

Abstract
Severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) neutralizing antibodies are shown to be effective therapeutics for providing coronavirus disease 2019 (COVID‐19) protection. However, recurrent variants arise and facilitate significant escape from current antibody therapeutics. Bispecific antibodies (bsAbs) represent a unique platform to increase antibody breadth and to reduce neutralization escape. Herein, a novel immunoglobulin G–variable domains of heavy‐chain‐only antibody (IgG–VHH) format bsAb derived from a potent human antibody R15‐F7 and a humanized nanobody P14‐F8‐35 are rationally engineered. The resulting bsAb SYZJ001 efficiently neutralizes wild‐type SARS‐CoV‐2 as well as the alpha, beta, gamma, and delta variants, with superior efficacy to its parental antibodies. Cryo‐electron microscopy structural analysis reveals that R15‐F7 and P14‐F8‐35 bind to nonoverlapping epitopes within the RBD and sterically hindered ACE2 receptor binding. Most importantly, SYZJ001 shows potent prophylactic and therapeutic efficacy against SARS‐CoV‐2 in three established mouse models. Collectively, the current results demonstrate that the novel bsAb format is feasible and effective, suggesting great potential as an inspiring antiviral strategy.
Keywords: bispecific antibody, IgG, novel format, SARS‐CoV‐2, VHH
A novel immunoglobulin G–variable domains of heavy‐chain‐only antibody (IgG–VHH) format bispecific antibody (bsAb) that confers protection against severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) and its variants are rationally developed. This universal platform combines the strengths of IgG and nanobody, provides a promising tool for the development of bsAbs, strongly complements and strengthens the existing SARS‐CoV‐2 antibody therapeutics.
1. Introduction
Coronavirus disease 2019 (COVID‐19) caused by the newly identified severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) has resulted in a public health crisis worldwide.[ 1 ] To date, SARS‐CoV‐2 has led to at least 552 million infections and 6.34 million deaths (https://covid19.who.int/). In addition to effective and safe vaccines, preclinical data and clinical trials demonstrate that neutralizing antibodies (nAbs) could be effectively deployed for preventing or treating COVID‐19 during the viremic phase of the disease.[ 2 ] However, COVID‐19 pandemic control is being hampered by continued SARS‐CoV‐2 evolution.
The emerging SARS‐CoV‐2 neutralization escape mutations, especially main target epitope mutations, pose a considerable challenge for traditional monoclonal antibody (mAb) therapy.[ 3 ] To avert viral escape, one potential alternative is to use nanobodies, also called variable domains of heavy‐chain‐only antibodies (VHHs). Previous studies indicated that nanobodies possess some unique features, such as a small size and a long protruding complementarity‐determining region 3 (CDR3) loop, which exposes a convex paratope allowing them to bind pocket, hidden, cryptic, and concave epitopes that are inaccessible to conventional antibodies.[ 4 ] Additionally, nanobodies can be readily humanized, appear safe, and have low immunogenicity.[ 5 ] To enhance their antiviral potency and extend their serum half‐life, nanobodies have been further engineered into different multivalent forms (affinity matured, fused to fragment crystallizable (Fc) domains, etc.).[ 6 ] Another potential strategy is the use of antibody cocktails, which comprise two or more antibodies specific for receptor‐binding domain (RBD) and other regions in the S protein, which may further improve the breadth and potency of nAbs against SARS‐CoV‐2 and its escape‐mutant strains compared to a single antibody.[ 7 ] Data from several clinical trials have shown that a single intravenous infusion of mAb cocktail significantly reduced the risk of COVID‐19‐related hospitalization and death in comparison to placebo.[ 8 ] However, this approach increases manufacturing costs and complicates formulation. Instead, a bispecific antibody (bsAb), formed by two monoclonal antibodies with two different binding epitopes on the RBD of the spike protein, embodying the advantages of a cocktail within a single molecule, might emerge as a new class of promising antibody therapeutics for antiviral therapy.
An efficient recombinant bsAb format is critical for maximizing the potency of antibody therapeutics. To date, several recombinant bsAb formats have been developed and are generally divided into two major classes according to whether they comprise or lack the Fc region. Traditionally, bsAbs are derived from the antigen‐binding sites of two antibodies of the same type and commonly result in immunoglobulin G (IgG)‐like as well as VHH–VHH format.[ 9 ] A novel bsAb format combining the advantages of both IgG and nanobodies may complement and strengthen the existing COVID‐19 antibody therapeutics.
Herein, we rationally engineered an IgG–VHH format bsAb based on a human antibody R15‐F7 and a humanized nanobody P14‐F8‐35, both with broad and potent in vitro and in vivo activities. The resulting bsAb SYZJ001 showed potent neutralization against SARS‐CoV‐2, and the major variants of concern and exhibited pronounced prophylactic and therapeutic efficacy in three well‐established SARS‐CoV‐2 mouse models. Additionally, cryo‐electron microscopy (cryo‐EM) structure analysis revealed that R15‐F7 Fab and P14‐F8‐35 bound to nonoverlapping epitopes within the RBD and sterically hindered ACE2 receptor binding. These results highlight that our novel IgG–VHH format is feasible and effective, suggesting great potential as an inspiring bsAb construction strategy for future antibody therapy.
2. Results
2.1. Generation and Characterization of the bsAb Partners R15‐F7 and P14‐F8‐35
To construct a bispecific neutralization antibody, we initially selected two monoclonal antibodies: human antibody R15‐F7 and single domain antibody P14‐F8. First, we measured the binding affinity of R15‐F7 and P14‐F8 to SARS‐CoV‐2 RBD by enzyme linked immunosorbent assay (ELISA). The results indicated that both R15‐F7 and P14‐F8 showed high affinity for the SARS‐CoV‐2 RBD, with a half maximal effective concentration (EC50) of ≈0.007 µg mL−1 (Figure 1a). Then, we analyzed the blocking effect of R15‐F7 and P14‐F8 antibodies on SARS‐CoV‐2 RBD with ACE2 binding. The results indicated that both R15‐F7 and P14‐F8 showed an efficient full blocking effect (Figure 1b). To reduce the possible immunogenicity risk, the single‐domain antibody P14‐F8 from alpaca was humanized to increase the similarity of the framework region with the human antibody as previously described.[ 10 ] We finally selected P14‐F8‐hVH10 as a candidate due to its 98.3% similarity with the human antibody germline IGHV3‐23 and its similar blocking effect compared to the parental antibody. As expected, ELISA binding and blocking assays both indicated that humanized antibody P14‐F8‐hVH10 possesses superior binding and blocking activities to the parental antibody (Figure 1c,d). Moreover, to further increase the affinity and blocking effects, we performed affinity maturation of the humanized antibody P14‐F8‐hVH10 by applying CDR walking mutagenesis and phage display technology, as previously described,[ 11 ] and finally, P14‐F8‐35 was acquired. As expected, the affinity of the engineered antibody P14‐F8‐35 (the equilibrium dissociation constant (KD) = 2.20E‐10) increased ≈5‐fold compared with that of the parental antibody P14‐F8‐hVH10 (KD = 1.05E‐09) (Figure 1e). Additionally, to illustrate whether they share different epitopes, we also applied the competition test using the surface plasmon resonance technology (SPR) method. The results indicated that R15‐F7 and P14‐F8‐35 recognize different epitopes, because when the RBD protein was saturated with one antibody, another antibody could still independently bind to the RBD (Figure 1f).
Figure 1.
