

Abstract
The COVID-19 pandemic has challenged the conventional diagnostic field and revealed the need for decentralized Point of Care (POC) solutions. Although nucleic acid testing is considered to be the most sensitive and specific disease detection method, conventional testing platforms are expensive, confined to central laboratories, and are not deployable in low-resource settings. CRISPR-based diagnostics have emerged as promising tools capable of revolutionizing the field of molecular diagnostics. These platforms are inexpensive, simple, and do not require the use of special instrumentation, suggesting they could democratize access to disease diagnostics. However, there are several obstacles to the use of the current platforms for POC applications, including difficulties in sample processing and stability. In this review, we discuss key advancements in the field, with an emphasis on the challenges of sample processing, stability, multiplexing, amplification-free detection, signal interpretation, and process automation. We also discuss potential solutions for revolutionizing CRISPR-based diagnostics toward sample-to-answer diagnostic solutions for POC and home use.
Keywords: SARS-CoV2, COVID-19, CRISPR-Cas systems, CRISPR, nucleic acid detection, biosensors, molecular diagnostics, POC diagnostics
1. Introduction
Disease detection during the early stages of an illness is becoming a prerequisite to disease treatment and containment.1 Several biomarkers can guide diagnostic development, but nucleic acid testing (NAT) is the gold standard for a wide array of chronic and acute conditions, particularly diseases caused by infectious agents.2 For pandemics such as COVID-19, the true challenge is to facilitate massive testing and screening to identify infected individuals, especially those with asymptomatic infections, to prevent the unintentional spread of the virus.3,4 Besides disease detection, NAT is also employed for food safety,5 agriculture,6 and environmental biosensing of disease-causing agents.7
Several nucleic acid detection platforms have been developed based on polymerase chain reaction (PCR) and isothermal amplification methods. PCR, which remains the gold standard method for detecting nucleic acids, is highly sensitive (down to one copy per microliter), specific, and less prone to errors than other methods, but it requires trained personnel, sophisticated infrastructure, expensive equipment, and a long turnaround time.8 These limitations confine PCR diagnostics to central laboratories and limit its widespread utility, especially in low-resource settings. By contrast, isothermal amplification methods operate at a single temperature and therefore do not require expensive thermocyclers, allowing them to be deployed for field diagnostics in resource-constrained environments. Isothermal amplification methods are extremely sensitive; however, they are prone to nonspecific amplification, which renders their specificity questionable. Several attempts to improve the specificity of isothermal amplification platforms have been described, including the use of molecular beacons,9 strand-displacement probes,10 and fluorescent probes.11
Current diagnostic development focuses on POC applications and aims to decentralize access for the timely detection of disease, with the goal of performing quality testing in resource-constrained communities. Overall, the goal is to create diagnostic solutions that meet the ASSURED criteria (affordable, sensitive, specific, user-friendly, rapid, equipment-free, delivered) set by the World Health Organization (WHO).12 Although no specific value for affordability has been proposed, Malaria and HIV rapid tests valued at 0.50–1 USD are widely accepted among stakeholders.13 However, there are always trade-offs between cost and accuracy. Low-cost tests, such as rapid antigen tests, have lower sensitivity and specificity than NATs performed in the laboratory with sophisticated equipment.13 There is a need to develop a new class of diagnostics that meets the ASSURED criteria with minimum trade-offs between affordability and accuracy.
CRISPR (clustered regularly interspaced short palindromic repeats) systems show great promise for creating a class of next-generation molecular diagnostics that can meet ASSURED criteria with minimum trade-offs. In their native microbial systems, CRISPR systems function in adaptive immunity14 by identifying foreign invading nucleic acid molecules and degrading them using CRISPR-associated (Cas) enzymes. Target recognition is based on sequence complementarity with CRISPR RNA (crRNA),15 allowing CRISPR systems to be programmed and engineered to target any DNA or RNA molecule. CRISPR/Cas systems comprise a highly diverse family of enzymes that have expanded significantly since their initial discovery.15−17
CRISPR systems are categorized into two classes and six types based on their evolutionary relationships.16 Class I is characterized by complex, multiple effector proteins, except for the single effector Cas7–11 RNA nuclease,18 whereas class II is characterized by the presence of a single effector protein. Several engineering systems have been developed based on the highly programmable nature of CRISPR and the simplicity of class II enzymes, including systems for genome,19 epigenome,20 and transcriptome21 editing, bioimaging,22 and nucleic acid detection.23 Of the class II systems, Cas12 and Cas13 enzymes (Type V and VI, respectively) are most often deployed in CRISPR-based diagnostics (Figure 1). Both types of effectors have a nonspecific transcleavage activity that is activated upon binding to the target nucleic acid. Both enzymes are also guided by RNA, but Cas13 recognizes single-stranded (ss)RNA and trans-cleaves ssRNA, while Cas12 effectors recognize both double-stranded (ds)DNA and ssDNA, with a PAM (protospacer adjacent motif) sequence limitation for dsDNA targets,24 and cleaves ssDNA in trans.15 Most diagnostic tools make use of this promiscuous transcleavage activity by designing fluorescently quenched reporter molecules (ssDNA or ssRNA) that are cleaved upon activation of the Cas effector with the correct target, releasing a fluorescent signal.25 Besides Cas12 and Cas13, Cas9 (Class II type 2) was also engineered to detect nucleic acids in various systems.26−29 In general, CRISPR systems are engineered for diagnostic purposes in order to create molecular detection platforms suitable for POC use to ensure fair and equitable access to disease diagnostics.
Figure 1.

Mechanisms of action of Cas12 and Cas13. (a) Cas13 is activated upon binding of the crRNA to ssRNA targets, leading to the activation of collateral cleavage activity by the HEPN (Higher Eukaryotes and Prokaryotes Nucleotide-binding) domain. Any bystander ssRNA molecules are cleaved, including fluorescently labeled reporters, releasing a fluorescent signal. (b) Cas12 can bind to ssDNA targets without the need for a PAM (left) or to dsDNA (right) targets, which requires a PAM. Target binding activates the RuvC nuclease domain, resulting in collateral cleavage of ssDNA reporters. Cis-cleavage of ssDNA occurs near the 3′-end (right side of crRNA binding), whereas trans-cleavage of the same target occurs near the 5′-end (left side of crRNA binding).30
In this review, we discuss CRISPR-based molecular diagnostics tools, with a focus on key challenges and potential strategies for their mitigation. In particular, we focus on sample processing and discuss methods for quick sample preparation. Additionally, we describe two potential pathways for sample-to-answer solutions by combining sample processing, nucleic acid detection, and readout in a single-step, one-pot reaction; the user would simply need to load the sample. The two pathways are (1) creating compatible chemistries and (2) automation. We also discuss the problem of sample storage and lyophilization. Although this industrial problem requires expertise in formulation development and process engineering, we describe a simple, rational design for the successful lyophilization and long-term storage of CRISPR systems (and potentially any protein-based system). Finally, we discuss POC signal interpretation and multiplexing, which could potentially be integrated with Digital Microfluidics (DMF).
2. Revolutionizing Disease Diagnostics with CRISPR
2.1. Approaching a Gold-Standard Test Using Preamplification
The clinical use of CRISPR systems to detect pathogens is often challenged by the low titer of the pathogen.25 Introducing a nucleic acid preamplification step is usually required to achieve a clinically relevant limit of detection (LoD). To date, the most accurate nucleic acid amplification method is PCR, which relies on an expensive thermocycler and trained personnel.8 In-field applications, however, favor equipment-free isothermal amplification methods. Although isothermal amplification methods are susceptible to false positives,31,32 adding a specificity layer (such as a CRISPR system) overcomes the problem of nonspecific amplification.25 Several isothermal amplification methods have been described,33 but Recombinase Polymerase Amplification (RPA) and Loop-mediated Isothermal Amplification (LAMP) have dominated the field of CRISPR-based diagnostics.25
Both RPA and LAMP operate in simple, single-step, single-temperature reactions. RPA utilizes a recombinase, a DNA polymerase, and single-strand binding proteins (SSBs) to amplify a target dsDNA at a constant temperature. First, the recombinase-primer complex scans the dsDNA and catalyzes the binding and formation of a primer-template junction (PTJ). SSBs then stabilize the ssDNA and prevent the ejection of the primer by branch migration. A strand-displacing DNA polymerase extends the PTJ, forming dsDNA that serves as a substrate for another cycle. RPA operates at a single temperature (37–42 °C) and amplifies the product in as little as 30 min.34 LAMP uses 4 to 6 primers and a strand displacing DNA polymerase to amplify a target DNA molecule. LAMP operates at 55–65 °C and utilizes special primers to create loop-containing dumbbell-shaped DNA structures. These loops are single-stranded regions, offering multiple sites for primer annealing and extension. Large concatemers of DNA molecules of different sizes eventually form, each containing multiple amplicons of the initial target. To amplify RNA targets, a reverse transcriptase step can be incorporated into the reaction.35
The CRISPR system was first used in diagnostics to detect viruses, bacterial species, and human single nucleotide polymorphisms (SNPs) using Cas13. This assay, termed Specific High-Sensitivity Enzymatic Reporter UnLOCKing (SHERLOCK), harnesses the collateral RNase activity of Cas13 to excise bystander RNA reporter molecules upon activation by the target ssRNA. The sensitivity of this system using the Cas13 variant LwaCas13a from Leptotrichia wadei was significantly enhanced by incorporating RPA as a preamplification step. In field diagnostics, nucleic acid is extracted and amplified by RPA. If the target ssRNA is present, the Cas13-crRNA ribonucleoprotein (RNP) complex is activated. Fluorescently quenched ssRNA reporter molecules are cleaved, releasing the fluorophore signal. SHERLOCK can differentiate between similar ssRNAs with up to a two-base difference. To enable SNP differentiation, a synthetic base is incorporated into the spacer sequence of the crRNA, generating two mismatches to prevent the activation of the RNP complex. This system was successfully used to detect bacterial pathogens, viruses, and SNPs at relevant clinical concentrations.23,36
SHERLOCK was improved by incorporating (1) multiplexing, (2) quantitative measurements, (3) signal amplification with Csm6, and (4) a simple lateral flow readout.37 The simultaneous detection of different targets requires unique reporters for each target. Since each Cas13 variant has a dinucleotide base preference for collateral activity, three Cas13 variants (LwaCas13a, PsmCas13b from Prevotella sp. MA2016, and CcaCas13b from Capnocytophaga canimorsus Cc5) and one Cas12 enzyme (AsCas12a from Acidaminococcus sp. BV3L6) were multiplexed to detect four different targets and cleave four different reporters. To enable quantitative measurements, a feature that is missing in the original SHERLOCK assay, the concentration of RPA primers was controlled to prevent saturation of the RPA product. In addition, the signal was enhanced by incorporating a signal amplification module to amplify weak or previously undetectable signals and improve the detection time (Figure 2b). The enzyme Csm6 is a dimer containing a CRISPR-associated Rossmann Fold (CARF) domain for activation and a HEPN domain for collateral RNA cleavage. The CARF domain is activated by hexa- or tetra-adenylate (A6 or A4) with a 3′ end containing 2′,3′-cyclic phosphate. Upon binding to the activator, the CARF domain activates the HEPN domain, which then cleaves bystander RNA (Figure 2b).
Figure 2.

Enhancing the sensitivity of the CRISPR/Cas13 detection assays by targeting different regions simultaneously and amplifying the signal with Csm6. (a) In targeting with multiple RNPs, different crRNAs are designed to target different regions of the target RNA molecule, allowing one molecule to activate multiple RNPs, thereby enhancing the signal generated by a single target molecule. (b) In cascading, a reporter is cleaved by Cas13 and activates Csm6, which then further cleaves reporter molecules.
