

Abstract
Force fields form the basis for classical molecular simulations and their accuracy is crucial for the quality of, for instance, protein-ligand binding simulations in drug discovery. The huge diversity of small molecule chemistry makes it a challenge to build and parameterize a suitable force field. The Open Force Field Initiative is a combined industry and academic consortium developing a state-of-the-art small molecule force field. In this report industry members of the consortium worked together to objectively evaluate the performance of the force fields (referred to here as OpenFF) produced by the initiative on a combined public and proprietary dataset of 19,653 relevant molecules selected from their internal research and compound collections. This evaluation was important because it was completely blind; at most partners, none of the molecules or data were used in force field development or testing prior to this work. We compare the Open Force Field “Sage” version 2.0.0 and “Parsley” version 1.3.0 with GAFF-2.11-AM1BCC, OPLS4 and SMIRNOFF99Frosst. We analyzed force field-optimized geometries and conformer energies compared to reference quantum mechanical data. We show that OPLS4 performs best, and the latest Open Force Field release shows a clear improvement compared to its predecessors. The performance of established force fields such as GAFF-2.11 was generally worse. While OpenFF researchers were involved in building the benchmarking infrastructure used in this work, benchmarking was done entirely in-house within industrial organizations and the resulting assessment is reported here. This work assesses the force field performance using separate benchmarking steps, external datasets, and involving external research groups. This effort may also be unique in terms of the number of different industrial partners involved, with 10 different companies participating in the benchmark efforts.
Graphical Abstract

Introduction
The computational modeling of chemical and biological systems relies on an accurate assessment of the energetics and geometries of the systems. Methods range from more accurate, higher-cost quantum mechanical (QM) techniques to more approximate but efficient methods such as classical mechanics-based calculations. The latter have the advantage of being applicable to larger systems over longer timescales.1–4 Extended simulation timescales are particularly relevant for the calculation of thermodynamic properties such as protein-ligand binding affinity where the accurate treatment of entropy, desolvation, and other factors requires ensemble-based free energy approaches. The classical mechanical calculations use a force field that gives the energy of the system as a function of the coordinates, given a number of empirical parameters fit to describe this and other properties accurately.5,6
Force field development has an extensive history, and the approaches taken vary with respect to the chemical space covered, the data used for training, and the approach for parameter optimization.7–10 It is common to fit force fields using data from both experimental physical property measurements and reference QM calculations carefully chosen to represent the systems for which the force field is designed. The performance of the resulting force field is then assessed by its ability to reproduce either experimental observables, or QM data such as geometries and relative energies. Given that force field development is complex due to the diversity of training data, various functional forms, and approaches to chemical perception,11 it is expected that the resulting force fields vary in how accurately they reproduce the properties of interest.12–16
Force fields for proteins, based on the 20 common amino acids, have been refined over many years and are widely used and continue to be updated.17–25 A high quality general force field suitable for small, organic and drug-like molecules represents a bigger challenge due to the vast chemical space that must be considered. Furthermore, inherent to innovation is the search for novel chemical matter and a general force field should be suitable for application to as-yet undiscovered compounds. Popular current small molecule force fields include the General AMBER force field (GAFF),26,27 the CHARMM General force field (CGenFF),28 and the Optimized Potentials for Liquid Simulations force field (OPLS).29–33 These approaches have undergone substantial improvement, with the latest versions of OPLS32,33 in particular showing impressive performance, but it is widely accepted that further improvements are possible.34
Begun in late 2018, the Open Force Field (OpenFF) Initiative is a relatively new effort to build and optimize a general force field using an automated and reproducible procedure, with all software, data, and workflows made freely available. Rather than traditional atom-typing, the approach builds on the SMIRKS-native Open Force Field (SMIRNOFF) parameter assignment formalism, which can incorporate increased chemical diversity without needlessly increasing the complexity of the underlying specification and parameterization. The first version of the new OpenFF started with SMIRNOFF99Frosst,35 consisting of direct chemical perception typing rules and parameters from the prior AMBER parm99 force field and Merck-Frosst’s parm@frosst.36 Since then, nearly all of the 500 valence parameters have been optimized to improve agreement with quantum chemical optimized geometries, energetics, and vibrational frequencies. The first generation OpenFF 1.X37–39 (”Parsley”) releases largely retained the Lennard-Jones and electrostatic parameters of SMIRNOFF99Frosst, whereas the more recent OpenFF 2.0.040 (”Sage”) release refit Lennard-Jones parameters as well. The OpenFF Initiative includes the OpenFF Consortium, a pre-competitive, network of academic and industry researchers working together to advance the required science and infrastructure. The shared goal is to develop automated and systematic data-driven techniques to parameterize and assess new generations of the force field.
In this report, academic and industrial partners of the OpenFF consortium worked together to assess the performance of recent OpenFF releases. The work is an extension of the recent article from Lim et al.41 Each industry partner selected compounds from their internal research or compound collections; a total set of 19,653 are studied. As part of this study, a large proportion of the compounds (10,121) were compiled and made publicly available on the QCArchive,42 while the rest of the compounds remained proprietary and were studied internally at each industry partner, with only overall performance statistics being released. The study was enabled by the development of a standard workflow that could be easily installed and run at the sites of each collaborator. The workflow enabled the running and analysis of both QM and force field-based calculations. The identical approach used by each partner allowed the sharing of analysis data without compromising the confidentiality of the proprietary sets of molecules.
Force fields belonging to three families were assessed: (i) the second generation General Amber Force Field GAFF-2.11-AM1BCC (hereafter simply referred as GAFF-2.1126,27); (ii) the latest version of OPLS (OPLS433); and (iii) the latest version of each generation of OpenFF force fields (SMIRNOFF99Frosst v1.135), OpenFF “Parsley” v1.3.039 and the newest release, OpenFF “Sage” v2.0.0.40 For a pruned dataset of 137,052 molecular conformations of 18,154 small molecules, we compared the structures and energetics of conformers optimized using force fields to those optimized using quantum mechanical methods. This work provides a general understanding of the strengths of different small molecule force fields and identifies areas of improvement for future force field development.
Methods
The dataset
Industry partners from the following companies were involved in this collaborative effort: BASF, Bayer, Bristol Myers Squibb, Boehringer Ingelheim, Janssen, Merck KGaA, Roche, Genentech, Vertex, and XtalPi. Partners were asked to choose the molecules most suited or best capturing their research interests, by selecting a set of molecules that could be made public, as well as proprietary compounds. As an example for the public set, one company chose compounds from recent patents being sure to remove intermediates, solvents, and reagents etc. Meanwhile, proprietary molecules from internal drug discovery projects were studied on-site at each partner organization. We believe that the internal data has added value because it shows the performance on the very latest chemistry of interest within the industry partners. The newest internal drug discovery projects may in some cases require novel chemistry and therefore shows the relevance of the force fields on the cutting edge of medicinal chemistry.
All partners contributed proprietary molecules, while six of them also contributed with public molecules. In both cases, we recommended to each partner to keep the number of heavy atoms below 30-35 to avoid overly time consuming QM calculations. Overall, the set of molecules is likely to be highly representative of current small molecule drug discovery efforts. The compounds within the public dataset were deposited to the QCArchive43 (see the Supporting Information for an example of how to extract optimized records of the public dataset from QCArchive).