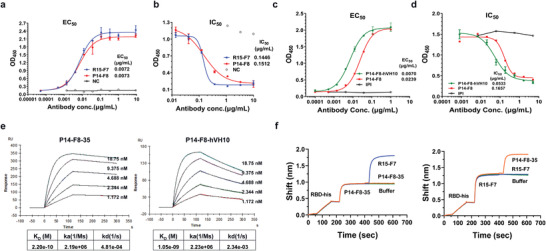
Characterization of a candidate partner, R15‐F7 and P14‐F8‐35. a) Binding efficacy of human antibody R15‐F7 and single domain antibody P14‐F8 to SARS‐CoV‐2 RBD by ELISA. Purified SARS‐CoV‐2 RBD was immobilized onto a 96‐well plate, and serially diluted antibodies were added to bind with SARS‐CoV‐2 RBD protein. b) Blocking efficacy of R15‐F7 and P14‐F8 antibodies against SARS‐CoV‐2 RBD and ACE2 binding by ELISA. The extracellular domain of the receptor ACE2 protein was immobilized onto a 96‐well plate at a reasonable concentration. Biotinylated Spike S1 protein was incubated with serially diluted antibodies and then added to ACE2‐coated 96‐well plates. c) Binding efficacy of humanized antibody P14‐F8‐hVH10 and parental antibody P14‐F8 by ELISA. d) Blocking assay of humanized antibody P14‐F8‐hVH10 and parental antibody P14‐F8 to SARS‐CoV‐2 RBD and ACE2 binding by ELISA. e) SPR kinetics of the engineered antibody P14‐F8‐35 and parental antibody P14‐F8‐hVH10. Data were analyzed with Biacore evaluation software. f) BLI kinetics of competitive binding of R15‐F7 and P14‐F8‐35 to the SARS‐CoV‐2 RBD. The SARS‐CoV‐2 RBD was immobilized onto the chip. One of the antibodies was first loaded, followed by another antibody. Data were analyzed using GraphPad Prism 8.0 software.
2.2. Engineering and Characterization of SYZJ001
We further designed and constructed a bsAb termed SYZJ001 by linking the IgG R15‐F7 and VHH of P14‐F8‐35 with the (G4S)3 linker shown in Figure 2a. Nonreduced sodium dodecyl sulphate‐polyacrylamide gel electrophoresis (SDS‐PAGE) analysis revealed that SYZJ001 had a molecular mass of 176 kDa (Figure S1, Supporting Information). Then, we compared the blocking effect of SYZJ001 with single antibody R15‐F7, P14‐F8‐35 and their combination via fluorescence‐activated cell sorting (FACS) assay. The results indicated that SYZJ001 had cooperative blocking efficacy with 50% maximal inhibitory concentration (IC50) of 0.022 µg mL−1; however, the IC50 values of R15‐F7, P14‐F8‐35, and their combination were 0.034, 0.020, and 0.031 µg mL−1, respectively (Figure 2b). We also measured the affinity of SYZJ001 and parental antibodies. The results showed that the KD of SYZJ001 was 1.48E‐10, with a significant increase of ≈10‐fold compared to parental R15‐F7 (1.48E‐09) (Figure 2c) and similar to parental P14‐F8‐35 (2.20E‐10) (Figure 1e).
Figure 2.
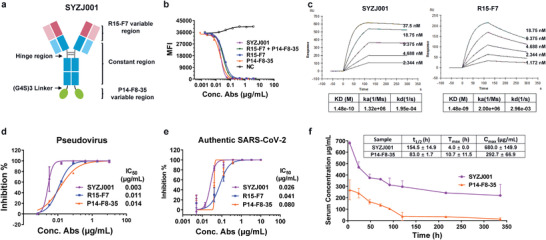
Design and characterization of SYZJ001. a) Format of the designed bispecific antibody. The format of SYZJ001 is IgG–VHH, which is composed of human antibody R15‐F7 and humanized single‐domain antibody P14‐F8‐35 linked by triple repeated GGGGS. b) An FACS blocking assay of antigen–receptor interactions was performed to evaluate the function of SYZJ001. Comparison with the single antibodies R15‐F7, P14‐F8‐35, and their combination. c) SPR kinetics of SYZJ001 and R15‐F7; the kinetics of P14‐F8‐35 are shown in Figure 1e. d) Neutralizing activity of SYZJ001 and its parental antibodies against SARS‐CoV‐2 WT pseudoviruses in huh7 cells. e) Neutralization activity of SYZJ001 and its parental antibodies against SARS‐CoV‐2 (BetaCoV/Beijing/IME‐BJ01/2020 strain) by PRNT in Vero cells. Neutralizing activities are represented as the mean ± SD. Experiments were repeated in triplicate. f) Pharmacokinetic (PK) activity of SYZJ001 and P14‐F8‐35 in the serum of C57BL/6 mice. At the indicated times post administration, the serum antibody concentration was measured by ELISA. The relevant PK parameter calculations were performed using WinNolin 6.1 software and are shown as the mean ± SD.
Importantly, pseudovirus (PDS) neutralization assays revealed that SYZJ001 possessed much stronger neutralizing activity with an IC50 of 0.003 µg mL−1 against SARS‐CoV‐2 wild‐type (WT) pseudovirus than the parental antibodies R15‐F7 (0.011 µg mL−1) and P14‐F8‐35 (0.014 µg mL−1) (Figure 2d). In addition, plaque reduction neutralization tests (PRNT) conducted against an authentic SARS‐CoV‐2 strain (BetaCoV/Beijing/IME‐BJ01/2020) also verified that SYZJ001 had a stronger neutralizing activity with an IC50 of 0.026 µg mL−1 than the parental antibodies R15‐F7 (0.041 µg mL−1) and P14‐F8‐35 (0.080 µg mL−1) (Figure 2e). Moreover, we evaluated the pharmacokinetic (PK) activity of SYZJ001 in the serum of C57BL/6 mice and obtained the mean PK parameters. The results showed that the serum concentration of SYZJ001 was superior to the parental nanobody P14‐F8‐35 (Figure 2f).
2.3. SYZJ001 Retained Neutralization Potency against SARS‐CoV‐2 Variants
We further evaluated the binding activity of SYZJ001 against a panel of emerging SARS‐CoV‐2 S1 or RBD mutants, including K417, L452, E484, and N501 mutations that have threatened the efficacy of most neutralizing antibodies developed thus far.[ 12 ] The results revealed that SYZJ001 maintained potent affinity with the majority of SARS‐CoV‐2 mutants with EC50 values lower than 0.042 µg mL−1, apart from the RBD‐E484K mutant with a decreased EC50 of 0.157 µg mL−1, which indicated that residue 484 may be located on or near the epitope of SYZJ001 and could affect the interaction between the RBD and SYZJ001 (Figure 3a). Then, we evaluated the neutralizing ability of SYZJ001 in vitro using pseudovirus neutralization assays. Notably, SYZJ001 retained potent neutralizing efficacy against alpha (B.1.1.7), beta (B.1.351), gamma (P.1), and delta (B.1.617.2) variant pseudoviruses (Figure 3b–e).
Figure 3.
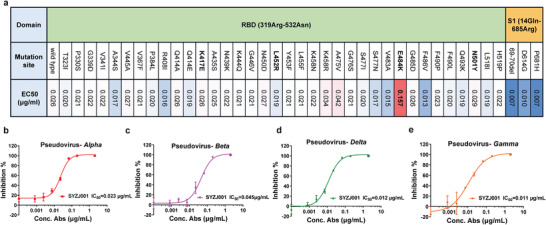
The efficacy of SYZJ001 against SARS‐CoV‐2 variant mutations. a) ELISA binding activity of SYZJ001 with a panel of emerging SARS‐CoV‐2 RBD or S1 mutants; wild‐type SARS‐CoV‐2 RBD was the positive control. b–e) Neutralization activity of SYZJ001 against SARS‐CoV‐2 alpha‐, beta‐, gamma‐, and delta‐variant pseudoviruses. Neutralizing activities are shown as the mean ± SD.
2.4. Prophylactic and Therapeutic Efficacy of SYZJ001 In Vivo
To assess the prophylactic and therapeutic efficacy of SYZJ001 in vivo, we investigated its ability to protect animals from infection and disease using three well‐established SARS‐CoV‐2 infection mouse models. First, we tested the prophylactic and therapeutic efficacy of SYZJ001 in a mouse model based on the SARS‐CoV‐2 mouse adapted strain MASCp6[ 13 ] (Figure 4a). As expected, upon MASCp6 intranasal challenge, aged BALB/c mice treated with phosphate buffered saline (PBS) sustained robust viral replication in the lungs with ≈1010.95 RNA copies g−1 at 5 d post inoculation. Remarkably, SYZJ001 treatment resulted in ≈105.37‐fold and ≈104.27‐fold reductions in viral titers in the lungs of the prophylactic plus therapeutic group and the therapeutic group at 5 d post inoculation, respectively (Figure 4b). RNA in situ hybridization (ISH) assays performed using RNAscope showed abundant SARS‐CoV‐2‐specific RNAs in mice from the PBS treatment group, while no viral RNA signals were detected in the SYZJ001 treatment group. Importantly, lung pathology analysis also showed that SARS‐CoV‐2 caused interstitial pneumonia, with the phenomenon of inflammatory cell infiltration (blue arrow), alveolar septal thickening (yellow arrow), and alveolar‐capillary barrier injury with hemorrhage (red arrow) upon PBS treatment. In contrast, no obvious lesions of alveolar epithelial cells or focal hemorrhage were observed in the lung sections from mice that received SYZJ001 treatment (Figure S2a, Supporting Information).
