

Summary
Immunological protection of transplanted stem cell-derived islet (SC-islet) cells is yet to be achieved without chronic immunosuppression or encapsulation. Existing genetic engineering approaches to produce immune-evasive SC-islet cells have so far shown variable results. Here, we show that targeting human leukocyte antigens (HLAs) and PD-L1 alone does not sufficiently protect SC-islet cells from xenograft (xeno)- or allograft (allo)-rejection. As an addition to these approaches, we genetically engineer SC-islet cells to secrete the cytokines interleukin-10 (IL-10), transforming growth factor β (TGF-β), and modified IL-2 such that they promote a tolerogenic local microenvironment by recruiting regulatory T cells (Tregs) to the islet grafts. Cytokine-secreting human SC-β cells resist xeno-rejection and correct diabetes for up to 8 weeks post-transplantation in non-obese diabetic (NOD) mice. Thus, genetically engineering human embryonic SCs (hESCs) to induce a tolerogenic local microenvironment represents a promising approach to provide SC-islet cells as a cell replacement therapy for diabetes without the requirement for encapsulation or immunosuppression.
Keywords: diabetes, cell replacement, immune evasion, immune tolerance, cell engineering, stem cells
Graphical abstract
Highlights
-
•
Stem cell-derived islets (SC-islets) possess an inhibitory NK cell ligand profile
-
•
Immune-evasive SC-islets are rapidly rejected upon xeno-transplantation
-
•
Immune-evasive SC-islets reverse diabetes and are slowly rejected in humanized mice
-
•
Immune-tolerizing SC-islets reverse diabetes and resist xeno-rejection in NOD mice
Currently, cell replacement therapy for type 1 diabetes is limited by the requirement for lifelong immunosuppression. Gerace et al. show that engineering stem cell-derived islets as vehicles of localized immune tolerance may be an approach to address this yet unanswered challenge.
Introduction
Type 1 diabetes (T1D) is an autoimmune disease that results in the destruction of the insulin-producing beta cells of the pancreas.1 Cadaveric whole pancreas or islet transplantation are successful treatments for T1D; however, these are hampered by the limited number of donors and the requirement for lifelong immunosuppression.2 To address the shortage of islet material, several protocols have been developed to steer the in vitro differentiation of human induced pluripotent stem cells (iPSCs) into functional stem cell-derived islets (SC-islets) that include glucose-response beta cells.3,4,5,6,7,8,9
Current strategies to protect allografted islet cells include systemic immunosuppression to modify the patient’s immune system10,11 and encapsulation.12,13 More recently, genetic engineering of SC-islets to evade immune recognition has been attempted.14,15,16 While this is a promising alternative, current engineering strategies are limited by transgene silencing and recurrent immune rejection.17,18,19 Here, we generated immune-evasive SC-islet cells that over-express PD-L1 and the human leukocyte antigen (HLA)-E long-chain fusion from the constitutively expressed GAPDH locus in both HLA-competent and -deficient settings.20,21 We also evaluated the induction of localized immune tolerance as a strategy to protect SC-islets. To achieve this, we engineered SC-islets to secrete a combination of tolerogenic cytokines including the interleukin-2 (IL-2) mutein (N88D),22,23 IL-10, and transforming growth factor β (TGF-β).24 Remarkably, immune-tolerizing SC-islets normalized hyperglycemia and provided protection from xenograft (xeno) rejection for up to 8 weeks after transplantation in non-obese diabetic (NOD) mice. Thus, immune-tolerizing SC-islet cells represent another step forward in providing a source of islets for the treatment of T1D with the long-term aim of eliminating the need for encapsulation or systemic immunosuppression.
Results
Engineering immune-evasive SC-islet cells
Since GAPDH is constitutively expressed in all cells of the human islet (Figures S1A and S1B), we targeted the expression of PD-L1 and the HLA-E long-chain fusion to the GAPDH locus of human embryonic SCs (hESCs) and used luminescence as a reporter of cell viability. We chose to over-express PD-L1 as it was previously shown to protect SC-islet cells from xeno-rejection,15 while the HLA-E long-chain fusion inhibits natural killer (NK) cells in an HLA-deficient context.25,26,27,28 GAPDH-targeting plasmids were modified to include PD-L1 or HLA-E (Figures S1C–S1E) and used to create five hESC lines (Figure 1A): wild type (WT), HLA-deficient (B2M−/−), PD-L1-expressing (PD-L1), HLA-deficient/PD-L1-expressing (B2P), and HLA-deficient/HLA-E-expressing (BEC). All five gene-modified hESC lines successfully differentiated into Nkx6.1+/C-peptide+ SC-β cells as assessed by flow cytometry (Figures 1B and S1F). After interferon γ (IFN-γ) stimulation, HLA class I and PD-L1 were upregulated in WT SC-islet cells,29 whereas B2P SC-islet cells constitutively over-expressed PD-L1 and lacked HLA class I expression (Figures 1C and S1G). A similar expression profile of HLA-E and HLA class I expression was observed in WT and BEC SC-islet cells (Figure 1D), with immunohistochemistry confirming the membrane localization of HLA-E on SC-β cells (Figure 1E).28
Figure 1.

Generation of immune-evasive SC-islet cells
(A) Schematic of the genetic engineering strategy to generate immune-evasive hESCs.
(B) Analysis of Nkx6.1+/C-peptide+ SC-β cells (S6d14). Data are presented as mean ± SD (n = 3 independent differentiations). p values were determined by one-way ANOVA, ∗p < 0.05.
(C) PD-L1 and HLA-ABC expression on SC-β cells. Data are presented as mean fluorescence intensity (MFI) normalized to mode.
(D) HLA-ABC and HLA-E expression on SC-β cells. Data are presented as MFI normalized to mode.
(E) Immunofluorescence staining of CD49a+-enriched SC-β cells (S6d14). White arrowheads highlight expression of Nkx6.1, C-peptide, and HLA-E, respectively. Scale bars: 100 μm.
(F) Schematic of in vivo SC-islet cell xeno-rejection model.
(G) Analysis of xenograft rejection. Data are presented as mean ± SEM (n = 5/group). p values were determined by two-way ANOVA between groups, ∗p < 0.05. Dashed line represents background luminescence.
To confirm that human PD-L1 expressed on the surface of SC-islet cells binds PD-1, we assessed binding of fluorescently labeled, soluble human and mouse PD-1-Fc on WT and B2P SC-islet cells (Figure S1H). Both soluble human and mouse PD-1 bind membrane-bound PD-L1 on B2P SC-islet cells, whereas WT SC-islet cells do not endogenously express sufficient levels of PD-L1 to detectably bind human or mouse PD-1. We also observed a decrease in the binding affinity of soluble mouse PD-1 to human PD-L1.30 We then transplanted WT, PD-L1, and B2P SC-islet cells under the kidney capsule of B6/albino mice to assess their ability to survive against xeno-rejection (Figure 1F), which had been previously demonstrated.15 We used bioluminescence imaging to track SC-islet cell survival and found that all gene-modified SC-islet cells were rejected within 10 days after transplantation (Figure 1G). These results suggest that PD-L1 over-expression is not sufficient to protect HUES8-derived SC-islet cells from xeno-rejection and that the additional ablation of HLA expression does not improve xenograft survival in our in vivo model.
HLA-deficient SC-islet cells are resistant to allogeneic immune cell destruction
Since xeno-rejection does not mimic allograft (allo) rejection, we next chose to assess the survival of our HLA engineered SC-islet cells in an allogeneic setting. In concordance with previous studies,31 we found that NK cells and CD4+ and CD8+ T cells represented ∼8%, 35%, and 10% of enriched peripheral blood mononuclear cells (PBMCs), respectively (Figures S2A and S2B). We then co-cultured IFN-γ-treated SC-islet cells with human PBMCs in vitro and showed that B2M−/− and BEC SC-islet cells possess significantly improved survival compared with WT (Figures 2A and 2B). HLA-E over-expression did not provide additional protective benefit. Additionally, similar survival patterns were observed when SC-islet cells were co-cultured with purified CD8+ and CD4+ T cells (Figures S2C–S2F). In all co-culture assays, there was no significant difference in the survival of all gene-modified SC-islet cells cultured with CD3/CD28 activated cells.
Figure 2.

In vitro co-culture of immune-evasive SC-islet cells with allogeneic human immune cells
(A and B) SC-islet cell survival when co-cultured with primary human PBMCs at 1:1 and 3:1 PBMC/SC-islet ratio. Data are presented as mean ± SD (n = 5 donors in technical triplicate). p values were determined by one-way ANOVA with Tukey’s post-hoc test, ∗p < 0.05.
(C) T cell ligand gene expression in IFN-γ-treated CD49a+ SC-β cells. Ligand expression is presented as fold change (log2).
(D and E) Differential expression of T cell co-inhibitory and co-activating ligands in IFN-γ-treated CD49a+ SC-β cells. Ligand expression is presented as fold-change (log2). p values were determined by Student’s t test.
(F) SC-β cell survival when co-cultured with primary human CD56+ NK cells at 1:1 and 10:1 NK:SC-β cell ratios. Data are presented as mean ± SD (n = 5 donors in technical triplicate). p values were determined by one-way ANOVA with Tukey’s post-hoc test, ∗p < 0.05.
(G) Schematic of in vivo NK cell cytotoxicity assay.
(H) Analysis of in vivo NK cell assay. NK and SC-islet cell mixtures (3:1) were transplanted subcutaneously in Scid/beige mice (n = 5/group), and all were monitored by bioluminescence imaging on days 1 and 5 post-transplantation. Data are presented as mean ± SEM. p values were determined by two-way ANOVA, ∗p < 0.05. Dashed line represents background luminescence.
Since expression of T cell co-activating and co-inhibitory ligands dictates T cell function and is regulated by various stimuli including IFN-γ stimulation,32 we performed bulk RNA sequencing on IFN-γ-stimulated WT CD49a+ SC-β cells and assessed T cell ligand expression (Figure 2C). As expected, IFN-γ stimulation upregulated expression of the T cell co-inhibitory ligands PD-L1 and LGALS9 (galectin 9) in SC-β cells (Figure 2D).33,34 Conversely, while we did not detect transcripts for many T cell co-activating ligands other than HLA genes, we found that IFN-γ stimulation upregulated members of the tumor necrosis factor (TNF) receptor superfamily HVEM and CD40 and the major histocompatibility complex (MHC)-associated gene butyrophilin subfamily 3 member A1 (BNT3A1) (Figure 2E).35,36,37 These results suggest that in the absence of classical HLA-TCR signaling due to HLA knockout, other co-stimulatory and co-inhibitory T cell ligands are expressed in SC-β cells that may influence T cell function.
HLA-deficient SC-islet cells are resistant to allogeneic NK cell destruction
We next assessed the effect of HLA deletion and HLA-E over-expression on NK cell function against SC-islet cells. When co-cultured with NK-92 MI cells, BEC SC-islet cells showed no significant difference in survival compared with WT SC-islet cells, whereas HLA-deficient SC-islet cells were susceptible to NK-92 MI cytotoxicity (Figure S3A).28 The strong protective effect of HLA-E against NK-92 MI cells is likely due to the high percentage (∼96.1%) of NKG2A+/NKG2C− cells (Figure S3B), which biases NK cell inhibition.
While NK cell lines are useful tools for assessing NK cell cytotoxicity, they do not accurately recapitulate primary NK cell receptor expression and function. Previous studies have shown that in the absence of IL-2 activation, human NK cells do not destroy HLA-deficient endothelial cells and platelets.38,39 Thus, prior to co-culture with gene-modified SC-islet cells, we pre-activated human NK cells with IL-2 for 5 days as previously described.38 Enriched NK cells consisted of ∼80% CD56dim and ∼5% CD56high NK cells (Figures S3C and S3D). Surprisingly, despite IL-2 pre-activation, we observed no significant difference in survival between gene-modified SC-islet cells (Figure 2F). Since HLA-E over-expression does not provide any additional protective benefit to HLA-deficient SC-islet cells against NK cell cytotoxicity, we chose to interrogate WT and B2M−/− SC-islet cells moving forward. We assessed the survival of WT and B2M−/− SC-islet cells after transplantation with IL-2 pre-activated NK cells in Scid/beige mice (Figure 2G). Again, there was no significant difference in WT and B2M−/− SC-islet cell survival in vivo (Figures 2H and S3E). Taken together, these results suggest that HLA-deficient SC-islet cells are intrinsically resistant to NK cell cytotoxicity and that their survival in vitro is recapitulated in vivo.
