

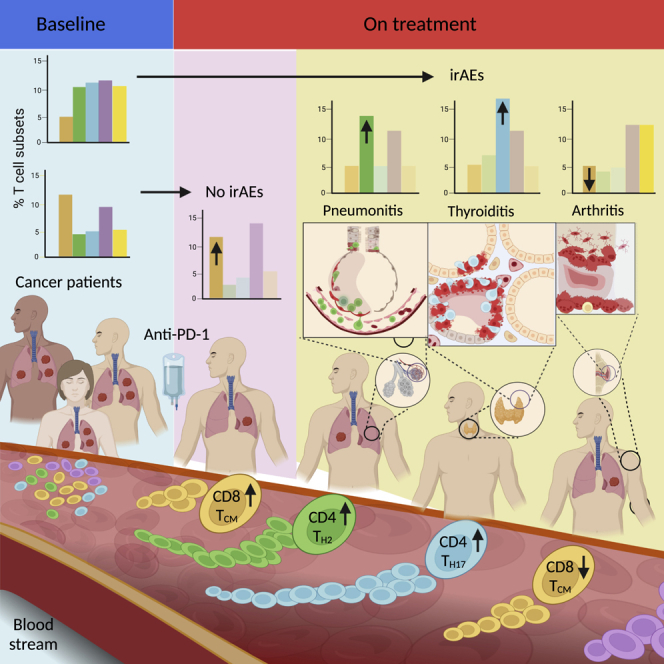
Summary
PD-1 is an inhibitory receptor in T cells, and antibodies that block its interaction with ligands augment anti-tumor immune responses. The clinical potential of these agents is limited by the fact that half of all patients develop immune-related adverse events (irAEs). To generate insights into the cellular changes that occur during anti-PD-1 treatment, we performed single-cell RNA sequencing of circulating T cells collected from patients with cancer. Using the K-nearest-neighbor-based network graph-drawing layout, we show the involvement of distinctive genes and subpopulations of T cells. We identify that at baseline, patients with arthritis have fewer CD8 TCM cells, patients with pneumonitis have more CD4 TH2 cells, and patients with thyroiditis have more CD4 TH17 cells when compared with patients who do not develop irAEs. These data support the hypothesis that different populations of T cells are associated with different irAEs and that characterization of these cells’ pre-treatment has the potential to serve as a toxicity-specific predictive biomarker.
Keywords: PD-1, T cells, single-cell RNA sequencing, irAEs, checkpoint inhibitor, autoimmunity, arthritis, pneumonitis, thyroiditis, immune-related adverse events
Graphical abstract
Highlights
-
•
Selected T cell subsets are associated with organ-specific immune-related adverse events
-
•
Patients with immune-related arthritis have lower levels of CD8 TCM cells at baseline
-
•
Patients with immune-related pneumonitis have more CD4 TH2 cells at baseline
-
•
Patients with immune-related thyroiditis have more CD4 TH17 cells at baseline
Bukhari et al. report that different subsets of T cells are associated with organ-specific immune-related adverse events of checkpoint inhibitors and that quantification and characterization of these populations of cells pre-treatment have the potential to serve as toxicity-specific predictive biomarkers.
Introduction
PD-1 is an inhibitory receptor expressed on T cells and exploited by tumor cells to evade immune detection. By overexpressing PD-L1, a PD-1 ligand, tumor cells engage PD-1 on T cells, blocking its activation and function. This pathway has been effectively thwarted by monoclonal antibodies (Abs) targeting either PD-1 or PD-L1, with great success in unleashing an anti-tumor response.1,2,3,4 Despite this powerful advance, there remain numerous challenges to immune checkpoint inhibitor (ICI) use that must be met to best advance the next generation of therapies. These challenges include increasing responsiveness to PD-1 and CTLA-4 blockade, uncovering additional targets to optimize pathway blockade, and, importantly, predicting and effectively managing immune-related adverse events (irAEs).5,6,7,8,9 Patients who have a prior history of autoimmune disease are thought to be at increased risk for developing irAEs, but aside from this, no predictive factors have been identified to guide patients regarding their individual risk of toxicity with ICI treatment, a major unmet need in clinical decision-making. One of the ways to achieve these goals is better understanding and characterization of T cell responses during treatment with ICIs.
ICIs are associated with the development of irAEs that affect various tissues and organ systems throughout the body.10 These acute and chronic inflammatory responses are thought to emerge because of the loss of the physiologic role played by immune checkpoints leading to unchecked T cell activation and loss of tolerance. irAEs are associated with significant morbidity and, in some cases, life-long disability. Some patients will require long-term immunosuppressive treatment, and others will have to withdraw from lifesaving anti-cancer therapy. Moreover, a particularly perplexing feature of irAEs is that they are often organ specific, with different organ systems being involved in different patients.11,12 Although the molecular pathways that predispose to and trigger irAEs are incompletely delineated, it is well established that irAEs are mediated by T cell responses that drive tissue- and organ-specific inflammation and assist B cells in autoantibody production.13,14 However, whether these T cell responses are similar in all irAEs, or whether distinctive responses are associated with different patterns of organ involvement, remains an unanswered question. Similarly, a related question is whether any features of the pre-treatment immune system might allow one to predict the development of an irAEs.
To better understand the biology of T cell responses that develop during the course of ICI treatment and through the onset of irAEs, we applied a single-cell RNA sequencing (scRNA-seq) methodology to peripheral blood T cells prior to and during irAE development. We combined these analyses with a cellular indexing of transcriptomes and epitopes sequencing (CITE-seq) approach to stratify CD4 and CD8 T cells by naive and effector phenotypes in order to increase the specificity and sensitivity of our downstream analyses. Using this approach, we defined the molecular and cellular changes associated with T cell transition from self-tolerant to sensitized effector cells. These data should provide the basis for additional mechanistic studies of organ-specific irAEs.
Results
Dimensionality reduction approach to visualize scRNA-seq data of patients with irAEs
We enrolled 40 patients with cancer into this study. These patients were divided into a discovery cohort, from which T cells were subjected to scRNA-seq, and a validation cohort, from which T cells were studied by flow cytometry (Figure 1A). The discovery group consisted of 24 patients from whom blood was collected before treatment with ICIs (at baseline) (Figure 1A). After 4–6 weeks of ICI treatment, 15 patients developed grade 2 or 3 irAEs, and from these patients, a second blood sample was collected (on treatment). Of the nine patients who did not develop AEs (no irAEs), the second matching blood sample was also collected after 4–6 weeks of the ICI treatment (total of 48 samples). The patients with no irAEs continued to have with no symptoms during the 12 month follow-up period. Out of the 24 patients, 18 had been diagnosed with lung adenocarcinoma, and 22 were treated with anti-PD-1 Abs (Figure 1B; Table S1A). Seven patients developed pneumonitis and were subsequently treated with high-dose corticosteroids, four patients presented with inflammatory polyarthritis, and four patients had thyroiditis, requiring hormonal replacement therapy. From each sample, a similar number of CD3 T cells were enriched using negative-selection sorting (Figures 1B and S1B). Initially, cells were subjected to CITE-seq with Abs tagged to CD4, CD8, CD27, and CD45RA (Figure S1A). Out of the 360,000 cells that were labeled, 135,287 CD3 T cells passed quality control (Figure S1C). Of these cells, 22,623 cells originated from patients with arthritis; 44,443 cells were collected from patients with pneumonitis; 12,913 cells came from patients with thyroiditis; and 51,177 cells were derived from patients with no irAEs.