Figure 4.
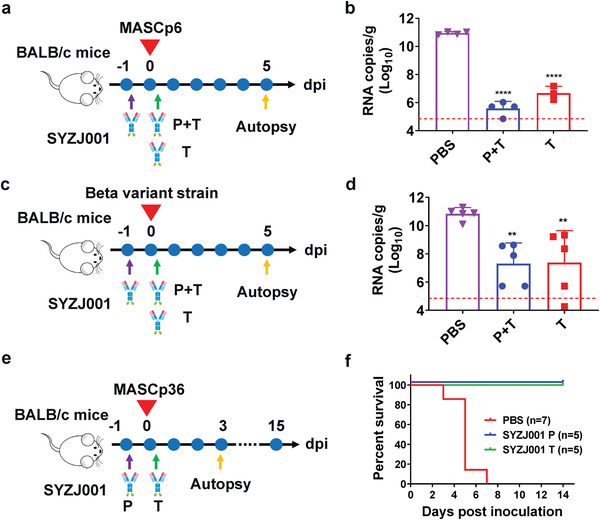
Prophylactic and therapeutic efficacy of SYZJ001 in three SARS‐CoV‐2 mouse models. a,c,e) Experimental design for the in vivo prophylactic and therapeutic efficacy of SYZJ001 were assessed using three well‐established mouse models based on a SARS‐CoV‐2 mouse‐adapted strain MASCp6, beta variant strain, and MASCp36, respectively. For the MASCp6 model and beta variant strain model, 8 month old female BALB/c mice were infected intranasally with 6 × 103 PFU of MASCp6 and 1 × 104 PFU of the SARS‐CoV‐2 beta variant strain as described previously. SYZJ001 (50 mg kg−1) was injected intraperitoneally 12 h before infection and 2 h after infection (the prophylactic plus therapeutic group, P + T) or 2 h after infection (the therapeutic group, T). All mice were monitored daily for morbidity and mortality. The lung tissues of mice were collected at 5 dpi for viral RNA load assays and pathological examination. For the MASCp36 model, 9 month old female BALB/c mice were infected intranasally with 12 000 PFU of MASCp36 as described previously. A dose of 20 mg kg−1 SYZJ001 was injected intraperitoneally 12 h before infection (the prophylactic group, P) or 2 h after infection (the therapeutic group, T). All mice were monitored daily for morbidity and mortality. The lung tissues of mice were collected at 3 dpi for subsequent and pathological examination. b,d) Virus RNA loads in the lungs at 5 dpi were measured by RT‐qPCR and are expressed as RNA copies g−1. Data are represented as the mean ± SD. Statistical significance was analyzed by one‐way ANOVA (**P < 0.01 and ****P < 0.0001). f) Survival curves of mice in the SYZJ001 and PBS treatment groups (n = 5 per group).
Recently, beta variant (so‐called 501Y.V2), which contains the E484K mutation in the RBD domain, has become predominant worldwide, and it was reported that beta variant could broadly reduce the neutralization activity of therapeutic antibodies and vaccine‐induced and SARS‐CoV‐2‐infected sera in vitro. Therefore, we also evaluated the in vivo protective efficacy of SYZJ001 to determine whether it could reduce the viral load in the lungs at 5 d after beta variant challenge[ 14 ] (Figure 4c). As expected, the results indicated that all aged mice that received PBS treatment had high levels of viral replication in the lungs, with ≈1010.84 RNA copies g−1. In contrast, SYZJ001 treatment resulted in ≈103.54‐fold and ≈103.46‐fold reductions in the prophylactic plus therapeutic and the therapeutic groups, respectively (Figure 4d). Then, the RNA ISH method was applied, and abundant SARS‐CoV‐2‐specific RNAs were detected in the mice from the PBS treatment group, while no viral RNA signals were detected in the prophylactic or prophylactic plus therapeutic groups. Furthermore, lung pathology analysis also showed that beta variant led to the development of pneumonia around the hilum, with the phenomenon of large quantities of desquamative necrotic epithelial cells in bronchioles, a large area of necrotic alveoli pneumocytes and alveoli structure collapse (yellow arrow), scattered hemorrhage (red arrow), and inflammatory cell infiltration (blue arrow) in the PBS treatment group. However, SYZJ001 treatment significantly prevented beta‐variant‐induced lung damage in the therapeutic group or the prophylactic and therapeutic groups (Figure S2b, Supporting Information).
Then, we tested the prophylactic and therapeutic efficacy of SYZJ001 in a newly established mouse model based on the SARS‐CoV‐2 mouse adapted strain MASCp36[ 15 ] (Figure 4e). As expected, the survival analysis results showed that all aged mice from the placebo group developed typical respiratory symptoms, and 100% eventually died from respiratory diseases within 5 d after challenge with MASCp36. In contrast, all mice survived (100%) in the prophylactic and therapeutic groups (P < 0.0001) (Figure 4f). Then, we compared lung pathology at 3 d after virus challenge using ISH assays, and the results indicated abundant SARS‐CoV‐2‐specific RNAs in the PBS‐treated mice, while no viral RNA signals were detected in the prophylactic group, and only a few viral RNA signals were detected in the therapeutic groups. Importantly, lung pathology analysis showed that MASCp36 could cause serious lung damage with a large area of fused alveoli walls, desquamative epithelial cells (green arrow), severe edema and scattered hemorrhage (red arrow) in the PBS treatment group. However, SYZJ001 treatment completely prevented MASCp36‐induced lung damage in the prophylactic group and significantly prevented lung damage with only minimal or very mild inflammatory cell infiltration in the therapeutic group (Figure S2c, Supporting Information).
Collectively, these results demonstrate that SYZJ001 is a promising prophylactic agent that is effective in providing protection in mice against various SARS‐CoV‐2 variants.
2.5. Structural Basis for Neutralization Mediated by P14‐F8‐35 and R15‐F7
Finally, to define the structural basis for neutralization mediated by P14‐F8‐35 and R15‐F7, we determined the cryo‐EM structure of a perfusion‐stabilized SARS‐CoV‐2 WT S trimer in complex with R15‐F7 Fab and P14‐F8‐35 at an overall resolution of 3.8 Å (Table S1, Supporting Information). Cryo‐EM of the complex showed full occupancy, where one P14‐F8‐35 and R15‐F7 were bound to each RBD of the homotrimeric S (Figure 5a; Figures S3 and S4, Supporting Information).
Figure 5.
