

Abstract
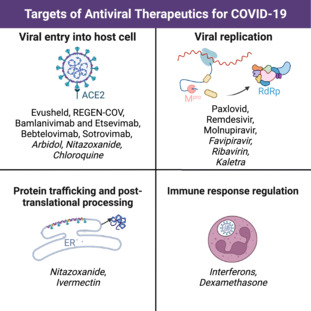
Antiviral therapies are integral in the fight against SARS‐CoV‐2 (i.e. severe acute respiratory syndrome coronavirus 2), the causative agent of COVID‐19. Antiviral therapeutics can be divided into categories based on how they combat the virus, including viral entry into the host cell, viral replication, protein trafficking, post‐translational processing, and immune response regulation. Drugs that target how the virus enters the cell include: Evusheld, REGEN‐COV, bamlanivimab and etesevimab, bebtelovimab, sotrovimab, Arbidol, nitazoxanide, and chloroquine. Drugs that prevent the virus from replicating include: Paxlovid, remdesivir, molnupiravir, favipiravir, ribavirin, and Kaletra. Drugs that interfere with protein trafficking and post‐translational processing include nitazoxanide and ivermectin. Lastly, drugs that target immune response regulation include interferons and the use of anti‐inflammatory drugs such as dexamethasone. Antiviral therapies offer an alternative solution for those unable or unwilling to be vaccinated and are a vital weapon in the battle against the global pandemic. Learning more about these therapies helps raise awareness in the general population about the options available to them with respect to aiding in the reduction of the severity of COVID‐19 infection. In this ‘A Guide To’ article, we provide an in‐depth insight into the development of antiviral therapeutics against SARS‐CoV‐2 and their ability to help fight COVID‐19.
Keywords: antiviral, Arbidol, bamlanivimab and etesevimab, bebtelovimab, chloroquine, COVID‐19, dexamethasone, Evusheld, favipiravir, interferons, ivermectin, Kaletra, molnupiravir, nitazoxanide, Paxlovid, REGEN‐COV, remdesivir, ribavirin, SARS‐CoV‐2, sotrovimab, therapeutics, treatment, virus
This review details antiviral therapeutics investigated for use in the fight against COVID‐19, classified by their targets in the SARS‐CoV‐2 (i.e. severe acute respiratory syndrome coronavirus 2) life cycle. Both therapeutics authorized for COVID‐19 treatment and unauthorized treatments (italicized in the image) are discussed. Four major target categories are examined: viral entry into host cell, viral replication (including protease inhibition and interactions with the replication‐transcription complex), protein trafficking and post‐translational processing, and immune response regulation.
Abbreviations
- 3CLpro
3‐chymotrypsin‐like protease
- ACE2
angiotensin‐converting enzyme 2
- BNE
bamlanivimab and etesevimab
- EUA
emergency use authorization
- FDA
United States Food and Drug Administration
- FRTP
favipiravir‐ribofuranosyl‐5′‐triphosphate
- IFN
interferon
- IgG1κ
human immunoglobulin G1
- IMP
inosine monophosphate
- MERS‐CoV
Middle East respiratory syndrome coronavirus
- Mpro
severe acute respiratory syndrome coronavirus 2 main protease
- NHC
β‐d‐N4‐hydroxycytidine
- NIH
National Institutes of Health
- NSP
non‐structural protein
- NTP
nucleotide triphosphate
- NTZ
nitazoxanide
- PDB
Protein Data Bank
- PDI
protein disulfide isomerase
- RBD
receptor binding domain
- RdRp
RNA‐dependent RNA polymerase
- RMP
remdesivir monophosphate
- RTP
remdesivir triphosphate
- SARS‐CoV‐2
severe acute respiratory syndrome coronavirus 2
- STAT
signal transducers and activators of transcription
- TIZ
tizoxanide
- WHO
World Health Organization
Introduction
With the emergence of the COVID‐19 pandemic in March 2020 came the challenge of synthesizing novel therapeutics to combat the spread and reduce the lethality of the virus that causes it: severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) [1]. Previous therapies used to treat HIV and Ebola virus disease show promise in targeting related mechanisms in SARS‐CoV‐2. Almost 180 vaccines against SARS‐CoV‐2 have undergone clinical development across the globe, yet only 11 have been approved for emergency use by the World Health Organization (WHO) [2]. Vaccine development is a lengthy and exhaustive process, relying on effective distribution in a systematic, regulated manner to reach as many individuals as possible. Without proper distribution methods, the vaccine ceases to accomplish its function of attaining herd immunity and protecting a community from disease outbreaks. It is also worth noting that vaccination of immunocompromised individuals is not as efficacious as in immunocompetent counterparts [3]. Therefore, antiviral treatments are actively being developed as another measure to fight COVID‐19.
Antiviral therapies have numerous benefits, including the ability to be distributed to wide populations and administered to individuals who are immunodeficient. Additionally, negative perception of vaccines has led to distrust and vaccine refusal [4]. Therefore, antiviral therapeutics broaden the options for treatments and preventative measures.
In this ‘A Guide To’ article, we describe the mechanisms that the SARS‐CoV‐2 virus exploits as it infects the human body. We then discuss how antiviral therapeutics are used to stop its progression at each of these crucial steps. Each category of antiviral therapeutic [entry obstruction via S protein targeting (Fig. 1.1), interference with viral replication (Fig. 1.2–5), protein trafficking and post‐translational modifications (Fig. 1.6–7), and immune response upregulation] will be discussed as we look at therapeutic treatments.
Fig. 1.
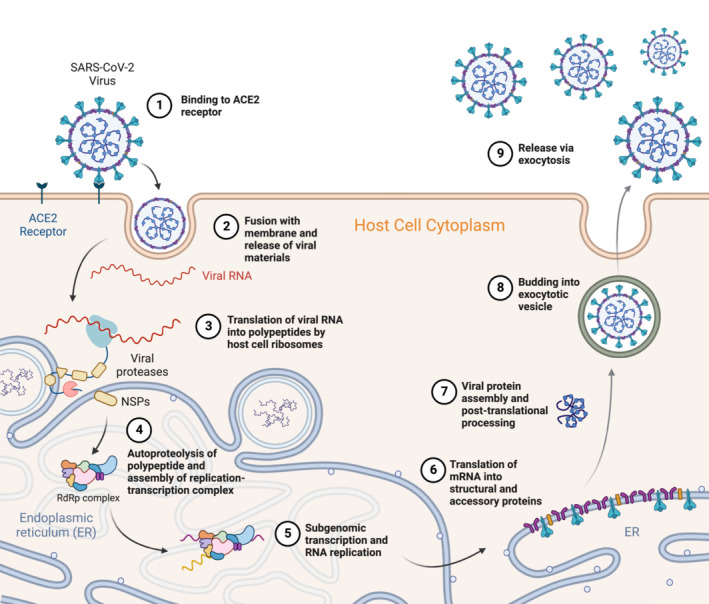
Summarized pathway of the entry into host cell, replication, maturation, and release of the SARS‐CoV‐2 virus. (1) Entry and binding of S protein to the ACE2 receptor. (2) Shedding of the virus's outer endosome coat and fusion with the cellular membrane releases viral materials. Host factors allow for fusion of the endosomal and cellular membranes. (3) Translation of viral mRNA into polypeptides and viral proteases by host cell occurs. The polyproteins are post‐translationally processed to form non‐structural proteins (NSPs). (4) Autoproteolysis of the virus's translated proteins and assembly of the replication‐transcription complex (RdRp complex). NSPs form the replication‐transcription complex after processing. (5) Subgenomic transcription of host cell DNA into RNA and RNA replication occurs. The integrated viral genetic material is also processed at this step. (6) Translation of host cell mRNA and the integrated viral cell mRNA into structural and accessory proteins occurs on the ribosomes of the RER. Translocation of proteins occurs here as well. (7) Viral proteins are assembled and processed post‐translation. This helps form secretory vesicles for exocytosis out of the host cell. (8) Viral RNA instructions cause the formation of exocytic vesicles. (9) Virus is released via exocytosis from the host cell [5]. Figure created with BioRender.com and adapted from ‘Life Cycle of Coronavirus’, by BioRender.com [6].
SARS‐CoV‐2 initiates infection using a viral spike protein (S protein) which binds to the angiotensin‐converting enzyme 2 (ACE2) receptor of a host cell, triggering endocytosis of the virus (Fig. 1.1) [7]. Once endocytosed, the outer protein coat of the virus is shed, releasing viral contents into the host cell (Fig. 1.2). The genomic RNA of the virus is then translated by host cell ribosomes, followed by processing of the resulting proteins with viral proteases (Fig. 1.3–4). The 3‐chymotrypsin‐like cysteine protease of SARS‐CoV‐2 processes large viral polypeptides into proteins necessary for viral replication, which then form elements of the replication‐transcription complex (Fig. 1.4) [5]. RNA‐dependent RNA polymerase (RdRp), a major component of the replication‐transcription complex, fulfills an essential role in the replication mechanism of SARS‐CoV‐2 (Fig. 1.5). The replicated RNA strands are then translated and structural and accessory viral proteins are processed, assembled, and receive post‐translational modifications at the Golgi apparatus (Fig. 1.7). Finally, the virus with its fully assembled proteins forms an exocytic vesicle when budding with the cell membrane and is released through exocytosis from the cell (Fig. 1.8–9) [5].
Viral entry into host cell
The treatments discussed in this section target the first step of the viral life cycle (i.e. entry into the host cell) through one of two mechanisms. The first mechanism involves spike protein targeting, in which the drug binds to the S protein of SARS‐CoV‐2 and prevents it from forming interactions with the ACE2 receptor that are necessary for viral uptake. The second mechanism is endosomal targeting that creates an alkaline environment within endosomes/lysosomes, lowering cytokine levels. This mechanism disrupts endocytosis and thus does not allow the virus to enter the cell [8]. A summary of these treatments and their timing is found in Table 1.
Table 1.
Summary of recommendations for viral entry targeted antivirals. A list of the antivirals that target viral entry is provided, as well as the recommended time of treatment during disease progression. Note that only one antiviral is recommended as preventative treatment (before infection has occurred); the majority are recommended for mild to moderate infection, none are recommended for severe infection, and two are not currently authorized for COVID‐19 treatment.
| Antiviral | Timing of treatment | References |
|---|---|---|
| Evusheld | Preventative | [9] |
| REGEN‐COV | Mild to moderate infection | [10] |
| BNE | Mild to moderate infection | [11, 12] |
| Bebtelovimab | Mild to moderate infection | [13] |
| Sotrovimab | Mild to moderate infection | [14] |
| Arbidol | Not authorized for COVID‐19 treatment | [15, 16, 17, 18, 19] |
| Chloroquine | Not authorized for COVID‐19 treatment | [20] |
Spike protein targeting
Evusheld
Evusheld was developed by AstraZeneca as a SARS‐CoV‐2 preventative treatment for adults and children (aged at least 12 years old and weighing 40 kg). The drug consists of two consecutive intramuscular injections of monoclonal antibodies tixagevimab and cilgavimab, preferably with one injection into each gluteal [9]. Originating from research carried out by the Vanderbilt University Medical Center in 2020, these antibodies were isolated from patients with COVID‐19 and neutralized the S protein of SARS‐CoV‐2 in vitro [21].
The antibodies are classified as SARS‐CoV‐2 S protein‐directed attachment inhibitors, comprising a type of human immunoglobulin G1 (IgG1κ) monoclonal antibody with amino acid substitutions to extend half‐life. These antibodies simultaneously bind to separate regions of the receptor‐binding domain (RBD) of the SARS‐CoV‐2 S protein to block its interaction with ACE2 (Fig. 2) [23]. Both monoclonal antibodies have a heavy and light chain, with the interface between the heavy/light chains of tixagevimab forming an aromatic cage. By contrast, cilgavimab has a long light chain and a heavy chain that is able to interact with the side opposite of tixagevimab on the RBD [21]. This means that, when used in tandem, the two antibodies provide complementary protection against the binding of the S protein to the host cell, thereby preventing entry.