SC-β cells possess an NK cell evasive ligand phenotype
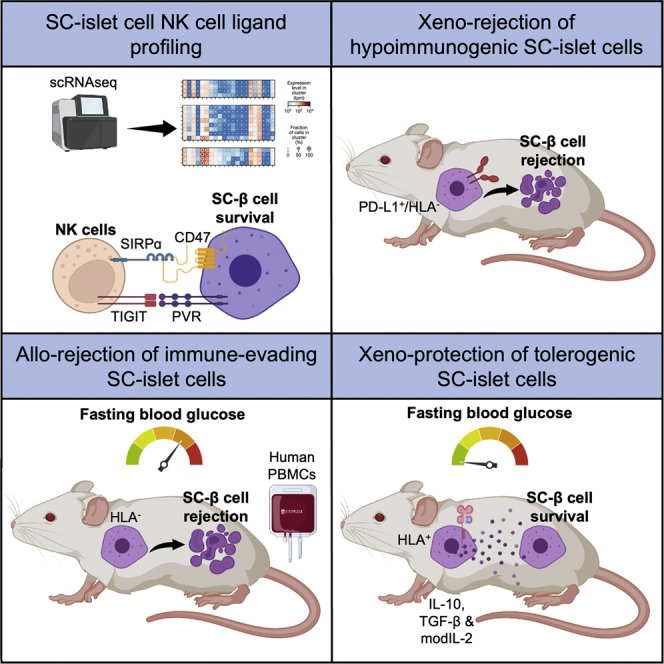
Since we expected B2M−/− SC-islet cells to be susceptible to IL-2 pre-activated NK cells,38 and considering NK cell function is dictated by a balance of inhibitory and activating signals, we hypothesized that the endogenous ligand receptor profile of SC-β cells may be conducive to an NK cell evasive phenotype. We therefore re-analyzed single-cell RNA sequencing (scRNA-seq) expression datasets from our existing in vitro SC-β cell differentiation,7 an in vivo SC-β cell transplantation,40 and primary human islets,41 focusing on the expression of NK cell ligands. We found that SC-β cells intrinsically lack expression of many non-HLA NK cell activating ligands such as NCR3LG1, MICA, MICB, UBLP1, ULBP2, CD48, and CD72, while they do express many non-HLA inhibitory ligands such as PVR, CD47, CDH2, NECTIN1, NECTIN2, and NECTIN3 (Figure 3A). Importantly, the expression of non-HLA ligands does not change in SC-β cells after transplantation. However, in contrast, HLA expression increased after transplantation almost exclusively in SC-α cells, except for HLA-E, which also increased in SC-β and SC-enterochromaffin cells. Furthermore, while the expression of non-HLA NK cell ligands in in vitro differentiated and 6-month post-transplantation SC-islets is like that in primary human islets, SC-islets remain somewhat less immunogenic.42 We then compared the expression of non-HLA ligands at the surface of WT and B2M−/− SC-β cells by flow cytometry and showed that HLA deletion does not alter their expression (Figure 3B).
Figure 3.

SC-islet cell NK cell ligand profile
(A) NK cell ligand expression in pre- and post-transplant SC-β cells and primary human islet cells. The shading displays mean expression as z-normalized transcripts per million mapped reads (z-norm, TPMs), and diameter denotes fractional expression.
(B) NK cell ligand expression on SC-β cells. Data are presented as MFI normalized to mode and are representative of three independent experiments.
(C and D) Analysis of NK cell activating and inhibitory receptor expression. Data are presented as mean fold change in MFI ±SD (n = 5 donors) normalized to isotype control (dashed line). p values were determined by two-way ANOVA, ∗p < 0.05.
(E) NK cell receptor antibody blocking assay. Data are presented as mean ± SD (n = 5 donors in technical triplicate). p values were determined by one-way ANOVA with Tukey’s post-hoc test, ∗p < 0.05.
(F) Schematic of SC-islet cell transplantation in diabetic, humanized mice.
(G) Non-fasting blood glucose concentrations after SC-islet transplantation in humanized mice. At week 10, each group of mice was divided, and half were humanized by intraperitoneal (i.p.) injection of 5 × 107 HLA-A2− human PBMCs. Data are presented as mean ± SD (n = 12/group). p values were determined by two-way ANOVA, ∗p < 0.05.
(H) In vivo GSIS pre- and 7 weeks post-humanization. Plasma insulin concentration was measured t = 0 and 30 min after glucose injection. Data are represented as mean ± SD (n = 5/group). p values were determined by unpaired t test, ∗p < 0.05.
While scRNA-seq and flow cytometry suggests that SC-β cells possess a diminished NK cell activating ligand phenotype, we chose to confirm this by comparing the expression of NK cell ligands on SC-β cells and endothelial cells. This is because it had been shown that HLA-deficient endothelial cells are susceptible to IL-2 pre-activated NK cells,38 and we hypothesized that the susceptibility to NK cell cytotoxicity may be due to the expression of activating ligands. Thus, we differentiated WT and B2M−/− hESCs into SC-endothelial cells and assessed differentiation efficiency by flow cytometry, which showed >95% CD31+ SC-endothelial cells derived from both cell lines (Figures S4A and S4B). A flow cytometric comparison of NK cell ligand expression on SC-β and SC-endothelial cells showed that the MIC and ULBP proteins were expressed in SC-endothelial cells, whilst they were either absent or less expressed on SC-β cells (Figures S4C and S4D). Next, we sought to understand how the expression of NK cell ligands is regulated by a partial pro-inflammatory stimulus. We performed bulk RNA-seq of IFN-γ-treated SC-β and SC-endothelial cells and found that the expression of most NK cell ligands is not IFN-γ regulated, except for the HLA molecules (Figure S4E). Thus, within an HLA-deficient context, the expression of non-HLA NK cell ligands is stable upon inflammatory stimulus.
To identify potential ligand-receptor pathways that may result in the survival of B2M−/− SC-islet cells when co-cultured with IL-2 pre-activated NK cells, we assessed the surface expression of NK cell activating and inhibitory receptors on freshly isolated and IL-2 pre-activated NK cells. In general, we found that the expression of the activating receptors NKp30, NKp46, DNAM-1, and NKG2D were upregulated upon IL-2 pre-activation in both CD56dim and CD56high NK cells, whereas NKp80 was downregulated (Figure 3C).43 Similarly, since we observed surface expression of the inhibitory ligands CD47, PVR, and the cadherins and nectins on SC-β cells, we assessed the expression of their cognate receptors SIRPα, TIGIT, KLRG1, and CD96, respectively, on NK cells. As reported by Deuse et al.,38 we observed significant upregulation of SIRPα on IL-2 pre-activated NK cells (Figure 3D). Additionally, TIGIT and CD96 were upregulated after IL-2 pre-activation. Next, we hypothesized that the CD47-SIRPα and PVR-TIGIT ligand-receptor pathways may be involved in the NK cell evasive phenotype of B2M−/− SC-islet cells. When B2M−/− SC-islet cells were co-cultured with pre-activated NK cells in the presence of anti-SIRPα and anti-TIGIT neutralizing antibodies, SC-islet cell survival decreased (Figure 3E). Taken together, these data suggest that SC-β cells intrinsically possess and sustain a diminished NK cell activating ligand profile and that this ligand phenotype may explain their resistance to IL-2 pre-activated NK cell cytotoxicity.
HLA-deficient SC-islet cells demonstrate delayed graft rejection and reverse diabetes in humanized mice
Having demonstrated that HLA-deficient SC-islet cells resist PBMC cytotoxicity in vitro and NK cell cytotoxicity in vitro and in vivo, we assessed the ability of HLA-deficient SC-islet cells to normalize hyperglycema in diabetic, NSG double knockout (DKO) mice prior to PBMC injection (Figure 3F). By 10 weeks post-transplantation, there was no significant difference in blood glucose levels of mice transplanted with WT and B2M−/− SC-islet cells (Figure 3G). Once blood glucose levels had been normalized, we injected HLA-A2− (mis-matched) PBMCs and assessed allo-rejection by monitoring blood glucose levels. WT SC-islet cells were destroyed within 2 weeks, whereas the rejection of B2M−/− SC-islet cells was delayed. At 7 weeks post-PBMC injection, in vivo glucose-stimulated insulin secretion (GSIS) assay showed that animals transplanted with WT SC-islet cells lost in vivo graft function, while some graft function was observed in animals transplanted with B2M−/− SC-islet cells (Figure 3H). These results suggest that B2M−/− SC-islet cells reverse diabetes and demonstrate delayed allo-rejection; however, it is likely that they would ultimately be completely rejected.
Cytokine-secreting SC-β cells are protected from xeno-rejection and reverse diabetes in NOD mice
As a result of the varying success of immune-evasive engineering to protect SC-islet cells, we pursued a complementary strategy by engineering SC-islet cells to secrete the cytokines IL-2 mutein, TGF-β, and IL-10 as vehicles of localized immune tolerance. The IL-2 mutein (N88D) possesses reduced affinity for the IL-2Rβγ receptor, resulting in the production of a regulatory T cell (Treg)-selective molecule that preferentially expands Tregs while having a minimal effect on CD4+ and CD8+ memory T cells.22,23 Since the immune-suppressive phenotype of Tregs requires IL-10 and TGF-β, we included these two cytokines in our immune-tolerizing approach.24 Consequently, we engineered human pluripotent stem cells to express modified IL-2, IL-10, and TGF-β from the GAPDH locus (Figure 4A). Differentiation of this genetically modified cell line, called 2B10, resulted in ∼30% Nkx6.1+/C-peptide+ SC-β cells as assessed by flow cytometry (Figure 4B). When stimulated with sequential low- and high-glucose challenges in an in vitro GSIS assay, 2B10 SC-islet cells demonstrated a similar insulin secretion profile as WT and significantly upregulated insulin secretion in response to glucose stimulation (Figure 4C). We also confirmed that 2B10 SC-islet cells secrete modified IL-2, TGF-β, and IL-10 and that the amount of cytokine secreted was SC-islet-cell-concentration dependent (Figure S4F). We next sought to determine if cytokine-secreting SC-islet cells were protected from allogeneic human PBMC destruction in vitro. When WT and 2B10 SC-islet cells were co-cultured with human PBMCs in vitro, 2B10 SC-islet cells showed significantly improved survival compared with their WT counterparts (Figure 4D).
Figure 4.

Immune-tolerizing SC-islet cells survive xeno-rejection
(A) Schematic of the genetic engineering strategy to generate immune-tolerizing hESCs.
(B) Analysis of Nkx6.1+/C-peptide+ SC-β cells derived from immune-tolerizing hESCs (S6d8).
(C) In vitro GSIS. WT (red), 2B10 (blue) SC-islets, and primary human islets (green) were challenged with consecutive low (2.8 mM) and high (20 mM) glucose stimulations, followed by depolarization with 30 mM KCl. Data are represented as mean ± SD (n = 4). p values were determined by unpaired t test, ∗p < 0.05.
(D) SC-islet cell survival when co-cultured with primary human PBMCs at a 1:1 ratio. Data are presented as mean ± SD (n = 5 donors in technical triplicate). p values were determined by Student’s t test.
(E) Analysis of in vivo graft survival in B6/albino mice. Data are presented as mean ± SD (n = 4/group). p values were determined by two-way ANOVA, ∗p < 0.05.
