

This randomized clinical trial evaluates matching efficacy, safety, and immunogenicity between biosimilar natalizumab and reference natalizumab in patients with relapsing-remitting multiple sclerosis.
Key Points
Question
Does proposed biosimilar natalizumab (biosim-NTZ) PB006 match reference natalizumab (ref-NTZ) in terms of efficacy, safety, and immunogenicity in patients with relapsing-remitting multiple sclerosis (RRMS)?
Findings
In this randomized, double-blind phase 3 clinical trial, 264 adult patients with RRMS received treatment with either biosim-NTZ or ref-NTZ. At week 24, the model-based mean difference in the cumulative number of new active lesions was similar between treatment groups.
Meaning
The study demonstrated that biosim-NTZ matches the efficacy, safety, and immunogenicity of ref-NTZ in patients with RRMS.
Abstract
Importance
Proposed biosimilar natalizumab (biosim-NTZ) PB006 is the first biosimilar monoclonal antibody therapy developed for multiple sclerosis (MS) treatment.
Objective
To evaluate matching efficacy, safety, and immunogenicity between biosim-NTZ and reference natalizumab (ref-NTZ) in patients with relapsing-remitting MS (RRMS).
Design, Setting, and Participants
The Antelope trial was a phase 3, parallel-group, randomized, active-controlled study, conducted between October 2019 and March 2021, with last patient follow-up visit on August 23, 2021. The study took place in 48 centers in 7 countries. Of 531 patients with RRMS aged 18 to 60 years screened, 266 were excluded before randomization in line with study criteria. Eligible participants had 1 or more documented relapse within the previous year and either 1 or more gadolinium-enhancing T1-weighted or 9 or more T2-weighted brain lesions, Kurtzke Expanded Disability Status Scale score of 0 to 5.0 (inclusive), and John Cunningham virus index of 1.5 or less at screening. One patient withdrew consent before dosing.
Interventions
Intravenous infusions every 4 weeks of biosim-NTZ, 300 mg, or ref-NTZ, 300 mg (1:1 randomization), from week 0 to week 44 (end-of-study visit: week 48). At week 24, the ref-NTZ group was rerandomized and 30 patients were switched to biosim-NTZ for the remainder of the study.
Main Outcomes and Measures
The primary end point was the cumulative number of new active lesions on magnetic resonance imaging (new gadolinium-enhancing T1-weighted lesions and new/enlarging T2-weighted lesions without double counting) over 24 weeks. Additional end points included further magnetic resonance imaging parameters, annualized relapse rate, and Kurtzke Expanded Disability Status Scale score. Safety, tolerability, and immunogenicity assessments included adverse events, laboratory evaluations, and positivity for anti–John Cunningham virus antibodies and antinatalizumab antibodies.
Results
A total of 264 participants (mean [SD] age, 36.7 [9.38] years; 162 [61.4%] female) received treatment with biosim-NTZ (n = 131) or ref-NTZ (n = 133). At week 24, the model-based mean difference in cumulative number of new active lesions between biosim-NTZ and ref-NTZ treatment groups was 0.17 (least square means [SE]: biosim-NTZ, 0.34 [0.34]; ref-NTZ, 0.45 [0.28]; 95% CI, –0.61 to 0.94 within the prespecified margins of ±2.1). No significant differences between treatment groups were observed across secondary efficacy end points, safety, tolerability, or immunogenicity assessments.
Conclusions and Relevance
Biosim-NTZ matched ref-NTZ in efficacy, safety, and immunogenicity for patients with RRMS in the tested setting. This phase 3 trial supports proposed biosim-NTZ as a biosimilar alternative to ref-NTZ for treating RRMS.
Trial Registration
ClinicalTrials.gov Identifier: NCT04115488
Introduction
Disease-modifying therapies (DMTs), particularly targeted biologic medicines, have revolutionized health care in the treatment of chronic conditions such as multiple sclerosis (MS).1,2 However, DMTs are the primary drivers of health care costs after MS diagnosis,3,4,5,6,7 particularly in relapsing-remitting MS (RRMS), which accounts for 85% of initial diagnoses and is associated with the highest treatment costs of all MS subtypes.5,8,9
Due to continuously increasing health care costs, uniform access to DMTs remains difficult.10,11,12 In other therapy areas, such as rheumatology and oncology, the introduction of biosimilars, defined as medicines highly similar and with no clinically meaningful differences to an already marketed reference biologic medicine, demonstrated significant cost savings and improved treatment access.13,14,15 Biosimilar medicines may thus open the door for not only effective and safe, but also affordable biologic treatments for MS.
The humanized monoclonal antibody natalizumab was the first targeted biologic therapy to be approved for RRMS.16,17,18 Natalizumab binds to the α4 subunit of α4β1 and α4β7 integrins, inhibiting leukocyte adhesion to vascular adhesion molecules and disrupting leukocyte transmigration across the blood–brain barrier.16,17 In Europe and the US, natalizumab is indicated as monotherapy in patients with RRMS with high disease activity despite previous DMT or in rapidly evolving severe RRMS.18,19 In the US, it is also approved for use in adults with active moderate to severe Crohn disease who have had an inadequate response to, or are unable to tolerate, other therapy lines.18
PB006 is a biosimilar natalizumab (biosim-NTZ) and was developed by Polpharma Biologics SA as a proposed biosimilar to the reference natalizumab (ref-NTZ) in accordance with US Food and Drug Administration (FDA) and European Medicines Agency (EMA) guidelines, which require biosimilars to match the reference medicine in analytical comparability, bioequivalence, safety, efficacy, and immunogenicity, confirming no clinically relevant differences in performance across approved indications.15,20,21
Previous pharmacokinetic/pharmacodynamic and safety studies in healthy participants provided evidence to support PB006 phase 3 studies involving patients with RRMS.22,23 The primary objective of the Antelope study was to confirm equivalent efficacy between biosim-NTZ and ref-NTZ in patients with RRMS by assessing magnetic resonance imaging (MRI)–based end points supported by clinical, safety, and immunogenicity secondary end point analyses. The potential impact of switching from ref-NTZ to biosim-NTZ on efficacy outcomes and immunogenicity was also assessed in a subpopulation analysis.
Methods
Study Design
Antelope was a phase 3, multicenter, double-blind, active-controlled, randomized, parallel-group trial conducted at 48 centers across Belarus, Croatia, Georgia, Moldova, Poland, Serbia, and Ukraine. A statistical equivalence design was chosen to assess similarity between biosim-NTZ and ref-NTZ in efficacy, safety, and immunogenicity. The study was conducted in accordance with International Council for Harmonisation Good Clinical Practice, the Declaration of Helsinki,24 and relevant ethics committee or regulatory agency procedures. Independent ethics committees and health authorities approved the study before initiation, and patients or their legally authorized representatives signed statements of informed consent before enrollment and reconsent during the trial.
