

Abstract
Selective death of midbrain dopaminergic neurons is a hallmark pathology of Parkinson’s disease (PD), but the molecular mechanisms that initiate the cascade of events resulting in neurodegeneration in PD remain unclear. Compelling evidence suggests that dysregulation of dopamine (DA) induces neuronal stress and damage responses that are operative processes in striatal degeneration preceding PD-like symptoms. Improper DA sequestration to vesicles raises cytosolic DA levels, which is rapidly converted into electrophilic dopaquinone species (DQs) that react readily with protein nucleophiles forming covalent modifications that alter the native structure and function of proteins. These so-called DA-protein adducts (DPAs) have been reported to play a role in neurotoxicity, and their abundance with respect to neurodegeneration has been linked to clinical and pathological features of PD that suggest that they play a causal role in PD pathogenesis. Therefore, characterizing DPAs is a critical first step in understanding the susceptibility of midbrain dopaminergic neurons during PD. To help achieve this goal, we report here a novel DA-mimetic (DAyne) containing a biorthogonal alkyne handle that exhibits a reactivity profile similar to DA in aqueous buffers. By linking DPAs formed with DAyne to a fluorescent reporter molecule, DPAs were visualized in fixed cells and within lysates. DAyne enabled global mapping of cellular proteins affected by DQ modification and their bioactive pathways through enrichment. Our proteomic profiling of DPAs in neuronal SH-SY5Y cells indicates that proteins susceptible to DPA formation are extant throughout the proteome, potentially influencing several diverse biological pathways involved in PD such as endoplasmic reticulum (ER) stress, cytoskeletal instability, proteotoxicity, and clathrin function. We validated that a protein involved in the ER stress pathway, protein disulfide isomerase 3 (PDIA3), which was enriched in our chemoproteomic analysis, is functionally inhibited by DA, providing evidence that dysregulated cellular DA may induce or exacerbate ER stress. Thus, DAyne provided new mechanistic insights into DA toxicity that may be observed during PD by enabling characterization of DPAs generated reproducibly at physiologically relevant quinone exposures. We anticipate our design and application of this reactivity-based probe will be generally applicable for clarifying mechanisms of metabolic quinone toxicity.
Graphical Abstract
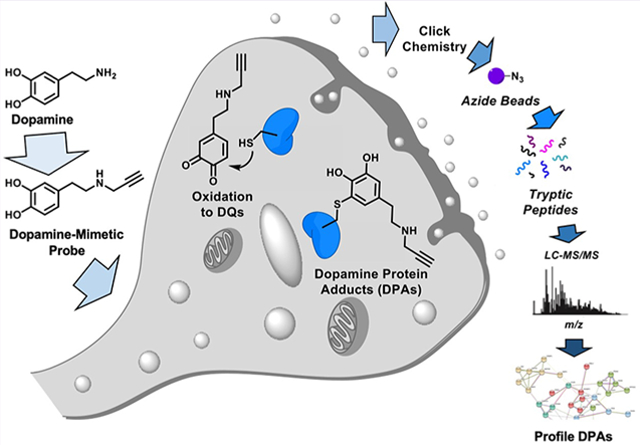
INTRODUCTION
Parkinson’s disease (PD) is the second most common neurodegenerative disease, which currently affects five million individuals worldwide.1 PD is characterized by a progressive decline in motor function due to the selective death of midbrain dopaminergic neurons in the striatum, and particularly in the substantia nigra pars compacta (SNc).2 The molecular events that initiate dopaminergic collapse in the SNc at the onset of PD occur up to a decade before clinical presentation. Other features of PD that precede symptom onsets include reductions in synaptic density, decreased synaptic dopamine (DA), and loss of DA-derived neuromelanin, suggesting that DA dysregulation is a key factor in PD pathophysiology.3 Indeed, DA loss correlates with motor dysfunction severity and DA metabolites have recently garnered attention as potential diagnostic biomarkers.4 Therefore, unraveling the mechanisms by which DA influences PD-related clinical and pathological changes will be critical for developing effective drugs and pre-symptomatic PD diagnostic assays.5 Clinical studies have associated PD incidence with impairments in cellular regulation of cytosolic DA levels. A large-scale genome-wide association study (GWAS) meta-analysis identified PD-linked mutations in genes that are involved in DA regulation, such as synaptic vesicle endocytosis.6 Genetic mutations or pharmacological agents that perturb DA homeostasis are also known to drive neuropathic phenotypes in vivo.7 Conversely, lowering cytosolic DA by increasing vesicular monoamine transporter (VMAT) activity enhances DA release and opposes PD-associated neurodegeneration in mice.8 This evidence suggests that DA dysregulation plays a significant role in PD etiology. Functional measures of DA transfer out of the cytosol by VMAT are 87–90% lower in autopsied brains of PD patients as compared to control donors.9 While the specific mechanisms underlying this observation have not been characterized, they implicate DA toxicity as an early and key component of PD-related neurodegeneration. Time-resolved mechanistic analyses of dopaminergic neurons derived from human induced pluripotent stem cells found that DQs accumulate in the neurons of PD donors, but not healthy donors.10 Additionally, these oxidized DA metabolites induced mitochondrial and lysosomal dysfunction, two cellular processes involved in early mechanisms of PD-associated cell death. Collectively, these studies support a causal role for DA in neuronal dysfunction and degeneration.
In addition to its receptor-binding activity, DA is also chemically reactive toward biomolecules under conditions that mirror cytosolic environments. The chemical reactivity of DA derives from its electron-rich catechol moiety, which renders DA prone to auto-oxidation outside of acidic vesicles.11 These oxidative processes produce a series of electrophilic dopaquinone (DQ) metabolites through sequential oxidative transformations (Figure 1). To prevent oxidative damage resulting from a surge in cellular DQ metabolites, DA levels are maintained through metabolic clearance, repackaging into synaptic vesicles and polymerization into neuromelanin.12
Figure 1.
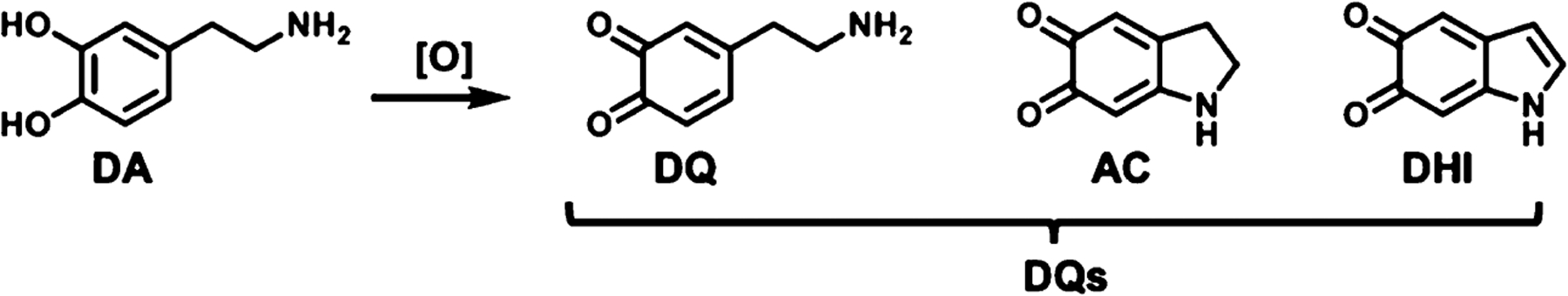
Simplified schematic of dopamine oxidation to electrophilic DQs metabolites. Shown are the 2 e−, 4 e−, and 6 e− oxidation products of DA; dopa-o-quinone (DQ), aminochrome (AC), and dihydroxyindole (DHI), respectively.
Dysfunction in any of these regulatory processes could result in high levels of DQs. DQs induce oxidative damage by increasing reactive oxygen species (ROS), a byproduct of DA-DQ redox cycling. Additionally, DQs nonenzymatically modify protein nucleophiles to form DA-protein adducts (DPAs).13 These modifications can occur at low-occupancy particularly at the active site of enzymes, resulting in alterations to the native protein structure and functions that affect critical cellular processes. Many PD-implicated proteins that are susceptible to DA modification have been identified using target-based approaches.14–25 For example, interactions between DQ and α-synuclein (αsyn) promote the self-assembly of toxic oligomers, leading to neurodegeneration in mouse models.14–16 DQ modification of parkin found in human brain tissue samples reportedly inhibits its E3 ubiquitin ligase function, which can lead to proteotoxic stress underlying PD-related neurodegeneration. Finally, covalent dimerization of DJ-1, a protein linked genetically to PD susceptibility, occurs in the presence of DQs, although its biological significance in vivo remains unclarified.17 These cases exemplify target-based approaches to identify DPAs, which rely on foreknowledge of well-established PD targets. Conversely, few studies have successfully applied unbiased approaches to characterize and validate novel DPAs at the proteome-wide level.26–28 This is largely because DA reactivity vitiates the sensitivity of analytical platforms capable of comprehensively profiling DPAs. Developing sensitive analytical characterization methods for reactive metabolites of DA guarantees to enhance our understanding of how DPAs contribute to aging and early stages of PD progression.
To access caches of candidate DPAs involved during early stages of PD pathology, we developed a chemo-proteomic probe that enables DPA profiling at low micromolar exposures of DA in human cells. In this proof-of-concept study, we synthesized a novel dopamine-mimetic probe, which we call DAyne, containing a bio-orthogonal alkyne handle for proteomic enrichment of DPAs. Herein, we show that DAyne is a suitable biochemical mimetic of DA and demonstrate the utility of DAyne as a visualization and enrichment probe enabled by the versatility of click chemistry modification. We highlight the generality and impact of our newly developed chemical probe by identifying DPAs and extracting cellular processes likely perturbed in the early stages of PD pathogenesis using chemoproteomics. Further, we provide validation of our findings by demonstrating that one of the proteins identified as a target of DAyne in our proteomic analyses, protein disulfide isomerase (PDI) involved in endoplasmic reticulum (ER) stress, is functionally inactivated by DA. These studies demonstrate that DAyne is a useful reactivity-based probe molecule that enables rapid discovery and characterization of cellular DPAs using a suite of qualitative and quantitative assays.
RESULTS
Synthesis of DAyne.
DAyne was designed to functionally mimic DA while presenting a bio-orthogonal alkyne handle distal to the o-quinone for click chemistry modification to enable fluorescence visualization and adduct enrichment preceding analytical characterization (Figure 2A). The DA mimetic probe was synthesized in seven steps as shown in Figure 2B (synthetic procedure described in the Supporting Information). Briefly, the catechol was protected with an acid-labile acetonide to enable elaboration of the amine.29 A pendant propargyl moiety was installed at the amine terminus through a Fukuyama-inspired route.30 Global acidic deprotection furnished DAyne as a HCl salt. The overall reaction sequence gave DAyne in a 32% yield.
Figure 2.