(F) Immunostaining of SC-islet grafts showing presence of INS+ cells and Treg recruitment at 5 weeks post-transplantation. Scale bars: 100 μm.
(G) Schematic of cytokine-secreting SC-β cell transplantation in diabetic NOD mice.
(H) Non-fasting blood glucose concentrations after SC-β cell transplantation in NOD mice. Data are presented as mean ± SD (n = 3/group). p values were determined by two-way ANOVA, ∗p < 0.05.
Because humanized mice bias the engraftment of CD3+ T cells,44 we chose to assess the survival of cytokine-secreting SC-islet cells in a xenogeneic mouse model. When 2B10 SC-islet cells were transplanted in B6/albino mice, they survived up to 9 weeks post-transplantation, while WT SC-islet cells were destroyed within 2 weeks (Figures 4E and S4G). We also found that WT grafts contained little to no remaining SC-islet cells, while 2B10 grafts contained surviving insulin-producing cells and Tregs localized within the graft (Figure 4F). Finally, to demonstrate that cytokine-secreting SC-islet cells can survive xeno-rejection and reverse diabetes, we transplanted CD49a+ SC-β cells in autoimmune diabetic NOD mice (Figures 4G and 4H). We found that WT SC-β cells temporarily survive and reverse diabetes for 2–3 weeks. Excitingly, 2B10 SC-β cells reversed diabetes for the period of the experiment (8 weeks) in the absence of any xeno-rejection. Collectively, these data suggest that SC-islet cells can be engineered to co-secrete immune-modulatory cytokines that induce a tolerogenic local microenvironment characterized by Treg infiltration that sustains xenograft survival while also reversing autoimmune diabetes in NOD mice.
Discussion
The utility of an immune-evasive/-tolerogenic islet cell replacement therapy relies on the ability to maintain transgene expression throughout cell differentiation and after transplantation. Here, we showed that engineering SC-islet cells to constitutively express tolerogenic molecules from the GAPDH locus resulted in persistent transgene expression. In contrast to a study by Yoshihara et al.,15 we also report the lack of a xeno-protective effect of PD-L1 over-expression in HUES8-derived SC-islet cells. This may be explained by species-specific differences in PD-L1/PD-1 binding.45,46 In fact, we observed decreased binding of soluble mouse PD-1 to human PD-L1 expressed on SC-islet cells. We also cannot exclude the possibility that the level of PD-L1 expression driven by the endogenous GAPDH promoter (∼50-fold increase) is not sufficient to provide immune protection. While our results suggest that over-expression of human PD-L1 is not sufficient to overcome xeno-rejection in our model, this strategy may still be useful in an allogeneic setting.
Diminishing the immune recognition of SC-islet cells by manipulating the expression of HLA molecules has also shown some promise as a potential immune-protective strategy.14 Thus, we chose to assess a universal HLA engineering strategy that would potentially allow for transplantation of SC-islets in any diabetic individual regardless of HLA type.28 In concordance with previous studies, HLA-deficient SC-islet cells were resistant to PBMC cytotoxicity in vitro.47,48 We also found that SC-β cells modulate their T cell ligand profile in response to partial inflammatory stimulus. Analysis of T cell ligand transcripts in SC-β cells suggests that exploiting the LGALS9/TIM-3 signaling axis may be of interest. In fact, like PD-L1, LGALS9 is frequently upregulated in cancer cells, where it contributes to tumor progression by inhibition of T cell function.49,50 The T cell activating ligands HVEM, CD40, and BTN3A1 are potential targets to knock out in SC-islet cells to further influence T cell function.
We also showed that HLA-deficient SC-islet cells are resistant to pre-activated NK cell cytotoxicity both in vitro and in vivo. This may be due to the lack of expression of NK cell activating ligands such as the MIC and ULBP proteins on SC-β cells. While treatment with IFN-γ did not alter the expression of non-HLA NK cell ligands on SC-β cells, other pro-inflammatory cytokines have been shown to regulate the expression of anti-viral and immune-associated genes in mouse islets.51 This observation may also extend to NK cell ligand expression, which would potentially alter the immunogenicity of SC-islet cells. However, since many immunodeficient mouse models lack critical components for NK cell survival and function such as SIRPα and IL-15,52 they do not support long-term engraftment of human NK cells, and we cannot exclude the possibility of HLA-deficient SC-islet cell destruction in a mouse model that better supports NK cell engraftment. Furthermore, while the transplantation of HLA-deficient SC-islet cells was able to normalize blood glucose levels in diabetic mice, after humanization, the cells were eventually destroyed with delayed rejection kinetics. This could be explained by the presence of other human immune cell subsets such as macrophages, monocytes, and dendritic cells that may play a role in indirect allo-rejection.53,54,55 Ultimately, this highlights the limitations of exclusively using in vitro immune cell co-culture assays to assess the effect of genetic modification on the protection of SC-islet cells from immune destruction.29,48
Finally, we show for the first time that SC-islet cells engineered to secrete modified IL-2, TGF-β, and IL-10 are protected against xeno-rejection and reverse autoimmune diabetes in NOD mice. This combination of cytokines resulted in the localization of Tregs within the SC-islet grafts, providing localized immune tolerance and sustained survival of the SC-islets. Similarly, several studies have attempted to induce localized immune tolerance of SC-islet grafts by engineering chimeric antigen receptor (CAR)-Tregs to recognize cell-type-specific antigens such that they migrate toward the grafted cells and protect them from immune rejection.56,57,58 However, while these studies demonstrate the potential utility of ex-vivo-engineered Tregs for protection against allo-rejection, the phenotypic stability of these cells is poorly understood, and patients would likely require repeated treatments to sustain immune tolerance. Thus, our unique approach of using SC-islet grafts to induce localized immune tolerance has several important implications. First, since β cells are professional secretory cells with a significant translatory demand, it demonstrates that SC-islet cells can be co-opted to secrete other proteins while maintaining their designed function.59 Second, the intrinsic ligand profile of the desired cell type should be considered when determining the set of genetic modifications required to generate immune-evasive cells, as some cell types may not require extensive genetic manipulation. Third, our results provide validation for the use of immune-tolerizing approaches (either alone or in conjunction with immune evasion) as a method to protect SC-islet cells from the immune system. A variation of this approach may be to include some, but not all, cells in the transplant that secrete these tolerizing molecules. Similarly, accessory cells that intrinsically possess immune-modulatory functions could also be included within the designer islets.60 Overall, our approach may eliminate the need for encapsulation or immunosuppression, a long-standing goal of the islet transplantation field.
Limitations of the study
Although HLA-deficient SC-islet cells possessed improved survival in in vitro PBMC co-culture assays (Figures 2A and 2B), we found that these results did not translate in vivo. Choosing an appropriate humanized mouse model is essential to evaluate immune-evasion/-tolerizing genetic engineering strategies. Many PBMC humanized mouse models such as that used in this study do not fully recapitulate human allo-rejection since they bias CD3+ cell engraftment and lack other immune cell subsets. Thus, while HLA-deficient SC-islets demonstrated delayed rejection in the PBMC humanized mouse model (Figure 3G), we cannot exclude the possibility that in CD34+ or BLT mice (humanized mouse models that better recapitulate human immune components) these cells would be rejected earlier. For these reasons, validating the protective effect of immune-evasion/-tolerizing genetic engineering strategies should be conducted in humanized mouse models that more accurately reflect allo-rejection and recapitulate human T1D.
In this study, we ultimately chose to use the NOD mouse as a xeno-rejection model to evaluate our immune-tolerizing strategy as it is an autoimmune model of diabetes and does not possess the immune cell bias of humanized mouse models. However, by using such a strong model of graft rejection, additional immune barriers are introduced that require complex genetic engineering approaches as a solution. This means combining gene knockouts and knockins to generate the desired immune-evasive/-tolerizing cell product. Knockin of multiple genes at a specific locus in the genome requires large homology directed repair templates, which is associated with poor integration efficiency and recovery of genetically unstable clones. Exploring the possibility of introducing tolerogenic molecules at multiple constitutively expressed loci could reduce the size of homology-directed repair (HDR) templates and result in the recovery of genetically stable homozygous knockin clones. Striking a balance between HDR length and the number of editing events may permit the introduction of more foreign genetic material into the genome without destabilizing effects.
Furthermore, since immune-tolerizing SC-islets secrete cytokines from all cells within the islet, there is a risk that the concentration of constitutively secreted cytokines may result in chronic immunosuppression. Thus, it is important to evaluate the immune status of mice receiving immune-tolerizing SC-islet cells as chronic immunosuppression should be avoided. This could have been partially evaluated by titrating the concentration of the individual cytokines in vitro with human PBMCs and conducting a high-resolution analysis of the composition of immune cells and their resulting phenotypes. However, if transplantation of immune-tolerizing SC-islets does result in chronic immunosuppression, the ability to enrich for specific endocrine cell populations may allow us to adjust the dose of cytokine-secretion by generating designer islets composed of cytokine-secreting and non-cytokine-secreting endocrine cells such that localized graft tolerance is achieved.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| PE Mouse monoclonal anti-human HLA-ABC | Biolegend | Cat# 311405; RRID: AB_314874 |
| PE Mouse monoclonal anti-human HLA-E | Biolegend | Cat# 342603; RRID: AB_1659250 |
