

Abstract
Purpose
Plasma circulating tumor DNA (ctDNA) could reflect the genetic alterations present in tumor tissues. However, there is little information about the clinical relevance of cell-free DNA genotyping in peripheral T-cell lymphoma (PTCL).
Materials and Methods
After targeted sequencing plasma cell-free DNA of patients with various subtypes of PTCL (n=94), we analyzed the mutation profiles of plasma ctDNA samples and their predictive value of dynamic ctDNA monitoring for treatment outcomes.
Results
Plasma ctDNA mutations were detected in 53 patients (56%, 53/94), and the detection rate of somatic mutations was highest in angioimmunoblastic T-cell lymphoma (24/31, 77%) and PTCL, not otherwise specified (18/29, 62.1%). Somatic mutations were detected in 51 of 66 genes that were sequenced, including the following top 10 ranked genes: RHOA, CREBBP, KMT2D, TP53, IDH2, ALK, MEF2B, SOCS1, CARD11, and KRAS. In the longitudinal assessment of ctDNA mutation, the difference in ctDNA mutation volume after treatment showed a significant correlation with disease relapse or progression. Thus, a ≥ 1.5-log decrease in genome equivalent (GE) between baseline and the end of treatment showed a significant association with better survival outcomes than a < 1.5-log decrease in GE.
Conclusion
Our results suggest the clinical relevance of plasma ctDNA analysis in patients with PTCL. However, our findings should be validated by a subsequent study with a larger study population and using a broader gene panel.
Keywords: Circulating tumor DNA, Peripheral T-cell lymphoma, Biomarker, Liquid biopsy
Introduction
Peripheral T-cell lymphoma (PTCL) is a rare heterogeneous group of non-Hodgkin lymphomas (NHLs) that accounts for approximately 10% of all cases of NHL [1]. In clinical practice, most patients with PTCL receive anthracycline-based chemotherapies such as cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) as first-line therapy and autologous stem cell transplantation (ASCT) as a consolidation treatment [2,3]. Nevertheless, a substantial number of patients do not achieve long-term disease-free survival because of a high incidence of relapse, and the disease ultimately has a poor prognosis because of treatment resistance [4,5]. Relapse in patients who achieve a complete response might be associated with the presence of residual disease that is invisible in imaging studies, and the emergence of treatment-resistant aggressive clones could result in refractoriness to subsequent salvage therapies. Tumor-tissue genotyping by next-generation sequencing (NGS) has become a common process to identify mutation profiles that represent tumor characteristics related to a poor prognosis. However, acquisition of tumor tissue through biopsy is an invasive procedure that is not always easy to perform, especially in older and frail patients.
Accordingly, analysis of circulating tumor DNA (ctDNA) in blood has received major attention because blood sampling is minimally invasive and the small nucleotides that are released from cells undergoing apoptosis or necrosis could harbor tumor-derived genetic alterations. Because ctDNA is defined as the fraction of cell-free DNA that carries mutations, genotyping of ctDNA could provide information concerning the genetic profiles of tumor tissues [6]. Indeed, ctDNA-based tumor genotyping has been suggested as a new molecular technology for evaluation of minimal residual disease and monitoring of disease status in lymphoma patients [7,8]. For example, assessment of ctDNA in blood was reported to serve as a biomarker to predict early treatment outcome in diffuse large B-cell lymphoma [9]. Recently, the feasibility of ctDNA genotyping was reported in patients with extranodal natural killer/T-cell lymphoma [10]. However, there is little information regarding whether genotyping and monitoring of ctDNA could have a role in predicting the outcomes of patients with PTCL. Recently, one study performed NGS-based monitoring of T-cell receptor (TCR) sequences in 45 patients with PTCLs [11]. Another study also reported the clinical relevance of high-throughput sequencing of TCR in 23 patients with PTCLs [12]. However, all these studies focused on sequencing of TCR rather than gene panel-based targeted sequencing. Only one retrospective study demonstrated the presence of disease-specific ctDNA mutations in 14 patients with angioimmunoblastic T-cell lymphoma (AITL) [13].
Thus, we performed targeted sequencing using a series of longitudinal plasma samples obtained from PTCL patients enrolled in our previously reported prospective cohort study [14,15]. We analyzed the mutation profiles of plasma ctDNA in various subtypes of PTCL and their association with clinical outcomes using serially collected plasma samples. We also validated the usefulness of plasma ctDNA analysis in PTCL, comparing its concordance with the mutation profiles of primary tumor tissues using paired tumor and plasma samples. Subsequently, we analyzed the predictive value of dynamic ctDNA monitoring and the association of detected emerging mutations with treatment outcomes.
Materials and Methods
1. Patients
All patients were enrolled in our single-center prospective cohort study (the Prospective Cohort Study for Lymphoma: NCT03117036), which was approved by the Institutional Review Board of Samsung Medical Center (No. 2016-11-040). In accordance with the approved guidelines, we collected and stored germline DNA and plasma cell-free DNA at −80°C after obtaining written informed consent. Plasma samples were collected at enrollment of the prospective cohort (baseline), during interim evaluations (interim), at the final response evaluation after primary treatment (end of treatment), and/or at the time of relapse or progression (progression). The staging workup in clinical practice included computed tomography (CT), 18F-fluorodeoxyglucose positron emission tomography/computed tomography (PET/CT), and bone marrow biopsy. The tumor pathology was confirmed by two lymphoma pathologists (Y.H.K. and J.C.). Newly diagnosed patients underwent primary treatment such as CHOP, and relapsed or refractory patients underwent salvage treatments. The response evaluations were performed using CT and PET/CT according to the Lugano classification [16]. After completion of the planned treatment, patients were followed every 3 to 6 months, and relapse or disease progression was confirmed by CT and PET/CT scans. The survival status of all enrolled patients was regularly updated, and the last update regarding survival and disease status was completed in August 2021.
