

Abstract
Objective.
To develop an experimental method for routine isolation and short-term culture of primary lymphatic endothelial cells from collecting vessels.
Methods.
Lymphatic endothelial cell tubes (LECTs) were isolated from micro-dissected collecting vessels. LECTs were allowed to attach and grow for ~3 weeks before being passaged. Non-purified cultures were partially characterized by immunofluorescence and RT-PCR at passages 1–2.
Results.
The method was validated in cultures of primary lymphatic endothelial cells (LECs) from male and female mice. After 1 or 2 passages, >60% of the LECs maintained expression of Prox1. Expression of 22 different genes was assessed using RT-PCR. Prox1, Vegfr3, eNos, Cdh5, Pecam1, Cx43, Cx37, and Cx47, among others, were expressed in these short-term cultured LECs, while Myh11, Cnn1, Desmin, and Cd11b were not detected. Prox1 expression, as determined by western blotting, was similar in cultured LECs from age-matched male and female mice. Confocal imaging of intracellular calcium in cultures of primary LECs from Cdh5-GCaMP8 mice demonstrated that a functional phenotype was maintained, similar to lymphatic endothelial cells in freshly isolated vessels.
Conclusions.
This method provides an innovative tool for routine isolation and study of primary LECs from specific collecting lymphatic vessels from any mouse, and in fact, from other species.
Keywords: Cell culture, lymphatic endothelial cells, primary endothelial cells, collecting lymphatic vessel, calcium imaging
INTRODUCTION
For over a century, the isolation and growth of cells for their study in-vitro under controlled conditions has been one of the most utilized experimental approaches in the field of biology. At present, a large variety of vascular cell lines, including lymphatic endothelial cells, are now commercially available. As with any other experimental technique, the culturing of cells has several limitations. In an attempt to make studies that utilize cell cultures both accessible and physiologically relevant, researchers have devoted significant efforts towards maintaining cell lines, establishing reproducible protocols, and minimizing limitations and artifacts from a variety of cell lines, including lymphatic endothelial cells (LECs)1–16. Commercially available cell lines undergo extensive characterization and are rigorously assessed for quality, stability, and performance; however, controlling important physiological factors such as sex17, age, and anatomical origin among others remains limited.
Mice continue to be utilized as models of disease due to the availability of a vast number of already existing genetically modified mice and the ability to introduce novel genetic mutations in specific cell types. Relevant to this study, when assessing the functional role of specific genes in regulating the function of lymphatic and vascular endothelial cells in-vitro, i.e., in cultures of commercial cell lines, the use of genetically modified mice is limited; instead, the use of interfering RNAs continue to be the gold standard18–20. This is primarily due to the difficulty of isolating and culturing primary endothelial cells. In the specific case of LECs, recent studies demonstrated important genetic profile differences between lymphatic capillaries and collecting lymphatic vessels, which highlights the importance for controlling the anatomical and tissue-specific origin of cells for in-vitro studies. In this regard, an important contribution, and significant advance to the field, was previously accomplished by Hayes H. et al., who developed an experimental approach for isolating and culturing primary LECs from rat mesenteric collecting lymphatic vessels14.
In this study, we present a novel experimental protocol for the isolation and short-term culture of primary LECs from specific collecting lymphatic vessels. This protocol incorporates lymphatic vessel micro-dissection techniques and experimental methods initially developed for the isolation of endothelial cell tubes from resistance arteries of the mouse,21 that were later adapted by us for their application towards lymphatic vessels22. We were able to partially characterize these cultures of primary LECs by passage 2, limiting potential changes in phenotype. Further, we present data that highlights a few of the major advantages of the protocol described herein. Importantly, while the present study focuses on LECs, this method may also be applied to isolate and culture primary endothelial cells from specific blood vessels.
MATERIALS AND METHODS
Animals
C57BL/6J (Cat. No. 000664) WT mice were purchased at 8 weeks of age from The Jackson Laboratory – Bar Harbor, MA, USA. Prox1-GFP mice23, expressing green fluorescence protein in Prox1 positive cells, were a gift from Dr. Young-Kwon Hong (University of Southern California). Cdh5-GCaMP8 (B20) male mice, expressing the genetically encoded Ca2+ indicator in endothelial cells, were obtained from CHROMus at Cornell University and bred in-house with C57BL/6J female mice from the Jackson Laboratory. Male and female mice were used for experiments between 2–6 months of age. Mice were housed under a 12-hour light/dark cycle at 22–25°C and had access to food and water at all times. Mice were anesthetized by means of isoflurane (at 25 g of body weight, induction ~3% at a rate of 400–500 mL/min, maintenance ~2–2.6% at 31–35 mL/min) using a SomnoSuite low-flow anesthesia machine (Kent Scientific, Co). For tissue dissection, mice were placed on a heated pad. Adequate levels of anesthesia were ensured by continuous monitoring for indicators of pain and by assessing loss of pedal and pinna reflexes. Following dissection, animals were euthanized via overdose of isoflurane at 5% and subsequent cervical dislocation. Rat and non-human primate mesenteric tissues were obtained from male Sprague-Dawley rats (4–6 months of age) and male and female Macaca Mulatta (10–15 years of age) monkeys through a tissue-sharing collaboration with the Tulane Department of Comparative Medicine and the Tulane National Primate Research Center respectively.
Solutions and Chemicals
Krebs-BSA Solution.
146.9 mM NaCl, 4.7 mM KCl, 2 mM CaCl2·2H2O, 1.2 mM MgSO4, 1.2 mM NaH2PO4·H2O, 3 mM NaHCO3, 1.5 mM Na-HEPES, 5 mM D-glucose, and 0.5% BSA (pH = 7.4). All chemicals were obtained from Sigma-Aldrich (St. Louis, MO).
Physiological Salt Solution (PSS).
140 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 10 mM Hepes, and 10 mM D-glucose (pH = 7.4). All chemicals were obtained from Sigma-Aldrich (St. Louis, MO).
Cell Culture Media.
Primary LECs were cultured in complete DMEM media (Gibco 11995-065) containing 25 mM D-glucose and 1 mM sodium pyruvate, and supplemented with FBS (Sigma F2442, 10%), Bovine Brain Extract (BBE, Lonza CC-4098, 10 μg/mL), Heparin (Sigma H3393-100KU, 50 ug/mL), Penicillin G and streptomycin (Gibco 15140-122, 1%), MEM-NEAA (Gibco 11140-050, 1%), and L-Glutamine (Gibco 25030-081, 1%).
RT-PCR on Primary Lymphatic Endothelial Cells
Total RNA was extracted from primary lymphatic endothelial cells using the Arcturus PicoPure RNA isolation kit (ThermoFisher Scientific, Waltham, MA) with on-column DNase I treatment (Qiagen, Valencia, CA) according to the manufacturer’s instructions. RNA was eluted with 30 μL nuclease-free water. Purified RNA was then used for cDNA synthesis via reverse transcription using random hexamer and oligod (T) primers, and SuperScript III First-Strand Synthesis System (ThermoFisher Scientific, Waltham, MA). All PCR reactions (20 μL) contained 1 μL of first-strand cDNA mixture as the template, 2 mM MgCl2, 0.25 μM primers, 0.2 mM deoxynucleotide triphosphates; and GoTaq Flexi DNA polymerase (Promega, Madison, WI). The PCR program comprised an initial denaturation step at 95°C for three minutes; followed by 35 cycles of denaturation for 30 seconds at 95°C, annealing for 30 seconds at 58°C, and elongation for 30 seconds at 72°C; and a final extension step at 72°C for five minutes. The expected size of PCR products and primer sequences are listed in Supplemental Table 1. PCR amplification products were separated on a 2% agarose gel by electrophoresis and the DNA bands were visualized by SYBR safe staining24–26.