Based on the observation that Cas13 can successfully cleave reporter molecules yielding 2′,3′-cyclic phosphate end, it was hypothesized that Csm6 could be used to further amplify the signal generated by SHERLOCK. The authors designed a “protected” reporter that can (1) generate a signal upon cleavage with Cas13, and (2) serve as an activator for Csm6 (Figure 2b). This design led to a marked increase in the signal and improved the kinetics of the SHERLOCK reaction. Finally, a lateral flow readout was incorporated into SHERLOCK to make it easier to interpret the signal visually. Altogether, SHERLOCK.v2 has 250-times greater sensitivity compared to the original SHERLOCK.37
Coupling amplification and detection in one pot was the next milestone. Both versions of SHERLOCK were performed in two pots: RPA in one, followed by detection using Cas13 in the other. SHINE (Streamlined Highlighting of Infections to Navigate Epidemics) was designed to reduce the number of handling steps by combining amplification and detection in a one-pot SHERLOCK assay to detect SARS-CoV-2.38 This one-pot setup was achieved by optimizing the reaction conditions, pH, temperature, and monovalent ion concentrations and creating an environment compatible with Cas13 and recombinase activity. Combining these steps in a single tube did not significantly compromise the sensitivity of the assay (Table 1).38 Similar to SHINE, we previously developed a system that combines reverse transcription LAMP (RT-LAMP) and Cas13 detection in a single tube.39 The assay, termed OPTIMA-Dx, utilizes a thermostable Cas13a (TccCas13) derived from Thermoclostridium caenicola and optimized buffer conditions to perform reverse transcription of SARS-CoV-2 RNA, followed by LAMP, transcription by hiT7 RNA polymerase, and recognition and detection by TccCas13. In under an hour, the OPTIMA-Dx assay achieved 95% sensitivity for RNA extracted from nasopharyngeal swabs (Table 1, Figure 3a).39
Table 1. One-Pot Amplification and Detection Modules.
| technique | preamplification | detection | temp (°C) | time (min) | LoD (copies/μL) | clinical sensitivity | ref |
|---|---|---|---|---|---|---|---|
| HOLMESv2 | LAMP | AacCas12b | 55 | 60 | 6 | NA | (42) |
| DETECTR | RPA | LbCas12a | 37 | 60 | 0.6 | 96%, Cta value not reported | (40) |
| iSCAN-v2 | RT-RPA | AapCas12b | 42 | 30 | 8 | 93.75%, Ct < 34, SARS-CoV-2 nasopharyngeal swab | (43) |
| STOPCovid | RT-LAMP | AapCas12b | 60 | 45 | 0.3 | 100%, Ct < 37 | (45) |
| SHINE | RT-RPA/T7 transcription | LwaCas13a | 37 | 50 | 10 | 94%, Ct < 27 | (38) |
| OPTIMA-Dx | RT-LAMP/T7 transcription | TccCas13a | 56 | 60 | 10 | 95%, Ct (14–34), SARS-CoV-2 nasopharyngeal swab | (39) |
Ct: cycle threshold.
Figure 3.

One-pot amplification and detection via OPTIMA-Dx and iSCAN-v2. (a) OPTIMA-Dx begins with reverse transcription of ssRNA targets, followed by LAMP, transcription of the LAMP product, and detection using TccCas13. All processes are performed in a one-pot, single-step reaction at 56 °C for 1 h. (b) ISCAN-V2 is used to detect ssRNA molecules by reverse transcription, RPA, and detection using Cas12 in a one-pot, single-step reaction at 42 °C for 30 min.
Cas12 effectors have also been utilized for diagnostic applications. Cas12a recognizes both dsDNA (requires a PAM) and ssDNA (does not require a PAM) targets and features ssDNA collateral cleavage activity. This collateral cleavage activity has a higher turnover rate than target-specific cleavage activity.40 The first assay to report Cas12-based diagnostics was termed HOLMES (a one-HOur Low-cost Multipurpose highly Efficient System). In HOLMES, target is first preamplified with PCR and then detected with Lbcas12 which cleaves ssDNA reporters. Challenged with different targets (DNA viruses and RNA viruses), HOLMES had a sensitivity similar to that of SHERLOCK and higher than that of PCR alone.41
Coupling amplification and detection in one-pot was the next milestone for Cas12-based diagnostics. In HOLMESv2, the detection setup was further simplified by replacing PCR amplification with LAMP-based amplification. To further simplify the reaction into a one-pot detection system, the authors coupled LAMP (which operates at high temperature) to a thermostable Cas12 effector, AacCas12b from Alicyclobacillus acidoterrestris. For RNA detection, a reverse transcriptase is usually incorporated into the reaction to make cDNA templates for LAMP amplification. In HOLMESv2, however, the authors utilized Bst 3.0 DNA polymerase which can recognize both RNA and DNA as a template, eliminating the need for a reverse transcription step. The one pot system had a limit of detection of 6 copies/μL for both DNA and RNA target.42 DETECTR is another one-pot assay that combines RPA with Cas12a detection and collateral cleavage of ssDNA reporters. The assay was validated using human papillomavirus (HPV), a dsDNA virus, with a LoD in the attomolar range (0.6 copy/μL).40 Whereas the main target in DETECTR is dsDNA, iSCAN-v2 adds a reverse transcription reaction to detect RNA molecules using Cas12 (Figure 3b). iSCAN-v2 combines amplification with RT-RPA and detection with AapCas12b from Alicyclobacillus acidiphilus in a single tube,43 simplifying the reaction setup compared to the previous version of iSCAN.44 Similarly, STOPCovid is a one-pot detection assay that combines thermostable Cas12b with RT-LAMP to detect SARS-CoV-2.45
The use of Cas12 or Cas13 combined with a preamplification step requires the addition of several enzymes and reagents, which affects the final price of the assay. However, the combination of isothermal amplification and detection in a single tube represents a significant milestone toward the development of a sample-to-answer CRISPR-based solution.
2.2. Amplification-Free CRISPR-based Detection
Independent of an error-prone nucleic acid preamplification step, Cas13 and Cas12 effectors can be harnessed to detect nucleic acids that are naturally present in the picomolar range in a sample.25 The picomolar LoD of some Cas effectors is attributed to the high turnover rate of the associated trans-cleavage activity.46 Besides detecting genomic DNA,47 microRNAs (miRNAs),48 and mRNA (mRNA),49 Cas effectors can detect SARS-CoV-2 mRNA at the early stages of symptom onset without a preamplification step.50 Here, we discuss attempts to enhance amplification-free detection using Cas12 and Cas13 effectors via targeting with multiple RNPs and cascading.
Combining several Cas effectors to detect different regions of the same target sequence can dramatically increase the sensitivity of detection (Figure 2a). The goal is to activate multiple Cas effectors using a single target molecule. Amplification-free detection of SARS-CoV-2 was achieved using triple LbuCas13a (from Leptotrichia buccalis) RNPs.50 LbuCas13a was chosen because it showed the highest collateral cleavage activity among several Cas13 variants tested.51 To enhance sensitivity, two Cas13-crRNA RNP complexes were used in tandem to detect two different regions of the SARS-CoV-2 genome, improving the sensitivity 10-fold. To further improve the sensitivity, a third RNP complex was added to generate triple RNP complexes. The triplet setup had a 30-fold greater sensitivity compared to that of a single RNP, reaching levels comparable to amplification-based detection modules. By removing the amplification layer, quantitative measurements were easily obtained by correlating the signal generated to a specific copy number using a standard curve.50,51 However, the clinical sensitivity was compromised—the triplet RNP reaction detected samples in the 14.37–22.13 Ct range, which is far below the Ct value of 37 reported in one amplification-enabled CRISPR-based study.45
Coupling Cas effectors to signal amplification modules (cascading) further enhances the LoD of amplification-free methods. The enzyme Csm6 is one such signal amplifier that is compatible with Cas13. In one instance, LbuCas13a was successfully coupled to the Csm6 variant TtCsm6 from Thermus thermophilus to detect SARS-CoV-2 RNA without the need to amplify the RNA input, enhancing the sensitivity compared to using Cas13 alone.52 In this study, Liu et al. combined eight LbuCas13a RNPs and coupled them to Csm6, attaining a relatively unprecedented LoD of 31 extracted SARS-CoV-2 genomic copies/μL.52 Coupling Csm6 not only enhanced the sensitivity of the assay but also decreased the detection time from 2 h to 20 min. Overall, targeting with multiple RNPs and cascading (via Csm6 signal amplification) achieved clinical validation similar to that reported for amplification-enabled modules (with a Ct value of 33 and below) and reduced the detection time without the need for amplification.
2.2.1. Amplification-Free Detection Using Class I CRISPR Systems
Csm6 was also harnessed in its native type III CRISPR/Cas complex to detect SARS-CoV-2 mRNA.53 The TtCsm CRISPR Complex is composed of the Cas10 polymerase, Cas10 DNase, Csm3 RNase, and Csm2, Csm4, and 5Csm. Ideally, upon target recognition, Cas10 polymerase is activated and converts ATP into three products: cA4 (cyclic tetra-adenylate), protons, and pyrophosphates (PPi). The authors designed three signal detection modules to detect these three products. First, cA4 activates the collateral RNase activity of Csm6, cleaving fluorescent probes. Second, the protons alter the pH of the solution, which is detected by observing the color change of litmus paper. Third, PPi is detected colorimetrically and fluorometrically using the metal indicator calcein.53 Overall, the TtCsm system is less sensitive than the Cas13 amplification-free detection modules.
2.2.2. A Single Cas Effector Detects Different Targets in an Amplification-Free Approach
Having a high detection limit in the picomolar range is beneficial and can be harnessed to detect a target that is naturally present at high concentrations. This high sensitivity has been exploited to detect mRNA,49 genomic DNA,47 and differentially expressed nucleic acids.48 Such a detection limit is particularly beneficial when an overexpressed gene is detectable while the same gene expressed at normal or below normal levels falls below the LoD. For example, a Cas13-based detection system was developed to detect only high levels of microRNA 19b (miR-19b), which acts as a biomarker for patients suffering from medulloblastoma, a type of brain cancer. Amplification-free detection of miR-19b was successfully performed by integrating Cas13a detection with glucose oxidase (GOx) signal generation.48 In this system, Cas13a detects miR-19b and cleaves a biotin-FAM labeled reporter RNA. FAM is attached to GOx; this enzyme oxidizes glucose to yield H2O2, which is detected using an electrochemical cell. Following 4 h of incubation, the reaction mixture (Cas13a, reporters, and miRNA) is applied to a microfluidic chip containing streptavidin, which captures biotin. If the reporter is not cleaved, it is fully immobilized on the chip, and GOx generates an H2O2 electric signal upon the addition of glucose. However, if the reporter is cleaved, GOx cannot bind and is washed off, and the addition of glucose does not yield a signal. Amplification-free detection could be utilized to detect medulloblastoma, since only high levels of mir-19b can be detected using the Cas13-GOx system, whereas normal levels fall below the LoD.
More recently, a new study offered a protein engineering strategy to improve the sensitivity of Cas13 in amplification-free detection systems. By fusing an RNA binding domain (RBD) to LwaCas13a (as a Cas13 model system), the activity of the enzyme was boosted owing to the enhanced binding to RNA species in the solution. After several rounds of protein engineering, particularly testing 7 different RBDs at various insertion sites, the group attained a super active LwaCas13a variant that is 518% higher fluorescence relative to the wild type version for one particular target. They further enhanced the RNA reporter by increasing its length to fit into the catalytic residues. The engineered LwaCas13a and reporters were deployed in an electrochemical cell, and, without preamplification, the approach was sensitive to 0.6 copies/μL of synthetic RNA and 12 copies/μL in clinical samples in 30 min. They attained a new benchmark for amplification-free detection that is very comparable, and probably better than some amplification-based detection modalities.54 However, more robust clinical validation is needed. The strategy outlined in the study can be applied to other Cas13 variants, particularly the insertion site and the selected RBDs. For thermostable Cas13 variants such as TccCas13a and HheCas13a, the choice of RBDs can be different accounting for the thermophilic nature of these effectors.