2. Sample preparation
As this study prospectively enrolled patients and collected peripheral blood samples according to treatment and response evaluation schedules, whole blood samples were collected in Cell-Free DNA BCT tubes (Streck Inc., Omaha, NE) for stabilization of plasma cell-free DNA at room temperature. After blood samples were collected from the enrolled patients, plasma was prepared using three centrifugation steps at room temperature with increasing force: 840 ×g for 10 minutes, 1,040 ×g for 10 minutes, and 5,000 ×g for 10 minutes. Peripheral blood leukocytes were collected from the initial centrifugation. After separation of the plasma in the initial centrifugation, the granulocytes were separated by Ficoll gradient centrifugation and isolated from the pelleted lymphocytes using red blood cell lysis buffer (Qiagen, Santa Clarita, CA). The DNA extracted from granulocytes was used for analysis of germline DNA mutations as well as exclusion of clonal hematopoiesis of indeterminate potential. The detailed methods for DNA extraction and library preparation are described in Supplementary Methods. The study population was 94 patients whose baseline plasma samples for ctDNA mutation analysis were collected during our prospective study. For evaluation of concordance between mutation profiles of plasma ctDNA and primary tumor DNA, formalin-fixed paraffin-embedded tissue samples were collected from 21 patients with available samples. Lastly, for longitudinal assessment of plasma ctDNA mutations, serially collected plasma samples from baseline to the end of treatment or disease relapse were analyzed in 69 of 94 patients with available samples. For the comparison of patients’ plasma cell-free DNA concentrations, patients were dichotomized into high and low cell-free DNA groups and the cutoff was determined by the median value of cell-free DNA concentrations of all patients (12.0 ng/dL).
3. Targeted sequencing and data processing
We selected 66 genes for ultra-deep targeted sequencing for plasma cell-free DNA obtained from NHL patients after a literature review of lymphoma-related genes including our 426-gene panel for targeted sequencing of primary tumor tissue. In our previous reports, genetic profiling in plasma cell-free DNA using the panel resulted in 88.0% sensitivity and > 99% specificity in detecting mutations present at a frequency greater than 20% in the tumor biopsies [14]. The sequenced ctDNA data were analyzed on a HiSeq 2500 system (Illumina, San Diego, CA). All sequencing data were aligned to the hg19 reference using the BWA-mem (v0.7.17, Wellcome Trust Sanger Institute, Cambridge, UK) algorithm. The sequence alignment map (SAM) format was converted into a binary alignment map (BAM) file using SAMTOOLS software (v1.9, Wellcome Trust Sanger Institute). MarkDuplicates software in the Picard package (v2.19.1, Broad Institute, Cambridge, UK) was used to identify polymerase chain reaction duplicates. Lab-built Python (v3.6.4) scripts were used to implement the integrated design environment method and process duplicate reads [17]. GATK (v4.1.0.0) [18], Picard (v2.19.1), and SAMTOOLS (v1.9) [19] were used for base quality recalibration and sorting of SAM and BAM files. During processing, discordant pairs and off-target reads were filtered out. The filtering steps and sequencing metrics are summarized in the Supplementary Methods and S1 Table. The levels of ctDNA were quantified as genome equivalents (GE) that were determined as the product of total cell-free DNA concentration and the maximal variant allele frequency of somatic mutations [20].
4. Statistical analysis
Descriptive statistics were determined as proportions and medians, and the intergroup comparisons for categorical variables were assessed by Fisher’s exact test. Progression-free survival (PFS) was calculated as the period from the date of enrollment to the date of a PFS event, which was designated as disease relapse or progression or death from any cause. Overall survival (OS) was defined as the period from the date of enrollment to death from any cause or the last date of follow-up. Survival curves were described using Kaplan-Meier estimates and were compared between groups using the log-rank test. Statistical analyses were performed using the PASW Statistics software program ver. 24.0 (IBM SPSS Inc., Armonk, NY).
Results
1. Patients
Peripheral blood samples were obtained from 94 patients with PTCL who were consecutively enrolled into our prospective cohort between March 2017 and November 2019. The median age of patients was 58 years (range, 19 to 82 years). Of these, 73 patients were newly diagnosed/treatment-naïve and 21 had relapsed or refractory disease at the time of enrollment. As patients with various subtypes of PTCL were included in this study, we categorized them into four groups: (1) T follicular helper cell (TFH) lymphomas (n=40) including AITL (n=31), follicular T-cell lymphoma (FTCL; n=5), and nodal PTCL with TFH phenotype (PTFH; n=4); (2) PTCL (n=33) including PTCL, not otherwise specified (PTCL-NOS; n=29) and monomorphic epitheliotropic intestinal T-cell lymphoma (n=4); (3) systemic anaplastic large cell lymphomas (ALCL; n=9) consisting of anaplastic lymphoma kinase (ALK)–positive (n=5) and ALK-negative ALCL (n=4); and (4) cutaneous T-cell lymphomas including cutaneous ALK-negative ALCL (n=5), transformed mycosis fungoides (MF; n=1), subcutaneous panniculitis-like T-cell lymphoma (SPTCL; n=3), and other cutaneous T-cell lymphoma (CTCL; n=3). The clinical characteristics of those four groups are compared in Table 1. At the median follow-up of 36.0 months (95% confidence interval, 31.4 to 40.6 months), disease relapse or progression had occurred in 56 patients. Among these, 37 patients had died, and the cause of death was lymphoma in all patients. In the OS analysis of newly diagnosed patients (n=73), patients with PTCL and TFH lymphomas showed significantly worse OS than patients with systemic ALCL and CTCLs. However, the OS was not significantly different among patients with relapsed or refractory disease (S2A and S2B Fig.).
Table 1.