| PE Mouse monoclonal anti-human CD274 (B7-H1, PD-L1) | Biolegend | Cat# 393607; RRID: AB_2749924 |
| Rat monoclonal anti-human C-peptide | DSHB | Cat# GN-ID4; RRID: AB_2255626 |
| Mouse monoclonal anti-human Nkx6.1 | DSHB | Cat# F55A12; RRID: AB_532379 |
| Goat polyclonal anti-Rat IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 647 | Life Technologies | Cat# A-21247; RRID: AB_141778 |
| Goat polyclonal anti-Mouse IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 405 | Life Technologies | Cat# A-31553; RRID: AB_221604 |
| PE Mouse monoclonal IgG2a, κ Isotype Ctrl (FC) | Biolegend | Cat# 400213; RRID: AB_2800438 |
| PE Mouse monoclonal IgG1, κ Isotype Ctrl | Biolegend | Cat# 400112; RRID: AB_2847829 |
| PE Mouse monoclonal anti-human CD49a | BD Biosciences | Cat# 559596; RRID: AB_397288 |
| Rabbit polyclonal anti-human HLA-E | Atlas Antibodies | Cat# HPA031454; RRID: AB_2673891 |
| Goat polyclonal anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 594 | Life Technologies | Cat# A-11032; RRID: AB_2534091 |
| Goat polyclonal anti-Rat IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | Life Technologies | Cat# A-11006; RRID: AB_2534074 |
| Goat polyclonal anti-Rabbit IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 647 | Life Technologies | Cat# A-21244; RRID: AB_2535812 |
| APC Mouse monoclonal anti-human CD3 | Biolegend | Cat# 300311; RRID: AB_314047 |
| PE Rat monoclonal anti-human CD4 | Biolegend | Cat# 357403; RRID: AB_2562035 |
| Pacific Blue Mouse anti-human CD8 | Biolegend | Cat# 344717; RRID: AB_10551616 |
| Pacific Blue Mouse monoclonal anti-human CD56 (NCAM) | Biolegend | Cat# 362519; RRID: AB_2564096 |
| Pacific Blue Mouse monoclonal IgG1, κ Isotype Ctrl | Biolegend | Cat# 400131; RRID: AB_2923473 |
| APC Mouse monoclonal IgG2a, κ Isotype Ctrl | Biolegend | Cat# 400221; RRID: AB_2891178 |
| PE Rat monoclonal IgG2b, κ Isotype Ctrl | Biolegend | Cat# 400607; RRID: AB_326551 |
| Ultra-LEAF Purified Mouse monoclonal anti-human TIGIT (VSTM3) | Biolegend | Cat# 372719; RRID: AB_2650966 |
| Pacific Blue Mouse monoclonal anti-human CD31 | Biolegend | Cat# 303113; RRID: AB_1877196 |
| APC Mouse monoclonal anti-human CD47 | Biolegend | Cat# 323123; RRID: AB_2716202 |
| APC Mouse monoclonal anti-human CD324 (E-Cadherin) | Biolegend | Cat# 324107; RRID: AB_756069 |
| PE Mouse monoclonal anti-human CD325 (N-Cadherin) | Biolegend | Cat# 350806; RRID: AB_10660824 |
| PE Mouse monoclonal anti-human CD112 (Nectin-2) | Biolegend | Cat# 337409; RRID: AB_2174163 |
| APC Mouse monoclonal anti-human CD155 (PVR) | Biolegend | Cat# 337617; RRID: AB_2565814 |
| PE Mouse monoclonal anti-human CD111 (Nectin-1) | Biolegend | Cat# 340404; RRID: AB_2174152 |
| APC Mouse monoclonal anti-human MICA/MICB | Biolegend | Cat# 320907; RRID: AB_493196 |
| Alexa Fluor 647 Mouse monoclonal anti-human/mouse/rat PCNA | Biolegend | Cat# 307912; RRID: AB_2267947 |
| PE Rabbit monoclonal anti-human BAT3/BAG-6 | Abcam | Cat# ab210838; RRID: AB_2923475 |
| APC Mouse monoclonal anti-human CD48 | Biolegend | Cat# 336713; RRID: AB_2810516 |
| PE Mouse monoclonal anti-human CD70 | Biolegend | Cat# 355103; RRID: AB_2561430 |
| Mouse monoclonal anti-human Nectin-3/PVRL3 (CD113) | Millipore-Sigma | Cat# MABT63; RRID: AB_10807983 |
| APC Mouse monoclonal anti-human ULBP1 | R&D Systems | Cat# FAB1380A; RRID: AB_2923476 |
| PE Mouse monoclonal anti-human ULBP2/5/6 | R&D Systems | Cat# FAB1298P; RRID: AB_2214693 |
| APC Mouse monoclonal anti-human OCIL/CLEC2D | R&D Systems | Cat# FAB3480A; RRID: AB_2044641 |
| PE Mouse monoclonal anti-human CD72 | Biolegend | Cat# 316207; RRID: AB_2819945 |
| APC Mouse monoclonal IgG1, κ Isotype Ctrl | Biolegend | Cat# 400121; RRID: AB_326443 |
| APC Mouse monoclonal IgG2a, κ Isotype Ctrl | Biolegend | Cat# 400221; RRID: AB_2891178 |
| PE Mouse monoclonal IgG2a, κ Isotype Ctrl | Biolegend | Cat# 400213; RRID: AB_2800438 |
| APC Rat monoclonal IgG1, λ Isotype Ctrl | Biolegend | Cat# 401903; RRID: AB_292347 |
| PE Rabbit (DA1E) mAb IgG XP Isotype Control | Cell Signalling Technology | Cat# 5742; RRID: AB_10694219 |
| Goat ployclonal anti-Mouse IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | Life Technologies | Cat# A-11001; RRID: AB_2534069 |
| PE Mouse monoclonal anti-human CD159c (NKG2C) | Biolegend | Cat# 375003; RRID: AB_2888871 |
| APC Mouse monoclonal anti-human CD159a (NKG2A) | Biolegend | Cat# 375107; RRID: AB_2888862 |
| APC Mouse monoclonal anti-human TIGIT (VSTM3) | Biolegend | Cat# 372705; RRID: AB_2632731 |
| PE Mouse monoclonal anti-human KLRG1 | Biolegend | Cat# 368609; RRID: AB_2572136 |
| APC Mouse monoclonal anti-human CD96 (TACTILE) | Biolegend | Cat# 338409; RRID: AB_2566141 |
| PE Mouse monoclonal anti-human CD172a (SIRPα) | Biolegend | Cat# 372103; RRID: AB_2650860 |
| PE/Cyanine7 Mouse monoclonal anti-human CD337 (NKp30) | Biolegend | Cat# 325213; RRID: AB_2716064 |
| APC Mouse monoclonal anti-human CD335 (NKp46) | Biolegend | Cat# 331917; RRID: AB_2561649 |
| PE Mouse monoclonal anti-human NKp80 | Biolegend | Cat# 346706; RRID: AB_1967147 |
| APC Mouse monoclonal anti-human CD226 (DNAM-1) | Biolegend | Cat# 338311; RRID: AB_2561951 |
| PE Mouse monoclonal anti-human CD314 (NKG2D) | Biolegend | Cat# 320805; RRID: AB_492961 |
| APC Mouse monoclonal IgG2a, κ Isotype Ctrl | Biolegend | Cat# 400221; RRID: AB_2891178 |
| PE/Cyanine7 Mouse monoclonal IgG1, κ Isotype Ctrl | Biolegend | Cat# 400125; RRID: AB_2861433 |
| Guinea Pig polyclonal anti-human Insulin | DAKO | Cat# A0564; RRID: AB_10013624 |
| Rat monoclonal anti-mouse CD8a | Biolegend | Cat# 100702; RRID: AB_312741 |
| Mouse monoclonal anti-mouse FOXP3 | Biolegend | Cat# 320002; RRID: AB_439746 |
| Goat polyclonal anti-Guinea Pig IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 647 | Life Technologies | Cat# A-21450; RRID: AB_2735091 |
| Bacterial and virus strains | ||
| One Shot™ Stbl3™ Chemically Competent E. coli | Thermofisher | Cat# C737303 |
| Biological samples | ||
| Aphaersis leukoreduction collars | BWH, Boston | N/A |
| Human islets | Prodo Laboratories | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| mTesR1 | Stemcell Technologies | Cat# 85850 |
| Alt-R® S.p. HiFi Cas9 Nuclease V3 | IDT | Cat# 1081059 |
| Y27632 | DNSK | Cat# DNSK-KI-15-02 |
| Corning® Matrigel® hESC-Qualified Matrix, LDEV-free | Corning | Cat# 354277 |
| rhIFN-γ | R&D Systems | Cat# 285-IF-100 |
| CloneR | Stemcell Technologies | Cat# 05888 |
| Accutase | Stemcell Technologies | Cat# 07920 |
| Phosphate Buffered Saline | Corning | Cat# 21040CV |
| Bovine Serum Albumin | Gibco | Cat# A10008-01 |
| Donkey Serum | Jackson Labs | Cat# 100181-234 |
| Paraformaldehyde | EMS | Cat# 15710 |
| OCT compound | Tissue Tek | Cat# 4583 |
| Histo-Clear | EMS | Cat# 64110-01 |
| Triton X-100 | Millipore-Sigma | Cat# 9036-19-5 |
| Vectashied with DAPI | Vector Laboratories | Cat# H-1200 |
| Lymphoprep | Stemcell Technologies | Cat# 07801 |
| CryoStor CS10 | Stemcell Technologies | Cat# 07930 |
| D-luciferin, Potassium Salt | Gold Biotechnology | Cat# 115144-35-9 |
| ImmunoCult™-XF T Cell Expansion Medium | Stemcell Technologies | Cat# 10981 |
| rhIL-2 | Peprotech | Cat# 200-02 |
| NK MACS medium | Milteny Biotec | Cat# 130-114-429 |
| Human AB serum | Valley Biomed | Cat# HP1022HI |
| HyClone FBS | GE Healthcare | Cat# SH30070.03 |
| Matrigel® Basement Membrane Matrix, Phenol Red-free, LDEV-free | Corning | Cat# 356237 |
| Streptozotocin | Millipore-Sigma | Cat# S0130-50MG |
| TrypLE Express | Thermofisher | Cat# 12604013 |
| Vectashield with DAPI | Vector Laboratories | Cat# H-1200 |
| Critical commercial assays | ||
| RosetteSep Human CD4+ T Cell Enrichment Cocktail | Stemcell Technologies | Cat# 15062 |
| RosetteSep CD8+ T Cell Enrichment Cocktail | Stemcell Technologies | Cat# 15063 |
| RosetteSep NK Cell Enrichment Cocktail | Stemcell Technologies | Cat# 15065 |
| STEMdiff™ Endothelial Differentiation Kit | Stemcell Technologies | Cat# 08005 |
| ImmunoCult™ Human CD3/CD28 T Cell Activator | Stemcell Technologies | Cat# 10991 |
| RNeasy Plus Mini Kit | Qiagen | Cat# 74134 |
| SMART-Seq v4 Ultra Low Input Kit for Sequencing | Clontech | Cat# 634894 |
| Illumina Nextera XT library | Illumina | Cat# FC-131-1096 |
| Human Ultrasensitive Insulin ELISA | ALPCO Diagnostics | Cat# 80-INSHUU-E01.1. |
| Legend Max IL-2 ELISA | Biolegend | Cat# 431807 |
| Legend Max TGF-β ELISA | Biolegend | Cat# 436707 |
| Legend Max IL-10 ELISA | Biolegend | Cat# 430607 |
| Deposited data | ||
| Bulk RNA sequencing | https://www.ncbi.nlm.nih.gov/geo/ | Accession no. GSE200021 |
| Experimental models: Cell lines | ||
| NK-92mi | ATCC | Cat# CRL-2408 |
| K562-Luc2 | Biocytogen | Cat# BCG-PS-015-luc |
| Raji-GFP-Luc2 | Biocytogen | Cat# BCG-PS-087-luc |
| HUES 8 hESC line (NIH approval number NIHhESC-09-0021) | HSCI iPS Core | hES Cell Line: HUES-8 |
| Experimental models: Organisms/strains | ||
| B6(Cg)-Tyrc-2J/J mice | Jackson Labs | Cat# 000058 |
| CB17.Cg-PrkdcscidLystbg-J/Crl mice | Charles River Labs | Cat# 250 |
| NSG-(KbDb)null (IA)null mice | Jackson Labs | Cat# 025216 |
| NOD/ShiLtJ mice | Jackson Labs | Cat# 001976 |
| Recombinant DNA | ||
| B2M gRNA 5′-GCTACTCTCTCTTTCTGGCC′3 | IDT | N/A |
| PD-L1 gBlock | Genscript, this paper | N/A |
| peptide::B2M::HLA-E fusion gBlock | Genscript, this paper | N/A |
| IL-2 mutein-TGFβ-IL-10 gBlock | Genscript, this paper | N/A |
| Software and algorithms | ||
| FlowJo 10.7.1 | BD Biosciences | https://www.flowjo.com/solutions/flowjo/downloads |
| Living Image | PerkinElmer | Cat# 128113 |
| MARS data analysis | BMG LABTECH | https://www.bmglabtech.com/en/microplate-reader-software/ |
| BD FACSDiva v9.0 | BD Biosciences | N/A |
| HiSeq Control Software | Illumina | N/A |
| bcl2fastq 2.17 Software | Illumina | N/A |
| Trimmomatic v.0.36 | USADELLAB | http://www.usadellab.org/cms/?page=trimmomatic |
| STAR aligner v.2.5.2b | N/A | https://github.com/alexdobin/STAR |
| DESeq2 | N/A | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| Prism v9 | Graphpad | https://www.graphpad.com/scientific-software/prism/ |
| Other | ||
| 4D Nucleofector | Lonza | Cat# AAF-1003B |
| FACS Aria II flow cytometer | BD Biosciences | N/A |
| LSR II flow cytometer | BD Biosciences | N/A |
| Zeiss Axio Imager Z2 with Apotome microscope | Zeiss | N/A |
| IVIS SpectrumCT | PerkinElmer | 128201 |
| CLARIOstar Plus microplate reader | BMG LABTECH | https://www.bmglabtech.com/en/clariostar-plus/ |
| Illumina HiSeq 2000 | Illumina | Cat# SY-401-1001 |
| Vi-CELL XR Cell Viability Analyser | Beckman Coulter | Cat# 731050 |
| Qubit Fluorometer | Thermofisher | N/A |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Douglas A. Melton (dmelton@harvard.edu).