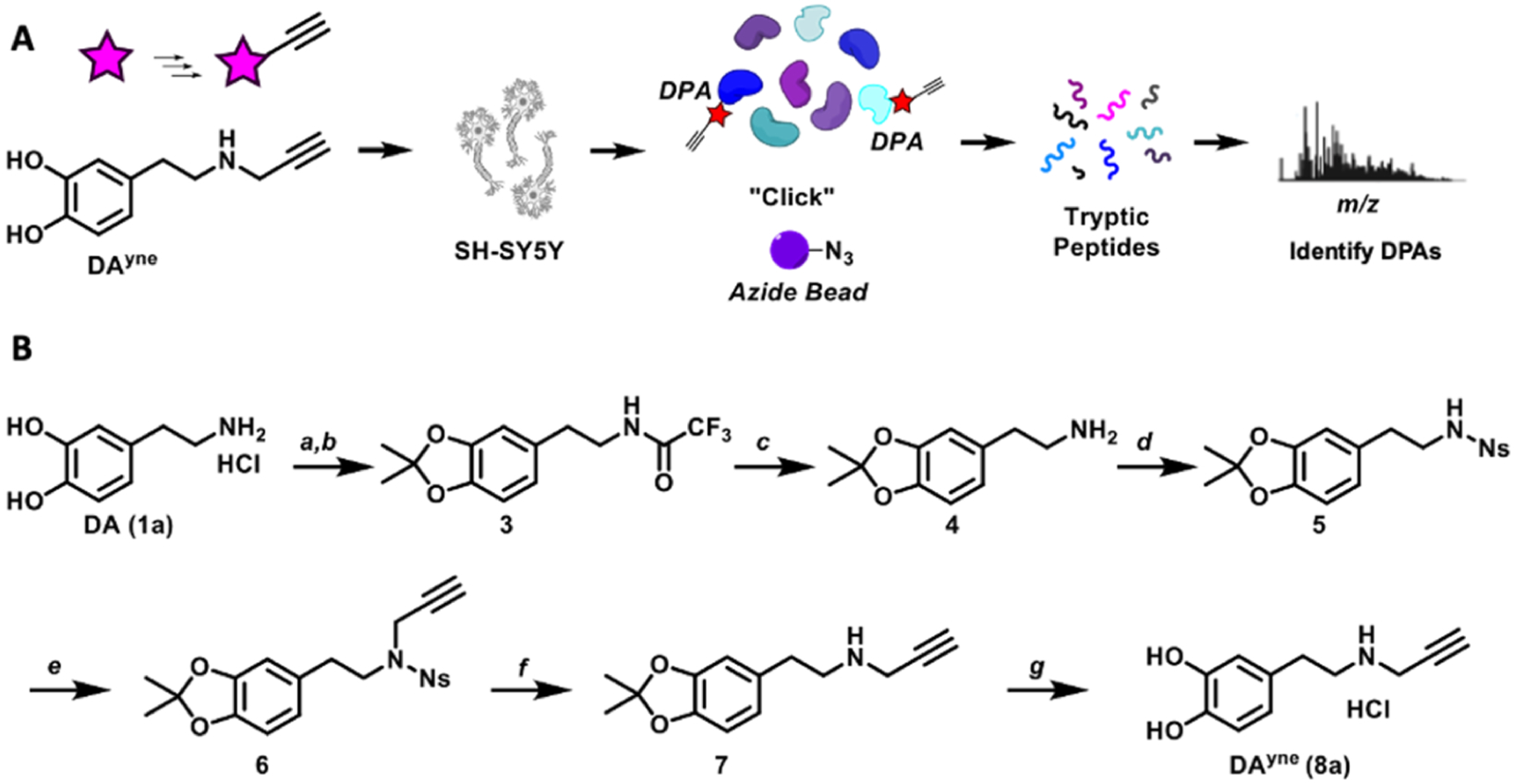
(A) Chemical biology strategy to visualize and identify DPAs. (B) Synthesis of DAyne. (a) Methyl trifluoroacetate, triethylamine, CH3OH; 93%. (b) pTSA, 2,2-dimethoxypropane, toluene; 79%. (c) LiOH, CH3OH; 98%. (d) Nosyl chloride, triethylamine, CH2Cl2; 97%. (e) Propargyl bromide, K2CO3, acetone; 96%. (f) Thiophenol, KOH, CH3CN; 50%. (g) Trifluoroacetic acid, CHCl3 then HCl, tetrahydrofuran; 96%.
DA and DAyne Oxidation Intermediates Form at Similar Rates.
The oxidation pathways of DAyne and DA were monitored spectroscopically to characterize the reaction intermediates (Figure 3A) and kinetics upon oxidation. We used UV–vis spectroscopy to monitor DQ generation over time. Initially, we monitored the oxidation of DA and DAyne in the presence of equimolar NaIO4 (Figure 3B,C) in argon-purged water. As shown in Figure 3B, an increase in absorbance at 395 nm was observed at the reaction onset, indicating the formation of the o-quinone 1b. This gave rise to the appearance of two new maxima at 300 and 475 nm, which are characteristic absorbance bands for aminochrome 1d.31 However, no o-quinone 8b was observed in the oxidation of DAyne as the reaction proceeded rapidly to the aminochrome 8d (Figure 3C). Kinetically, DAyne cyclized to the aminochrome 8d five times faster than DA cyclized to 1d in the presence of NaIO4 (Figure 3D), which we attribute to the increased nucleophilicity of the amine due to alkyl substitution. NaIO4 oxidation in phosphate-buffered saline (PBS) (pH 7.4) led to DA and DAyne oxidation products that were unstable under these conditions, which was evident by the rapid decay of the respective aminochrome species (Figure 3E). In the absence of NaIO4, the rates of DA and DAyne auto-oxidation at pH 7.4 were comparable and auto-oxidation could be inhibited in the presence of an antioxidant, N-acetylcysteince (NAC), (Supporting Information 1) when aminochrome absorbance was monitored at 475 nm.
Figure 3.
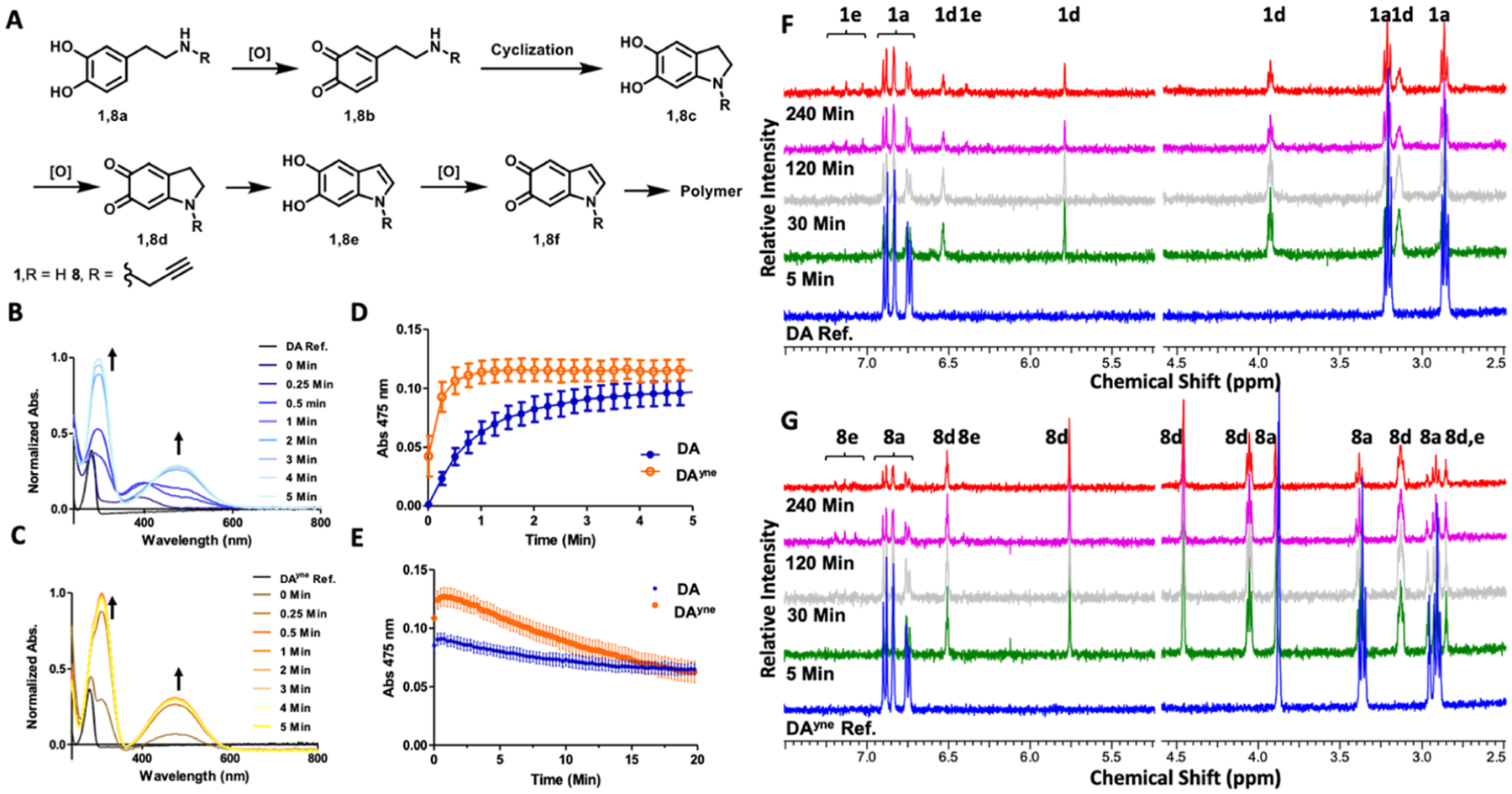
Spectroscopic characterization of DA and DAyne oxidation. (A) Oxidation products of DA and DAyne. (B/C) Absorbance spectra time course of 50 μM DA/DAyne respectively and 50 μM NaIO4 in H2O. (D/E) Change in absorbance at 475 nm of 50 μM DA/DAyne and 50 μM NaIO4 in H2O or PBS pH 7.4 respectively. 475 nm absorption is characteristic of aminochrome formation. Data are represented as mean ± SEM from three replicates. (F/G) NMR spectra time course of 1 mM DA/DAyne, respectively, and 1 mM NaIO4 in PBS pH 7.4. Labels above proton resonances denote corresponding compounds. Water solvent peak was removed for clarity.
Given the instability of DA metabolites observed in PBS using NaIO4 as an oxidant, we confirmed that DAyne is a substrate for the enzyme tyrosinase (TY), which is used routinely in DA oxidation studies.32,33 TY oxidizes DA to DQs and can be conveniently separated from resultant DQs by molecular weight centrifugal filters. Therefore, TY is an attractive oxidant as it obviates the use of chemical oxidizing agents such as NaIO4 which, due to its strong and promiscuous oxidation potential and ability to chelate vicinal oxygens, may create artifacts in downstream cellular experiments. The absorbance of DA and DAyne monitored at 300 and 475 nm over a 1 h incubation (Supporting Information 2) shows that DA and DAyne oxidize at similar rates under treatment with TY. Collectively, these UV–vis experiments indicate that DA and DAyne are oxidized at comparable rates in the presence of TY and during auto-oxidation in PBS. The ability of TY to bind and oxidize DAyne also demonstrates the suitability of DAyne as a DA-mimetic as a protein binding substrate.
As an additional confirmation that DA and DAyne feature comparable reaction directionalities, oxidation intermediates of both were characterized structurally using time-resolved nuclear magnetic resonance (NMR) spectroscopy (Figure 3F,G). The NMR spectral fingerprints of intermediates 1d and 1e were observed upon DA oxidation using NaIO4 in deuterated PBS as the oxidant. Proton NMR shifts were consistent with literature values.34 Oxidation of DAyne produced the metabolites 8d and 8e, mirroring those generated by DA (see Supporting Information 3 for detailed NMR peak assignments). Taken together, our NMR and UV–vis spectroscopic data show that DA and DAyne are oxidized to chemically analogous electrophilic species, namely, 1d and after long exposures to 1e, supporting the application of DAyne as a DA surrogate in a cellular context.
DAyne and DA are Equitoxic at Low Concentrations.
We compared the biological activity of DA and DAyne in undifferentiated neuronal SH-SY5Y cells to determine whether similarities in their chemical reactivity parallel toxicity.35 Cells were dosed with various concentrations of DA or DAyne, and their viability was measured with an alamarBlue assay, which indirectly reports oxidative stress.36 As shown in Figure 4A,B, DAyne and DA toxicity were dependent on both dose and exposure time. At physiologically relevant concentrations of 0.1–1 μM, no statistical differences were observed between DA and DAyne toxicity and after 48 h, with most cells remaining viable. At 100 μM, a concentration that exceeds physiological levels of DA by 100–1000×, a significant impact on cell viability was observed for DAyne when compared to DA. At this concentration, 24 h exposure to DAyne reduced the cell viability to 6%, whereas equivalent treatment with DA left 41% of cells viable. While DAyne is slightly more toxic at high concentrations, the inherent toxicity of native DA at high doses and relatively short exposure times underscores the necessity to develop probes that enable sub-micromolar detection limits for use in standard analytical platforms.
Figure 4.