Assigning force field parameters
For the OpenFF force field family (SMIRNOFF99Frosst, Parsley and Sage) and GAFF-2.11, we assigned AM1 Mulliken-type partial charges with bond-charge corrections (AM1-BCC charges).44,45 Partial charges were generated using the antechamber software package provided within the AmberTools.27 Parameters for the OpenFF family were assigned using the Open Force Field toolkit, whereas for GAFF-2.11 force field, we used tleap27 via open-moltools.46
For OPLS4 charge and parameter assignment was performed using Schrödinger Maestro.47 Available pre-computed general purpose (default) parameters were applied. Also, molecule specific custom parameters were derived using the default approach with the ff-builder tool. This approach checks for missing parameters and if necessary derives new ones based on QM calculations (B3LYP/6-31G* level geometry optimization followed by single-point M06-2X/cc-pVTZ(-f) calculations). Custom OPLS4 parameters were derived for 9057 dihedrals for the public set of 10,121 molecules, indicating the overall set of molecules is rather diverse. OpenFF 2.0.0 only contains 174 torsion parameters, whereas OPLS4 uses a library of 147K or more diverse torsions as reported for OPLS3e, before addition of these custom parameters. Upon recommendation of scientists from Schrödinger, we used the ffld_server command line tool to perform OPLS4 optimizations (see Supporting Information for more details). This tool contains the latest version of the force field and includes features such as virtual sites. The command used was $SCHRODINGER/utilities/ffld_server -imae <input_structures> -omae <output_structures> -opt -OPLSDIR <path/to/oplsdir> -cutoff 999 -min_verbose 1 to perform the optimization with custom OPLS4 parameters and $SCHRODINGER/utilities/ffld_server -imae <input_structures> -omae <output_structures> -opt -cutoff 999 -min_verbose 1 to perform the optimization with default parameters. All other geometry optimizations were completed in OpenMM48 with the same specifications previously used by Lim et al.41
Corresponding files containing QM geometries and energies, SMILES strings and depictions of the public dataset are deposited on GitHub49 (See section Data and code availability)
Automation of our approach
Figure 1 depicts the automated workflow which was deployed at all partners and permitted consistent benchmarking of proprietary molecules between partners and with the public set. During the production runs, it was found that iodine-containing molecules gave unreliable QM results for our choice of basis set with density fitting. This has since been fixed in psi4 as of version 1.450 but they were removed from this study. An additional filter was applied to remove silicon- and boron-containing molecules, as these elements are currently unsupported by OpenFF.
Figure 1:

The automated benchmark workflow was deployed in-house by all partners and allowed consistent benchmarking of proprietary datasets. After checking that the molecules are compatible with the OpenFF force field (”Validation”), it generates up to 10 conformers per molecule (”Conformer Generator”), optimizes the conformers first with a QM method (”QM Minimization (Psi4)”) and then with various MM methods (”MM Minimization”). Finally, the non-proprietary data is extracted and plots are generated for the comparison of results (”Analysis”).
The first step in the protocol performs the validation of the chosen molecules. This step checks that the OpenFF Toolkit can sufficiently interpret each molecule, and for molecules that it can, a unique identifier is then assigned to each. Conformers are aligned during this step, and if a subsequently-loaded conformer has an RMSD of less than 0.5 Å from an existing conformer, it will be discarded as a duplicate. In the Public OpenFF Industry Dataset 85 out of 10,226 initial molecules were filtered by validation, hence 10,141 successfully passed this step.
The second step in the protocol generates additional conformers beyond those provided in the input set of molecules. This step attempts to generate up to 10 conformers per molecule in total, with selection based on minimum inter-conformer RMSD of 0.5 Å. Conformers provided by the user in the previous step are preserved.
The third step in the protocol creates a coverage report that gives the number of times each parameter in OpenFF 1.3.0 is exercised by the molecules in the dataset. Any molecules that could not be parameterized are discarded. All molecule conformers that could successfully be parameterized are exported as SDFs and used for the following geometry optimization. In this step, a total of 60 molecular structures (of which 40 from the proprietary and 20 from the public set) were not successfully parameterized, and were therefore removed from the combined dataset. Our pruned set going into QM minimization contained 19,653 molecules, of which 10,121 were from the public dataset, with unique chemical connectivity.
The fourth step in the protocol executes the optimizations required for benchmarking. There are two stages to this step: the first generates QM geometry-optimized structures and energies at the gas-phase, B3LYP-D3BJ/DZVP level of theory51–55 using psi4.56 This method and basis set were chosen by the Open Force Field initiative to provide reasonably accurate conformational energies and geometries at moderate computational cost37,57,58 and are consistent with the method used previously.41 Molecular modeling approaches typically rely upon accurate assessment of low energy minima but there are cases where it is useful to predict higher-energy structures,.59,60 The protocol used here does not exclude conformers based on a cut-off for relative QM energy (ΔE) and indeed some higher-energy local minima were retrieved, < 0.06% for ΔE >20 kcal/mol. The second stage performs gas-phase MM optimizations using the different forcefields, starting from the minimized QM structures for each molecule conformation. Publicly available compounds on the QCArchive were minimized with the latest generation of each force field family, namely GAFF-2.11, OPLS4 with both custom (OPLS4CST) and default (OPLS4DEF) parameters, and OpenFF “Sage” v2.0.0.40 Proprietary compounds were minimized using each OpenFF generation (SMIRNOFF, “Parsley”v1.3.0, “Sage”v2.0.0) and GAFF-2.11, as OPLS4 was not available at all the industry partners. The SMIRNOFF99Frosst version used here is SMIRNOFF99Frosst-1.1.0.offxml. Because QM geometry optimization can, in rare cases, change the connectivity of a molecule, the final QM geometries were assessed to ensure that their interatomic distances remained consistent with the connectivity of the input molecule, and any conformer that deviated from their original connectivity were discarded from further analysis.
From the pruned set, 18,154 molecular structures were successfully optimized during step (i) and subsequently went through step (ii), producing a different success rate in MM optimization than QM (see Table S2 and S1).
Once all optimizations are finished, in the final step the data produced are analyzed and the corresponding plots subsequently generated.
Energies and optimized geometries with respect to QM reference are finally compared by relative energy difference (ΔΔE), root-mean-square deviation of atomic positions (RMSD) and torsion fingerprint deviation (TFD),61–63 similar to our previous benchmark assessment.41 The ΔΔE energy accounts for the energy difference (ΔE MM) between each MM optimized conformer and the MM conformer with minimum energy, relative to the energy difference (ΔE QM) between each corresponding QM optimized conformer and the QM conformer with minimum energy (compare-forcefields).
In addition, to address any potential low agreement between force field and QM energies due to change in conformation after MM energy minimization, we performed a conformer matching process (match-minima) for each MM structure which considered the final optimized geometries and energy differences. In this case, the ΔΔE takes into account the energy difference between each MM optimized conformer and the MM conformer with lowest RMSD with respect to the QM minimum, relative to (ΔE QM). The equations used in compare-forcefields and match-minima to compute the ΔΔE between the MM and QM energy for the ith conformer of a specific molecule are reported in the SI Eq S1 and Eq S2, respectively).
The complete Python code used for the setup, minimizations, and analysis of this work is open source and available on GitHub 49 and the protocol used to run minimization on Confluence (See section Data and code availability).
Results and Discussion
Here, we present and discuss our results comparing several general small molecule force fields against reference QM data. We are interested in two major categories of comparison – energetic agreement and geometric agreement. An ideal force field will yield the same energy minima or optimized geometries as the QM energy landscape, with no additional or missed minima, and the energies of those minima will agree between QM and MM. However, with even minor energy errors, the relative ordering of the QM and MM minima could be different even if all minima are present in both landscapes. Thus, to assess performance in these two categories, we computed relative conformer energies and compared these between MM and QM, as well as assessed geometric agreement of MM optimized geometries with those from QM.