Fig. 2.
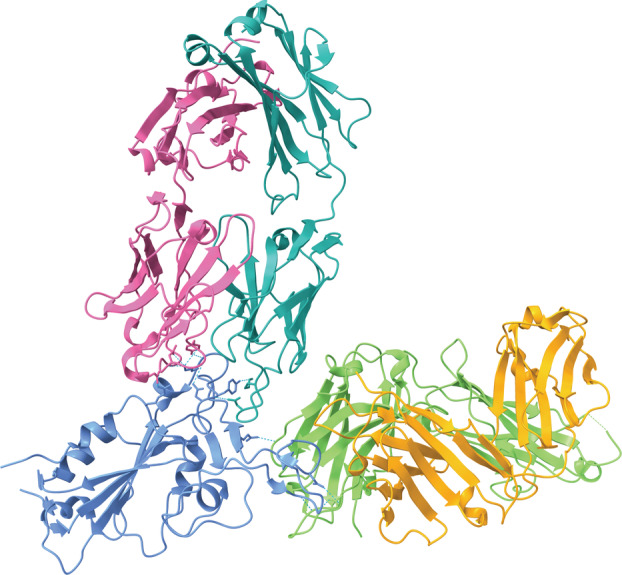
The component antibodies of Evusheld simultaneously bind to separate regions of the RBD of the SARS‐CoV‐2 S protein (blue) [Protein Data Bank (PDB) ID: 7L7E, visualized with ucsf chimerax, version 1.4] [21, 22]. Tixagevimab consists of a heavy chain (green) and a light chain (orange) that form an aromatic cage. Cilgavimab consists of a heavy chain (pink) that is able to interact with the RBD and a light chain (teal). Hydrogen bonds between each of the antibodies and the RBD are represented by dotted cyan lines.
There are two ongoing phase 3 clinical trials: PROVENT [24] and STORM CHASER [25]. PROVENT used a randomized, double‐blind trial with 5172 subjects who tested negative for SARS‐CoV‐2, with 3441 individuals receiving Evusheld and another 1731 receiving a placebo. Data collected after 83 days of treatment demonstrated a 77% risk reduction for contracting SARS‐CoV‐2, which was statistically significant compared to the placebo. In a follow‐up 6.5 months after treatment, there was an 83% risk reduction for symptomatic illness [24]. Similarly, the STORM CHASER trial treated its 1121 subjects at a 2 : 1 ratio of Evusheld to saline placebo, resulting in a 33% relative risk reduction [25]. Observed adverse effects include increased cardiovascular abnormalities, including myocardial infarctions and heart failure; however, no causal relationship has been shown between these effects and Evusheld treatment [9].
The Food and Drug Administration (FDA) then issued an Emergency Use Authorization (EUA) with an authorized dose of 150 mg of each antibody on 16 December 2021, later increasing the approved dose to 300 mg on 24 February 2022 [11]. More recently, the European Union also approved Evusheld on 25 March 2022 [26]. Evusheld is only recommended for those who are not infected with COVID‐19 at the time of administration and for those unable to receive COVID‐19 vaccinations because of allergies to COVID‐19 vaccines or a compromised immune system [27, 28]. Evusheld is authorized for pre‐exposure prophylaxis of COVID‐19 infection, even showing reduced viral load for all Omicron variants [29]. Evusheld is not authorized for post‐exposure prophylaxis [23, 26].
REGEN‐COV
REGEN‐COV is a IgG1κ monoclonal antibody mixture composed of casirivimab and imdevimab [30]. These antibodies target the RBD of the S protein and prevent viral entry of the SARS‐CoV‐2 virus into the host cell. The two antibodies work as a combination treatment rather than as an individual monotherapy because they bind to non‐overlapping epitopes on the S protein to increase the efficacy of inhibition of viral entry (Fig. 3) [33]. The simultaneous binding to distinct sites of the RBD was intended to protect against the emergence of new variants of the virus and circulating variants. In vitro studies have shown that the cocktail of the two antibodies was effective at presenting the rapid escape of the virus from binding that occurs with single antibodies [34].
Fig. 3.
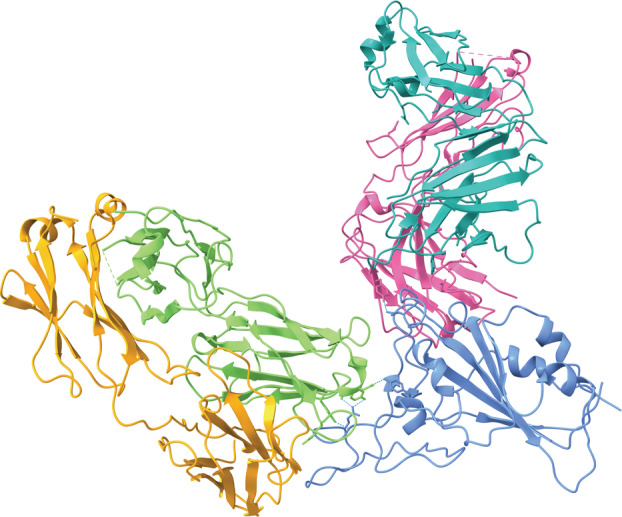
Casirivimab (REGN 10933, heavy chain: green, light chain: orange) and imdevimab (REGN 10987, heavy chain: pink, light chain: teal) bind mutually exclusive epitopes on a portion of the RBD of the SARS‐CoV‐2 S protein (blue) (PDB ID: 6XDG, visualized with ucsf chimerax, version 1.4) [31, 32]. Casirivimab binds the top of the RBD of the virus and overlaps greatly with the binding site for ACE2. The epitope for imdevimab is located away from the epitope for casirivimab and has little overlap with the ACE2 binding site. The non‐overlapping binding sites allow for increased efficiency when binding to the virus due to the multiple binding locations. Hydrogen bonds between each of the antibodies and the RBD are represented by dotted cyan lines.
On 21 November 2020, the FDA issued an EUA for REGEN‐COV to treat mild to moderate COVID‐19 cases in adults and children (aged at least 12 years old and weighing 40 kg) who are at high risk of disease progression to severe COVID‐19 infection. The treatment is not preventative in nature and is not recommended for treatment of severe COVID‐19 infections [10]. The authorized dosage of REGEN‐COV is 600 mg of casirivimab and 600 mg of imdevimab administered intravenously in conjunction 10 days after COVID‐19 onset. Efforts were made to increase global supply of REGEN‐COV with Regeneron's collaboration with Roche, which is primarily responsible for development and distribution outside the USA. The goal was to increase access in developing countries through drug donations to public health organizations [35].
REGEN‐COV is still being evaluated as an antiviral treatment for COVID‐19. In a previously conducted trial, 799 non‐hospitalized adults with mild to moderate COVID‐19 symptoms were evaluated. Two hundred and sixty‐six patients received 2400 mg of the combination drugs, 267 received 8000 mg of the combination drugs, and 266 received a placebo within 3 days of a positive COVID‐19 test. Researchers noted a statistically significant drop of 16.6% in hospitalizations for patients treated with REGEN‐COV compared to those receiving a placebo, notably in patients with no previous immune response initiation or with a viral load at baseline measurements [33]. Another study in 2021 showed that REGEN‐COV was associated with decreases in hospitalization for any cause or death from any cause, including COVID‐19. Eighteen of 1355 patients treated with 2400 mg of REGEN‐COV (1.3%) experienced COVID‐19‐related hospitalization or death, whereas 62 of 1341 patients in the placebo group who underwent randomization (4.6%) experienced such complications. Similarly, such outcomes occurred in seven of 736 patients in the REGEN‐COV 1200‐mg group (1.0%) and in 24 of 748 patients in the placebo group who underwent randomization. Additionally, five individuals died during the efficacy assessment period, including one in the REGEN‐COV 2400‐mg group, one in the REGEN‐COV 1200‐mg group, and three in the placebo group [30]. Other studies have shown that, in cases where one of the antibodies in the cocktail is impacted, the combination treatment retains full neutralization potency of the virus. Furthermore, REGEN‐COV is described to retain neutralization capabilities against SARS‐CoV‐2 variants, including those with S protein mutants with a D614G substitution, the B.1.1.7 variant, and other variants associated with reduced vaccine efficacy (e.g. B.1.351) [34].
Although adverse effects are still being studied, observed effects include symptoms associated with allergic reactions and worsening COVID‐19 symptoms after treatment [36, 37]. As a result of intravenous administration of the drug, bleeding and bruising of the skin and soreness at the infusion site are also possible.
REGEN‐COV is stated to be ineffective against the Omicron variant because of markedly reduced neutralization activity [38]. The specificity of REGEN‐COV against the S protein of the virus was reduced as a result of mutations in the virus structure with the rise of Omicron and was stated to be an insufficient treatment. Because of the inability to test for the exact virus variant that each person was infected with, REGEN‐COV was limited in its distribution and is no longer authorized for distribution in the US because of the prevalence of Omicron subvariants [39].
Bamlanivimab and etesevimab
Administered as a single intravenous infusion, bamlanivimab and etesevimab (BNE) are two monoclonal antibodies developed by Lilly Investors that, once combined, target the S protein of SARS‐CoV‐2. The antibodies respectively bind two distinct overlapping sites on the RBD of the S protein, interfering with the ability of the S protein to attach to the ACE2 receptor on the human cell (Fig. 4) [44]. Bamlanivimab was observed to bind to an epitope overlapping the ACE2 binding site: seven of the approximate 25 side chains in the RBD interact with ACE2. This epitope is fully accessible on both the up and down conformations of the binding domain. This property is similar to the binding of the Ebola virus‐specific monoclonal antibody 114 that binds the Ebola virus glycoprotein binding domain in both the preactivation and activated states. Etesevimab can only bind to the RBD when it is open, unlike Bamlanvimab [45, 46].
Fig. 4.
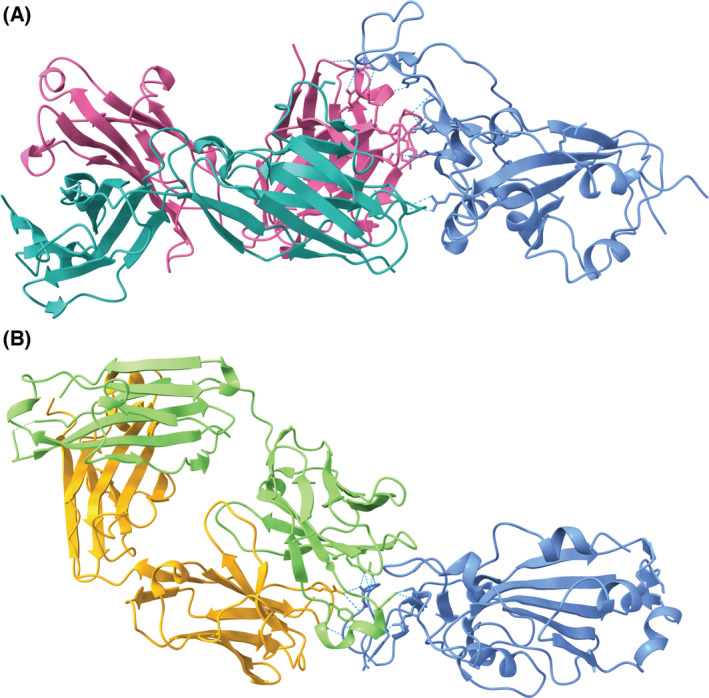
(A) Etesevimab (heavy chain: pink, light chain: teal) [40] binds to an epitope on a portion of the RBD of the SARS‐CoV‐2 S protein (blue) overlapping the ACE2 binding site (PDB ID: 7C01, visualized with ucsf chimerax, version 1.4) [41]. (B) Bamlanivimab (heavy chain: green, light chain: orange) [42] binds to an epitope on the RBD that overlaps with the binding site of etesevimab (PDB ID: 7KMG) [43]. Bamlanivimab is able to bind the epitope with the up and down conformations of its monomers [42]. Hydrogen bonds between each of the antibodies and the RBD are represented by dotted cyan lines.