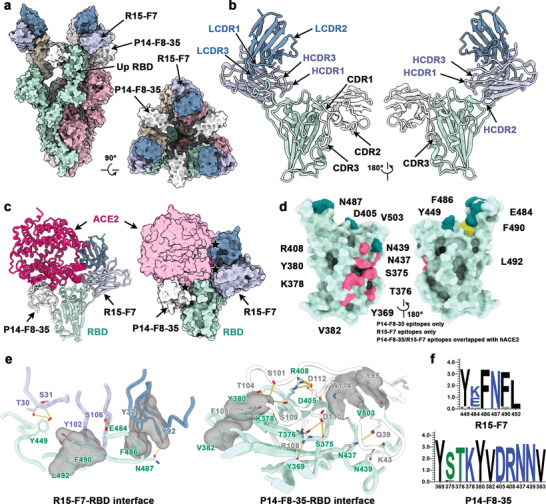
Structural basis of SYZJ001 neutralization. a) Surface representations of the structures of the SARS‐CoV‐2 S trimer in complex with P14‐F8‐35 and R15‐F7 with different colors for each S monomer (light green, pink, and brown), P14‐F8‐35 (white), and R15‐F7 (heavy chain: purple; light chain: blue). b) Cartoon representations show different views for binding interfaces to illustrate the binding modes of P14‐F8‐35 and R15‐F7. c) Superimposition of ACE2 upon the RBD/P14‐F8‐35/R15‐F7 complex. ACE2 is colored magenta, and other components are shown in the same color scheme as in panel (a). The clash area between ACE2 and antibodies is highlighted by a pentagram. d) Surface representations of the SARS‐CoV‐2 RBD. Residues colored pink and yellow are the residues recognized by P14‐F8‐35 and R15‐F7, respectively. Among these, residues overlapping with binding sites for ACE2 are colored in dark green. e) Details of the interactions between P14‐F8‐35 and R15‐F7 and the SARS‐CoV‐2 RBD. Residues involved in the formation of hydrogen bonds are shown as sticks and labeled. Hydrophobic patches are shown in gray surface representation. Hydrogen bonds are depicted as yellow dots. f) Analysis of sequence conservation on epitopes involved in R15‐F7 (top) and P14‐F8‐35 (bottom) binding with RBD by WEBLOGO. The logo plots represent the conversation of epitopes for these two antibodies from 18 SARS‐CoV‐2, including wild‐type, VOCs (alpha, beta, gamma, delta, and omicron), VOIs (lambda and mu), and other variants (delta plus, eta, iota, kappa, theta, iota, B.1.1.318, B.1.620, C.1.2, and C.363).
To further improve the density around interfaces between R15‐F7 or P14‐F8‐35 and RBD, local refinement was performed, enabling reliable analysis of the interfaces. R15‐F7 binds to the epical tip of the RBD, where the epitope largely overlaps with that of human ACE2. R15‐F7 interacts with RBD mainly through four CDR loops (HCDR1, HCDR3, LCDR1, and LCDR3) by both hydrophobic and hydrophilic interactions, while P14‐F8‐35 interacts with RBD through one CDR3 loop (Figure 5b; Figure S5, Supporting Information). Consistent with the higher affinity of P14‐F7‐35 for RBD than R15‐F8, as we described previously (Figures 1e and 2c), P14‐F7‐35 also possesses more interactions, especially hydrophilic interactions (salt bridge and hydrogen bonds) (Figure S5, Supporting Information). Superimposition of ACE2 upon RBD in complex with P14‐F8‐35 and R15‐F7 revealed steric clashes between both antibodies and ACE2 (Figure 5c). The epitopes of R15‐F7 include only six residues, and five of six are implicated in extensive interactions with ACE2 (Figure 5d; Table S2, Supporting Information). Tight binding is made through hydrophobic interactions formed by F486, F490, and L492 on the receptor‐binding motif, Y102 of the heavy chain, and Y32 of the light chain. In addition, extensive hydrophilic interactions further reinforce the interaction between RBD and R15‐F7. Both hydrophilic interactions, including 11 hydrogen bonds, and hydrophobic interactions (V503 on RBD and Y95, W114 on P14‐F8‐35) are involved in the binding of P14‐F8‐35 to RBD (Figure 5e). Notably, superimposition of beta and omicron variants, which both contain E484 mutations (beta: E484K; omicron: E484A) upon RBD/R15‐F7/P14‐F8‐35 complex indicates that hydrogen bond is completely abolished between omicron RBD and R15‐F7, but persists in beta, which consists with the potency neutralization against beta variant (Figure S6, Supporting Information). In addition, compared with R15‐F7, the residues of the P14‐F8‐35 epitopes are relatively conserved, with several mutations among all circulating SARS‐CoV‐2 variants (Figure 5f). In conclusion, cryo‐EM structural analysis revealed that R15‐F7 and P14‐F8‐35 bound to nonoverlapping epitopes within the RBD and sterically hindered ACE2 access to the RBD.
3. Discussion
The continuing emergence of SARS‐CoV‐2 mutations has become a major challenge in COVID‐19 pandemic control. As numerous mutations in SARS‐CoV‐2 continue to emerge, viral phenotypes such as transmissibility, virulence, and immune evasion may change accordingly.[ 16 ] For the therapeutic antibody approach, potential escape is a major concern.[ 17 ] It has been demonstrated that several mutations alter S1, especially in RBD epitopes, and can escape neutralization.[ 18 ] However, to date, most of the reported SARS‐CoV‐2 antibody therapeutics have been RBD‐targeted. Many SARS‐CoV‐2 nAbs, including some clinically approved antibodies, have been shown to exhibit strongly reduced or even abrogated neutralizing activity against SARS‐CoV‐2 immune escape variants.[ 19 ] To prevent the risk of nAb potency reduction due to viral escape, it is vitally important to choose broadly neutralizing antibodies with highly conserved epitopes or use antibody combinations.
Currently, SARS‐CoV‐2 bsAbs based on human‐derived IgGs and Camelidae‐derived/humanized nanobodies with either IgG‐like, VHH–Fc, or VHH–VHH format have been reported.[ 20 ] Distinct from these bsAb formats, herein, we first reported a novel SARS‐CoV‐2 bsAb construction strategy combining both the nanobody and IgG with the IgG–VHH format and developed the potent human bispecific neutralizing antibody SYZJ001. SYZJ001 is based on the nanobody P14‐F8‐35 and the human antibody R15‐F7, both of which can bind the SARS‐CoV‐2 RBD with high affinity by recognizing different epitopes in the RBD and blocking the binding of the SARS‐CoV‐2 RBD with ACE2. Compared with its parental antibodies, SYZJ001 exhibited superior IC50 and EC50 and neutralizing activity. Recently, similar to our research, Li et al. reported an engineered bsAb against SARS‐CoV‐2 with IgG single‐chain variable fragment (ScFv) format (IgG–ScFv). As both the IgG–VHH and IgG–ScFv formats have similar strategies with variable fragments linked to the C‐terminus of IgG Fc, the binding between Fc and FcγR may interfere with this format, thus possibly contributing to reducing the possibility of ADE.[ 21 ]
In the current study, we evaluated our bsAb's ability to engage SARS‐CoV‐2 spike with a panel of mutations frequently detected in clinically relevant SARS‐CoV‐2 variants, especially those affecting ACE2‐spike interactions and closely related to resistance to neutralizing antibodies, for instance, mutations at residues K417, L452, E484, and N501.[ 12 ] Satisfyingly, SYZJ001 exhibited high potency against the majority of SARS‐CoV‐2 mutants except the RBD‐E484K mutant. In addition, SYZJ001 maintained potent neutralizing efficacy against most SARS‐CoV‐2 variants of concern (alpha, beta, gamma, and delta) pseudoviruses. Furthermore, we evaluated the prophylactic and therapeutic efficacy of SYZJ001 in SARS‐CoV‐2‐infected mice using three well‐established mouse models based on a SARS‐CoV‐2 mouse adapted strain MASCp6, a beta variant strain, and MASCp36. Notably, SYZJ001 showed potent protective efficacy against various SARS‐CoV‐2 variants in vivo.
Cryo‐EM structural analysis revealed P14‐F8‐35 and R15‐F7 occupying nonoverlapping epitopes within the SARS‐CoV‐2 RBD and sterically hindering the interaction between ACE2 and the RBD. In addition, both binding and structural‐based epitope analyses indicate that the E484 residue plays a significant role in the escape of variants, possibly because mutations (E484A and E484K) introduce short side chains or distinct electric charges, which could lead to a reduction in the interactions between the mAb and RBD. The E484 mutation has been identified as an escape mutation that emerges in several SARS‐CoV‐2 variants of concern.[ 22 ] Moreover, it has been proven that the E484 site tends to have a striking effect on binding and neutralization, especially on RBD‐targeted antibodies, which commonly use heavy‐chain germline IGHV3‐53/3‐66 and are often E484‐targeted.[ 23 ] Thus, subsequent SARS‐CoV‐2 neutralizing antibody therapeutic and vaccine strategies should be engineered to circumvent the escape of variants caused by mutations in these important interaction sites, such as E484.
Taken together, our current results indicate that the novel bsAb format IgG–VHH is an effective and promising strategy for the development of antibody therapeutics. The bsAb involving broad‐spectrum SARS‐CoV‐2 IgG and nanobodies warrants further study.