Figure 1.

Dimensionality reduction approach to visualize single-cell RNA sequencing data of patients with immune-related adverse events
(A) Schematic workflow of study design.
(B) Clinical characterization of the patients.
(C and D) UMAP (C) and KNetL projection (D) of 135,287 T cells from patients with and without irAEs.
(E) Percentage variation in T cells between the baseline and on treatment within the cohorts of patients with no irAEs and with irAEs (paired t test).
Statistical significance for paired comparisons was performed by Student’s t test applying Wilcoxon matched-pairs signed rank test, p value reported, one-tailed. Data are presented as mean ± SD. p value, exact, two-tailed. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Uniform manifold approximation and projection (UMAP) was used to display the data (Figure 1C). A K-nearest-neighbor-based network graph-drawing layout (KNetL) plot was generated to increase the resolution of the data and to better separate and characterize the 25 cellular clusters (Figure 1D). Overall, the distribution of the cells into clusters across individual patients was comparable, mitigating bias secondary to variation in the numbers of the sequenced cells (Figures S1B and S1D). While several populations of cells were enriched across all patients, other clusters were dominant among the patients that developed irAEs (Figures S1B and S1D). The relative distribution of some populations of cells through the different clusters was similar in samples collected at baseline and on treatment with ICIs (Figure 1E). However, few clusters showed statistically significant changes in their relative percentages between baseline and on treatment (Figure 1E). No major cluster distribution differences were observed between cells collected from male or from female patients (Figures S1F and S1G).
Gene-marker-based cluster annotation identifies effector, regulatory, and memory subsets of peripheral T cells
Twenty-five clusters were annotated using the iCellR pipeline (Figure S1E).15 Fourteen clusters expressed the gene CD3E, of which six were dominated by CD8A and eight revealed high levels of the CD4 transcript (Figures 2A, 2D, and S1E). Based on reported gene markers (Table S2), within the CD8 T cells, naive cells (cluster 21) were characterized by the expression of CD45RA, CCR7, SELL (CD62L), and LEF1 (Figures 2A and S2A). The cells with the features of central memory CD8 T cells (cluster 4), beside containing CCR7, SELL, and CD27, expressed CD44, CXCR3, FAS, and high levels of CD28 transcripts. A subset of effector CD8 T cells (cluster 16) expressed TBX21 and cytotoxic markers such as GZMB, KLRD1, and PRF1 mRNA, along with the highest expression of CD3E. Complementary to the central memory CD8 T cells, the cells with the features of effector memory CD8 T cells (cluster 19) displayed EMOS, GZMK, and IFNG, highlighting their dampened, yet potential, cytotoxic activity. Interestingly, mucosal-associated invariant T cells (cluster 18) were discovered in the blood of all patients (Figure S2A). A cluster of cells expressing GATA3 suggested a commitment to CD8 T helper cells (cluster 24).
Figure 2.

Gene-marker-based cluster annotation identifies effector, regulatory, and memory subsets of peripheral T cells
(A and B) KNetL plot schematic of clusters according to the expression levels of CD8A with a corresponding heatmap showing the expression of selected markers in CD8A clusters (CD45RA in red fonts indicates the CITE-seq-based protein expression) (A) and CD4 clusters (B).
(C) Heatmap-dot plot indicating the association between T cell states and clusters based on the relative signature score variabilities.
(D) Dot plot showing the comparison of T cell states between the cohorts of patients with no irAEs and with irAEs based on the marker gene scores (unpaired t test).
For unpaired comparisons, statistical significance was performed by Student’s t test applying Mann-Whitney test. Data are presented as mean ± SD. p value, exact, two-tailed, the center lines denote the mean of SD. ∗∗∗∗p < 0.0001.
Among the CD4 T cell lineages, 8 clusters were annotated based on published markers (Table S2; Figure 2B). Three meta clusters comprised of cells expressing genes associated with TH1 and TH2 cells (cluster 25), TH17 cells (cluster 14), and naive cells (cluster 22) occupied most of the CD4 projections. The hybrid population of TH1 and TH2 cells was characterized by the expression of AREG, GATA3, PTGDR2, and CXCR3, known features of helper CD4 T cells (Figures 2A and S2A).16,17,18,19 Cluster 14 expressed genes that were mainly involved in TH17 responses (further details in Figure 4). As expected, the majority of the peripheral CD4 T cells resembled naive cells (cluster 22), characterized also by the expression of the transcription factors TCF7 and LEF1 and the chemokine receptor CCR7. Surprisingly, a cytotoxic CD4 cluster expressing GZMB, GZMH, PRF1, and TBX21 was noted (cluster 15). While the classic regulatory T cells (clusters 17 and 11) showed high levels of FOXP3, IL2RA, and CTLA4, cluster 12 was described as regulatory-like cells based on the expression of shared regulatory genes (Figure S2B) while having relatively lower levels of FOXP3 and IL2RA expression.20 Minor subsets of cells such as gamma delta and double-positive T cells were detected as well (Figure S3D). To confirm the rigor of our cluster annotations, we performed nearest-neighbor analysis for each cluster (Figure S2C). Through this analysis, we calculated the distance between the clusters obtained by each gene set used for cluster annotation and ranked the subsets based on that score (Figure S2C).
Figure 4.

Selected subsets of CD4 helper T cells are associated with organ-specific irAEs
(A) Representative KNetL plot of cluster 14 and mean percentage differences in TH RORC+ IL21+ cells per patient and across the disease groups: arthritis, pneumonitis, and thyroiditis.
(B) Subclustering of C14 cells into 6 sub clusters: sC1–sC6. Representative genes are shown separately.
(C) Heatmap showing the expression of markers associated with different subclusters among TH2 and TH17 families of cells. Black arrows indicate the subset defining markers.
(D) Differential clustering among the clinical irAE groups at baseline. Quantification of the percentages of cells in three clusters (sC3, sC4, and sC2) among irAE and no-irAE groups. The patients elected for each of the no-irAE groups were selected to match the type of underlying tumor.
(E) Correlation and RNA expression plots highlighting the association of candidate gene markers with TH2 and TH1723 clusters based on enrichment score. Representative KNetL plots showing the expression of candidate gene markers KLF6, S100B, and SIGLEC14 specifying the predictive cell populations present in pneumonitis and thyroiditis.
Statistical significance for unpaired comparisons was performed by Student’s t test applying Mann-Whitney test. Data are presented as mean ± SEM. p value, exact, two-tailed, the center lines denote the mean of SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
To further investigate the variability in the T cells between the patients with and without irAEs, we performed gene set signature analysis. Functional annotation (based on the T cell states and gene signatures) revealed association of irAEs’ T cells with genes associated with T cell activation and cytokine secretion compared with no irAEs’ T cells (Figure 2C). Cluster 4 (central memory T [TCM] CXCR3+), 16 (effector T [TE] TBX21+), 15 (cytotoxic T [TC] PRF1+), and 23 (helper T [TH] JUND+) had more cytotoxic and proinflammatory genes in the patients with irAEs compared with patients with no irAEs, while clusters 11 (regulatory T [TREG] SELL+MKI67+), 12 (TREG SELL+MKI67+), and 18 (mucosal-associated invariant T [TMAIT] SLCA410+) showed higher expression of activation genes in the patients with no irAEs compared with the irAE group. Further, we show that genes associated with T cell states (such as HLA-DRB1, TNF, IFNG, CCL4, CCL3, and PRF1) were highly expressed in the patients with irAEs even after normalization to baseline expression levels (Figure 2D). Altogether, these findings suggest that the population of T cells in the irAE groups were more likely to be associated with effector functions, while the cells of the no irAE groups were more likely to be associated with regulatory functions secondary to PD-1 blockade.