Our study relies on the assumption that force field accuracy can be evaluated using gas phase energies and geometries. One of the greater goals of force field science, such as that of the Open Force Field Initiative, is building force fields that will work well in the condensed phase (e.g., small molecules in solution or binding to biomolecules). That being said, we make our assumption based on two key observations. First, force fields, especially those in the AMBER family, are usually fitted to reproduce gas phase conformational energies and geometries.26 This means that we are testing these force fields on properties they are fitted to reproduce. Second, bonded parameters are not expected to change significantly on transfer to the condensed phase. Rather, non-bonded interactions are particularly important in condensed phase simulations. Regarding the non-bonded interactions, electrostatics could be over polarized beyond what would be expected in the gas phase in order to reproduce condensed-phase properties, and Lennard-Jones parameters can be tuned to reproduce condensed phase properties (as has been a particular focus of the OPLS force fields30,64). Even when these are done, force fields retain bonded terms parameterized to reproduce QM geometries and energetics, further emphasizing the importance of testing in such a context. We therefore believe our assumption is reasonable and that this work warrants investigation.
We start our force field benchmark analysis by comparing MM energies to QM energies of the two different datasets, namely (i) the Public OpenFF Industry Dataset and (ii) the Proprietary OpenFF Industry Dataset. All the optimizations were performed consistently, using the same software installed and running identical workflows.
We found that 99.94% of the relative conformer energies of the molecular structures in the two datasets with the six force fields were within −55< ΔΔE <45 kcal/mol, according to Equation S1. However, 62 conformers in (i) and 24 in (ii) that had outlying energies beyond this range were treated as outliers and removed from the two datasets (Table S3 and S4).
After excluding these outliers, the ΔΔE energy histograms for datasets (i) and (ii) are shown in Figure 2a and Figure 2b, respectively. In density histograms, bin height is normalized so that the total area of the histogram is 1 and the unity of density is 1/kcal mol−1 for ΔΔE (whereas for RMSD plots it is 1/Å). OPLS4 results were generated with the ffld_server tool, not macromodel as in previous work,41 (see section Methods and Supporting Information for more details). OPLS4 could not be calculated for the proprietary dataset (ii) because that force field was not available in house for some of the industry collaborators in this study. The difference between MM relative conformer energies and QM relative conformer energies exhibits very similar distributions for all force fields. All distributions appear asymmetric, having a skew towards more negative ΔΔE values than positive ones, indicating that the conformer energy differences may be underpredicted by MM compared to QM, though we have no clear explanation of this behaviour. In (i) the comparison between OPLS4, OpenFF-2.0.0 and GAFF-2.11 shows that the qualitative ordering of force fields from lowest to highest agreement with QM energies goes as GAFF-2.11 < OpenFF-2.0.0 < OPLS4DEF < OPLS4CST. In other words, the peak size around ΔΔE = 0 kcal/mol (the fraction of conformations with good agreement between QM and MM relative energies) is greatest for OPLS4CST, closely followed by OPLS4DEF, then by OpenFF-2.0.0 and GAFF-2.11. OPLS4CST and OPLS4DEF predict 34.0±0.4% and 31.6 ± 0.4% of conformers within 1 kcal/mol of QM, respectively. OpenFF-2.0.0 predicts 26.2 ± 0.4% and GAFF-2.11 24.6 ± 0.3% (standard error with 95% CI calculated with 2000 bootstrap iterations).
Figure 2:
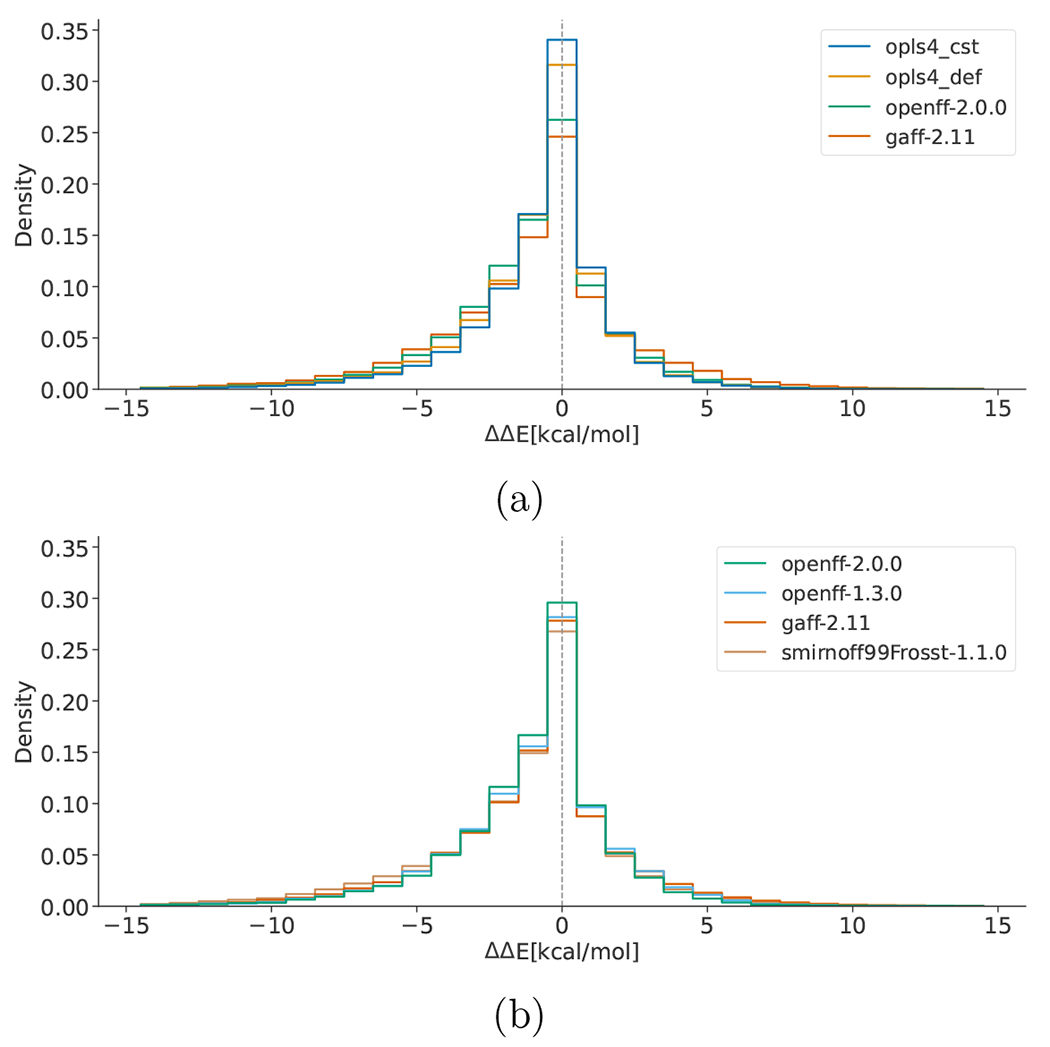
Histograms of the relative conformer energy differences as computed for compare-forcefields (equation S1) for each force field relative to QM. Each molecular structure, including different conformers of the same molecule, is counted separately. Since the global minimum molecular structures were set to zero deliberately and add a constant offset to the central bin, they are removed from the counts. A force field having higher agreement with QM would have a higher bin centered at ΔΔE = 0 kcal/mol. (a) compares the latest release of all three force field families over the public dataset. (b) shows the three histograms belonging to the OpenFF family of force fields and GAFF-2.11 over the proprietary set. OpenFF-1.3.0 (cyan) and GAFF-2.11 (orange) slightly overlap in the central bin of (b)
Figure 2b illustrates the progress made within the OpenFF family of force fields with respect to GAFF-2.11 in the benchmark of dataset (ii). Smirnoff99Frosst and GAFF-2.11 almost overlap, performing worse than all other investigated force fields. Improvements can be noticed in OpenFF-1.3.0 and more so in the most recent release OpenFF-2.0.0, which clearly performs better than its predecessors. Indeed, OpenFF-2.0.0 predicts 29.6 ± 0.4% of conformers within 1 kcal/mol of QM, OpenFF-1.3.0 28.2 ± 0.4%, GAFF-2.11 27.8 ± 0.4% and Smirnoff99Frosst 26.8 ± 0.4%
We next examine agreement between MM-optimized geometries and those from QM, as calculated by each molecule’s root-mean-square deviation of atomic positions (RMSD) and Torsion Fingerprint Deviation (TFD) scores with reference to the parent QM-optimized geometry. While RMSD is the more common metric, it depends on the molecule size, complicating interpretation.65,66 In contrast, TFD was designed to be more independent of molecule size in order to compare molecular conformations more meaningfully.61 The TFD score between two molecular structures is evaluated by computing, normalizing, and Gaussian weighting the (pseudo-)torsion deviation for each bond and ring system. While TFD is normalized from 0 to 1, RMSD is unbounded. Both RMSD and TFD are similar in that a higher value signifies lower agreement between the geometries of two molecules. A FF which yields optimized geometries closer to those of QM would have generally smaller RMSD and TFD values. We calculated RMSD and TFD scores for all MM optimized geometries with respect to QM geometries. We plotted this data in histograms,in Figure 3.