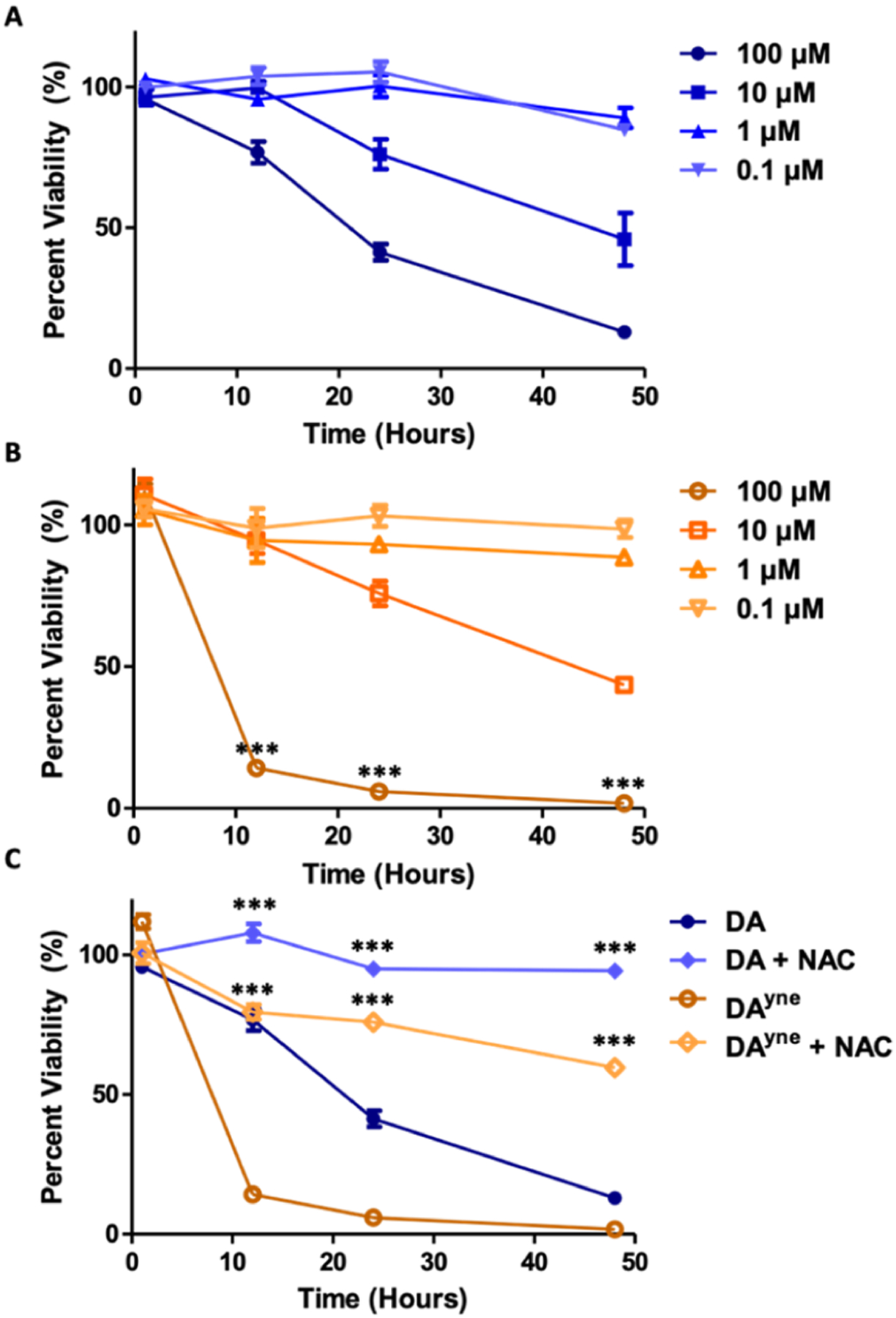
DA and DAyne reduce SH-SY5Y cell viability. Cell viability was determined through an alamarBlue assay. Data are represented as mean ± SEM from four replicates and normalized to DMSO control. All treatments of DA and DAyne at analogous times and concentrations were analyzed via a one-way ANVOA and an unpaired t-test (***p < 0.001). Percent viability of cells was determined following treatment with (A) DA or (B) DAyne alone. (C) Cells were treated with 100 μM DA or DAyne in the presence or absence of 2.5 mM NAC, which showed that cell viability was rescued under these conditions.
To ensure that the observed DA and DAyne toxicity was caused by oxidation of the catechol to a quinone, we co-treated cells with DA or DAyne and an antioxidant, NAC. Addition of 2.5 mM NAC significantly rescued viability in cells treated with 100 μM of DA and DAyne (Figure 4C). Co-treatment with NAC for 24 h improved cell viability to 95 and 76% for DA and DAyne, respectively, compared with 41% for DA and 6% for DAyne when NAC is absent. Rescue due to other processes influenced by NAC cannot be ruled out by our experiments.37 Nevertheless, our in vitro data reported here support DA and DAyne quinone electrophiles as drivers of cellular toxicity.
DAyne Aids in Visualizing DPAs.
Having demonstrated conditions wherein cells tolerate the DAyne probe, we focused on characterizing DPAs in SH-SY5Y cells. We first quantified the relative number of DPAs generated in undifferentiated cells treated with DAyne under a variety of conditions that we knew to be favorable for generating DPAs in vivo. Notably, the cells were treated with the reduced probe, which auto-oxidized in situ. Following DAyne administration and washing off excess probe, the cells were lysed, and a biotin handle was conjugated to DPAs using copper-catalyzed azide–alkyne cycloaddition (CuAAC) click chemistry.38 The resultant lysates were separated using sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) and transferred to a nitrocellulose membrane, where DPAs were visualized with streptavidin (SA) conjugated to an 800 nm fluorescent dye, denoted as SA800 (Figure 5A). Protein concentrations were normalized following staining with the REVERT total protein stain, and the relative amounts of DPAs per treatment condition were quantified (Figure 5B, additional representative blots found in Supporting Information 4). DAyne concentration and duration of treatment influenced the quantity of DPAs observed per treatment group. Incubation for 1 h proved to be insufficient to produce detectable levels of DPAs by Western blotting. Treatment for 12 h with 10 μM DAyne provided optimal results, as DPAs were detectable at 2.5 times above background (Figure 5B), and relatively little toxicity was observed. Treatment with any concentration of DAyne for 24 h resulted in decreased levels of detectable DPAs compared to a 12 h incubation. In the case of 10 μM DAyne treatment, the normalized DPA signal for 24 h exposure was 3.3-fold lower than the 12 h treatment group. Co-incubation of 100 μM DAyne and 2.5 mM NAC for 12 h resulted in a 25-fold reduction of DPA formation compared to DAyne alone, which is consistent with results in Figure 4C that show that NAC mediates rescue cell viability upon DQ exposure.
Figure 5.
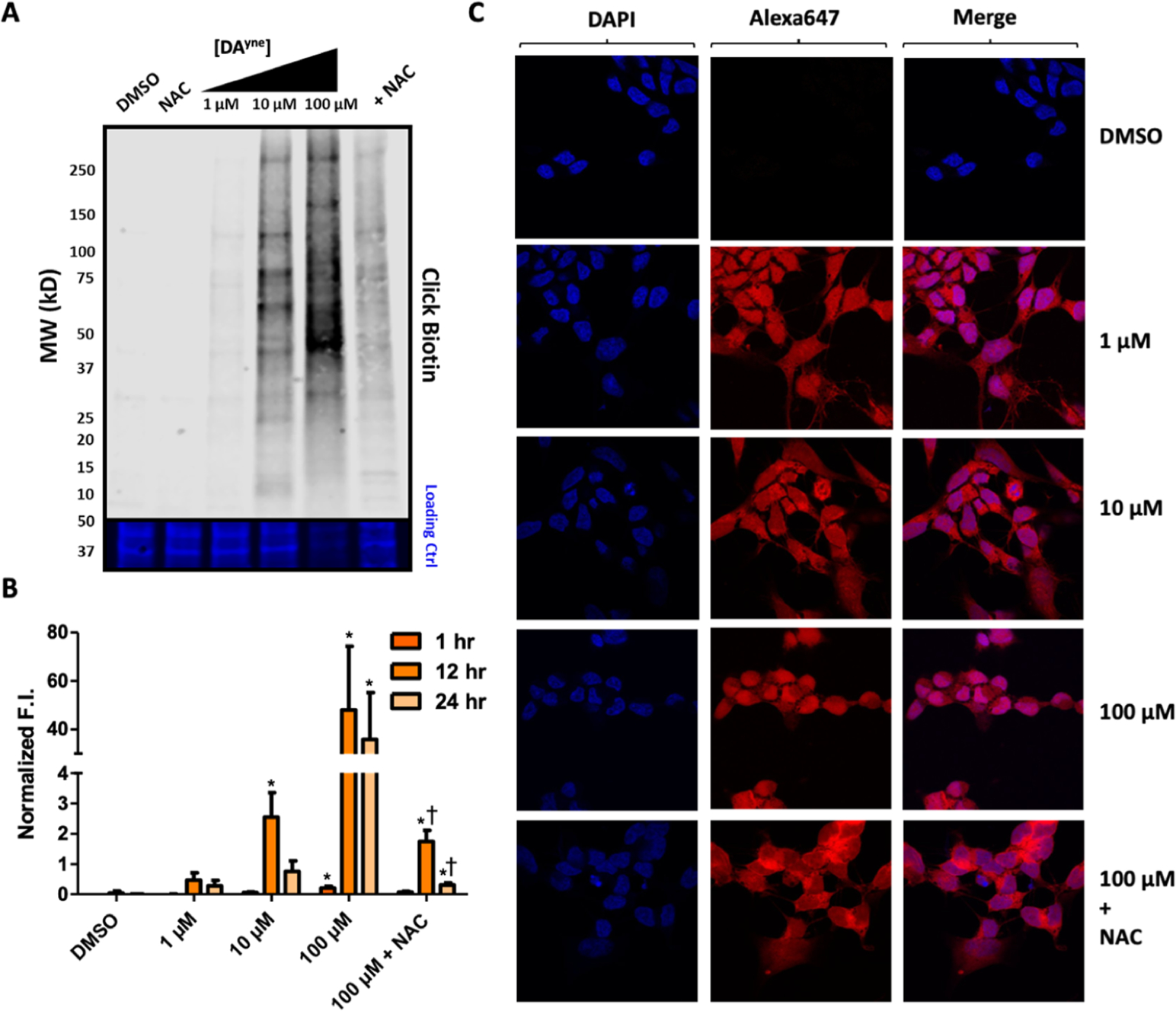
Visualization of DPAs by DAyne. (A) Representative nitrocellulose blot following incubation of SH-SY5Y cells with DMSO, 2.5 mM NAC, 1 μM, 10 μM, 100 μM, or 100 μM DAyne and 2.5 mM NAC for 12 h, lysis and ensuing CuAAC click reaction with biotin azide. Top: 800 nm channel following incubation with SA800. Bottom: 700 nm channel following REVERT total protein stain. (B) Quantification of DPAs. Fluorescence intensity (F.I.) of SA800 for each treatment was normalized by total protein through the REVERT stain. Data are represented as mean ± SEM from three replicates. Data were analyzed via a two-way ANOVA and an unpaired t-test. (Comparisons between: DMSO *p < 0.05; 100 μM DAyne †p < 0.05 for given treatment duration). (C) Confocal imaging of SH-SY5Y cells following treatment with DMSO, 1 μM, 10 μM, 100 μM, or 100 μM DAyne and 2.5 mM NAC, respectively for 12 h, fixation with paraformaldehyde, ensuing CuAAC click reaction with Alexa Fluor 647 azide and staining with DAPI. Images were acquired at 60× magnification.
Reduction in visualized DPAs at extended exposure times was expected when considering the toxicity of DQs. For example, only 75% of the cells remained viable following 10 μM treatment with DA for 24 h. The presence of melanin-like aggregates in cellular debris pellets at higher DAyne treatments suggests that DPAs polymerize and aggregate, which likely sequester DPAs, reducing their levels in western blots (Supporting Information 5). This activation of DA regulatory or DPA stress responses may account for time-dependent reductions in DPA concentrations.
Finally, we wished to determine whether the rate of DAyne oxidation was causing low detection limits at 1 μM doses. We treated SH-SY5Y cells with DAyne in the presence of TY to generate DQs more rapidly than auto-oxidation. Using TY, 1 h exposure of DAyne was sufficient to produce detectable DPAs at doses as low as 10 nM (Supporting Information 6). Thus, DAyne can be used to improve the sensitivity in DPA visualization assays requiring physiologically relevant concentrations of the DA-mimetic.
To verify that DAyne forms adducts with the same proteins that bind DA, we performed a competition binding experiment in which SH-SY5Y cells were pre-incubated with DA and TY for 30 min prior to the addition of DAyne (Supporting Information 7). A dose-dependent decrease in DAyne labeling upon increasing concentration of DA pre-treatment was observed. Incomplete competition at the highest concentration of DA competitor (50 μM) suggests that DQs binds to target proteins at low occupancy, which is consistent with previous reports of similarly functionalized quinone methides.39 Since only a fraction of a particular protein population is modified in the initial DA treatment, the subsequent DAyne exposure results in additional partial labeling. Nevertheless, low levels of DQ protein modification can still drive robust cellular responses,40 supporting the need for biological probes that enhance analytical sensitivity so that DPAs can be characterized in cell models treated with physiological levels of DA-mimetic probes.