The trial initiated in October 2019 and completed in March 2021, with the last patient follow-up visit (24 weeks after last dosing) on August 23, 2021. A sensitivity analysis on a pre–COVID-19 per-protocol population was planned to account for any major deviations related to COVID-19. However, this analysis is not reported as there were no missing data to impute. The full trial protocol is available in Supplement 1. The study followed the Consolidated Standards of Reporting Trials (CONSORT) reporting guideline.
Participants
Eligible participants were male and female individuals aged 18 to 60 years diagnosed with RRMS, as defined by the revised 2010 McDonald criteria.25 At screening, inclusion criteria included 1 or more documented relapse within the previous year and either 1 or more gadolinium-enhancing T1-weighted or 9 or more T2-weighted brain lesions on MRI and a Kurtzke Expanded Disability Status Scale (EDSS) score of 0 to 5.0 (inclusive).26
Eligible participants included those undergoing and not undergoing disease therapy. The use of corticosteroids for 3 to 5 days to treat relapses and symptomatic MS, if they were stable for at least 3 months before screening, was also allowed as concomitant therapy during the study. However, patients who exhibited a relapse within the 30 days before screening and until administration of the first dose of the study drug were excluded.
Progressive multifocal leukoencephalopathy (PML), a rare, potentially fatal, demyelinating disease of the central nervous system arising from infection with the John Cunningham human polyomavirus (JCV), is a known safety concern associated with some DMTs, notably ref-NTZ.18,27,28,29 Key exclusion criteria therefore included JCV index more than 1.5 at screening and past or current PML diagnosis, as well as prior treatment with natalizumab, alemtuzumab, ocrelizumab, daclizumab, rituximab, cladribine, or other B- and T-cell–targeting therapies.
Patients positive for low-titer JCV (JCV index <1.5) were included in the study as the level of PML risk in patients who did not previously receive immunosuppressive therapies could be quantified by association with the anti-JCV antibody index.30 Detailed exclusion criteria can be found in the trial protocol in Supplement 1.
Randomization and Masking
Eligible consenting participants were randomly assigned to 1 of 2 treatment groups 1:1 to receive intravenous infusions every 4 weeks of 300 mg of biosim-NTZ or 300 mg of ref-NTZ, starting at visit 1 (week 0) through to visit 12 (week 44) for a total of 12 infusions. At week 24, patients in the ref-NTZ group underwent rerandomization, whereby 30 patients were switched from ref-NTZ to biosim-NTZ for the remainder of their infusions (total 6 infusions each of ref-NTZ and biosim-NTZ) (Figure 1).
Figure 1. Overall Study Design.
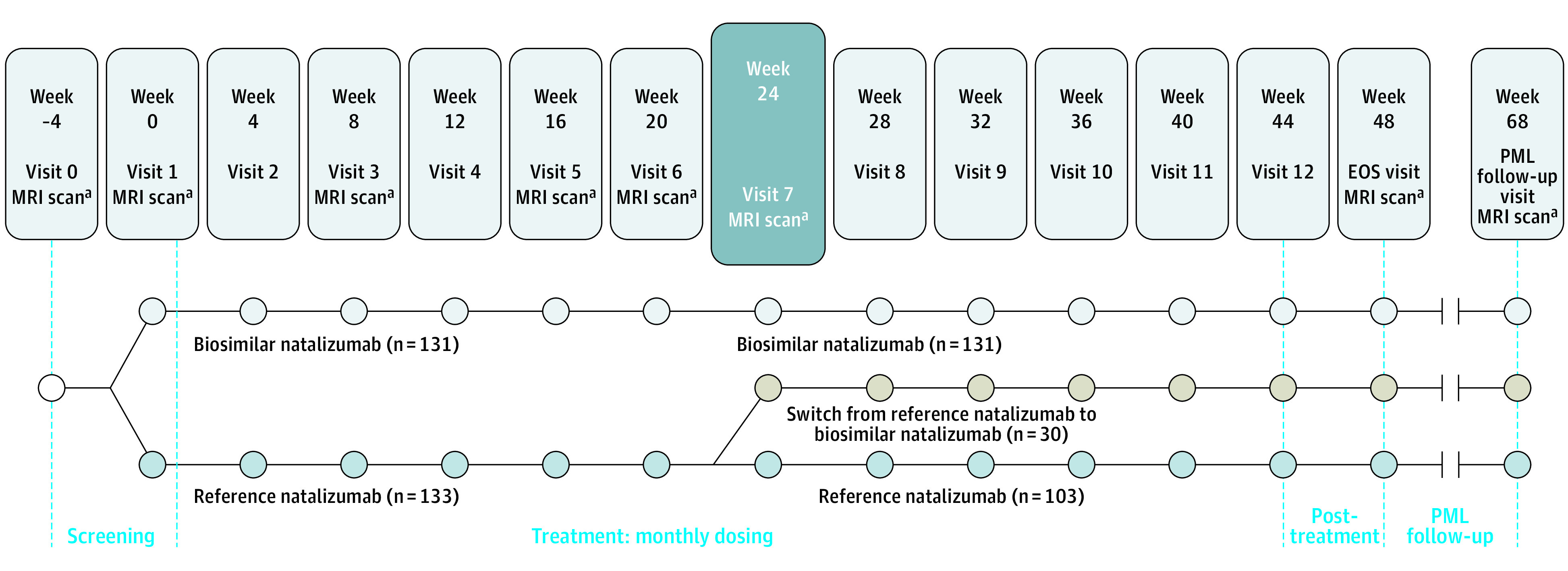
EOS indicates end of study; MRI, magnetic resonance imaging; PML, progressive multifocal leukoencephalopathy.
aAssessed by the central reading center.
Patients were randomized dynamically using Medidata Randomization and Trial Supply Management integrated with electronic data capture (Rave), carried out in a double-blind fashion. An unblinded pharmacist/designee was responsible for maintaining accountability, blinding, and dispensing the study drugs according to the handling instructions. All other study center personnel remained blinded to the study drug until database lock. Randomization was stratified at screening by absence/presence of gadolinium-enhancing lesions (0 and >0), presence of T2 lesions (≤15 and >15), and JCV status (negative and positive).
Study Procedures and End Points
The primary efficacy end point assessed the cumulative number of new active lesions on MRI of the brain (new gadolinium-enhancing T1-weighted lesions and new/enlarging T2-weighted lesions) over 24 weeks in the per-protocol population. The use of week 24 as the readout for the primary end point was agreed with the health authorities and is supported by the treatment effect observed at early time intervals in natalizumab studies.31
A comprehensive range of secondary radiologic outcomes were also assessed (eAppendix 1 in Supplement 2). Central review of lesion assessment was performed by a trained and certified radiology reviewer using MRI with a macrocyclic gadolinium-enhancing–based contrast agent.