Figure 3:
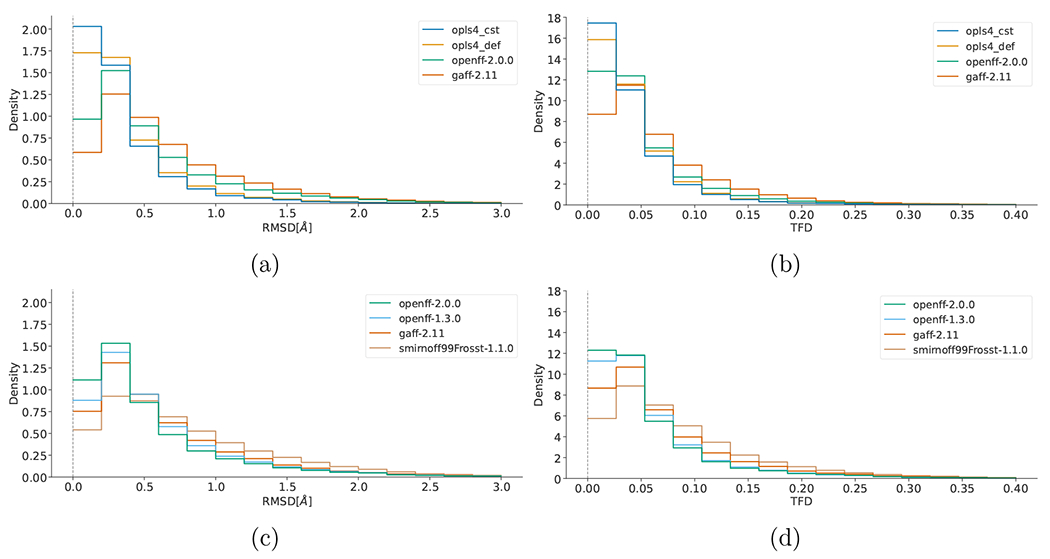
Histograms of the RMSD (a, c) and TFD (b, d) values between force field structures as compared to QM structures. Values closer to zero indicate higher geometric similarity for both RMSD and TFD. Panels (a) and (b) compare the families of force fields (GAFF-2.11, OPLS4, and OpenFF-2.0.0) over the public dataset. Panels (c) and (d) compare the force fields of the OpenFF family (Smirnoff99Frosst, OpenFF-1.3.0, and OpenFF-2.0.0) and GAFF-2.11 over the proprietary set.
In terms of geometry agreement, we observed similar results between the RMSD and TFD plots. The ranking of the force fields is mostly the same as with the ΔΔE rankings above, with OPLS4CST performing best, followed by OPLS4DEF, the latest Open Force Field release OpenFF-2.0.0 and finally GAFF-2.11. The use of the custom parameters made a notable improvement for OPLS4 compared to the default parameters in both the energetic and geometric comparisons. The OpenFF force fields show clear improvement with newer generations having higher densities close to zero and also by having tails successively reduced.
To understand how each forcefield scored in both energetics and geometries we reported in Figure 4 the percentage of conformers within certain threshold values of both |ΔΔE| and RMSD with respect to QM reference. The trend in scoring both metrics is consistent with that reported separately in Figure 2, 3a and 3c, showing that the qualitative ordering of force fields from highest agreement to both QM energies and geometries goes as OPLS4CST > OPLS4DEF > OpenFF-2.0.0 > GAFF-2.11 for the public dataset (i), and OpenFF-2.0.0 > OpenFF-1.3.0 > GAFF-2.11 > Smirnoff99Frosst for the proprietary dataset (ii).
Figure 4:
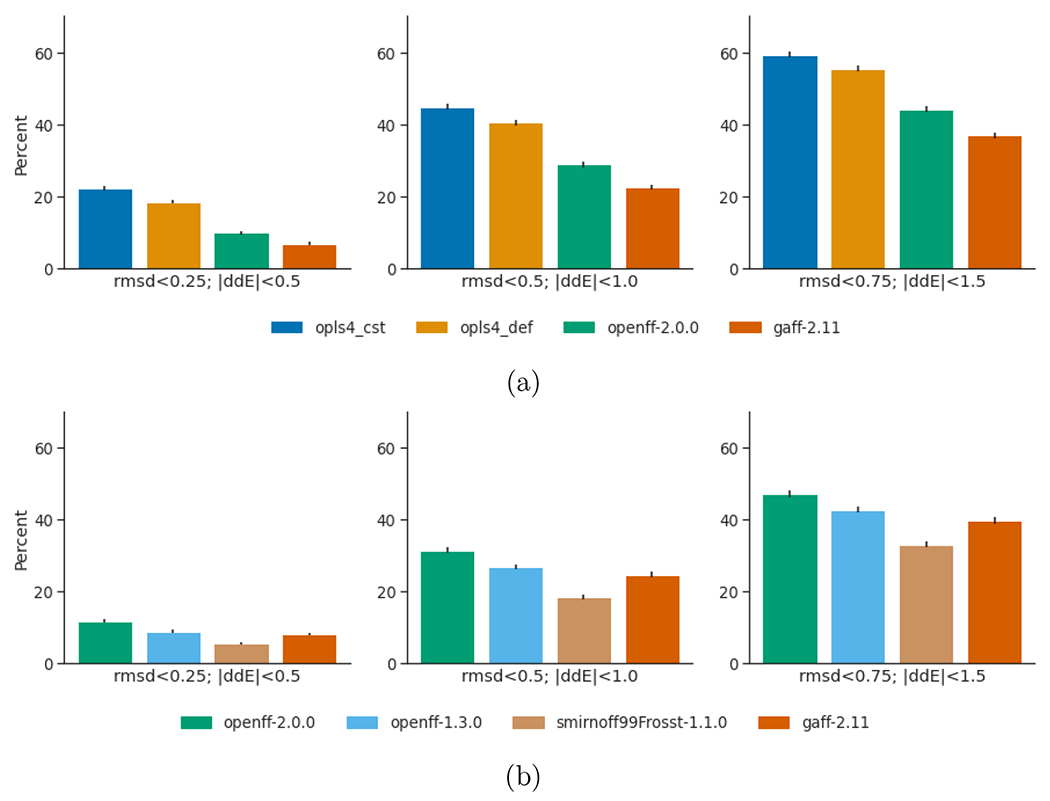
Bar plots with percentages of conformations predicted by the different force fields matching both |ΔΔE| and RMSD given thresholds. (a) compares the force fields assessed on the public dataset. (b) compares the force fields assessed on the proprietary dataset.
