1999. AAA+: A class of chaperone-like ATPases associated with assembly, operation, and disassembly of protein complexes. Genome Res. 9(1):27–43. [PubMed] [Google Scholar]
- Ogura T, Wilkinson AJ. 2001. AAA+ superfamily ATPases: common structure-diverse function. Genes Cells. 6(7):575–597. [DOI] [PubMed] [Google Scholar]
- Olivares AO, Kotamarthi HC, Stein BJ, Sauer RT, Baker TA. 2017. Effect of directional pulling on mechanical protein degradation by ATP-dependent proteolytic machines. Proc Natl Acad Sci USA. 114(31):E6306–E6313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivares AO, Baker TA, Sauer RT. 2018. Mechanical Protein Unfolding and Degradation. Annu Rev Physiol. 80:413–429. [DOI] [PubMed] [Google Scholar]
- Ortega J, Lee HS, Maurizi MR, Steven AC. 2002. Alternating translocation of protein substrates from both ends of ClpXP protease. EMBO J. 21(18):4938–4949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park EY, Lee BG, Hong SB, Kim HW, Jeon H, Song HK. 2007. Structural basis of SspB-tail recognition by the zinc binding domain of ClpX. J Mol Biol. 367(2):514–526. [DOI] [PubMed] [Google Scholar]
- Pavelka A, Sebestova E, Kozlikova B, Brezovsky J, Sochor J, Damborsky J. 2016. CAVER: algorithms for analyzing dynamics of tunnels in macromolecules. IEEE/ACM Trans Comput Biol Bioinform. 13(3):505–517. [DOI] [PubMed] [Google Scholar]
- Puchades C, Rampello AJ, Shin M, Giuliano CJ, Wiseman RL, Glynn SE, Lander GC. 2017. Structure of the mitochondrial inner membrane AAA+ protease YME1 gives insight into substrate processing. Science. 358(6363):eaao0464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puchades C, Sandate CR, Lander GC. 2020. The molecular principles governing the activity and functional diversity of AAA+ proteins. Nat Rev Mol Cell Biol. 21(1 ):43–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripstein ZA, Vahidi S, Houry WA, Rubinstein JL, Kay LE. 2020b. A processive rotary mechanism couples substrate unfolding and proteolysis in the ClpXP degradation machinery. Elife. 9:e52158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripstein ZA, Vahidi S, Rubinstein JL, Kay LE. 2020a. A pH-dependent conformational switch controls N. meningitidis ClpP protease function. J Am Chem Soc. 142(49):20519–20523. [DOI] [PubMed] [Google Scholar]
- Roche ED, Sauer RT. 1999. SsrA-mediated peptide tagging caused by rare codons and tRNA scarcity. EMBO J. 18(16):4579–4589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Aliaga P, Ramirez L, Kim F, Bustamante C, Martin A. 2016. Substrate-translocating loops regulate mechanochemical coupling and power production in AAA+ protease ClpXP. Nat Struct Mol Biol. 23(11 ):974–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- San Martín Á, Rodriguez-Aliaga P, Molina JA, Martin A, Bustamante C, Baez M. 2017. Knots can impair protein degradation by ATP-dependent proteases. Proc Natl Acad Sci USA. 114(37):9864–9869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer RT, Baker TA. 2011. AAA+ proteases: ATP-fueled machines of protein destruction. Annu Rev Biochem. 80:587–612. [DOI] [PubMed] [Google Scholar]
- Saunders RA, Stinson BM, Baker TA, Sauer RT. 2020. Multistep substrate binding and engagement by the AAA+ ClpXP protease. Proc Natl Acad Sci USA. 117(45):28005–28013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz KR, Carney DW, Sello JK, Sauer RT. 2014. Crystal structure of Mycobacterium tuberculosis ClpP1P2 suggests a model for peptidase activation by AAA+ partner binding and substrate delivery. Proc Natl Acad Sci USA. 111(43):E4587–4595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen M, Maillard RA, Nyquist K, Rodriguez-Aliaga P, Pressé S, Martin A, Bustamante C. 2013. The ClpXP protease unfolds substrates using a constant rate of pulling but different gears. Cell. 155(3):636–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiqui SM, Sauer RT, Baker TA. 2004. Role of the processing pore of the ClpX AAA+ ATPase in the recognition and engagement of specific protein substrates. Genes Dev. 18(4):369–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh SK, Grimaud R, Hoskins JR, Wickner S, Maurizi MR. 2000. Unfolding and internalization of proteins by the ATP-dependent proteases ClpXP and ClpAP. Proc Natl Acad Sci USA. 97(16):8898–8903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh SK, Rozycki J, Ortega J, Ishikawa T,Lo J, Steven AC, Maurizi MR. 2001. Functional domains of the ClpA and ClpX molecular chaperones identified by limited proteolysis and deletion analysis. J Biol Chem. 276(31 ):29420–29429. [DOI] [PubMed] [Google Scholar]