To confirm that the levels of DPA formation were similar between DAyne and DA treatment, two previously described methods of detecting quinone-protein adducts, nitro blue tetrazolium (NBT) staining and near infrared (nIR) fluorescence, were used to test the levels of DPAs produced by DAyne and DA.41,42 Qualitative comparison of stained nitrocellulose membranes between DA and DAyne generated DPAs revealed similar amounts of adduct formation (Supporting Information 8), providing confidence that our probe is a relevant mimetic of DA. However, in our hands, these methods proved less sensitive at detecting DPAs compared to our bio-orthogonal visualization strategy using DAyne, which may be due to the lower concentration of DQs used in the current study. Despite increased protein loading to 50 μg per lane to improve signal, quantitative analysis between DA and DAyne produced DPAs could not be performed using these adduct detection methods.
Finally, we demonstrated that fluorescently modified DPAs generated using DAyne could be mapped cellularly using confocal microscopy. Following treatment with DAyne, cells were fixed with paraformaldehyde, and DPAs were conjugated with Alexa Fluor 647 azide using an in-cell click reaction. The cell’s nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI) and imaged on a confocal microscope. In this manner, we verified that DAyne permeates the cell membrane and forms adducts homogeneously throughout the cytosol (Figure 5C). The use of DAyne to visualize DPAs subcellularly demonstrates the versatility of a DA mimetic with alkyne functionality.
Proteomic Identification of DPAs Modified with DAyne.
After demonstrating that cellular DPAs formed using DAyne were similar to those generated with DA, we performed a chemoproteomic experiment to enrich and characterize the proteins that are modified by oxidized metabolites of DAyne. A 12 h incubation period with 10 μM DAyne was selected based on our earlier labeling results (see Figures 4B and 5B). After exposing SH-SY5Y cells to DAyne, DPAs were immobilized onto azide-functionalized agarose beads using CuAAC click chemistry. This approach is advantageous compared to traditional biotin-streptavidin enrichment as it eliminates the need to remove excess biotin and click reagents prior to immobilization. It also precludes interference from naturally biotinylated proteins, avoids streptavidin contamination, and enables stringent washing steps to remove proteins that nonspecifically bind the Sepharose column without removing DPAs. Using this approach, we verified that DPAs were captured by the azide beads through Western blot (Supporting Information 9). Following capture, the DPA bound beads were washed extensively and subjected to on-bead trypsin digestion. The resultant peptides were desalted and analyzed mass spectrometrically and quantified using the MaxQuant software package.
SH-SY5Y cells were treated with DAyne or dimethyl sulfoxide (DMSO) across three biological replicates. 38 proteins were significantly enriched by DAyne using the methods outlined above (Figure 6A, and Supporting Information Table S1 for a full listing of proteins). We ensured that enrichment with DAyne was consistent between treatment groups by correlating the label-free quantitation (LFQ) intensity values of identified proteins between different treatment conditions and replicates (Supporting Information 10). We observed a correlation of 0.91 ± 0.02 in our DAyne-enriched samples, indicating reproducible enrichment. Additionally, the correlation within the DMSO treatment group was also high, 0.79 ± 0.06; whereas the correlation between DAyne and DMSO groups was low, 0.38 ± 0.06, verifying that DAyne enrichment was robust.
Figure 6.
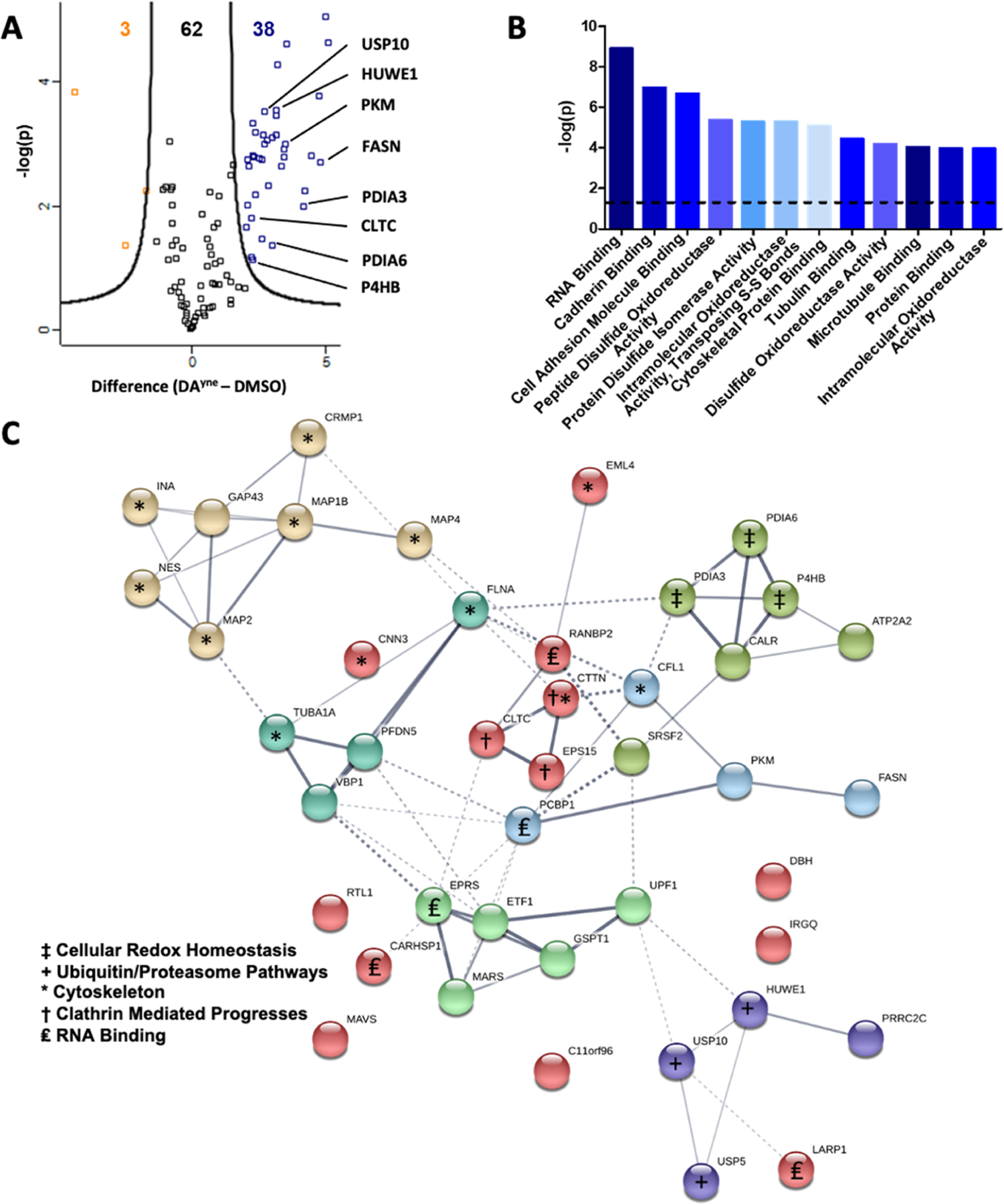
Proteomic enrichment and bioinformatic analysis of DPAs. (A) Volcano plot for quantitative analysis of proteins identified following 12 h treatment of SH-SY5Y cells with DMSO or 10 μM DAyne and enrichment at a FDR of 0.05 and a minimal coefficient of variation (S0) of 2.0. Proteins highlighted in orange or blue were significantly enriched in the DMSO or DAyne treatment, respectively. Data are representative of 3 biological replicates from each treatment condition. (B) Molecular function of DAyne enriched proteins. Analysis performed with PANTHER overrepresentation test (released 20190711); annotation version: GO Ontology database (released 20190703), using Fisher’s exact test with FDR correction. The dashed line represents a statistical cutoff of p = 0.05. (C) Pathway analysis of DAyne enriched proteins was performed with the STRING (Search Tool for the Retrieve of Interacting Genes, version 11) database. Confidence of interaction represented by line thickness. Proteins involved in enriched processes are labeled.
Gene ontology (GO) analysis of these enriched proteins (http://www.pantherdb.org/) revealed a variety of over-represented molecular functions, such as RNA binding, disulfide oxidoreductase activity, and cytoskeleton regulation (Figure 6B).43 Other notable DPAs found in our proteomic results were pyruvate kinase and fatty acid synthase, which play important roles in cellular energy homeostasis. The DPA interactome network of enriched proteins was analyzed using the STRING (Search Tool for the Retrieve of Interacting Genes) database (https://string-db.org/) to visualize the pathways and functions that might be impacted by DPA formation (Figure 6C).44 Notably, cell redox homeostasis and ubiquitin/proteosome pathways were enriched, suggesting DPAs may interfere with these processes. Enriched proteins featured a high prevalence of sulfhydryl-containing active sites, such as those within the oxidoreductase pathway. Pathways such as cellular energy homeostasis and clathrin-mediated processes identified in our STRING analysis were unexpected as proteins involved in these processes have not yet been characterized as DPAs. Such protein adducts, once validated, would represent novel targets as potential contributors to neurotoxicity during PD.
PDI Activity is Inhibited by DQ Modification.
Our next objective was to validate candidate DPAs from our proteomic analysis. We focused on the PDIs because three isoforms of PDIs (PDIA3, PDIA6 and P4BH) were significantly enriched by DAyne. PDIs are critical in a variety of cell stress responses, including five molecular functions: peptide disulfide oxidoreductase activity, PDI activity, transposing S–S bonds, disulfide oxidoreductase activity, and intramolecular oxidoreductase activity (shown in Figure 6B) that were significantly altered upon treatment with DAyne. Notably, there is evidence that PDIs play an important role in preventing neurodegeneration.
We initially verified that DAyne covalently modifies recombinant PDIA3 using a simple binding assay by exposing expressed PDIA3 to DAyne and monitoring the appearance of adduct formation by SDS-PAGE analysis. To do so, DAyne was oxidized with TY and incubated with recombinant PDIA3 for 1 h, then Alexa Fluor 647 azide was conjugated to DAyne in the mixture, and conjugate formation was visualized as a band at 58 kDa corresponding to the DPA. As shown in Figure 7A, DAyne labeled PDIA3, and binding was competed upon exposure of PDIA3 to oxidized DA for 30 min prior to DAyne treatment. These data support labeling of PDIA3 by DAyne at sites also occupied natively by DA.
Figure 7.