Annualized relapse rate (ARR) and change from baseline EDSS score after 24 and 48 weeks were secondary clinical end points. Relapse was defined as the appearance of a new neurologic abnormality or worsening of previously stable or improving preexisting neurologic abnormality, present for more than 24 hours and occurring in the absence of fever (temperature <37.5 °C) or infection and separated by 30 or more days from onset of a preceding clinical demyelinating event.9,25
Safety and tolerability were assessed via local and systemic adverse events (AEs) and serious AEs after 24 and 48 weeks; incidence rate of antinatalizumab antibodies (hereafter referred to as antidrug antibodies [ADAs]) and persistent antibodies after 24 and 48 weeks and after switching; incidence rate of antinatalizumab-neutralizing antibodies (NAbs) after 24 and 48 weeks and after switching; and lowest plasma concentration of natalizumab (trough concentration) over time. Assessments included physical examinations, electrocardiogram, vital signs, laboratory evaluations, the Columbia-Suicide Severity Rating Scale, and anti-JCV antibodies.
Blood samples for anti-JCV antibodies were collected at screening, weeks 24 and 48, and evaluated using a cell-based validated assay. The incidence rates of positive anti-JCV antibody samples by JCV index level (≤0.9, >0.9 to ≤1.5, and >1.5) were summarized descriptively by visit and for the study duration.
An ADA assay was developed for use in the biosim-NTZ study program according to applicable guidelines for ADA testing in biosimilar studies32,33 and taking into account the highly sensitive tailored immunogenicity evaluation required.34 ADA measurements were conducted using an electrochemiluminescence immunoassay assay incorporating a sample pretreatment step to optimize drug tolerance. Based on the signal for the positive control antibody, the ADA assay had a lower limit of detection of 3.88 ng/mL for screening and 2.55 ng/mL for confirmation. Adequate sensitivity and drug tolerance of the biosim-NTZ ADA assay was determined for detection of the ADA signal in the treated population.
NAbs were defined as ADAs specifically inhibiting binding of natalizumab to α4-integrin and measured by a flow cytometry assay. ADA and NAb sampling occurred predose and at weeks 4, 8, 16, 24, 28, 32, and 48.
AE assessment occurred at screening, at each of the 12 treatment visits, the end of study/early discontinuation visit, and the PML follow-up visits. AEs were graded using version 4.03 of the National Cancer Institute Common Terminology Criteria for Adverse Events.35 AEs were considered treatment-emergent if they started or worsened in severity after the first infusion, regardless of their relation to the study drug.
Statistical Analysis
A total of 230 evaluable patients (115 per group) were required to achieve 90% power for the similarity assessment with respect to the primary end point, assuming a common SD of 4.0 lesions and no difference between groups. To account for up to 10% of potential dropouts and nonevaluable patients, approximately 260 patients were to be randomized.
The primary efficacy analysis was based on the per-protocol population. Similarity was analyzed using a negative binomial generalized linear model with fixed effects for the treatment group and stratification factors with logarithmic link function. A biosimilarity margin for the mean difference of ±2.1 lesions was chosen based on the US FDA guidelines for noninferiority trials to ensure 50% of the treatment effect was preserved, based on the lower bound of the 95% CI of the pooled effect size, as estimated by Miller et al.31,36 To address different regulatory requirements, similarity was tested based on the corresponding 90% CIs (US FDA) and 95% CIs (EMA). The full statistical analysis plan is included in Supplement 1.
Results
Baseline Characteristics
Of 531 patients screened for enrollment, 266 (50.1%) were excluded before randomization in line with trial criteria. A total of 265 patients (49.9%) progressed to randomization and 264 (biosim-NTZ, 131 [49.6%]; ref-NTZ, 133 [50.4%]) received treatment; 1 patient in the biosim-NTZ group withdrew consent during screening. Of 264 individuals (162 [61.4%] female; 102 [38.6%] male; mean [SD] age, 36.7 [9.38] years), 247 (93.6%) completed the 24-week primary treatment period (biosim-NTZ, 122 [49.4%]; ref-NTZ, 125 [50.6%]).
At week 24, all 125 patients in the ref-NTZ group who completed primary treatment were rerandomized (ref-NTZ, 95 [76%]; ref-NTZ/biosim-NTZ switch, 30 [24%]). Overall, 239 patients (90.5%) completed the 48-week study (biosim-NTZ, 117 [49.0%]; biosim-NTZ/ref-NTZ switch, 29 [12.1%]; ref-NTZ, 93 [38.9%]) (Figure 2).
Figure 2. CONSORT Diagram of Enrollment and Follow-up of Study Patients.
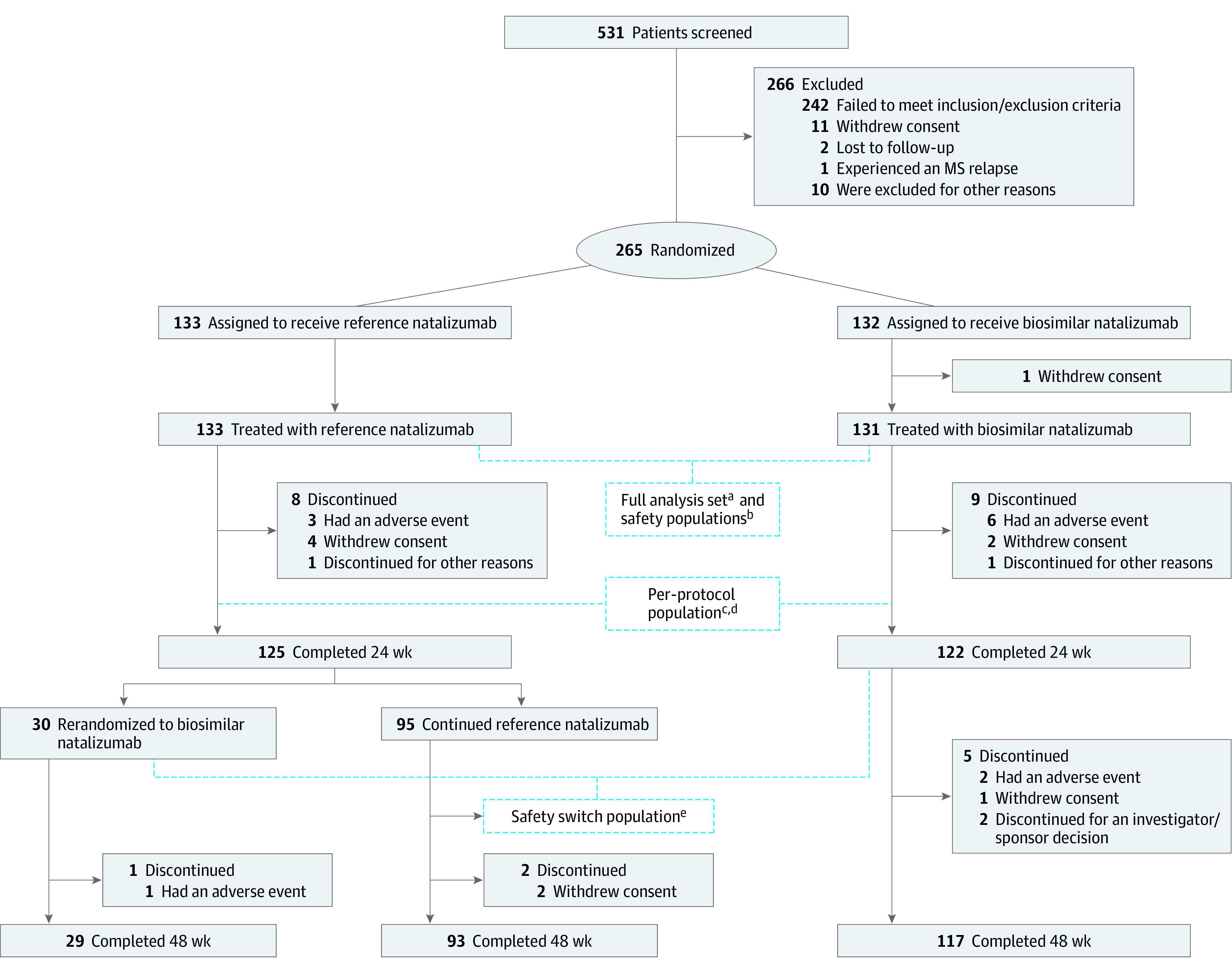
MS indicates multiple sclerosis.