Patients with immune-related arthritis have higher percentages of CD4 TH cells at baseline
Among the eight CD4 clusters previously annotated (Figure 3A), no major differences in the total percentages of CD4 T cells were noted (Figure S3Ai). The patients in the irAE-negative groups were selected to match the cancer types to patients in the arthritis, pneumonitis, and thyroiditis groups (Figure S3Aii). Although patients with thyroiditis had higher levels of CD4 T cells compared with patients with pneumonitis and arthritis, these differences were not significant (Figure S3Aii). Interestingly, patients with multiple organs afflicted by irAEs had lower levels of CD4 T cells than patients with no clinical toxicities (Figure S3Aiii), and by multiple comparison analysis, we show that this was statistically significant in one of the comparative components (Figure S3Aiv).
Figure 3.

Patients with immune-related arthritis have higher TH1/2 cells and lower percentages of naive CD4 T cells at baseline
(A) Representative KNetL plot of annotated CD4 T cell clusters.
(B) Mean percentage of cells in six CD4 T cell subsets across the different clinical outcomes: arthritis, pneumonitis, and thyroiditis at baseline (unpaired t test).
(C) Volcano plot highlighting the up-regulated and down-regulated genes in cluster 25 of patients with arthritis, with paired-dot plots comparing the RNA expression of upregulated genes between no-irAE and arthritis groups.
(D and E) Gene Ontology (GO) analysis for differential genes present in cluster 25 from patients with arthritis for enriched terms from Immune.MSigDB (D) and from the GWAS Catalog 2019 (E).
(F) Pathway analysis of differentially expressed genes in cluster 25 from patients with arthritis using integrative KEGG-String platform.
Statistical significance for unpaired comparisons was performed by Student’s t test applying Mann-Whitney test. Data are presented as mean ± SEM. p value, exact, two-tailed, the center lines denote the mean of SEM. ∗p < 0.05, ∗∗∗∗p < 0.0001.
Analyzing the distribution of T cells in the different CD4 clusters revealed no differences at baseline between patients with irAEs and no irAEs (Figure S3B). However, stratifying the data by organ-specific irAEs, it became clear that patients with arthritis had significantly more TH1/2 CXCR3+GATA3+ cells (cluster 25), more TREG SELL+ cells (cluster 11), fewer TH JUND+ cells (cluster 23), and fewer naive T (TN) TCF7+LEF1+ cells (cluster 22) at baseline than patients from the corresponding irAE-negative group (Figure 3B). Differences in the percentages of these clusters did not hold for patients with pneumonitis and thyroiditis. These differences in subset percentages were not due to the gender or type of ICIs used (Figure S3C). These data suggest that enhanced T cell subset transition from naive to effector cells, specifically in patients who developed arthritis, can serve as evidence of pervasive immune responsiveness, as reflected by the predominance of later developmental stages.
The most significant baseline cluster that differentiated patients with arthritis from patients that did not develop irAEs was 25, TH1/2 CXCR3+GATA3+. Further analysis of the genes that defined this specific cluster revealed significant upregulation of inflammatory genes such as IL32, IL2RG, and AIRE (Figure 3C). Performing ontology analysis on upregulated genes in TH1/2 CXCR3+ GATA3+ of patients with arthritis revealed a significant overlap with genes related to autoimmune and other activated CD4 T cell conditions (Figure 3D). Moreover, through browsing a genome-wide association study (GWAS) catalog, we discovered that the same upregulated genes that characterized cluster 25 were overrepresented also in T cells from patients with other autoimmune diseases (Figure 3E), suggesting a potential mechanistic role in the pathogenesis of irAEs. Finally, through KEGG-Strings network analysis, we discovered that most the genes that defined cluster 25 were associated with specific signaling pathways that were previously shown to be involved in inflammatory arthritis (Figure 3F).
Selected subsets of CD4 TH cells are associated with organ-specific irAEs
The analysis of cluster 14 (Figure S4A) revealed that in patients with thyroiditis, a higher percentage of CD4 TH cells expressed high levels of RORC and interleukin-21 (IL-21) (Figure 4A). This expression pattern suggested that cluster 14 was a meta-cluster that contained cells with interesting features other than RORC expression (Figure S4B). To gain a better insight into this population, we subclustered the cells of cluster 14 into 6 additional subclusters (Figure 4B). Subclusters sC3, sC4, sC5, and sC6 expressed genes that were strongly associated with TH2 cells (Figures 4B and 4C). Likewise, subclusters sC1 and sC2 conveyed genes correlated with TH17 cells and genes associated with pathogenic features highlighted by the expression of IL-23R and CSF2 (Figures 4B and 4C).21,22 Most striking was the fact that while patients with arthritis had low sC3 cells (TH2 TCF7+) at baseline, patients with pneumonitis collectively had more sC4 T cells (TH2 CCR7+), and patients with thyroiditis had more sC1 T (TH17 SLAMF8+) and sC2 T cells (TH17 KRT27+), suggesting that different cellular subclusters are associated with distinct organ-specific irAEs (Figure 4D, S4A, and S4B). In terms of marker gene association, KLF6, a gene distinctive to the TH2 JUN+ cells, correlated to some degree with the canonical TH2 signature score (CD4, GATA3, CXCR4, and AREG). The correlation coefficient was not strong, but nevertheless, higher mRNA expression KLF6 was noted in patients with pneumonitis (Figure 4E). Similarly, S100B and SIGLEC14, marker genes characteristic of TH17 KRT27+ cells in the thyroiditis group, correlated with the TH17 signature score (CD4, RORC, and IL-23R) (Figure 4F). The ability to genetically differentiate between the types of irAEs is demonstrated through a heatmap and a Venn diagram showing 1,011, 1,162, and 628 genes distinctive to thyroiditis, pneumonitis, and arthritis, respectively (Figure S5A) This finding is also validated through principal-component analysis (PCA). Further, the set of differentially expressed genes between the three clinical groups was found to collectively enrich for CD4 subsets associated with activation phenotype (Figure S5B).
Patients with inflammatory arthritis have lower levels of CD8 TCM cells at baseline
Six CD8 T cell clusters were annotated using the KNetL plot (Figure 5A). There were no differences in the proportion of total CD8 T cells between the baseline and the on-treatment groups (Figure S6Ai). The patients in the irAE-negative group were selected to match cancer types with the arthritis, pneumonitis, and thyroiditis groups (Figure S6Aii). Patients with pneumonitis and thyroiditis had lower, but not significant, levels of CD8 T cells at baseline and on treatment compared with patients with arthritis (Figure S6Aii). When analyzing patients with multi-organ irAEs, the levels of CD8 T cells were higher both at baseline and on treatment with ICIs compared with patients with single-organ irAEs (Figures S6Aiii and S6Aiv).