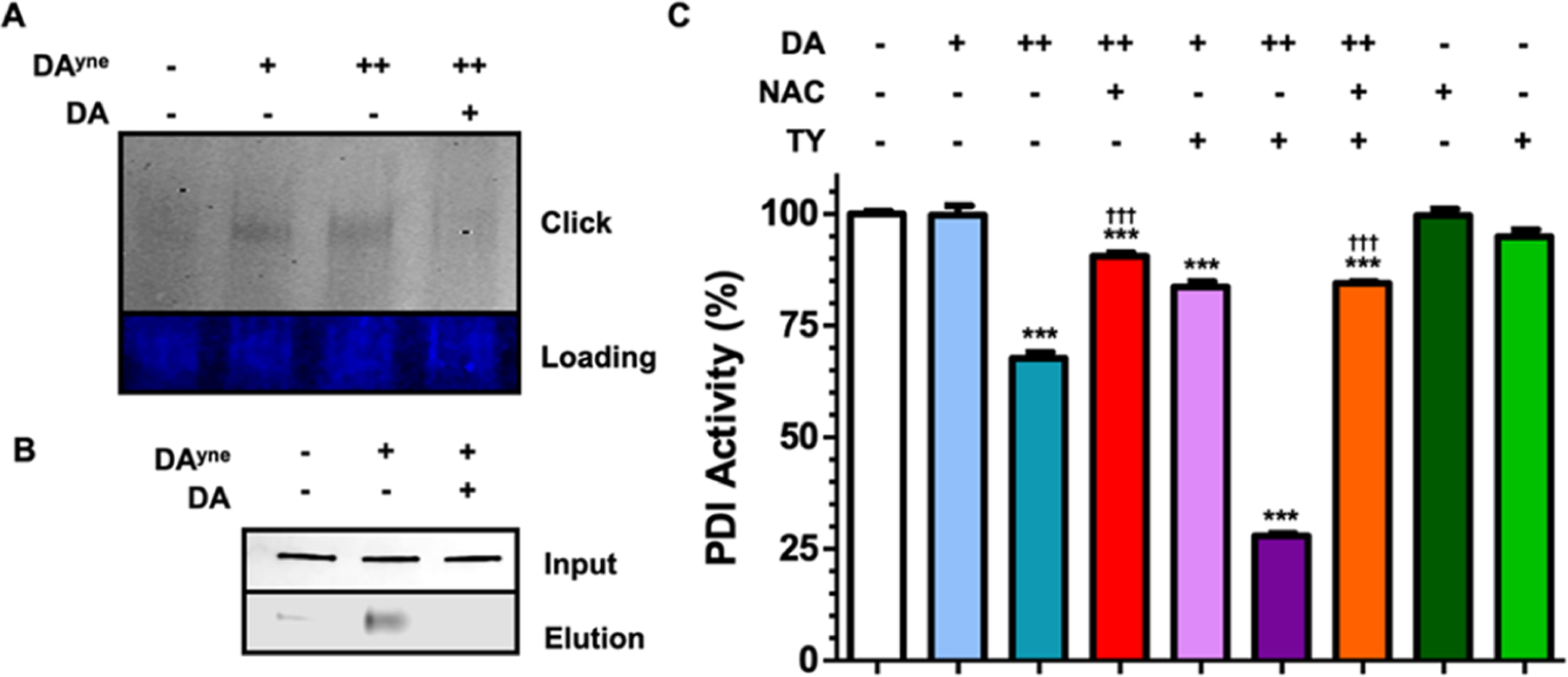
PDIA3 is covalently modified and functionally inhibited by DQs. (A) Representative SDS-PAGE gel after incubating PDIA3 for 1 h with DMSO, 10 μM, 150 μM DAyne preoxidized with TY, or with 150 μM DAyne after a 30 min 150 μM DA pre-treatment (both preoxidized with TY), and ensuing CuAAC click reaction with Alexa Fluor 647 azide. Top: Alexa Fluor 647 fluorescence intensity. Bottom: Imperial protein stain. (B) Representative nitrocellulose blot probed for PDIA3 following capture with DAyne. SH-SY5Y lysate was incubated with DMSO, 150 μM DAyne, or with 150 μM DAyne after a 30 min 150 μM DA pre-treatment lysis and subjected to a CuAAC click reaction with biotin azide. Samples were then incubated with streptavidin agarose beads to capture DPAs. Top: sample lysate prior to streptavidin enrichment. Bottom: elution of DPAs following streptavidin agarose enrichment. (C) Percent PDI activity following a 30 min incubation with the indicated treatment. Final concentrations of DA used were 10 and 150 μM. TY was used to oxidize DA to DQs. The final NAC concentration was 1 mM. Data are represented as mean ± SEM from four replicates and normalized to DMSO control. Data were analyzed via a two-way ANOVA and a tukey post hoc test. (Comparisons between: DMSO ***P < 0.0001; 150 μM DA †††P < 0.0001) for given treatment duration.
Native PDIA3 in non-transfected SH-SY5Y cells was also captured by DAyne following 1 h exposures of the lysates to oxidized DAyne. In these experiments, DPAs were modified with biotin azide using click chemistry and captured on streptavidin agarose beads. After DPA capture, non-specifically bound proteins were washed off and DPAs were eluted at 95 °C in gel-loading buffer. Immunoblots shown in Figure 7B revealed that DAyne-modified PDIA3 was successfully captured in our pulldown. DA also competed with DAyne binding to PDIA3 (Figure 7B). Attempts to identify the specific site(s) of DA modification on PDIA3 by liquid chromatography (LC)–mass spectrometry (MS)/MS analysis of tryptic peptides were unsuccessful. These results are consistent with previous studies that have reported difficulties detecting site specific DA adducted peptides.45–47
Finally, we explored the influence of DA on PDI enzymatic activity using a functional assay that measures the protein folding activity of PDI. We incubated PDI with DA and preoxidized DA (DQ), both in the presence or absence of NAC, and measured enzymatic activity through a fluorescence-based assay as shown in Figure 7C (raw data shown in Supporting Information 11). We found that treatment with 150 μM DA and 150 μM DQ significantly lowered PDI activity to 67 and 29%, respectively, and that NAC and TY treatment alone had no effect on activity. However, cotreatment of DA and DQ with 1 mM NAC to intercept DQs, significantly rescued PDI activity to 90 and 84%, respectively, suggesting that covalent modification of PDI by oxidized DA metabolites inhibits the function of PDI. Collectively, our results validate PDIA3 as an extravesicular target of oxidized DA and that DPA formation results in comprised enzymatic activity.
DISCUSSION
Therapeutic interventions capable of halting or reversing PD progression will require a complete understanding of the initial molecular events that predispose dopaminergic neurons to degenerate and the subsequent biochemistry that results in clinical symptoms. There is substantial evidence indicating that oxidized dopamine metabolites, DQs, are involved in early mechanisms of neurodegeneration during PD, including their spatiotemporal tissue distribution and their increased abundance observed in tissue samples from patients suffering from more severe forms of PD.5 These studies are consistent with a model of PD progression that is highly dependent on or exacerbated by DA metabolites in early stages of cellular dysfunction. As these reactive metabolites that are measurable before PD symptoms are detected clinically through movement tests, they are enticing biomarkers that predict PD.5 DPAs represent surrogates of DQs that could be measured in patients in a clinical setting and form the basis for early biochemical tests that predict both the probability of developing PD symptoms and their severity. Moreover, DQ-modification leads to generation of DPAs that result in neurotoxic outcomes and many DPAs are expected to represent novel targets for therapeutic design and development.
Given the heterogeneity and complexity of PD pathogenesis, unbiased screening for DPAs on a proteome-wide scale using mass spectrometry technologies is ideal for comprehensively enumerating the DA-modified proteome without foreknowledge of disease etiology. Such DPAs can later be validated for disease-relevance using standard biochemical assays, which we demonstrated for PDIA3. However, owing to the complex reactivity profile of DA, few studies have reported the development of such unbiased techniques for DA.48 Several limitations have prevented such technologies from reaching fruition, including the poor sensitivity of analytical methods used for the detection of incipient DPAs in cell models. These include the low DPA levels expected natively in cell and in vivo models due to low occupancy binding of DA and regulatory mechanisms that maintain low DPA and cytosolic DA levels, such as neuromelanin formation (Figure 5). Given that DPA levels and detection sensitivities in the presence of such regulatory mechanisms must be low, we reasoned that DPA detection sensitives might be increased by enriching adducts. Thus, a DA-mimetic proteomic probe (DAyne) was designed with an alkynyl enrichment handle included to extract DPAs and improve proteomic and fluorometric sensitivity. Reduction of ostensible off-targets in these enrichment studies could then be reduced by modifying DA exposure to cells without compromising analytical sensitivity. These principles led to the design of DAyne.
Our DA-mimetic reactivity-based probe DAyne, includes an amino-modified propargyl enrichment tag on the DA scaffold, which makes it unique among DA-mimetic probes. This modification does not prevent our probe from cyclizing into an aminochrome-mimetic upon oxidation in a reaction trajectory that mirrors DA oxidation both kinetically and thermodynamically. Developing a probe that shares a similar kinetic and thermodynamic profile to DA is of paramount importance to ensure DQ metabolite lifetimes and protein bindings mirror native DQ metabolite binding when profiling representative DPAs in cell models. Indeed, we show this to be the case spectroscopically in Figure 3, which demonstrates that the oxidation of DA and DAyne leads to the formation of DQ (1b) and aminochrome species (1d) at similar apparent rates. Similar to previous studies, we observed the aminochrome species to be the most thermodynamically stable under our in vitro conditions for both DA and DAyne and likely the primary DQ metabolites involved in DPA generation. Alkylating the amine did not drastically alter DA-induced cytotoxicity at low doses in vitro and enabled visualization of DPAs by fluorescent microscopy. These latter studies confirm, at least qualitatively, that the functional profile of DA is represented faithfully by DAyne due to kinetic and thermodynamic properties among their chief metabolites. Therefore, amino-tagged DA offers the potential to comprehensively profile DPAs generated from all DA oxidative metabolites.
Probe molecules designed previously to mimic DA have taken advantage of nonreactive functional groups to prevent cyclization of DQ, increasing the abundance of DQ-modified DPAs but preventing aminochrome (1d) formation and their DPA counterparts. For example, functionalized hydroxydopamine (6-OHDA) quinone-based reactivity probes have incorporated enrichment handles via an amide bond at nitrogen.48 However, these probes rely on aminoacyl installation chemistries for progargyl incorporation. Such probes bear polarized amide carbonyls that significantly alter the electrostatic potential of the DA scaffold, preventing it from traversing the membrane of live cells, limiting its broad utility. Additionally, 6-OHDA based probes can only generate DQ-like oxidation products and not cyclic intermediates like 1d, 1e, and 1f, limiting the scope of potential DPAs detected to those generated with DA. Given the short lifetime of DQ-like metabolites (1b) and the long lifetime of aminochrome (1d) metabolites observed for DA, the biological pathways of these product metabolites may be different, and DPAs identified using 6-OHDA probes may not represent PD-specific DPAs. The failure of 6-OHDA to cyclize into 1d and 1e might account for differences in pathologies observed between 6-OHDA rat models and clinical PD.2 However, DQ-based probes should complement the use of probes like DAyne as they can aid in characterization efforts that distinguish the relative importance of each class of DPA modification in disease etiology. DPA enrichment should serve as a complement to other powerful characterization methods that use native DA as a probe, such as 14C autoradiography and isotopic tandem orthogonal proteolysis-activity based protein profiling (iso-TOP-ABPP) methodology.26,27,49
There are several advantages to using DAyne as a DPA characterization that cannot be achieved using state-of-the-art analytical platforms discussed above. For example, the use of a DA mimic to identify native DPAs eliminates the use of toxic radioactive 14C detection isotopes to visualize low-abundant DPAs. Alkyne–azide pull-downs also provide higher selectivity than boronic acid resins used previously to enrich catechol-containing DPAs, but that are unable to distinguish catechols from other vicinal alcohols. Additionally, enrichment chemistries for DAyne are robust, allowing exposures at physiological concentrations. Distinct protein bands were observed in our immunoblots after exposing SH-SY5Y cells to concentrations as low as 10 nM after oxidation with TY (Figure S6). DAyne enrichment also proved advantageous for carrying out studies requiring long-term exposures. The amount of external DA— or, as in the case here, DAyne—added to cells for the detection of DPAs are more than ten times lower and fall within physiologically relevant concentrations than many previous studies.26,27 These lower concentrations enabled long time-resolved in vitro experiments to be conducted without significant cell death or compromising detection of DPAs. Parallel multidimensional analyses, such as fluorescence microscopy and proteomic analyses (Figures 5 and 6) also enhance the utility of this probe molecule, enabling reproducible discovery and validation of DPA targets. Collectively, DAyne proved to be a versatile probe molecule to generate reproducible adducts that may begin to shape our understanding of DA’s contribution to neurodegeneration during the entire aging spectrum.
One of the useful features of DAyne highlighted here is the high sensitivity realized in chemoproteomic enrichment experiments, which revealed that DQs form adducts with proteins contained within several diverse families that influence myriad functions. Proteins that are inactivated or structurally altered within these families and pathways have the potential to significantly compromise neuronal integrity and influence age-related neurodegeneration or PD progression. Within our collection of candidate DPAs are various classes of proteins that have been identified in previous studies, including cytoskeletal proteins, ubiquitin proteosome machinery and protein folding.26–28 PDIs were among the major classes of enriched DPAs identified in our GO analysis (Figure 6B). PDIs assist in protein folding and rely on thiol disulfide oxidoreductase activity to create S–S bonds. Impairment of that function by DQ modification is expected to activate the unfolded protein response, elicit ER stress and perturb cell redox homeostasis, which were identified as major pathways affected by DAyne exposure in a STRING analysis (Figure 6C). We confirmed that both DA and DAyne modify PDIA3 through competitive experiments with recombinant protein and in cellular lysates (Figure 7A,B). We also showed that DQs functionally inhibit PDI activity (Figure 7C). Collectively, the data presented here are consistent with reports from other laboratories that demonstrate the initiation of ER stress due to quinone exposure and, specifically, inactivation of PDI family proteins upon quinone modification.48,50 For example, Farzam et al. identified that PDIA1 oxidoreductase activity was inhibited by 6-OHDA.48 They also show that this inhibition is rescued in the presence of cysteine, but not methionine, suggesting that inhibition is a direct result of PDI modification and not the generation of ROS. The results from our study, therefore, provide conclusive evidence that PDI proteins are modified and functionally inactivated by cytosolic DA metabolites.