aThe full analysis set included patients who were randomized and received ≥1 (complete or partial) infusion of the study drug. Patients were to be analyzed according to the treatment group to which they were randomized.
bThe safety population included patients who received ≥1 (complete or partial) infusion of the study drug. Patients in this group were to be analyzed as treated.
cThe per-protocol population included only patients who completed the 24-week treatment period without major protocol deviation that may have influenced the analysis of the primary end point and for whom sufficient postbaseline magnetic resonance imaging data were available.
dThe per-protocol population comprised 229 patients who completed the 24-week treatment period without major protocol deviations and for whom sufficient postbaseline magnetic resonance imaging data were available (biosimilar natalizumab: n = 111; reference natalizumab: n = 118).
eThe safety-switch population included patients in the safety population who received ≥1 infusion of the study drug after the rerandomization time point, independent of switching status. Patients in this group were to be analyzed as treated after rerandomization, also considering treatment before rerandomization.
Baseline characteristics were similar between treatment groups for the full analysis set (Table 1) and all other planned cohorts, including patients with confirmed COVID-19 (n = 22) in the safety population (biosim-NTZ, 11 [50%]; ref-NTZ, 11 [50%]).
Table 1. Baseline Characteristics (Full Analysis Set).
| Characteristic | No. (%) | |
|---|---|---|
| Biosim-NTZ (n = 131) | Ref-NTZ (n = 133) | |
| Positive JCV status | 51 (38.9) | 55 (41.4) |
| Positive for ADAsa | 9 (7) | 8 (6) |
| Age, mean (SD), y | 36.8 (9.1) | 36.6 (9.7) |
| Male | 47 (35.9) | 55 (41.4) |
| Femaleb | 84 (64.1) | 78 (58.6) |
| Childbearing potential | 73 (86.9) | 70 (89.7) |
| Whitec | 131 (100) | 133 (100) |
| Absence/presence of gadolinium-enhancing lesions | ||
| 0 | 68 (51.9) | 72 (54.1) |
| >0 | 63 (48.1) | 61 (45.9) |
| Presence of T2 lesions | ||
| ≤15 | 4 (3.1) | 5 (3.8) |
| >15 | 127 (96.9) | 128 (96.2) |
| Time since diagnosis, y | ||
| Mean (SD) | 5.3 (4.7) | 5.3 (4.8) |
| Median (range) | 4.2 (0.1-21.8) | 4.4 (0.1-20.5) |
| Time since last relapse, mo | ||
| Mean (SD) | 5.1 (2.9) | 5.9 (3.0) |
| Median (range) | 4.3 (1.7-12.7) | 5.6 (1.7-12.6) |
| No. of relapses in the year prior to screening | ||
| 1 | 86 (65.6) | 91 (68.4) |
| 2 | 37 (28.2) | 38 (28.6) |
| ≥3 | 8 (6.1) | 4 (3.0) |
| Mean (SD) | 1.4 (0.7) | 1.4 (0.6) |
| Baseline EDSS score | ||
| Mean (SD) | 3.4 (1.1) | 3.2 (1.2) |
| Median (range) | 3.5 (2-5) | 3.5 (1-5) |
Abbreviations: ADAs, anti-drug antibodies; biosim-NTZ, biosimilar natalizumab; EDSS, Kurtzke Expanded Disability Status Scale; JCV, John Cunningham virus; ref-NTZ, reference natalizumab.
The reported proportion of patients with positive ADA results is part of the safety analysis performed in the safety population, which was identical to the full analysis set population.
A serum pregnancy test was conducted to confirm nonpregnancy in those of childbearing potential. Female participants of childbearing potential sexually active with a fertile male partner and male participants sexually active with female partners of childbearing potential agreed to use adequate contraception from screening until 90 days after the last dose of the study drug.
Race and ethnicity data were collected per trial protocol. The choices were American Indian or Alaska Native, Asian, Black or African American, Native Hawaiian or Other Pacific Islander, White, not reported, and other.
Overall, the disease activity at baseline was similar for patients in the biosim-NTZ and ref-NTZ treatment groups in the full analysis set, as demonstrated by the mean time since diagnosis and time since last relapse (Table 1). Both the number of relapses in the year before screening and the mean baseline EDSS score were also similar between treatment groups. Similar disease activity at baseline was reported for the per-protocol, safety population, and safety-switch populations. Detailed disease activity data at baseline can be found in Table 1.
Additionally, 158 patients (59.8%) in the full analysis set and 158 (59.8%) in the safety population were negative for anti-JCV antibodies at baseline. Patients who were anti-JCV–positive were similarly distributed between treatment groups, stratified by a positive index of 0.9 or less (biosim-NTZ, 35 [26.7%]; ref-NTZ, 37 [27.8%]) and 0.9 to 1.5 (biosim-NTZ, 16 [12.2%]; ref-NTZ, 18 [13.5%]).
Efficacy
At week 24, the model-based mean difference in the cumulative number of new active lesions between treatment groups in the per-protocol population was 0.17 (least square means [SE]: biosim-NTZ, 0.34 [0.34]; ref-NTZ, 0.45 [0.28]), with 95% CI, –0.61 to 0.94 with the point estimate within the prespecified margins of ±2.1 (Figure 3A). Sensitivity analysis of the cumulative number of new active lesions at week 24 in the full analysis set population confirmed the primary end point, showing a model-based mean difference (SE) of 0.23 (0.43) between ref-NTZ and biosim-NTZ, with 95% CI, –0.61 to 1.08 within the specified margins of ±2.1.
Figure 3. Primary Study End Point and Secondary Efficacy End Point.

A, Exponentiated difference in cumulative number of new active lesions at week 24 between the biosimilar natalizumab and reference natalizumab groups (per-protocol population). B, Cumulative number of new active lesions over 48 weeks in the biosimilar natalizumab and reference natalizumab groups by primary randomization (full analysis set population).