Western Blotting
In preparation for lysing, cell suspensions were prepared from cultures of primary LECs between passages 1–2 using the Cell Passage Reagent System (Cell Systems, Cat. No. 4Z0-840) according to the manufacturer’s instructions. Cells were transferred to 5 mL centrifuge tubes, where they were spun down by centrifugation at 500 × g for 10 min at 4°C. Cell pellets were then rinsed by resuspension in sterile PBS followed by spinning down at 500 × g for 10 min at 4°C. Finally, PBS was carefully removed, and cell lysates were then prepared in 200 μL of 1X Western Lysis Buffer (Phosphosolutions, Cat.No. 100-LYS) following the manufacturer’s instructions. Cell lysates were thawed, analyzed for total protein content (Thermo Pierce BCA Assay Kit), and fresh 10 μg aliquots of lysate proteins were separated on 4–12% Bis-Tris Plus Gels (Invitrogen) in MES buffer (Invitrogen) for 20 minutes at 200 V. The gel was washed for 10 minutes with 20% ethanol. Proteins were then transferred to a PVDF membrane with the iBlot2 Gel Transfer Device (Invitrogen) using program P0, blocked with iBind Solution (Invitrogen), and incubated at room temperature with primary and secondary antibodies with the iBind Western Device (Invitrogen) for 2.5 hours or overnight. The primary antibodies used in this study were anti-Prox1 rabbit polyclonal antibody (Proteintech, Cat. No. 11067-2-AP) at 1:1000 dilution, and anti-Cyclophilin B rabbit polyclonal antibody (Proteintech, Cat. No. 11607-1-AP) at a dilution of 1:5000. The secondary antibody was Donkey anti-Rabbit IgG(H+L)HRP (Life Technologies #A16037) at a dilution of 1:4000. The membrane was developed with SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific #34577) and the membrane was imaged with the BioRad ChemiDoc Imaging System.
Confocal Fluorescence Microscopy
Equipment.
For confocal fluorescence microscopy, we used a Leica DMI8 inverted fluorescence microscope and an Andor Dragonfly 202 high-speed confocal imaging platform equipped with a 40-μm pinhole disk, Borealis™ enhanced illumination, simultaneous imaging with dual camera capability, and solid state 405, 488, 561, and 637 nm diode laser lines.
Immunofluorescence.
At passages 1–2, primary LECs were seeded onto the culturing surface of collagen coated, glass bottom, 35-mm petri dishes (Mattek, Cat. No. P35GCOL-1.5-14-C) at a concentration of 100,000–200,000 cells per dish. Cells were then incubated in complete DMEM media at 37°C in a 5% CO2 atmosphere of a cell incubator until an even cell monolayer was formed, usually 3–5 days later. Primary LEC-monolayers were then rinsed with warm PBS and fixed in 4% PFA for 15 min, followed by rinsing with PBS. Fixed LECs were permeabilized with a 0.1% Triton X-100 solution in PBS for 20 minutes, then rinsed with PBS, and blocked for non-specific binding using a 5% donkey serum (Sigma, Cat. No. D9663) in PBS for 2 hours at 4°C. Cells were incubated overnight with primary antibodies (anti-Prox1 [1:500] Proteintech Cat. No. 11067-2-AP, anti-Cd144 (VE-cadherin) [1:500]: BD Pharmingen Cat. No. 550548, anti-GFP [1:300]: Invitrogen Cat. No. A-11122) at 4°C, then washed with PBS for 1–2 hours on a rocker platform, replacing the PBS solution every ~30 minutes. Cells were then incubated overnight with secondary antibodies at 1:500 at 4°C. Finally, cells were washed with PBS for 1–2 hours on a rocker platform replacing the PBS solution every ~30 minutes, mounted with ProLong Glass antifade mountant with NucBlue stain (Invitrogen, Cat. No. P36980), and imaged with the confocal microscope described above, using the 405-nm, 488-nm, and 647-nm excitations. Images were acquired using a Zyla PLUS 4.2 Megapixel sCMOS camera and an HCX PL APO 40x/1.10W CORR objective.
Intracellular Ca2+ Imaging.
Characterization of LEC-specific intracellular Ca2+ dynamics was performed on cultures of primary LECs isolated from inguinal axillary collecting lymphatic vessels from Cdh5-GCaMP8 mice. As described in the previous section, at passage 1–2, primary LECs were seeded in glass bottom petri dishes and incubated until forming even cell monolayers. A petri dish heater was utilized to maintain adequate temperature during experimentation. Imaging was performed using the confocal microscope described above, exciting with the 488-nm laser. Events were recorded at 20–30 fps with an iXon Life 888 EMCCD camera (1024 × 1024 pixels, pixel size=13×13 μm). A Leica HC FLUOTAR L 25x/0.95 objective was used for Ca2+ imaging.
Generation of Space Time Maps (STMs).
Fluorescence videos of lymphatic endothelial cells expressing the Ca2+ indicator GCaMP8 were processed and analyzed. Ca2+ STMs representing the time-dependent fluorescence intensity from a line-scan analysis at every position along the field of view were generated as previously described24. For displaying purposes, the resulting 16-bit grayscale STMs were color-coded using the ImageJ’s physics LUT (with red indicating the highest detected fluorescence intensity over a blue background in which the fluorescence intensity was minimum). All video processing and 2-dimensional analyses were performed using custom-written Python-based programs developed by our laboratory.
RESULTS
Isolation, Short-Term Culturing, and Passaging of Primary Lymphatic Endothelial Cells
Vessel Isolation.
Inguinal axillary and mesenteric lymphatic vessels from mice were excised as previously described27–30 (Figure 1 Step 1, P1, and Supplemental Video 1). Short segments of 40 μm stainless steel wire were used to pin down each lymphatic tissue onto a Sylgard-coated dissection chamber filled with room temperature Krebs-BSA buffer. Using micro-dissection techniques, the majority of adipose and connective tissues surrounding the lymphatic vessels were cleared.
Figure 1. Isolation and Culture of Lymphatic Endothelial Cell Tubes (LECTs) from collecting lymphatic vessels.
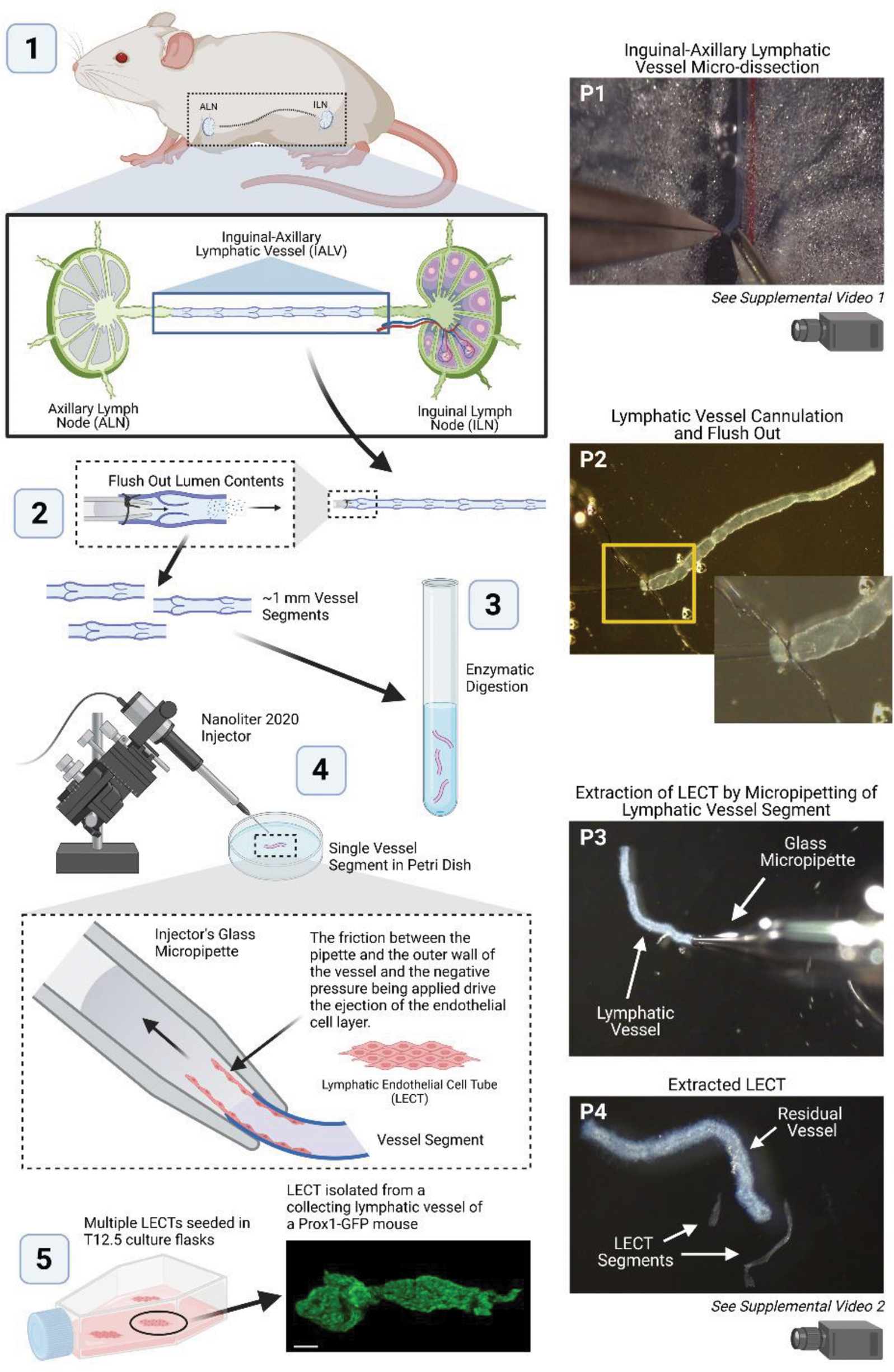
Stepwise depiction of the experimental methodology for isolating LECTs from inguinal axillary lymphatic vessels of a mouse. Pictures of key steps along the process, denoted P1-P4, are shown on the right-side panels. Additional supporting videos can be found in the supplemental materials. This figure was created with BioRender.com.