Figure 5.

Patients with inflammatory arthritis have lower levels of central memory CD8 T cells at baseline
(A) Representative KNetL plot of annotated CD8 T cell clusters.
(B) Mean percentage of cells in six CD8 T cell subsets across the different clinical outcomes: arthritis, pneumonitis, and thyroiditis at baseline (unpaired t test).
(C) Volcano plot highlighting the up-regulated and down-regulated genes in TCM CXCR3+ cells in patients with no irAEs.
(D) GO analysis for differential genes present in TCM CXCR3+ cells from patients with no irAEs for enriched terms from Immune.MSigDB.
(E) Line graph showing the comparison of T cell states between cohorts of patients with no irAEs and arthritis based on the T suppressor cell signature score.
(F) Pathway analysis of differentially expressed genes in TCM CXCR3+ cells from patients with no irAEs using integrative KEGG-String platform.
(G) Quantification of flow cytometry data of 19 patients, showing percentages of naive, central memory (CM), effector memory (EM), and terminally differentiated T (TEMRA) cells at baseline.
Statistical significance for unpaired comparisons was performed by Student’s t test applying Mann-Whitney test. Data are presented as mean ± SEM. p value, exact, two-tailed, the center lines denote the mean of SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
To better identify CD8 T cell clusters that could differentiate between patients with or without irAEs, we analyzed the distribution of cells within each cluster compared with the recruitment parameters. Patients with irAEs had less CXCR3-expressing central memory-like T cells (cluster 4) at baseline and on treatment (Figure S6B). No major differences in the cluster distribution were observed with the other population of cells. However, patients with arthritis had extremely low levels of TCM cells (cluster 4) at baseline, suggesting that the evaluation of this cluster could serve as a clinical predictive biomarker (Figure 5B). Interestingly, the same patients had more effector (cluster 16) and effector memory (cluster 19) T cells at baseline. The patients with pneumonitis had fewer cells with features of effector memory T cells (cluster 19) than the patients who did not develop toxicity. The patients who developed thyroiditis had non-significant lower levels of effector cells (cluster 16) at baseline (Figure 5B). No other differences in the distribution of the CD8 T cell clusters were observed in the on-treatment group (Figure S6B). There were no differences in cluster C16 and cluster C4 based on the type of drug given or the gender of the patients (Figure S6C).
To better understand cluster 4, we show by volcano plot that most of the differentially expressed genes derived from patients did not developed arthritis (Figure 5C). These genes were subjected to Gene Ontology analysis and, interestingly, were associated with regulatory functions (Figure 5D). The contribution of this cluster to overall T cell suppression phenotype is further demonstrated through comparison of T cell suppressor signature scores between patients with arthritis and no irAEs (Figure 5E). Like other central memory populations, the genes in this cluster were associated with IL-3 and IL-5 secretion24 and class I signaling25 (Figure 5F).
To validate the observation that patients with arthritis had low CD8 TCM cells at baseline at the protein level, we analyzed by flow cytometry T cells collected for a second cohort of 16 patients treated with PD-1 blockade (Figures 1A and 5G). Consistent with the analysis based on gene transcription, patients who developed arthritis had less naive and CD8 TCM cells and more effector memory and terminally differentiated CD8 T cells, suggesting an enhanced CD8 maturation in patients who developed this toxicity (Figure 5G).
Patients with immune-related pneumonitis have distinctive distributions of T cell populations
Because pneumonitis is one of the most morbid complications of ICI treatment and our cohort included a relatively high proportion of patients who went on to develop this AE, we wondered whether distinct baseline T cell features might associate with distinct subtypes of pneumonitis. Careful radiological analysis of serial chest computed tomography (CT) scans performed on the nine patients with pneumonitis (Figures S7A and S7B) revealed two clear subtypes: chronic hyperintensity pneumonitis (CHP), characterized by traction bronchiectasis and honeycombing, and organized pneumonia (OP), defined by patchy consolidation with a predominantly subpleural and peri-bronchial distribution (Figure 6A).26 Moreover, the T cell cluster distribution (Figure 6B) of patients with CHP (Figure 6C) was distinct from that of patients with OP (Figure 6D). At baseline, unlike the patients with OP, patients with CHP had low levels of cells in clusters 4 (characterized by features of CD8 TCM CXCR3+ cells), 3 (double-positive T cells), and 10 (gamma delta T cells) and high levels of cells in clusters 22 (naïve-like CD4 TN TCF7+LEF1+) and 25 (CD4 TH1/2 CXCR3+GATA3+) (Figure 6E). Evaluation of the expression levels of genes exclusive to patients with CHP versus OP over the same subset of cells further supports the ability of this signature to differentiate between the two clinical presentations (Figure 6F). These findings suggest that the two types of pneumonitis were likely mediated by different populations of T cells. Moreover, this strong radiographical-immunological correlation supports the ability of scRNA-seq approach not just to annotate different subsets of T cells among patients with irAEs but also to uncover the potential contribution of specific T cell populations to clinical pathogenesis.
Figure 6.

Patients with immune-related pneumonitis have distinctive distributions of T cell populations
(A) Representative chest CT scans of patients with chronic hypersensitive pneumonitis (CHP) and organized pneumonia (OP) at baseline and on treatment.
(B) Representative KNetL plot indicating key clusters to be used to distinguish the pneumonitis groups.
(C) Cells of patients with CHP are enriched in clusters C10, C3, and C4.
(D) Cells of patients with OP are depleted in clusters of C22 and C25.
(E) Quantification of the mean percentage differences in CD3 T cell subsets among the different pneumonitis groups.
(F) Heatmap showing the RNA expression of differentially expressed genes between CHP and OP groups within the clusters C10, C22, and C25.
Statistical significance for unpaired comparisons was performed by Student’s t test applying Mann-Whitney test. Data are presented as mean ± SEM. p value, exact, two-tailed, the center lines denote the mean of SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Discussion
Immune checkpoints are T cell-surface-expressed inhibitory receptors that prevent excessive T cell responses. Tumor cells have evolved to usurp those inhibitory mechanisms to prevent T cell mediated tumor killing. Initially, the immune system recognizes and eliminates transformed cancerous cells prior to their development into tumors. However, during the process of carcinogenesis, tumor cells progressively express multiple inhibitory receptor ligands that will prevent T cell recognition. Consequently, therapeutic blockade of these checkpoints or their ligands with ICIs helps recover anti-tumor immunity. Since 2013, ICIs have been increasingly considered as targets for cancer immunotherapies due to the effectiveness of drugs blocking the inhibitory receptors CTLA-4, PD-1, and PD-L1.27,28,29,30 The PD-1-PD-L1 interaction directly inhibits anti-tumor T cells, promotes peripheral TE cell exhaustion, and supports the conversion of TE cells into suppressive TREG cells. Based on prolonged overall survival in clinical trials, Abs inhibiting PD-1 and PD-L1 have been approved for multiple clinical indications, including melanoma, non-small cell lung cancer, head and neck squamous cell carcinoma, urothelial carcinoma, renal cell carcinoma, Hodgkin’s lymphoma, and other adult and pediatric solid tumors.