Materials availability
Gene-modified HUES8 lines used in this study are available upon request; additionally, the reagents used in this study are available from the lead contact with a completed Materials Transfer Agreement.
Experimental model and subject details
Mice
Male (6–8 weeks of age) B6(Cg)-Tyrc-2J/J (B6/abino; Jackson Labs, #000058), CB17.Cg-PrkdcscidLystbg-J/Crl (Scid/beige; Charles River Labs, #250), NSG-(KbDb)null (IA)null (NSG-MHC Class I/II KO; Jackson Labs, #025216) and female (10 weeks of age) NOD/ShiLtJ (NOD; Jackson Labs, #001976) were purchased. Mice were housed in specific pathogen-free conditions at Harvard University. All animal research was conducted under Harvard IACUC approval.
Cell lines
Human ES maintenance and differentiation was carried out as previously described.3,5 SC-islet cell differentiations were initiated 72 h after initial passage by aspirating mTeSR1 (STEMCELL Technologies, 85850) and replenished with stage and day-specific media supplemented with the appropriate small molecules or growth factors as previously described.7 All cell lines (GAPluc, B2M−/−, GAP-PD, GAP-B2P, GAP-BEC and 2B10) were routinely tested for mycoplasma and were mycoplasma-free. All experiments involving human ES cells were approved by the Harvard University ESCRO (#E00024) committee.
Human samples
The study was carried out in accordance with the Harvard University guidelines and was approved by the University of Massachusetts (UMASS) institutional review board ethics (IRB14-4650) committee. Informed consent was obtained from the patients and healthy donors before blood donation. All blood samples were received as de-identified, therefore, the information on the age and/or gender of the donors is not available.
Primary immune cells
PBMCs, CD8 and CD4 T cells were isolated and maintained in ImmunoCult™-XF T Cell Expansion Medium (STEMCELL Technologies, 10981) + 100U/mL rhIL-2 (Peprotech, 200-02). NK cells were isolated and maintained in NK cell medium (NK MACS medium (Milteny Biotec, 130-114-429) + 5% Human AB serum (Valley Biomed, HP1022HI) + 5% HyClone FBS (GE Healthcare) + 0.5 ng/mL rhIL-2). Details of specific culture conditions can be found in the STAR Methods details.
Method details
Generation of hypo-immunogenic hESCs
The existing GAPluc (WT) hESC line served as starting material for the generation of HLA-deficient hESCs.20 To generate HLA-deficient hESCs, 1 × 106 WT hESCs were nucleofected using the 4D-Nucleofector (Lonza) with RNP complexed with 120 pmol B2M gRNA (5′-GCTACTCTCTCTTTCTGGCC′3)61 (IDT) and 104 pmol Alt-R® S.p. HiFi Cas9 Nuclease V3 (IDT) according to the manufacturer’s instructions. Nucleofected cells were resuspended in mTesR1 + 10 μM Y27632 (DNSK International, DNSK-KI-15-02) and plated in a matrigel-coated tissue culture plate. After 48 h cells were treated with 10 ng/mL IFN-γ (R&D, 285-IF-100) for 24 h and then stained with APC anti-human HLA-ABC (W6/32, 1:100) (Biolegend, 311409). HLA-ABC−/− cells were sorted on a FACS Aria II (BD Biosciences) and plated in a matrigel-coated tissue culture plate containing mTesR1 + CloneR (STEMCELL Technologies, 05888), with single colonies picked for expansion. The resulting HLA-deficient line was named B2M−/−.
To generate GAP-PD and GAP-BEC hESCs, human PD-L1 and the peptide::B2M::HLA-E fusion sequences were synthesized as gBlocks (Genscript) and cloned into our existing GAPluc targeting plasmid downstream of the Luc2 gene. The sequence of the covalently attached peptide (VMAPRTLLL) in the peptide::B2M::HLA-E fusion is derived from the HLA-Cw7 molecule.62 hESCs were nucleofected with the GAP-PD or GAP-BEC targeting plasmids and GAPDH-targeting RNP as previously described.20 For GAP-B2P and GAP-BEC lines, a polyclonal population of puromycin-selected cells was subsequently nucleofected with B2M-targeting RNP, and single-colonies were picked for expansion after FACS sorting as described above. The gating strategy for sorting HLA-ABC−/− hESCs is described in Figure S1I.
Flow cytometry
Differentiated WT, B2M−/−, BEC, PD-L1 and B2P SC-islet cells were both treated and untreated with 10 ng/mL IFN-γ for 24 h prior to staining. Cells were dissociated with Accutase (STEMCELL Technologies, 07920), washed twice with PBS + 0.1% BSA (Gibco, A10008-01) and blocked for 30 min on ice with PBS + 5% donkey serum (Jackson Labs; 100181-234). Cells were then stained for 30 min on ice in blocking buffer with PE or APC anti-human HLA-ABC (Biolegend, 311405), PE anti-human HLA-E (Biolegend, 342603) and PE anti-human PD-L1 (Biolegend, 393607). Cells were then washed three times and fixed in 4% PFA (EMS, 15710) for 15 min at 4°C. Fixed cells were then incubated in blocking buffer with rat anti-human C-peptide (DHSB; GN-ID4) and mouse anti-human Nkx6.1 (DHSB; F55A12) (overnight at 4°C), washed three times with blocking buffer, incubated with goat anti-rat 647 (Life Technologies, A-21247; 1:300) and goat anti-mouse 405 (Life Technologies, A-31553; 1:300) in blocking solution (1 h at room temperature), washed three times and resuspended in PBS +0.1% BSA. Samples were captured on the LSR II (BD) flow cytometer and analyzed using BD FACSDiva v9.0 (BD Biosciences) and FlowJo 10.7.1 (BD). All antibodies were used at 1:100 unless otherwise stated. The gating strategy for identifying SC-β cells is described in Figure S1J. PE mouse IgG2a (Biolegend, 400213) and PE mouse IgG1 (Biolegend, 400111) served as isotype controls.
Magnetic enrichment using CD49a and reaggregation
WT and BEC SC-β cells were magnetically enriched from SC-islet clusters as previously described.7 Enriched cells were resuspended in S6 medium and plated at 5 × 103 cells/well in low-attachment 96-well v-bottom tissue-culture plates (Thermo Scientific, 277143), centrifuged at 300 g for 1 min and incubated at 37°C for 4–7 days. CD49a enriched SC-β cell clusters were fed fresh S6 medium every 2 days.
SC-β cell immunohistochemistry
CD49a+ enriched SC-β cell clusters were fixed in 4% PFA for 1 h at room temperature, washed and frozen in OCT (Tissue-Tek, 4583) and sectioned to 14 μm. For staining, slides were incubated in blocking buffer (PBS + 0.1% Triton-X + 5% donkey serum) for 1 h at room temperature, incubated in PBS + 5% donkey serum containing rat anti-human C-peptide (1:300), mouse anti-human Nkx6.1 (1:100) and rabbit anti-human HLA-E (Atlas Antibodies, HPA031454, 1:100) for 1 h at room temperature, washed three times, incubated in goat anti-mouse 594 (Life Technologies, A-11032; 1:500), goat anti-rat 488 (Life Technologies, A-11006; 1:500) and goat anti-rabbit 647 (Life technologies, A-21244; 1:500) for 2 h at room temperature, washed, mounted in Vectashield with DAPI (Vector Laboratories; H-1200), covered with coverslips and sealed with clear nail polish. Representative regions were imaged using Zeiss.Z2 with Apotome microscope (Zeiss) and analyzed using Zen Blue v2 (Zeiss).
Immune cell isolation and identification
PBMCs, CD8 and CD4 T cells, and NK cells were isolated from apheresis leukoreduction collars (n = 5 donors) obtained from Brigham and Women’s Hospital in compliance with our IRB approval. PBMCs were isolated by density gradient centrifugation with lymphoprep (STEMCELL Technologies, 07801) SepMate™-50 (IVD) tubes (STEMCELL Technologies, 85450) according to the manufacturer’s instructions. CD4, CD8 and NK cells were isolated by density gradient centrifugation with lymphoprep and SepMate™-50 (IVD) tubes following the addition of RosetteSep Human CD4+ T Cell Enrichment Cocktail (STEMCELL Technologies, 15062), CD8+ T Cell Enrichment Cocktail (STEMCELL Technologies, 15063) and NK Cell Enrichment Cocktail (STEMCELL Technologies, 15065) respectively. Enriched immune cells were cryopreserved in CryoStor CS10 (STEMCELL Technologies, 07930) at a concentration of 10M cells/vial.
Flow cytometric analysis of enriched immune cell subpopulations was performed by staining cells in blocking buffer containing APC anti-human CD3 (Biolegend, 300311), PE anti-human CD4 (Biolegend, 357403), Pacific Blue anti-human CD8 (Biolegend, 344717) and Pacific Blue anti-human CD56 (Biolegend, 362519). Isotype controls were Pacific Blue mouse IgG1 (Biolegend, 400131), APC mouse IgG2a (Biolegend, 400221) and PE rat IgG2b (Biolegend, 400607). All antibodies were used at 1:100 or unless otherwise stated. Representative gating strategies are demonstrated in Figures S2G–S2I.
Xenotransplantation of SC-islets in B6/albino mice
WT, PD-L1 and B2P SC-islet cell clusters (5 × 106 cells) were transplanted under the kidney capsule of B6/albino mice (n = 5/group) and graft survival was monitored for 10 days following i.p. injection of D-luciferin (Gold Biotechnology, 115144-35-9) by bioluminescence imaging on the IVIS SpectrumCT (PerkinElmer, 128201) and analyzed using Living Image Software (PerkinElmer, 128113).
In vitro T cell cytotoxicity assays
WT, B2M−/− and BEC SC-islet cells were seeded in S3 medium at 5 × 104 cells/well of a matrigel-coated 96-well black flat-bottom plate (Corning, 3916) in the presence or absence of IFN-γ (10 ng/mL). After 24 h, the medium was switched to T cell medium (ImmunoCult™-XF T Cell Expansion Medium (STEMCELL Technologies, 10981) + 100U/mL rhIL-2 (Peprotech, 200-02)) for immune cell co-culture. Primary human PBMCs, CD4 or CD8 cells (n = 5 donors) cultured in T cell medium were added to SC-islet cells at 1:1 and 3:1 effector:target ratios with and without the addition of ImmunoCult™ Human CD3/CD28 T Cell Activator (STEMCELL Technologies, 10991). All T cell cytotoxicity assays were co-cultured for 72 h, after which luminescence was measured following the addition of 150 μg/mL D-luciferin on the CLARIOstar Plus microplate reader (BMG LABTECH) and analyzed using MARS data analysis (BMG LABTECH). SC-islet cell survival was calculated as a percentage relative to luminescence in the absence of T cells (test SC-islet cell luminescence/no T cell SC-islet cell luminescence x 100).
In vitro NK cell cytotoxicity assays
WT, B2M−/− and BEC SC-islet cells were seeded in NK cell medium (NK MACS medium (Milteny Biotec, 130-114-429) + 5% Human AB serum (Valley Biomed, HP1022HI) + 5% HyClone FBS (GE Healthcare) + 0.5 ng/mL rhIL-2) at 2 × 104 cells/well of a 96-well black round-bottom ultra-low attachment plate (Corning, 4591). Primary NK cells (n = 5 donors) or NK-92 MI cells (ATCC, CRL-2408) cultured in NK cell media for 5 days were added to SC-islet cells at 1:1 and 10:1 effector:target ratios. K562-Luc2 (Biocytogen, BCG-PS-015-luc) and Raji-GFP-Luc2 (Biocytogen, BCG-PS-087-luc) cells were used as positive and negative controls respectively. In some experiments, primary NK cells were pre-treated with 50 ug/mL anti-human TIGIT (Biolegend, 372719) or anti-SIRPα blocking peptide (MyBiosource, MBS822365) for 2 h and during the assay. All NK cell cytotoxicity assays were co-cultured for 5 h, after which luminescence and SC-islet cell survival was measured as described above.