Ultimately, we assessed the performances of OpenFF-2.0.0, GAFF-2.11 and OPLS4 on charged and neutral molecules of the public dataset (i) (Figure S4 and Table S5). Overall, charged molecules feature a more negative ΔΔE mean distribution than neutral molecules according to all FFs. In OPLS4DEF this difference was largest (ΔΔE charged = −2.54±0.12 kcal/mol, ΔΔE neutral = −0.78±0.02 kcal/mol). Training custom parameters in OPLS4CST reduces the discrepancy (ΔΔE charged = −2.08±0.11 kcal/mol, ΔΔE neutral = −0.63±0.02 kcal/mol) to a comparable level with OpenFF-2.0.0 and GAFF-2.11 (ΔΔE charged = −2.07± 0.12 kcal/mol, ΔΔE neutral = −1.00±0.03 kcal/mol and ΔΔE charged = −1.51±0.12 kcal/mol, ΔΔE neutral = −0.98± 0.03 kcal/mol, respectively). No major geometric differences were seen for charged versus neutral molecules, the greatest divergence in the RMSD mean distribution is only 0.1 Å for OPLS4DEF (Table S5).
Analysis of OpenFF-2.0.0 shortcomings
As mentioned in section Automation of our approach, our FF benchmark was performed running the MM optimization on top of QM-optimization and comparing results. Ideally, the conformer which is the global minimum on the QM potential energy surface should still be found as global minimum on the MM surface. Nevertheless, in practice, the MM optimization could lead to a structurally different conformer which is local rather than global MM minimum. Thus, to objectively identify these types of FF shortcomings, we computed the relative energy difference (ΔE) between the MM reference conformer with lowest RMSD with respect to the QM global minimum (MM,ref) and the MM conformer with the lowest energy (MM,min) according to Equation 1:
| (1) |
Large energy differences in this metric are thereby indicative of suboptimal FF behaviour in terms of the MM force field’s ability to identify the same global minimum as QM does. We ran this analysis on the proprietary Roche set and inspected molecular structures with ΔE > 2 kcal/mol. According to OpenFF-2.0.0, a total of 40 out of 809 molecules were identified. The low number of identified problematic issues indicates that Sage performed generally well in the Roche dataset. A selection of the problematic torsions compared to the QM reference geometry are shown in Figure 5 and Figure S5, including incorrect intramolecular hydrogen (Figure 5a) and chalcogen (5b) bonds, tendencies to form flipped ureas (5c), cis-amides (5d) and -amines (5e,f).
Figure 5:

Molecular fragments of the Roche dataset containing concerning torsions. Global minima conformers optimized with QM and MM are shown with the concerning torsion(s) marked in brackets. Relative (ΔE) energies calculated according to equation 1 and torsion parameter(s) associated with corresponding concerning torsion(s) are reported.
Motivated to identify systematic issues in torsion parameters, we developed a workflow to detect any dihedral deviation from a threshold value for each i-th MM optimized conformer with respect to the same i-th conformer optimized with QM. We performed the analysis on the public dataset (i) and counted all the torsion violations in MM structures that were off by more than 30° threshold for any dihedral bond angle (Figure 6). Some torsion parameters are more common than others, therefore we weighted by the number of times that it was used in the dataset. Most of the problematic parameters found by this analysis were also identified in the previous RCH set (Figure 5), namely t17, t64, t66, t67, and t74. Interestingly, other parameters that appeared more frequently among the most concerning cases (>50 counted violations and >1 weighted violations) include t18 (torsion comprised by a tetra- and a trivalent C, Figure S6) and t105 (torsion formed by a trivalent C and bivalent O). Improvements resulting from this and further analysis, which are currently being performed, are expected to be incorporated in the next OpenFF force field reselase and will be part of our future report on this topic.
Figure 6:
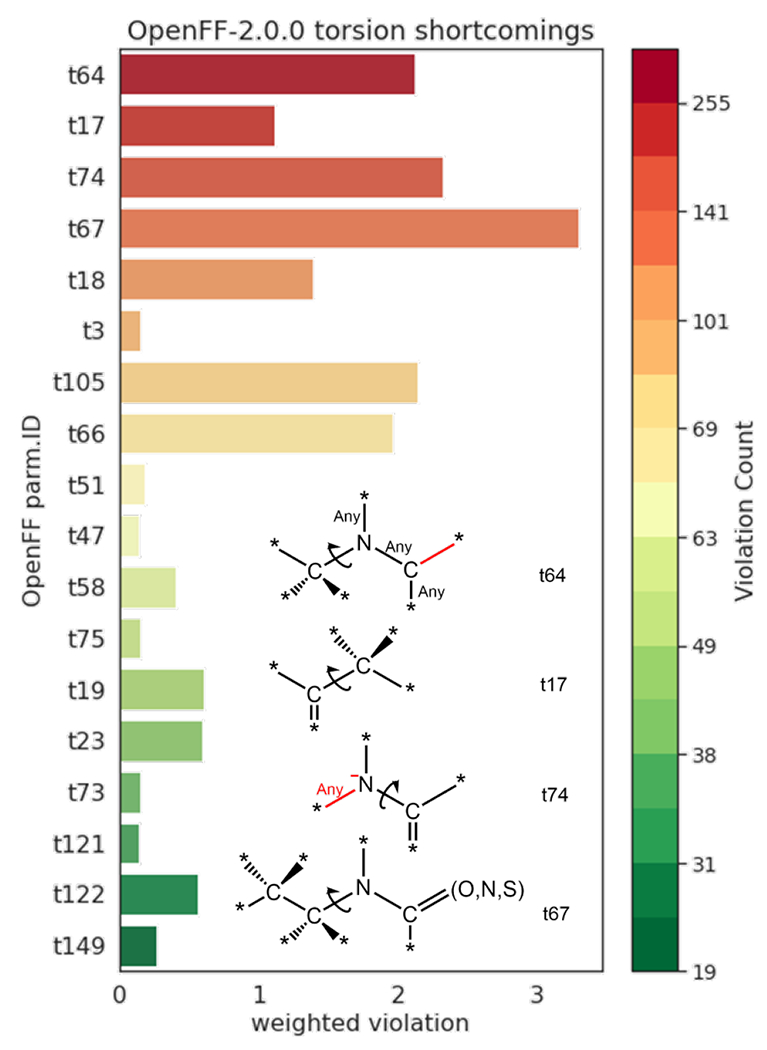
Analysis of torsion violations in the Public OpenFF Industry Dataset. Inset: 2D sketch chemistry match of selected torsion parameters. Elements in red color (bond, charge) may or may not exist, meaning that the corresponding atom can be either tri- or dicoordinated, respectively.