PD is a multifactorial and heterogeneous degenerative disease and several possible pathways involved in protein folding (e.g., ER stress), oxidative stress, and cellular organization (e.g., cytoskeletal and endosomal dysfunction) have been proposed as primary mechanisms leading to neurodegeneration.51 Therefore, multiple pathways are likely involved in PD pathogenesis and an unbiased proteomics approach for identifying protein targets perturbed by the presence of DQs may help elucidate precise mechanisms by which DA contributes to neurodegeneration. PDI is upregulated during ER stress, for example, which is highly associated with protein misfolding and oxidative stress responses. Members of the PDI family are highly expressed during PD and are found in Lewy bodies from PD donor SNc.52 Thus, our data suggest that PDI inactivation by DA contributes to ER dysfunction that may contribute to protein misfolding and Lewy body pathology. Interrogating the role of DA and PDI within these specific pathways may uncover molecular-level targets for future therapeutic discovery.
Modification of cytoskeletal proteins by DA has been proposed as another molecular mechanism for DPA-induced toxicity,53 and several proteins involved in these pathways were identified as DA targets in our study. The microtubule associated proteins (MAPs) were identified here as proteomic hits are particularly interesting due to their interactions with the tau protein that forms neurofibrillary tangles during Alzheimer’s disease pathogenesis. MAPs organize subcellular components, organelles, and synaptic vesicles. We also found proteins impacted by DPAs that are involved in proteasomal degradation pathways, such as UPS10 and HUWE3. Other studies have postulated that DA modification of these proteolytic enzymes contribute to DA derived toxicity, as cells exposed to DA lose the ability to break down damaged proteins.17,24 The identification of DPAs here that have been reported elsewhere further validates the utility of our probe technology to characterize DPAs.26–28
In addition to the identification of DPAs featured in PD-related pathogeneses, our chemo-proteomic experiments also identified novel DPAs that have not been reported previously. Notably, we found a significant enrichment of clathrin heavy chain 1 (CLTC), which affects synaptic vesicle formation.54 This result is notable given recent GWAS data associating mutations in CLTC with impaired synaptic vesicle endocytosis and PD incidence.55 Our discovery that DA modifies CLTC requires more thorough investigation to validate how DA modification specifically influences synaptic vesicle endocytosis.
It is important to note significant differences between our study and prior works, which likely account for the absence of PD-related DPAs enriched in our proteomics data set, including the redox sensor protein DJ-1 and the E3 ubiquitin ligase parkin. We attribute this difference to our use of undifferentiated SH-SY5Y cells and our dosing of cells at lower concentrations for longer exposures. Our cell-based experiments were conducted at 10 μM DAyne to reduce the level of nonspecific or off-target adducts, while other studies have employed up to 150 μM DA.26,27 Low probe concentrations also enabled us to expose cells for 24 h. We hypothesize that these longer treatment conditions allow for study of time-resolved PD models, which should enable interrogation of the earliest mechanisms of synaptic density decline during aging and PD.
Controlling the complex oxidation chemistry of DA has been a major hurdle in understanding the consequences of dysregulated DA during normal aging and PD. While the development of a chemoproteomic DA-mimetic represents a significant advance in characterizing DPAs generated from oxidized DA, the rapid kinetics of AC formation under our conditions may limit enriched adducts to those generated with AC and, potentially, DHI. It is possible that cellular conditions favoring DA dysregulation may enhance DQ levels, in which case probes designed with amido-alkyne linkages or 6-substituted catechols may be more suitable to characterize these quinoproteins. The rate constant for ortho-quinones modification by thiols is approximately 10,000-fold higher than for amines and several studies have demonstrated that cysteine sidechains are the most susceptible protein residues towards DQ adduction, so we assume that the proteins identified in this current study are generated through cysteine residues.11,56 We foresee utility in developing conditions that permit DAyne to map specific sites of DQ modification on DPAs by enriching DQ-adducted peptides for mass spectrometric analyses to validate this and aid in determining motifs with high propensities for DQ-modification.57–60 Until this problem is solved, future studies could explore a suite of DA mimetic probes that each represent a specific DA oxidation state (DQ, AC, DHI) to access any differential reactivity between these metabolites.
METHODS
Materials.
All chemicals required for the synthesis of DAyne, preparation of buffers, mushroom tyrosinase (Lot#: SLBZ0022), NBT, Amicon ultra 0.5 mL centrifugal filters (3k) and click reactions were purchased from Sigma-Aldrich unless otherwise stated. NMR solvents were obtained from Cambridge Isotope Laboratories Inc. SH-SY5Y neuroblastoma cells were acquired from ATCC. Cell culture media, additives, and consumables were purchased from Corning. High capacity streptavidin agarose resin, Zeba Spin Desalting Columns (7K MWCO, 0.5 mL), PDIA3 (ERp57) antibody (Cat. no. CL2444), and DAPI were obtained from Thermo Scientific. alamarBlue HS reagent and ProLong Glass were purchased from Invitrogen. BCA kits and C-18 spin columns were bought from Pierce. Mini-PROTEAN TGX Precast Gels, 0.2 μm nitrocellulose, and filter paper transfer stacks were obtained from BioRad. Biotin azide, THPTA, Alexa Fluor 647 azide, and azide agarose beads were purchased from Click Chemistry Tools. REVERT total protein stain kit, Odyssey TBS blocking buffer, and IRDye800CW streptavidin were purchased from LI-COR. Recombinant PDIA3 and PDI inhibitor screening kit (fluorometric) were obtained from BioVision (Cat. no. 7601–100 and K840–100, respectively).
alamarBlue Cell Viability Assay.
Undifferentiated SH-SY5Y cells were cultured in 1:1 MEM:F12 media containing 10% FBS and Pen/Strep 100 U/mL under a 5% CO2 atmosphere at 37 °C. Cells were seeded into 96 well plates at a density of 20,000 cells per well. 24 h after seeding, the media were removed and replaced with the indicated treatment condition in 1:1 MEM:F12 media absent of FBS, 0.2% DMSO. Following the allotted incubation time, the plates were spun at 1000g for 5 min and the media were removed and replaced with 100 μL of a 10% alamarBlue solution in FBS free 1:1 MEM:F12. The plates were incubated for 4 h in the dark at 37 °C and then fluorescence was measured at Ex/Em 560/590 nm on a BioTek Synergy H1 plate reader. Cell viability for a given treatment condition was calculated by normalizing the fluorescence signal to the DMSO treated control.
UV–vis Spectroscopy.
Fresh stock solutions of DA, DAyne, NaIO4, and TY were prepared in Millipore Milli-Q water or PBS (pH 7.4) that had been purged for 15 min under argon. The oxidation reactions were initiated by combining the reactants in a 2 mL Beckman quartz cuvette prior to reading. The spectra were acquired every 30 s for 30 min on a Thermo Scientific NanoDrop One.
NMR Spectroscopy.
Spectra were obtained on a Varian 400 MHz spectrometer. Fresh stock solutions of DA, DAyne, and NaIO4 were prepared in deuterated PBS (pH 7.4, pD uncorrected) that had been purged for 15 min with argon. The oxidation reactions were initiated by combining the reactants in an NMR tube. Reactions were allowed to proceed for 4 h at 25 °C and monitored using a data accumulation of eight scans for each spectrum.
Click Reaction and Western Blot.
Undifferentiated SH-SY5Y cells were cultured in 6 well plates to 90% confluency with 1:1 MEM:F12 media containing 10% FBS and Pen/Strep 100 U/mL under a 5% CO2 atmosphere at 37 °C. The media was removed and replaced with the indicated treatment condition in 1:1 MEM:F12 media absent of FBS, 0.2% DMSO. Following the allotted incubation time, the cells were washed three times with PBS and lysed in 200 μL of MPER containing HALT protease and phosphatase inhibitor cocktail. The lysates were centrifuged at 17,000g for 12 min and the supernatant was incubated with 10 μL of high capacity streptavidin agarose resin (Cat: 20359; Lot; TK27535) at 4 °C for 16 h to remove naturally biotinylated proteins. Samples were then centrifuged at 1000g for 5 min and the supernatant was carefully aspirated. Protein concentrations were determined through a BCA assay per manufacturer’s protocol.
Click reactions were performed in 200 μL of MPER at a protein concentration of 100 μg/mL with 25 μM biotin azide, 1.5 mM THPTA, 1 mM CuSO4, and 3 mM Na ascorbate. The reactions were agitated at 25 °C for 1 h in the dark. Proteins were precipitated with 800 μL cold acetone overnight at −20 °C. Following 20 min of centrifugation at 17,000g, acetone was carefully aspirated, and the protein pellets were dissolved into 15 μL of 1× LI-COR loading buffer with 10% BME and denatured for 5 min at 95 °C. Each sample was loaded using 13 μL of sample into 4–20% gradient Mini-PROTEAN TGX Precast Gels for SDS-PAGE. The gels were then blotted onto 0.2 μm nitrocellulose with a BioRad Trans-Blot Turbo apparatus. Blots were stained with REVERT total protein stain per manufacturer’s protocol and imaged by a LI-COR Odyssey CLX. This was followed by a 1 h block at RT in Odyssey TBS blocking buffer and incubated with IRDye800CW streptavidin (Lot#: C80918–11) at a 1:10,000 dilution for 1 h. The membranes were then washed and imaged by a LI-COR Odyssey CLX. Western blot quantification was performed with Image Studio Software following the manufacturer’s protocol for normalization with REVERT total protein stain.
Confocal Microscopy.
Undifferentiated SH-SY5Y cells were cultured on Neuvitro GC-25–1.5-Laminin coverslips to 90% confluency with 1:1 MEM:F12 media containing 10% FBS and Pen/Strep 100 U/mL under a 5% CO2 atmosphere at 37 °C. The media were removed and replaced with the indicated treatment condition in 1:1 MEM:F12 media absent of FBS, 0.2% DMSO. Following the allotted incubation time, the cells were washed three times with 2 mL PBS and fixed with 2 mL 4% PFA (molecular biology grade) in PBS and washed an additional three times with PBS. In-cell click reactions were performed by incubating the cells in 0.1% Triton X-100 PBS (PBT) solution containing 10 μM Alexa Fluor 647 azide, 1.5 mM THPTA, 1 mM CuSO4 and 50 mM Na ascorbate for 1 h. The cells were washed three times with PBT, stained with 300 nM of DAPI for 30 min, and washed an additional three times with PBT. A drop of ProLong Glass anti-photobleaching solution was placed on the coverslips and was allowed to cure for 48 h. Coverslips were mounted to glass slides, sealed with nail polish and stored at 4 °C until imaging. Images were acquired on an Olympus FluoView FV1000 BX2 upright confocal microscope using a PLAPON 60 × O NA:1.42 objective. Laser Ex/Em were set at 405:461 nm and 635:668 nm for DAPI and Alexa Fluor 647, respectively. Acquisition parameters for example voltage, gain, offset, ect were optimized for 10 μM DAyne treatment and kept constant when imaging other conditions.
Proteomics.
Probe Treatment.
SH-SY5Y cells in 1:1 MEM:F12 media containing 10% FBS and Pen/Strep 100 U/mL were grown to 90% confluency in T75 flasks. The cells were then washed with PBS and incubated with 1:1 MEM:F12 media (no FBS) containing 10 μM DAyne or vehicle for 12 h at 37 °C. The cells were then washed with PBS, suspended in a trypsin-containing buffer, pelleted at 500g for 5 min, and further washed three times in PBS. The cells were lysed with 200 μL of MPER containing HALT protease and phosphatase inhibitors for 10 min at RT per the manufacture’s protocol. The samples were subjected to centrifugation at 17,000g for 12 min to remove cellular debris and the protein concentration was determined by BCA.
On-Bead Click Reaction.
Click Chemistry Tools azide agarose beads (100 μL of Cat: 31038–2; Lot: 2333) were washed twice with 1 mL of water. The click reaction was conducted by adding a 1 mL solution of 100 μg of protein lysate, 1.5 mM THPTA, 1 mM CuSO4, 3 mM Na ascorbate in MPER to the beads. The reaction was allowed to proceed for 16 h before the beads were pelleted by centrifugation and the supernatant was aspirated.
Washing of Agarose Beads.
The beads were washed once with water and the proteins bound to the beads were reduced by addition of 100 mM dithiothreitol (DTT) in 1 mL of 1% SDS, 5 mM ethylenediaminetetraacetic acid (EDTA), 250 mM NaCl, 100 mM Tris pH 8.0 and incubation at 70 °C for 15 min. After pelleting the beads and removing the supernatant, alkylation was performed with 40 mM iodoacetamide in the dark for 30 min in the same buffer as the previous step. Following centrifugation and removal of the supernatant, the beads were washed five times with 1 mL of 1% SDS, 5 mM EDTA, and 100 mM NaCl in PBS; then, 10 times with 8 M urea in 100 mM Tris pH 8.0; and finally, 10 times with 20% CH3CN.