Under a 19 February 2021, EUA, the FDA approved BNE for the treatment of mild‐to‐moderate COVID‐19 in children (aged at least 12 years old and weighing 40 kg) and adults at high risk for severe COVID‐19 [11, 12]. BNE is not authorized for treating patients with severe COVID‐19 or underlying COVID‐19 comorbidities. The drugs are not approved as a preventative treatment, potentially as a result of an inability to protect against new and upcoming variants [44]. A procurement agreement allowed multiple European countries to purchase the antibody cocktail directly from Lilly for approved use. The combination package with the cocktail comes with one vial of bamlanivimab (700 mg) and two vials of etesevimab (700 mg) [47].
A phase 2–3 clinical trial was conducted to evaluate drug efficacy and a greater reduction from baseline in the log viral load was observed among patients already testing positive for Covid‐19 who received bamlanivimab plus etesevimab compared to those who received placebo. In phase 2, 612 adult patients received a single infusion of the combination drugs of 2800 mg each, an infusion of bamlanivimab alone of 700, 2800 or 7000 mg, and a placebo. On day 11, a 4.8% reduction in hospitalization and death was observed in the group administered the combination drugs compared to the group who received the placebo. In the second trial of the experiment, similar results were obtained [12]. In another study, 1035 patients underwent randomization and received an infusion of bamlanivimab‐etesevimab or placebo. By day 29, 11 out of 518 patients (2.1%) in the bamlanivimab‐etesevimab group experienced COVID‐19‐related hospitalization or death from any cause compared with 36 of 517 patients (7.0%) in the placebo group. No deaths occurred in the bamlanivimab‐etesevimab group, whereas 10 occurred in the placebo group.
Adverse effects of the drug include hypersensitivity reactions, anaphylaxis, and the possible worsening of COVID‐19 symptoms. Worsening symptoms may be a result of the progression of COVID‐19 or the combination treatment, and neither has been ruled out as a potential cause. Other possible adverse effects associated with intravenous administration include bleeding, skin bruising, soreness, swelling, and infection at the infusion site [44].
Similar to REGEN‐COV, the efficacy of BNE is noted to be markedly reduced with the Omicron variant because of increased resistance of the virus as a result of changes in the S protein and binding domains. The specificity of the binding capabilities of BNE is thus reduced against such variants and it is considered to be an insufficient treatment. BNE was minimally distributed during the height of the Omicron pandemic for this reason and is authorized for distribution in the USA in most states and territories [39]. It has remained effective against other global variants.
Bebtelovimab
Developed by Eli Lilly, bebtelovimab (also known as LY‐CoV1404) is a human IgG1 monoclonal antibody that targets the RBD of the SARS‐CoV‐2 S protein, preventing the virus from binding with ACE2 to trigger endocytic entry into the cell (Fig. 5). Both the up and down conformers of the RBD on the SARS‐CoV‐2 S protein can be bound by the fragment antigen‐binding domain of the antibody. The epitope, being exposed in both conformations, allows bebtelovimab to bind to the S protein regardless of conformation, which is not a feature of many other monoclonal antibodies targeting the RBD. Multiple molecules of bebtelovimab can bind to the 3‐RBD‐up conformation of the S protein (which has all three RBDs on the trimeric S protein exposed for binding to the ACE2 receptors), although only one molecule of bebtelovimab can bind to the 3‐RBD‐down conformation (in which the S protein cannot bind to ACE2, on account of the RBDs being partially buried). Binding affinities were determined to be relatively high for bebtelovimab binding to the S protein, ranging from a binding constant (K D) of 75 to 220 pm. Furthermore, in vitro viral neutralization studies revealed bebtelovimab to have a higher potency than the comparable monoclonal antibody bamlanivimab because the half maximal inhibitory concentration (IC50) of bebtelovimab was consistently two to three times lower than that of bamlanivimab [48, 50].
Fig. 5.
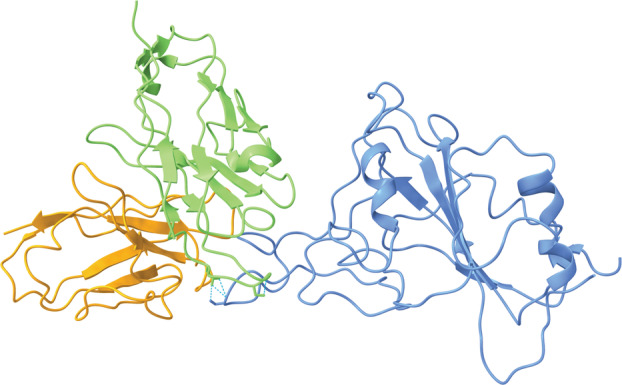
Bebtelovimab interacting with a component of the RBD of the SARS‐CoV‐2 S protein (PDB ID: 7MMO, visualized with ucsf chimerax, version 1.4) [48, 49]. The S protein is blue, the light chain of bebtelovimab is orange, and the heavy chain of bebtelovimab is green. Hydrogen bonds between bebtelovimab and the RBD are represented by dotted cyan lines.
Bebtelovimab was authorized for use in the treatment of mild‐to‐moderate COVID‐19 infection on 11 February 2022, by the FDA under an EUA [13]. Bebtelovimab is able to bind and neutralize all current variants of concern as of June 2022, including the Omicron variant and its subvariants (BA.1, BA.2, BA.4, and BA.5). No other monoclonal antibody has been shown to be effective against all variants of concern, making bebtelovimab a promising therapeutic [48, 50, 51, 52].
The population intended for treatment with bebtelovimab under the FDA's EUA is adults and children (aged at least 12 years old and weighing 40 kg) who test positive for COVID‐19 and are at high risk for progression to severe COVID‐19. Bebtelovimab is administered via a single 175‐mg intravenous injection, providing an advantage over therapeutics administered over multiple days [13]. As of July 2022, bebtelovimab remains only authorized for use in the USA and no applications for authorization or approval have been submitted in any other country [53].
A phase 2 clinical trial tested the ability of bebtelovimab individually and in conjunction with BNE to reduce the proportion of patients with persistently high viral load from baseline to day 11. Bebtelovimab administered with BNE in low‐risk patients produced a 36% relative risk reduction, whereas bebtelovimab administered on its own in low‐risk patients produced a 40% relative risk reduction and reduced the time for sustained symptom resolution by a median of 2 days compared to placebo; these results were statistically significant at a level of α = 0.01 or below [54]. Nausea and vomiting were the most common adverse effects observed in patients who received bebtelovimab, alone or in conjunction with BNE, at 0.8% and 0.7%, respectively [55].
The epitope on the S protein targeted by bebtelovimab is notably more conserved than epitopes targeted by other monoclonal antibodies. This fact, in conjunction with its ability to tolerate mutations in the S protein at the less conserved N439 and N501 sites, explains its ability to effectively inhibit all current variants of concern; N439K and N501Y mutations that have been observed in current SARS‐CoV‐2 strains have no effect on bebtelovimab binding or neutralization efficiency. However, individual substitution mutations at positions 444 (K444Q), 445 (V445A), and 499 (P499R, P449S) did lead to a notable decrease in ACE2 competition, binding, and neutralization activity, although these mutations are not common in the global population of SARS‐CoV‐2 strains; resistance to bebtelovimab may evolve if any of these mutations occur in a future variant of concern [48].
Sotrovimab
Developed by GlaxoSmithKline and Vir Biotechnology, sotrovimab is a monoclonal antibody that was developed from the antibody S309 (Fig. 6). S309 was originally isolated from the memory B cells of a patient who recovered from severe acute respiratory syndrome coronavirus (SARS‐CoV) in 2003 [58]. This was carried out to demonstrate how the epitope of the antigen was assumed to be highly conserved.
Fig. 6.
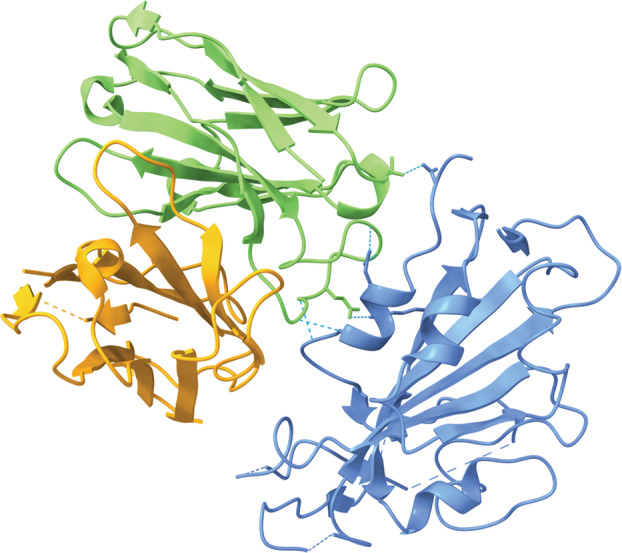
S309, the parent antibody of Sotrovimab, interacting with a portion of the RBD of the SARS‐CoV‐2 S protein (PDB ID: 6WPS, visualized with ucsf chimerax, version 1.4) [56, 57]. S309 consists of a heavy chain (green) and a light chain (orange). Hydrogen bonds between S309 and the RBD are represented by dotted cyan lines.
Sotrovimab is a IgG1κ monoclonal antibody composed of two identical light chain polypeptides (214 amino acids) and two identical heavy chain polypeptides (457 amino acids). The drug binds to a conservative epitope on the S protein RBD of SARS‐CoV‐2. Sotrovimab inhibits an unknown step between virus attachment and the fusion of viral and cell membranes [59]. Further work is needed to elucidate the mechanism of inhibition.
Sotrovimab was approved under a September 2021 EUA for mild‐to‐moderate COVID‐19 cases in adults and children (aged at least 12 years old and weighing 40 kg), but it is currently not authorized for use in the US due to Omicron prevalence. The drug should be administered within 7 days to COVID‐19‐positive individuals who do not require hospitalization or respiratory support. It is given in the form of 500 mg of solution diluted via intravenous diffusion over the course of 30 min, followed by a monitoring period of 1 h [14].
In the COMET‐ICE trial, 1057 subjects were given a sotrovimab or placebo infusion. After 29 days, the adjusted relative risk reduction was 79% compared to the placebo [60]. In a similar clinical trial called COMET‐TAIL, 385 subjects received the sotrovimab infusion. After 29 days, only 1.3% of patients had a progression that led to hospitalization over 24 h. Adverse effects of this treatment include anaphylaxis or infusion‐related reactions such as fevers, difficulty breathing, arrhythmia, fatigue, and chest pain [61].
However, because sotrovimab only targets one viral epitope, this means that mutations can lead to resistance to the drug, as shown by a study using patients from the Western Sydney Local Health District in New South Wales, Australia. Analyzing the first 100 patients who received treatment of sotrovimab at the facility, it was discovered that eight patients had remained positive for the virus when PCR testing was performed. The genomic analysis then confirmed that mutations occurred in four of the eight patients that had led to viral resistance against the drug [62].
Arbidol
Umifenovir, brand‐name Arbidol, is a hydrophobic, indole‐derived antiviral agent manufactured by Russian pharmaceutical company OTCPharm [63]. Arbidol has been used for approximately 25 years in Russia and for 16 years in China as a prophylactic treatment for influenza and other respiratory infections [64], but has not been approved anywhere for use against COVID‐19. It has been well established that Arbidol acts to inhibit viral fusion with the host‐cell membrane by binding to the virus and the membrane simultaneously, thus preventing viral endocytosis [63, 65, 66].