The elimination of a DNA amplification step, reverse transcription in case of RNA targets, and transcription event in case of using Cas13 effectors has reduced the complexity of the detection assays, thereby reducing the price of reagents and the time needed to complete the assay.
3. Challenges and Potential Solutions
Designing a POC assay is a challenging task, even with CRISPR-based systems. Although the SHERLOCK assay was designed for use in POC settings,23 the FDA-approved version is only approved for use in Clinical Laboratory Improvement Amendments (CLIA)-certified laboratories capable of performing complex testing.55 Two major challenges limit the use of CRISPR-based diagnostics for POC and home-use settings. First, sample processing requires lengthy protocols and, in most cases, depends on instrumentation. Second, reaction components must be stored and transported at ultralow temperatures. The ultimate goal is to create a single-step assay that can be widely deployed without depending on a cold-chain supply. Additionally, creating a POC assay that meets all the ASSURED criteria does not necessarily guarantee that the assay can be used in home-based settings. The assay must be simple and easy to perform by a typical untrained person with a minimal chance of obtaining erroneous results.56 Multiplexing and data sharing represent additional desirable features that can be incorporated in home use diagnostic kits. Given these challenges, the Lucira Check It COVID-19 test kit is currently the only FDA-approved molecular diagnostic test kit for home use.
3.1. Sample Processing: A Key Challenge for Most Diagnostic Applications
Two approaches could potentially solve the challenge of sample processing and provide a sample-to-answer solution: automation by creating a simple device and/or assay simplification by creating compatible chemistries. The goal is to combine all reactions into a single-step assay or to encapsulate a multistep assay in a simple device.
3.1.1. Recent Innovations in Reaction Chemistry to Simplify Sample Processing
Minimizing the steps required to perform an assay is an essential step toward designing POC and home-use kits. A kit that provides a sample-to-answer solution with minimal handling from the end-user would be ideal for the home-use diagnostics market. Although amplification-free and amplification-based detection can be performed in a single tube, sample processing remains a bottleneck.
Traditional sample processing technologies require several liquid handling steps and special instruments.57 Sample processing varies depending on the sample type, nucleic acid category (DNA or RNA), and downstream application. Nonetheless, nucleic acid extraction generally requires three steps: sample lysis, inactivation of nucleases, and purification. Traditional nucleic acid extraction platforms employ liquid-phase or solid-phase extraction methods.57 In liquid-phase extraction, the sample is lysed in a phenol-based solution, which forms a density gradient, and nucleic acids are separated from the rest of the solution by centrifugation.58 Solid-phase extraction utilizes the chemical properties of nucleic acids to bind to an affinity matrix such as cellulose,59 silica,60 or functionalized magnetic beads.61 Highly pure nucleic acids can be extracted using these platforms. However, several liquid handling steps are required for washing and elution, and all techniques (except functionalized magnetic beads) require a centrifuge. Additionally, the recovery efficiency is compromised at the expense of obtaining pure samples.
Simpler, rapid, equipment-free sample processing approaches can be employed for field diagnostics.25 The sample is processed (but not purified) in the presence of a lysis buffer that disrupts the cells and releases and stabilizes nucleic acids, with little or no effect on downstream analysis. In a typical direct lysis protocol, the sample is incubated in buffer for a certain period of time under a specific condition (such as high temperature) and is directly used for downstream analysis.62 Even though the nucleic acids are not purified, the majority of nucleic acids is recovered using the quick extraction approach (Figure 4).
Figure 4.

Current and envisioned sample processing techniques. Traditional nucleic acid extraction: Column-based method: Many steps are required, from sample collection to lysis, washing, and elution. As a result, the final product is highly purified. One-step extraction followed by detection: After sample collection, only a lysis step is required, after which a portion of the sample is loaded into the detection mix. Envisioned one-step sample-to-answer: Simple diagnostics tests are needed to meet the growing demand for home-based kits. Such kits would require minimal handling from the user, such as self-collection of the sample and loading it into a one-step assay system.
Several quick extraction protocols are now available (Table 2), and some have been used directly for CRISPR applications, such as the HUDSON protocol (Heating Unextracted Diagnostic Samples to Obliterate Nucleases).62 Besides merging SHERLOCK amplification and detection in a one-pot reaction, SHINE utilized HUDSON as a quick extraction protocol. The sample is processed in only two steps (heat denaturation and chemical reduction), releasing nucleic acids that are ready for downstream SHINE detection.38,62 HUDSON further expanded sample types that are amenable for quick extraction. Virus lysis and RNase inactivation were achieved by heat denaturation and chemical reduction using TCEP (tris(2-carboxyethyl)phosphine) and EDTA. TCEP is a reducing agent that denatures proteins and Rnases and decreases the viscosity of saliva by denaturing mucins by breaking disulfide bonds; EDTA chelates divalent ions, which are cofactors of Rnases. Briefly, the heating steps were optimized for different samples. For example, inactivation of Zika virus particles and Rnases was achieved in blood, plasma, serum, and saliva via two heating steps in the presence of 100 mM TCEP and 1 mM EDTA: (1) heating at 50 °C for 5 min followed by (2) increasing the temperature to 64 °C for an additional 5 min. The resulting HUDSON products were directly used for downstream detection with SHERLOCK without the need for purification steps.62
Table 2. Quick Extraction Methods.
| sample processing module | steps | time (min) | temperature (°C) | compatible with | reference |
|---|---|---|---|---|---|
| HUDSON | Two steps (Heat treatment; Chemical denaturation) | 10 | 50–64 | SHINE (RT-RPA/Cas13) | (62) |
| DISCoVER | Two steps (Heat treatment; Chemical denaturation) | Not reported | 75 | DISCoVER (RT-LAMP/T7 transcription/Cas13) | (63) |
| QuickExtract | One-step (Heat treatment) | 5 | 95 | RT-LAMP/Cas12 | Lucigen45 |
| SHINE-v2 | One-step (Mixing sample and buffer) | 5 | 25 | SHINE (RT-RPA/Cas13) | (64) |
| Vre | One-step (Mixing sample and buffer) | 5 | 25 | LAMP | Sigma-Aldrich |
DISCoVER (Diagnostics with CoronaVirus Enzymatic Reporting) represents another simple approach to sample processing. In DISCoVER, sample processing is simple but requires two manual steps: (1) lysis and RNA stabilization are mediated by an initial heat treatment at 75 °C, followed by (2) the addition of TCEP/EDTA. Even though these reagents should interfere with downstream reactions, low concentrations of TCEP/EDTA were shown to lyse samples and protect RNAs without later interference.63 Despite the simplicity of HUDSON and DISCoVER, they still require two liquid handling steps to process the samples.
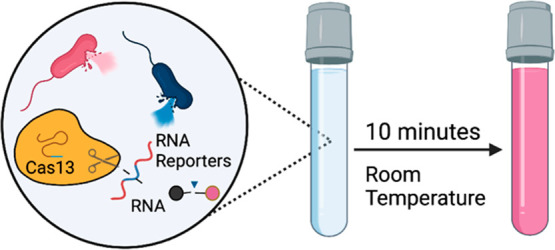
To further reduce the number of steps involved in sample processing, several one-step quick extraction protocols have been developed. QuickExtract (Lucigen) was developed to lyse viral particles via a one-step heat treatment in lysis buffer at 95 °C for 5 min. The inactivated sample is compatible with One-step RT-LAMP/Cas12 detection.45 A simpler, 5 min, room temperature lysis protocol was developed in SHINEv2.64,65 The authors reasoned that the commercial lysis buffer FastAmp (Intact Genomics), which functions at room temperature and is compatible with isothermal amplification, might be compatible with their downstream isothermal amplification and CRISPR detection module. First, they tested if the lysis method could inactivate RNases. After incubating a saliva sample with FastAmp and RNaseAlert (ThermoFisher), a significant amount of the RNA reporter was degraded, indicating that the commercial FastAmp lysis buffer did not successfully inactivate nucleases in the sample. Addition of RNase inhibitors complemented the activity of FastAmp, allowing the samples to be inactivated at room temperature after only 5 min. Following sample inactivation, 10% (v/v) of the inactivated sample was added to the SHINE reaction. Increasing the amount of sample decreased the performance of the assay, indicating that lysis reagents must be maintained at a certain ratio to prevent them from inhibiting downstream reactions. The approach taken in SHINEv2 is simple; nonetheless, the assay is still far from sample-to-answer. The assay separates sample processing from detection and incorporates a lateral flow readout, which requires an additional dilution step.
3.1.2. Innovating New Chemistries toward a Single-Step Sample-to-Answer Solution
There are several challenges to assay development to reaching the sample-in-answer-out stage. First, lysis reagents are usually incompatible with downstream processing. The strategies discussed above involve optimizing the concentrations and ratios of lysis reagents and choosing the proper buffer components. Notably, all sample processing techniques described above were designed for viral samples, which are difficult to lyse and require very harsh conditions to degrade the envelope and capsid. Since viral capsids are primarily composed of proteins,66 lysis reagents that denature viral capsids might denature proteins used in downstream analysis. The capsids can be degraded chemically using strong reducing agents such as TCEP,64 physically by heat denaturation,26 or biologically by protease treatment.67 An innovative method is needed to develop a lytic agent that does not interfere with downstream detection.
The ultimate goal of CRISPR molecular diagnostics is to combine sample processing, detection, and readout in a single tube (Figure 4). One attempt to combine these steps in a single tube was recently reported.68 Li et al. developed a colorimetric RT-LAMP assay to detect SARS-CoV-2 in nasopharyngeal swabs using a single-step sample-to-answer approach. Since RT-LAMP buffer typically contains low concentrations of surfactants (Triton X-100), the authors reasoned that the RT-LAMP reaction on its own might lyse the sample. Although this approach failed to lyse the viral particle, by adding 6% (v/v) formamide (a mild detergent) along with Triton X-100, the authors successfully performed one-pot detection of SARS-CoV-2 from NP samples in viral transport medium (VTM). Spiking a pseudovirus in VTM, the LoD was approximately 8 copies/μL, which is highly comparable to that of RT-qPCR. For clinical validation, 45 NP swabs (30 negative and 15 positive) were incubated in one-pot master mix, which achieved 100% sensitivity. However, the Ct value of the positive samples was not reported. Still, the choice of a specific ratio of mild surfactants led to the successful lysing of virus particles without interfering with the RT-LAMP reagents.68 The use of such an approach with CRISPR systems could in theory generate a one-pot sample-to-answer assay.
Another strategy is to detect easier-to-burst targets such as bacterial cells. In protein research, detergents are used to break open cells and release and stabilize proteins.69 Detergents are categorized as ionic (denaturing) or nonionic (nondenaturing).69 Denaturing detergents such as SDS can solubilize membranes, disrupt protein–protein interactions, and denature proteins. By contrast, nondenaturing detergents can solubilize hydrophobic membranes and proteins but do not denature proteins.69 In principle, nondenaturing detergents can degrade membranes without affecting proteins. Optimizing detergent concentrations and lysis conditions, such as temperature, pH, and ionic salt contents, could eventually lead to the desired equipment-free one-step solution.
This approach can be utilized to detect bacterial pathogens. Although most bacterial pathogens are surrounded by a cell wall that provides an extra layer of defense, breaking the cell wall is not as challenging as breaking viral capsids. Lysozymes are typical lytic components that are used routinely for protein purification from Escherichia coli.23 These enzymes degrade the cell wall by catalyzing the breakage of peptidoglycan bonds, leading to cell death. After the cell wall is degraded by lysozymes, the cell membrane is still intact. A combination of lysozymes and detergents can however be used to break open bacteria, potentially without interfering with downstream analysis.