Preparation of Vessel Segments for Enzymatic Digestion.
In order to minimize contamination from other cell types in our LEC cultures, freshly isolated and cleaned lymphatic vessels were flushed through the lumen. Briefly, using a glass micropipette attached to a syringe containing Krebs-BSA buffer, the vessel lumen was opened at the output end, then cannulated and tied at the input end. Next, by gently lifting the syringe about 5 cm, the pressure at the cannulated end of the vessel was raised and the lumen contents of the vessel were flushed (Figure 1 Step 2 and P2). Importantly, cannulation of the vessel to flush out the vessel lumen contents is recommended, but this step is not critical for the overall completion and success of the protocol. Finally, the vessels were cut into ~2-mm long segments. In average, about 5–8 vessel segments were obtained from a given animal.
Vessel Digestion.
Cleaned vessel segments were transferred to a 4-mL borosilicate glass tube containing ~2 mL of filtered (use a 0.22-μm pore size syringe filter) PSS containing 0.1 mM CaCl2 and 0.5% BSA and then spun down at 4°C and 900 × g for 2–3 minutes. An enzyme cocktail of collagenase A, DTE, and papain (1 mg/mL final concentration for each enzyme), was prepared in a second 4-mL borosilicate glass tube and pre-warmed to 37°C21, 22. The solution from the tube containing the spun down vessel segments was carefully removed and replaced with the pre-warmed enzymes-containing solution. Gentle pipetting allowed for vessels to resuspend. Lymphatic segments were incubated in the digesting enzyme solution for a period of 16–25 minutes at 37°C in a shaking water bath. The vessels were then spun down at 4°C and 900 × g for 2 minutes, the enzyme solution was subsequently carefully removed, and the vessels were then resuspended in cold filtered PSS containing 0.1 mM CaCl2 and 0.5% BSA (Figure 1 Step 3).
Endothelial Cell Tube Extraction.
Next, the partially digested vessels were transferred, one-by-one, to a 35-mm cell culture plastic petri dish filled with filtered PSS containing 0.1 mM CaCl2 and 0.5% BSA. The lymphatic endothelial cell tube (LECT) was then extracted from each vessel segment by means of controlled automated micro-pipetting using a Nanoliter 2020 Injector (World Precision Instruments) under a stereo-microscope as previously described21, 22. Release of LECTs from digested vessels was accomplished by means of adequate pipetting-mediated friction between the inner wall of the glass micropipette and the adventitial/smooth muscle cell layer of the vessel segment. LECTs separated from the smooth muscle layer and were ejected from the inside of the vessel (Figure 1 Step 4, P3 and P4, and Supplemental Video 2). Often, only part of the endothelium was successfully extracted from a vessel, in average, each LECT was ~1 mm long. Intact isolated LECTs were then rinsed twice by pipetting them in and out from petri dishes containing filtered PSS containing CaCl2 and BSA. This rinsing step minimized contamination from non-LECs that may have detached from the lymphatic wall during enzymatic digestion and LECT extraction.
LECT Seeding and Culturing.
A 12.5 cm2 cell culture flask, or a 35-mm cell culture petri dish, was pre-coated with Attachment Factor (Cat. No. 4Z0-210) according to the manufacturer’s instructions, complete culture media was added (2.5 mL or 1 mL respectively), and then the flask, or petri dish, was placed in a cell incubator at 37°C in a 5% CO2 atmosphere and 90% relative humidity. Isolated and rinsed LECTs were then spun down at 900 × g at 4°C for 5 minutes. In a biosafety cabinet, PSS solution was carefully removed, and LECTs were gently resuspended in 1 mL of pre-warmed complete cell culture media (LECTs were not visible by naked eye). Finally, the cell culture media containing the LECTs was transferred to the previously prepared flask, or petri dish, and incubated at 37°C in a 5% CO2 atmosphere (Figure 1 Step 5 and Figure 2A).
Figure 2. Fluorescence Images at Each Stage of LECT Seeding and Expansion.
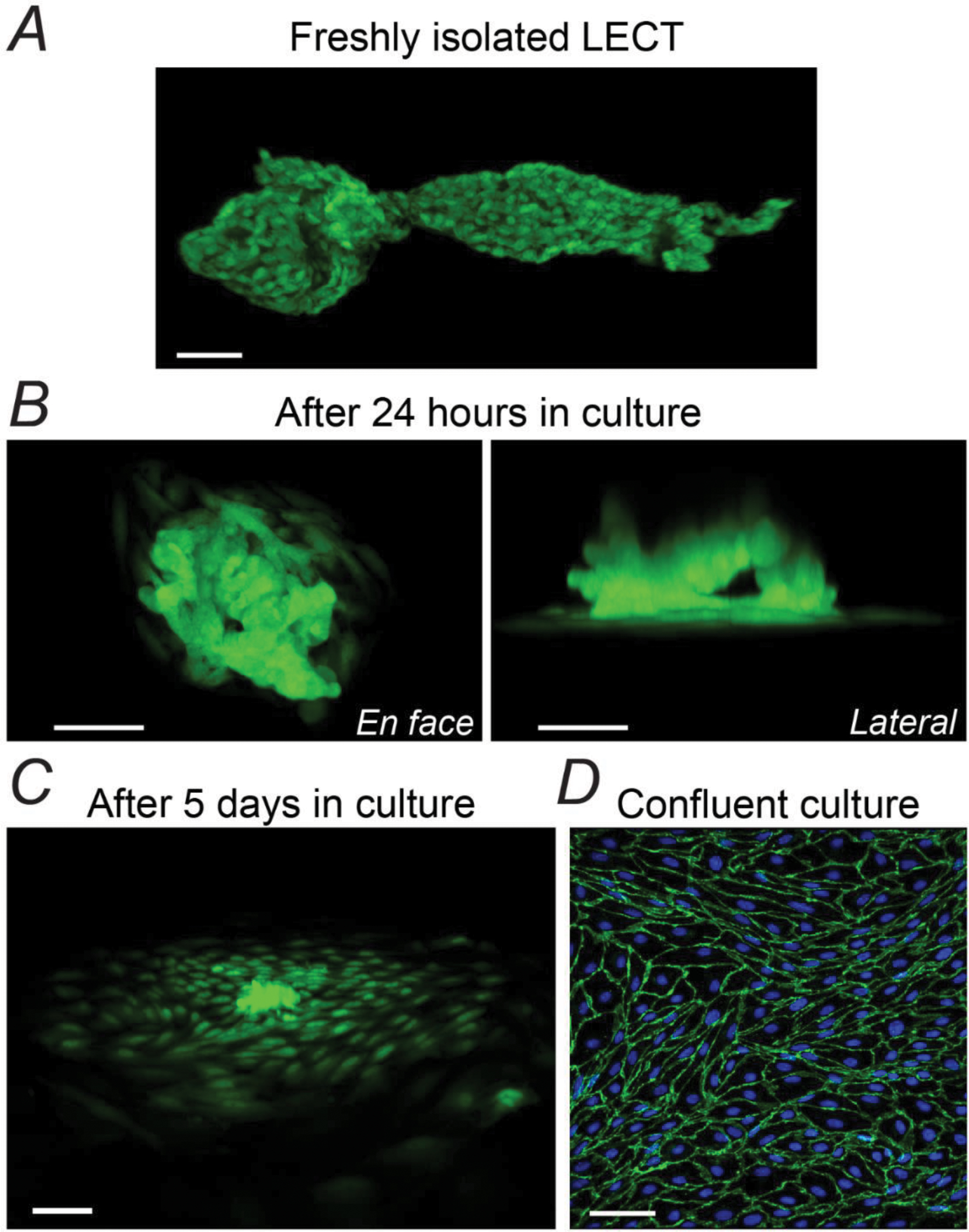
(A-C) Progressive cell attachment and proliferation beginning with an intact LECT isolated from a Prox1-GFP mouse and after 24 hours or 5 days in culture, respectively. GFP fluorescence was imaged on live cells. (D) Immunofluorescent image of a ~70–90% confluent culture of primary LECs after one passage (~6 weeks after LETC isolation and seeding). Fixed culture was co-stained for VE-Cadherin and DAPI. Scale bar equals 50 μm in all panels.