After database lock, it was determined that 1 or more of the 3 stratification factors (absence/presence of gadolinium-enhancing lesions, presence of T2 lesions, JCV status) were incorrectly recorded at randomization for 62 of 265 patients (23.4%). Following a thorough root-cause analysis and impact assessment, the immediate risk for imbalanced strata was reported as low. Additionally, since the study was blinded and the randomization performed according to the entered strata, the integrity of the data was not affected. A further sensitivity analysis was performed based on the corrected stratification factors to assess the impact of the erroneous stratification. This analysis confirmed the primary end point in the per-protocol population showing a model-based mean difference (SE) of 0.06 (0.08) between ref-NTZ and biosim-NTZ, with 90% CI, –0.07 to 0.20 and 95% CI, –0.10 to 0.23 within the specified margins of ±2.1 (see eAppendix 2 in Supplement 2 for detailed results of the sensitivity analysis).
The secondary efficacy analysis in the full analysis set showed no clinically relevant difference in the mean cumulative number of new active lesions observed between treatment groups (Figure 3B). At week 48, a mean (SD) cumulative number of new active lesions of 1.5 (3.72) was observed in the biosim-NTZ group vs 2.3 (5.68) in the ref-NTZ group. A similar pattern of outcomes was reported for the per-protocol and safety-switch populations.
Clinical End Points
ARR
In the full analysis set population, ARR (relapses/y) was similar at 24 weeks (biosim-NTZ, 0.21; ref-NTZ, 0.15) and 48 weeks (biosim-NTZ, 0.17; ref-NTZ, 0.13). A similar pattern of outcomes was reported for the per-protocol population. ARR was also similar between all treatment groups in the safety-switch population at rerandomization at 24 weeks (biosim-NTZ, 0.19; ref-NTZ, 0.11; ref-NTZ/biosim-NTZ switch, 0.14) and 48 weeks (biosim-NTZ, 0.17; ref-NTZ, 0.11; ref-NTZ/biosim-NTZ switch, 0.15).
EDSS Score
At baseline, EDSS scores by primary randomization were similar between treatment groups (mean [SD]: biosim-NTZ, 3.36 [1.07]; ref-NTZ, 3.20 [1.21]) in the full analysis set population. Distribution of EDSS scores at baseline can be found in eAppendix 3 in Supplement 2.
Change from baseline was minimal and similar in both treatment groups at 24 weeks (mean [SD]: biosim-NTZ, –0.03 [0.21]; ref-NTZ, 0.00 [0.35]) and at 48 weeks (biosim-NTZ, –0.14 [0.54]; ref-NTZ, –0.05 [0.44]). A similar pattern of outcomes was reported for the per-protocol population. EDSS scores at rerandomization at 24 weeks were also similar between all treatment groups in the safety-switch population (mean [SD]: biosim-NTZ, 3.37 [1.13]; ref-NTZ, 3.19 [1.30]; ref-NTZ/biosim-NTZ switch, 3.15 [1.13]). Change from baseline (week 24) to week 48 was minimal and similar between all treatment groups (mean [SD]: biosim-NTZ, –0.10 [0.50]; ref-NTZ, –0.02 [0.31]; ref-NTZ/biosim-NTZ switch, –0.03 [0.33]).
Safety and Tolerability
Treatment-emergent AE (TEAE) rates (biosim-NTZ, 85 [64.9%]; ref-NTZ, 71 [68.9%]; ref-NTZ/biosim-NTZ switch, 22 [73.3%]; Table 2) and event rates per 100 patient-years were similar across all treatment groups (biosim-NTZ, 221/192.3; ref-NTZ, 176/194.7; ref-NTZ/biosim-NTZ switch, 60/219.6) (eAppendix 4 in Supplement 2). The most reported TEAEs among all treatment groups were nervous system disorders (biosim-NTZ, 33 [25.2%]; ref-NTZ, 24 [23.3%]; ref-NTZ/biosim-NTZ switch, 8 [26.7%]) and infections and infestations (biosim-NTZ, 39 [29.8%]; ref-NTZ, 34 [33.0%]; ref-NTZ/biosim-NTZ switch, 15 [50.0%]). See eAppendix 4 in Supplement 2 for a full breakdown of reported TEAEs.
Table 2. Summary of AEs by Treatment Sequence and ADA/NAb Results to Week 48a.
| Safety population | No. (%) | ||
|---|---|---|---|
| Biosim-NTZ (n = 131) | Ref-NTZ/biosim-NTZ switch (n = 30) | Ref-NTZ (n = 103) | |
| Any TEAE | 85 (64.9) | 22 (73.3) | 71 (68.9) |
| Any related TEAE | 31 (23.7) | 8 (26.7) | 22 (21.4) |
| Any TEAE ≥grade 3 | 4 (3.1) | 0 | 1 (1.0) |
| Investigations | 2 (1.5) | 0 | 0 |
| Musculoskeletal and connective tissue disorders | 0 | 0 | 1 (1.0) |
| Respiratory, thoracic, and mediastinal disorders | 1 (0.8) | 0 | 0 |
| Skin and subcutaneous tissue disorders | 1 (0.8) | 0 | 0 |
| Any TEAE of special interest | 6 (4.6) | 2 (6.7) | 6 (5.8) |
| Immune system disorders | 0 | 1 (3.3) | 0 |
| Infections and infestations | 2 (1.5) | 1 (3.3) | 5 (4.9) |
| Investigations | 1 (0.8) | 0 | 0 |
| Skin and subcutaneous tissue disorders | 3 (2.3) | 0 | 1 (1.0) |
| Any TEAE leading to permanent study drug discontinuation | 8 (6.1)b | 1 (3.3)c | 3 (2.9)d |
| Any TEAE leading to withdrawal from studye | 0 | 0 | 0 |
| ADA prevalence at baseline | 9 (7) | 1 (3) | 7 (7) |
| Treatment-emergent ADA positivity, total incidence | 104 (79) | 23 (77) | 76 (74) |
| Neutralizing ADA (NAb) | 90 (69) | 20 (67) | 69 (67) |
| % of ADA positive | 87 | 87 | 91 |
Abbreviations: ADA, antidrug antibody; AE, adverse event; biosim-NTZ, biosimilar natalizumab; NAb, neutralizing antibody; ref-NTZ, reference natalizumab; TEAE, treatment-emergent adverse event.
The number and percentage of patients with TEAEs, AEs of special interest (ie, progressive multifocal leukoencephalopathy, John Cunningham virus granule cell neuronopathy, opportunistic infections, liver injury, hypersensitivity, encephalitis, meningitis, and acute retinal necrosis), and serious AEs that occurred after the start of the first infusion and through 4 weeks after the last infusion date of the study drug (visit 13, end of study visit), were summarized by MedDRA system organ class and preferred term overall, by severity, and by relationship to study drug for each treatment group.