4. Experimental Section
Ethics Statement
All animal studies were performed in strict accordance with the guidelines set by the Chinese Regulations of Laboratory Animals and Laboratory Animal Requirements of Environment and Housing Facilities. All animal procedures were reviewed and approved by the Animal Experiment Committee of the Laboratory Animal Center, AMMS, China (approval number: IACUC‐DWZX‐2020‐045).
Cells and Viruses
African green monkey kidney Vero (ATCC, #CCL‐81) and Vero E6 cells were maintained in Dulbecco's minimal essential medium (DMEM; Thermo Fisher Scientific) supplemented with 10% fetal bovine serum (FBS; Thermo Fisher Scientific), penicillin (100 U mL−1) and streptomycin (100 µg mL−1) (Thermo Fisher Scientific). SARS‐CoV‐2 was passaged in Vero cells, and the virus stock was aliquoted and titrated to PFU mL−1 in Vero cells by plaque assay. All experiments involving infectious SARS‐CoV‐2 were performed at the biosafety level 3 facility.
R15‐F7 and P14‐F8 Antibody Screening and Identification
The candidate antibody R15‐F7 was screened from a phage‐display fully human naïve antibody library (simplified as ST‐HuNAL, constructed by Sanyoubio with a capacity of 2.2 × 1011, Fab format) by a solid‐phase immunotube screening method using recombinant RBD of SARS‐CoV‐2 as a target described previously.[ 24 ] The P14‐F8 single‐domain antibody was screened from the phage‐display Alpaca naïve single domain antibody library (constructed by Sanyoubio with a capacity of 2 × 1010, VHH format), fused with human IgG1 Fc. R15‐F7 and P14‐F8 were, respectively, named CoVIC‐039 and CoVIC‐038 previously.[ 25 ] Antibodies were prepared by the ExpiCHO system (Thermo Fisher Scientific). Briefly, the plasmids harboring the heavy chain and light chain of R15‐F7 and heavy‐chain‐only P14‐F8 were mixed in ExpiFectamine CHO and then added to the prepared cell suspension and mixed with gentle shaking. The cell culture was then transferred and incubated at 37 °C in a shaker with 7% CO2. Approximately 18–22 h after transfection, ExpiCHO Enhancer and ExpiCHO Feed were added, and the cell culture was transferred to another shaker for incubation at 32 °C supplied with 5% CO2. 7 to 15 days after transfection, the culture supernatant containing antibody was collected and centrifuged at 12 000 g × for 10 min, and the resulting supernatant was subjected to affinity purification with MabSelect SuRe LX (GE Healthcare). The antibody was eluted with 100 mm glycine–HCl (pH 3.0) and then neutralized with 1 m Tris‐HCl. Finally, the purified antibody was exchanged into PBS buffer using a centrifugal filter unit (Millipore). Aliquots were made and stored at −80 °C.
SDS‐PAGE Analysis of the Purified Antibodies
For validation of SYZJ001, P14‐F8‐35, and R15‐F7, 1 µg of the above purified antibodies was, respectively, separated on an SDS–polyacrylamide gel (4–20% gradient) under nonreduced condition, and then stained with Coomassie blue staining solution (Beyotime) according to the manufacturer's instructions. 1 µg of the human monoclonal IgG1 antibody (Ipilimumab) with a purity of over 95% was used as isotype control.
Binding and Blocking Assay by ELISA
For the ELISA binding assay, a 96‐well plate was coated with SARS‐CoV‐2 spike RBD‐His protein (2 µg mL−1) at 4 °C overnight. The 96‐well plate was then blocked with PBS containing 5% nonfat milk powder for 1 h. Serial dilutions of antibodies were added to the plate and incubated for 2 h at room temperature. Then, the horseradish peroxidase (HRP)‐labeled goat antihuman Fc secondary antibody (1:5000, Abcam, ab98624) was added and incubated for 1 h. The plate was washed with phosphate‐buffered saline with tween 20 (PBST) with 0.1% Tween 20 9 times, incubated with HRP substrate 3,3′,5,5′‐tetramethylbenzidine (TMB) for color development and quenched by adding 40 µL of 2 m HCl. Finally, the binding ability of the candidate antibody was revealed by the absorbance at 450 nm wavelength using a plate reader (SurModics, TMBS‐1000‐01).
For the ELISA‐blocking assay, a 96‐well ELISA plate was coated with the extracellular domain of the receptor ACE2 (ACE2‐huFc, 8 µg mL−1) at 4 °C overnight. Then, the plate was blocked with PBS containing 5% nonfat milk powder for 1 h. Meanwhile, in another 96‐well plate, serial dilutions of antibodies were incubated with an equal volume of the biotinylated Spike S1‐huFc protein at room temperature for 1 h. Then, the mixture was transferred to a 96‐well ELISA plate containing coated ACE2‐huFc and incubated for 2 h. The plate was washed three times with PBST buffer containing 0.1% Tween‐20, and then 30 µL of NeutrAvidin protein‐HRP (1:5000, Thermo Fisher, 31 001) was added to the plate. An hour later, the plate was washed nine times with PBST. After 20 min of incubation with 30 µL of TMB (SurModics, TMBS‐1000‐01), the reaction was stopped by adding 30 µL of 2 m HCl. Finally, the absorbance of the plate was read at a wavelength of 450 nm using a plate reader (SurModics, TMBS‐1000‐01).
Affinity and Kinetics of Candidate Antibodies Measured by SPR
The affinity between candidates and SARS‐CoV‐2 RBD was determined using a Biacore T200 system (GE Healthcare). All experiments were performed at 25 °C. Briefly, candidate antibodies were diluted and immobilized on Protein A chips. SARS‐CoV‐2 RBD‐His protein was diluted to 37.5, 18.75, 9.375, 4.688, 2.344, and 1.172 nm, which were then injected at a flow rate of 30 µL min−1 (association 120 s, dissociation 180 s). Data were analyzed with Biacore evaluation software.
Competition Assay by BLI
Epitope binning was performed by using the Fortebio octet instrument. Briefly, RBD‐His of the SARS‐CoV‐2 spike protein was first prepared at 10 µg mL−1, while tested antibodies, including R15‐F7 and P14‐F8, were prepared at a concentration of 200 nm in 10× kinetics buffer (10× PBS, pH 7.4, 1% bovine serum albumin and 0.5% Tween 20). First, RBD‐His was loaded onto the Anti‐Penta‐HIS biosensor (HIS1K, Fortebio) and incubated for 200 s. After equilibrating in 10× kinetics buffer for 30 s, the biosensor was first incubated with a saturated concentration of 200 nm R15‐F7 or P14‐F8. After equilibrating in 10× kinetics buffer for another 30 s, 200 nm P14‐F8 or R15‐F7 was added. Binding signals were recorded to deduce the epitope difference between the two antibodies. Data were analyzed using GraphPad Prism 8.0 software.
Antibody PK Testing in C57BL/6 Mice
To test the antibody PK activity, 6–8 week old female C57BL/6 mice were given a single dose of 40 mg kg−1 SYZJ001 and P14‐F8‐35, with a dose volume of 10 mL kg−1 per mouse, via intraperitoneal inoculation. Serum samples were collected at 4 h, 1 d, 2 d, 3 d, 4 d, 5 d, 9 d, and 14 d postadministration. The serum antibody concentration (µg mL−1) was determined by ELISA, and the relevant PK parameters were analyzed using WinNonlin 6.1 software.
FACS Blocking Assay of Antigen–Receptor Interactions with Antibodies
ACE2‐overexpressing HEK293 cells (ACE2‐HEK293) validated for their confluence and viability were detached by trypsin‐ethylenediaminetetraacetic acid. After centrifugation at 4 °C and 300 × g for 2 min, the cells were resuspended in FACS buffer (1× PBS + 2% FBS) at a density of 2 × 106 cells mL−1, and 100 µL of the cells was added to each well of a 96‐well round bottom plate. Additionally, in another 96‐well plate, serial dilutions of antibodies were mixed with an equal volume of 0.1 µg mL−1 RBD– mouse Fc (mFc). After incubation at 4 °C for 1 h, the mixture was transferred to a plate containing ACE2‐HEK293 cells, mixed well and incubated at 4 °C for half an hour. Then, the plate was subjected to centrifugation at 4 °C and 300 × g for 2 min, and the cells were resuspended and washed with FACS buffer three times. Then, phycoerythrin‐labeled goat antimouse Fc secondary antibody (1:200, Jackson, 115‐115‐164) was added to the plate and incubated at 4 °C for half an hour. After centrifugation at 4 °C and 300 × g for 2 min, the cells were resuspended and washed with FACS buffer three times. Finally, the binding of RBD–mFc to the cells was detected and analyzed by flow cytometry (Beckman, CytoFLEX, AOO‐1‐1102).