Overnight, LECTs (usually 3–4) attached to the cell-growing surface of the culture flask (or petri dish) and began to slowly expand (Figure 2B). These sites were carefully identified and marked. The seeded LECTs then formed clusters that continued to grow as cells proliferated (Figure 2C). Based on the fluorescence images of LECTs isolated from Prox1-GFP mice and our previously reported calculation of number of LECs per unit of length of lymphatic vessel31, we estimated that each LECT, and initial LEC-clusters, contained approximately 750 cells per cluster, at a density of 20,000–30,000 cells/mm2. Depending on the number of LECTs initially seeded, these clusters were allowed to slowly expand until LECs occupied an area of ~2.5 cm2, i.e., ~20% of the culturing surface of the 12.5 cm2 flask, or ~30% of the culturing surface of a 35-mm petri dish. This usually took ~3 weeks. A pilot set of experiments showed that it would take 6–8 weeks for these initial clusters to reach 75% confluency. Instead, for the experiments included in this study, initial cultures were routinely passaged at ~3 weeks. Attempting to passage clusters of cells before this period of time resulted in significantly slowed cell proliferation or even loss of the culture.
Although efforts were made to minimize contamination, clusters of cells occasionally appeared over time in places where attached LECTs had not been initially identified. These clusters were scraped off upon initial discovery and before initial passage. During the initial passage, all cells were transferred onto a fresh 12.5-cm2 cell culture flask (pre-coated with Attachment Factor as described above), where cells were allowed to expand until reaching 70–90% confluency, usually ~2 weeks later (Figure 2D). Cells were passaged using the Cell Passage Reagent System (Cell Systems, Cat. No. 4Z0-840) following the manufacturer’s protocol. During passage 2, the 70–90% confluent cell culture from the initial passage was split into 1:3, and from this passage on, cells were reseeded into 25-cm2 flasks, or into glass-bottom petri dishes at a concentration of ~100,000–200,000 cells per dish (i.e., ~16,000–32,000 cells/mm2) for calcium imaging or immunofluorescence experiments. For a research study, this strategy would allow for 1-flask or 3-petri dish replicates from the same animal using primary LECs at passage 1, or 3-flask or 4–5-petri dish replicates at passage 2.
Characterization of Primary Lymphatic Endothelial Cell Cultures
Endothelial cells, including those of lymphatic origin, are known to undergo phenotypic transformation in culture. In order to assess whether our primary cultures of LECs would maintain their phenotype, we established cultures of LECs isolated from inguinal axillary collecting lymphatic vessels from WT and Prox1-GFP 4–6-month-old mice (n=6, including male and female mice). Prox1-GFP mice are a useful research tool for studies of the lymphatic vasculature as Prox1 is a known marker of LECs and its function is required for the development of the lymphatic system23, 32. At passages 1–2, we performed immunofluorescence staining for GFP, in the case of LECs from Prox1-GFP mice; or Prox1, in the case of LECs from WT mice, and VE-Cadherin and DAPI (Figure 3). We determined the percentage of LECs that maintained expression of Prox1 by counting and comparing the total number of cells (i.e., DAPI-stained nuclei) present in a given region of interest versus the number of Prox1-expressing cells. For a given culture, data from 3 different regions of interest was collected and values were averaged. Cultures from six different animals at passages 1–2 were characterized. Our analysis showed that the mean percentage of Prox1-expressing cells in our non-purified cultures was 78.3±3.7% (Figure 3O). The residual number of non-Prox1-expressing cells was relatively stable up to passage 2, then, a slow but sustained loss of Prox1 was observed as cells continued to be expanded and/or passaged.
Figure 3. Characterization of Prox1 Expression in Short-Term Cultured LECs from Collecting Vessels.
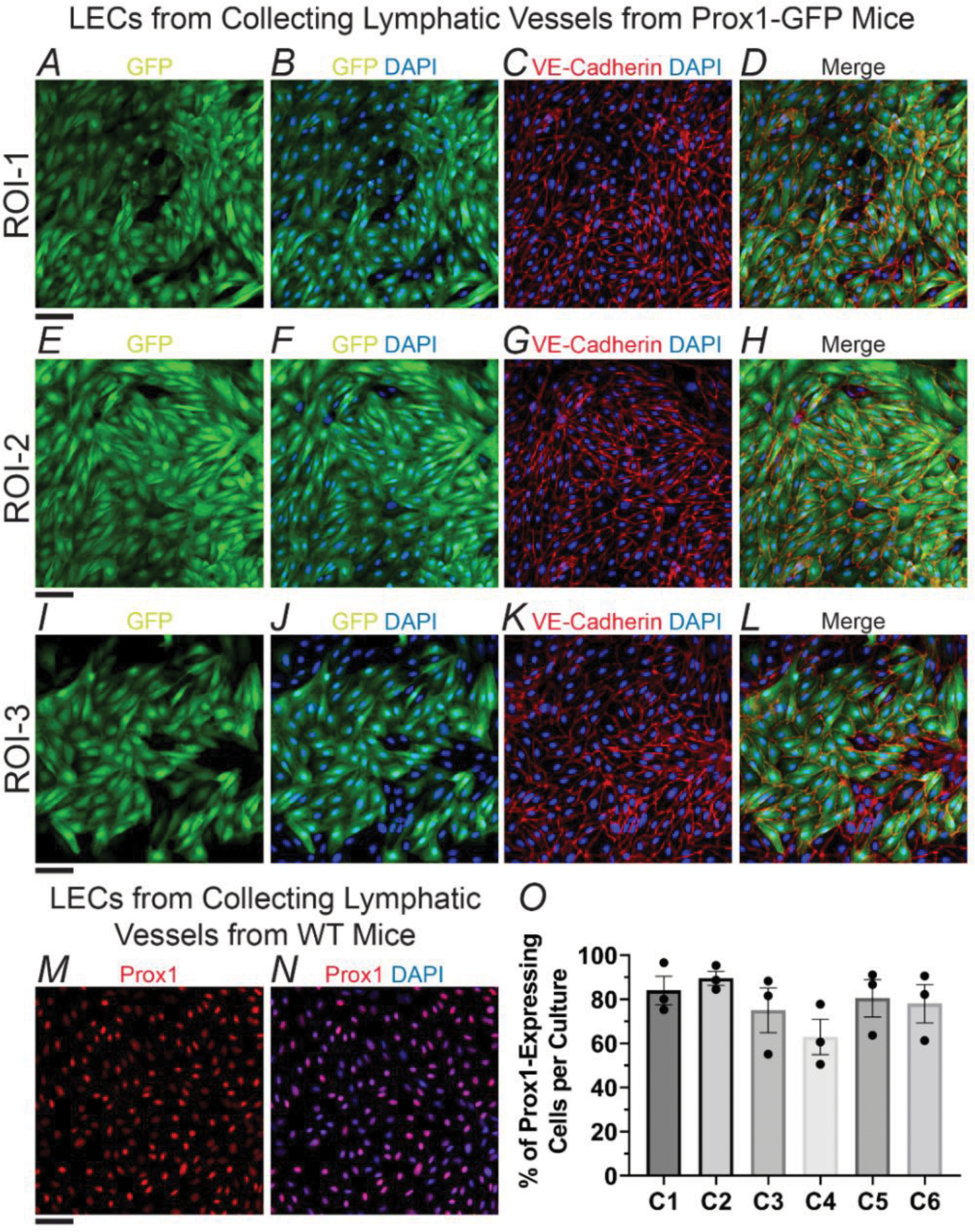
(A-L) Immunofluorescence images collected from three different regions of interest (ROIs) in a representative culture of primary LECs from inguinal axillary collecting lymphatic vessels from a Prox1-GFP mouse. LECs were stained with anti-GFP and anti-VE-Cadherin antibodies, as well as DAPI. (M, N) Representative immunofluorescence images of a culture of primary LECs from inguinal axillary collecting lymphatic vessels from a WT mouse. LECs were stained with an anti-Prox1 antibody and DAPI. Scale bar is equal to 50 μm in all panels, i.e., A-N. (O) Percent of cells expressing Prox1 after 1–2 passages for n=6 different cultures. The representative example shown in panels A-L corresponds with culture C6. Data is presented as the mean±SEM. Means were calculated from values measured from 3 different regions of interest.