3.1.3. Engineering Automation: Performing Multistep Assays in a Single Step
Encapsulating reagents in a compact device and processing these reagents by automation provide another approach to creating a user-friendly diagnostic kit. Several well-established platforms can serve this purpose, including tube-based devices, paper-based platforms, and microfluidics modules. These platforms provide several sample-to-answer solutions, which have been comprehensively reviewed.57 Here we discuss attempts to automate CRISPR-based methods and provide successful examples of FDA-approved molecular diagnostic devices for related LAMP-based and PCR-based assays.
A simple test, miSHERLOCK (minimally instrumented SHERLOCK), was designed to minimize the liquid handing steps in SARS-CoV-2 detection.70 In two user-friendly steps, sample processing (including nucleic acid purification and concentration), amplification, detection, and readout are performed in under an hour. First, the user loads saliva into a collector containing the lysis reagents and turns on the heater. After 6 min, the nucleases are deactivated, and the saliva (along with the lysis reagents) is filtered through a polyethersulfone (PES) membrane via gravity and capillary forces from a downstream cellulose absorbent filter, which concentrates the nucleic acids onto the PES membrane. Second, the user transfers the collector into the SHERLOCK reaction chamber and presses a plunger to puncture a water reservoir, which allows the PES membrane along with prestored water to enter the reaction chamber and rehydrate the lyophilized SHERLOCK reagents. After one hour, the device yields a readable fluorometric signal. The device costs only 15 USD, and although it is not completely automated, the assay requires only two simple steps.
Moving one step toward semiautomated systems, DISCoVER is an emerging sample-to-answer platform that combines lysis, amplification, and detection in microfluidic modules.63,65 The platform does not require an extraction step and relies instead on sample denaturation and chemical reduction to release nucleic acids. Amplification is mediated by RT-LAMP and T7 transcription, followed by Cas13 detection in a separate compartment. The assay requires two separate steps: manual sample processing and automated detection reactions.
The detection reaction (amplification, detection, and readout) is fully automated in microfluidic modules. The inactivated saliva sample is loaded into a compact microfluidic device. The device is composed of air displacement pumps, a separate heater for the LAMP reaction, a Thermoelectric Heater/Cooler (TEC) to maintain the Cas13 chamber at 25 °C, and a custom fluorescence detector. First, lysed saliva is dispensed into the LAMP chamber and heated at 65 °C for 30 min. At the end of the LAMP reaction, a valve opens and 4 μL of the mixture is metered into to the Cas13 chamber (which is protected from heat by the TEC). The Cas13 mixture is then automatically transferred to the detector.63
It is worth noting that the transition from a wet lab assay to a microfluidic DISCoVER solution led to reduction in sensitivity. Whereas a wet lab assay achieved 93% positive percent agreement (PPA) for samples ranging from 15 to 35 Ct (n = 33),63 validation of the device on clinical samples revealed a marked drop in sensitivity. The group only reported validation of clinical samples below 21 Ct values, likely because the sensitivity was reduced due to insufficient mixing of the reagents. Nonetheless, the complete assay takes 35 min to complete. Importantly, this assay requires separate amplification and detection modules. The authors would have created a simpler device by utilizing one of the recently developed one-pot amplification/detection module with thermostable Cas13.39 In addition, the authors did not provide an automated sample denaturation module: sample processing required two steps: heating, followed by the addition of TCEP and EDTA.
3.1.4. Innovations in Automation in Other Diagnostic Applications
Full automation represents another approach to performing multistep assays in a user-friendly manner. However, full automation incurs additional costs, which might limit the use of such systems for POC diagnostics in low-resource settings. Check It Covid (Lucira) is an example of a fully automated device and is currently the only molecular test that has been granted FDA approval for home use.71 This assay is based on colorimetric RT-LAMP chemistry, which changes the color of a halochromic substance upon a change in pH resulting from target amplification by LAMP. The device is extremely easy to use; the user simply takes a self-swab and mixes it with a lysis buffer that works at room temperature. The user presses on the vial to allow a portion of the lysed sample to enter the embedded fluidic chambers and rehydrate the lyophilized RT-LAMP reagents. The device contains an internal heater that turns on once the fluidic chambers are filled, as well as an electronic processor and sensors for signal detection. LAMP reactions are known to have high false-positive rates.31,32 Nevertheless, the Check It Covid text shows high specificity (98% negative percent agreement) and high sensitivity (100% for samples below Ct values of 37.5). Moreover, the device does not require cold-chain storage during transport, since the company successfully lyophilized RT-LAMP reagents and developed room-temperature lysis reagents (both storage and lysis occur at room temperature). Despite its simplicity, the Check It Covid test currently costs 75 USD per test (https://checkit.lucirahealth.com), which limits its use in low-resource settings. Furthermore, in terms of pricing this test is not competitive with PCR tests, which can cost as little as 43 USD per test.72 Nonetheless, Check It Covid provides decentralized access to PCR-quality molecular diagnostics at home.
Accula is another platform for POC molecular diagnostics. This device performs sample processing and PCR amplification in three simple steps. First, room temperature lysis of a nasal swab sample is performed by mixing the swab with room-temperature lysis buffer. Second, droplets of the lysed sample are loaded onto the reaction cartridge using a simple preprogrammed disposable pipet. The reaction cartridge contains prestored lyophilized amplification reagents in separate compartments. Third, the user loads the reaction cartridge onto the Accula Dock, a nondisposable device containing pressure pumps, heaters, sensors, and other electronic parts required for the translocation of reagents, PCR amplification, and detection. Unlike the Lucira Check It test kit, Accula uses a multiuse Dock device and disposable reaction units, ultimately reducing the cost of electronic components. The Dock device costs 350 USD (https://www.fishersci.com/shop/products/accula-dock/D2000), and the reaction units are purchased in bulk for about 63 USD per test (https://www.fishersci.com/shop/products/accula-sars-cov-2-test-kit/COV4100). Accula was granted FDA Emergency Use Authorization (EUA) for POC use at CLIA-waived laboratories that meet some requirements for performing simple tests. Clinical validation revealed that the Accula test is 100% specific and 95.8% sensitive.73 Although quite expensive, a much simpler device can be adapted for a one-pot CRISPR-based assay and would potentially be less costly.
Encapsulating liquid handling steps in automated devices is a multidisciplinary problem that involves collaboration between biologists, engineers, and material scientists. Automated POC devices such as Check It Covid and Accula provide solutions for decentralizing disease diagnostics, but they do not ensure equitable access, especially in resource-constrained environments. There is still much room for innovation to bring the price of automated devices down to a suitable range. However, devising sample processing chemistries that are compatible with downstream analysis should eliminate the extra costs arising from the high level of automation required.
3.2. Lyophilization: A Rational Approach for Achieving a Successful Process
An ideal diagnostic platform would require simple storage and would avoid cold-chain transport in order to reduce final costs. A long shelf life would also reduce much of the burden of timely manufacturing. The majority of CRISPR-based platforms are reported in the solution format, and these require ultralow temperature to preserve the components, especially the guide RNAs and Cas enzymes.25
Proteins in solution are susceptible to physical degradation (precipitation and aggregation) and chemical degradation (e.g., oxidation, and deamidation).74,75 Although storage at ultralow temperatures can slow chemical degradation, the proteins are often exposed to harsh conditions during freezing, which can lead to aggregation and chemical denaturation due to a phenomenon called “freeze-concentrate”, as described below. Stabilizers can be added to protect the protein solution.74,75 Nonetheless, the cold-chain requirement during transport and storage poses constraints on large-scale deployment, especially in low-resource communities. On the other hand, RNAs tend to degrade in solution due to two main factors: RNases and hydrolysis,76 mandating the aliquoting and ultralow storage of guide RNAs at −80 °C for long-term use. Lyophilization provides a solution to the storage and transport problem, since successful lyophilization prevents or minimizes degradation of both proteins and RNA species. Lyophilization can also simplify the reaction setup. A lyophilized reaction format would only require a single reconstitution step by the end-user.
However promising, lyophilization exposes biologics to harsh conditions that can in principle render proteins inactive. Understanding the lyophilization process is essential for pinpointing, troubleshooting, and finding solutions to these issues. Lyophilization, i.e., dehydrating a solution to minimize reactions that can inactivate the reagents,77 involves three major steps: (1) snap freezing: freezing the solution below its triple point; (2) primary drying: sublimating the ice crystals to evaporate 95% of water contents by lowering the pressure and increasing the temperature; and (3) secondary drying (or desorption): removing the remaining 5% of water (hydration shell) bound to the reagents by further increasing the temperature.
Excipients (chemical additives) can minimize the damage and cushion biologics and active ingredients against harsh conditions during and after lyophilization.78 Lyophilization of RNAs in nuclease-free water lacking excipients reduces the rate of recovery. RNAs lyophilized in the presence of 10% trehalose retained high recovery rate when stored at 4 °C for 10 months.79 Most protein formulations are partially or fully denatured in the absence of appropriate excipients due to the stress encountered during freezing and dehydration. Practical advice and the rational design of formulation for protein lyophilization are discussed in Carpenter et al.,80 and the mechanism behind several excipients is detailed in Satoshi et al.78 Multiple parameters must be optimized to ensure successful lyophilization, which can be overwhelming. However, some general rules apply in most cases.
To protect the protein from freezing stress, three general approaches can be followed: (1) increasing the protein concentration; (2) decreasing the rate of cooling to prevent surface-induced denaturation; and (3) using a buffer (such as a Tris-based buffer) that does not change in pH during freezing.78,80 Denaturation usually occurs at the ice–water interface. Assuming that a finite number of proteins will occupy the ice–water interface, increasing the protein concentration reduces the fraction of degraded proteins. However, a high cooling rate results in small ice crystals, which increases the surface area of the ice–water interface. Accordingly, decreasing the rate of cooling should generate larger ice crystals with minimal surface area.
Protecting the protein only during freezing does not ensure successful lyophilization. Several approaches can be undertaken to protect the protein during drying and subsequent storage.78,80 A bulking agent such as mannitol or glycine can form a structure that prevents the protein from escaping. Among all excipients tested, nonreducing disaccharides provided proteins with the most stability during drying and storage, as they form hydrogen bonds with the protein to minimize damage from desorbing the hydration shell. Successfully employed nonreducing disaccharides include sucrose and trehalose.81 Proteins are also susceptible to aggregation during freezing and reconstitution (rehydration). Aggregation can be inhibited by including a nonionic surfactant such as Tween 20.82 Optimizing these variables should yield a stable formulation.
Still, designing a formulation for successful lyophilization does not guarantee a stable protein with a long shelf life.64 Some problems might arise due to contaminants present in the excipients. For example, some surfactants contain minute amounts of peroxides,83 which can facilitate the oxidation of proteins.84 It is therefore imperative to consider the purity of the additives.
The majority of the CRISPR-enabled diagnostics are described in a solution format and only a few attempted to lyophilize the reaction. In SHINEv2, lyophilization was improved by the using certain excipients and removing destabilizing agents. The activity of the master mix (including Cas13, crRNA, and RT/RPA proteins) strongly decreased after lyophilization. However, when excipients (sucrose as a stabilizer and mannitol as a bulking agent) were added, or when destabilizers (polyethylene glycol and potassium chloride) were removed, slight activity was retained, and when both approaches were combined, most of the activity of SHINEv2 was retained. However, most of the activity was lost for low input targets. To test the shelf life, the final lyophilized master mix retained activity for only 1 week when stored at room temperature,64 likely because the proteins aggregated due to the lack of nonionic surfactants in the master mix. In other attempts to lyophilize CRISPR systems, the reagents (including Cas nucleases and crRNAs) maintained either robust sensitivity85 or higher sensitivity than the control.86 Although no excipients were added to the solution before lyophilization, Lee et al. reported that the sensitivity of the SHERLOCK assay increased after lyophilization,86 perhaps because the sample volume input was much higher (more targets), as the reaction was resuspended with the processed sample input.