SARS‐CoV‐2 Pseudovirus Neutralization
Construction of SARS‐CoV‐2 PSV and PSV‐based neutralization assays were performed as described previously.[ 26 ] Briefly, threefold serially diluted antibodies were incubated with 1.3 × 104 TCID50 SARS‐CoV‐2 pseudovirus at 37 °C for 1 h. Then, the mixtures were used to infect Huh7 cells or ACE2‐HEK293 cells in 96‐well plates at 37 °C for 24 h. After incubation, 100 µL of supernatant was removed, and equal volumes of luciferase reagent were added to each well and incubated for 2 min. Luciferase luminescence (RLU) was measured using a luminescence microplate reader. The neutralization percentage was calculated using the formula: Inhibition (%) = [1 − (sample RLU − Blank RLU)/(Virus control RLU − Blank RLU)] (%). Neutralization titers were presented as the IC50. Data were analyzed using GraphPad Prism 8.0 software.
Plaque Reduction Neutralization Tests
Standard PRNT against SARS‐CoV‐2 were performed to test the neutralization activity of mAbs in Vero cells. Briefly, fivefold serial dilutions of mAbs were added to ≈100 PFU of SARS‐CoV‐2 (BetaCoV/Beijing/IME‐BJ01/2020 strain) and incubated at 37 °C for 1 h. Then, the mixture was added to Vero‐E6 cell monolayers in a 12‐well plate in duplicate and incubated at 37 °C for 1 h. The mixture was removed, and 1 mL of 1.0% (w/v) low melting point agarose (Promega) in DMEM plus 4% (v/v) FBS was layered onto the infected cells. After further incubation at 37 °C for 2 d, the wells were stained with 1% (w/v) crystal violet dissolved in 4% (v/v) formaldehyde to visualize the plaques. The PRNT50 values were determined using nonlinear regression analysis by GraphPad Prism 8.0 software.
Protection against SARS‐CoV‐2 Challenge in Mice
The in vivo protection efficacy of SYZJ001 was assessed by using three well‐established mouse models based on a SARS‐CoV‐2 mouse‐adapted strain MASCp6, beta variant strain, and MASCp36, respectively. Briefly, for the MASCp6 model and beta variant strain model, 8 month old female BALB/c mice were infected intranasally with 6 × 103 PFU of MASCp6 and 1 × 104 PFU of the SARS‐CoV‐2 beta variant strain. SYZJ001 (50 mg kg−1) was injected intraperitoneally 12 h before infection and 2 h after infection or 2 h after infection. All mice were monitored daily for morbidity and mortality. The lung tissues of mice were collected at 5 dpi for viral RNA load assays and pathological examination. For the MASCp36 model, 9 month old female BALB/c mice were infected intranasally with 12 000 PFU of MASCp36 as described previously. A dose of 20 mg kg−1 SYZJ001 was injected intraperitoneally 12 h before infection or 2 h after infection. All mice were monitored daily for morbidity and mortality. The lung tissues of mice were collected at 3 dpi for and pathological examination.
Viral RNA Quantification
Viral RNA quantification was performed by real‐time quantitative reverse transcription polymerase chain reaction (RT‐qPCR) using a One Step PrimeScript RT‐PCR Kit (Takara, Japan) as described previously.[ 27 ] The primers and probe targeting the SARS‐CoV‐2‐S gene used for RT‐qPCR were CoV‐F3 (5′‐TCCTGGTGATTCTTCTTCAGGT‐3′), CoV‐R3 (5′‐TCTGAGAGAGGGTCAAGTGC‐3′), and CoV‐P3 (5′‐FAM‐AGCTGCAGCACCAGCTGTCCA‐BHQ1‐3′).
Histology and RNA ISH
Lung tissues from mice were fixed with perfusion fixative (formaldehyde) for 48 h and embedded in paraffin according to standard histological assays. For histopathology, lung tissues were stained with hematoxylin and eosin (H&E). Images were captured using an Olympus BX51 microscope equipped with a DP72 camera. RNA ISH assays were performed with an RNAscope 2.5 (Advanced Cell Diagnostics) according to the manufacturer's instructions. Briefly, formalin‐fixed paraffin‐embedded tissue sections of 5 µm were deparaffinized by incubation for 60 min at 60 °C. Endogenous peroxidases were quenched with hydrogen peroxide for 10 min at room temperature. Slides were then boiled for 15 min in RNAscope Target Retrieval Reagents and incubated for 30 min in RNAscope Protease Plus before probe hybridization. The probe targeting SARS‐CoV‐2 RNA was designed and synthesized by Advanced Cell Diagnostics (catalog no. 848 561). Tissues were counterstained with Gill's hematoxylin and visualized with standard bright‐field microscopy at 10× magnification.
Cryo‐EM Sample Preparation and Data Collection
The purified S trimer was mixed with P14‐F8‐35 and R15‐F7 at a molar ratio of 1:1.5:1.5 for 10 s in ice incubation. Then, 3 µL of the mixture was dropped onto the preglow‐discharged gold grid (C‐flat, 300 mesh, 1.2/1.3, Protochips In.), blotted for 6 s with no force in 100% relative humidity and immediately plunged into the liquid ethane using Vitrobot (FEI). Cryo‐EM data were collected by an FEI Titan Krios microscope (FEI) at 300 kV. Movies (32 frames, each 0.2 s, total dose of 60 e− Å‐2) were recorded using a K3 Summit direct detector with a defocus range between 1.5 and 2.7 µm. Automated single particle data acquisition was carried out by SerialEM, with a calibrated magnification of 22 500 yielding a final pixel size of 1.07 Å.
Cryo‐EM Data Processing
A total of 11 991 movies of the S/P14‐F8‐35/R15‐F7 complex were recorded. All movies were subjected to beam‐induced motion correction using MotionCorr in the RELION3.0 package. The defocus value of the motion‐corrected microscopic images was estimated by Gctf. A total of 4510 707 particles were autopicked and extracted. Particles were applied to 2D classification and 3D classification by RELION3.0. After that, 372 940 particles were selected and processed by nonuniform autorefinement and postprocessing in cryoSPARC to generate the final cryo‐EM density. To improve the resolution of the interfaces of the S‐P14‐R15 complex, local refinement was performed. The resolution of each structure was determined based on the gold‐standard Fourier shell correlation (threshold = 0.143) and evaluated by ResMap.
Model Fitting and Refinement
The atomic models of the complex were generated by first fitting the chains of the SARS‐CoV‐2 S trimer (PDB number of 6VYB) and Fabs (predicted by alphafold2) into the cryo‐EM densities of the complex by Chimera, followed by manual adjustment according to the densities in Coot. Real space refinement by Phenix was performed on the atomic models.
R15‐F7/P14‐F8‐35‐RBD Interactions’ Analysis
To analyze the analogies and connections of R15‐F7/P14‐F8‐35‐RBD interactions, schematic representations of amino acids involved in interactions between R15‐F7/P14‐F8‐35 and SARS‐CoV‐2 Spike RBD were analyzed using LigPlot+ (version 2.2.5).
Statistical Analysis
Data were plotted with individual values and presented as means ± standard deviation (SD). Comparisons among groups (n = 5 per group) were conducted by one‐way analysis of variance (ANOVA) in virus challenging study. P‐values of **<0.01 and ****<0.0001 were considered statistically significant. Statistical analyses were carried out using GraphPad Prism version 8.0 (GraphPad software).
Conflict of Interest
The authors declare no conflict of interest.