To further, but partially, characterize our primary cultures of LECs, we performed RT-PCR to determine the expression of known markers of LECs, ECs, and SMCs, among other cell types. On a 22-target panel, results from RT-PCR assays showed that our isolated mouse primary LECs expressed the known endothelial cell markers eNos, Cdh5, and Pecam1, as well as the characteristic LEC markers Prox1 and Vegfr3. Importantly, expression of the smooth muscle and/or non-LEC genes Myh11, Cnn1, Desmin, NG2, and Cd11b were not detected (Figure 4). Our cultures of primary LECs consistently showed expression of α-actin, CaV1.2, Pdgfrα, Pdgfrβ, Kit, Vimentin, Cd146, and Cd34 (Figure 4). Mouse brain was used as a positive control for these assays (Figure 4B). We also assessed expression of major vascular endothelial connexins, our results showed that short-term cultured LECs expressed Cx43, Cx37, Cx47, and in, 2 out of 4 experiments, mRNA for Cx40 was also detected (Figure 4C). By employing the same experimental protocols, we isolated endothelial cell tubes from mesenteric arterioles and utilized them as a negative control. We found that, in contrast to primary cultures of LECs, arteriolar endothelial cells did not show expression of Prox1 nor Cx47, but consistently showed expression of Cx40 (Figure 4C–D).
Figure 4. Characterization of Gene Expression in Cultures of Primary LECs from Collecting Lymphatic Vessels.
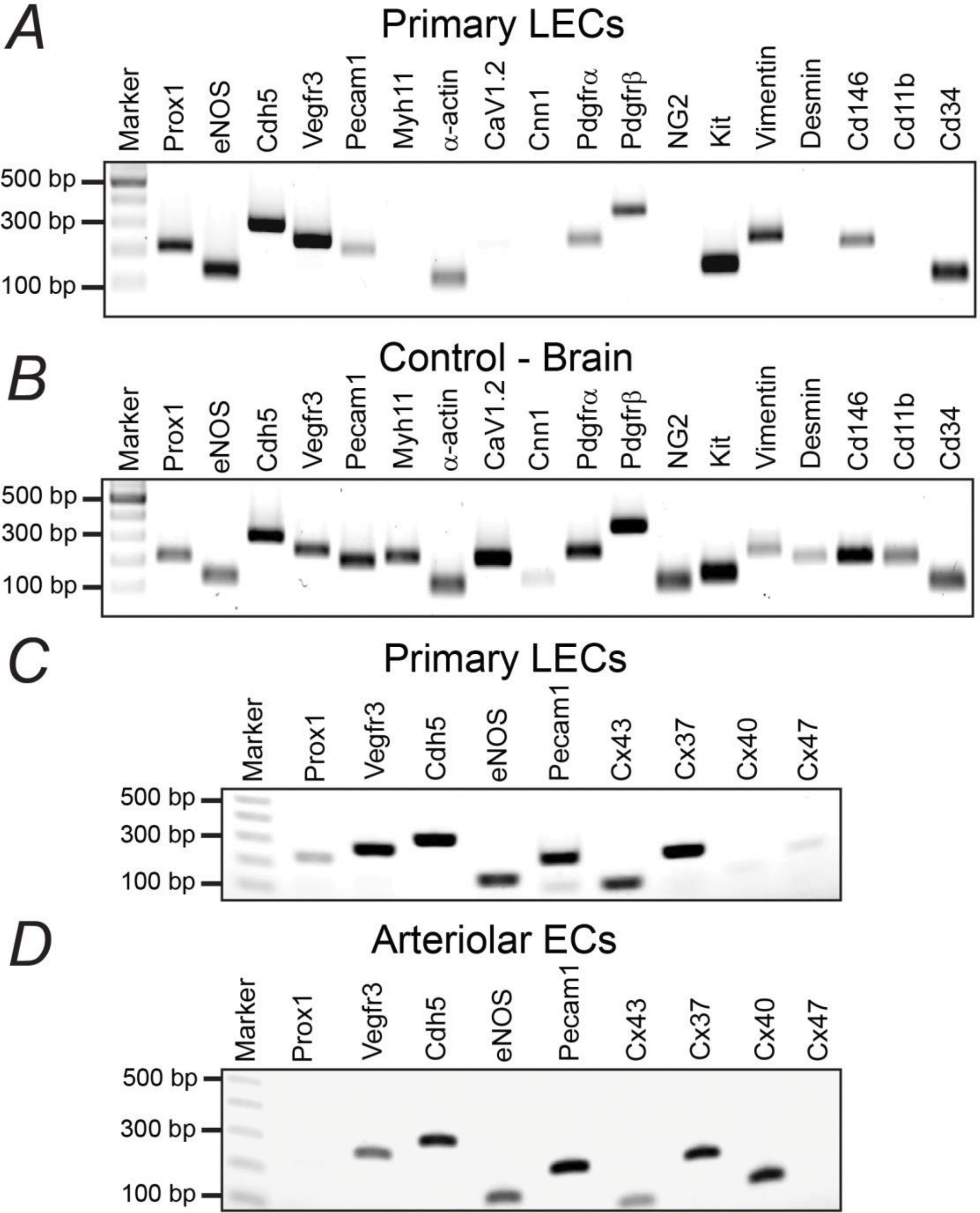
Representative gels from RT-PCR. (A, B) 18-gene panel including some known markers for lymphatic endothelial, vascular endothelial, and smooth muscle cells among others. Mouse brain was used as a positive control. (C, D) Expression of known markers for vascular and/or lymphatic endothelial cells, including major endothelial connexins (Cxs), in short-term cultured primary LECs or arteriolar endothelial cells respectively.
Proof-of-Concept Experiments Highlighting Potential Applications of Short-Term Cultures of Primary LECs from Collecting Vessels
To determine if our primary LECs maintained a functional phenotype regarding intracellular calcium in culture comparable to that observed and previously reported by us24 in freshly isolated lymphatic vessels, we applied our method for isolating and short-term culturing of primary LECs from inguinal axillary lymphatic vessels to evaluate Ca2+ signaling using mice expressing the genetically encoded calcium indicator GCaMP8 in endothelial cells, i.e., Cdh5-GCaMP8. This also allowed us to further demonstrate the functionality of our protocol to establish cultures of primary LECs from mice carrying genetic modifications. Utilizing high-speed confocal microscopy, we characterized the intracellular calcium activity in LECs under control conditions (Figure 5A, B) and upon stimulation with 100 nM ACh (Figure 5C, D). As anticipated, LECs were quiescent in control conditions, with most cells displaying low frequency spontaneous calcium events, as indicated by the dominant, blue-colored areas in the STMs for all LECs within a field of view (Figure 5A) and within a single-LEC (Figure 5B), in comparison with the rare areas displaying calcium activity. In contrast, upon stimulation with 100 nM ACh, a rapid and robust increase in intracellular calcium activity was observed in all LECs, as indicated by the significant presence of brighter colors (e.g., yellow, and red among others) over an initial blue background in the STMs for all LECs in the field of view (Figure 5C), as well as for single-LEC (Figure 5D). In addition, the minimal presence of areas not displaying GFP fluorescence in the field of view for ACh-stimulated cells (Figure 5C, left panel) indicates that the great majority of the LECs remained functional.