One in every two patients with cancer treated with ICIs targeting PD-1 will develop a side effect fitting within a spectrum termed irAEs. This broad array of inflammatory AEs affects different organ systems, including the skin, gastrointestinal tract, liver, endocrine organs, lungs, and joints. irAEs can develop at any time during the treatment, even months after discontinuing the treatment with the checkpoint blockade. The absence of predictive factors makes pre-treatment risk stratification extremely challenging on an individual patient level, as scant information exists to inform the type and severity of toxicity he or she may experience with treatment. Additionally, since the onset of irAEs is often sudden, and even fatal toxicities may occur, it is essential that clinicians recognize and manage the events early. The frequency of irAEs can be broken down by the type of ICIs received. A recent meta-analysis of irAE frequencies over multiple trials revealed that irAEs occurred in 74% of patients with cancer treated with anti-PD-1 or PD-L1 Abs; 89% of patients treated with anti-CTLA-4; 90% of patients receiving ICI combinations; and 89% of patients receiving ICIs with chemotherapy.30 irAEs with grade 3 (severe) or grade 4 (life threatening) were reported in 14% of patients treated with PD-1 or PD-L1 inhibitors; 34% of patients treated with anti-CTLA-4 Abs; 55% of patients treated with ICI combinations; and 46% of patients treated with ICIs + chemotherapy agents. The rates of irAEs leading to treatment withdrawal were 6% after using the PD-1 or PD-L1 inhibitors; 21% for anti-CTLA-4 Abs; 38% for ICI combinations; and 13% for combinations with chemotherapy.
We identified that at baseline, patients with arthritis had significantly less cells with features of CD8 TCM cells, patients with pneumonitis had more CD4 TH2 cells, and patients with thyroiditis had more CD4 TH17 cells when compared with patients who did not develop irAEs. These data support our hypothesis that different populations of T cells are associated with different irAEs and that quantification and characterization of these populations of T cell pre-treatment could serve as a toxicity-specific predictive biomarker. Quantification of these population of cells by means of larger studies using flow cytometry and RT-PCR are needed to further validate and translate our exploratory findings to the clinic.
In a previous study, gene expression profiling was performed on whole-blood samples from patients with melanoma to discover gene expression differences at baseline between patients who developed gastrointestinal toxicity following anti-CTLA-4 Ab treatment.31 A more recent study documented T cell characteristics associated with irAEs in patients with melanoma using variety of single-cell technologies.32 This study found that the pre-treatment levels of activated CD4 memory T cell and TCR diversity were associated with irAE development regardless of organ system involvement. This study complements our data and further support the concept that pre-treatment T cell characterization should predict irAE onset. This could be cancer-type dependent, where memory CD4 T cells are predictive in patients with melanoma while CD8 effecter T cells are associated with a lung cancer population. A recent work applied scRNA-seq to study peripheral blood samples from patients with melanoma treated with ICIs and discovered that pre-treatment-activated memory CD4 T cell abundance was associated with severe irAEs.32 Another, more recent study analyzed peripheral blood from patients with arthritis irAEs and uncovered a correlation between the arthritis and a population of CD8 CX3CR1hi T cells. Furthermore, and like our study, a subset of patients with irAEs had enhanced TH1 and TH17 gene signatures.33 These studies, as well as our own work, support the concept that circulating T cells are associated with ICI-induced toxicity, with potential implications for improved diagnostics and clinical management.
To summarize, we discovered correlations between the presence of specific subsets of T cells at baseline and the development of arthritis, pneumonitis, and thyroiditis AEs. These exciting findings suggest that quantification of these populations of T cells before the treatment has the potential to predict who will develop irAEs and position the clinical team to initiate earlier immunomodulatory therapy. The ability to predict irAEs is critically important to improve the effectiveness and the usability of cancer immunotherapies. Through our ongoing efforts, we will translate our findings to the clinic by developing a personalized flow cytometry- and RT-PCR-based tool to prove the premise that quantification of these population of cells could accurately predict irAEs, altogether enabling many more patients with cancer to safely receive immunotherapies.
Limitations of the study
Our study is not free of limitations.34 One limitation is the fact that this was a single-center, retrospective, case control study of 40 patients, and the incidence and types of irAEs in our institute might be different from other locations. Another limitation is the relatively small number of patients with each type of irAE. With these numbers, we discovered T cell populations and subsets that were associated with specific irAEs. In order to validate these subsets of T cells as a biomarker to predict irAEs, additional patients must be recruited. One of the interesting findings of our study was the lack of differences in naive, TH, or TE cell population percentages between the patients that were treated with anti-PD-1 or anti-PD-L1. On the one hand, this is not unexpected since the type, incidence, and severity of irAEs are similar between these groups. On the other hand, it is unlikely that these drugs utilize the same molecular mechanism to trigger irAEs.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| APC/Cyanine7 anti-human CD3 (clone HIT3A) | BioLegend | Cat# 300318; RRID: AB_314054 |
| Alexa Fluor® 700 anti-human CD4 (clone RPA-T4) | BioLegend | Cat# 300526; RRID: AB_493743 |
| PE/Cyanine7 anti-human CD8a (clone RPA-T8) | BioLegend | Cat# 301012; RRID: AB_314130 |
| Brilliant Violet 605™ anti-human CD45RA (clone HI100) | BioLegend | Cat# 304134; RRID: AB_11126164 |
| Alexa Fluor® 488 anti-human CD27 Recombinant antibody | BioLegend | Cat# 393204; RRID: AB_2750088 |
| BD Horizon™ BUV496 Mouse Anti-Human CD8 | BD Biosciences | Cat# 612942; RRID: AB_2870223 |
| BD Horizon™ BUV805 Mouse Anti-Human CD3 | BD Biosciences | Cat# 612895; RRID: AB_2870183 |
| BD Horizon™ BV750 Mouse Anti-Human CD4 | BD Biosciences | Cat# 566355; RRID: AB_2744426 |
| BD OptiBuild™ BUV395 Mouse Anti-Human CD45RA | BD Biosciences | Cat# 740298; RRID: AB_2740037 |
| BD Horizon™ BV786 Rat Anti-Human CCR7 (CD197) | BD Biosciences | Cat# 563710; RRID: AB_2738384 |
| CITEseq antibodies | ||
| TotalSeq™-C0072 anti-human CD4 Antibody | BioLegend | Cat# 300567; RRID: AB_2800725 |
| TotalSeq™-C0046 anti-human CD8 Antibody | BioLegend | Cat# 344753; RRID: AB_2800922 |
| TotalSeq™-C0154 anti-human CD27 Antibody | BioLegend | Cat# 302853; RRID: AB_2800747 |
| TotalSeq™-C0063 anti-human CD45RA Antibody | BioLegend | Cat# 304163; RRID: AB_2800747 |
| Chemicals, peptides, and recombinant proteins | ||
| DNase I | STEMCELL Technologies | Cat# 7470 |
| Zombie UV™ Fixable Viability Kit | BioLegend | Cat# 423107 |
| Critical commercial assays | ||
| Chromium Single Cell 5′ Library & Gel Bead Kit | 10x Genomics | Cat# 1000006 |
| RNA Isolation Kit | Qiagen | Cat# 74104 |
| Software and algorithms | ||
| FlowJo 10.1r7 | https://www.flowjo.com | SCR_008520 |
| Single cell RNAseq analysis | https://github.com/rezakj/iCellR | N/A |
| GraphPad Prism 9 | https://www.graphpad.com | SCR_002798 |
| InnateDB | https://www.innatedb.com | SCR_006714 |
| ShinyGO v0.76 | http://bioinformatics.sdstate.edu/go | SCR_019213 |
| Enrichr | https://maayanlab.cloud/Enrichr | SCR_001575 |
| Cell Ranger | https://support.10xgenomics.com/single-cell-vdj/software/pipelines/latest/what-is-cell-ranger | SCR_017344 |
| Deposited data | ||
| RNA sequencing data | https://www.ncbi.nlm.nih.gov/geo | GSE159774 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be direct to and will be fulfilled by the lead contact, Adam Mor, M.D., Ph.D. (am5121@cumc.columbia.edu).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
The expedient model in this work was limited to human subjects. The average age of the patients was 70 years old, 45% were males and 55% were females. All the patients were diagnosed with solid malignancies and were treated with immune checkpoint inhibits in our institute. This study protocol was approved and oversighted by our institute (IRB protocol IRB#AAA-O5706).