Characteristics of patients at enrollment
| TFH lymphomas (n=40, %) | PTCL (n=33, %) | ALCL (n=9, %) | Cutaneous T-cell (n=12, %) | p-value | |
|---|---|---|---|---|---|
| Disease status | |||||
| Newly diagnosed | 31 (77.5) | 25 (75.6) | 7 (77.8) | 10 (83.3) | 0.962 |
| Relapsed or refractory | 9 (22.5) | 8 (24.4) | 2 (22.2) | 2 (16.7) | |
| Diagnosis | |||||
| AITL/FTCL/PTFH | 31/5/4 | - | - | - | - |
| PTCL-NOS/MEITL | - | 29/4 | - | - | - |
| ALK-negative/positive ALCL | - | - | 4/5 | - | - |
| Cutaneous ALCL/SPTCL | - | - | - | 5/3 | - |
| Transformed MF/CTCL | - | - | - | 1/3 | - |
| Age (yr) | |||||
| ≤ 60 | 22 (55.0) | 15 (45.5) | 9 (100) | 10 (83.3) | 0.007 |
| > 60 | 18 (45.0) | 18 (54.5) | 0 ( | 2 (16.7) | |
| Sex | |||||
| Male | 23 (57.5) | 22 (66.7) | 8 (88.9) | 8 (66.7) | 0.348 |
| Female | 17 (42.5) | 11 (33.3) | 1 (11.1) | 4 (33.3) | |
| Serum LDH (n) | |||||
| Normal | 14 (35.0) | 18 (54.5) | 4 (44.4) | 5 (41.7) | 0.419 |
| Increased | 26 (65.0) | 15 (45.5) | 5 (55.6) | 7 (58.3) | |
| Bone marrow involvement (n) | |||||
| Absent | 29 (72.5) | 24 (72.7) | 8 (88.9) | 10 (83.3) | 0.659 |
| Present | 11 (27.5) | 9 (27.3) | 1 (11.1) | 2 (16.7) | |
| Ann Arbor stage (n) | |||||
| I/II | 5 (12.5) | 12 (36.4) | 1 (11.1) | 7 (58.3) | 0.005 |
| III/IV | 35 (87.5) | 21 (63.6) | 8 (88.9) | 5 (41.7) | |
| IPI (n) | |||||
| Low/Low-intermediate risk | 25 (62.5) | 19 (57.6) | 7 (77.8) | 12 (100) | 0.044 |
| High-intermediate/High risk | 15 (37.5) | 14 (42.4) | 2 (22.2) | 0 | |
| Cell-free DNA a) | |||||
| Low (≤ 12.0 ng/dL) | 17 (42.5) | 18 (54.5) | 3 (33.3) | 9 (75.0) | 0.160 |
| High (> 12.0 ng/dL) | 23 (57.5) | 15 (45.5) | 6 (66.7) | 3 (25.0) | |
| ctDNA mutation | |||||
| Not detected | 12 (30.0) | 14 (42.4) | 5 (55.6) | 10 (83.3) | 0.010 |
| Detected | 28 (70.0) | 19 (57.6) | 4 (44.4) | 2 (16.7) | |
AITL, angioimmunoblastic T-cell lymphoma; ALCL, anaplastic large cell lymphoma; CTCL, cutaneous T-cell lymphoma; ctDNA, circulating tumor DNA; FTCL, follicular helper T-cell lymphoma; IPI, International Prognostic Index; LDH, lactate dehydrogenase; MEITL, monomorphic epitheliotropic intestinal T-cell lymphoma; MF, mycosis fungoides; PTCL, peripheral T-cell lymphoma; PTCL-NOS, peripheral T-cell lymphoma, not otherwise specified; PTFH, peripheral T-cell lymphoma with TFH phenotype; SPTCL, subcutaneous panniculitis-like T-cell lymphoma; TFH, T follicular helper.
The cutoff value for dichotomization was the median value of 94 patients’ cell-free DNA concentration (12.0 ng/dL).
2. Detection of ctDNA mutation at baseline prior to treatment
Plasma cell-free DNA was extracted from 94 baseline samples, and targeted sequencing with 66 genes identified at least one somatic mutation in 53 patients (56%, 53/94). The detection rate of somatic mutations was highest in subtypes AITL (24/31, 77%) and PTCL-NOS (18/29, 62.1%). The ctDNA detection was relatively low in systemic ALCL (4/9, 44.4%), especially in ALK-positive ALCL (1/4, 25%), even though the number of samples was small. The ctDNA mutation was not detected in patients with CTCLs except in a case of transformed MF and SPTCL (Fig. 1A). Accordingly, ctDNA mutation was more frequently detected in the groups with TFH lymphomas (70%, 28/40) and PTCL (58%, 19/33) than in those with systemic ALCL (44%, 4/9) and cutaneous lymphomas (17%, 2/12) (Fig. 1A). However, the median plasma cell-free DNA concentration of patients with TFH lymphomas (13.3 ng/dL; range, 2.4 to 648.0 ng/dL) did not differ significantly from those with PTCL-NOS (10.5 ng/dL; range, 2.5 to 299.6 ng/dL) and ALCL (15.4 ng/dL; range, 3.1 to 44.1 ng/dL; p > 0.05). As the study population consisted of newly diagnosed (n=73) and relapsed or refractory disease (n=21) patients, the ctDNA mutations in these groups were analyzed separately (S2C and S2D Fig.), and the detection of ctDNA mutation was higher in relapsed or refractory lymphomas (76%, 16/21) than in newly diagnosed lymphomas (51%, 37/73, p=0.047) even though the median cell-free DNA concentration did not differ between them (12.8 ng/dL vs. 10.5 ng/dL, p=0.133). However, patients with detected ctDNA mutations at diagnosis did not show significantly poorer OS than patients without detected ctDNA mutations in newly diagnosed TFH lymphomas and PTCL (S2E and S2F Fig.). The detection of ctDNA mutation itself also failed to show an association with the OS in patients with CTCLs because ctDNA mutation was detected in only two patients (data not shown).
Fig. 1.

The detection of plasma ctDNA mutations from 94 baseline samples and mutation profiles of plasma cell-free DNA in 53 patients with peripheral T-cell lymphoma obtained from the baseline samples of our prospective cohort. (A) The detection rate of somatic mutations was highest in two subtypes: AITL (24/31, 77%) and PTCL-NOS (18/29, 62.1%). The ctDNA mutation was detected more frequently in the groups with TFH lymphomas (70%, 28/40) and peripheral T-cell lymphomas (58%, 19/33) than in those with systemic ALCL (44%, 4/9) and cutaneous lymphomas (17%, 2/12). (B) Somatic mutations in RHOA, CREBBP, KMT2D, TP53, and IDH2 were most common across the subtypes. A red box represents the detected mutation of each gene. AITL, angioimmunoblastic T-cell lymphoma; ALCL, anaplastic large cell lymphoma; ALK, anaplastic lymphoma kinase; CTCL, cutaneous T-cell lymphoma; ctDNA, circulating tumor DNA; FTCL, follicular helper T-cell lymphoma; MEITL, monomorphic epitheliotropic intestinal T-cell lymphoma; MF, mycosis fungoides; PTCL-NOS, peripheral T-cell lymphoma, not otherwise specified; PTFH, peripheral T-cell lymphoma with T follicular helper type; SPTCL, subcutaneous panniculitis-like T-cell lymphoma; TFH, T follicular helper cell.