In vivo NK cell cytotoxicity
2.5 × 105 WT and B2M−/− SC-islet cell clusters were resuspended either alone or with 7.5 × 105 primary human NK cells (pre-treated with 0.5 ng/mL rhIL-2 for 12 h) in phenol-free Matrigel (Corning, 356237) and transplanted subcutaneously in Scid/beige mice (n = 5). Bioluminescence was measured 10 min after i.p injection of 10 μL/g D-luciferin (15 mg/mL) on days 1 and 5 following cell transplantation on the IVIS SpectrumCT as previously described.20
Single cell RNA-sequencing analyses
For RNA expression analysis, datasets from41 and7 were downloaded from GSE114412, and for40 from GSE151117. Data was re-analyzed using Scanpy.63 For Baron et al.41 and Veres et al.,7 pseudobulk TPM values were used as previously published. For Augsornworawat et al.,40 pseudobulk values were computed using the original cell type labels provided by the authors, by summing raw counts across labeled cells, followed by TPM normalization.
Endothelial cell differentiation
WT and B2M−/− hESCs were seeded in mTesR1 + 10 μM Y27632 at a concentration of 7.5 × 104 cells/well of a Matrigel-coated 6-well tissue-culture plate (Corning, 3516). Thereafter, cells were differentiated into endothelial cells using STEMdiff™ Endothelial Differentiation Kit (STEMCELL Technologies, 08005) according to the manufacturer’s instructions. Differentiation efficiency was quantified by FACS analysis after staining endothelial cells with Pacific Blue anti-human CD31 (Biologend, 303113). Pacific Blue mouse IgG1 (Biolegend, 400131) was used as an isotype control. All antibodies were used at 1:100 unless otherwise stated.
Bulk RNA sequencing
Magnetically-enriched CD49a+ SC-β cells and SC-Endothelial cells were seeded in 6-well plates at 1 × 106 cells/well (in triplicate) and treated with and without 10 ng/mL IFN-γ for 24 h. Cells were then harvested using Accutase and the pellets snap-frozen on dry-ice. RNA extractions, library preparations and sequencing reactions were conducted at GENEWIZ, LLC. (South Plainfield, NJ, USA). Total RNA was extracted from cell pellet samples using RNeasy Plus Mini Kit (Qiagen, Germantown, MD, USA) and quantified using Qubit Fluorometer (Life Technologies, Carlsbad, CA, USA). SMART-Seq v4 Ultra Low Input Kit for Sequencing was used for full-length cDNA synthesis and amplification (Clontech, 634894), and Illumina Nextera XT library (Illumina, FC-131-1096) was used for sequencing library preparation.
Multiplexed sequencing libraries were loaded on the Illumina HiSeq 2000 instrument (Illumina, SY-401-1001) and sequenced using a 2 × 150 Paired End (PE) configuration. Image analysis and base calling were conducted by the HiSeq Control Software (HCS). Raw sequence data (.bcl files) generated from Illumina HiSeq was converted into FASTQ files and de-multiplexed using Illumina’s bcl2fastq 2.17 software. One mismatch was allowed for index sequence identification.
After investigating the quality of the raw data, sequence reads were trimmed to remove possible adapter sequences and nucleotides with poor quality using Trimmomatic v.0.36. The trimmed reads were mapped to the Homo sapiens reference genome available on ENSEMBL using the STAR aligner v.2.5.2b. The STAR aligner is a splice aligner that detects splice junctions and incorporates them to help align the entire read sequences. BAM files were generated as a result of this step. Unique gene hit counts were calculated by using feature Counts from the Subread package v.1.5.2. Only unique reads that fell within exon regions were counted.
After extraction of gene hit counts, the gene hit counts table was used for downstream differential expression analysis. Using DESeq2, a comparison of gene expression between the groups of samples was performed. The Wald test was used to generate p values and Log2 fold changes. Genes with adjusted p values <0.05 and absolute log2 fold changes >1 were called differentially expressed genes for each comparison.
NK cell ligand analysis
WT and B2M−/− SC-islet cells and SC-endothelial cells were washed twice with washing buffer and blocked for 30 min on ice with blocking buffer. Cells were then stained for 30 min on ice in blocking buffer with APC anti-human CD47 (Biolegend, 323123), APC anti-human CD324 (E-Cadherin) (Biolegend, 324107), PE anti-human CD325 (N-Cadherin) (Biolegend, 350806), PE anti-human CD112 (Nectin-2) (Biolegend, 337409), APC anti-human CD155 (PVR) (Biolegend, 337617), PE anti-human CD111 (Nectin-1) (Biolegend, 340404), APC anti-human MICA/MICB (Biolegend, 320907), AF647 anti-human PCNA (Biolegend, 307912), PE anti-human BAG6 (Abcam, ab210838), APC anti-human CD48 (Biolegend, 336713), PE anti-human CD70 (Biolegend, 355103), mouse anti-human CD113 (Nectin-3) (Millipore-Sigma, MABT63), APC anti-human ULBP1 (R&D, FAB1380A), PE anti-human ULBP2/5/6 (R&D, FAB1298P), APC anti-human CLEC2D (R&D, FAB3480A) and PE anti-human CD72 (Biolegend, 316207). PE Mouse IgG1 (Biolegend, 400112), APC mouse IgG1 (Biolegend, 400121), APC mouse IgG2a (Biolegend, 400221), PE Mouse IgG2a (Biolegend, 400213), APC rat IgG1 (Biolegend, 401903), PE rabbit IgG (Cell Signaling technology, 5742S) and goat anti-mouse 488 (Thermofisher, A-11001, 1:300) served as isotype controls. All antibodies were used at 1:100 unless otherwise stated.
NK cell receptor analysis
Primary NK or NK92mi cells were washed twice with washing buffer and blocked for 30 min on ice with blocking buffer. Cells were then stained for 30 min on ice in blocking buffer with Pacific Blue anti-human CD56 (Biolegend, 362519, 1:100), PE anti-human NKG2C (Biolegend, 375003, 1:100) and APC anti-human NKG2A (Biolegend, 375107, 1:100), APC anti-human TIGIT (Biolegend, 372705), PE anti-human KLRG1 (Biolegend, 368609), APC anti-CD96 (Biolegend, 338409), PE anti-human SIRPα (Biolegend, 372103), PE/Cy7 anti-human NKp30 (Biolegend, 325213), APC anti-human NKp46 (Biolegend, 331917), PE anti-human NKp80 (Biolegend, 346706), APC anti-human DNAM-1 (Biolegend, 338311) and PE anti-human NKG2D (Biolegend, 320805). Cells were then washed three times and resuspended in PBS + 0.1% BSA for analysis. Pacific Blue mouse IgG1 (Biolegend, 400131), PE mouse IgG1 (Biolegend, 400111), APC mouse IgG1 (Biolegend, 400121), APC mouse IgG2a (Biolegend, 400221), PE mouse IgG2a (Biolegend, 400213) and PE/Cy7 mouse IgG1 (Biolegend, 400125) served as isotype controls. All antibodies were used at 1:20 unless otherwise stated.
Transplantation of HLA-deficient SC-islet cells in humanized mice
Diabetes was induced in NSG-MHC Class I/II KO mice by multiple low-dose (40 mg/kg) streptozotocin (STZ) (Millipore-Sigma, S0130-50MG) i.p. injection as previously described.64 Once animals reached a blood glucose of >500 mg/dL, 5 × 106 WT or B2M−/− SC-islet cells were transplanted under the kidney capsule (n = 12) as previously described.3,5 Blood glucose and body weight was measured twice a week after transplantation.
Ten weeks after transplantation (before PBMC injection) and seven weeks after PBMC injection the function of transplanted cells was assessed by performing in vivo peritoneal glucose-stimulated insulin secretion (IPGTT) as previously described.3,5 Insulin secretion was quantified using the Human Ultrasensitive Insulin ELISA (ALPCO Diagnostics; 80-INSHUU-E01.1.)
Generation of immune-tolerizing SC-islet cells
The cDNAs of human IL-2 mutein, TGF-β and IL-10 were synthesized as a polycistronic gBlock (Genscript) and cloned into the existing GAPluc targeting plasmid as described above to generate the 2B10 plasmid. The human IL-2 sequence was modified by substituting a single amino acid (N88D) as previously described.22 2B10 hESCs were generated by co-nucleofection of the 2B10 targeting plasmid and the GAPDH-targeting RNP as described above. 2B10 SC-islet cells were differentiated as previously described.3,5
In vitro glucose-stimulated insulin secretion
In vitro function was assessed by measuring glucose-stimulated insulin secretion (GSIS) as previously described.3,5 SC-islet clusters and primary human islets (Prodo Laboratories) were washed twice in Krebs buffer (KRB), and preincubated at 37°C for 1 h in KRB containing 2.8 mM glucose (low glucose). Clusters were then challenged with three sequential treatments of alternating low-high-low KRB containing glucose (high; 20 mM), followed by depolarization with low KRB containing 30 mM KCl. Each treatment lasted 30 min, after which 100 μL of supernatant was collected and human insulin quantified using the Human Ultrasensitive Insulin ELISA. Human insulin measurements were normalized by viable cell counts that were acquired by dispersing clusters with TrypLE Express (Thermofisher, 12604013) and counted using a ViCell (Beckman Coulter).
In vitro SC-islet cell cytokine secretion and PBMC cytotoxicity assays
2B10 SC-islet cell clusters were dispersed with TrypLE Express and seeded in 96-well matrigel-coated plates in S3 media at a linear concentration (1, 2, 4, 6, 8 × 104, and 1 × 105 cells/well) in duplicate. After 24 h, supernatants were collected, centrifuged at 3000g for 5 min and the cytokines IL-2, TGF-β and IL-10 quantified using Legend Max ELISAs (IL-2, Biolegend, 431807; TGF-β, Biolegend, 436707; IL-10, Biolegend, 430607) according to the manufacturer’s instructions. For cytotoxicity assays, WT and 2B10 SC-islet cells were co-cultured with human PBMCs as described above.
Xenotransplantation of 2B10 SC-islet cells in B6/albino mice
WT and 2B10 SC-islet cell clusters (5 × 106 cells) were transplanted under the kidney capsule of B6/albino mice (n = 4/group; of which one animal per group was sacrificed at 5 weeks post-transplantation for immunohistochemistry) and graft survival was monitored weekly for nine weeks by bioluminescence imaging following i.p. injection of D-luciferin as described above. At five weeks post-transplantation SC-islet grafts were removed for immunohistochemical analysis of surviving INS+ cells and CD8+ T and FOXP3+ Treg cells.
Immunohistochemistry of SC-islet grafts
The kidneys of B6/Albino mice transplanted with WT and 2B10 SC-islets were excised, fixed overnight in 4% paraformaldehyde (PFA) at room temperature and embedded in paraffin. Sections were pre-cleared with Histo-Clear, rehydrated using an ethanol gradient and antigen fixed by incubating in boiling antigen retrieval reagent (10 mM sodium citrate, pH 6.0) for 50 min. Slides were then blocked in 5% donkey serum for 1h and stained with Guinea pig anti-human Insulin (DAKO, A0564), Rat anti-mouse CD8α (Biolegend, 100702) and Mouse anti-mouse FOXP3 (Biolegend, 320002) overnight at 4°C. The slides were then washed three times, incubated in secondary antibodies goat anti-mouse 594 (Life Technologies, A-11032), goat anti-rat 488 (Life Technologies, A-11006) and goat anti-guinea pig 647 (Life technologies, A-21450) for 2 h at room temperature, washed, mounted in Vectashield with DAPI (Vector Laboratories; H-1200), covered with coverslips and sealed with clear nail polish. Representative regions were imaged using Zeiss.Z2 with Apotome microscope. All primary and secondary antibodies were used at dilution of 1:200 and 1:500 respectively.