Improvements resulting from this and further analyses, which are currently being performed, are expected to be incorporated in the next OpenFF force field reselase and will be part of our future report on the topic
Conclusions
In this work, we presented a large-scale analysis of five small molecule force fields in terms of their relative conformer energies and geometries compared to QM data. Amongst the force fields (GAFF-2.11, OPLS4, SMIRNOFF99Frosst, OpenFF-1.3.0 and OpenFF-2.0.0), OPLS4CST performed best in terms of reproducing QM conformer energies and geometries. However, there is a higher computational cost to perform the DFT torsion fitting for generating the custom OPLS4 parameters (likely in part due to the diversity of the present molecule set), whereas with the other force fields, including OPLS4DEF, parameter assignment is immediate, because no new quantum chemical calculations are required.
As previously reported,41 the OpenFF showed improvements in both energetic and geometric metrics with each new version. We herein show that the latest OpenFF-2.0.0 appears to be positioned as the best open source/free small molecule force field in this study.
In the view of the industry collaborators performing this benchmarking work, this study highlights the progress the Open Force Field Initiative has made towards its goal of producing high quality public, open force fields built with infrastructure which enables rapid parameterization. Particularly, the series of OpenFF force fields presented here demonstrate marked improvements in accuracy over a relatively short time, and these improved force fields are available to everyone. One key challenge going forward will be to continue improving the treatment of problematic areas of chemical space and expanding coverage. Future OpenFF updates are planned to include improved treatment of torsions (e.g. via Wiberg bond order-based parameter interpolation67 which was recently implemented in the OpenFF Toolkit), off-site charges and better handling of trivalent nitrogen geometries68 (which we anticipate will boost performance further). Additionally, a tool for fitting bespoke torsion parameters for specific molecules/chemistries of interest is now available,69 likely further improving accuracy. In parallel, a biopolymer force field and an OpenFF software stack that will enable the conversion from OpenMM objects to file formats understood by other molecular simulation engines, like AMBER and GROMACS (OpenFF Interchange70), will soon be released.
Beyond these specific conclusions, we believe the general strategies employed here for benchmarking force field performance will be useful far more broadly than this specific study. Particularly, comparing performance by both geometric and energetic measures is particularly important, as the analysis we have done demonstrates. Additionally, the availability of a large amount of public data in QCArchive facilitates straightforward large scale benchmarking of force fields in a way it has not been done previously.
Supplementary Material
Acknowledgement
The authors thank the Open Force Field Consortium and Initiative for financial and scientific support; the Open Molecular Software Foundation (OMSF) for its support of the Open Force Field Initiative; Ed Harder and Rob Abel from Schrödinger Inc. for their helpful comments on the manuscript. We acknowledge funding from NIH grants GM098973, 1R01GM108889-01 and 1R01GM124270-01A1 (to DLM).
Footnotes
The Supporting Information contains (1) equations used to compute ΔΔE energies; (2) tables with the number of molecules selected by each industry partner and optimized with QM and MM for the public and the proprietary dataset; (3) table with outliers (defined as ΔΔE <−55 or ΔΔE >45 kcal/mol) of the public and proprietary datasets; (4) plots similar to those of Figure 2 and Figure 3 comparing OPLS4 using both ffld_server and macromodel obtained with compare-forcefields and the conformer matching process match-minima; (5) table with mean ΔΔE and RMSD values of charged and neutral molecules and corresponding scatter plots for charged and neutral molecules, (6) molecular fragments of the Roche dataset containing concerning torsions not shown in Figure 5, (7) code to extract optimized records from QCArchive for the public datasets hosted there.
Data and code availability
QM geometries and energies, SMILES strings and depictions of the public dataset are deposited on GitHub: https://github.com/openforcefield/qca-dataset-submission/tree/master/submissions/2021-07-28-OpenFF-Industry-Benchmark-Season-1-MM-v1.1.
The Python code used for the setup, minimizations, and analysis of this work is open source and available on GitHub at https://github.com/openforcefield/openff-benchmark; the protocol used to run minimization is available on Confluence at https://openforcefield.atlassian.net/wiki/spaces/FF/pages/971898891/Optimization+Benchmarking+Protocol+-+Season+1.
References
- (1).González MA Force Fields and Molecular Dynamics Simulations. JDN 2011, 12, 169–200. [Google Scholar]
- (2).Cole DJ; Vilseck JZ; Tirado-Rives J; Payne MC; Jorgensen WL Biomolecular Force Field Parameterization via Atoms-in-Molecule Electron Density Partitioning. J. Chem. Theory Comput 2016, 12, 2312–2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Lane TJ; Shukla D; Beauchamp KA; Pande VS To Milliseconds and beyond: Challenges in the Simulation of Protein Folding. Current Opinion in Structural Biology 2013, 23, 58–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Lange OF; van der Spoel D; de Groot BL Scrutinizing Molecular Mechanics Force Fields on the Submicrosecond Timescale with NMR Data. Biophysical Journal 2010, 99, 647–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Riniker S Fixed-Charge Atomistic Force Fields for Molecular Dynamics Simulations in the Condensed Phase: An Overview. J. Chem. Inf. Model 2018, 58, 565–578. [DOI] [PubMed] [Google Scholar]
- (6).Ponder JW; Case DA Advances in Protein Chemistry; Protein Simulations; Academic Press, 2003; Vol. 66; pp 27–85. [DOI] [PubMed] [Google Scholar]
- (7).Nerenberg PS; Head-Gordon T New Developments in Force Fields for Biomolecular Simulations. Current Opinion in Structural Biology 2018, 49, 129–138. [DOI] [PubMed] [Google Scholar]
- (8).Monticelli L; Tieleman DP In Biomolecular Simulations: Methods and Protocols; Monticelli L, Salonen E, Eds.; Methods in Molecular Biology; Humana Press: Totowa, NJ, 2013; pp 197–213. [Google Scholar]
- (9).Hagler AT Force Field Development Phase II: Relaxation of Physics-Based Criteria… or Inclusion of More Rigorous Physics into the Representation of Molecular Energetics. J Comput Aided Mol Des 2018, [DOI] [PubMed] [Google Scholar]