Urticaria (n = 2); pruritus (n = 2); asthenia, hyperhidrosis, blood pressure fluctuations, and dizziness (n = 1); trigeminal neuralgia, herpes simplex, and ear infection (n = 1); COVID-19 (n = 1); hypotension (n = 1).
Hypersensitivity (n = 1).
Urinary tract infection (n = 1); pharyngitis (n = 1); urticaria and angioedema (n = 1).
A TEAE was considered to lead to withdrawal from the study only if the patient did not proceed to progressive multifocal leukoencephalopathy follow-up because of this event.
Few AEs of special interest were reported with similar proportions across all treatment groups up to week 48 (biosim-NTZ, 6 [4.6%]; ref-NTZ, 6 [5.8%]; ref-NTZ/biosim-NTZ switch, 2 [6.7%]). See Table 2 for a full breakdown of AEs of special interest.
No fatal or National Cancer Institute Common Terminology Criteria for Adverse Events grade 4 TEAEs were reported. The majority of reported TEAEs were grade 1. In the biosim-NTZ group, 4 patients (3.1%) (3.48/100 patient-years) had any TEAEs of grade 3 up to week 48 (increased alanine aminotransferase, increased blood triglycerides, nasal septum deviation, and urticaria (each: 1 [0.8%]; 0.87/100 patient-years). In the ref-NTZ group, 1 patient (1.0%) (1.11/100 patient-years) had a grade 3 TEAE of pain in extremity. No grade 3 TEAEs were reported in the ref-NTZ/biosim-NTZ switch group (Table 2). Twelve patients experienced TEAEs leading to study drug discontinuation (biosim-NTZ, 8 [5.1%]; ref-NTZ, 4 [3.0%]).
Per the study exclusion criteria, no patient had JCV-positive index more than 1.5 at baseline and the proportions of patients positive for anti-JCV antibodies were similarly distributed between treatment groups throughout the study (eAppendix 5 in Supplement 2). At week 24, 5 patients (4.9%) in the biosim-NTZ and 5 (4.5%) in the ref-NTZ groups reported a JCV-positive index more than 1.5. At week 48, an index more than 1.5 was reported for 7 patients (6.0%) and 7 patients (5.9%) in each group, respectively. No patients with PML were identified during the treatment period (48 weeks) or PML follow-up visit (24 weeks [±2 weeks] after the last study drug infusion).
Regarding immunogenicity, at week 24, 104 patients (79.4%) and 98 patients (74.0%) were confirmed positive for treatment-emergent ADAs in the biosim-NTZ and ref-NTZ treatment groups, respectively; 90 participants (69.0%) in the biosim-NTZ group were NAb-positive vs 88 (66.2%) for ref-NTZ. Overall, ADA and NAb responses to biosim-NTZ mirrored that of ref-NTZ throughout the 48-week treatment period (Table 2).
No patients who were negative for ADA or NAb at week 24 seroconverted following the switch from ref-NTZ to biosim-NTZ, ie, switching from ref-NTZ to biosim-NTZ was not associated with an enhanced antinatalizumab humoral immune response in these patients.
Discussion
The Antelope trial assessed the matching efficacy, safety, and immunogenicity between biosim-NTZ and ref-NTZ in patients with RRMS. Comparative phase 3 trials are a key component of the stringent evaluations required by health authorities to confirm no clinically relevant differences between a proposed biosimilar and its reference medicine.15,32 Both the US FDA and EMA recommend using an equivalence design with predefined upper and lower margins based on knowledge of efficacy with the reference medicine and clinical judgment.15,32
Biosim-NTZ demonstrated similarity to ref-NTZ in the primary efficacy end point of cumulative number of new active lesions over 24 weeks, with 95% CI values of the point estimate within the prespecified margins (±2.1). These results were confirmed in all sensitivity analyses, as well as analysis with corrected stratification factors to assess the impact of the erroneous stratification and analysis using multiple imputation (data not reported) as described in eAppendix 2 in Supplement 2.
Furthermore, no significant differences between biosim-NTZ, ref-NTZ/biosim-NTZ switch, and ref-NTZ treatment were observed across the secondary MRI and clinical outcomes. The ARR observed for the biosim-NTZ group at week 24 (0.21) was comparable with that seen in the AFFIRM phase 3 trial of natalizumab (0.26), which had similar population baseline characteristics.27
The safety profile reported in the Antelope trial was as expected for patients with RRMS receiving natalizumab, characterized by similar percentages of TEAEs in both treatment groups. The number of patients experiencing TEAEs leading to study drug discontinuation was comparable with the results from the AFFIRM trial, wherein 6% of patients receiving natalizumab discontinued the study drug due to AEs.27 The infections and infestations rate per 100 patient-years observed here (biosim-NTZ, 54/47.00; ref-NTZ, 44/48.66) was also similar to the infection rate of 1 per patient-year in both the natalizumab and placebo groups in the AFFIRM trial.27
Notably, the overall AE profile for patients who switched from ref-NTZ to biosim-NTZ was similar to patients continuing ref-NTZ treatment and did not indicate any new or increased risks associated with switching to biosim-NTZ.
Strong concordance was reported in this study between the biosim-NTZ and ref-NTZ treatment groups in ADA (biosim-NTZ, 79.4%; ref-NTZ, 74.0%) and NAb (biosim-NTZ, 69.0%; ref-NTZ, 66.2%) response dynamics. The clinical program for ref-NTZ reported that 57 patients (9%) had detectable antibodies at some time during the study; persistent antibodies to natalizumab developed in 37 of 57 patients (6%).27 The consistently higher ADA-positive values reported across all treatment groups in the Antelope trial are considered to be due to the higher sensitivity of the assay used vs the reference clinical program, which applied an ADA assay known to have relatively low sensitivity due to drug interference.18,27 Similarly higher ADA-positive values have been reported in another study of ref-NTZ that detected higher ADA levels in patients receiving natalizumab when using a more sensitive radioimmunoassay assay.37
Antelope was conducted in a patient population with JCV-positive index less than 1.5 at baseline. At week 48, a positive seroconversion rate (index >1.5) was reported for 6.0% and 5.9% of patients in the biosim-NTZ and ref-NTZ groups, respectively. This value is not dissimilar to results from a 2018 meta-analysis of 17 independent natalizumab-treated cohorts of patients with MS, which reported a mean positive seroconversion annualized rate of 10.8% (positivity threshold: index >0.4).38 A 2021 registry substudy also reported a similar durable positive seroconversion of 7.3% per year (mean JCV index >1.6) among initially JCV-negative patients treated with natalizumab for RRMS.39
Even though no biosimilar has been approved for treating MS, off-label rituximab and its biosimilars are extensively used.40,41 In Sweden, off-label prescriptions of rituximab and its biosimilars for MS treatment have been reported at more than 50% compared with initiations of other new DMTs, showing that biosimilars may provide an effective and affordable treatment option in MS.42
Limitations
The small sample size in the ref-NTZ/biosim-NTZ switch subgroup may be considered a limitation of the current study. However, the equivalence design used in biosimilar phase 3 clinical trials aims to show clinical equivalence with the reference medicine.32 The Antelope trial reported equivalence between biosim-NTZ and ref-NTZ treatment across efficacy, safety, and secondary MRI and clinical outcomes. The ref-NTZ/biosim-NTZ switch subgroup further confirmed this equivalence in a practical setting.