Correction of autoimmune diabetes by 2B10 SC-β cells in NOD mice
Once female NOD mice had blood glucose concentrations >250 mg/dL (∼12 weeks of age), CD49a+ magnetically enriched and reaggregated WT and 2B10 SC-β cell clusters (2 × 106 cells) were transplanted under the kidney capsule. Graft survival was monitored by measuring blood glucose levels weekly for 8 weeks.
Quantification and statistical analysis
All data are presented as means ± SD and were analyzed by GraphPad Prism 9 (GraphPad Software). Statistically significant differences were determined either by one-way or two-way ANOVA, with Tukey’s and Sidak’s post-hoc test for multiple comparisons, and two-tailed t test for pairwise comparisons. p values are indicated in the figures as ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.005 and ∗∗∗∗p < 0.001.
Acknowledgments
D.A.M. is the Distinguished Research Fellow at Vertex Pharmaceuticals. This research was performed while D.A.M. was on the faculty at Harvard University. This work was supported by grants from the Harvard Stem Cell Institute (DP-0180-18-02), JDRF (5-COE-2020-967-M-N), and the JPB Foundation (award no. 1094). We would like to thank Ramona Pop for discussions on manuscript content. Some illustrations in the figures were made using BioRender.
Author contributions
Conceptualization, original hypothesis, and design of the study, D.G. and D.A.M.; methodology, D.G., Q.Z., J.H.-R.K., and D.A.M.; investigation, D.G., Q.Z., J.H.-R.K., A.V., E.S., X.W., K.R.B., H.L., and D.A.M.; writing – original draft, D.G.; writing – review & editing, all authors; resources, D.G., Q.Z., J.H.-R.K., and D.A.M.; supervision, D.G., Q.Z., J.H.-R.K., and D.A.M.
Declaration of interests
D.G., Q.Z., and D.A.M. are named on US provisional patent application serial no. 63/340,453, which is related to this work. D.G. is an employee of Inventia Life Sciences. D.A.M. and Q.Z. are employees of Vertex Pharmaceuticals. These companies were not involved in any aspect of the work.
Published: January 3, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2022.100879.
Supplemental information
Data and code availability
Bulk SC-β and SC-Endothelial cell RNA sequencing data has been submitted to the NIH Gene Expression Omnibus (GEO) repository and is available under accession GSE200021. This manuscript analyzes existing, publicly available data. The accession numbers for the datasets are listed in the key resources table. This paper does not report original code. Any additional information required to reanalyze the data reported in the work paper is available from the lead contact upon request.
References
- 1.Atkinson M.A., Maclaren N.K. The pathogenesis of insulin-dependent diabetes mellitus. N. Engl. J. Med. 1994;331:1428–1436. doi: 10.1056/NEJM199411243312107. [DOI] [PubMed] [Google Scholar]
- 2.Shapiro, A.M.J., Ricordi, C., Hering, B.J., Auchincloss, H., Lindblad, R., Robertson, R.P., Secchi, A., Brendel, M.D., Berney, T., Brennan, D.C., et al. International trial of the Edmonton protocol for islet transplantation. N. Engl. J. Med. 2006; 355: 1318-1330. [DOI] [PubMed]
- 3.Millman J.R., Xie C., Van Dervort A., Gürtler M., Pagliuca F.W., Melton D.A. Generation of stem cell-derived beta-cells from patients with type 1 diabetes. Nat. Commun. 2016;7:12379. doi: 10.1038/ncomms11463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nair G.G., Liu J.S., Russ H.A., Tran S., Saxton M.S., Chen R., et al. Recapitulating endocrine cell clustering in culture promotes maturation of human stem-cell-derived beta cells. Nat. Cell Biol. 2019;21:263–274. doi: 10.1038/s41556-018-0271-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pagliuca F.W., Millman J.R., Gürtler M., Segel M., Van Dervort A., Ryu J.H., et al. Generation of functional human pancreatic beta cells in vitro. Cell. 2014;159:428–439. doi: 10.1016/j.cell.2014.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rezania A., Bruin J.E., Arora P., Rubin A., Batushansky I., Asadi A., et al. Reversal of diabetes with insulin-producing cells derived in vitro from human pluripotent stem cells. Nat. Biotechnol. 2014;32:1121–1133. doi: 10.1038/nbt.3033. [DOI] [PubMed] [Google Scholar]
- 7.Veres A., Faust A.L., Bushnell H.L., Engquist E.N., Kenty J.H.R., Harb G., et al. Charting cellular identity during human in vitro beta-cell differentiation. Nature. 2019;569:368–373. doi: 10.1038/s41586-019-1168-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Russ H.A., Parent A.V., Ringler J.J., Hennings T.G., Nair G.G., Shveygert M., Guo T., Puri S., Haataja L., Cirulli V., et al. Controlled induction of human pancreatic progenitors produces functional beta-like cells in vitro. EMBO J. 2015;34:1759–1772. doi: 10.15252/embj.201591058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.D'Amour K.A., Bang A.G., Eliazer S., Kelly O.G., Agulnick A.D., Smart N.G., Moorman M.A., Kroon E., Carpenter M.K., Baetge E.E. Production of pancreatic hormone-expressing endocrine cells from human embryonic stem cells. Nat. Biotechnol. 2006;24:1392–1401. doi: 10.1038/nbt1259. [DOI] [PubMed] [Google Scholar]
- 10.Hartemann A., Bensimon G., Payan C.A., Jacqueminet S., Bourron O., Nicolas N., Fonfrede M., Rosenzwajg M., Bernard C., Klatzmann D. Low-dose interleukin 2 in patients with type 1 diabetes: a phase 1/2 randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol. 2013;1:295–305. doi: 10.1016/S2213-8587(13)70113-X. [DOI] [PubMed] [Google Scholar]
- 11.Herold K.C., Bundy B.N., Long S.A., Bluestone J.A., DiMeglio L.A., Dufort M.J., Gitelman S.E., Gottlieb P.A., Krischer J.P., Linsley P.S., et al. An anti-CD3 antibody, teplizumab, in relatives at risk for type 1 diabetes. N. Engl. J. Med. 2019;381:603–613. doi: 10.1056/NEJMoa1902226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alagpulinsa D.A., Cao J.J.L., Driscoll R.K., Sîrbulescu R.F., Penson M.F.E., Sremac M., Engquist E.N., Brauns T.A., Markmann J.F., Melton D.A., Poznansky M.C. Alginate-microencapsulation of human stem cell-derived β cells with CXCL12 prolongs their survival and function in immunocompetent mice without systemic immunosuppression. Am. J. Transplant. 2019;19:1930–1940. doi: 10.1111/ajt.15308. [DOI] [PubMed] [Google Scholar]
- 13.Bochenek M.A., Veiseh O., Vegas A.J., McGarrigle J.J., Qi M., Marchese E., Omami M., Doloff J.C., Mendoza-Elias J., Nourmohammadzadeh M., et al. Alginate encapsulation as long-term immune protection of allogeneic pancreatic islet cells transplanted into the omental bursa of macaques. Nat. Biomed. Eng. 2018;2:810–821. doi: 10.1038/s41551-018-0275-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Parent A.V., Faleo G., Chavez J., Saxton M., Berrios D.I., Kerper N.R., Tang Q., Hebrok M. Selective deletion of human leukocyte antigens protects stem cell-derived islets from immune rejection. Cell Rep. 2021;36:109538. doi: 10.1016/j.celrep.2021.109538. [DOI] [PubMed] [Google Scholar]
- 15.Yoshihara E., O’Connor C., Gasser E., Wei Z., Oh T.G., Tseng T.W., Wang D., Cayabyab F., Dai Y., Yu R.T., et al. Immune-evasive human islet-like organoids ameliorate diabetes. Nature. 2020;586:606–611. doi: 10.1038/s41586-020-2631-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sintov E., Nikolskiy I., Barrera V., Hyoje-Ryu Kenty J., Atkin A.S., Gerace D., Ho Sui S.J., Boulanger K., Melton D.A. Whole-genome CRISPR screening identifies genetic manipulations to reduce immune rejection of stem cell-derived islets. Stem Cell Rep. 2022;17:1976–1990. doi: 10.1016/j.stemcr.2022.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Monti P., Scirpoli M., Maffi P., Ghidoli N., De Taddeo F., Bertuzzi F., Piemonti L., Falcone M., Secchi A., Bonifacio E. Islet transplantation in patients with autoimmune diabetes induces homeostatic cytokines that expand autoreactive memory T cells. J. Clin. Invest. 2008;118:1806–1814. doi: 10.1172/JCI35197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stegall M.D., Lafferty K.J., Kam I., Gill R.G. Evidence of recurrent autoimmunity in human allogeneic islet transplantation. Transplantation. 1996;61:1272–1274. doi: 10.1097/00007890-199604270-00027. [DOI] [PubMed] [Google Scholar]
- 19.Abou-Daya K.I., Tieu R., Zhao D., Rammal R., Sacirbegovic F., Williams A.L., Shlomchik W.D., Oberbarnscheidt M.H., Lakkis F.G. Resident memory T cells form during persistent antigen exposure leading to allograft rejection. Sci. Immunol. 2021;6:eabc8122. doi: 10.1126/sciimmunol.abc8122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gerace D., Boulanger K.R., Hyoje-Ryu Kenty J., Melton D.A. Generation of a heterozygous GAPDH-Luciferase human ESC line (HVRDe008-A-1) for in vivo monitoring of stem cells and their differentiated progeny. Stem Cell Res. 2021;53:102371. doi: 10.1016/j.scr.2021.102371. [DOI] [PubMed] [Google Scholar]
- 21.Sintov E., Gerace D., Melton D.A. A human ESC line for efficient CRISPR editing of pluripotent stem cells. Stem Cell Res. 2021;57:102591. doi: 10.1016/j.scr.2021.102591. [DOI] [PubMed] [Google Scholar]
- 22.Peterson L.B., Bell C.J.M., Howlett S.K., Pekalski M.L., Brady K., Hinton H., Sauter D., Todd J.A., Umana P., Ast O., et al. A long-lived IL-2 mutein that selectively activates and expands regulatory T cells as a therapy for autoimmune disease. J. Autoimmun. 2018;95:1–14. doi: 10.1016/j.jaut.2018.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Khoryati L., Pham M.N., Sherve M., Kumari S., Cook K., Pearson J., Bogdani M., Campbell D.J., Gavin M.A. An IL-2 mutein engineered to promote expansion of regulatory T cells arrests ongoing autoimmunity in mice. Sci. Immunol. 2020;5:eaba5264. doi: 10.1126/sciimmunol.aba5264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Horwitz D.A., Zheng S.G., Wang J., Gray J.D. Critical role of IL-2 and TGF-beta in generation, function and stabilization of Foxp3+CD4+ Treg. Eur. J. Immunol. 2008;38:912–915. doi: 10.1002/eji.200738109. [DOI] [PubMed] [Google Scholar]
- 25.Riolobos L., Hirata R.K., Turtle C.J., Wang P.R., Gornalusse G.G., Zavajlevski M., Riddell S.R., Russell D.W. HLA engineering of human pluripotent stem cells. Mol. Ther. 2013;21:1232–1241. doi: 10.1038/mt.2013.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang D., Quan Y., Yan Q., Morales J.E., Wetsel R.A. Targeted disruption of the β2-microglobulin gene minimizes the immunogenicity of human embryonic stem cells. Stem Cells Transl. Med. 2015;4:1234–1245. doi: 10.5966/sctm.2015-0049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mattapally S., Pawlik K.M., Fast V.G., Zumaquero E., Lund F.E., Randall T.D., Townes T.M., Zhang J. Human leukocyte antigen class I and II knockout human induced pluripotent stem cell-derived cells: universal donor for cell therapy. J. Am. Heart Assoc. 2018;7:e010239. doi: 10.1161/JAHA.118.010239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gornalusse G.G., Hirata R.K., Funk S.E., Riolobos L., Lopes V.S., Manske G., Prunkard D., Colunga A.G., Hanafi L.A., Clegg D.O., et al. HLA-E-expressing pluripotent stem cells escape allogeneic responses and lysis by NK cells. Nat. Biotechnol. 2017;35:765–772. doi: 10.1038/nbt.3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Castro-Gutierrez R., Alkanani A., Mathews C.E., Michels A., Russ H.A. Protecting stem cell derived pancreatic beta-like cells from diabetogenic T cell recognition. Front. Endocrinol. 2021;12:707881. doi: 10.3389/fendo.2021.707881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Freeman G.J., Long A.J., Iwai Y., Bourque K., Chernova T., Nishimura H., Fitz L.J., Malenkovich N., Okazaki T., Byrne M.C., et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 2000;192:1027–1034. doi: 10.1084/jem.192.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kleiveland C.R. In: The Impact of Food Bioactives on Health: in vitro and ex vivo models. Verhoeckx K., Cotter P., López-Expósito I., Kleiveland C., Lea T., Mackie A., Requena T., Swiatecka D., Wichers H., editors. Springer International Publishing; Cham: 2015. Peripheral blood mononuclear cells. [PubMed] [Google Scholar]