Several lyophilized reagents, as well as Lyo-ready reagents, are commercially available. Lyo-ready reagents and master mixes contain the necessary additives and are optimized for efficient lyophilization. Lyo-ready reagents relevant to CRISPR-based diagnostics include Lyo-ready reverse transcriptase and isothermal DNA polymerases (ThermoFisher), as well as Lyo-ready RT-LAMP master mix (Meridian Bioscience). These reagents contain almost no glycerol (an antifreezing agent) and contain the required classes of chemicals to allow lyophilization. These Lyo-ready reagents and master mixes are in principle ready to be lyophilized without optimization.
3.3. Multiplexing—Toward a Comprehensive Syndromic Approach to Molecular Diagnostics
Ideally, a diagnostic kit would feature surveillance capabilities for the simultaneous detection of several disease targets and allow the differentiation of distinct pathogenic strains. Multiplexed PCR from BioFire can detect 22 respiratory pathogens in a single sample,87 enabling a comprehensive syndromic approach and providing objective data to guide treatment and reduce antibiotic use. However, very few diagnostic methods for COVID-19 can detect all variants in one run.88−90
CRISPR-based diagnostics offer the opportunity for multiplexing at the POC. The collateral cleavage of Cas13 effectors is not completely promiscuous, i.e., they show a dibase preference for cleavage (Table 3). Characterization of several Cas effectors for reporter cleavage preference allowed four targets to be detected simultaneously in SHERLOCK.V2. The reporters used in this assay had three features: (1) susceptibility to cleavage by a single Cas effector, (2) resistance to cleavage by the rest of the Cas panel, and (3) fluorescence at a signature (distinct) wavelength.37 Adding Cas12a, which cleaves ssDNA, to the panel enabled further multiplexing: SHERLOCK.V2 was ultimately deployed using LwaCas13a, PsmCas13b, CcaCas13b, and AsCas12a with reporters tagged with FAM, TEX, Cy5, and HEX, respectively. Similarly, OPTIMA-Dx, which operates at 56 °C, achieved multiplexing with thermostable TccCas13 and AapCas12b in a single-pot, single-temperature reaction.39
Table 3. Dibase Preferences of Different Cas13 Effectors for Collateral Cleavage Activity.
Screening for Cas enzymes with orthogonal collateral cleavage activity can in theory increase the potential for multiplexing. However, multiplexing beyond four targets with SHERLOCK.v2 is limited by RPA preamplification.37 It is even harder to multiplex in a CRISPR-based modality with LAMP preamplification, which requires the use of 4–6 primers.39 However, further multiplexing can be unleashed in amplification-free modalities.
3.3.1. Engineering Fluidic Platforms for Multiplexing
Multiplexing (multiple targets detected per sample) and high throughput (allowing many samples to be processed) are important features that allow massive testing at the population level, especially during pandemics. There is always a trade-off between multiplexing and throughput; for instance, some RT–qPCR platforms are high throughput (testing 88 samples at the same time) but are not suitable for multiplexing (testing one to three targets at a time), whereas highly multiplexed platforms often suffer from low throughput: Cepheid Xpert Xpress detects four respiratory viral targets in 16 samples at a time, and BioFire multiplexed PCR detects a panel of 22 respiratory pathogens but only in one sample.87 CARMEN (Combinatorial Arrayed Reactions for Multiplexed Evaluation of Nucleic acids) is a new approach that combines high-throughput testing with multiplexing. CARMEN-Cas utilizes minute amounts of reagents and self-assembly of nanoliter-sized emulsion droplets to increase the multiplexing potential as well as the throughput of SHERLOCK. Whereas SHERLOCK can multiplex four different targets in the same reaction, CARMEN can perform 4500 reactions in one array and was clinically validated on a panel of 169 human-associated viruses in eight samples. In the CARMEN-Cas system, preprocessed and amplified samples (by PCR or RPA), as well as Cas13 detection mixes, are color-coded for optical identification. The color-coded samples and detection mixes are emulsified into nanoliter-sized oil droplets, pooled in a single tube, and loaded into the detection chip. Droplets of samples and detection mixes self-organize on the chip, which is designed to accommodate two droplets per well. Upon induction using an electric current, the emulsion droplets are mixed and the detection reaction starts.
This system was successfully used for massive multiplexing due to the use of color-coding technology. Using only four liquid fluorescent dyes, 1050 different color codes were generated by mixing the four color codes in different ratios. CARMEN-Cas detected targets in the attomolar range (1 copy/μL) and was validated using clinical samples. Although CARMEN exhibits excellent multiplexing and high throughput capabilities, a single CARMEN reaction is labor-intensive and can take up to 24 h to complete.91 To further simplify the setup, mCARMEN was developed using a Fluidigm integrated fluidic circuit (IFC) to eliminate the need for dropletization and color-coding in CARMEN.v1.92 The Fluidigm IFC chip can simultaneously detect 24 targets in 192 samples or 96 targets in 96 samples on specially separated reaction spots. The elimination of color-coding and dropletization, together with the use of an automated sample prep protocol, decreased the time needed for detection from >8 h to <5 h per run. However, the significant advances in multiplexing and throughput obtained using CARMEN and mCARMEN were achieved at the expense of increased time.
3.3.2. Multiplexing and Automation with Digital Microfluidics
Digital microfluidics (DMF) is a disruptive technology capable of controlling discrete, minute volumes of reagents. DMF enables extensive multiplexing and parallelization by controlling droplets, each acting as a biological or chemical reactor. The technology features all the advantages of traditional microfluidic platforms such as volume miniaturization, improved sensitivity and reaction time, decreased cross-contamination, portability, and full automation. Moreover, DMF features accurate control over discrete droplets, a great multiplexing potential, easy integration with signal detection modules, and elimination of propulsion devices for reagent movement.93,94 The movement of reagents on a DMF board is reviewed ref (95), but here we highlight the high multiplexing potential of DMF. By controlling the movement of reagents on spatially separated arrays and spots, several reactions can operate on the same chip. The multiplexing potential can be further increased when used in conjunction with orthogonal Cas effectors (Figure 5). The 4-plex potential of a single SHERLOCK.v2 reaction may be coupled with DMF in an automated manner, potentially yielding a one-for-all diagnostic kit in a portable POC device. Notably, sample processing is automated via DMF in the FINDER 1.5 RT-PCR COVID test (Baebies). DMF could potentially be combined with multiplexing for POC sample-to-answer comprehensive syndromic testing.
Figure 5.

Envisioned multiplexing with orthogonal Cas variants and DMF. Different Cas13 effectors have different dibase preferences for cleaving reporter molecules. Building upon this orthogonality, different reporter molecules carrying different fluorophores can be designed for a multiplexed reaction, with each Cas variant detecting one marker. Multiplexing can be expanded using a DMF board in which each spot would correspond to the same Cas variants, but with crRNAs for different targets other than the previous spot.
3.4. Signal Acquisition, Readout, and Data Sharing
Several approaches have been designed to detect the signal from CRISPR-based assays in a user-friendly manner, including fluorescent, lateral flow, and electrochemical-based methods. Fluorescence readouts interpret the fluorescent signal generated by the separation of a fluorophore from a quencher by collateral cleavage of the reporter. The signal can be read in the laboratory using a fluorometer or in the field using an inexpensive portable device such as a P51 molecular fluorescence viewer (Figure 6b).43 For lateral flow assay (LFA) readouts (Figure 6a), the commercially available system from Millenia 1T is widely used for CRISPR-based diagnostics.37 The LFA strip contains three lanes: gold nanoparticles coated with antifluorescein isothiocyanate (FITC) antibodies (which bind to fluorescein amidite [FAM]), streptavidin (which binds to biotin), and anti-FITC antibodies. The reporter molecule carries both FAM and biotin. After completing the CRISPR detection assay, the mixture is loaded onto the LFA strip and migrates through the three lanes. FAM molecules bind to FITC antibodies and carry them as they continue to migrate. If the reporter is intact, it will completely stop migrating further at the second lane where streptavidin binds to biotin, developing color at that lane. If the reporter is cleaved, the FITC-FAM complex migrates further and binds to the anti-FITC antibody, revealing color at the third lane. In electrochemical-based signal generation, a change in the electric current due to a chemical reaction is sensed using widely available electrochemical cells. Amplification-free detection of miRNA was achieved using a portable electrochemical cell,48 as discussed in detail above (Amplification-free CRISPR-based detection).
Figure 6.

Signal detection and data sharing. (a) LFA readout: A reporter labeled with FAM and biotin can be detected on the commercial Millenia 1T LFA strip. FAM has an affinity for the anti-FITC antibody, and biotin binds streptavidin. If the reporter is not cleaved, migration stops at the streptavidin lane. If the reporter is cleaved, the FITC-bound FAM reporter migrates further toward the anti-FITC antibody lane, giving a positive test result. (b) Fluorescent readout: A fluorescence signal can be visually interpreted using portable, affordable devices such as P51. (c) Digital signal acquisition and data sharing: Smartphone apps enabled by artificial intelligence can acquire a signal generated by a device such as P51, interpret the signal, and share it with concerned public health institutions.
Coupling signal acquisition with a data-sharing feature is quite important, especially during pandemics. Aman et al.43 developed a user-friendly artificial intelligence (AI)-enabled smartphone application for the digital acquisition of test signals and data sharing with centralized public health institutes (Figure 6c). This feature is particularly important for infectious diseases to guide preventative measures against the further spread of the infection.
4. Conclusion and Final Remarks
The field of CRISPR-based diagnostics began with the development of the SHERLOCK assay in 2017 and has greatly expanded since then. This technology could provide a new gold standard for molecular diagnostics and could potentially democratize disease detection. The challenges outlined in this article may be solved in the near term, possibly creating disruptive technologies for the rapidly growing molecular diagnostics market. Specifically, the sample processing challenge, which affects most molecular diagnostic approaches, requires innovations to create a sample processing chemistry that is compatible with downstream analysis. Alternatively, innovations in the area of automation could potentially yield a simple device that requires only sample loading from the end-user. The issue of long-term storage and transport could be solved with lyophilization, and the choice of the appropriate excipients could result in a successful lyophilization process.
Ideally, the final product should be resistant to harsh conditions, be independent from cold-chain storage and handling, be easy to use with zero-to-minimal training by the end-user, and be inexpensive. Although most CRISPR-based diagnostic platforms require a preamplification step at high temperature, inexpensive commercial heaters are widely available.70 Finally, most readouts are reported as fluorimetric signals, but several commercial innovations can translate a fluorimetric signal into a visual signal,43 and smartphone cameras were recently used to detect the fluorimetric signal.50 We believe that the rapid pace of innovation in this field will lead to the development of simple assays or inexpensive devices (Figure 7) that could reshape the field of point-of-care molecular diagnostics.
Figure 7.

Envisioned CRISPR-based platforms for POC and home testing. Simple diagnostics tests are needed to meet the growing demand for home-based kits. Such kits would require minimal handling by the user, such as self-collection of the sample (1) and loading the sample into a simple device (2a) or a one-step assay (2b).
Acknowledgments
This work was supported, in part, by the Smart Health Initiative at King Abdullah University of Science and Technology (KAUST) and Impact Acceleration Fund (IAF) and (Near Term Grand Challenge) NTGC grants from the KAUST (Innovation and Economic Development) IED to MM. All figures were created using Bioreneder.com.