Because of the success of Arbidol against a wide array of viral pathogens, the drug has been investigated as a therapeutic agent against SARS‐CoV‐2. Much like in influenza, Arbidol binds dually to the RBD of the SARS‐CoV‐2 S protein and ACE2 receptors on the host‐cell membrane, forming stronger interactions with the RBD than ACE2 (Fig. 7) [67, 68]. This higher binding affinity of Arbidol to the RBD is the result of multiple residues including Arg403, Asp405, Glu406, Gln409, Gly416, Lys417, Ile418, and Tyr505 [67]. Arbidol is proposed to interact with three identical binding sites on each of the three subunits of the S protein trimer [67, 68, 69]. The triple binding in this region is suggested to prevent trimerization of the S protein, a necessary step in viral entry [68]. Specifically, Arbidol increases the affinity of the RBD to ACE2, but induces rigidity in the RBD‐ACE2 complex, preventing it from forming the conformation necessary for the assembly of endosomes [67]. This interaction is sufficient for inhibition of viral entry [69]. Additionally, an in vitro study has also shown that Arbidol may also impede release of the virus from intracellular vesicles [70], suggesting that the drug may additionally disrupt S‐activation impeding an interaction between the S protein and cathepsin L in the endolysosome. However, studies must be performed to test this hypothesis and elucidate the mechanism by which Arbidol inhibits the release of SARS‐CoV‐2 from intracellular vesicles.
Fig. 7.
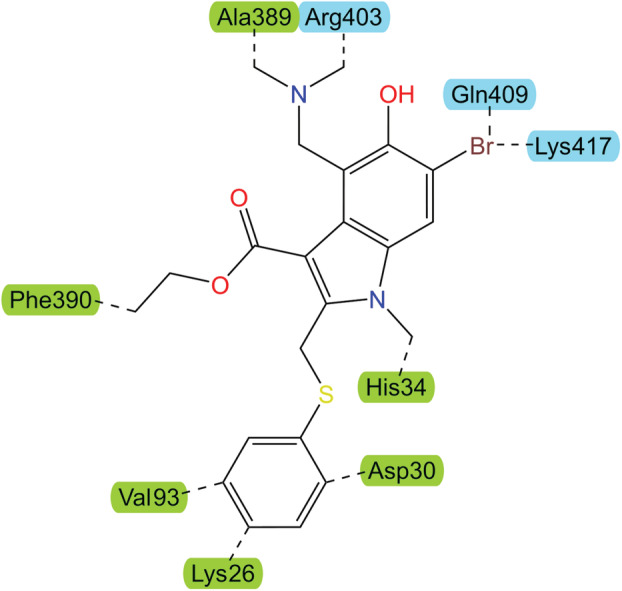
The chemical structure of Arbidol, an indolic carboxylic acid. Dashed lines indicate interactions between dually‐bound Arbidol and residues on ACE2 (green label) and the RBD of the SARS‐CoV‐2 S protein (light blue label). The ability of Arbidol to bind dually with the RBD and ACE2 is assumed to prevent the S‐protein from forming the necessary configuration for viral uptake into the host‐cell. Adapted from Padhi et al. [67].
In vitro studies established Arbidol as having very promising antiviral effects on SARS‐CoV‐2, demonstrating statistically significant decreases in binding efficiency, inhibition of viral entry, and inhibition of post‐viral events [70]. However, clinical trials have been inconclusive. One study of 100 individuals with COVID‐19 in a teaching hospital found that Arbidol significantly contributed to clinical and laboratory improvements, including peripheral oxygen saturation, hospitalization duration, white blood‐cell count, erythrocyte sedimentation rate, intensive care unit admissions, and chest computed tomography involvements [71]. However, a study of 81 COVID‐19 patients in a non‐intensive care unit ward, as well as another study of 101 moderate‐severe COVID‐19, found no significant difference in SARS‐CoV‐2 clearance or prognosis [15, 16]. Another study shows that the use of Arbidol as a preventative measure led to significantly decreased infection rates in health professionals (N = 164), although the same study reports no significant difference in hospitalization rate in infected individuals [17]. Furthermore, one study even reported increased in‐hospital mortality in severe COVID‐19 patients when Arbidol was administered alone or in combination with Oseltamivir [18]. This study used a total of 109 patients with 23 receiving Arbidol monotherapy, 10 receiving the Arbidol and Oseltamivir combination, and the remainder receiving other antiviral therapeutics, none of which resulted in the same increased in‐hospital mortality [18]. The discrepancy between Arbidol efficacy in vitro versus clinical trials could be a result of the dosage administered to patients [15]. In vitro studies utilize high dosages of the drug to obtain the inhibitory effects but, because of concerns with side effects, high doses of Arbidol cannot be prescribed to patients [15, 19]. The efficacy of Arbidol remains controversial and thus more studies are needed to show its safety and efficacy in vivo.
Arbidol is orally administered three times daily in doses of 200 mg [15]. Additionally, Arbidol has been shown to have mild adverse effects, including nausea, diarrhea, dizziness, and elevated serum transaminase, and it should be used cautiously in patients with liver dysfunction [72]. No data have been reported regarding the development of Arbidol resistance in SARS‐CoV‐2. One study exploring new potential antiviral compounds reports a binding affinity of Arbidol to the Omicron strain S protein as −20.88 kJ·mol−1, which is weaker than the reported binding affinity of Arbidol to the 2019 strain of −105.95 ± 14.25 kJ·mol−1, indicating a weaker interaction with the newer strain [73]. However, the reasons for this reduced affinity, as well as the consequences, have not been reported.
Endosomal targeting
Chloroquine
Originally introduced as an antimalarial drug and used as an antiviral since the 1960s, chloroquine is now being used to treat autoimmune diseases such as lupus and rheumatoid arthritis in clinical trials (Fig. 8). Chloroquine is continually aiding the global healthcare system by being widely accessible and low‐cost [74].
Fig. 8.
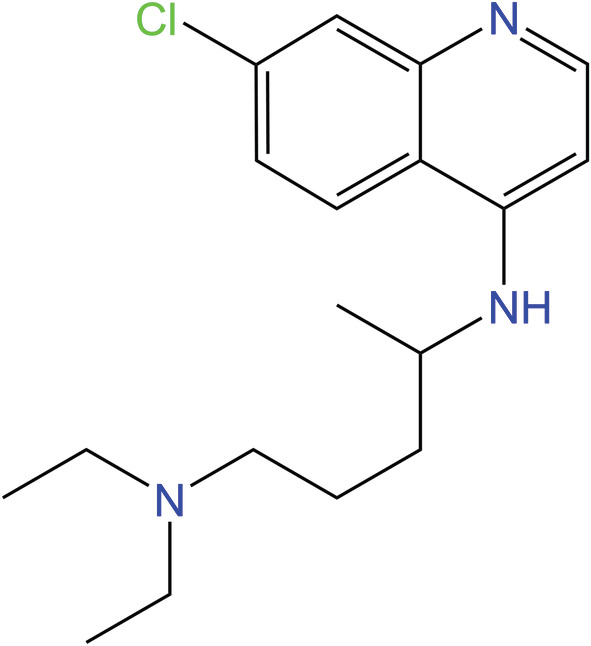
Chemical structure of chloroquine, an aminoquinoline that increases the pH of endosomes. Chloroquine has been used to treat malaria, viral diseases, and autoimmune diseases and was investigated for use against SARS‐CoV‐2 by preventing endosomal entry in the early stages of the pandemic [8, 74].
Chloroquine alters the pH of endosomes/lysosomes, creating an alkaline environment that prevents binding and endocytosis in vitro [8]. Additionally, chloroquine disrupts the glycosylation of the cellular receptors of severe acute respiratory syndrome coronavirus (SARS‐CoV) [75]. Specifically, the number of Toll‐like receptor signaling molecules is reduced, which eventually lowers cellular cytokine levels. The lowering of cytokine levels eventually leads to the blocking of viral activity within the cell. This reduction mitigates the cytokine storm that develops in the cell as a result of COVID‐19 [75]. Because of its in vitro success with SARS‐CoV, chloroquine was investigated for use against SARS‐CoV‐2 [20].
In March 2020, the FDA issued an EUA to administer chloroquine for severe cases of COVID‐19 [74]. However, the use of chloroquine for COVID‐19 has spurred controversy because of a lack of well‐designed clinical studies supporting the efficacy of chloroquine in treating COVID‐19. There are clinical trials testing the efficacy of chloroquine (23 trials in China alone), yet none have produced sufficient evidence of the benefits of chloroquine for COVID‐19 [20]. The WHO views chloroquine use for COVID‐19 as solely experimental, and so it must be designated as off‐label to be prescribed. Chloroquine also has a narrow margin between beneficial and toxic doses. For short periods of use, the adverse effects of chloroquine are considered mild, with mainly gastrointestinal reactions, dizziness, and headaches. The most severe adverse effects from short‐term use can be seizures or psychosis, although these quickly dissipate after treatment [74].
Long‐term use of chloroquine has been linked to serious adverse effects. Over time, the toxicity of chloroquine can lead to retinopathy, circular defects, and retinal diameter defects. These are permanent conditions that are caused by the accumulation of chloroquine in the eyes. Chloroquine poisoning is also linked to cardiovascular disorders. Overall, the grave adverse effects that arise from long‐term use have called into question the benefits of this drug as a therapeutic for COVID‐19 [20].
Viral replication
The treatments in this section target the maturation process of the virus once it is already in a cell. Here, we focus on treatments targeting protease inhibition, RdRp stalling, and error catastrophe. A summary of these treatments and their timing is found in Table 2.
Table 2.
Summary of recommendations for viral replication targeted antivirals. A list of all of the antivirals discussed in the viral replication section is provided, as well as an indication of when in the progression of the disease they are recommended. Note that no antivirals are recommended as preventative treatment (before one is infected); the majority are recommended for mild to moderate infection, only one is recommended for individuals who might develop severe infection, and three are not currently authorized for COVID‐19 treatment.
| Antiviral | Timing of treatment | References |
|---|---|---|
| Paxlovid | Mild to moderate infection | [76, 77] |
| Kaletra | Not authorized for COVID‐19 treatment | [78] |
| Remdesivir | Infection with high risk to develop severe infection | [79] |
| Ribavirin | Not authorized for COVID‐19 treatment | [48, 49] |
| Favipiravir | Not authorized for COVID‐19 treatment | [80] |
| Molnupiravir | Mild to moderate infection | [81] |
Protease inhibition
The main protease of SARS‐CoV‐2 (Mpro) is a 3‐chymotrypsin‐like cysteine protease (3CLpro). Following the translation of viral mRNA by ribosomes in the host cell, Mpro produces non‐structural proteins (NSPs) by cleaving peptide bonds in 11 distinct sites on the two viral polyproteins (polyproteins 1a and 1ab), which are coded for by overlapping sections in open reading frame 1ab of the viral genome. The catalytic dyad of the His41 and Cys145 residues form the catalytic site of Mpro. Additionally, the substrate binding sites of Mpro are highly conserved among coronaviruses. NSP5‐NSP16 are produced by the actions of Mpro and NSP4 is produced by the combined actions of Mpro (which is NSP5) and the papain‐like protease (NSP3) of SARS‐CoV‐2 (which is also responsible for cleaving these polyproteins to produce NSP1‐NSP3). Many of these NSPs are involved in the formation of the replication‐transcription complex of SARS‐CoV‐2; for example, NSP12 is RdRp, with NSP7 and NSP8 being cofactors of RdRp, whereas NSP13 is a helicase and nucleoside‐triphosphatase. Anchored in place by NSP3, NSP4, and NSP6, the replication‐transcription complexes are localized to convoluted membrane structures in the rough endoplasmic reticulum. Therefore, inhibiting Mpro impedes viral replication because the necessary proteins that form the replication‐transcription complex will not be produced, making it a promising candidate for targeting by therapeutics. The downstream targeting of RdRp, a product produced by the enzymatic activity of Mpro, is also be discussed later in this section [82, 83, 84, 85, 86, 87, 88].
Paxlovid
Paxlovid, developed specifically in response to the COVID‐19 pandemic by Pfizer, is a combination treatment of nirmatrelvir (also known as PF‐07321332) and ritonavir (Fig. 9A,B) [90, 91].