- 32.Chen L., Flies D.B. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 2013;13:227–242. doi: 10.1038/nri3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Garcia-Diaz A., Shin D.S., Moreno B.H., Saco J., Escuin-Ordinas H., Rodriguez G.A., Zaretsky J.M., Sun L., Hugo W., Wang X., et al. Interferon receptor signaling pathways regulating PD-L1 and PD-L2 expression. Cell Rep. 2017;19:1189–1201. doi: 10.1016/j.celrep.2017.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Imaizumi T., Kumagai M., Sasaki N., Kurotaki H., Mori F., Seki M., Nishi N., Fujimoto K., Tanji K., Shibata T., et al. Interferon-gamma stimulates the expression of galectin-9 in cultured human endothelial cells. J. Leukoc. Biol. 2002;72:486–491. [PubMed] [Google Scholar]
- 35.Benci J.L., Xu B., Qiu Y., Wu T.J., Dada H., Twyman-Saint Victor C., Cucolo L., Lee D.S.M., Pauken K.E., Huang A.C., et al. Tumor interferon signaling regulates a multigenic resistance program to immune checkpoint blockade. Cell. 2016;167:1540–1554.e12. doi: 10.1016/j.cell.2016.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wagner A.H., Gebauer M., Pollok-Kopp B., Hecker M. Cytokine-inducible CD40 expression in human endothelial cells is mediated by interferon regulatory factor-1. Blood. 2002;99:520–525. doi: 10.1182/blood.v99.2.520. [DOI] [PubMed] [Google Scholar]
- 37.Rigau M., Ostrouska S., Fulford T.S., Johnson D.N., Woods K., Ruan Z., McWilliam H.E.G., Hudson C., Tutuka C., Wheatley A.K., et al. Butyrophilin 2A1 is essential for phosphoantigen reactivity by γδ T cells. Science. 2020;367:eaay5516. doi: 10.1126/science.aay5516. [DOI] [PubMed] [Google Scholar]
- 38.Deuse T., Hu X., Agbor-Enoh S., Jang M.K., Alawi M., Saygi C., Gravina A., Tediashvili G., Nguyen V.Q., Liu Y., et al. The SIRPα-CD47 immune checkpoint in NK cells. J. Exp. Med. 2021;218:e20200839. doi: 10.1084/jem.20200839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Suzuki D., Flahou C., Yoshikawa N., Stirblyte I., Hayashi Y., Sawaguchi A., Akasaka M., Nakamura S., Higashi N., Xu H., et al. iPSC-derived platelets depleted of HLA class I are inert to anti-HLA class I and natural killer cell immunity. Stem Cell Rep. 2020;14:49–59. doi: 10.1016/j.stemcr.2019.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Augsornworawat P., Maxwell K.G., Velazco-Cruz L., Millman J.R. Single-cell transcriptome profiling reveals β cell maturation in stem cell-derived islets after transplantation. Cell Rep. 2020;32:108067. doi: 10.1016/j.celrep.2020.108067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Baron M., Veres A., Wolock S.L., Faust A.L., Gaujoux R., Vetere A., Ryu J.H., Wagner B.K., Shen-Orr S.S., Klein A.M., et al. A single-cell transcriptomic map of the human and mouse pancreas reveals inter- and intra-cell population structure. Cell Syst. 2016;3:346–360.e4. doi: 10.1016/j.cels.2016.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.van der Torren C.R., Zaldumbide A., Duinkerken G., Brand-Schaaf S.H., Peakman M., Stangé G., Martinson L., Kroon E., Brandon E.P., Pipeleers D., Roep B.O. Immunogenicity of human embryonic stem cell-derived beta cells. Diabetologia. 2017;60:126–133. doi: 10.1007/s00125-016-4125-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Klimosch S.N., Bartel Y., Wiemann S., Steinle A. Genetically coupled receptor–ligand pair NKp80-AICL enables autonomous control of human NK cell responses. Blood. 2013;122:2380–2389. doi: 10.1182/blood-2013-01-479790. [DOI] [PubMed] [Google Scholar]
- 44.Brehm M.A., Kenney L.L., Wiles M.V., Low B.E., Tisch R.M., Burzenski L., Mueller C., Greiner D.L., Shultz L.D. Lack of acute xenogeneic graft- versus-host disease, but retention of T-cell function following engraftment of human peripheral blood mononuclear cells in NSG mice deficient in MHC class I and II expression. FASEB J. 2019;33:3137–3151. doi: 10.1096/fj.201800636R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Viricel C., Ahmed M., Barakat K. Human PD-1 binds differently to its human ligands: a comprehensive modeling study. J. Mol. Graph. Model. 2015;57:131–142. doi: 10.1016/j.jmgm.2015.01.015. [DOI] [PubMed] [Google Scholar]
- 46.Finger L.R., Pu J., Wasserman R., Vibhakar R., Louie E., Hardy R.R., Burrows P.D., Billips L.G. The human PD-1 gene: complete cDNA, genomic organization, and developmentally regulated expression in B cell progenitors. Gene. 1997;197:177–187. doi: 10.1016/s0378-1119(97)00260-6. [DOI] [PubMed] [Google Scholar]
- 47.Han X., Wang M., Duan S., Franco P.J., Kenty J.H.-R., Hedrick P., Xia Y., Allen A., Ferreira L.M.R., Strominger J.L., et al. Generation of hypoimmunogenic human pluripotent stem cells. Proc. Natl. Acad. Sci. USA. 2019;116:10441–10446. doi: 10.1073/pnas.1902566116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Leite N.C., Pelayo G.C., Melton D.A. Genetic manipulation of stress pathways can protect stem-cell-derived islets from apoptosis in vitro. Stem Cell Rep. 2022;17:766–774. doi: 10.1016/j.stemcr.2022.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang R., Sun L., Li C.-F., Wang Y.-H., Yao J., Li H., Yan M., Chang W.C., Hsu J.M., Cha J.H., et al. Galectin-9 interacts with PD-1 and TIM-3 to regulate T cell death and is a target for cancer immunotherapy. Nat. Commun. 2021;12:832. doi: 10.1038/s41467-021-21099-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Heusschen R., Griffioen A.W., Thijssen V.L. Galectin-9 in tumor biology: a jack of multiple trades. Biochim. Biophys. Acta. 2013;1836:177–185. doi: 10.1016/j.bbcan.2013.04.006. [DOI] [PubMed] [Google Scholar]
- 51.Stancill J.S., Kasmani M.Y., Khatun A., Cui W., Corbett J.A. Single-cell RNA sequencing of mouse islets exposed to proinflammatory cytokines. Life Sci. Alliance. 2021;4:e202000949. doi: 10.26508/lsa.202000949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Herndler-Brandstetter D., Shan L., Yao Y., Stecher C., Plajer V., Lietzenmayer M., Strowig T., de Zoete M.R., Palm N.W., Chen J., et al. Humanized mouse model supports development, function, and tissue residency of human natural killer cells. Proc. Natl. Acad. Sci. USA. 2017;114:9626–9634. doi: 10.1073/pnas.1705301114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wyburn K.R., Jose M.D., Wu H., Atkins R.C., Chadban S.J. The role of macrophages in allograft rejection. Transplantation. 2005;80:1641–1647. doi: 10.1097/01.tp.0000173903.26886.20. [DOI] [PubMed] [Google Scholar]
- 54.Oberbarnscheidt M.H., Zeng Q., Li Q., Dai H., Williams A.L., Shlomchik W.D., Rothstein D.M., Lakkis F.G. Non-self recognition by monocytes initiates allograft rejection. J. Clin. Invest. 2014;124:3579–3589. doi: 10.1172/JCI74370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhuang Q., Liu Q., Divito S.J., Zeng Q., Yatim K.M., Hughes A.D., Rojas-Canales D.M., Nakao A., Shufesky W.J., Williams A.L., et al. Graft-infiltrating host dendritic cells play a key role in organ transplant rejection. Nat. Commun. 2016;7:12623. doi: 10.1038/ncomms12623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Muller Y.D., Ferreira L.M.R., Ronin E., Ho P., Nguyen V., Faleo G., Zhou Y., Lee K., Leung K.K., Skartsis N., et al. Precision engineering of an anti-HLA-A2 chimeric antigen receptor in regulatory T cells for transplant immune tolerance. Front. Immunol. 2021;12:686439. doi: 10.3389/fimmu.2021.686439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hu M., Rogers N.M., Li J., Zhang G.Y., Wang Y.M., Shaw K., O'Connell P.J., Alexander S.I. Antigen specific regulatory T cells in kidney transplantation and other tolerance settings. Front. Immunol. 2021;12:717594. doi: 10.3389/fimmu.2021.717594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wagner J.C., Tang Q. CAR-tregs as a strategy for inducing graft tolerance. Curr. Transplant. Rep. 2020;7:205–214. doi: 10.1007/s40472-020-00285-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lim D., Sreekanth V., Cox K.J., Law B.K., Wagner B.K., Karp J.M., Choudhary A. Engineering designer beta cells with a CRISPR-Cas9 conjugation platform. Nat. Commun. 2020;11:4043. doi: 10.1038/s41467-020-17725-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang X., Wang K., Yu M., Velluto D., Hong X., Wang B., Chiu A., Melero-Martin J.M., Tomei A.A., Ma M. Engineered immunomodulatory accessory cells improve experimental allogeneic islet transplantation without immunosuppression. Sci. Adv. 2022;8:eabn0071. doi: 10.1126/sciadv.abn0071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mandal P.K., Ferreira L.M.R., Collins R., Meissner T.B., Boutwell C.L., Friesen M., Vrbanac V., Garrison B.S., Stortchevoi A., Bryder D., et al. Efficient ablation of genes in human hematopoietic stem and effector cells using CRISPR/Cas9. Cell Stem Cell. 2014;15:643–652. doi: 10.1016/j.stem.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kaiser B.K., Barahmand-pour F., Paulsene W., Medley S., Geraghty D.E., Strong R.K. Interactions between NKG2x immunoreceptors and HLA-E ligands display overlapping affinities and thermodynamics. J. Immunol. 2005;174:2878–2884. doi: 10.4049/jimmunol.174.5.2878. [DOI] [PubMed] [Google Scholar]
- 63.Wolf F.A., Angerer P., Theis F.J. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 2018;19:15. doi: 10.1186/s13059-017-1382-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Furman B.L. Streptozotocin-induced diabetic models in mice and rats. Curr. Protoc. 2021;1:e78. doi: 10.1002/cpz1.78. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Bulk SC-β and SC-Endothelial cell RNA sequencing data has been submitted to the NIH Gene Expression Omnibus (GEO) repository and is available under accession GSE200021. This manuscript analyzes existing, publicly available data. The accession numbers for the datasets are listed in the key resources table. This paper does not report original code. Any additional information required to reanalyze the data reported in the work paper is available from the lead contact upon request.