Method details
Patient recruitment
We collected blood from cancer patients prior to anti-PD-1 therapy (Nivolumab or Pembrolizumab), anti-PD-L1 (Atezolizumab), and anti-CTLA-4 (Ipilimumab) initiation and at the time of irAEs onset according to our experimental workflow (Figure 1). Twenty-four immunotherapy-naïve cancer patients were consented according to IRB protocol IRB#AAA-O5706/L5871. We collected information about treatment regimens, duration of therapy, progression-free survival, and overall survival for all patients. We additionally collected information on documented irAE emergence and treatment. Inclusion criteria included histologically confirmed cancer, immunotherapy-naïve, and planned initiation of immunotherapy as an immediate next step in therapy. Major exclusion criteria were active infection, history of autoimmune disease, immune suppressive therapy, active pneumonitis or pulmonary fibrosis, or recent administration of live-virus vaccines. We collected 10 mL of blood from each patient in EDTA vacutainers prior to initiation of the treatment. Additional 10 mL peripheral blood were collected at the time of first irAEs presentation. For our control group, cancer patients who did not develop irAEs, the timing of the second blood collection was matched to the number of weeks following anti-PD-1 therapy initiation at which blood was collected from the irAEs patients.
General reagents
RPMI 1640 medium, Dulbecco’s PBS, and FBS were purchased from Life Technologies.
Cell isolation and preparation for sequencing
Red blood cells were lysed by resuspending cell pellets in ACK Lysis Buffer (Gibco) for 2 min, followed by washing with cold PBS. We used RosetteSep T cell enrichment kit (StemCell) to isolate untouched CD3 T cells. Each sample was washed once with RPMI and 3 times with PBS +2% BSA before labeling and pooling. Specimens were then filtered through 70 um strainers (Fisher). Cell concentration, singularity, and viability were confirmed with a hematocytometer before submission for scRNA-Seq (10X Genomics). Cells were processed for scRNAseq by employing CITEseq methodology using multiplexed cell surface markers using labeled with a cocktail of oligonucleotide tagged Total-Seq anti-human antibodies (BioLegend) against CD4, CD8, CD45RA and CD27. 17,000 antibody labeled cells, from each sample, were loaded on Chromium microfluidics chip for single cell/gel bead encapsulation, barcoding and reverse transcription according to the manufacturer’s recommendations. After cDNA amplification, <180bp nucleotide fragments containing antibody-derived tags (ADTs) and >300bp nucleotide fragments containing mRNA-derived cDNA will be size separated as previously described. 5′ single cell sequencing library was generated using a tagmentation-based approach and according to manufacturer’s recommendations. ADTs and cDNA libraries were merged and subjected to Illumina 150bp paired-end sequencing.
Data analysis
Quality controls included calculation of the number of genes, UMIs, and the proportion of mitochondrial genes for each cell. Cells with a low number of covered genes (gene-count <500) or high mitochondrial counts (mt-genes >0.2) were filtered out, and the matrix was normalized based on library size. A general statistical test was performed to calculate gene dispersion, base mean, and cell coverage. iCellR, an R package (v1.5.5) (https://CRAN.R-project.org/package=iCellR) and genes with high dispersion and coverage (2000 genes) were used to perform Principal Component Analysis (PCA) and batch alignment. Uniform Manifold Approximation and Projection (UMAP) and K-nearest-neighbor-based Network graph drawing Layout (KNetL) were then performed. KNetL map has a zoom option which allowed us to see variable levels of detail (more or fewer sub-populations in cell communities). In the study, we used a zoom of 500. The network layout used in KNetL map was force-based, and the zoom option changed the force in the system. Force-directed graph drawing algorithms assign attractive (analogous to spring force) and repulsive forces (usually described as analogous to the forces in atomic particles) to separate all pairs of nodes. PhenoGraph clustering was then performed on the KNetL map results, and marker genes were found for each cluster and visualized on heatmaps, bar plots, and boxplots as indicate. Marker genes were then used to assign cell types. Imputation was used for data visualizations only and not for the analysis. Signature enrichment scoring was generated by calculating z-scores relative to naive T cells or by the scoring method described by Tirosh et al.,32 using i.score function on the main data. Gene Ontology analysis of immune cell signatures and disease catalogs were collectively performed using innateDB (https://www.innatedb.com), ShinyGO v0.76 (http://bioinformatics.sdstate.edu/go) and Enrichr (https://maayanlab.cloud/Enrichr). Overlayed network analysis and pathway analysis was performed by innateDB (https://www.innatedb.com).
Flow cytometry
For protein expression analysis following isolation cells were collected and stained with the following antibodies for surface protein expression: CD3 APC-Cy7 (BioLegend), CD4 AF-700 (BioLegend), CD8 PE-Cy7 (BioLegend), CD45RA BV-605 (BioLegend), CD27 AF-488 (BioLegend), CD8 BUV-496 (BD Biosciences), CD3 BUV-805 (BD Biosciences), CD4 BV-750 (BD Biosciences), CD45RA BUV-395 (BD Biosciences), and CCR7 BV-786 (BD Biosciences). Dead cells were excluded from the analysis by using Zombie-UV (Biolegend). Doublets and double-positive CD4/CD8 cells were removed through sequential gating. Flow cytometry acquisition was done using the BD LSRII with BD FACSDiva. Data was analyzed by FlowJo 10.1r7 and GraphPad Prism 9.
CT scan
Computed-tomography scans were performed per standard of care before and during immunotherapy administration. Scans from patients who experienced pneumonitis were reviewed and annotated by an experienced thoracic radiologist.
Quantification and statistical analysis
Unless otherwise specified, the data are presented as mean ± standard error of the mean. Statistical significance was determined using Student’s t test, 1-way ANOVA, or log rank test, as indicated. Statistical analyses were performed using Prism 9 (GraphPad Software). Significance was set at p = 0.05. To measure the variance between each patient at baseline to on treatment condition, un-paired non-parametric test was applied wherever indicated. To compute the correlation between signature enrichment scores, a non-parametric spearman correlation was applied choosing two-tailed p value with a confidence interval of 95%, where indicated. For this observational study, our sample size calculation of n = 24 subjects per time point was based on published RNA-seq data assuming that the average read count among the prognostic genes in the control group is 100, the maximum dispersion is 0.15, and the ratio of the geometric mean of normalization factors is 1. Our minimum effect size was 2 and we were able to reject the null hypothesis that the population means of the two groups are equal with an estimated power of 0.98. The FDR associated with this test is 0.01.