Fig. 9.
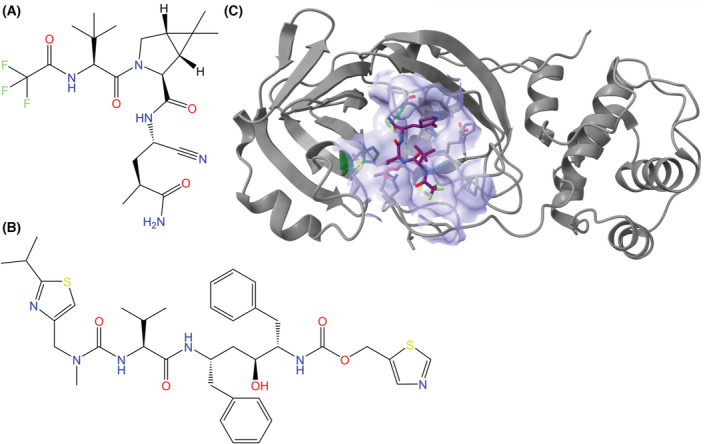
Chemical structures of (A) nirmatrelvir and (B) ritonavir, the two active components of Paxlovid. (C) Nirmatrelvir in complex with the Mpro of SARS‐CoV‐2 (PDB ID: 7SI9, visualized with ucsf chimerax, version 1.4) [89], with the binding pocket (lavender, composed of atoms within 5 Å of the inhibitor) visualized [82]. Nirmatrelvir is represented by a stick model with a purple carbon backbone. The peptide backbone of Mpro is represented in gray, with secondary structures displayed in cartoon format. In the binding pocket, stick models of amino acid residues are shown, and the catalytic dyad of Mpro (His41 and Cys145) is emphasized with carbon atoms in green. Hydrogen bonds between nirmatrelvir and Mpro are represented by dotted cyan lines, and heteroatom colors correspond to those in the 2D visualization.
Ritonavir is a pharmacokinetic enhancer that increases the half‐life of nirmatrelvir and other pharmaceuticals by inhibiting cytochrome P450 3A4, a major enzyme involved in human drug metabolism. The N‐terminus thiazole group of ritonavir irreversibly binds to heme iron within the cytochrome P450 3A4 active site to inhibit the enzyme [92, 93]. Because ritonavir inhibits drug metabolism by cytochrome P450 3A4, Paxlovid is not advised to be administered in tandem with pharmaceuticals relying on this enzyme, which limits the population that can benefit from treatment with Paxlovid [76]. Ritonavir also induces other cytochrome P isoenzymes and uridine diphosphate glucuronosyltransferases, a class of drug‐metabolizing enzymes, at the same time as inhibiting the efflux transporter P‐glycoprotein and organic anion transporting polypeptides 1B1 and 1B3 [93].
Nirmatrelvir inhibits the main protease (Mpro) of SARS‐CoV‐2, which is a 3‐chymotrypsin‐like cysteine protease [90, 94, 95, 96]. When not inhibited, Mpro proteolytically processes proteins that are critical for viral replication, including RdRp and other instances of Mpro [94]. Nirmatrelvir inhibits Mpro via reversible covalent bonding between the nitrile warhead of nirmatrelvir and the catalytic cysteine (Cys145) of Mpro, which causes a thioimidate group to form (Fig. 9C). Prior to this occurring, nirmatrelvir forms a non‐covalent complex with Mpro [95, 96].
Computational models suggest that the non‐covalent complex between Mpro and nirmatrelvir and the subsequent steps for covalent bond formation occur via the following interactions. The P1 γ‐lactam ring of nirmatrelvir mimics interactions of the enzyme with glutamine, which occurs before cleavage sites in the SARS‐CoV‐2 polyproteins. The bicycloproline component of nirmatrelvir also mimics a leucine residue, allowing it to fit into the S2 site of Mpro, and it has a stacking interaction with the catalytic His41. The P3 position of nirmatrelvir contains a tert‐butyl group that maintains similar interactions to valine residues at cleavage sites on the polyproteins, and nitrogen and oxygen atoms in this position hydrogen bond with Glu166, which plays a major role in the binding of both the intended substrate peptides and nirmatrelvir. P4 has the largest difference between nirmatrelvir and the serine residue with respect to peptide substrate interactions with the enzyme because the fluorine atoms in nirmatrelvir form hydrogen bonds with Glu192 opposed to Glu166, Gln189, and Thr190 for the serine residue in the peptide substrate. The formation of the covalent bond between nirmatrelvir and Mpro results in the electrophilic carbon atom of nirmatrelvir's nitrile group being attacked by a nucleophilic thiol group on the cysteine residue, which is first activated via proton transfer to the catalytic histidine (His41). Once the nucleophilic attack has taken place, a second proton transfer from His41 to nirmatrelvir occurs to complete the reaction [95, 96].
On 22 December 2021, the FDA issued an EUA for the use of Paxlovid in the treatment of COVID‐19 in adults and children (aged at least 12 years old and weighing 40 kg) who have tested positively for SARS‐CoV‐2 and display mild‐to‐moderate symptoms with a high risk of advancing to severe COVID‐19 symptoms. Paxlovid is not authorized for preventative use by the FDA [76, 77]. Standard treatment protocols for Paxlovid call for 300 mg of nirmatrelvir and 100 mg of ritonavir to be administered orally, twice daily for five consecutive days [77, 90].
Paxlovid has also been authorized for use in the treatment of COVID‐19 across the European Union and other countries such as the UK, Israel, and China. The WHO currently recommends the use of Paxlovid in COVID‐19 patients a high risk for hospitalization, and Pfizer has entered a Medicines Patent Pool agreement with the WHO to allow for the production of generic therapeutics that are bioequivalent to Paxlovid in 95 low‐ and middle‐income countries [97, 98].
In the EPIC‐HR phase 2–3 clinical trial, Paxlovid treatment within 5 days of symptom onset reduced COVID‐19 related hospitalizations and death by 89.1% among patients who had not received monoclonal antibody treatments for COVID‐19 compared to placebo [77, 90]. In this trial, 7.8% of patients treated with Paxlovid experienced one or more adverse effects, only two of which affected over 1% of patients treated with Paxlovid: 4.5% experienced an impaired sense of taste (dysgeusia) and 1.3% experienced diarrhea [90].
Pfizer recently announced early findings from the Phase 2/3 EPIC‐PEP clinical trial that analyzed the post‐exposure prophylactic potential of Paxlovid; against placebo, the trial found a 32% and 37% risk reduction in adults who took Paxlovid for 5 and 10 days, respectively, after being exposed to a symptomatic household contact who tested positive for SARS‐CoV‐2, but these results lacked statistical significance. Additionally, in the Phase 2/3 EPIC‐SR trial, a lack of statistically significant findings in risk reduction for standard risk patients and a low observed risk of hospitalization and death caused Pfizer to cease enrollment, affirming the decision by the FDA to limit the use of Paxlovid under the current EUA to only patients who are at a high risk of progression to severe COVID‐19 symptoms. Full data and results from the EPIC‐PEP and EPIC‐SR trials will be published in the coming months [99, 100, 101, 102].
Additionally, cases of ‘Paxlovid rebound’ have recently been reported in the media and literature, with mild COVID‐19 symptoms recurring after initial recovery post‐treatment with Paxlovid; early research suggests that insufficient drug exposure is a likely cause of this phenomenon because infected cells not exposed to the drug would allow for continued viral replication and a reemergence of symptoms [103, 104, 105]. However, these rebound cases remain rare because, in the Paxlovid clinical trial, < 1% of 5287 patients were hospitalized (six patients) or required care in an emergency department for COVID‐19 symptoms (39 patients) 5–15 days after treatment with Paxlovid [103, 106].
Nirmatrelvir has been found to have a similar level of efficacy across variants with missense mutations affecting Mpro, including C.37 (Lambda), B.1.1.318, B.1.2, B.1.351 (Beta), B.1.1.529 (Omicron), and P.2 (Zeta). However, none of the Mpro missense mutations in the current variants take place at the active site of the enzyme, meaning that there are still many questions about how such a mutation would affect the inhibition potential of nirmatrelvir [107, 108]. Because nirmatrelvir shares many of the same interactions with Mpro that the peptide substrate uses, mutations that prevent nirmatrelvir from inhibiting Mpro will likely also decrease the binding efficiency of the substrate; however, the low conservation of the flexible amino acid 45–51 region may allow for the evolution of resistance, and mutations in other regions that directly interact with nirmatrelvir, such as 163–169 and 186–192, may also impart resistance [109].
Indeed, resistance to nirmatrelvir has been reported for individual Mpro mutants with at least one of the following amino acid substitutions, many of which occur in the aforementioned regions: L50F, S144M/F/A/G/Y, M165T, E166A/Q/V, L167F, H172Q/F, and Q192T/S/V. Computationally, an Mpro mutant with two substitutions, L50F and E166V, conferred up to an 80‐fold resistance to nirmatrelvir in homologous infectious cell culture and, in vitro, a triple mutant (L50F, E166A, and L167F) was reported to have a 72‐fold increase in resistance compared to wild‐type Mpro, although at the sacrifice of only having 5.3% of the enzymatic activity of the wild‐type [110, 111, 112]. This finding implies that mutations in the active site of Mpro, although imparting nirmatrelvir resistance, may reduce the overall fitness of the virus because of the many shared protein–inhibitor and protein–substrate interactions, potentially reducing its capacity for infection. However, because Mpro has a highly conserved structure and a significantly lower mutation rate than other SARS‐CoV‐2 proteins (such as the S protein), resistance to nirmatrelvir may be slow to evolve for variants in the wild [109, 113].
Kaletra
Kaletra is a combination treatment of lopinavir and ritonavir (Fig. 10), originally developed by Abbott Laboratories to treat HIV‐1 infections [115, 116, 117]. It was initially approved for use in this manner by the FDA in September of 2000 [118]. Lopinavir inhibits the 3CLpro of HIV by targeting the C2‐symmetric pocket of the active site. Ritonavir, as in Paxlovid, acts as a pharmacokinetic enhancer to increase lopinavir half‐life [119]. Because lopinavir–ritonavir was also proposed to inhibit the 3CLpro of SARS‐CoV and Middle East respiratory syndrome coronavirus (MERS‐CoV) based on the results of in vitro, in vivo, computational, and clinical studies, it was logical to test its efficacy against SARS‐CoV‐2 in the initial stages of the pandemic despite a lack of evidence for a specific mechanism of inhibition of the prior coronaviruses [114, 120].
Fig. 10.
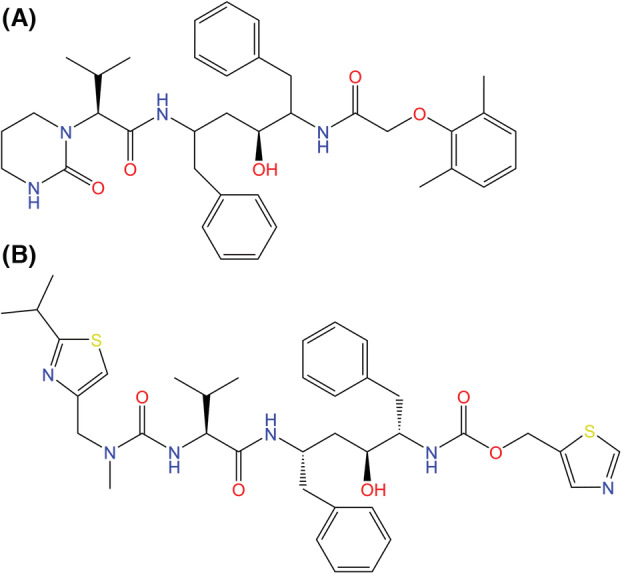
Chemical structures of (A) lopinavir and (B) ritonavir, the two components of Kaletra. Kaletra was originally developed to treat HIV‐1 infections and its potency in fighting SARS‐CoV‐2 infections was investigated during the early stages of the COVID‐19 pandemic as a potential protease inhibitor [114].