3. Comparison of ctDNA mutations based on subtype
Somatic mutations were detected in 51 of 66 genes that were sequenced, including the following top 10 ranked genes: RHOA, CREBBP, KMT2D, TP53, IDH2, ALK, MEF2B, SOCS1, CARD11, and KRAS (Fig. 1B). In addition, somatic mutations involving TCR signaling genes such as FYN and CD28 and JAK/STAT signaling genes such as STAT3 and STAT6 were found in PTCL-NOS and AITL patients, although their frequencies were lower than those in the top 10 ranked genes (Fig. 1B). The most commonly identified mutation, in RHOA, was detected mainly in TFH-origin lymphomas such as AITL (n=16), FTCL (n=1), and PTFH (n=1) and infrequently in PTCL-NOS (n=3). Most cases (n=20) harbored the RHOA mutation encoding p.Gly17Val, and only one case (PTFH) had the RHOA mutation encoding p.Thr19Ile (Fig. 2A). IDH2 mutations, all of which affected residue R172 (e.g., p.Arg172Ser), were more frequent in TFH-origin lymphomas including AITL (n=8) and FTCL (n=1) than in PTCL-NOS (n=1) (Fig. 2A). Somatic mutations in CREBBP, TP53, and KM2TD were identified across all subtypes (Fig. 2A). The dominant mutation in CREBBP was detected at position 2384, including p.His2384Thr; however, the mutation sites of TP53 and KM2TD did not overlap (Fig. 2A). The results of baseline ctDNA were compared with the mutation profiles of primary tumor tissue in 21 patients for whom paired tumor and plasma samples were available. Concordance of the somatic mutations in plasma ctDNA and tumor tissue was found for the aforementioned top 10 ranked genes (Fig. 2B). For example, concordance of the RHOA mutation was observed in the plasma ctDNA and tumor tissue of AITL (n=7), FTCL (n=1), and PTFH (n=1), whereas concordance of the TP53 mutation was found in PTCL-NOS (n=4), AITL (n=1), and ALCL (n=1) (Fig. 2B).
Fig. 2.
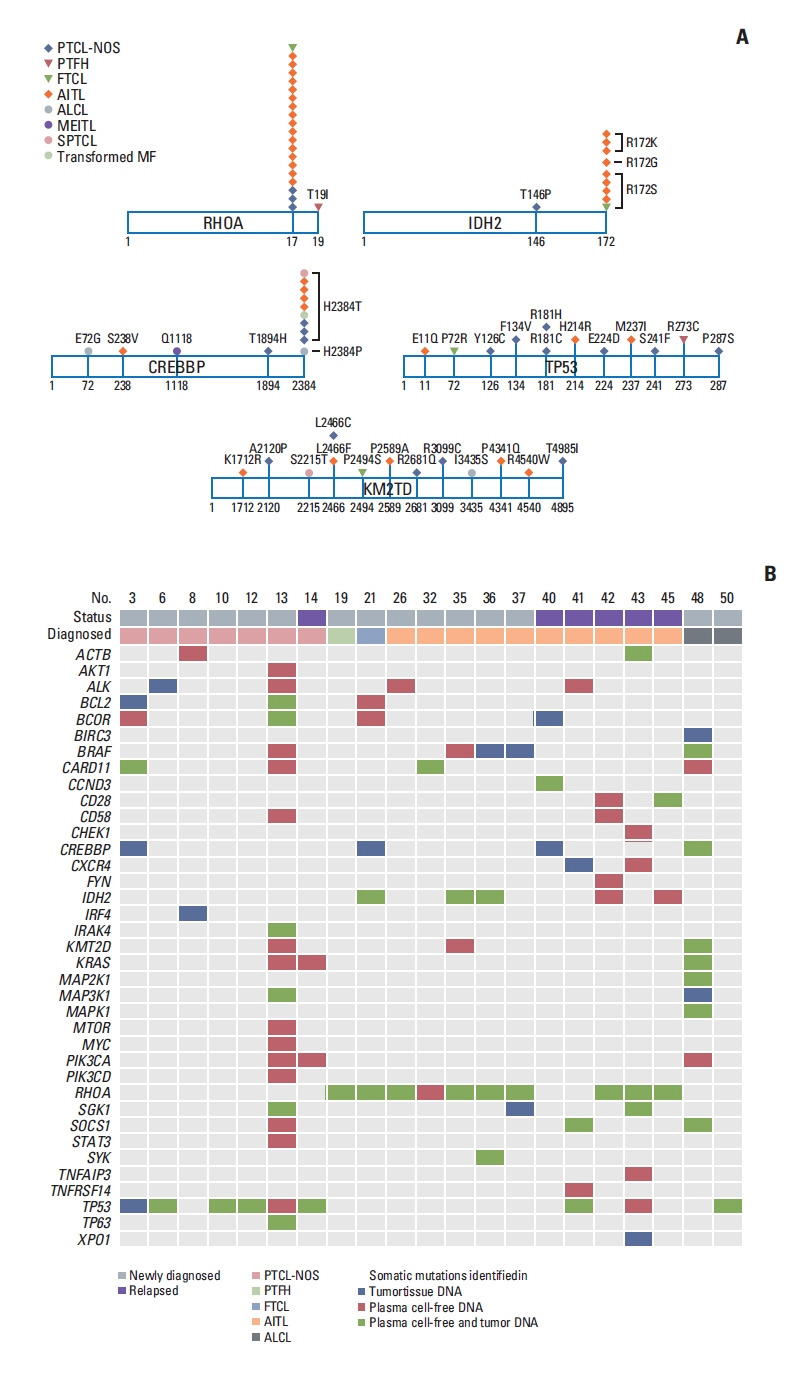
Mutation sites of most common cell-free DNA mutations and the comparison of plasma ctDNA and tumor tissue DNA mutations. (A) Most cases harbored the RHOA mutation encoding p.Gly17Val, and the IDH2 mutations predominantly affected residue R172. The dominant mutation in CREBBP was at position 2384, including p.His2384Thr, whereas the mutation sites of TP53 and KM2TD did not overlap. (B) A paired analysis using 21 cases shows concordance of the somatic mutations in plasma cell-free DNA and tumor tissue. AITL, angioimmunoblastic T-cell lymphoma; ALCL, anaplastic large cell lymphoma; ctDNA, circulating tumor DNA; FTCL, follicular helper T-cell lymphoma; MEITL, monomorphic epitheliotropic intestinal T-cell lymphoma; MF, mycosis fungoides; PTCL-NOS, peripheral T-cell lymphoma, not otherwise specified; PTFH, peripheral T-cell lymphoma with T follicular helper type; SPTCL, subcutaneous panniculitis-like T-cell lymphoma.