Authors Contribution
H.C., L.W., C.J.L., X.H.C., and H.L.Z. contributed equally to this work. H.C., Y.Q.D., C.J.L., X.H.C., H.L.Z., L.L.L., Y.C.T., N.N.Z., S.Q.Z., M.W., D.L., H.Y.Q., J.Z., X.C.X., Y.F.Z., S.L.Z., Q.C., Q.Y., X.T.W., Y.H.H., Z.Z., R.Y., J.E.Y., P.L., and W.J.W. performed the experiments. L.W., R.F., and W.J.F. performed the cryo‐EM analysis. H.C., L.W., and Y.Q.D. carried out the data analysis. G.J.L, Y.Q.D., X.X.W., and C.F.Q. conceived and supervised the study. H.C., Y.Q.D., and L.W. wrote the manuscript. All authors reviewed the manuscript.
Supporting information
Supporting Information
Acknowledgements
This work was supported by the National Key Plan for Scientific Research and Development of China (Grant No. 2020YFC0848500), the National Natural Science Foundation of China (Grant Nos. 82174055 and 82151222), and Key‐Area Research and Development Program of Guangdong Province (Program No. 2022B1111020002).
Chi H., Wang L., Liu C., Cheng X., Zheng H., Lv L., Tan Y., Zhang N., Zhao S., Wu M., Luo D., Qiu H., Feng R., Fu W., Zhang J., Xiong X., Zhang Y., Zu S., Chen Q., Ye Q., Yan X., Hu Y., Zhang Z., Yan R., Yin J., Lei P., Wang W., Lang G., Shao J., Deng Y., Wang X., Qin C., An Engineered IgG–VHH Bispecific Antibody against SARS‐CoV‐2 and Its Variants. Small Methods 2022, 6, 2200932. 10.1002/smtd.202200932
Contributor Information
Guojun Lang, Email: guojun.lang@sanyoubio.com.
Junbin Shao, Email: junbin_shao@liferiver.com.cn.
Yongqiang Deng, Email: dengyongqiang@bmi.ac.cn.
Xiangxi Wang, Email: xiangxi@ibp.ac.cn.
Chengfeng Qin, Email: qincf@bmi.ac.cn.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Hu B., Guo H., Zhou P., Shi Z. L., Nat. Rev. Microbiol. 2021, 19, 141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.a) Du L., Yang Y., Zhang X., Cell Mol. Immunol. 2021, 18, 2293; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Klasse P. J., Moore J. P., Elife 2020, 9, e57877; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) DeFrancesco L., Nat. Biotechnol. 2020, 38, 1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Khateeb J., Li Y., Zhang H., Crit. Care 2021, 25, 244; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chen R. E., Winkler E. S., Case J. B., Aziati I. D., Bricker T. L., Joshi A., Darling T. L., Ying B., Errico J. M., Shrihari S., VanBlargan L. A., Xie X., Gilchuk P., Zost S. J., Droit L., Liu Z., Stumpf S., Wang D., Handley S. A., Stine W. B. Jr., Shi P. Y., Davis‐Gardner M. E., Suthar M. S., Knight M. G., Andino R., Chiu C. Y., Ellebedy A. H., Fremont D. H., Whelan S. P. J., Crowe J. E. Jr., et al., Nature 2021, 596, 103; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Thakur S., Sasi S., Pillai S. G., Nag A., Shukla D., Singhal R., Phalke S., Velu G. S. K., Front. Med. 2022, 9, 815389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a) Voss J. E., Nature 2021, 595, 176; [DOI] [PubMed] [Google Scholar]; b) Nambulli S., Xiang Y., Tilston‐Lunel N. L., Rennick L. J., Sang Z., Klimstra W. B., Reed D. S., Crossland N. A., Shi Y., Duprex W. P., Sci. Adv. 2021, 7, eabh0319; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Xu J., Xu K., Jung S., Conte A., Lieberman J., Muecksch F., Lorenzi J. C. C., Park S., Schmidt F., Wang Z., Huang Y., Luo Y., Nair M. S., Wang P., Schulz J. E., Tessarollo L., Bylund T., Chuang G. Y., Olia A. S., Stephens T., Teng I. T., Tsybovsky Y., Zhou T., Munster V., Ho D. D., Hatziioannou T., Bieniasz P. D., Nussenzweig M. C., Kwong P. D., Casellas R., Nature 2021, 595, 278; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Huo J., Mikolajek H., Bas A. L., Clark J. J., Sharma P., Kipar A., Dormon J., Norman C., Weckener M., Clare D. K., Harrison P. J., Tree J. A., Buttigieg K. R., Salguero F. J., Watson R., Knott D., Carnell O., Ngabo D., Elmore M. J., Fotheringham S., Harding A., Moynié L., Ward P. N., Dumoux M., Prince T., Hall Y., Hiscox J. A., Owen A., James W., Carroll M. W., et al., Nat. Commun. 2021, 12, 5469; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Chen F., Liu Z., Jiang F., Front. Immunol. 2021, 12, 690742; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Li T., Cai H., Yao H., Zhou B., Zhang N., van Vlissingen M. F., Kuiken T., Han W., GeurtsvanKessel C. H., Gong Y., Zhao Y., Shen Q., Qin W., Tian X. X., Peng C., Lai Y., Wang Y., Hutter C. A. J., Kuo S. M., Bao J., Liu C., Wang Y., Richard A. S., Raoul H., Lan J., Seeger M. A., Cong Y., Rockx B., Wong G., Bi Y., et al., Nat. Commun. 2021, 12, 4635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a) Pillay T. S., Muyldermans S., Ann. Lab. Med. 2021, 41, 549; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Elverdi T., Eskazan A. E., Drug Des., Dev. Ther. 2019, 13, 1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Obeng E. M., Dzuvor C. K. O., Danquah M. K., Nano Today 2022, 42, 101350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.a) Jia L., Liu Y. P., Tian L. F., Xiong C., Xu X., Qu H., Xiong W., Zhou D., Wang F., Liu Z., Yan X. X., Xu W., Tang L., MedComm (2020) 2021, 2, 442; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Liang K. H., Chiang P. Y., Ko S. H., Chou Y. C., Lu R. M., Lin H. T., Chen W. Y., Lin Y. L., Tao M. H., Jan J. T., Wu H. C., J. Biomed. Sci. 2021, 28, 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Corti D., Purcell L. A., Snell G., Veesler D., Cell 2021, 184, 4593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kontermann R. E., Brinkmann U., Drug Discovery Today 2015, 20, 838. [DOI] [PubMed] [Google Scholar]
- 10. Waldmann H., Methods Mol. Biol. 2019, 1904, 2. [DOI] [PubMed] [Google Scholar]
- 11.a) Glaser S. M., Yelton D. E., Huse W. D., J. Immunol. 1992, 149, 3903; [PubMed] [Google Scholar]; b) Yang W. P., Green K., Pinz‐Sweeney S., Briones A. T., Burton D. R., Barbas C. F. 3rd, J. Mol. Biol. 1995, 254, 392. [DOI] [PubMed] [Google Scholar]