Author Contributions
All authors contributed to conception and critical revision of the article; AG wrote the initial manuscript; AG, AM, TM, RA, and MM revised and approved the final manuscript.
The authors declare no competing financial interest.
References
- Neal R. D.; Tharmanathan P.; France B.; Din N. U.; Cotton S.; Fallon-Ferguson J.; Hamilton W.; Hendry A.; Hendry M.; Lewis R.; Macleod U.; Mitchell E. D.; Pickett M.; Rai T.; Shaw K.; Stuart N.; Torring M. L.; Wilkinson C.; Williams B.; Williams N.; Emery J. Is increased time to diagnosis and treatment in symptomatic cancer associated with poorer outcomes? Systematic review. Br. J. Cancer 2015, 112 (Suppl 1), S92–S107. 10.1038/bjc.2015.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S.; Rothman R. E. PCR-based diagnostics for infectious diseases: uses, limitations, and future applications in acute-care settings. Lancet Infect Dis 2004, 4 (6), 337–48. 10.1016/S1473-3099(04)01044-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissleder R.; Lee H.; Ko J.; Pittet M. J. COVID-19 diagnostics in context. Sci. Transl. Med. 2020, 10.1126/scitranslmed.abc1931. [DOI] [PubMed] [Google Scholar]
- Burki T. K. Testing for COVID-19. Lancet Respir. Med. 2020, 8 (7), e63 10.1016/S2213-2600(20)30247-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L.; Magnani M.; Saunders N.; Harms C.; Bruce I. J. Current molecular techniques for the detection of microbial pathogens. Sci. Prog. 2007, 90 (1), 29–50. 10.3184/003685007780440521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aman R.; Mahas A.; Marsic T.; Hassan N.; Mahfouz M. M. Efficient, Rapid, and Sensitive Detection of Plant RNA Viruses With One-Pot RT-RPA-CRISPR/Cas12a Assay. Front Microbiol 2020, 11, 610872. 10.3389/fmicb.2020.610872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bej A. K. Molecular based methods for the detection of microbial pathogens in the environment. J. Microbiol Methods 2003, 53 (2), 139–40. 10.1016/S0167-7012(03)00035-6. [DOI] [PubMed] [Google Scholar]
- Mahony J. B.; Blackhouse G.; Babwah J.; Smieja M.; Buracond S.; Chong S.; Ciccotelli W.; O’Shea T.; Alnakhli D.; Griffiths-Turner M.; Goeree R. Cost analysis of multiplex PCR testing for diagnosing respiratory virus infections. J. Clin Microbiol 2009, 47 (9), 2812–7. 10.1128/JCM.00556-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Baar M. P.; Timmermans E. C.; Bakker M.; de Rooij E.; van Gemen B.; Goudsmit J. One-tube real-time isothermal amplification assay to identify and distinguish human immunodeficiency virus type 1 subtypes A, B, and C and circulating recombinant forms AE and AG. J. Clin Microbiol 2001, 39 (5), 1895–902. 10.1128/JCM.39.5.1895-1902.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips E. A.; Moehling T. J.; Bhadra S.; Ellington A. D.; Linnes J. C. Strand Displacement Probes Combined with Isothermal Nucleic Acid Amplification for Instrument-Free Detection from Complex Samples. Anal. Chem. 2018, 90 (11), 6580–6586. 10.1021/acs.analchem.8b00269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori Y.; Hirano T.; Notomi T. Sequence specific visual detection of LAMP reactions by addition of cationic polymers. BMC Biotechnol. 2006, 6, 3. 10.1186/1472-6750-6-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mabey D.; Peeling R. W.; Ustianowski A.; Perkins M. D. Diagnostics for the developing world. Nat. Rev. Microbiol 2004, 2 (3), 231–40. 10.1038/nrmicro841. [DOI] [PubMed] [Google Scholar]
- Land K. J.; Boeras D. I.; Chen X. S.; Ramsay A. R.; Peeling R. W. REASSURED diagnostics to inform disease control strategies, strengthen health systems and improve patient outcomes. Nat. Microbiol 2019, 4 (1), 46–54. 10.1038/s41564-018-0295-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mojica F. J.; Diez-Villasenor C.; Garcia-Martinez J.; Soria E. Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J. Mol. Evol 2005, 60 (2), 174–82. 10.1007/s00239-004-0046-3. [DOI] [PubMed] [Google Scholar]
- Makarova K. S.; Haft D. H.; Barrangou R.; Brouns S. J.; Charpentier E.; Horvath P.; Moineau S.; Mojica F. J.; Wolf Y. I.; Yakunin A. F.; van der Oost J.; Koonin E. V. Evolution and classification of the CRISPR-Cas systems. Nat. Rev. Microbiol 2011, 9 (6), 467–77. 10.1038/nrmicro2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova K. S.; Wolf Y. I.; Alkhnbashi O. S.; Costa F.; Shah S. A.; Saunders S. J.; Barrangou R.; Brouns S. J.; Charpentier E.; Haft D. H.; Horvath P.; Moineau S.; Mojica F. J.; Terns R. M.; Terns M. P.; White M. F.; Yakunin A. F.; Garrett R. A.; van der Oost J.; Backofen R.; Koonin E. V. An updated evolutionary classification of CRISPR-Cas systems. Nat. Rev. Microbiol 2015, 13 (11), 722–36. 10.1038/nrmicro3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova K. S.; Wolf Y. I.; Iranzo J.; Shmakov S. A.; Alkhnbashi O. S.; Brouns S. J. J.; Charpentier E.; Cheng D.; Haft D. H.; Horvath P.; Moineau S.; Mojica F. J. M.; Scott D.; Shah S. A.; Siksnys V.; Terns M. P.; Venclovas C.; White M. F.; Yakunin A. F.; Yan W.; Zhang F.; Garrett R. A.; Backofen R.; van der Oost J.; Barrangou R.; Koonin E. V. Evolutionary classification of CRISPR-Cas systems: a burst of class 2 and derived variants. Nat. Rev. Microbiol 2020, 18 (2), 67–83. 10.1038/s41579-019-0299-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozcan A.; Krajeski R.; Ioannidi E.; Lee B.; Gardner A.; Makarova K. S.; Koonin E. V.; Abudayyeh O. O.; Gootenberg J. S. Programmable RNA targeting with the single-protein CRISPR effector Cas7–11. Nature 2021, 597 (7878), 720–725. 10.1038/s41586-021-03886-5. [DOI] [PubMed] [Google Scholar]
- Cong L.; Ran F. A.; Cox D.; Lin S.; Barretto R.; Habib N.; Hsu P. D.; Wu X.; Jiang W.; Marraffini L. A.; Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339 (6121), 819–23. 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilton I. B.; D’Ippolito A. M.; Vockley C. M.; Thakore P. I.; Crawford G. E.; Reddy T. E.; Gersbach C. A. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat. Biotechnol. 2015, 33 (5), 510–7. 10.1038/nbt.3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox D. B. T.; Gootenberg J. S.; Abudayyeh O. O.; Franklin B.; Kellner M. J.; Joung J.; Zhang F. RNA editing with CRISPR-Cas13. Science 2017, 358 (6366), 1019–1027. 10.1126/science.aaq0180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B.; Gilbert L. A.; Cimini B. A.; Schnitzbauer J.; Zhang W.; Li G. W.; Park J.; Blackburn E. H.; Weissman J. S.; Qi L. S.; Huang B. Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell 2013, 155 (7), 1479–91. 10.1016/j.cell.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellner M. J.; Koob J. G.; Gootenberg J. S.; Abudayyeh O. O.; Zhang F. SHERLOCK: nucleic acid detection with CRISPR nucleases. Nat. Protoc 2019, 14 (10), 2986–3012. 10.1038/s41596-019-0210-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolotin A.; Quinquis B.; Sorokin A.; Ehrlich S. D. Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiology 2005, 151 (8), 2551–2561. 10.1099/mic.0.28048-0. [DOI] [PubMed] [Google Scholar]
- Kaminski M. M.; Abudayyeh O. O.; Gootenberg J. S.; Zhang F.; Collins J. J. CRISPR-based diagnostics. Nat. Biomed Eng. 2021, 5 (7), 643–656. 10.1038/s41551-021-00760-7. [DOI] [PubMed] [Google Scholar]
- Pardee K.; Green A. A.; Takahashi M. K.; Braff D.; Lambert G.; Lee J. W.; Ferrante T.; Ma D.; Donghia N.; Fan M.; Daringer N. M.; Bosch I.; Dudley D. M.; O’Connor D. H.; Gehrke L.; Collins J. J. Rapid, Low-Cost Detection of Zika Virus Using Programmable Biomolecular Components. Cell 2016, 165 (5), 1255–1266. 10.1016/j.cell.2016.04.059. [DOI] [PubMed] [Google Scholar]
- Jiao C.; Sharma S.; Dugar G.; Peeck N. L.; Bischler T.; Wimmer F.; Yu Y.; Barquist L.; Schoen C.; Kurzai O.; Sharma C. M.; Beisel C. L. Noncanonical crRNAs derived from host transcripts enable multiplexable RNA detection by Cas9. Science 2021, 372 (6545), 941–948. 10.1126/science.abe7106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali Z.; Sanchez E.; Tehseen M.; Mahas A.; Marsic T.; Aman R.; Sivakrishna Rao G.; Alhamlan F. S.; Alsanea M. S.; Al-Qahtani A. A.; Hamdan S.; Mahfouz M. Bio-SCAN: A CRISPR/dCas9-Based Lateral Flow Assay for Rapid, Specific, and Sensitive Detection of SARS-CoV-2. ACS Synth. Biol. 2022, 11 (1), 406–419. 10.1021/acssynbio.1c00499. [DOI] [PubMed] [Google Scholar]