Although an in vitro study did find lopinavir to have a 50% effective concentration of 26.63 μm against SARS‐CoV‐2 in Vero E6 cells [115], SARS‐CoV‐2 Mpro and other coronavirus proteases (which are also 3CLpros) do not contain the C2‐symmetric pocket, and so questions remain about a potential mechanism for antiviral action against SARS‐CoV‐2 [119].
Conversely, a randomized control trial of 199 patients in China reported no significant difference in recovery time, 28‐day mortality or viral load for lopinavir–ritonavir treatment compared to the control [121]. Another study obtained similar results with a larger sample size of 1616 patients in the UK, finding that treatment with lopinavir–ritonavir was not associated with a reduced risk of progressing to mechanical ventilation or death, 28‐day mortality, or hospital stay duration [122].
Diarrhea was a common adverse drug reaction for patients taking lopinavir–ritonavir, even outside of its use in the early stages of the COVID‐19 pandemic, because approximately 50% of patients in clinical trials for HIV were reported to experience this adverse event; a retrospective study in one hospital found this combination treatment to be the therapy most implicated in the occurrence of adverse effects among those initially prescribed in the early days of the COVID‐19 pandemic, outclassing even hydroxychloroquine [123].
Given the lack of evidence for the efficacy of lopinavir as an antiviral therapeutic for SARS‐CoV‐2, further large‐scale clinical trials of lopinavir–ritonavir have not been pursued at this time and the National Institutes of Health (NIH) COVID‐19 Treatment Guidelines Panel strongly recommends against the use of lopinavir–ritonavir in the treatment of SARS‐CoV‐2 infection [78].
RNA‐dependent RNA polymerase stalling
A class of COVID‐19 drugs that have proven to be effective in recent clinical trials comprises the RdRp inhibitors. RdRp enzymes are instrumental in the replication and transcription mechanisms of coronaviruses such as COVID‐19. In SARS‐CoV‐2 specifically, the RdRp complex is composed of an NSP12 core catalytic subunit, an NSP7–NSP8 heterodimer, and another NSP8 subunit. These components work in conjunction to allow incoming nucleoside triphosphate (NTP) substrates bind to the +1 site, whereas the 3′‐terminal nucleotide of the RNA product remains in the −1 site. Additional ribonucleotides are incorporated via the catalytic cycle, thereby growing the RNA product strand [124].
Therapeutics such as remdesivir, ribavirin, and favipiravir act as nucleoside analogs: they compete with nucleosides to form strong hydrogen bonds with mRNA, thus disrupting RNA replication via a stalling mechanism [125].
Remdesivir
Remdesivir is a phosphoramidate prodrug that produces the active NTP analog remdesivir triphosphate (RTP) when metabolized. When RdRp uses RTP as a substrate, remdesivir monophosphate (RMP) is incorporated into the RNA product strand (Fig. 11). As a result of the function of remdesivir as an adenosine analog, RMP is able to base pair with uridine monophosphate and thus replace AMP in the product RNA. Kokic et al. [127] found that RdRp was able to overcome the stalling mechanism of remdesivir at higher concentrations of NTP, whereas RdRp inhibition was strong with higher concentrations of RMP.
Fig. 11.
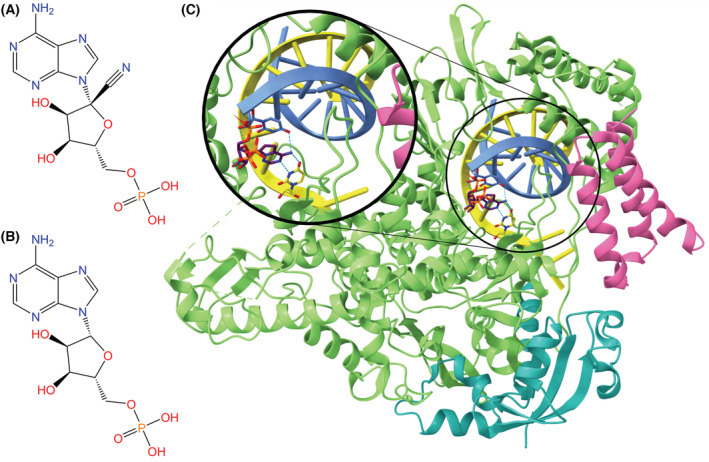
(A) Structures of remdesivir monophosphate and (B) AMP. Remdesivir functions as an adenosine analog, such that RMP can replace AMP in the product RNA, thereby inhibiting the RdRp viral replication mechanism. (C) RdRp complex (NSP12: green, NSP7: pink, NSP8: teal) bound to template (yellow) and primer (blue) RNA strands and remdesivir triphosphate (purple backbone) (PDB ID: 7BV2, visualized with ucsf chimerax, version 1.4) [124, 126]. Hydrogen bonds between remdesivir triphosphate and RdRp or the RNA strands are represented by dotted cyan lines, and heteroatom colors correspond to those in the 2D visualization.
The RdRp stalling mechanism is specific to SARS‐CoV‐2 in that RMP incorporation by the RdRp results in a three nucleotide extension of the product strand, as opposed to the five nucleotide extension seen in Ebola virus [128]. This limit between position –3 and –4 is known as the translocation barrier, meaning that additional RMPs cannot be incorporated into the RNA product past position –3. The translocation barrier is caused by steric hindrance at position –4 between the C1′‐cyano group in the ribose part of remdesivir and a S861 side chain in NSP12.
Stalling the RdRp replication and transcription mechanism in turn inhibits the proliferation of SARS‐CoV‐2 in vitro and in vivo. This impaired growth eventually leads to degradation of the viral RNA genome [127].
On 22 October 2020, remdesivir (known commercially as Veklury) became the first drug approved by the FDA for COVID‐19 treatment, in patients aged at least 12 years and weighing 40 kg. On 21 January 2022, its use was expanded to include COVID‐19 treatment in non‐hospitalized patients with positive results of direct viral testing who are at high risk for progression to severe COVID‐19 [79]. On 25 April 2022, remdesivir was additionally approved for COVID‐19 treatment in hospitalized or high‐risk pediatric patients aged at least 28 days and weighing 3.0 kg by the FDA under a supplemental new drug application (replacing the EUA), making it the first approved treatment for children under 12 years of age [129].
The original decision by the FDA to approve remdesivir for use on hospitalized patients was supported by a randomized, placebo‐controlled trial in which 1062 hospitalized patients with moderate‐to‐severe COVID‐19 received either remdesivir or a placebo. The dosing procedure involved 200 mg of remdesivir as a loading dose on day 1, followed by 100‐mg doses each day for as many as additional 9 days or until recovery. The patients who received remdesivir had a median 10‐day recovery period and a 6.7% mortality rate compared to a 15‐day recovery period and 11.9% mortality rate for those who received the placebo [130]. The decision to expand remdesivir for use in non‐hospitalized COVID‐19 patients was informed by a randomized trial of 562 patients that showed an 87% reduction in risk of hospitalization or death among high‐risk, non‐hospitalized patients who received a 3‐day course of remdesivir compared to those who received the placebo [131].
In a press release dated 25 June 2020, announced by the European Medicines Agency, remdesivir became the first medicine recommended for authorization in the European Union, and later became the first antiviral fully licensed for COVID‐19 treatment in Europe [132, 133]. The European Commission expanded remdesivir approval on 21 December 2021 to include adults who do not require supplemental oxygen but are still at a high risk of progressing to severe COVID‐19. However, recent trials undertaken by Gilead and WHO involving patients hospitalized for COVID‐19 showed no significant difference in symptom improvement between those who received remdesivir and those who received no study drug [134, 135].
In March 2022, the first remdesivir‐resistant variant of SARS‐CoV‐2 was identified in an immunocompromised patient. The resistance arose as a result of a mutation, E802D, in NSP12. Similar resistance has been demonstrated in the E802A mutation. Although researchers are still investigating the underlying mechanisms of this resistance, a possible explanation is that mutations at 802 cause conformational changes to the RdRp active site that alleviate RMP‐induced steric hindrance and thus allow for continued elongation of the RNA product strand [136].
Ribavirin
Ribavirin functions as a guanosine analog that produces broad‐spectrum antiviral activity, primarily for hepatitis C and viral hemorrhagic fevers when used in conjunction with interferons (IFNs) (Fig. 12). Ribavirin monophosphate competes with inosine monophosphate (IMP) for binding to IMP dehydrogenase, which, when bound to IMP, synthesizes intracellular GMP and GTP [137]. The inhibition of IMP binding by ribavirin monophosphate leads to decreased GMP and GTP levels that, when low, indirectly inhibit RdRp enzyme activity by causing nucleotide imbalance. The downstream effects of this imbalance in turn interfere with mRNA capping, ribosomal activity, G‐protein signaling, and other post‐transcriptional processes [138].
Fig. 12.
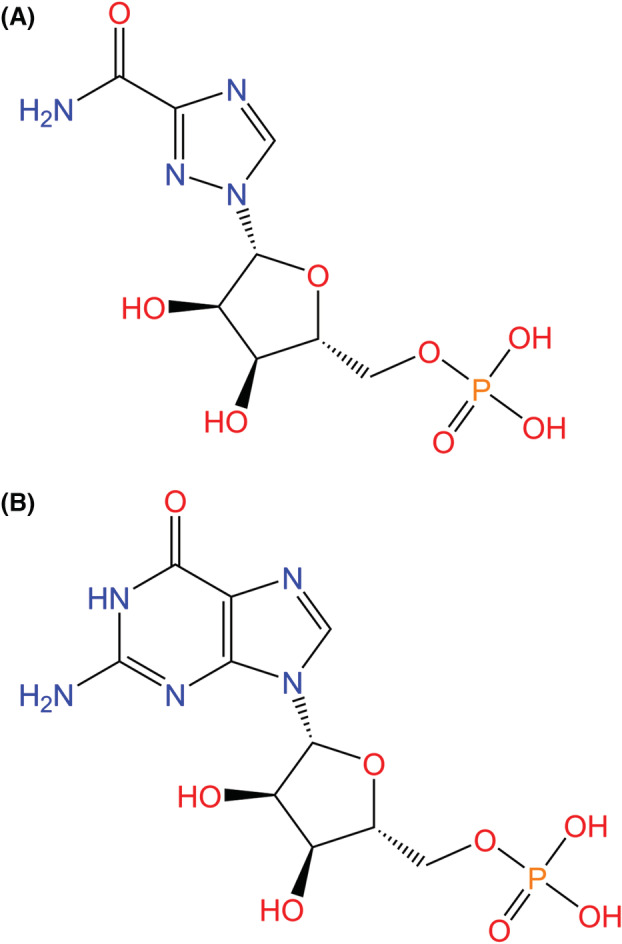
Chemical structures of (A) ribavirin monophosphate and (B) GMP. Due to its function as a guanosine analog, ribavirin monophosphate is a broad‐spectrum antiviral because it is able to compete with IMP for binding to IMP dehydrogenase, which in turn inhibits RdRp enzyme activity as a result of the creation of nucleotide imbalance [137, 138].
Studies have demonstrated successful ribavirin activity against SARS‐CoV replication in vitro when administered in combination with IFN‐β [139, 140]. However, ribavirin was shown to be ineffective against SARS‐CoV in vivo [141]. When ribavirin and IFN‐α‐2a were used to treat patients infected with severe MERS‐CoV, the treatment improved survival at 14 days but not at 28 days [142].
A retrospective study involving 115 patients with severe SARS‐CoV‐2 found that ribavirin therapy is not associated with improved health outcomes or mortality rates, with 17.1% of ribavirin patients and 24.6% of control group patients dying as a result of COVID‐19 progression [48]. A larger, more recent retrospective study of 2037 COVID‐19 patients confirmed the ineffectiveness of ribavirin/IFN‐α in improving clinical health outcomes [143].