4. Longitudinal assessment of ctDNA mutations
Out of 69 patients that were analyzed using serially collected plasma cell-free DNA, the longitudinal assessment of ctDNA mutation was performed in 45 patients consisting of 33 newly diagnosed and 12 relapsed patients. In the remaining patients, the longitudinal comparison could not be performed because ctDNA mutation was not detected. The longitudinal difference in GE between baseline and end of treatment, representing the difference in ctDNA mutation volume after treatment, showed a significant correlation with disease relapse or progression. Thus, among eight patients with a ≥ 1.5-log decrease of GE after treatment, six maintained their response and only two relapsed (2/8, 25%), whereas 11 of 14 patients with a < 1.5-log decrease of GE experienced relapse (11/14, 79%). Likewise, 19 (19/23, 83%) among 23 patients showing an increase in GE after treatment showed relapse (log change < 0). Accordingly, this difference ≥ 1.5-log decrease in GE between baseline and the end of treatment showed a significant association with better PFS and OS than the difference < 1.5-log decrease in GE (Fig. 3A and B). For example, of two patients with newly diagnosed PTCL-NOS who received CHOP chemotherapy, one who showed rapid clearance of the TP53 mutation at the interim evaluation was alive with a complete response, whereas the other patient who showed persistence of the TP53 mutation had relapsed (Fig. 3C and D). Among the patients who underwent upfront ASCT, one patient with PTCL-NOS who initially harbored a high GE level of ctDNA mutations, especially in EZH2, showed a > 1.5-log decrease in the level of mutations at the end of treatment and maintained a complete response (Fig. 3E). By contrast, the other patient with AITL failed to show clearance of baseline mutations in CREBBP and MEF2B and developed an additional mutation in MEF2B, even though radiology indicated a complete response after ASCT. Eventually, he relapsed with persistent detection of mutations (Fig. 3F). Among relapsed cases, one patient with relapsed AITL who received salvage chemotherapy showed an increase in TP53 and SOCS1 mutations (Fig. 3G), and another patient with relapsed ALK-positive ALCL showed disease progression with an increase of CREBBP and STAT6 mutations after salvage treatment with brentuximab vedotin (Fig. 3H).
Fig. 3.
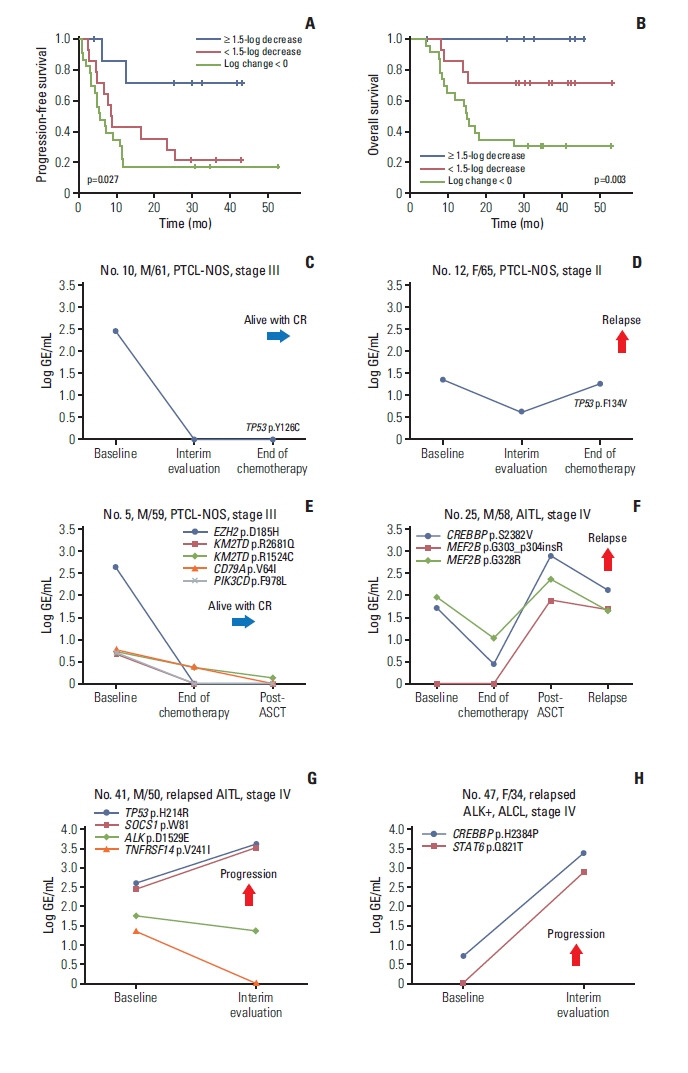
Longitudinal analysis of plasma cell-free DNA mutations shows the correlation with treatment outcomes. (A, B) Patients with a decrease in mutation volume at the end of treatment (log change in genome equivalents ≥ 1.5) showed better progression-free and overall survival than patients showing increased mutation volume (log change in genome equivalents < 0) or a small decrease in mutation volume (log change in genome equivalents < 1.5). (C) A 61-year-old male with PTCL-NOS maintained a CR with loss of a TP53 mutation. (D) A 65-year-old female with PTCL-NOS showing an increase in a TP53 mutation at the end of treatment finally relapsed. (E) A 59-year-old male with PTCL-NOS maintained a complete response with a decrease in initially detected mutations including EZH2. (F) A 58-year-old male with AITL showed increases in CREBBP and MEF2B mutations after autologous stem cell transplantation and relapsed during follow-up. (G) A 50-year-old male with relapsed AITL showed increases in TP53 and SOCS1 mutations after salvage treatment during disease progression. (H) A 34-year-old female with ALK-positive ALCL showed increases in CREBBP and STAT6 mutation volumes after salvage therapy. AITL, angioimmunoblastic T-cell lymphoma; ALCL, anaplastic large cell lymphoma; ALK, anaplastic lymphoma kinase; ASCT, autologous stem cell transplantation; CR, complete response; GE, genome equivalent; PTCL-NOS, peripheral T-cell lymphoma, not otherwise specified.
5. Emergence of new somatic mutations during disease progression
During disease progression, new somatic mutations with an increase in initially detected mutations were identified. For example, a 56-year-old man with ALK-negative ALCL showed an increase in the initially detected SOCS1, MAPK1, MAP2K1i, and KM2TD mutations. The emergence of new mutations in genes including PIM1, MTOR, and CARD11 was observed, whereas BRAF p.D594G increased; however, BRAF p.V600G and p.V600M decreased. This patient became refractory to subsequent treatment with brentuximab vedotin (Fig. 4A). A 57-year-old man with PTCL-NOS achieved a complete response after six cycles of fractionated ifosfamide, carboplatin, and etoposide chemotherapy and then underwent ASCT. However, mutations involving CREBBP, KMT2D, MEF2B, BRAF, IRK1, and GATA3 were detected at the time of his complete response, and he eventually relapsed with a further increase in these emergent mutations (Fig. 4B). When the mutation profiles of 45 longitudinally assessed patient serial samples were compared, KMT2D, CREBBP, and MEF2B were the most common genes showing increases in mutation volumes during disease progression or relapse (Fig. 4C).