- 12.a) Sun C., Kang Y. F., Liu Y. T., Kong X. W., Xu H. Q., Xiong D., Xie C., Liu Y. H., Peng S., Feng G. K., Liu Z., Zeng M. S., Signal Transduction Targeted Ther. 2022, 7, 42; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Yuan M., Huang D., Lee C. D., Wu N. C., Jackson A. M., Zhu X., Liu H., Peng L., van Gils M. J., Sanders R. W., Burton D. R., Reincke S. M., Prüss H., Kreye J., Nemazee D., Ward A. B., Wilson I. A., Science 2021, 373, 818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gu H., Chen Q., Yang G., He L., Fan H., Deng Y. Q., Wang Y., Teng Y., Zhao Z., Cui Y., Li Y., Li X. F., Li J., Zhang N. N., Yang X., Chen S., Guo Y., Zhao G., Wang X., Luo D. Y., Wang H., Yang X., Li Y., Han G., He Y., Zhou X., Geng S., Sheng X., Jiang S., Sun S., et al., Science 2020, 369, 1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen Q., Huang X. Y., Sun M. X., Li R. T., Gu H., Tian Y., Zhang R. R., Luo D., Zhou C., Zhang Y., Cao T., Zhang N. N., Deng Y. Q., Li X. F., Qin C. F., Natl. Sci. Rev. 2021, 8, nwab167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sun S., Gu H., Cao L., Chen Q., Ye Q., Yang G., Li R. T., Fan H., Deng Y. Q., Song X., Qi Y., Li M., Lan J., Feng R., Guo Y., Zhu N., Qin S., Wang L., Zhang Y. F., Zhou C., Zhao L., Chen Y., Shen M., Cui Y., Yang X., Wang X., Tan W., Wang H., Wang X., Qin C. F., Nat. Commun. 2021, 12, 5654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cosar B., Karagulleoglu Z. Y., Unal S., Ince A. T., Uncuoglu D. B., Tuncer G., Kilinc B. R., Ozkan Y. E., Ozkoc H. C., Demir I. N., Eker A., Karagoz F., Simsek S. Y., Yasar B., Pala M., Demir A., Atak I. N., Mendi A. H., Bengi V. U., Cengiz Seval G., Gunes Altuntas E., Kilic P., Demir‐Dora D., Cytokine Growth Factor Rev. 2022, 63, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chen J., Gao K., Wang R., Wei G. W., J. Mol. Biol. 2021, 433, 167155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Güttler T., Aksu M., Dickmanns A., Stegmann K. M., Gregor K., Rees R., Taxer W., Rymarenko O., Schünemann J., Dienemann C., Gunkel P., Mussil B., Krull J., Teichmann U., Groß U., Cordes V. C., Dobbelstein M., Görlich D., EMBO J. 2021, 40, 107985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.a) Cao Y., Wang J., Jian F., Xiao T., Song W., Yisimayi A., Huang W., Li Q., Wang P., An R., Wang J., Wang Y., Niu X., Yang S., Liang H., Sun H., Li T., Yu Y., Cui Q., Liu S., Yang X., Du S., Zhang Z., Hao X., Shao F., Jin R., Wang X., Xiao J., Wang Y., Xie X. S., Nature 2022, 602, 657; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Liu L., Iketani S., Guo Y., Chan J. F., Wang M., Liu L., Luo Y., Chu H., Huang Y., Nair M. S., Yu J., Chik K. K., Yuen T. T., Yoon C., To K. K., Chen H., Yin M. T., Sobieszczyk M. E., Huang Y., Wang H. H., Sheng Z., Yuen K. Y., Ho D. D., Nature 2022, 602, 676; [DOI] [PubMed] [Google Scholar]; c) Hoffmann M., Krüger N., Schulz S., Cossmann A., Rocha C., Kempf A., Nehlmeier I., Graichen L., Moldenhauer A. S., Winkler M. S., Lier M., Dopfer‐Jablonka A., Jäck H. M., Behrens G. M. N., Pöhlmann S., Cell 2022, 185, 447; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Shah M., Woo H. G., Front. Immunol. 2021, 12, 830527; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Cao Y., Yisimayi A., Jian F., Song W., Xiao T., Wang L., Du S., Wang J., Li Q., Chen X., Yu Y., Wang P., Zhang Z., Liu P., An R., Hao X., Wang Y., Wang J., Feng R., Sun H., Zhao L., Zhang W., Zhao D., Zheng J., Yu L., Li C., Zhang N., Wang R., Niu X., Yang S., et al., Nature 2022, 602, 657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.a) De Gasparo R., Pedotti M., Simonelli L., Nickl P., Muecksch F., Cassaniti I., Percivalle E., Lorenzi J. C. C., Mazzola F., Magrì D., Michalcikova T., Haviernik J., Honig V., Mrazkova B., Polakova N., Fortova A., Tureckova J., Iatsiuk V., Di Girolamo S., Palus M., Zudova D., Bednar P., Bukova I., Bianchini F., Mehn D., Nencka R., Strakova P., Pavlis O., Rozman J., Gioria S., et al., Nature 2021, 593, 424; [DOI] [PubMed] [Google Scholar]; b) Wu X., Cheng L., Fu M., Huang B., Zhu L., Xu S., Shi H., Zhang D., Yuan H., Nawaz W., Yang P., Hu Q., Liu Y., Wu Z., Cell Rep. 2021, 37, 109869; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Cho H., Gonzales‐Wartz K. K., Huang D., Yuan M., Peterson M., Liang J., Beutler N., Torres J. L., Cong Y., Postnikova E., Bangaru S., Talana C. A., Shi W., Yang E. S., Zhang Y., Leung K., Wang L., Peng L., Skinner J., Li S., Wu N. C., Liu H., Dacon C., Moyer T., Cohen M., Zhao M., Lee F. E., Weinberg R. S., Douagi I., Gross R., et al., Sci. Transl. Med. 2021, 13, eabj5413; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Lim S. A., Gramespacher J. A., Pance K., Rettko N. J., Solomon P., Jin J., Lui I., Elledge S. K., Liu J., Bracken C. J., Simmons G., Zhou X. X., Leung K. K., Wells J. A., mAbs 2021, 13, 1893426; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Wang Y., Liu M., Shen Y., Ma Y., Li X., Zhang Y., Liu M., Yang X. L., Chen J., Yan R., Luan D., Wang Y., Chen Y., Wang Q., Lin H., Li Y., Wu K., Zhu T., Zhao J., Lu H., Wen Y., Jiang S., Wu F., Zhou Q., Shi Z. L., Huang J., Cell Discovery 2022, 8, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Li Z., Li S., Zhang G., Peng W., Chang Z., Zhang X., Fan Z., Chai Y., Wang F., Zhao X., Li D., Zhang R., He Z., Zou W., Xu K., Lei W., Liu P., Hao J., Zhang J., Sun L., Wu G., Tan S., Gao G. F., Gao F., Wu Y., Nat. Immunol. 2022, 23, 423. [DOI] [PubMed] [Google Scholar]
- 22.a) Yang W. T., Huang W. H., Liao T. L., Hsiao T. H., Chuang H. N., Liu P. Y., Infect. Drug Resist. 2022, 15, 373; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Beeraka N. M., Sukocheva O. A., Lukina E., Liu J., Fan R., Rev. Med. Virol. 2022, 32, e2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.a) Greaney A. J., Loes A. N., Crawford K. H. D., Starr T. N., Malone K. D., Chu H. Y., Bloom J. D., Cell Host Microbe 2021, 29, 463; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Weisblum Y., Schmidt F., Zhang F., DaSilva J., Poston D., Lorenzi J. C., Muecksch F., Rutkowska M., Hoffmann H. H., Michailidis E., Gaebler C., Agudelo M., Cho A., Wang Z., Gazumyan A., Cipolla M., Luchsinger L., Hillyer C. D., Caskey M., Robbiani D. F., Rice C. M., Nussenzweig M. C., Hatziioannou T., Bieniasz P. D., Elife 2020, 9, e61312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yao H., Sun Y., Deng Y. Q., Wang N., Tan Y., Zhang N. N., Li X. F., Kong C., Xu Y. P., Chen Q., Cao T. S., Zhao H., Yan X., Cao L., Lv Z., Zhu D., Feng R., Wu N., Zhang W., Hu Y., Chen K., Zhang R. R., Lv Q., Sun S., Zhou Y., Yan R., Yang G., Sun X., Liu C., Lu X., et al., Cell Res. 2021, 31, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hastie K. M., Li H., Bedinger D., Schendel S. L., Dennison S. M., Li K., Rayaprolu V., Yu X., Mann C., Zandonatti M., Diaz Avalos R., Zyla D., Buck T., Hui S., Shaffer K., Hariharan C., Yin J., Olmedillas E., Enriquez A., Parekh D., Abraha M., Feeney E., Horn G. Q., Aldon Y., Ali H., Aracic S., Cobb R. R., Federman R. S., Fernandez J. M., Glanville J., et al., Science 2021, 374, 472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nie J., Li Q., Wu J., Zhao C., Hao H., Liu H., Zhang L., Nie L., Qin H., Wang M., Lu Q., Li X., Sun Q., Liu J., Fan C., Huang W., Xu M., Wang Y., Emerging Microbes Infect. 2020, 9, 680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang N. N., Li X. F., Deng Y. Q., Zhao H., Huang Y. J., Yang G., Huang W. J., Gao P., Zhou C., Zhang R. R., Guo Y., Sun S. H., Fan H., Zu S. L., Chen Q., He Q., Cao T. S., Huang X. Y., Qiu H. Y., Nie J. H., Jiang Y., Yan H. Y., Ye Q., Zhong X., Xue X. L., Zha Z. Y., Zhou D., Yang X., Wang Y. C., Ying B., et al., Cell 2020, 182, 1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.