Figure 5. Proof-of-Concept Experiments Highlighting Potential Applications of Short-Term Cultures of Primary LECs from Collecting Vessels.
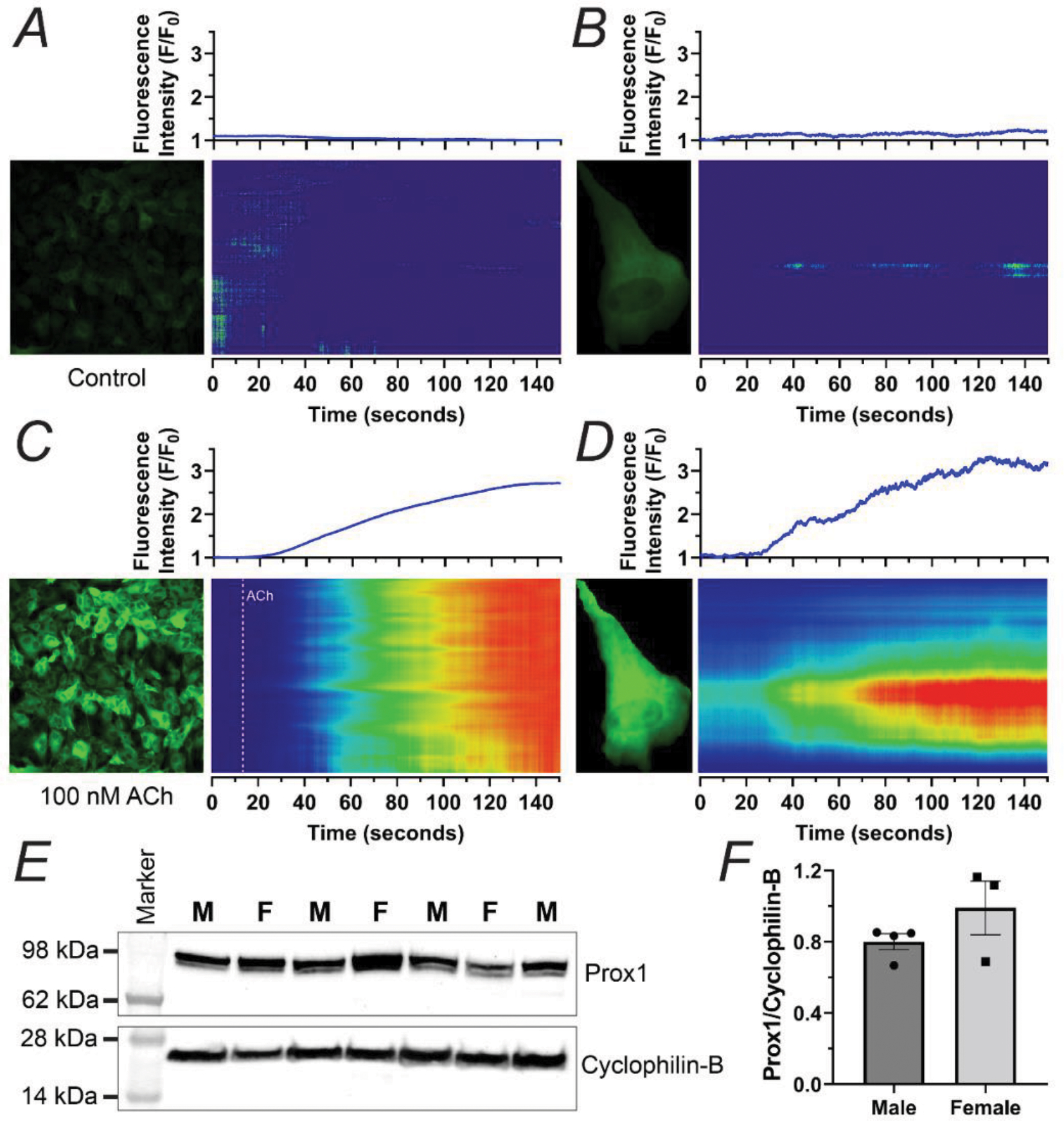
Intracellular calcium imaging on primary LECs from inguinal axillary collecting lymphatic vessels from a Cdh5-GCamP8 mouse under control conditions (A, B) or upon exposure to 100 nM ACh (C, D). The intensity of the fluorescence along each position on the vertical axis and over time is encoded in a colored scale where blue represents the lowest level of fluorescence intensity and red represents the highest. The integrated normalized fluorescence intensity per frame, i.e., F/F0, where F0 is the baseline fluorescence, is displayed in the blue traces on top of each of the corresponding two-dimensional maps of fluorescence intensity, i.e., space time maps (STMs). Representative data consistent with n=4 replicates. (E) Western blot showing expression of Prox1 in protein lysates prepared from primary cultures of LECs from mouse inguinal axillary collecting vessels from age-matched male (n=4) and female (n=3) mice. Male and Female samples are identified with the labels M or F respectively. Cyclophilin B was used as a loading control. (F) Summary data of Prox1 protein expression, normalized by Cyclophilin B. Data is presented as mean±SEM.
Finally, to exemplify additional applications of the method here presented for LEC isolation and short-term culture, we established cultures of primary LECs from inguinal axillary collecting lymphatic vessels from the mouse (n=7). Protein lysates were prepared from primary cultures of LECs established from lymphatic collecting vessels and used at passage 2, and proof-of-concept experiments utilizing western blotting for protein quantification of Prox1 were performed. Prox1 expression was similar between male and female (Figure 5E,F). In addition to the mouse, the methodology here described was successfully employed to isolate and short-term culture LECs from mesenteric collecting vessels from rat and monkeys (Supplemental Figure 1), illustrating the potential for this method to be applied to a variety of species.
DISCUSSION
In this study, we have reported a novel protocol for isolating and establishing short-term cultures of primary LECs from collecting lymphatic vessels. We generated proof-of-concept data that highlights some of the major advantages and applications of this protocol versus the use of standard commercial cell lines. Furthermore, we have validated our protocol in various species, including mice, rats, and monkeys. Although demonstrated for LECs, this method can be applied with minor modifications to establish cultures of primary endothelial cells from different blood vascular beds.
At present, for established lines of immortalized or primary cells, the originating species and originating tissue are known; however, limited information is provided about other describing factors including the sex and age of the originating subject(s). Within the vascular field, it is known that important anatomical regional differences may be observed along the vasculature. For instance, we have previously shown that lymphatic vessels display a regional dichotomy in pumping behavior that was partially explained by differences in the activity of L-type Ca2+ channels between peripheral and visceral lymphatic vessels of the mouse30. In addition to regional differences, within a specific anatomical region, molecular and functional differences between LECs from capillaries, pre-collecting, and collecting lymphatic vessels are likely present. This was in fact demonstrated in a recent study using single-cell transcriptome profiling on LECs in the mesenteric lymphatic vasculature33. Within collecting lymphatic vessels, some genes/proteins are known to be differentially expressed in LECs at valve areas, i.e., leaflets34–37. Here, we have demonstrated that by micro-dissecting and isolating specific vessels, not only can we control for a specific vascular bed and anatomical origin, but also, we can control other important factors, including sex, age, and genetic background among others. Whether sex, age, and/or genetic background genotypic and/or phenotypic differences will be maintained after short- or long-term culture, was not assessed, and remains to be determined.
Another major advantage of our protocol is its application towards establishing cultures of primary LECs from any genetically modified animal model. This allows us to take advantage of mice carrying specific gene modifications, without the need for transfection with siRNAs to partially down-regulate a gene of interest.
Intracellular calcium is known to be a critical component and major regulator of signal transduction and cellular function. The mainstream approach for the imaging of intracellular calcium in cultures requires the use of cell-permeable indicators (e.g., Fluo-4, Fura-2, Calbryte). Although these dyes have proven to be an exceptional tool for researchers, major limitations are associated with loading variability between experiments and between different cell types, in addition to the stress induced as cells are exposed to these agents. Here, we presented an alternative approach, by applying our protocol for isolating and establishing cultures of primary LECs from mice expressing a genetically encoded calcium indicator. Confocal fluorescence imaging of these cultures showed that our primary LECs maintained their intracellular calcium quiescence under control conditions, and also remained functional, as they displayed a rapid response upon stimulation with 100 nM ACh. These findings are consistent with observations in isolated, cannulated, and pressurized lymphatic vessels studied ex-vivo24, suggesting the main phenotype of these cells is not significantly altered, after 1–2 passages.