Fig. 4.
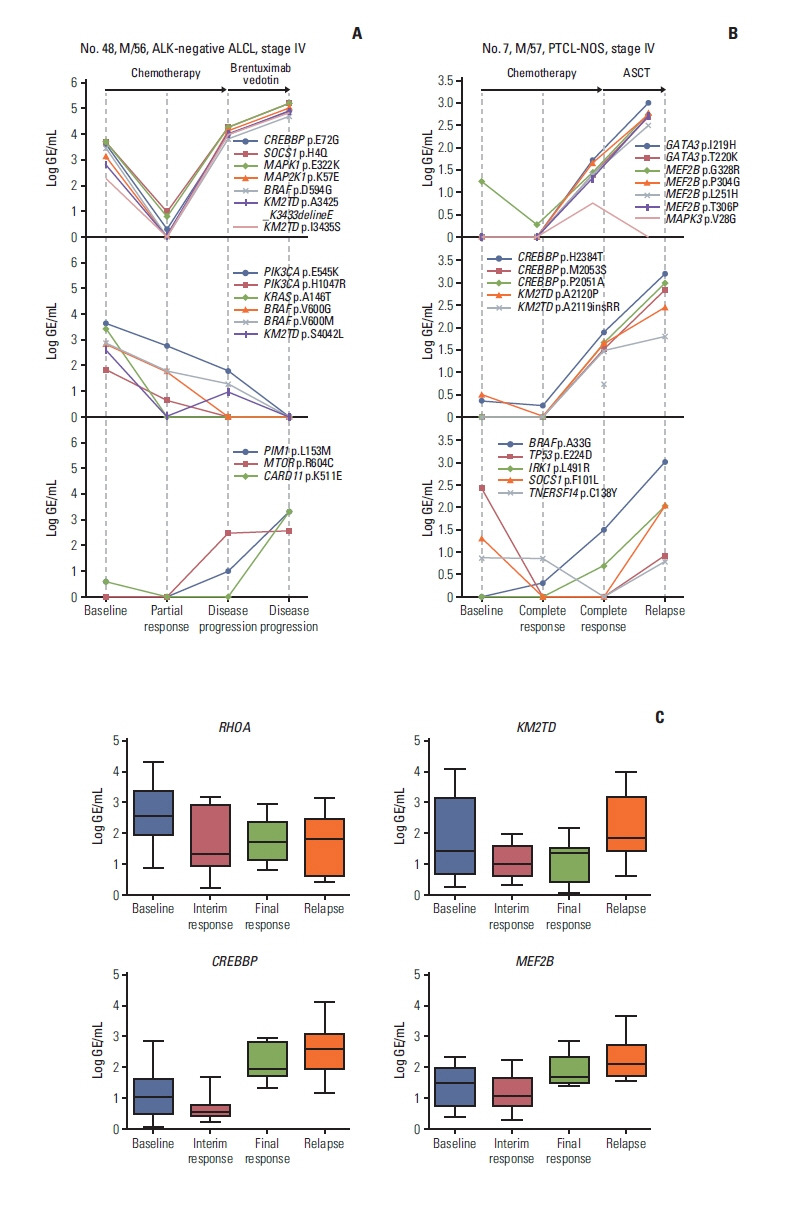
Monitoring of plasma cell-free DNA harboring somatic mutations revealed the emergence of new somatic mutations during disease relapse or progression. (A) A 56-year-old male with ALK-negative ALCL showed increases in CREBBP, SOCS1, MAPK1, and MAP2K1 mutations at the time of disease progression even after salvage therapy with brentuximab vedotin, whereas the mutations of KMT2D and BRAF showed different patterns according to the sites of mutations (top and middle). The mutations of PIM1, MTOR, and CARD11 increased as disease progressed, reflecting the emergence of new mutations (bottom). (B) A 57-year-old male with PTCL-NOS showed elevation of mutation volume in multiple genes such as CREBBP, KMT2D, GATA3, and MEF2B at the time of complete response and finally showed disease relapse together with further increases in those mutations after autologous stem cell transplantation (top and middle). As the patient exhibited relapse, new mutations such as BRAF and IRK1 appeared (bottom). (C) The mutation profiles of longitudinally assessed serial samples from 45 patients showed the increases in mutation volumes during disease progression or relapse predominantly in KMT2D, CREBBP, and MEF2B. ALCL, anaplastic large cell lymphoma; ALK, anaplastic lymphoma kinase; ASCT, autologous stem cell transplantation; GE, genome equivalent; PTCL-NOS, peripheral T-cell lymphoma, not otherwise specified.
Discussion
This study evaluated the analytical performance and clinical utility of our targeted sequencing platform for detecting plasma ctDNA mutations in patients with PTCL using 94 patient-derived plasma cell-free DNA samples. Our targeted sequencing revealed somatic mutations in the plasma cell-free DNA of 53 patients (56%, 53/94), and mutations were more frequently detected in patients with TFH lymphomas and PTCL (Fig. 1A). Furthermore, genotyping of baseline plasma ctDNA showed that the RHOA mutation was most common (n=21), and that its frequency was highest in AITL (16/31, 51.6%) (Fig. 1B). In fact, the recurrent mutation Gly17Val in the GTP/GDP binding domain of RHOA is considered a genetic alteration specific to AITL. A previous Japanese study demonstrated the presence of the p.Gly17Val mutation in RHOA of tumor tissue in 68% of patients with AITL [21]. They reported that this RHOA mutation was identified more specifically in tumor cells than were the TET2 mutations that were found in both tumor cells and non-tumor hematopoietic cells. Our previous study using exome and transcriptome sequencing of tumor samples identified the RHOA mutation encoding p.Gly17Val in 53.3% of patients with AITL [22]. The RHOA mutation was identified in patients with PTCL-NOS, especially in those with AITL-like features, although at a lower frequency (18%, 8/44) than that found in AITL [23]. In our study, all patients had the RHOA mutation encoding p.Gly17Val, except for one patient (PTFH) who had the RHOA mutation encoding p.Thr17Ile, which also was reported in PTCL, albeit far less frequently than the p.Gly-17Val mutation (Fig. 2A) [23]. A comparison of genotypes during paired sample analysis also found a concordance of somatic mutations between plasma ctDNA and tumor DNA (Fig. 2B). In particular, the RHOA mutation was detected most frequently in AITL (n=9), FTCL (n=1), and PTFH (n=1), which were introduced to the 2016 WHO classification as nodal lymphomas of TFH origin [24]. In addition to the RHOA mutation, IDH2 mutations were frequent in these nodal lymphomas of TFH origin, which is consistent with a recent study that reported frequent mutations of IDH2 affecting residue R172 in patients with AITL (Fig. 2A) [25]. We also found that TP53 mutations occurred commonly in PTCL-NOS, similar to the results of previous studies reporting the role of genetic alterations including TP53 in these tumors [26]. Furthermore, other genes related to histone methylation and acetylation, such as KMT2D and CREBBP, showed frequent somatic mutations, consistent with the findings of a study reporting histone-modifier gene mutations in PTCL-NOS [27].