- (10).Dauber-Osguthorpe P; Hagler AT Biomolecular Force Fields: Where Have We Been, Where Are We Now, Where Do We Need to Go and How Do We Get There? J Comput Aided Mol Des 2018, [DOI] [PubMed] [Google Scholar]
- (11).Mobley DL; Bannan CC; Rizzi A; Bayly CI; Chodera JD; Lim VT; Lim NM; Beauchamp KA; Slochower DR; Shirts MR; Gilson MK; Eastman PK Escaping Atom Types in Force Fields Using Direct Chemical Perception. J. Chem. Theory Comput 2018, 14, 6076–6092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Cailliez F; Pernot P Statistical Approaches to Forcefield Calibration and Prediction Uncertainty in Molecular Simulation. J. Chem. Phys 2011, 134, 054124. [DOI] [PubMed] [Google Scholar]
- (13).Geballe MT; Guthrie JP The SAMPL3 Blind Prediction Challenge: Transfer Energy Overview. J Comput Aided Mol Des 2012, 26, 489–496. [DOI] [PubMed] [Google Scholar]
- (14).Hopkins CW; Roitberg AE Fitting of Dihedral Terms in Classical Force Fields as an Analytic Linear Least-Squares Problem. J. Chem. Inf. Model 2014, 54, 1978–1986. [DOI] [PubMed] [Google Scholar]
- (15).Köster A; Spura T; Rutkai G; Kessler J; Wiebeler H; Vrabec J; Kühne TD Assessing the Accuracy of Improved Force-Matched Water Models Derived from Ab Initio Molecular Dynamics Simulations. J. Comput. Chem 2016, 37, 1828–1838, _eprint: https://onlinelibrary.wiley.com/doi/pdf/10.1002/jcc.24398. [DOI] [PubMed] [Google Scholar]
- (16).Mishra SK; Calabró G; Loeffler HH; Michel J; Koča J Evaluation of Selected Classical Force Fields for Alchemical Binding Free Energy Calculations of Protein-Carbohydrate Complexes. J. Chem. Theory Comput 2015, 11, 3333–3345. [DOI] [PubMed] [Google Scholar]
- (17).Cornell WD; Cieplak P; Bayly CI; Gould IR; Merz KM; Ferguson DM; Spellmeyer DC; Fox T; Caldwell JW; Kollman PA A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules. Journal of the American Chemical Society 1995, 117, 5179–5197. [Google Scholar]
- (18).MacKerell AD et al. All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. The Journal of Physical Chemistry B 1998, 102, 3586–3616, [DOI] [PubMed] [Google Scholar]
- (19).Foloppe N; MacKerell AD Jr. All-atom empirical force field for nucleic acids: I. Parameter optimization based on small molecule and condensed phase macromolecular target data. Journal of Computational Chemistry 2000, 21, 86–104. [Google Scholar]
- (20).MacKerell AD Jr.; Banavali NK All-atom empirical force field for nucleic acids: II. Application to molecular dynamics simulations of DNA and RNA in solution. Journal of Computational Chemistry 2000, 21, 105–120. [Google Scholar]
- (21).Hornak V; Abel R; Okur A; Strockbine B; Roitberg A; Simmerling C Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins: Structure, Function, and Bioinformatics 2006, 65, 712–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Lindorff-Larsen K; Piana S; Palmo K; Maragakis P; Klepeis JL; Dror RO; Shaw DE Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins: Structure, Function, and Bioinformatics 2010, 78, 1950–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Wang L-P; McKiernan KA; Gomes J; Beauchamp KA; Head-Gordon T; Rice JE; Swope WC; Martínez TJ; Pande VS Building a More Predictive Protein Force Field: A Systematic and Reproducible Route to AMBER-FB15. The Journal of Physical Chemistry B 2017, 121, 4023–4039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Oostenbrink C; Villa A; Mark AE; Van Gunsteren WF A biomolecular force field based on the free enthalpy of hydration and solvation: The GROMOS force-field parameter sets 53A5 and 53A6. Journal of Computational Chemistry 2004, 25, 1656–1676. [DOI] [PubMed] [Google Scholar]
- (25).Shi Y; Xia Z; Zhang J; Best R; Wu C; Ponder JW; Ren P Polarizable Atomic Multipole-Based AMOEBA Force Field for Proteins. Journal of Chemical Theory and Computation 2013, 9, 4046–4063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Wang J; Wolf RM; Caldwell JW; Kollman PA; Case DA Development and Testing of a General Amber Force Field. J. Comput. Chem 2004, 25, 1157–1174, _eprint: https://onlinelibrary.wiley.com/doi/pdf/10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- (27).Case DA, Betz RM, Cerutti DS, Cheatham TE III, Darden TA, Duke RE, Giese TJ, Gohlke H, Goetz AW, Homeyer N, Izadi S, Janowski P, Kaus J, Kovalenko A, Lee TS, LeGrand S, Li P, Lin C, Luchko T, Luo R, Madej B, Mermelstein D, Merz KM, Monard G, Nguyen H, Nguyen HT, Omelyan I, Onufriev A, Roe DR, Roitberg A, Sagui C, Simmerling CL, Botello-Smith WM, Swails J, Walker RC, Wang J, Wolf RM, Wu X, Xiao L and Kollman PA, AMBER 2016, University of California, San Francisco. 2016, [Google Scholar]
- (28).Vanommeslaeghe K; Hatcher E; Acharya C; Kundu S; Zhong S; Shim J; Darian E; Guvench O; Lopes P; Vorobyov I; Mackerell AD CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. Journal of Computational Chemistry 2010, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Jorgensen WL; Tirado-Rives J The OPLS [optimized potentials for liquid simulations] potential functions for proteins, energy minimizations for crystals of cyclic peptides and crambin. Journal of the American Chemical Society 1988, 110, 1657–1666. [DOI] [PubMed] [Google Scholar]
- (30).Jorgensen WL; Maxwell DS; Tirado-Rives J Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc 1996, 118, 11225–11236. [Google Scholar]
- (31).Harder E et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. Journal of Chemical Theory and Computation 2016, 12, 281–296. [DOI] [PubMed] [Google Scholar]
- (32).Roos K; Wu C; Damm W; Reboul M; Stevenson JM; Lu C; Dahlgren MK; Mondal S; Chen W; Wang L; Abel R; Friesner RA; Harder ED OPLS3e: Extending Force Field Coverage for Drug-Like Small Molecules. J. Chem. Theory Comput 2019, 15, 1863–1874. [DOI] [PubMed] [Google Scholar]
- (33).Lu C; Wu C; Ghoreishi D; Chen W; Wang L; Damm W; Ross GA; Dahlgren MK; Russell E; Von Bargen CD; Abel R; Friesner RA; Harder ED OPLS4: Improving Force Field Accuracy on Challenging Regimes of Chemical Space. Journal of Chemical Theory and Computation 2021, 17, 4291–4300. [DOI] [PubMed] [Google Scholar]
- (34).Riniker S Fixed-Charge Atomistic Force Fields for Molecular Dynamics Simulations in the Condensed Phase: An Overview. Journal of Chemical Information and Modeling 2018, 58, 565–578. [DOI] [PubMed] [Google Scholar]
- (35).Mobley DL; Bannan CC; Wagner JR; Rizzi A; Lim NM; Henry M open-forcefield/smirnoff99Frosst: Version 1.1.0. 2019; 10.5281/zenodo.3351714. [DOI] [Google Scholar]
- (36).Bayly C; McKay D; Truchon J An Informal AMBER Small Molecule Force Field: Parm@ Frosst. http://www.ccl.net/cca/data/parm_at_Frosst/, 2010.