Prior to the aforementioned studies demonstrating the ineffectiveness of ribavirin‐based therapies against SARS‐CoV‐2, the recommended dosage of ribavirin was 500 mg two or three times per day for no longer than a 10‐day period, often in combination with IFN‐α or lopinavir/ritonavir [144].
Favipiravir
Developed in 2014 as a potential treatment for influenza, favipiravir (also known as Favilavir) functions as a nucleoside analog and disrupts RdRp activity in SARS‐CoV‐2, similar to remdesivir and ribavirin [145, 146]. Favipiravir undergoes phosphoribosylation upon entrance into the body, transforming favipiravir into its active form favipiravir‐ribofuranosyl‐5′‐triphosphate (FRTP) [147, 148]. Upon entrance into infected cells, FRTP mimics a substrate of RdRp. From there, FRTP incorporates itself into the viral RNA strand where it is mistaken for a purine nucleotide by RdRp (Fig. 13). Incorporation halts viral protein synthesis [150]. Favipiravir contains a strong binding affinity with RdRp, which causes a reduction in new viral RNA [151].
Fig. 13.
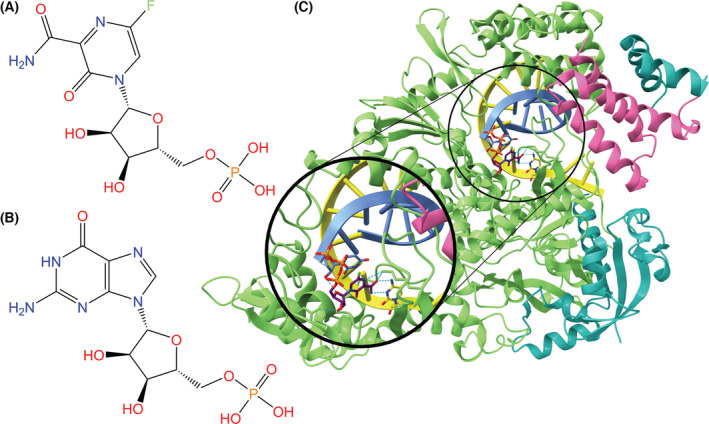
Chemical structures of (A) favipiravir‐ribofuranosyl‐5′‐monophosphate, a purine analog, and (B) guanosine monophosphate for comparison. (C) RdRp complex (NSP12: green, NSP7: pink, NSP8: teal) bound to template (yellow) and primer (blue) RNA strands and FRTP (purple backbone) (PDB ID: 7AAP, visualized with ucsf chimerax, version 1.4) [148, 149]. Hydrogen bonds between FRTP and RdRp or the RNA strands are represented by dotted cyan lines, and heteroatom colors correspond to those in the 2D visualization.
A study conducted in China compared the effectiveness of favipiravir and Arbidol with respect to shortening the recovery rate of individuals infected with SARS‐CoV‐2. No significant recovery rate differences were found between the two groups at day 7, but earlier resolution of fever and cough was seen in favipiravir‐treated individuals [152]. A separate study carried out in Japan found over 80% of individuals with mild and moderate cases recovered after 14 days, whereas only 60% of COVID‐19 patients with severe symptoms recovered following treatment with favipiravir [150]. Thus, favipiravir was concluded to be effective against non‐severe COVID‐19 but has subsequently been deemed non‐essential for treatment of COVID‐19 [80].
Favipiravir is an oral therapeutic that has an ability to treat many other RNA viruses. The typical treatment is 1800 mg twice a day for 1 day, then 800 mg twice a day for up to an additional 4 days. Common side effects of favipiravir include uric acid elevations, psychiatric symptom reactions, and increase of aspartate aminotransferase. The drug is already commercialized in several countries including Russia, Jordan, and Saudi Arabia [151].
Error catastrophe
Error catastrophe involves increasing the mutation rate in the viral genome to the extent of genomic instability, leading to the extinction of the virus. A notable mechanism in error catastrophe is tautomerization, where tautomers change DNA from their original form into their isomeric form. This reaction in therapeutics can cause mutagenesis in the creation of enzymes and thus makes the enzymes non‐functional [153].
Molnupiravir
Initially considered a potential treatment for influenza virus, molnupiravir is an orally administered COVID‐19 treatment developed by Drug Innovation Ventures at Emory University and later acquired by Merck and Ridgeback [153]. It was issued an EUA on 23 December 2021, for non‐hospitalized patients experiencing mild‐to‐moderate COVID‐19 in adults, but is notably not approved for children because of potential effects on bone and cartilage growth [81]. On 23 March 2022, the FDA extended molnupiravir's EUA to include COVID‐19‐positive adults for whom other FDA‐approved treatments are not accessible [154].
Molnupiravir utilizes error catastrophe to inhibit RdRp through a two‐step process. Upon entering the body, molnupiravir begins the process of mutagenesis. Molnupiravir enters the body as an isopropyl ester prodrug, where it is cleaved by the esterases of a host cell, forming the active nucleoside analog β‐d‐N4‐hydroxycytidine (NHC), also known as EIDD‐1931 (Fig. 14A,B). Active kinases convert the analog to NHC 5′ triphosphate (EIDD‐1931‐triphosphate). NHC‐triphosphates are used as substrates for RdRp enzymes over cytidine and uridine triphosphates; adenine or guanine are then added to the active centers of RdRp to maintain RNA stability (Fig. 14C–E). The subsequent copies of RdRp are thus composed of these mutated RNA substrates that inhibit normal function. Molnupiravir is more prone to be an electron donor as opposed to an electron acceptor, which alters the conditions for viral infection [153, 157].
Fig. 14.
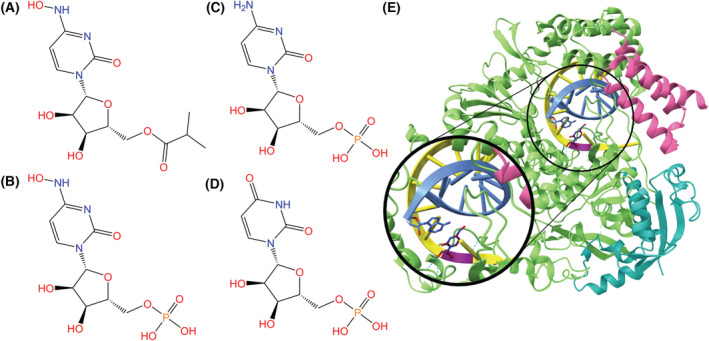
(A) Chemical structure of molnupiravir, an isopropylester prodrug of β‐d‐N4‐hydroxycytidine (NHC). (B) Chemical structure of NHC monophosphate. The active 5′‐triphosphate form of the drug is incorporated into the RdRp and induces viral activity. (C) Chemical structures of cytidine monophosphate and (D) uridine monophosphate, which NHC/Molnupiravir imitates. (E) RdRp complex (NSP12: green, NSP7: pink, NSP8: teal) bound to template (yellow) and primer (blue) RNA strands. NHC (purple backbone) is incorporated in the template strand and is base paired with adenosine (PDB ID: 7OZU, visualized with ucsf chimerax, version 1.4) [155, 156]. Hydrogen bonds between NHC and RdRp or the RNA strands are represented by dotted cyan lines, and heteroatom colors correspond to those in the 2D visualization.
In non‐human primates, NHC has poor bioavailability and is rapidly metabolized in enterocytes. Molnupiravir prolongs antiviral effects by increasing the bioavailability of NHC. Molnupiravir has a wide therapeutic window with a quick onset of action. The drug has a half‐life of 7 h and a maximum serum concentration of 13.2 ng·mL−1 [157].
Molnupiravir has been observed to target the upper respiratory tract. In a clinical trial using immunodeficient mice with human lung tissue, molnupiravir reduced the number of infectious particles in human lung tissue by 25 000‐fold, with no adverse effects on the white blood cell or platelet counts. In a clinical trial with 741 patients with mild COVID‐19 symptoms, molnupiravir significantly lowered the hospitalization rate of unvaccinated adults. A two phase, double‐blind clinical study found that 1600 mg over 5.5 days is the most tolerable amount, with 800 mg taken twice a day. Molnupiravir showed a hospitalization risk of 7.1% compared to 14.1% with placebo. Additionally, the drug showed a significant decrease in hospital admissions and death by 50%; however, this was only observable in patients with mild cases of COVID‐19. Current clinical trials in India do not show molnupiravir effectively treating moderate cases of COVID‐19. As a result of its recent discovery, there are still many clinical trials testing the efficacy of molnupiravir [153]. Molnupiravir contains a risk of carcinogenesis although these observations were only at the cellular level. In phase 1 clinical trials, a greater proportion of participants reported more adverse effects with the placebo than molnupiravir (43.8% and 35.4%) [158]. Pre‐clinical trials have also demonstrated the effectiveness of molnupiravir with respect to treating remdesivir‐resistant viruses [158].
Few clinical studies have investigated the resistance of SARS‐CoV‐2 to molnupiravir. Because of molnupiravir's quick treatment, the viral error possibly blocks resistance, creating a high genetic barrier. In studies that exposed the influenza virus to molnupiravir multiple times, no allele dominant mutations or strong resistance were demonstrated. Another study exposed two lineages of MERS‐CoV to increasing concentrations of NHC; however, there was little development of resistance [158].
Protein trafficking and post‐translational modifications
Following cell entry, SARS‐CoV‐2 leverages host‐cell ribosomes to create viral proteins necessary for replication. These viral proteins have become a target of multiple antiviral therapeutics, which are discussed in this section. Through either protein degradation or inhibition of intracellular transport, proper protein assembly is prevented, and subsequent viral functionality is lost. Here, we look at two antivirals that have shown some initial promise in the fight against COVID‐19, but neither are approved for medicinal use against COVID‐19 (Table 3). However, these therapeutics are unlikely to be used for COVID‐19 treatment because of negative side effects (nitazoxanide) and lack of efficacy (ivermectin).
Table 3.
Summary of recommendations for protein trafficking and post‐translational modifications targeted antivirals. Both antivirals in this section are not authorized for COVID‐19 treatment.
Nitazoxanide
Nitazoxanide (NTZ) is the first compound discovered in a class of antiviral agents known as thiazolides [161]. The thiazolide class contains numerous nitazoxanide derivatives possessing antibacterial, antiparasitic, antiprotozoal, and antiviral properties [161, 162, 163]. Thiazolides were first developed and tested in the mid‐1970s as an antiparasitic agent, but NTZ became increasingly popular in 1993 as an antiprotozoal treatment against Cryptosporidium parvum infections [164, 165]. After being approved by the FDA in 2002 for treatment against C. parvum, NTZ has been explored as a remedy for numerous infections: influenza A, Ebola, hepatitis C, and MERS‐CoV [166, 167]. As such, the NIH instituted the first phases of trials for NTZ in August of 2020 as a potential treatment for COVID‐19 [159]. Experiments and trials assessing the effectiveness of NTZ against COVID‐19 in diverse populations are ongoing, and research continues to focus on both the potential mechanisms of action and efficiency of NTZ in a clinical setting [168, 169, 170].
NTZ is readily metabolized, after consumption, into its active metabolite tizoxanide (TIZ) (Fig. 15). TIZ forms via rapid deacetylation through hydrolysis by plasma esterases [171, 172]. As a result of the versatility of TIZ, various mechanisms targeting viral entry, replication, and post‐translational modification have been proposed and identified in the previously mentioned viruses [164, 173, 174, 175]. There are various proposed mechanisms for TIZ in response to COVID‐19, but three primary mechanisms exist: two regarding inhibition of viral entry inhibition and one involving inhibition of post‐translational processing inhibition [168].
Fig. 15.