Based on the quantitative levels of ctDNA mutations, patients with a > 1.5-log decrease of mutation volume at the end of treatment had better PFS and OS than patients with a < 1.5-log decrease or an increase (Fig. 3A and B). Consistent with this significant association between posttreatment ctDNA changes and survival outcomes, patients without detected ctDNA mutations were alive and maintaining their complete response at the time of analysis, whereas patients with persistent ctDNA mutations after completion of treatment eventually relapsed, even though they achieved radiologically complete responses (Fig. 3C–F). Likewise, relapsed patients who were refractory to salvage therapies showed increased mutation volumes after treatment (Fig. 3G and H). Furthermore, additional new somatic mutations in plasma ctDNA were detected during treatment in patients undergoing aggressive clinical courses, and these ctDNA mutations could be a sign of the development of resistant clones because those patients became refractory to subsequent salvage chemotherapies (Fig. 4A and B). The longitudinal assessment of plasma ctDNA mutation profiles showed increased mutation volumes in KMT2D, CREBBP, and MEF2B at the time of relapse (Fig. 4C). KMT2D and CREBBP are related to epigenetic regulation and are well known for their association with PTCL-NOS, AITL, and ALCL [28]. Indeed, detection of a CREBBP mutation was reported to represent a residual clone with clonal revolution in relapsed or refractory lymphomas [29]. Furthermore, myocyte enhancer factor 2B (MEF2B) is a transcription factor, and MEF2B mutations were reported to dysregulate cell migration in NHL [30]. Thus, these genes could be useful for monitoring plasma ctDNA mutations during and after treatment in patients with PTCL. This ability of detecting newly occurred mutations during treatment or follow-up could provide additional value to physicians because these new mutations-related changes could be potential therapeutic targets. This could be advantage of panel-based sequencing compared to the high-throughput sequencing of TCR. Taken together, the results of our study demonstrate the clinical relevance of plasma ctDNA-based genotyping and monitoring in patients with PTCL.
However, our results should be interpreted with caution due to several limitations. First, this was a single-center study with a relatively small number of patients with heterogeneous disease subtypes. Second, several epigenetic regulating genes crucial in T-cell lymphomas, such as TET2 and DNMT3A, were omitted because our study used a pilot panel with a limited number of genes for analysis of cell-free DNA mutations in patients with NHL. As a result, our platform only identified somatic mutations in 56% of 94 patients. Thus, a subsequent study with a larger study population and using a gene panel including a greater number of genes is required to validate our results. Lastly, the feasibility of ctDNA evaluation, especially the longitudinal assessment as a tool of monitoring the disease status still remains as a problematic issue considering its technical limitation as well as cost and time-effectiveness. Thus, the analytical performance of this procedure in terms of its detection rate of ctDNA mutations and clinical application should be improved.
Acknowledgments
This research was supported by a grant from the Korea Health Technology R&D Project through the Korea Health Industry Development Institute, funded by the Ministry of Health & Welfare of the Republic of Korea (grant number: HR20C0025) and by a National Research Foundation of Korea grant funded by the Korean government (2021R1A2C1007531).
Footnotes
Ethical Statement
The Institutional Review Board at Samsung Medical Center approved this study (SMC 2016-11-040), and all methods were carried out in accordance with the approved guidelines. All patients were registered to the prospective cohort study after written informed consents.
Author Contributions
Conceived and designed the analysis: Kim SJ, Kim WS.
Collected the data: Kim SJ, Yoon SE, Ryu KJ, Park B, Cho D, Kim HY, Cho J, Ko YH, Kim WS.
Contributed data or analysis tools: Kim SJ, Kim YJ, Ryu KJ, Park B, Park D, Cho D, Kim HY, Cho J, Ko YH, Park WY.
Performed the analysis: Kim SJ, Kim YJ, Yoon SE, Park D, Park WY, Kim WS.
Wrote the paper: Kim SJ, Kim YJ, Yoon SE
Conflicts of Interest
Conflict of interest relevant to this article was not reported.
Electronic Supplementary Material
Supplementary materials are available at Cancer Research and Treatment website (https://www.e-crt.org).