- (37).Qiu Y et al. Development and Benchmarking of Open Force Field v1.0.0—the Parsley Small-Molecule Force Field. Journal of Chemical Theory and Computation 2021, 17, 6262–6280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Jang H Update on Parsley Minor Releases (Openff-1.1.0, 1.2.0). 2020. [Google Scholar]
- (39).Wagner J; Thompson M; Dotson D; Jang H; SimonBoothroyd,; Rodríguez-Guerra J openforcefield/openff-forcefields: Version 1.3.1 ”Parsley” Update (1.3.1). 2021. [Google Scholar]
- (40).Wagner J; Thompson M; Dotson D; Jang H; SimonBoothroyd,; Rodríguez-Guerra J openforcefield/openff-forcefields: Version 2.0.0 ”Sage”. 2021. [Google Scholar]
- (41).Lim VT; Hahn DF; Tresadern G; Bayly CI; Mobley D Benchmark Assessment of Molecular Geometries and Energies from Small Molecule Force Fields. F1000Research 2020, 17, 1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Smith D; Altarawy D; Burns L; Welborn M; Naden LN; Ward L; Ellis S; Crawford T The MolSSI QCArchive Project: An Open-Source Platform to Compute, Organize, and Share Quantum Chemistry Data. 2020, [Google Scholar]
- (43).Smith DGA; Altarawy D; Burns LA; Welborn M; Naden LN; Ward L; Ellis S; Pritchard BP; Crawford TD The MolSSI QCArchive project: An open-source platform to compute, organize, and share quantum chemistry data. WIREs Computational Molecular Science 2021, 11, e1491. [Google Scholar]
- (44).Jakalian A; Bush BL; Jack DB; Bayly CI Fast, Efficient Generation of High-Quality Atomic Charges. AM1-BCC Model: I. Method. J. Comput. Chem 2000, 21, 132–146. [DOI] [PubMed] [Google Scholar]
- (45).Jakalian A; Jack DB; Bayly CI Fast, Efficient Generation of High-Quality Atomic Charges. AM1-BCC Model: II. Parameterization and Validation. J. Comput. Chem 2002, 23, 1623. [DOI] [PubMed] [Google Scholar]
- (46).Beauchamp K; Rustenburg A; Rizzi A; Behr J; Matos G; Wang L; McGibbon R; Mobley D; Chodera J OpenMolTools. [Google Scholar]
- (47).Schrödinger, Schrödinger Release 2021-1: Maestro. 2021. [Google Scholar]
- (48).Eastman P; Swails J; Chodera JD; McGibbon RT; Zhao Y; Beauchamp KA; Wang L-P; Simmonett AC; Harrigan MP; Stern CD; Wiewiora RP; Brooks BR; Pande VS OpenMM 7: Rapid Development of High Performance Algorithms for Molecular Dynamics. PLOS Computational Biology 2017, 13, e1005659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Dotson D; Wagner J; Hahn DF; Horton J; Thompson M; Rodríguez-Guerra J; D’Amore L BenchmarkFF. https://github.com/openforcefield/openff-benchmark, 2022.
- (50).Wang LP; Kraus P; Simmonett A; Kruse H; Burns LA DF bases for dzvp. https://github.com/psi4/psi4, 2021.
- (51).Becke AD Density-functional Thermochemistry. III. The Role of Exact Exchange. The Journal of Chemical Physics 1993, 98, 5648–5652. [Google Scholar]
- (52).Lee C; Yang W; Parr RG Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [DOI] [PubMed] [Google Scholar]
- (53).Vosko SH; Wilk L; Nusair M Accurate Spin-Dependent Electron Liquid Correlation Energies for Local Spin Density Calculations: A Critical Analysis. Can. J. Phys 1980, 58, 1200–1211. [Google Scholar]
- (54).Stephens PJ; Devlin FJ; Chabalowski CF; Frisch MJ Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem 1994, 98, 11623–11627. [Google Scholar]
- (55).Godbout N; Salahub DR; Andzelm J; Wimmer E Optimization of Gaussian-Type Basis Sets for Local Spin Density Functional Calculations. Part I. Boron through Neon, Optimization Technique and Validation. Can. J. Chem 1992, 70, 560–571. [Google Scholar]
- (56).Turney JM et al. Psi4: an open-source ab initio electronic structure program. WIREs Computational Molecular Science 2012, 2, 556–565. [Google Scholar]
- (57).íezáč J; Bím D; Gutten O; Rulíšek L Toward Accurate Conformational Energies of Smaller Peptides and Medium-Sized Macrocycles: MPCONF196 Benchmark Energy Data Set. J. Chem. Theory Comput 2018, 14, 1254–1266. [DOI] [PubMed] [Google Scholar]
- (58).Kesharwani MK; Karton A; Martin JML Benchmark Ab Initio Conformational Energies for the Proteinogenic Amino Acids through Explicitly Correlated Methods. Assessment of Density Functional Methods. J. Chem. Theory Comput 2016, 12, 444–454. [DOI] [PubMed] [Google Scholar]
- (59).Hermann JC; Ghanem E; Li Y; Raushel FM; Irwin JJ; Shoichet BK Predicting Substrates by Docking High-Energy Intermediates to Enzyme Structures. Journal of the American Chemical Society 2006, 128, 15882–15891. [DOI] [PubMed] [Google Scholar]
- (60).Peach ML; Cachau RE; Nicklaus MC Conformational energy range of ligands in protein crystal structures: The difficult quest for accurate understanding. Journal of Molecular Recognition 2017, 30, e2618, e2618 JMR-16-0070.R2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Schulz-Gasch T; Schärfer C; Guba W; Rarey M TFD: Torsion Fingerprints As a New Measure To Compare Small Molecule Conformations. J. Chem. Inf. Model 2012, 52, 1499–1512. [DOI] [PubMed] [Google Scholar]
- (62).Ehrman JN; Bannan CC; Lim VT; Thi N; Kyu DY; Mobley DL Improving Force Fields by Identifying and Characterizing Small Molecules with Parameter Inconsistencies. 2019; 10.5281/zenodo.3385278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Ehrman J; Lim VT; Bannan CC; Thi N; Kyu D; Mobley D Improving Small Molecule Force Fields by Identifying and Characterizing Small Molecules with Inconsistent Parameters. ChemRxiv 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Dodda LS; Cabeza de Vaca I; Tirado-Rives J; Jorgensen WL LigParGen Web Server: An Automatic OPLS-AA Parameter Generator for Organic Ligands. Nucleic Acids Res 2017, 45, W331–W336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Reva BA; Finkelstein AV; Skolnick J What Is the Probability of a Chance Prediction of a Protein Structure with an Rmsd of 6 å? Folding and Design 1998, 3, 141–147. [DOI] [PubMed] [Google Scholar]
- (66).Sargsyan K; Grauffel C; Lim C How Molecular Size Impacts RMSD Applications in Molecular Dynamics Simulations. J. Chem. Theory Comput 2017, 13, 1518–1524. [DOI] [PubMed] [Google Scholar]
- (67).Stern C Capturing Non-Local through-Bond Effects When Fragmenting Molecules for QC Torsion Scans. 2020. [Google Scholar]
- (68).Mobley DL Current Status of OpenFF and Our Near-Term Roadmap. 2020. [Google Scholar]
- (69).Horton J; Boothroyd S; Wagner J; Mitchell J; Trevor G; Dotson D; Behara P; Ramaswamy V; Mackey M; Chodera J; Anwar J; Mobley D; Cole D Open Force Field BespokeFit: Automating Bespoke Torsion Parametrization At Scale. ChemRxiv 2022, [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Thompson M e. a. Interchange https://github.com/openforcefield/openff-interchange, 2022.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
QM geometries and energies, SMILES strings and depictions of the public dataset are deposited on GitHub: https://github.com/openforcefield/qca-dataset-submission/tree/master/submissions/2021-07-28-OpenFF-Industry-Benchmark-Season-1-MM-v1.1.
The Python code used for the setup, minimizations, and analysis of this work is open source and available on GitHub at https://github.com/openforcefield/openff-benchmark; the protocol used to run minimization is available on Confluence at https://openforcefield.atlassian.net/wiki/spaces/FF/pages/971898891/Optimization+Benchmarking+Protocol+-+Season+1.