Acknowledgments
We would like to acknowledge Dr. Michael Peled and Dr. Jair Bar (Tel Aviv University) for providing blood samples and Dr. Faizan Danish for assisting with the statistical analysis. We would like to thank the expertise and assistance of Darwin D'Souza and Travis Dawson of the Human Immune Monitoring Center (HIMC) at Mount Sinai Icahn School of Medicine. This work was supported by grants from the NIH (AI125640, CA231277, and AI150597); the Cancer Research Institute; the Lisa Baker Autoimmunity Innovation Fund; and the V Foundation. We would like to thank the Genome Technology Center (GTC) for expert library preparation and sequencing and the Applied Bioinformatics Laboratories (ABL) for providing bioinformatics support and helping with the analysis and interpretation of the data. The GTC and ABL are shared resources partially supported by Cancer Center Support Grant P30CA016087 at the Laura and Isaac Perlmutter Cancer Center. This work has used computing resources at the NYU School of Medicine High Performance Computing (HPC) Facility. Research reported in this publication was also performed in the CCTI Flow Cytometry Core, supported in part by the Office of the Director, National Institutes of Health under awards S10RR027050 and S10OD020056.
Author contributions
Conception and design, S.B., N.R., and A.M.; development of methodology, S.B., K.A., R.J.W., A.T., S.L., and A.M.; acquisition of data, S.B., R.M., S.L.R., and A.M.; analysis and interpretation of data, S.B., R.J.W., Z.L., A.K.-J., A.T., G.G.L., M.M.S., and A.M.; writing – review and/or revision of the manuscript, S.B. and A.M.; technical or material support, B.S.H., M.M.S., G.G.L., M.C.D., M.M., N.R., M.S., and K.A.; study supervision, B.S.H. and A.M.
Declaration of interests
The authors declare no competing interests.
Published: December 12, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2022.100868.
Supplemental information
Data and code availability
Datasets generated in this study have been uploaded to GEO database: accession number GSE216329. This study reports original code: https://github.com/rezakj/iCellR. Any additional information required to reanalyze the data reported in this study is available from the lead contact upon request.
References
- 1.Hamid O., Robert C., Daud A., Hodi F.S., Hwu W.-J., Kefford R., Wolchok J.D., Hersey P., Joseph R.W., Weber J.S., Ribas A. Safety and tumor responses with lambrolizumab (Anti–PD-1) in melanoma. N. Engl. J. Med. 2013;369:134–144. doi: 10.1056/NEJMoa1305133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rizvi N.A., Mazières J., Planchard D., Stinchcombe T.E., Dy G.K., Antonia S.J., Horn L., Lena H., Minenza E., Mennecier B., et al. Activity and safety of nivolumab, an anti-PD-1 immune checkpoint inhibitor, for patients with advanced, refractory squamous non-small-cell lung cancer (CheckMate 063): a phase 2, single-arm trial. Lancet Oncol. 2015;16:257–265. doi: 10.1016/S1470-2045(15)70054-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hodi F.S., O'Day S.J., McDermott D.F., Weber R.W., Sosman J.A., Haanen J.B., Gonzalez R., Robert C., Schadendorf D., Hassel J.C., et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010;363:711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Postow M.A., Chesney J., Pavlick A.C., Robert C., Grossmann K., McDermott D., Linette G.P., Meyer N., Giguere J.K., Agarwala S.S., et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N. Engl. J. Med. 2015;372:2006–2017. doi: 10.1056/NEJMoa1414428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weber J.S., D'Angelo S.P., Minor D., Hodi F.S., Gutzmer R., Neyns B., Hoeller C., Khushalani N.I., Miller W.H., Jr., Lao C.D., et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2015;16:375–384. doi: 10.1016/S1470-2045(15)70076-8. [DOI] [PubMed] [Google Scholar]
- 6.Borcoman E., Kanjanapan Y., Champiat S., Kato S., Servois V., Kurzrock R., Goel S., Bedard P., Le Tourneau C. Novel patterns of response under immunotherapy. Ann. Oncol. 2019;30:385–396. doi: 10.1093/annonc/mdz003. [DOI] [PubMed] [Google Scholar]
- 7.de Miguel M., Calvo E. Clinical challenges of immune checkpoint inhibitors. Cancer Cell. 2020;38:326–333. doi: 10.1016/j.ccell.2020.07.004. [DOI] [PubMed] [Google Scholar]
- 8.Postow M.A., Sidlow R., Hellmann M.D. Immune-related adverse events associated with immune checkpoint blockade. N. Engl. J. Med. 2018;378:158–168. doi: 10.1056/NEJMra1703481. [DOI] [PubMed] [Google Scholar]
- 9.Dolladille C., Ederhy S., Sassier M., Cautela J., Thuny F., Cohen A.A., Fedrizzi S., Chrétien B., Da-Silva A., Plane A.F., et al. Immune checkpoint inhibitor rechallenge after immune-related adverse events in patients with cancer. JAMA Oncol. 2020;6:865–871. doi: 10.1001/jamaoncol.2020.0726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chang C.Y., Park H., Malone D.C., Wang C.Y., Wilson D.L., Yeh Y.M., Van Boemmel-Wegmann S., Lo-Ciganic W.H. Immune checkpoint inhibitors and immune-related adverse events in patients with advanced melanoma: a systematic review and network meta-analysis. JAMA Netw. Open. 2020;3 doi: 10.1001/jamanetworkopen.2020.1611. e201611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martins F., Sofiya L., Sykiotis G.P., Lamine F., Maillard M., Fraga M., Shabafrouz K., Ribi C., Cairoli A., Guex-Crosier Y., et al. Adverse effects of immune-checkpoint inhibitors: epidemiology, management and surveillance. Nat. Rev. Clin. Oncol. 2019;16:563–580. doi: 10.1038/s41571-019-0218-0. [DOI] [PubMed] [Google Scholar]
- 12.Da L., Teng Y., Wang N., Zaguirre K., Liu Y., Qi Y., Song F. Organ-specific immune-related adverse events associated with immune checkpoint inhibitor monotherapy versus combination therapy in cancer: a meta-analysis of randomized controlled trials. Front. Pharmacol. 2019;10:1671. doi: 10.3389/fphar.2019.01671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oh D.Y., Cham J., Zhang L., Fong G., Kwek S.S., Klinger M., Faham M., Fong L. Immune toxicities elicted by CTLA-4 blockade in cancer patients are associated with early diversification of the T-cell repertoire. Cancer Res. 2017;77:1322–1330. doi: 10.1158/0008-5472.CAN-16-2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yasuda Y., Iwama S., Sugiyama D., Okuji T., Kobayashi T., Ito M., Okada N., Enomoto A., Ito S., Yan Y., et al. CD4+ T cells are essential for the development of destructive thyroiditis induced by anti-PD-1 antibody in thyroglobulin-immunized mice. Sci. Transl. Med. 2021;13 doi: 10.1126/scitranslmed.abb7495. eabb7495. [DOI] [PubMed] [Google Scholar]