In a similar way, we utilized Prox1-GFP mice to demonstrate the advantage of using reporter mice as a tool to partially characterize our cultures of primary LECs. Furthermore, we showed that, after 1–2 passages and without a purification step, our cultures of mouse LECs displayed a Prox1-expression level of ~78%, based on the number of Prox1-expressing cells (i.e., Prox1-driven GFP nuclear and cytoplasmic fluorescence and/or Prox1 expression by direct antibody-staining). Interestingly, our RT-PCR results obtained in murine cultures showed no detectable expression for known non-LEC markers Myh11, Cnn1, Desmin, NG2, and Cd11b. In our cultures, primary LECs will undergo phenotypic transformation, with LECs transitioning perhaps into mesenchymal cells38. No purification step was included for the acquisition of the proof-of-concept data presented herein; however, a purification step could easily be incorporated by means of super-paramagnetic beads, e.g., Dynabeads, immuno-conjugated with LEC-specific antibodies as previously described1. Alternatively, LECs from reporter mice such as Prox1-GFP, as demonstrated here, and tamoxifen-induced Prox1-CreERT2;Rosa26mT/mG could be FACS-purified without immunolabeling. In our cultures, FACS-purification after 1–2 passages would certainly improve their quality, i.e., purity; however, it is important to consider that, if these primary LECs are undergoing phenotypical transitions in culture overtime, loss of signature genes, including Prox1 and/or Vegfr3, would likely reappear in purified cultures, if these were subjected to further passaging.
We developed this protocol with the objective of providing an experimental tool that would allow for routine isolation and short-term culture of specific primary lymphatic endothelial cells. In effect, the ability to study LECs after minimal culturing (i.e., between passages 1–2) would presumably maintain the physiological relevance of the resulting observables and minimize artifacts from culture. It is also important to mention that, to demonstrate the various advantages and applications of our protocol, we used traditional 2D cultures and specific cell culture conditions; however, choosing the most physiologically relevant type of culture, optimal substrate, culture media, added growth factors, among other culture conditions was not a goal for this study. For instance, work by colleagues in the field recently reported on a model that optimized the conditions for in-vitro 3-dimensional collagen-based culturing of LECs and demonstrated these cultures better recapitulated the physiological formation of lymphatic capillaries39. Future work could evaluate the formation of LEC tube-like structures, i.e., lumen-forming, in this 3-dimensional model with primary LECs isolated from specific collecting lymphatics.
In conclusion, we have developed a new experimental protocol that allows for the isolation and short-term culture of primary lymphatic endothelial cells from specific collecting lymphatic vessels and from various species. It is important to note that the degree of difficulty for the isolation of endothelial cells from different lymphatic and/or blood vessels will vary. We have highlighted how our protocol overcomes several limitations associated with current cell cultures. A critical step in this protocol is the dissection of micro-vessels (i.e., lymphatic or blood vessels). This involves a certain degree of technical skill; however, with adequate training, this protocol is highly reproducible and could be an important addition to every laboratory’s repertoire of experimental techniques. Our laboratory is open to providing technical guidance and training. Major limitations of current LEC cultures, including those in this study, include genotypic and phenotypic transformations; therefore, identifying the ideal culture conditions necessary to maintain the physiological relevance of studying LECs after a higher number of passages remains to be elucidated. A multi-laboratory collaborative effort would certainly help advance and improve the short-term cultures of LECs presented herein.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Dr. Michael J. Davis and Dr. Steven S. Segal from the University of Missouri for their support and contributions during the initial stages of this project.
Funding:
Research reported in this publication was supported by the National Institutes of Health (NIH). Award number: R00-HL141143 to JC-G.
LIST OF ABBREVIATIONS
- ACh
Acetylcholine
- BBE
Bovine brain extract
- FBS
Fetal bovine serum
- FACS
Fluorescence-activated cell sorting
- LEC
Lymphatic endothelial cell
- EC
Endothelial cell
- SMC
Smooth muscle cell
- LMC
Lymphatic muscle cell
- LECTs
Lymphatic endothelial cell tubes
- MEM-NEAA
Minimum essential media – non-essential amino acids
- STMs
Space-time maps
- PBS
Phosphate buffered saline
- PSS
Physiological salt solution
Footnotes
Disclosures: No conflicts of interest, financial or otherwise, are declared by the authors.
Ethics Statement: All animal protocols and procedures were approved by the Institutional Animal Care and Use Committees of Tulane University under protocol number 997. These were performed in accordance with the NIH Office of Laboratory Animal Welfare’s Public Health Service Policy on Humane Care and Use of Laboratory Animals and the Guide for the Care and Use of Laboratory Animals.
Data Availability Statement:
All datasets and protocols are included and described within this manuscript and the Supplemental Materials. Any additional information will be provided by the corresponding author as requested.
REFERENCES
- 1.Martinez-Corral I, Stanczuk L, Frye M, Ulvmar MH, Dieguez-Hurtado R, Olmeda D, Makinen T and Ortega S. Vegfr3-CreER (T2) mouse, a new genetic tool for targeting the lymphatic system. Angiogenesis. 2016;19:433–45. [DOI] [PubMed] [Google Scholar]
- 2.Martinez-Corral I, Zhang Y, Petkova M, Ortsater H, Sjoberg S, Castillo SD, Brouillard P, Libbrecht L, Saur D, Graupera M, Alitalo K, Boon L, Vikkula M and Makinen T. Blockade of VEGF-C signaling inhibits lymphatic malformations driven by oncogenic PIK3CA mutation. Nat Commun. 2020;11:2869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Makinen T, Veikkola T, Mustjoki S, Karpanen T, Catimel B, Nice EC, Wise L, Mercer A, Kowalski H, Kerjaschki D, Stacker SA, Achen MG and Alitalo K. Isolated lymphatic endothelial cells transduce growth, survival and migratory signals via the VEGF-C/D receptor VEGFR-3. EMBO J. 2001;20:4762–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Clasper S, Royston D, Baban D, Cao Y, Ewers S, Butz S, Vestweber D and Jackson DG. A novel gene expression profile in lymphatics associated with tumor growth and nodal metastasis. Cancer Res. 2008;68:7293–303. [DOI] [PubMed] [Google Scholar]
- 5.Datar SA, Gong W, He Y, Johengen M, Kameny RJ, Raff GW, Maltepe E, Oishi PE and Fineman JR. Disrupted NOS signaling in lymphatic endothelial cells exposed to chronically increased pulmonary lymph flow. Am J Physiol Heart Circ Physiol. 2016;311:H137–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rogic A, Auger F and Skobe M. Isolation of Human Skin Lymphatic Endothelial Cells and 3D Reconstruction of the Lymphatic Vasculature In Vitro. Methods Mol Biol. 2018;1846:279–290. [DOI] [PubMed] [Google Scholar]
- 7.Hirakawa S, Hong YK, Harvey N, Schacht V, Matsuda K, Libermann T and Detmar M. Identification of vascular lineage-specific genes by transcriptional profiling of isolated blood vascular and lymphatic endothelial cells. Am J Pathol. 2003;162:575–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee S, Kang J, Yoo J, Ganesan SK, Cook SC, Aguilar B, Ramu S, Lee J and Hong YK. Prox1 physically and functionally interacts with COUP-TFII to specify lymphatic endothelial cell fate. Blood. 2009;113:1856–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shin JW, Min M, Larrieu-Lahargue F, Canron X, Kunstfeld R, Nguyen L, Henderson JE, Bikfalvi A, Detmar M and Hong YK. Prox1 promotes lineage-specific expression of fibroblast growth factor (FGF) receptor-3 in lymphatic endothelium: a role for FGF signaling in lymphangiogenesis. Mol Biol Cell. 2006;17:576–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Selahi A, Chakraborty S, Muthuchamy M, Zawieja DC and Jain A. Intracellular calcium dynamics of lymphatic endothelial and muscle cells co-cultured in a Lymphangion-Chip under pulsatile flow. Analyst. 2022;147:2953–2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Selahi A, Fernando T, Chakraborty S, Muthuchamy M, Zawieja DC and Jain A. Lymphangion-chip: a microphysiological system which supports co-culture and bidirectional signaling of lymphatic endothelial and muscle cells. Lab Chip. 2021;22:121–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Si H, Wang J, Meininger CJ, Peng X, Zawieja DC and Zhang SL. Ca(2+) release-activated Ca(2+) channels are responsible for histamine-induced Ca(2+) entry, permeability increase, and interleukin synthesis in lymphatic endothelial cells. Am J Physiol Heart Circ Physiol. 2020;318:H1283–H1295. [DOI] [PubMed] [Google Scholar]