- Marsic T.; Ali Z.; Tehseen M.; Mahas A.; Hamdan S.; Mahfouz M. Vigilant: An Engineered VirD2-Cas9 Complex for Lateral Flow Assay-Based Detection of SARS-CoV2. Nano Lett. 2021, 21 (8), 3596–3603. 10.1021/acs.nanolett.1c00612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S. Y.; Cheng Q. X.; Liu J. K.; Nie X. Q.; Zhao G. P.; Wang J. CRISPR-Cas12a has both cis- and trans-cleavage activities on single-stranded DNA. Cell Res. 2018, 28 (4), 491–493. 10.1038/s41422-018-0022-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D. G.; Brewster J. D.; Paul M.; Tomasula P. M. Two methods for increased specificity and sensitivity in loop-mediated isothermal amplification. Molecules 2015, 20 (4), 6048–59. 10.3390/molecules20046048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong Y. P.; Othman S.; Lau Y. L.; Radu S.; Chee H. Y. Loop-mediated isothermal amplification (LAMP): a versatile technique for detection of micro-organisms. J. Appl. Microbiol. 2018, 124 (3), 626–643. 10.1111/jam.13647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y.; Chen F.; Li Q.; Wang L.; Fan C. Isothermal Amplification of Nucleic Acids. Chem. Rev. 2015, 115 (22), 12491–545. 10.1021/acs.chemrev.5b00428. [DOI] [PubMed] [Google Scholar]
- Piepenburg O.; Williams C. H.; Stemple D. L.; Armes N. A. DNA detection using recombination proteins. PLoS Biol. 2006, 4 (7), e204 10.1371/journal.pbio.0040204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notomi T.; Okayama H.; Masubuchi H.; Yonekawa T.; Watanabe K.; Amino N.; Hase T. Loop-mediated isothermal amplification of DNA. Nucleic Acids Res. 2000, 28 (12), E63 10.1093/nar/28.12.e63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gootenberg J. S.; Abudayyeh O. O.; Lee J. W.; Essletzbichler P.; Dy A. J.; Joung J.; Verdine V.; Donghia N.; Daringer N. M.; Freije C. A.; Myhrvold C.; Bhattacharyya R. P.; Livny J.; Regev A.; Koonin E. V.; Hung D. T.; Sabeti P. C.; Collins J. J.; Zhang F. Nucleic acid detection with CRISPR-Cas13a/C2c2. Science 2017, 356 (6336), 438–442. 10.1126/science.aam9321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gootenberg J. S.; Abudayyeh O. O.; Kellner M. J.; Joung J.; Collins J. J.; Zhang F. Multiplexed and portable nucleic acid detection platform with Cas13, Cas12a, and Csm6. Science 2018, 360 (6387), 439–444. 10.1126/science.aaq0179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arizti-Sanz J.; Freije C. A.; Stanton A. C.; Petros B. A.; Boehm C. K.; Siddiqui S.; Shaw B. M.; Adams G.; Kosoko-Thoroddsen T. F.; Kemball M. E.; Uwanibe J. N.; Ajogbasile F. V.; Eromon P. E.; Gross R.; Wronka L.; Caviness K.; Hensley L. E.; Bergman N. H.; MacInnis B. L.; Happi C. T.; Lemieux J. E.; Sabeti P. C.; Myhrvold C. Streamlined inactivation, amplification, and Cas13-based detection of SARS-CoV-2. Nat. Commun. 2020, 11 (1), 5921. 10.1038/s41467-020-19097-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahas A.; Marsic T.; Lopez-Portillo Masson M.; Wang Q.; Aman R.; Zheng C.; Ali Z.; Alsanea M.; Al-Qahtani A.; Ghanem B.; Alhamlan F.; Mahfouz M. Characterization of a thermostable Cas13 enzyme for one-pot detection of SARS-CoV-2. Proc. Natl. Acad. Sci. U. S. A. 2022, 119 (28), e2118260119 10.1073/pnas.2118260119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J. S.; Ma E.; Harrington L. B.; Da Costa M.; Tian X.; Palefsky J. M.; Doudna J. A. CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science 2018, 360 (6387), 436–439. 10.1126/science.aar6245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S. Y.; Cheng Q. X.; Wang J. M.; Li X. Y.; Zhang Z. L.; Gao S.; Cao R. B.; Zhao G. P.; Wang J. CRISPR-Cas12a-assisted nucleic acid detection. Cell Discovery 2018, 4, 20. 10.1038/s41421-018-0028-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L.; Li S.; Wu N.; Wu J.; Wang G.; Zhao G.; Wang J. HOLMESv2: A CRISPR-Cas12b-Assisted Platform for Nucleic Acid Detection and DNA Methylation Quantitation. ACS Synth. Biol. 2019, 8 (10), 2228–2237. 10.1021/acssynbio.9b00209. [DOI] [PubMed] [Google Scholar]
- Aman R.; Marsic T.; Sivakrishna Rao G.; Mahas A.; Ali Z.; Alsanea M.; Al-Qahtani A.; Alhamlan F.; Mahfouz M. iSCAN-V2: A One-Pot RT-RPA-CRISPR/Cas12b Assay for Point-of-Care SARS-CoV-2 Detection. Front. Bioeng. Biotechnol. 2022, 9, 800104. 10.3389/fbioe.2021.800104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali Z.; Aman R.; Mahas A.; Rao G. S.; Tehseen M.; Marsic T.; Salunke R.; Subudhi A. K.; Hala S. M.; Hamdan S. M.; Pain A.; Alofi F. S.; Alsomali A.; Hashem A. M.; Khogeer A.; Almontashiri N. A. M.; Abedalthagafi M.; Hassan N.; Mahfouz M. M. iSCAN: An RT-LAMP-coupled CRISPR-Cas12 module for rapid, sensitive detection of SARS-CoV-2. Virus Res. 2020, 288, 198129. 10.1016/j.virusres.2020.198129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joung J.; Ladha A.; Saito M.; Kim N. G.; Woolley A. E.; Segel M.; Barretto R. P. J.; Ranu A.; Macrae R. K.; Faure G.; Ioannidi E. I.; Krajeski R. N.; Bruneau R.; Huang M. W.; Yu X. G.; Li J. Z.; Walker B. D.; Hung D. T.; Greninger A. L.; Jerome K. R.; Gootenberg J. S.; Abudayyeh O. O.; Zhang F. Detection of SARS-CoV-2 with SHERLOCK One-Pot Testing. N Engl J. Med. 2020, 383 (15), 1492–1494. 10.1056/NEJMc2026172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran A.; Santiago J. G. CRISPR Enzyme Kinetics for Molecular Diagnostics. Anal. Chem. 2021, 93 (20), 7456–7464. 10.1021/acs.analchem.1c00525. [DOI] [PubMed] [Google Scholar]
- Hajian R.; Balderston S.; Tran T.; deBoer T.; Etienne J.; Sandhu M.; Wauford N. A.; Chung J. Y.; Nokes J.; Athaiya M.; Paredes J.; Peytavi R.; Goldsmith B.; Murthy N.; Conboy I. M.; Aran K. Detection of unamplified target genes via CRISPR-Cas9 immobilized on a graphene field-effect transistor. Nat. Biomed Eng. 2019, 3 (6), 427–437. 10.1038/s41551-019-0371-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruch R.; Baaske J.; Chatelle C.; Meirich M.; Madlener S.; Weber W.; Dincer C.; Urban G. A. CRISPR/Cas13a-Powered Electrochemical Microfluidic Biosensor for Nucleic Acid Amplification-Free miRNA Diagnostics. Adv. Mater. 2019, 31 (51), e1905311 10.1002/adma.201970365. [DOI] [PubMed] [Google Scholar]
- East-Seletsky A.; O’Connell M. R.; Knight S. C.; Burstein D.; Cate J. H.; Tjian R.; Doudna J. A. Two distinct RNase activities of CRISPR-C2c2 enable guide-RNA processing and RNA detection. Nature 2016, 538 (7624), 270–273. 10.1038/nature19802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fozouni P.; Son S.; Diaz de Leon Derby M.; Knott G. J.; Gray C. N.; D’Ambrosio M. V.; Zhao C.; Switz N. A.; Kumar G. R.; Stephens S. I.; Boehm D.; Tsou C. L.; Shu J.; Bhuiya A.; Armstrong M.; Harris A. R.; Chen P. Y.; Osterloh J. M.; Meyer-Franke A.; Joehnk B.; Walcott K.; Sil A.; Langelier C.; Pollard K. S.; Crawford E. D.; Puschnik A. S.; Phelps M.; Kistler A.; DeRisi J. L.; Doudna J. A.; Fletcher D. A.; Ott M. Amplification-free detection of SARS-CoV-2 with CRISPR-Cas13a and mobile phone microscopy. Cell 2021, 184 (2), 323–333. 10.1016/j.cell.2020.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- East-Seletsky A.; O’Connell M. R.; Burstein D.; Knott G. J.; Doudna J. A. RNA Targeting by Functionally Orthogonal Type VI-A CRISPR-Cas Enzymes. Mol. Cell 2017, 66 (3), 373–383. 10.1016/j.molcel.2017.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T. Y.; Knott G. J.; Smock D. C. J.; Desmarais J. J.; Son S.; Bhuiya A.; Jakhanwal S.; Prywes N.; Agrawal S.; Diaz de Leon Derby M.; Switz N. A.; Armstrong M.; Harris A. R.; Charles E. J.; Thornton B. W.; Fozouni P.; Shu J.; Stephens S. I.; Kumar G. R.; Zhao C.; Mok A.; Iavarone A. T.; Escajeda A. M.; McIntosh R.; Kim S.; Dugan E. J.; Consortium I. G. I. T.; Pollard K. S.; Tan M. X.; Ott M.; Fletcher D. A.; Lareau L. F.; Hsu P. D.; Savage D. F.; Doudna J. A.; et al. Accelerated RNA detection using tandem CRISPR nucleases. Nat. Chem. Biol. 2021, 17 (9), 982–988. 10.1038/s41589-021-00842-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santiago-Frangos A.; Hall L. N.; Nemudraia A.; Nemudryi A.; Krishna P.; Wiegand T.; Wilkinson R. A.; Snyder D. T.; Hedges J. F.; Cicha C.; Lee H. H.; Graham A.; Jutila M. A.; Taylor M. P.; Wiedenheft B. Intrinsic signal amplification by type III CRISPR-Cas systems provides a sequence-specific SARS-CoV-2 diagnostic. Cell Rep. Med. 2021, 2 (6), 100319. 10.1016/j.xcrm.2021.100319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J.; Song Y.; Deng X.; Vanegas J. A.; You Z.; Zhang Y.; Weng Z.; Avery L.; Dieckhaus K. D.; Peddi A.; Gao Y.; Zhang Y.; Gao X. Engineered LwaCas13a with enhanced collateral activity for nucleic acid detection. Nat. Chem. Biol. 2022, 10.1038/s41589-022-01135-y. [DOI] [PubMed] [Google Scholar]
- ref.FDA-report , SHERLOCK TM CRISPR SARS-CoV-2 kit. https://www.fda.gov/media/137746/download (accessed on July 31, 2022).
- ref.FDA-report , Design Considerations for Devices Intended for Home Use. https://www.fda.gov/media/84830/download (accessed on July 31, 2022).