- 15.Tang K.H., Li S., Khodadadi-Jamayran A., Jen J., Han H., Guidry K., Chen T., Hao Y., Fedele C., Zebala J.A., et al. Combined inhibition of SHP2 and CXCR1/2 promotes antitumor T-cell response in NSCLC. Cancer Discov. 2021;12:47–61. doi: 10.1158/2159-8290.CD-21-0369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ho I.C., Tai T.S., Pai S.Y. GATA3 and the T-cell lineage: essential functions before and after T-helper-2-cell differentiation. Nat. Rev. Immunol. 2009;9:125–135. doi: 10.1038/nri2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Singh S.S., Chauhan S.B., Ng S.S., Corvino D., de Labastida Rivera F., Engel J.A., Waddell N., Mukhopadhay P., Johnston R.L., Koufariotis L.T., et al. Increased amphiregulin expression by CD4+ T cells from individuals with asymptomatic Leishmania donovaniinfection. Clin. Transl. Immunology. 2022;11 doi: 10.1002/cti2.1396. e1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.García-Sánchez A., Estravís M., Martin M.J., Pérez-Pazos J., Martín-García C., Gil-Melcón M., Ramos-González J., Eguiluz-Gracia I., Triviño J.C., Isidoro-García M., et al. PTGDR2 expression in peripheral blood as a potential biomarker in adult patients with asthma. J. Pers. Med. 2021;11:827. doi: 10.3390/jpm11090827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koetzier S.C., van Langelaar J., Wierenga-Wolf A.F., Melief M.J., Pol K., Musters S., Lubberts E., Dik W.A., Smolders J., van Luijn M.M. Improving glucocorticoid sensitivity of brain-homing CD4+ T helper cells by steroid hormone crosstalk. Front. Immunol. 2022;13 doi: 10.3389/fimmu.2022.893702. 893702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lam A.J., Lin D.T.S., Gillies J.K., Uday P., Pesenacker A.M., Kobor M.S., Levings M.K. Optimized CRISPR-mediated gene knockin reveals FOXP3-independent maintenance of human Treg identity. Cell Rep. 2021;36 doi: 10.1016/j.celrep.2021.109494. 109494. [DOI] [PubMed] [Google Scholar]
- 21.Lee Y., Awasthi A., Yosef N., Quintana F.J., Xiao S., Peters A., Wu C., Kleinewietfeld M., Kunder S., Hafler D.A., et al. Induction and molecular signature of pathogenic TH17 cells. Nat. Immunol. 2012;13:991–999. doi: 10.1038/ni.2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gaublomme J.T., Yosef N., Lee Y., Gertner R.S., Yang L.V., Wu C., Pandolfi P.P., Mak T., Satija R., Shalek A.K., et al. Single-cell genomics unveils critical regulators of Th17 cell pathogenicity. Cell. 2015;163:1400–1412. doi: 10.1016/j.cell.2015.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tirosh I., Izar B., Prakadan S.M., Wadsworth M.H., 2nd, Treacy D., Trombetta J.J., Rotem A., Rodman C., Lian C., Murphy G., et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science. 2016;352:189–196. doi: 10.1126/science.aad0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Selleri S., Deola S., Pos Z., Jin P., Worschech A., Slezak S.L., Rumio C., Panelli M.C., Maric D., Stroncek D.F., et al. GM-CSF/IL-3/IL-5 receptor common beta chain (CD131) expression as a biomarker of antigen-stimulated CD8+ T cells. J. Transl. Med. 2008 Apr 15;6:17. doi: 10.1186/1479-5876-6-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sasaki K., Moussawy M.A., Abou-Daya K.I., Macedo C., Hosni-Ahmed A., Liu S., Juya M., Zahorchak A.F., Metes D.M., Thomson A.W., et al. Activated-memory T cells influence naïve T cell fate: a noncytotoxic function of human CD8 T cells. Commun. Biol. 2022;5:634. doi: 10.1038/s42003-022-03596-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Raghu G., Meyer K.C. Cryptogenic organising pneumonia: current understanding of an enigmatic lung disease. Eur. Respir. Rev. 2021;30 doi: 10.1183/16000617.0094-2021. 210094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hayashi H., Nakagawa K. Combination therapy with PD-1 or PD-L1 inhibitors for cancer. Int. J. Clin. Oncol. 2020;25:818–830. doi: 10.1007/s10147-019-01548-1. [DOI] [PubMed] [Google Scholar]
- 28.Mahoney K.M., Freeman G.J., McDermott D.F. The next immune-checkpoint inhibitors: PD-1/PD-L1 blockade in melanoma. Clin. Ther. 2015;37:764–782. doi: 10.1016/j.clinthera.2015.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gide T.N., Quek C., Menzies A.M., Tasker A.T., Shang P., Holst J., Madore J., Lim S.Y., Velickovic R., Wongchenko M., et al. Distinct immune cell populations define response to anti-PD-1 monotherapy and anti-PD-1/anti-CTLA-4 combined therapy. Cancer Cell. 2019;35:238–255.e6. doi: 10.1016/j.ccell.2019.01.003. [DOI] [PubMed] [Google Scholar]
- 30.Hussaini S., Chehade R., Boldt R.G., Raphael J., Blanchette P., Maleki Vareki S., Fernandes R. Association between immune-related side effects and efficacy and benefit of immune checkpoint inhibitors - a systematic review and meta-analysis. Cancer Treat Rev. 2021;92 doi: 10.1016/j.ctrv.2020.102134. 102134. [DOI] [PubMed] [Google Scholar]
- 31.Shahabi V., Berman D., Chasalow S.D., Wang L., Tsuchihashi Z., Hu B., Panting L., Jure-Kunkel M., Ji R.R. Gene expression profiling of whole blood in ipilimumab-treated patients for identification of potential biomarkers of immune-related gastrointestinal adverse events. J. Transl. Med. 2013;11:75. doi: 10.1186/1479-5876-11-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lozano A.X., Chaudhuri A.A., Nene A., Bacchiocchi A., Earland N., Vesely M.D., Usmani A., Turner B.E., Steen C.B., Luca B.A., et al. T cell characteristics associated with toxicity to immune checkpoint blockade in patients with melanoma. Nat. Med. 2022;28:353–362. doi: 10.1038/s41591-021-01623-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim S.T., Chu Y., Misoi M., Suarez-Almazor M.E., Tayar J.H., Lu H., Buni M., Kramer J., Rodriguez E., Hussain Z., et al. Distinct molecular and immune hallmarks of inflammatory arthritis induced by immune checkpoint inhibitors for cancer therapy. Nat. Commun. 2022;13:1970. doi: 10.1038/s41467-022-29539-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mor A., Strazza M. Bridging the gap: connecting the mechanisms of immune-related adverse events and autoimmunity through PD-1. Front. Cell Dev. Biol. 2021;9 doi: 10.3389/fcell.2021.790386. 790386. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Datasets generated in this study have been uploaded to GEO database: accession number GSE216329. This study reports original code: https://github.com/rezakj/iCellR. Any additional information required to reanalyze the data reported in this study is available from the lead contact upon request.