On Bead Digestion.
Digestion was performed by suspending the beads in 200 μL of 100 mM Tris, 2 mM CaCl2 pH 8.0 and the addition 1 μg of trypsin. The reaction was rotated at 37 °C overnight. The supernatant was transferred to a new tube and combined with two additional 300 μL water 0.1% formic acid washes. Samples were desalted with Pierce C-18 spin columns (Cat: 69725; Lot: TJ276015) per the manufacturer’s instruction with MS grade water, CH3CN, and formic acid. Samples were concentrated by vacuum evaporation and stored at −20 °C until LC–MS/MS analysis.
LC–MS/MS Analysis.
Tryptic peptides from each biological replicate were analyzed by HPLC-electrospray ionization + -MS/MS using a Thermo Scientific Q Exactive Hybrid Quadrupole-Orbitrap Mass Spectrometer in line with an Eksigent NanoLC-Ultra 2D HPLC system, a nanospray source, and Xcalibur 4.1.31.9 software for instrument control. Dried peptide samples were reconstituted in 20 μL of 2% CH3CN; 0.1% formic acid and 4 μL were separated on an in-house packed C18 column (15 cm × 75 μm, Luna C18) over a 90 min gradient with degassed HPLC buffer; buffer A (0.1% formic acid); buffer B (CH3CN; 0.1% formic acid). Source parameters detailed a spray voltage of 4.0 kV, a capillary temperature of 320 °C, and an S-lens RF level of 55%. MS analysis was performed in a top 12 data-dependent mode. The survey scan was performed using a resolution setting of 70,000 at 200 m/z with an AGC target of 1 × 106. Dynamic exclusion was enabled with an exclusion duration for 30 s. The 12 most intense precursor ions (excluding singly charged species) were selected for higher-energy C-trap dissociation fragmentation using a collision energy of 30%. The fragmentation scan was performed using a resolution of 17,500 with a AGC target of 5 × 104 or maximum injection time of 50 ms and an isolation window of 2.0 m/z.
Data Analysis.
MaxQuant (version 1.6.5.0) was used to search the raw mass spectrometry data. Default parameters61 were applied and the human Uniprot database (downloaded on 2019/05/15, with 74,349 sequences) was searched against. Trypsin was selected as the protease with allowance for a maximum of two missing cleavages. Variable modifications of methionine oxidation, protein N-terminal acetylation and the fixed modification of cysteine carbamidomethylation were included. The precursor ion and fragmentation mass tolerance of 4.5 ppm and 0.5 Da were selected, respectively. The false discovery rate (FDR) was set to 0.01 at the protein, peptide, and site levels with a minimum peptide length of 6 and a minimum Andromeda score of 40. A minimum of two peptides were required for protein identification. To obtain label-free protein quantification, default LFQ parameters were selected with normalized protein intensities as the output. The Perseus software suite (version 1.6.6.0) was used to process the resultant LFQ intensities.62 The data were Log2 transformed and filtered following previously described methods.63 A two-tailed, two-sample t-test was performed to compare protein abundance between groups.63 Statistically significant enrichment was determined with a Benjamini-Hochberg corrected FDR of 0.05 and a minimal coefficient of variation (S0) of 2.0.
PDIA3 Modification by DAyne and DA.
Click Reaction with Recombinant PDIA3 Protein.
Aliquots of PDIA3 (10 μL of a 1 mg/mL solution in 50 mM Tris buffer, pH 8) were treated with the indicated concentration of preoxidized DAyne for 1 h at 37 °C, the final reaction volume was 50 μL. For the DA competition experiments, PDIA3 was incubated with preoxidized DA for 30 min prior the addition of DAyne. DAyne and DA oxidation was conducted in 500 μL Tris buffer with 600 units of TY and 0.5 mM of DA or DAyne where TY was removed after a 30 min incubation at RT with an amicon ultra 0.5 mL centrifugal filter (3k). Following PDIA3 treatment with DA and DAyne, excess unreacted small molecules were removed with Zeba spin desalting columns (7K MWCO, 0.5 mL), and the samples were subjected to a CuAAC click reaction with Alexa Fluor 647 azide under the same conditions described above. Excess click reaction reagents were removed with another Zeba desalting column and samples were analyzed through SDS-PAGE.
DAyne Meditated Capture of PDIA3 in Lysates.
Aliquots of SH-SY5Y lysate (50 μL of a 2 mg/mL solution) were treated with the indicated concentration of preoxidized DAyne for 1 h at 37 °C, the final reaction volume was 100 μL. For the DA competition experiments, lysate was incubated with preoxidized DA for 30 min prior to the addition of DAyne. The oxidation procedure for DA and DAyne is described above. Following incubation, excess DA and DAyne were removed with Zeba spin desalting columns, a CuAAC click reaction with biotin azide was preformed, and the excess click reaction reagents were removed with another Zeba desalting column. The samples were then incubated with 50 μL of high capacity streptavidin agarose resin for 15 min at RT to capture DPAs. The beads were then washed five times with PBS and then boiled in 1× gel loading buffer to release DPAs. Samples were separated by SDS-PAGE, transferred to nitrocellulose membranes and blotted for PDIA3.
PDI Activity Assay.
The PDI inhibitor screening kit obtained from BioVision was used following the manufacturer’s instructions. Briefly, 2× solutions of the indicated treatments were prepared in the provided assay buffer. 50 μL of these solutions were placed into a black 96 well plate. 45 μL of additional assay buffer and 5 μL of PDI enzyme stock solution was then added and the plate was incubated at 37 °C for 30 min. Next, a 45 μL PDI substrate solution containing oxidized insulin, DTT, and fluorescence turn on probe molecule that binds to reduced insulin was added. The plate was then placed in a plate reader at 37 °C where the fluorescence was measured at Ex/Em 440/490 (indicative of reduced insulin formation) every minute for 30 min. PDI activity was calculated by normalizing the slope of a given treatment to DMSO control.
Supplementary Material
ACKNOWLEDGMENTS
The authors would like to thank the University of Minnesota Imaging Center for training and usage of microscopes and L. Masterson for his helpful discussions and advice.
Funding
This work was supported by the NIH Chemistry and Biology Interface Training grant T32 GM132029 and University of Minnesota Startup Funds. L.N.E. is supported by NIEHS grants ES023350 and ES029911-01.
ABBREVIATIONS
- CLTC
clathrin heavy chain 1
- CuAAC
copper-catalyzed azide alkyne cycloaddition
- DA
dopamine
- DAPI
4′,6-diamidino-2-phenylindole
- DPA
dopamine-protein adduct
- DTT
dithiothreitol
- DQ
dopa-quinone
- EDTA
ethylenediaminetetraacetic acid
- ER
endoplasmic reticulum
- isoTOP-ABPP
isotopic tandem orthogonal proteolysis-activity-based protein profiling
- NAC
N-acetylcysteine
- NBT
nitro blue tetrazolium
- nIR
near infrared
- NMR
nuclear magnetic resonance
- SA
streptavidin
- SNc
substantia nigra pars compacta
- THPTA
tris-hydroxypropyltriazolylmethylamine
- TY
tyrosinas
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschembio.1c00629.
NBT and nIR detection assay methodology, synthetic procedures, supplementary figures, NMR spectra, and proteomic datasets (PDF)
The authors declare no competing financial interest.
Complete contact information is available at: https://pubs.acs.org/10.1021/acschembio.1c00629
Contributor Information
Alexander K. Hurben, Department of Medicinal Chemistry, University of Minnesota, Minneapolis, Minnesota 55455, United States
Luke N. Erber, Department of Medicinal Chemistry and Masonic Cancer Center, University of Minnesota, Minneapolis, Minnesota 55455, United States
Natalia Y. Tretyakova, Department of Medicinal Chemistry and Masonic Cancer Center, University of Minnesota, Minneapolis, Minnesota 55455, United States
Todd M. Doran, Department of Medicinal Chemistry and Institute for Translational Neuroscience, University of Minnesota, Minneapolis, Minnesota 55455, United States
REFERENCES
- (1).GBD 2016 Parkinson’s Disease Collaborators. Global, regional, and national burden of Parkinson’s disease, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2018, 17, 939–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Poewe W; Seppi K; Tanner CM; Halliday GM; Brundin P; Volkmann J; Schrag A-E; Lang AE Parkinson disease. Nat. Rev. Dis. Primers 2017, 3, 17013. [DOI] [PubMed] [Google Scholar]
- (3).Zecca L; Fariello R; Riederer P; Sulzer D; Gatti A; Tampellini D The absolute concentration of nigral neuromelanin, assayed by a new sensitive method, increases throughout the life and is dramatically decreased in Parkinson’s disease. FEBS Lett. 2002, 510, 216–220. [DOI] [PubMed] [Google Scholar]
- (4).Cheng H-C; Ulane CM; Burke RE Clinical progression in Parkinson disease and the neurobiology of axons. Ann. Neurol 2010, 67, 715–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Monzani E; Nicolis S; Dell’Acqua S; Capucciati A; Bacchella C; Zucca FA; Mosharov EV; Sulzer D; Zecca L; Casella L Dopamine, Oxidative Stress and Protein-Quinone Modifications in Parkinson’s and Other Neurodegenerative Diseases. Angew. Chem., Int. Ed. Engl 2019, 58, 6512–6527. [DOI] [PubMed] [Google Scholar]
- (6).Chang D; Nalls MA; Hallgrímsdóttir IB; Hunkapiller J; van der Brug M; Cai F; Kerchner GA; Ayalon G; Bingol B; Sheng M; Hinds D; Behrens TW; Singleton AB; Bhangale TR; Graham RR; International Parkinson’s Disease Genomics Consortium; 23andMe Research Team. A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat. Genet 2017, 49, 1511–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Herrera A; Muñoz P; Steinbusch HWM; Segura-Aguilar J Are Dopamine Oxidation Metabolites Involved in the Loss of Dopaminergic Neurons in the Nigrostriatal System in Parkinson’s Disease? ACS Chem. Neurosci 2017, 8, 702–711. [DOI] [PubMed] [Google Scholar]
- (8).Lohr KM; Bernstein AI; Stout KA; Dunn AR; Lazo CR; Alter SP; Wang M; Li Y; Fan X; Hess EJ; Yi H; Vecchio LM; Goldstein DS; Guillot TS; Salahpour A; Miller GW Increased vesicular monoamine transporter enhances dopamine release and opposes Parkinson disease-related neurodegeneration in vivo. Proc. Natl. Acad. Sci. U. S. A 2014, 111, 9977–9982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Pifl C; Rajput A; Reither H; Blesa J; Cavada C; Obeso JA; Rajput AH; Hornykiewicz O Is Parkinson’s disease a vesicular dopamine storage disorder? Evidence from a study in isolated synaptic vesicles of human and nonhuman primate striatum. J. Neurosci 2014, 34, 8210–8218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Burbulla LF; Song P; Mazzulli JR; Zampese E; Wong YC; Jeon S; Santos DP; Blanz J; Obermaier CD; Strojny C; Savas JN; Kiskinis E; Zhuang X; Krüger R; Surmeier DJ; Krainc D Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson’s disease. Science 2017, 357, 1255–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Tse DCS; McCreery RL; Adams RN Potential oxidative pathways of brain catecholamines. J. Med. Chem 1976, 19, 37–40. [DOI] [PubMed] [Google Scholar]
- (12).Zecca L; Zucca FA; Costi P; Tampellini D; Gatti A; Gerlach M; Riederer P; Fariello RG; Ito S; Gallorini M; Sulzer D The neuromelanin of human substantia nigra: structure, synthesis and molecular behaviour. Journal of Neural Transmission. Supplementa; Springer, 2003; pp 145–155. [DOI] [PubMed] [Google Scholar]