- Sivertsson EM, Jackson SE, Itzhaki LS. 2019. The AAA+ protease ClpXP can easily degrade a 31 and a 52-knotted protein. Sci Rep. 9(1 ):2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprangers R, Gribun A, Hwang PM, Houry WA, Kay LE. 2005. Quantitative NMR spectroscopy of supramolecular complexes: dynamic side pores in ClpP are important for product release. Proc Natl Acad Sci USA. 102(46): 16678–16683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sriramoju MK, Chen Y, Hsu SD. 2020. Protein knots provide mechano-resilience to an AAA+ protease-mediated proteolysis with profound ATP energy expenses. Biochim Biophys Acta Proteins Proteom. 1868(2): 140330. [DOI] [PubMed] [Google Scholar]
- Stinson BM, Nager AR, Glynn SE, Schmitz KR, Baker TA, Sauer RT. 2013. Nucleotide binding and conformational switching in the hexameric ring of a AAA+ machine. Cell. 153(3):628–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Striebe IF, Kress W, Weber-Ban E. 2009. Controlled destruction: AAA+ ATPases in protein degradation from bacteria to eukaryotes. Curr Opin Struct Biol. 19(2):209–217. [DOI] [PubMed] [Google Scholar]
- Thompson MW, Maurizi MR. 1994. Activity and specificity of Escherichia coli ClpAP protease in cleaving model peptide substrates. J Biol Chem. 269(27): 18201–18208. [PubMed] [Google Scholar]
- Too PH, Erales J, Simen JD, Marjanovic A, Coffino P. 2013. Slippery substrates impair function of a bacterial protease ATPase by unbalancing translocation versus exit. J Biol Chem. 288(19): 13243–13257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay CY, Vass RH, Vachet RW, Chien P. 2020. The cleavage profile of protein substrates by ClpXP reveals deliberate starts and pauses. Biochemistry. 59(44):4294–4301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vass RH, Chien P. 2013. Critical clamp loader processing by an essential AAA+ protease in Caulobacter crescentus. Proc Natl Acad Sci USA. 110(45): 18138–18143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Hartling JA, Flanagan JM. 1997. The structure of ClpP at 2.3 Å resolution suggests a model for ATP-dependent proteolysis. Cell. 91(4):447–456. [DOI] [PubMed] [Google Scholar]
- Wojtyra UA, Thibault G, Tuite A, Houry WA. 2003. The N-terminal zinc binding domain of ClpX is a dimerization domain that modulates the chaperone function. J Biol Chem. 278(49):48981–48990. [DOI] [PubMed] [Google Scholar]
- Yien YY, Ducamp S, van der Vorm LN, Kardon JR, Manceau H, Kannengiesser C, Bergonia HA, Kafina MD, Karim Z, Gouya L, Baker TA, Puy H, Phillips JD, Nicolas G, Paw BH. 2017. Mutation in human CLPX elevates levels of δ-aminolevulinate synthase and protoporphyrin IX to promote erythropoietic protoporphyria. Proc Natl Acad Sci USA. 114: E8045–E8052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeiler E, Korotkov VS, Lorenz-Baath K, Böttcher T, Sieber SA. 2012. Development and characterization of improved β-lactone-based anti-virulence drugs targeting ClpP. Bioorg Med Chem. 20(2):583–591. [DOI] [PubMed] [Google Scholar]
- Zhang J, Ye F, Lan L, Jiang H, Luo C, Yang CG. 2011. Structural switching of Staphylococcus aureus Clp protease: a key to understanding protease dynamics. J Biol Chem. 286(43):37590–37601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Mao Y. 2020. AAA+ ATPases in protein degradation: Structures, functions and mechanisms. Biomolecules. 10(4):629. [DOI] [PMC free article] [PubMed] [Google Scholar]