- 13.Suntravat M, Cromer WE, Marquez J, Galan JA, Zawieja DC, Davies P, Salazar E and Sanchez EE. The isolation and characterization of a new snake venom cysteine-rich secretory protein (svCRiSP) from the venom of the Southern Pacific rattlesnake and its effect on vascular permeability. Toxicon. 2019;165:22–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hayes H, Kossmann E, Wilson E, Meininger C and Zawieja D. Development and characterization of endothelial cells from rat microlymphatics. Lymphat Res Biol. 2003;1:101–19. [DOI] [PubMed] [Google Scholar]
- 15.Motawe ZY, Abdelmaboud SS and Breslin JW. Involvement of Sigma Receptor-1 in Lymphatic Endothelial Barrier Integrity and Bioenergetic Regulation. Lymphat Res Biol. 2021;19:231–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Herrera M, Molina P and Souza-Smith FM. Ethanol-induced lymphatic endothelial cell permeability via MAP-kinase regulation. Am J Physiol Cell Physiol. 2021;321:C104–C116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shah K, McCormack CE and Bradbury NA. Do you know the sex of your cells? Am J Physiol Cell Physiol. 2014;306:C3–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Choi D, Park E, Jung E, Cha B, Lee S, Yu J, Kim PM, Lee S, Hong YJ, Koh CJ, Cho CW, Wu Y, Li Jeon N, Wong AK, Shin L, Kumar SR, Bermejo-Moreno I, Srinivasan RS, Cho IT and Hong YK. Piezo1 incorporates mechanical force signals into the genetic program that governs lymphatic valve development and maintenance. JCI Insight. 2019;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cha B, Ho YC, Geng X, Mahamud MR, Chen L, Kim Y, Choi D, Kim TH, Randolph GJ, Cao X, Chen H and Srinivasan RS. YAP and TAZ maintain PROX1 expression in the developing lymphatic and lymphovenous valves in response to VEGF-C signaling. Development. 2020;147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Geng X, Yanagida K, Akwii RG, Choi D, Chen L, Ho Y, Cha B, Mahamud MR, Berman de Ruiz K, Ichise H, Chen H, Wythe JD, Mikelis CM, Hla T and Srinivasan RS. S1PR1 regulates the quiescence of lymphatic vessels by inhibiting laminar shear stress-dependent VEGF-C signaling. JCI Insight. 2020;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Socha MJ and Segal SS. Isolation of microvascular endothelial tubes from mouse resistance arteries. J Vis Exp. 2013:e50759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Behringer EJ, Scallan JP, Jafarnejad M, Castorena-Gonzalez JA, Zawieja SD, Moore JE, Jr., Davis MJ and Segal SS. Calcium and electrical dynamics in lymphatic endothelium. J Physiol. 2017;595:7347–7368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Choi I, Chung HK, Ramu S, Lee HN, Kim KE, Lee S, Yoo J, Choi D, Lee YS, Aguilar B and Hong YK. Visualization of lymphatic vessels by Prox1-promoter directed GFP reporter in a bacterial artificial chromosome-based transgenic mouse. Blood. 2011;117:362–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Castorena-Gonzalez JA, Zawieja SD, Li M, Srinivasan RS, Simon AM, de Wit C, de la Torre R, Martinez-Lemus LA, Hennig GW and Davis MJ. Mechanisms of Connexin-Related Lymphedema. Circ Res. 2018;123:964–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Davis MJ, Kim HJ, Zawieja SD, Castorena-Gonzalez JA, Gui P, Li M, Saunders BT, Zinselmeyer BH, Randolph GJ, Remedi MS and Nichols CG. Kir6.1-dependent KATP channels in lymphatic smooth muscle and vessel dysfunction in mice with Kir6.1 gain-of-function. J Physiol. 2020;598:3107–3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zawieja SD, Castorena JA, Gui P, Li M, Bulley SA, Jaggar JH, Rock JR and Davis MJ. Ano1 mediates pressure-sensitive contraction frequency changes in mouse lymphatic collecting vessels. J Gen Physiol. 2019;151:532–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Scallan JP, Wolpers JH and Davis MJ. Constriction of isolated collecting lymphatic vessels in response to acute increases in downstream pressure. J Physiol. 2013;591:443–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Scallan JP, Zawieja SD, Castorena-Gonzalez JA and Davis MJ. Lymphatic pumping: mechanics, mechanisms and malfunction. J Physiol. 2016;594:5749–5768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zawieja SD, Castorena-Gonzalez JA, Dixon B and Davis MJ. Experimental Models Used to Assess Lymphatic Contractile Function. Lymphat Res Biol. 2017;15:331–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zawieja SD, Castorena-Gonzalez JA, Scallan JP and Davis MJ. Differences in L-type Ca(2+) channel activity partially underlie the regional dichotomy in pumping behavior by murine peripheral and visceral lymphatic vessels. Am J Physiol Heart Circ Physiol. 2018;314:H991–H1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hald BO, Castorena-Gonzalez JA, Zawieja SD, Gui P and Davis MJ. Electrical Communication in Lymphangions. Biophys J. 2018;115:936–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wigle JT and Oliver G. Prox1 function is required for the development of the murine lymphatic system. Cell. 1999;98:769–78. [DOI] [PubMed] [Google Scholar]
- 33.Gonzalez-Loyola A, Bovay E, Kim J, Lozano TW, Sabine A, Renevey F, Arroz-Madeira S, Rapin A, Wypych TP, Rota G, Durot S, Velin D, Marsland B, Guarda G, Delorenzi M, Zamboni N, Luther SA and Petrova TV. FOXC2 controls adult lymphatic endothelial specialization, function, and gut lymphatic barrier preventing multiorgan failure. Sci Adv. 2021;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bazigou E, Xie S, Chen C, Weston A, Miura N, Sorokin L, Adams R, Muro AF, Sheppard D and Makinen T. Integrin-alpha9 is required for fibronectin matrix assembly during lymphatic valve morphogenesis. Dev Cell. 2009;17:175–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kazenwadel J, Betterman KL, Chong CE, Stokes PH, Lee YK, Secker GA, Agalarov Y, Demir CS, Lawrence DM, Sutton DL, Tabruyn SP, Miura N, Salminen M, Petrova TV, Matthews JM, Hahn CN, Scott HS and Harvey NL. GATA2 is required for lymphatic vessel valve development and maintenance. J Clin Invest. 2015;125:2979–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sabine A, Bovay E, Demir CS, Kimura W, Jaquet M, Agalarov Y, Zangger N, Scallan JP, Graber W, Gulpinar E, Kwak BR, Makinen T, Martinez-Corral I, Ortega S, Delorenzi M, Kiefer F, Davis MJ, Djonov V, Miura N and Petrova TV. FOXC2 and fluid shear stress stabilize postnatal lymphatic vasculature. J Clin Invest. 2015;125:3861–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Scallan JP, Knauer LA, Hou H, Castorena-Gonzalez JA, Davis MJ and Yang Y. Foxo1 deletion promotes the growth of new lymphatic valves. J Clin Invest. 2021;131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Alvandi Z and Bischoff J. Endothelial-Mesenchymal Transition in Cardiovascular Disease. Arterioscler Thromb Vasc Biol. 2021;41:2357–2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kumaravel S, Abbey CA, Bayless KJ and Chakraborty S. The beta1-integrin plays a key role in LEC invasion in an optimized 3-D collagen matrix model. Am J Physiol Cell Physiol. 2020;319:C1045–C1058. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All datasets and protocols are included and described within this manuscript and the Supplemental Materials. Any additional information will be provided by the corresponding author as requested.