Conclusions
The proposed biosim-NTZ PB006 is the first biosimilar monoclonal antibody therapy to be developed for MS. The clinical efficacy, safety, and immunogenicity of the proposed biosimilar natalizumab matched the reference natalizumab in the tested setting, with no clinically relevant differences observed. The results from this phase 3 trial support biosimilarity of proposed biosimilar natalizumab PB006 to its reference medicine in RRMS.
Trial Protocol
eAppendix 1. Magnetic Resonance Imaging Secondary Endpoints in Full Analysis Set Population
eAppendix 2. Cumulative Number of New Active Lesions Over 24 Weeks by Corrected Stratification Variables – Sensitivity Analysis (Per-Protocol Population)
eAppendix 3. Kurtzke Expanded Disability Status Scale Score at Baseline by Primary Randomization (Full Analysis Set)
eAppendix 4. Common TEAEs (>1% of Patients in any Treatment Group) by System Organ Class and Preferred Term by Treatment Sequence (Safety population)
eAppendix 5. Anti-John Cunningham Virus Antibodies by Visit by Primary Randomization (Safety Population)
Data Sharing Statement
References
- 1.Dutta B, Huys I, Vulto AG, Simoens S. Identifying key benefits in European off-patent biologics and biosimilar markets: it is not only about price! BioDrugs. 2020;34(2):159-170. doi: 10.1007/s40259-019-00395-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cohen M, Morrow T, Penna P. Managing the expanded use of biologics across therapeutic areas: an example from b-cell targeted therapies. Am J Manag Care. 2006;12(2)(suppl):S24-S37. [PubMed] [Google Scholar]
- 3.Hartung DM. Economics and cost-effectiveness of multiple sclerosis therapies in the USA. Neurotherapeutics. 2017;14(4):1018-1026. doi: 10.1007/s13311-017-0566-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dahham J, Rizk R, Kremer I, Evers SMAA, Hiligsmann M. Economic burden of multiple sclerosis in low- and middle-income countries: a systematic review. Pharmacoeconomics. 2021;39(7):789-807. doi: 10.1007/s40273-021-01032-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cortesi PA, Cozzolino P, Capra R, Cesana G, Mantovani LG. The economic burden of different multiple sclerosis courses: analysis from Italian administrative and clinical databases. Farmeconomia. 2020;21(1):49-58. doi: 10.7175/fe.v21i1.1476 [DOI] [Google Scholar]
- 6.Moccia M, Palladino R, Lanzillo R, et al. Healthcare costs for treating relapsing multiple sclerosis and the risk of progression: a retrospective Italian cohort study from 2001 to 2015. PLoS One. 2017;12(1):e0169489. doi: 10.1371/journal.pone.0169489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gyllensten H, Kavaliunas A, Murley C, et al. Costs of illness progression for different multiple sclerosis phenotypes: a population-based study in Sweden. Mult Scler J Exp Transl Clin. 2019;5(2):2055217319858383. doi: 10.1177/2055217319858383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Müller S, Heidler T, Fuchs A, et al. Real-world treatment of patients with multiple sclerosis per MS subtype and associated healthcare resource use: an analysis based on 13,333 patients in Germany. Neurol Ther. 2020;9(1):67-83. doi: 10.1007/s40120-019-00172-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.National Institute for Health and Care Excellence . Multiple sclerosis in adults: management (CG186). Accessed May 5, 2022. https://www.ncbi.nlm.nih.gov/books/NBK552607/pdf/Bookshelf_NBK552607.pdf [PubMed]
- 10.European Commission . What you need to know about biosimilar medicinal products. Accessed May 5, 2022. http://www.medicinesforeurope.com/wp-content/uploads/2016/03/biosimilars_report_en.pdf.
- 11.McCamish M, Yoon W, McKay J. Biosimilars: biologics that meet patients’ needs and healthcare economics. Am J Manag Care. 2016;22(13)(suppl):S439-S442. [PubMed] [Google Scholar]
- 12.Mulcahy AW, Hlavka JP, Case SR. Biosimilar cost savings in the United States: initial experience and future potential. Rand Health Q. 2018;7(4):3. [PMC free article] [PubMed] [Google Scholar]
- 13.Gascón P, Tesch H, Verpoort K, et al. Clinical experience with Zarzio® in Europe: what have we learned? Support Care Cancer. 2013;21(10):2925-2932. doi: 10.1007/s00520-013-1911-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smolen JS, Goncalves J, Quinn M, Benedetti F, Lee JY. Era of biosimilars in rheumatology: reshaping the healthcare environment. RMD Open. 2019;5(1):e000900. doi: 10.1136/rmdopen-2019-000900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.European Medicines Agency . Biosimilars in the EU: information guide for healthcare professionals. Accessed May 5, 2022. https://www.ema.europa.eu/en/documents/leaflet/biosimilars-eu-information-guide-healthcare-professionals_en.pdf
- 16.Sheremata WA, Minagar A, Alexander JS, Vollmer T. The role of alpha-4 integrin in the aetiology of multiple sclerosis: current knowledge and therapeutic implications. CNS Drugs. 2005;19(11):909-922. doi: 10.2165/00023210-200519110-00002 [DOI] [PubMed] [Google Scholar]
- 17.Schneider-Hohendorf T, Rossaint J, Mohan H, et al. VLA-4 blockade promotes differential routes into human CNS involving PSGL-1 rolling of T cells and MCAM-adhesion of TH17 cells. J Exp Med. 2014;211(9):1833-1846. doi: 10.1084/jem.20140540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tysabri (natalizumab). US highlights of prescribing information. Revised 2021. Accessed May 5, 2022. https://www.tysabri.com/content/dam/commercial/tysabri/pat/en_us/pdf/tysabri_prescribing_information.pdf
- 19.Tysabri (natalizumab). Summary of Product Characteristics. Updated 2022. Accessed May 5, 2022. https://www.ema.europa.eu/en/medicines/human/EPAR/tysabri#product-information-section
- 20.Skingle D. Biosimilars: what do patients need to consider? RMD Open. 2015;1(1):e000141. doi: 10.1136/rmdopen-2015-000141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.US Food and Drug Administration . FDA’s overview of the regulatory guidance for the development and approval of biosimilar products in the US. Accessed May 5, 2022. https://www.fda.gov/files/drugs/published/FDA%E2%80%99s-Overview-of-the-Regulatory-Guidance-for-the-Development-and-Approval-of-Biosimilar-Products-in-the-US.pdf
- 22.Grabowski T, Leuschner J, Gad S. 4-Week toxicity study of biosimilar natalizumab in comparison to Tysabri® by repeated intravenous infusion to cynomolgus monkeys. Drug Chem Toxicol. 2022;45(2):499-506. doi: 10.1080/01480545.2020.1722155 [DOI] [PubMed] [Google Scholar]
- 23.Roth K, Wessels H, Höfler J, et al. Phase I PK/PD similarity study of proposed biosimilar natalizumab PB006: a single dose study in healthy individuals. EAN Congress, June 25-28, 2022. Accessed July 22, 2022. https://www.ean.org/fileadmin/user_upload/ean/congress-2022/EAN2022AbstractBook.pdf.