- (13).Meiser J; Weindl D; Hiller K Complexity of dopamine metabolism. Cell Commun. Signal 2013, 11, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Mor DE; Tsika E; Mazzulli JR; Gould NS; Kim H; Daniels MJ; Doshi S; Gupta P; Grossman JL; Tan VX; Kalb RG; Caldwell KA; Caldwell GA; Wolfe JH; Ischiropoulos H Dopamine induces soluble α-synuclein oligomers and nigrostriatal degeneration. Nat. Neurosci 2017, 20, 1560–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Lee H-J; Baek SM; Ho D-H; Suk J-E; Cho E-D; Lee S-J Dopamine promotes formation and secretion of non-fibrillar alpha-synuclein oligomers. Exp. Mol. Med 2011, 43, 216–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Norris EH; Giasson BI; Hodara R; Xu S; Trojanowski JQ; Ischiropoulos H; Lee VM-Y Reversible inhibition of alpha-synuclein fibrillization by dopaminochrome-mediated conformational alterations. J. Biol. Chem 2005, 280, 21212–21219. [DOI] [PubMed] [Google Scholar]
- (17).LaVoie MJ; Ostaszewski BL; Weihofen A; Schlossmacher MG; Selkoe DJ Dopamine covalently modifies and functionally inactivates parkin. Nat. Med 2005, 11, 1214–1221. [DOI] [PubMed] [Google Scholar]
- (18).Girotto S; Sturlese M; Bellanda M; Tessari I; Cappellini R; Bisaglia M; Bubacco L; Mammi S Dopamine-derived quinones affect the structure of the redox sensor DJ-1 through modifications at Cys-106 and Cys-53. J. Biol. Chem 2012, 287, 18738–18749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Hauser DN; Dukes AA; Mortimer AD; Hastings TG Dopamine quinone modifies and decreases the abundance of the mitochondrial selenoprotein glutathione peroxidase 4. Free Radic. Biol. Med 2013, 65, 419–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Armarego WLF; Waring P Inhibition of human brain dihydropteridine reductase by the oxidation products of catecholamines, the aminochromes. Biochem. Biophys. Res. Commun 1983, 113, 895–899. [DOI] [PubMed] [Google Scholar]
- (21).Belluzzi E; Bisaglia M; Lazzarini E; Tabares LC; Beltramini M; Bubacco L Human SOD2 modification by dopamine quinones affects enzymatic activity by promoting its aggregation: possible implications for Parkinson’s disease. PLoS One 2012, 7, No. e38026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Xu Y; Stokes AH; Roskoski R Jr.; Vrana KE Dopamine, in the presence of tyrosinase, covalently modifies and inactivates tyrosine hydroxylase. J. Neurosci. Res 1998, 54, 691–697. [DOI] [PubMed] [Google Scholar]
- (23).Whitehead RE; Ferrer JV; Javitch JA; Justice JB Reaction of oxidized dopamine with endogenous cysteine residues in the human dopamine transporter. J. Neurochem 2001, 76, 1242–1251. [DOI] [PubMed] [Google Scholar]
- (24).Zhou ZD; Lim TM Dopamine (DA) induced irreversible proteasome inhibition via DA derived quinones. Free Radic. Res 2009, 43, 417–430. [DOI] [PubMed] [Google Scholar]
- (25).Martinez-Vicente M; Talloczy Z; Kaushik S; Massey AC; Mazzulli J; Mosharov EV; Hodara R; Fredenburg R; Wu DC; Follenzi A; Dauer W; Przedborski S; Ischiropoulos H; Lansbury PT; Sulzer D; Cuervo AM Dopamine-modified alpha-synuclein blocks chaperone-mediated autophagy. J. Clin. Invest 2008, 118, 777–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Van Laar VS; Dukes AA; Cascio M; Hastings TG Proteomic analysis of rat brain mitochondria following exposure to dopamine quinone: implications for Parkinson disease. Neurobiol. Dis 2008, 29, 477–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Van Laar VS; Mishizen AJ; Cascio M; Hastings TG Proteomic identification of dopamine-conjugated proteins from isolated rat brain mitochondria and SH-SY5Ycells. Neurobiol. Dis 2009, 34, 487–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Yu G; Liu H; Zhou W; Zhu X; Yu C; Wang N; Zhang Y; Ma J; Zhao Y; Xu Y; Liao L; Ji H; Yuan C; Ma J In vivo protein targets for increased quinoprotein adduct formation in aged substantia nigra. Exp. Neurol 2015, 271, 13–24. [DOI] [PubMed] [Google Scholar]
- (29).Liu Z; Hu B-H; Messersmith PB Acetonide Protection of Dopamine for the Synthesis of Highly Pure N-docosahexaenoyldopamine. Tetrahedron Lett. 2010, 51, 2403–2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Fukuyama TJC; Jow TJC; Cheung M 2- and 4-Nitrobenzenesulfonamides: Exceptionally versatile means for preparation of secondary amines and protection of amines. Tetrahedron Lett. 1995, 36, 6373–6374. [Google Scholar]
- (31).Senoh S; Witkop B Formation and Rearrangements of Aminochromes from a New Metabolite of Dopamine and Some of its Derivatives. J. Am. Chem. Soc 1959, 81, 6231–6235. [Google Scholar]
- (32).Asanuma M; Miyazaki I; Ogawa N Dopamine- or L-DOPA-induced neurotoxicity: the role of dopamine quinone formation and tyrosinase in a model of Parkinson’s disease. Neurotox Res. 2003, 5, 165–176. [DOI] [PubMed] [Google Scholar]
- (33).Jimenez M; Garcia-Carmona F; Garcia-Canovas F; Iborra JL; Lozano JA; Martinez F Chemical intermediates in dopamine oxidation by tyrosinase, and kinetic studies of the process. Arch. Biochem. Biophys 1984, 235, 438–448. [DOI] [PubMed] [Google Scholar]
- (34).Bisaglia M; Mammi S; Bubacco L Kinetic and structural analysis of the early oxidation products of dopamine: analysis of the interactions with alpha-synuclein. J. Biol. Chem 2007, 282, 15597–15605. [DOI] [PubMed] [Google Scholar]
- (35).Xie HR; Hu LS; Li GY SH-SY5Y human neuroblastoma cell line: in vitro cell model of dopaminergic neurons in Parkinson’s disease. Chin. Med. J 2010, 123, 1086–1092. [PubMed] [Google Scholar]
- (36).Rampersad SN Multiple applications of Alamar Blue as an indicator of metabolic function and cellular health in cell viability bioassays. Sensors 2012, 12, 12347–12360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Monti DA; Zabrecky G; Kremens D; Liang T-W; Wintering NA; Cai J; Wei X; Bazzan AJ; Zhong L; Bowen B; Intenzo CM; Iacovitti L; Newberg AB N-Acetyl Cysteine May Support Dopamine Neurons in Parkinson’s Disease: Preliminary Clinical and Cell Line Data. PLoS One 2016, 11, No. e0157602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Kolb HC; Finn MG; Sharpless KB Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew. Chem., Int. Ed. Engl 2001, 40, 2004–2021. [DOI] [PubMed] [Google Scholar]
- (39).Pierce EN; Piyankarage SC; Dunlap T; Litosh V; Siklos MI; Wang Y-T; Thatcher GRJ Prodrugs Bioactivated to Quinones Target NF-κB and Multiple Protein Networks: Identification of the Quinonome. Chem. Res. Toxicol 2016, 29, 1151–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Parvez S; Long MJC; Poganik JR; Aye Y Redox Signaling by Reactive Electrophiles and Oxidants. Chem. Rev 2018, 118, 8798–8888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Paz MA; Flückiger R; Boak A; Kagan HM; Gallop PM Specific detection of quinoproteins by redox-cycling staining. J. Biol. Chem 1991, 266, 689–692. [PubMed] [Google Scholar]
- (42).Mazzulli JR; Burbulla LF; Krainc D; Ischiropoulos H Detection of Free and Protein-Bound ortho-Quinones by Near-Infrared Fluorescence. Anal. Chem 2016, 88, 2399–2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Thomas PD; Campbell MJ; Kejariwal A; Mi H; Karlak B; Daverman R; Diemer K; Muruganujan A; Narechania A PANTHER: a library of protein families and subfamilies indexed by function. Genome Res. 2003, 13, 2129–2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Szklarczyk D; Franceschini A; Wyder S; Forslund K; Heller D; Huerta-Cepas J; Simonovic M; Roth A; Santos A; Tsafou KP; Kuhn M; Bork P; Jensen LJ; von Mering C STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Bruning JM; Wang Y; Oltrabella F; Tian B; Kholodar SA; Liu H; Bhattacharya P; Guo S; Holton JM; Fletterick RJ; Jacobson MP; England PM Covalent Modification and Regulation of the Nuclear Receptor Nurr1 by a Dopamine Metabolite. Cell Chem. Biol 2019, 26, 674–685.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Hastings TG The role of dopamine oxidation in mitochondrial dysfunction: implications for Parkinson’s disease. J. Bioenerg. Biomembr 2009, 41, 469–472. [DOI] [PubMed] [Google Scholar]
- (47).Kuhn DM; Arthur RE Jr.; Thomas DM; Elferink LA Tyrosine hydroxylase is inactivated by catechol-quinones and converted to a redox-cycling quinoprotein: possible relevance to Parkinson’s disease. J. Neurochem 1999, 73, 1309–1317. [DOI] [PubMed] [Google Scholar]
- (48).Farzam A; Chohan K; Strmiskova M; Hewitt SJ; Park DS; Pezacki JP;Özcelik D A functionalized hydroxydopamine quinone links thiol modification to neuronal cell death. Redox Biol. 2020, 28, 101377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Weerapana E; Wang C; Simon GM; Richter F; Khare S; Dillon MBD; Bachovchin DA; Mowen K; Baker D; Cravatt BF Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature 2010, 468, 790–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Wang X; Thomas B; Sachdeva R; Arterburn L; Frye L; Hatcher PG; Cornwell DG; Ma J Mechanism of arylating quinone toxicity involving Michael adduct formation and induction of endoplasmic reticulum stress. Proc. Natl. Acad. Sci. U. S. A 2006, 103, 3604–3609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Perri ER; Thomas CJ; Parakh S; Spencer DM; Atkin JD The Unfolded Protein Response and the Role of Protein Disulfide Isomerase in Neurodegeneration. Front. Cell Dev. Biol 2016, 3, 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Conn KJ; Gao W; McKee A; Lan MS; Ullman MD; Eisenhauer PB; Fine RE; Wells JM Identification of the protein disulfide isomerase family member PDIp in experimental Parkinson’s disease and Lewy body pathology. Brain Res. 2004, 1022, 164–172. [DOI] [PubMed] [Google Scholar]
- (53).Segura-Aguilar J; Paris I; Muñoz P; Ferrari E; Zecca L; Zucca FA Protective and toxic roles of dopamine in Parkinson’s disease. J. Neurochem 2014, 129, 898–915. [DOI] [PubMed] [Google Scholar]
- (54).Kasprowicz J; Kuenen S; Miskiewicz K; Habets RLP; Smitz L; Verstreken P Inactivation of clathrin heavy chain inhibits synaptic recycling but allows bulk membrane uptake. J. Cell Biol 2008, 182, 1007–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Ebanks K; Lewis PA; Bandopadhyay R Vesicular Dysfunction and the Pathogenesis of Parkinson’s Disease: Clues From Genetic Studies. Front. Neurosci 2020, 13, 1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Li Y; Jongberg S; Andersen ML; Davies MJ; Lund MN Quinone-induced protein modifications: Kinetic preference for reaction of 1,2-benzoquinones with thiol groups in proteins. Free Radical Biol. Med 2016, 97, 148–157. [DOI] [PubMed] [Google Scholar]
- (57).Bateman LA; Zaro BW; Miller SM; Pratt MR An alkyne-aspirin chemical reporter for the detection of aspirin-dependent protein modification in living cells. J. Am. Chem. Soc 2013, 135, 14568–14573. [DOI] [PubMed] [Google Scholar]
- (58).Tsukidate T; Li Q; Hang HC Targeted and proteome-wide analysis of metabolite-protein interactions. Curr. Opin. Chem. Biol 2020, 54, 19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Counihan JL; Ford B; Nomura DK Mapping proteome-wide interactions of reactive chemicals using chemoproteomic platforms. Curr. Opin. Chem. Biol 2016, 30, 68–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Yang Y; Fonović M; Verhelst SHL Cleavable Linkers in Chemical Proteomics Applications. Methods Mol. Biol 2017, 1491, 185–203. [DOI] [PubMed] [Google Scholar]
- (61).Cox J; Mann M MaxQuant enables high peptide identification rates, individualized p. p. b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol 2008, 26, 1367. [DOI] [PubMed] [Google Scholar]
- (62).Tyanova S; Temu T; Sinitcyn P; Carlson A; Hein MY; Geiger T; Mann M; Cox J The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [DOI] [PubMed] [Google Scholar]
- (63).Cox J; Hein MY; Luber CA; Paron I; Nagaraj N; Mann M Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction termed MaxLFQ. Mol. Cell. Proteomics 2014, 13, 2513–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.