References
- 1.Vose J, Armitage J, Weisenburger D International T-Cell Lymphoma Project. International peripheral T-cell and natural killer/T-cell lymphoma study: pathology findings and clinical outcomes. J Clin Oncol. 2008;26:4124–30. doi: 10.1200/JCO.2008.16.4558. [DOI] [PubMed] [Google Scholar]
- 2.Briski R, Feldman AL, Bailey NG, Lim MS, Ristow K, Habermann TM, et al. The role of front-line anthracycline-containing chemotherapy regimens in peripheral T-cell lymphomas. Blood Cancer J. 2014;4:e214. doi: 10.1038/bcj.2014.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ellin F, Landstrom J, Jerkeman M, Relander T. Real-world data on prognostic factors and treatment in peripheral T-cell lymphomas: a study from the Swedish Lymphoma Registry. Blood. 2014;124:1570–7. doi: 10.1182/blood-2014-04-573089. [DOI] [PubMed] [Google Scholar]
- 4.Phan A, Veldman R, Lechowicz MJ. T-cell lymphoma epidemiology: the known and unknown. Curr Hematol Malig Rep. 2016;11:492–503. doi: 10.1007/s11899-016-0353-y. [DOI] [PubMed] [Google Scholar]
- 5.Ma H, Marchi E, O’Connor OA. The peripheral T-cell lymphomas: an unusual path to cure. Lancet Haematol. 2020;7:e765–71. doi: 10.1016/S2352-3026(20)30207-6. [DOI] [PubMed] [Google Scholar]
- 6.Cirillo M, Craig AFM, Borchmann S, Kurtz DM. Liquid biopsy in lymphoma: molecular methods and clinical applications. Cancer Treat Rev. 2020;91:102106. doi: 10.1016/j.ctrv.2020.102106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dogliotti I, Drandi D, Genuardi E, Ferrero S. New molecular technologies for minimal residual disease evaluation in B-cell lymphoid malignancies. J Clin Med. 2018;7:288. doi: 10.3390/jcm7090288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Camus V, Jardin F. Cell-free DNA and the monitoring of lymphoma treatment. Pharmacogenomics. 2019;20:1271–82. doi: 10.2217/pgs-2019-0099. [DOI] [PubMed] [Google Scholar]
- 9.Kurtz DM, Scherer F, Jin MC, Soo J, Craig AF, Esfahani MS, et al. Circulating tumor DNA measurements as early outcome predictors in diffuse large B-cell lymphoma. J Clin Oncol. 2018;36:2845–53. doi: 10.1200/JCO.2018.78.5246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Qi F, Cao Z, Chen B, Chai Y, Lin J, Ye J, et al. Liquid biopsy in extranodal NK/T-cell lymphoma: a prospective analysis of cell-free DNA genotyping and monitoring. Blood Adv. 2021;5:2505–14. doi: 10.1182/bloodadvances.2020001637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miljkovic MD, Melani C, Pittaluga S, Lakhotia R, Lucas N, Jacob A, et al. Next-generation sequencing-based monitoring of circulating tumor DNA reveals clonotypic heterogeneity in untreated PTCL. Blood Adv. 2021;5:4198–210. doi: 10.1182/bloodadvances.2020003679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang W, Wang W, Han X, Gan Y, Qian L, Zhang Y, et al. Circulating tumor DNA by high-throughput sequencing of T cell receptor monitored treatment response and predicted treatment failure in T cell lymphomas. Int J Lab Hematol. 2021;43:1041–9. doi: 10.1111/ijlh.13498. [DOI] [PubMed] [Google Scholar]
- 13.Sakata-Yanagimoto M, Nakamoto-Matsubara R, Komori D, Nguyen TB, Hattori K, Nanmoku T, et al. Detection of the circulating tumor DNAs in angioimmunoblastic T-cell lymphoma. Ann Hematol. 2017;96:1471–5. doi: 10.1007/s00277-017-3038-2. [DOI] [PubMed] [Google Scholar]
- 14.Shin SH, Kim YJ, Lee D, Cho D, Ko YH, Cho J, et al. Analysis of circulating tumor DNA by targeted ultra-deep sequencing across various non-Hodgkin lymphoma subtypes. Leuk Lymphoma. 2019;60:2237–46. doi: 10.1080/10428194.2019.1573998. [DOI] [PubMed] [Google Scholar]
- 15.Hur JY, Kim YJ, Yoon SE, Son DS, Park WY, Kim SJ, et al. Plasma cell-free DNA is a prognostic biomarker for survival in patients with aggressive non-Hodgkin lymphomas. Ann Hematol. 2020;99:1293–302. doi: 10.1007/s00277-020-04008-3. [DOI] [PubMed] [Google Scholar]
- 16.Cheson BD, Fisher RI, Barrington SF, Cavalli F, Schwartz LH, Zucca E, et al. Recommendations for initial evaluation, staging, and response assessment of Hodgkin and non-Hodgkin lymphoma: the Lugano classification. J Clin Oncol. 2014;32:3059–68. doi: 10.1200/JCO.2013.54.8800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Newman AM, Lovejoy AF, Klass DM, Kurtz DM, Chabon JJ, Scherer F, et al. Integrated digital error suppression for improved detection of circulating tumor DNA. Nat Biotechnol. 2016;34:547–55. doi: 10.1038/nbt.3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011;27:2987–93. doi: 10.1093/bioinformatics/btr509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Avanzini S, Kurtz DM, Chabon JJ, Moding EJ, Hori SS, Gambhir SS, et al. A mathematical model of ctDNA shedding predicts tumor detection size. Sci Adv. 2020;6:eabc4308. doi: 10.1126/sciadv.abc4308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sakata-Yanagimoto M, Enami T, Yoshida K, Shiraishi Y, Ishii R, Miyake Y, et al. Somatic RHOA mutation in angioimmunoblastic T cell lymphoma. Nat Genet. 2014;46:171–5. doi: 10.1038/ng.2872. [DOI] [PubMed] [Google Scholar]
- 22.Yoo HY, Sung MK, Lee SH, Kim S, Lee H, Park S, et al. A recurrent inactivating mutation in RHOA GTPase in angioimmunoblastic T cell lymphoma. Nat Genet. 2014;46:371–5. doi: 10.1038/ng.2916. [DOI] [PubMed] [Google Scholar]
- 23.Palomero T, Couronne L, Khiabanian H, Kim MY, Ambesi-Impiombato A, Perez-Garcia A, et al. Recurrent mutations in epigenetic regulators, RHOA and FYN kinase in peripheral T cell lymphomas. Nat Genet. 2014;46:166–70. doi: 10.1038/ng.2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Swerdlow SH, Campo E, Pileri SA, Harris NL, Stein H, Siebert R, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127:2375–90. doi: 10.1182/blood-2016-01-643569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lemonnier F, Safar V, Beldi-Ferchiou A, Cottereau AS, Bachy E, Cartron G, et al. Integrative analysis of a phase 2 trial combining lenalidomide with CHOP in angioimmunoblastic T-cell lymphoma. Blood Adv. 2021;5:539–48. doi: 10.1182/bloodadvances.2020003081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heavican TB, Bouska A, Yu J, Lone W, Amador C, Gong Q, et al. Genetic drivers of oncogenic pathways in molecular subgroups of peripheral T-cell lymphoma. Blood. 2019;133:1664–76. doi: 10.1182/blood-2018-09-872549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ji MM, Huang YH, Huang JY, Wang ZF, Fu D, Liu H, et al. Histone modifier gene mutations in peripheral T-cell lymphoma not otherwise specified. Haematologica. 2018;103:679–87. doi: 10.3324/haematol.2017.182444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xie C, Li X, Zeng H, Qian W. Molecular insights into pathogenesis and targeted therapy of peripheral T cell lymphoma. Exp Hematol Oncol. 2020;9:30. doi: 10.1186/s40164-020-00188-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Melchardt T, Hufnagl C, Weinstock DM, Kopp N, Neureiter D, Trankenschuh W, et al. Clonal evolution in relapsed and refractory diffuse large B-cell lymphoma is characterized by high dynamics of subclones. Oncotarget. 2016;7:51494–502. doi: 10.18632/oncotarget.9860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pon JR, Wong J, Saberi S, Alder O, Moksa M, Grace Cheng SW, et al. MEF2B mutations in non-Hodgkin lymphoma dysregulate cell migration by decreasing MEF2B target gene activation. Nat Commun. 2015;6:7953. doi: 10.1038/ncomms8953. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.