- 24.World Medical Association . World Medical Association Declaration of Helsinki: ethical principles for medical research involving human subjects. JAMA. 2013;310(20):2191-2194. doi: 10.1001/jama.2013.281053 [DOI] [PubMed] [Google Scholar]
- 25.Polman CH, Reingold SC, Banwell B, et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol. 2011;69(2):292-302. doi: 10.1002/ana.22366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kurtzke JF. Rating neurologic impairment in multiple sclerosis: an expanded disability status scale (EDSS). Neurology. 1983;33(11):1444-1452. doi: 10.1212/WNL.33.11.1444 [DOI] [PubMed] [Google Scholar]
- 27.Polman CH, O’Connor PW, Havrdova E, et al. ; AFFIRM Investigators . A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med. 2006;354(9):899-910. doi: 10.1056/NEJMoa044397 [DOI] [PubMed] [Google Scholar]
- 28.Major EO, Yousry TA, Clifford DB. Pathogenesis of progressive multifocal leukoencephalopathy and risks associated with treatments for multiple sclerosis: a decade of lessons learned. Lancet Neurol. 2018;17(5):467-480. doi: 10.1016/S1474-4422(18)30040-1 [DOI] [PubMed] [Google Scholar]
- 29.Schwab N, Schneider-Hohendorf T, Melzer N, Cutter G, Wiendl H. Natalizumab-associated PML: challenges with incidence, resulting risk, and risk stratification. Neurology. 2017;88(12):1197-1205. doi: 10.1212/WNL.0000000000003739 [DOI] [PubMed] [Google Scholar]
- 30.Ho PR, Koendgen H, Campbell N, Haddock B, Richman S, Chang I. Risk of natalizumab-associated progressive multifocal leukoencephalopathy in patients with multiple sclerosis: a retrospective analysis of data from four clinical studies. Lancet Neurol. 2017;16(11):925-933. doi: 10.1016/S1474-4422(17)30282-X [DOI] [PubMed] [Google Scholar]
- 31.Miller DH, Khan OA, Sheremata WA, et al. ; International Natalizumab Multiple Sclerosis Trial Group . A controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med. 2003;348(1):15-23. doi: 10.1056/NEJMoa020696 [DOI] [PubMed] [Google Scholar]
- 32.US Food and Drug Administration . Scientific considerations in demonstrating biosimilarity to a reference product. Published April 2015. Accessed May 5, 2022. https://www.fda.gov/media/82647/download
- 33.European Medicines Agency . Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: non-clinical and clinical issues. Published December 2014. Accessed October 27, 2022. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-similar-biological-medicinal-products-containing-biotechnology-derived-proteins-active_en-2.pdf
- 34.Corsaro B, Yang TY, Murphy R, et al. 2020 White paper on recent issues in bioanalysis: vaccine assay validation, qPCR assay validation, QC for CAR-T flow cytometry, NAb assay harmonization and ELISpot validation (part 3: recommendations on immunogenicity assay strategies, NAb assays, biosimilars and FDA/EMA immunogenicity guidance/guideline, gene & cell therapy and vaccine assays). Bioanalysis. 2021;13(6):415-463. doi: 10.4155/bio-2021-0007 [DOI] [PubMed] [Google Scholar]
- 35.National Cancer Institute . Common Terminology Criteria for Adverse Events (CTCAE), version 4.03. US Department of Health and Human Services (DHHS). Published June 14, 2010. Accessed May 5, 2022. https://evs.nci.nih.gov/ftp1/CTCAE/CTCAE_4.03/CTCAE_4.03_2010-06-14_QuickReference_8.5x11.pdf
- 36.US Food and Drug Administration . Non-inferiority clinical trials to establish effectiveness. guidance for industry. Published November 2016. Accessed May 6, 2022. https://www.fda.gov/media/78504/download
- 37.Vennegoor A, Rispens T, Strijbis EMM, et al. Clinical relevance of serum natalizumab concentration and anti-natalizumab antibodies in multiple sclerosis. Mult Scler. 2013;19(5):593-600. doi: 10.1177/1352458512460604 [DOI] [PubMed] [Google Scholar]
- 38.Schwab N, Schneider-Hohendorf T, Hoyt T, et al. Anti-JCV serology during natalizumab treatment: review and meta-analysis of 17 independent patient cohorts analyzing anti-John Cunningham polyoma virus sero-conversion rates under natalizumab treatment and differences between technical and biological sero-converters. Mult Scler. 2018;24(5):563-573. doi: 10.1177/1352458517728814 [DOI] [PubMed] [Google Scholar]
- 39.Dwyer CM, Jokubaitis VG, Stankovich J, et al. High rates of JCV seroconversion in a large international cohort of natalizumab-treated patients. Ther Adv Neurol Disord. 2021;14:1756286421998915. doi: 10.1177/1756286421998915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Torgauten HM, Myhr KM, Wergeland S, Bø L, Aarseth JH, Torkildsen Ø. Safety and efficacy of rituximab as first- and second line treatment in multiple sclerosis: a cohort study. Mult Scler J Exp Transl Clin. 2021;7(1):2055217320973049. doi: 10.1177/2055217320973049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brancati S, Gozzo L, Longo L, Vitale DC, Drago F. Rituximab in multiple sclerosis: are we ready for regulatory approval? Front Immunol. 2021;12:661882. doi: 10.3389/fimmu.2021.661882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Berntsson SG, Kristoffersson A, Boström I, Feresiadou A, Burman J, Landtblom AM. Rapidly increasing off-label use of rituximab in multiple sclerosis in Sweden: outlier or predecessor? Acta Neurol Scand. 2018;138(4):327-331. doi: 10.1111/ane.12963 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Trial Protocol
eAppendix 1. Magnetic Resonance Imaging Secondary Endpoints in Full Analysis Set Population
eAppendix 2. Cumulative Number of New Active Lesions Over 24 Weeks by Corrected Stratification Variables – Sensitivity Analysis (Per-Protocol Population)
eAppendix 3. Kurtzke Expanded Disability Status Scale Score at Baseline by Primary Randomization (Full Analysis Set)
eAppendix 4. Common TEAEs (>1% of Patients in any Treatment Group) by System Organ Class and Preferred Term by Treatment Sequence (Safety population)
eAppendix 5. Anti-John Cunningham Virus Antibodies by Visit by Primary Randomization (Safety Population)
Data Sharing Statement