- Ali N.; Rampazzo R. C. P.; Costa A. D. T.; Krieger M. A. Current Nucleic Acid Extraction Methods and Their Implications to Point-of-Care Diagnostics. Biomed Res. Int. 2017, 2017, 9306564. 10.1155/2017/9306564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chomczynski P.; Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 1987, 162 (1), 156–9. 10.1016/0003-2697(87)90021-2. [DOI] [PubMed] [Google Scholar]
- Choi E. H.; Lee S. K.; Ihm C.; Sohn Y. H. Rapid DNA extraction from dried blood spots on filter paper: potential applications in biobanking. Osong Public Health Res. Perspect 2014, 5 (6), 351–7. 10.1016/j.phrp.2014.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esser K.-H.; Marx W. H.; Lisowsky T. maxXbond: first regeneration system for DNA binding silica matrices. Nat. Methods 2006, 3 (1), i–ii. 10.1038/nmeth845. [DOI] [Google Scholar]
- Archer M. J.; Lin B.; Wang Z.; Stenger D. A. Magnetic bead-based solid phase for selective extraction of genomic DNA. Anal. Biochem. 2006, 355 (2), 285–97. 10.1016/j.ab.2006.05.005. [DOI] [PubMed] [Google Scholar]
- Myhrvold C.; Freije C. A.; Gootenberg J. S.; Abudayyeh O. O.; Metsky H. C.; Durbin A. F.; Kellner M. J.; Tan A. L.; Paul L. M.; Parham L. A.; Garcia K. F.; Barnes K. G.; Chak B.; Mondini A.; Nogueira M. L.; Isern S.; Michael S. F.; Lorenzana I.; Yozwiak N. L.; MacInnis B. L.; Bosch I.; Gehrke L.; Zhang F.; Sabeti P. C. Field-deployable viral diagnostics using CRISPR-Cas13. Science 2018, 360 (6387), 444–448. 10.1126/science.aas8836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agrawal S.; Fanton A.; Chandrasekaran S. S.; Charrez B.; Escajeda A. M.; Son S.; McIntosh R.; Bhuiya A.; de Leon Derby M. D.; Switz N. A.; Armstrong M.; Harris A. R.; Prywes N.; Lukarska M.; Biering S. B.; Smock D. C. J.; Mok A.; Knott G. J.; Dang Q.; Van Dis E.; Dugan E.; Kim S.; Liu T. Y.; Consortium I. G. I. T.; Harris E.; Stanley S. A.; Lareau L. F.; Tan M. X.; Fletcher D. A.; Doudna J. A.; Savage D. F.; Hsu P. D.. Rapid, point-of-care molecular diagnostics with Cas13. medRxiv, April 5, 2021. 10.1101/2020.12.14.20247874. [DOI]
- Arizti-Sanz J.; Bradley A. D.; Zhang Y. B.; Boehm C. K.; Freije C. A.; Grunberg M. E.; Kosoko-Thoroddsen T. F.; Welch N. L.; Pillai P. P.; Mantena S.; Kim G.; Uwanibe J. N.; John O. G.; Eromon P. E.; Kocher G.; Gross R.; Lee J. S.; Hensley L. E.; Happi C. T.; Johnson J.; Sabeti P. C.; Myhrvold C.. Equipment-free detection of SARS-CoV-2 and Variants of Concern using Cas13. medRxiv, November 2, 2021. 10.1101/2021.11.01.21265764. [DOI]
- Ghouneimy A.; Mahfouz M. Streamlined detection of SARS-CoV-2 via Cas13. Nat. Biomed Eng. 2022, 6 (8), 925–927. 10.1038/s41551-022-00926-x. [DOI] [PubMed] [Google Scholar]
- Hasan S. S.; Sevvana M.; Kuhn R. J.; Rossmann M. G. Structural biology of Zika virus and other flaviviruses. Nat. Struct Mol. Biol. 2018, 25 (1), 13–20. 10.1038/s41594-017-0010-8. [DOI] [PubMed] [Google Scholar]
- Azmi I.; Faizan M. I.; Kumar R.; Raj Yadav S.; Chaudhary N.; Kumar Singh D.; Butola R.; Ganotra A.; Datt Joshi G.; Deep Jhingan G.; Iqbal J.; Joshi M. C.; Ahmad T. A Saliva-Based RNA Extraction-Free Workflow Integrated With Cas13a for SARS-CoV-2 Detection. Front Cell Infect Microbiol 2021, 11, 632646. 10.3389/fcimb.2021.632646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Hu X.; Wang X.; Yang J.; Zhang L.; Deng Q.; Zhang X.; Wang Z.; Hou T.; Li S. A novel One-pot rapid diagnostic technology for COVID-19. Anal. Chim. Acta 2021, 1154, 338310. 10.1016/j.aca.2021.338310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linke D.Chapter 34: Detergents. In Guide to Protein Purification, 2nd ed.; Elsevier, 2009; pp 603–617. [Google Scholar]
- de Puig H.; Lee R. A.; Najjar D.; Tan X.; Soenksen L. R.; Angenent-Mari N. M.; Donghia N. M.; Weckman N. E.; Ory A.; Ng C. F.; Nguyen P. Q.; Mao A. S.; Ferrante T. C.; Lansberry G.; Sallum H.; Niemi J.; Collins J. J. Minimally instrumented SHERLOCK (miSHERLOCK) for CRISPR-based point-of-care diagnosis of SARS-CoV-2 and emerging variants. Sci. Adv. 2021, 10.1126/sciadv.abh2944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ref.FDA-report , Lucira Check It COVID-19 test kit. https://www.fda.gov/media/147494/download (accessed on July 31, 2022).
- Kurani N.; Pollitz K.; Cotliar D.; Ramirez G.; Cox C.. COVID-19 test prices and payment policy https://www.healthsystemtracker.org/brief/covid-19-test-prices-and-payment-policy/ (accessed on August 20, 2022).
- ref.FDA-report , Accula test FDA approval letter. https://www.fda.gov/media/136355/download (accessed on July 31, 2022).
- Arakawa T.; Prestrelski S. J.; Kenney W. C.; Carpenter J. F. Factors affecting short-term and long-term stabilities of proteins. Adv. Drug Deliv Rev. 2001, 46 (1–3), 307–26. 10.1016/S0169-409X(00)00144-7. [DOI] [PubMed] [Google Scholar]
- Manning M. C.; Chou D. K.; Murphy B. M.; Payne R. W.; Katayama D. S. Stability of protein pharmaceuticals: an update. Pharm. Res. 2010, 27 (4), 544–75. 10.1007/s11095-009-0045-6. [DOI] [PubMed] [Google Scholar]
- Xu Y.; Zhu T. F. Mirror-image T7 transcription of chirally inverted ribosomal and functional RNAs. Science 2022, 378 (6618), 405–412. 10.1126/science.abm0646. [DOI] [PubMed] [Google Scholar]
- Franks F.; Auffret T.. Freeze-Drying of Pharmaceuticals and Biopharmaceuticals; Royal Society of Chemistry, 2007. [Google Scholar]
- Ohtake S.; Kita Y.; Arakawa T. Interactions of formulation excipients with proteins in solution and in the dried state. Adv. Drug Deliv Rev. 2011, 63 (13), 1053–73. 10.1016/j.addr.2011.06.011. [DOI] [PubMed] [Google Scholar]
- Jones K. L.; Drane D.; Gowans E. J. Long-term storage of DNA-free RNA for use in vaccine studies. Biotechniques 2007, 43 (5), 675–81. 10.2144/000112593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter J. F.; Chang B. S.; Randolph T. W.. Rational Design of Stable Lyophilized Protein Formulations: Some Practical Advice; Springer, 1997. [DOI] [PubMed] [Google Scholar]
- Liu B.; Zhou X. Freeze-drying of proteins. Methods Mol. Biol. 2015, 1257, 459–76. 10.1007/978-1-4939-2193-5_23. [DOI] [PubMed] [Google Scholar]
- Chang B. S.; Kendrick B. S.; Carpenter J. F. Surface-induced denaturation of proteins during freezing and its inhibition by surfactants. J. Pharm. Sci. 1996, 85 (12), 1325–30. 10.1021/js960080y. [DOI] [PubMed] [Google Scholar]
- Ashani Y.; Catravas G. N. Highly reactive impurities in Triton X-100 and Brij 35: partial characterization and removal. Anal. Biochem. 1980, 109 (1), 55–62. 10.1016/0003-2697(80)90009-3. [DOI] [PubMed] [Google Scholar]
- Langer C. A.Formulation and Delivery of Proteins and Peptides; American Chemical Society, 1994. [Google Scholar]
- Abudayyeh O. O.; Gootenberg J. S.; Konermann S.; Joung J.; Slaymaker I. M.; Cox D. B.; Shmakov S.; Makarova K. S.; Semenova E.; Minakhin L.; Severinov K.; Regev A.; Lander E. S.; Koonin E. V.; Zhang F. C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science 2016, 353 (6299), aaf5573. 10.1126/science.aaf5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee R. A.; Puig H.; Nguyen P. Q.; Angenent-Mari N. M.; Donghia N. M.; McGee J. P.; Dvorin J. D.; Klapperich C. M.; Pollock N. R.; Collins J. J. Ultrasensitive CRISPR-based diagnostic for field-applicable detection of Plasmodium species in symptomatic and asymptomatic malaria. Proc. Natl. Acad. Sci. U. S. A. 2020, 117 (41), 25722–25731. 10.1073/pnas.2010196117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckbo E. J.; Locher K.; Caza M.; Li L.; Lavergne V.; Charles M. Evaluation of the BioFire(R) COVID-19 test and Respiratory Panel 2.1 for rapid identification of SARS-CoV-2 in nasopharyngeal swab samples. Diagn Microbiol Infect Dis 2021, 99 (3), 115260. 10.1016/j.diagmicrobio.2020.115260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konings F.; Perkins M. D.; Kuhn J. H.; Pallen M. J.; Alm E. J.; Archer B. N.; Barakat A.; Bedford T.; Bhiman J. N.; Caly L.; Carter L. L.; Cullinane A.; de Oliveira T.; Druce J.; El Masry I.; Evans R.; Gao G. F.; Gorbalenya A. E.; Hamblion E.; Herring B. L.; Hodcroft E.; Holmes E. C.; Kakkar M.; Khare S.; Koopmans M. P. G.; Korber B.; Leite J.; MacCannell D.; Marklewitz M.; Maurer-Stroh S.; Rico J. A. M.; Munster V. J.; Neher R.; Munnink B. O.; Pavlin B. I.; Peiris M.; Poon L.; Pybus O.; Rambaut A.; Resende P.; Subissi L.; Thiel V.; Tong S.; van der Werf S.; von Gottberg A.; Ziebuhr J.; Van Kerkhove M. D. SARS-CoV-2 Variants of Interest and Concern naming scheme conducive for global discourse. Nat. Microbiol 2021, 6 (7), 821–823. 10.1038/s41564-021-00932-w. [DOI] [PubMed] [Google Scholar]
- Huang H. S.; Tsai C. L.; Chang J.; Hsu T. C.; Lin S.; Lee C. C. Multiplex PCR system for the rapid diagnosis of respiratory virus infection: systematic review and meta-analysis. Clin Microbiol Infect 2018, 24 (10), 1055–1063. 10.1016/j.cmi.2017.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacky L.; Yurk D.; Alvarado J.; Belitz P.; Fathe K.; MacDonald C.; Fraser S.; Rajagopal A. Robust Multichannel Encoding for Highly Multiplexed Quantitative PCR. Anal. Chem. 2021, 93 (9), 4208–4216. 10.1021/acs.analchem.0c04626. [DOI] [PubMed] [Google Scholar]
- Ackerman C. M.; Myhrvold C.; Thakku S. G.; Freije C. A.; Metsky H. C.; Yang D. K.; Ye S. H.; Boehm C. K.; Kosoko-Thoroddsen T. F.; Kehe J.; Nguyen T. G.; Carter A.; Kulesa A.; Barnes J. R.; Dugan V. G.; Hung D. T.; Blainey P. C.; Sabeti P. C. Massively multiplexed nucleic acid detection with Cas13. Nature 2020, 582 (7811), 277–282. 10.1038/s41586-020-2279-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch N. L.; Zhu M.; Hua C.; Weller J.; Mirhashemi M. E.; Nguyen T. G.; Mantena S.; Bauer M. R.; Shaw B. M.; Ackerman C. M.; Thakku S. G.; Tse M. W.; Kehe J.; Uwera M. M.; Eversley J. S.; Bielwaski D. A.; McGrath G.; Braidt J.; Johnson J.; Cerrato F.; Moreno G. K.; Krasilnikova L. A.; Petros B. A.; Gionet G. L.; King E.; Huard R. C.; Jalbert S. K.; Cleary M. L.; Fitzgerald N. A.; Gabriel S. B.; Gallagher G. R.; Smole S. C.; Madoff L. C.; Brown C. M.; Keller M. W.; Wilson M. M.; Kirby M. K.; Barnes J. R.; Park D. J.; Siddle K. J.; Happi C. T.; Hung D. T.; Springer M.; MacInnis B. L.; Lemieux J. E.; Rosenberg E.; Branda J. A.; Blainey P. C.; Sabeti P. C.; Myhrvold C. Multiplexed CRISPR-based microfluidic platform for clinical testing of respiratory viruses and identification of SARS-CoV-2 variants. Nat. Med. 2022, 28 (5), 1083–1094. 10.1038/s41591-022-01734-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthier J.Micro-Drops and Digital Microfluidics; William Andrew, 2012. [Google Scholar]
- Millington D. S.; Sista R.; Eckhardt A.; Rouse J.; Bali D.; Goldberg R.; Cotten M.; Buckley R.; Pamula V. Digital microfluidics: a future technology in the newborn screening laboratory?. Semin Perinatol 2010, 34 (2), 163–9. 10.1053/j.semperi.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coelho B.; Veigas B.; Fortunato E.; Martins R.; Aguas H.; Igreja R.; Baptista P. V. Digital Microfluidics for Nucleic Acid Amplification. Sensors 2017, 17 (7), 1495. 10.3390/s17071495. [DOI] [PMC free article] [PubMed] [Google Scholar]