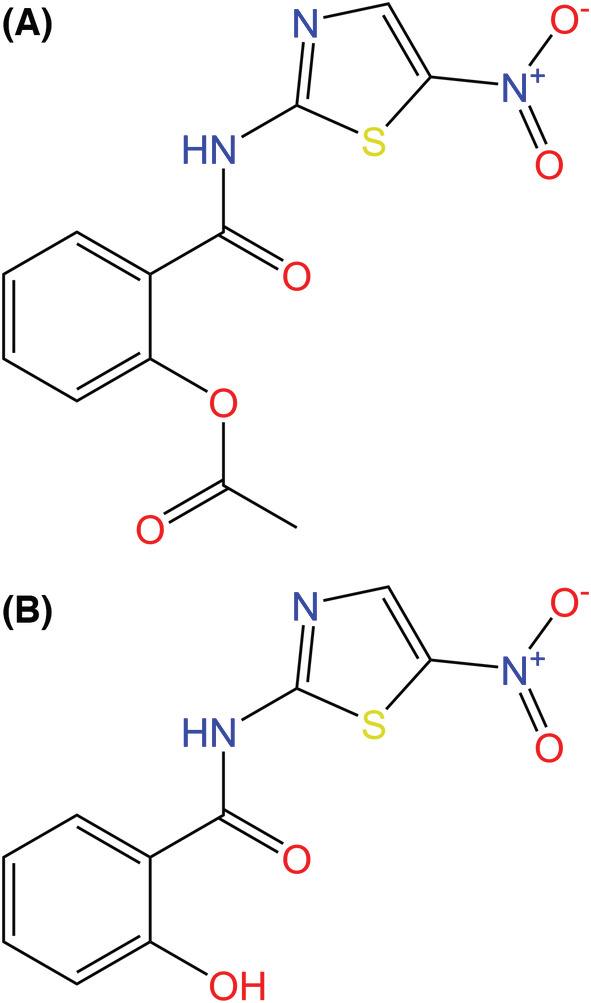
(A) Chemical structures of nitazoxanide (NTZ) and (B) tizoxanide (TIZ), the active metabolite of NTZ, which is formed through rapid hydrolysis of NTZ's acetyl group by plasma esterases. NTZ and its derivatives have been found to have antimicrobial, antiparasitic, and antiviral properties. NTZ is theorized to have antiviral activity against SARS‐CoV‐2 by inhibiting viral entry and/or post‐translational processing [161, 168].
The first mechanism involves inhibition of the endosomal entry pathway of SARS‐CoV‐2 [176]. The S1 subunit of SARS‐CoV‐2 S protein exchanges disulfide bonds with ACE2 through a thiol‐sulfide redox system, which is assisted by protein disulfide isomerase (PDI) activity, initiating endosomal entry. Consequently, inhibition of PDI prevents proper disulfide exchange, providing an inhibitory target for TIZ to act upon. TIZ allosterically inhibits PDI through S‐nitrosylation, preventing proper association of the S1 subunit with ACE2 and subsequent viral entry [168].
The second mechanism prevents non‐endosomal entry of SARS‐CoV‐2. Furin and transmembrane‐protease‐serine‐2, two host cell proteases, cleave S proteins, exposing the S2 subunit for fusion with the host cell membrane, allowing viral entry. The cleavage is dependent on thiol‐disulfide exchange, catalyzed through PDI activity. Transmembrane‐protease‐serine‐2 ectodomains, containing cysteine‐rich residues, are regulated by PDI and provide disulfide exchange functionality. Similar to the endosomal mechanism, TIZ allosterically deactivates PDI via s‐nitrosylation, preventing S2 exposure and viral entry [168].
The third involves post‐translational processing inhibition of SARS‐CoV‐2, which also works through an autophagic mechanism. When the virus is infecting the host cell, AMP‐protein activated kinases are inactivated, preventing autophagic degradation [168, 177, 178]. NTZ utilizes TIZ metabolites, which activate AMP‐protein activated kinases by allosterically increasing the ratio of AMP bound to the γ regulatory subunit [173]. This in turn activates Unc‐51‐like kinase 1/2, leading to autophagosome formation and post‐translational degradation of SARS‐CoV‐2 [179, 180].
In vivo studies have been conducted to assess the COVID‐19 symptom reduction ability of NTZ and TIZ. One study of 392 patients (194 treated with NTZ, 198 treated with placebo) found no significant difference in the reduction of symptoms when comparing placebo and NTZ patients. However, secondary data showed statistically significant viral load reduction in NTZ‐treated patients compared to placebo 5 days post‐treatment [170]. A second study assessed symptom reduction in pregnant individuals, similarly, uncovering no significant correlation between treatment and symptom reduction [169]. Any adverse effects mostly parallel the symptoms of COVID‐19 because NTZ has not shown clinical repression of symptoms [169, 170].
The primary limitation of NTZ originates from the highly bound plasma membrane property of TIZ. Highly bound plasma membrane treatments penetrate cell membranes worse than minimally bound counterparts [181]. However, the high membrane affinity of TIZ limits combinatorial treatment prospects involving NTZ. One study assessing the compatibility of warfarin and TIZ found TIZ greatly reduced the outcompeted warfarin for membrane binding, inhibiting proper Warfarin function and potentially risking internal bleeding [182].
Ivermectin
Ivermectin is an antiparasitic drug derived from macrocyclic 22,23‐dihydroavermectin B, which is obtained by bacterium in the Streptomyces genus (Fig. 16) [183]. Ivermectin was first commercialized to treat parasitic illness in animals. In 1987, ivermectin would later be cleared for antiparasitic treatment in humans [184, 185]. In recent years, ivermectin has also demonstrated antimicrobial activity against dengue fever, influenza, HIV, and other RNA viruses. The ability of ivermectin to inhibit viral reproduction promoted research on whether the same mechanism could work to combat SARS‐CoV‐2 [183, 186].
Fig. 16.
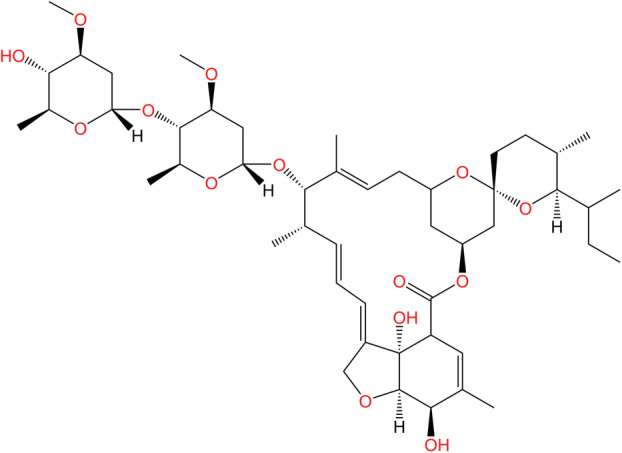
Chemical structure of ivermectin. Ivermectin is commonly used as an antiparasitic drug for animals and humans. The drug has also demonstrated antiviral characteristics and as such was investigated for action against COVID‐19 in the early stages of the pandemic, although the mechanism of action for these observed antiviral characteristics has yet to be determined [183].
The mechanism in which ivermectin works in stifling transmission of the COVID‐19 is relatively unknown. The most accepted mode of action is via the suppression of importin α/β1 dependent transport of viral proteins into and out of the nucleus. Previous in vitro studies on ivermectin have indicated its ability to limit nuclear import of viral infections by binding to the armadillo complex on importins [186]. Blocking nuclear import of viral infections has been shown to inhibit virus proliferation of HIV‐1 and dengue fever [187]. Other proposed mechanisms include competitive binding of ivermectin with the S protein of the virus, ionophore formation, anti‐inflammatory effects, etc. [183, 188].
There is conflicting information regarding the effectiveness of ivermectin for treating mild cases of SARS‐COV‐2. Some clinical trials earlier in the pandemic indicated a reduction in mortality rate and shorter recovery time post‐infection after treatment with ivermectin [189]. However, the lack of standardized concentrations for ivermectin and standardized sample sizes for clinical trials at the time made it difficult to determine the efficacy of the drug for treating COVID‐19 [160, 184].
A significant number of recent studies have concluded that ivermectin has little to no effect on patient outcomes for SARS‐CoV‐2. In A 2020 double‐blind study in Brazil, 1358 adults who had COVID‐19 symptoms for at most 7 days were randomly assigned 400 μg·kg−1 of ivermectin for 3 days or a placebo. The conclusion of the clinical trial indicated that early treatment of ivermectin had no significant difference between the placebo and ivermectin group in terms of hospitalization rates (16.3% and 14.7%) and mortality rates (3.5% and 3.1%) [190]. Other recent clinical trials with the same dosage also concluded no apparent difference between ivermectin and the placebo group [186, 190, 191].
An increasing number of clinicals trials have indicated that the ivermectin is not effective in improving patient outcomes. In addition, adverse effects associated with ivermectin have also lowered the benefits of its usage. Applying ivermectin at concentrations above 150 μg·kg−1 can cause ataxia, nausea, and comas [186]. Overall, the adverse effects coupled with the lack of clear efficacy of ivermectin has called into question its viability in treating SARS‐CoV‐2.
Immune response regulation
The treatments in this section focus on enhancing the body's natural immune defense system, rather than targeting the SARS‐CoV‐2 virus. The amplification of the body's response can better prepare the immune system to combat the negative symptoms that the virus will cause. Both drugs within this category target the inflammation response to improve COVID‐19 outcomes. They both have shown initial promise, although as of yet there is insufficient clinical evidence to allow treatments to be approved in the fight against COVID‐19 (Table 4).
Table 4.
Summary of recommendations for antivirals targeted towards immune response regulation in response to COVID‐19 infection. Both antivirals in this section are not authorized for COVID‐19 treatment.
Interferons
Consisting of three types (I, II, III), IFNs are cytokines naturally secreted by the body upon viral infection. IFNs act by binding to cell‐surface receptors and activating the Janus kinase signal transducer and activator of transcription via receptor tyrosine kinases [194]. This regulates the transcription of genes related to innate and adaptive immunity, which can increase an inflammatory response and drive immune cell differentiation to fight infection [195].
Interferon immune response properties have prompted research to determine the efficacy of IFNs as a therapeutic option for COVID‐19. Of the three types, type I (IFN‐β‐1a and IFN β‐1b) has been studied most extensively and show the best results in improving COVID‐19 outcomes (Fig. 17). Type II (IFN‐γ) has been cleared for safety, although more research is needed to determine their efficacy. Type III has not been cleared and the literature points to inconclusive data regarding its efficacy [198].
Fig. 17.
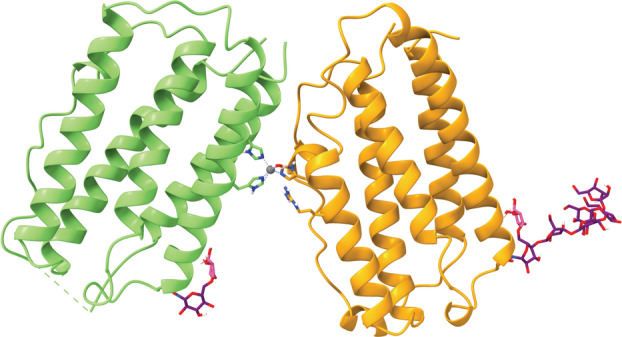
Structure of an interferon‐β dimer (PDB ID: 1AU1, visualized with ucsf chimerax version 1.4) [196, 197]. Interferons are natural cytokines, which regulate adaptive and innate immune responses [194]. The two monomer chains (orange and green) of the interferon‐β dimer are coordinated by a zinc molecule (gray, with metal coordination bonds in lavender). Both chains possess one or more beta‐d‐glucopyranose residues (purple backbone) and one 6‐tert‐butylsulfonyl‐N‐(5‐fluoro‐1H‐indazol‐3‐yl) quinolin‐4‐amine residue (pink backbone).
Type I IFNs are secreted by cells upon viral infection and stifle the proliferation of the virus. Type 1 IFNs act by binding to a heterodimer of IFN‐α receptors 1 and 2. Binding of the type I IFN leads to the phosphorylation of Janus kinase and tyrosine kinase, which phosphorylates the tyrosine residues on the heterodimer. Phosphorylation of the tyrosine residue recruits signal transducers and activators of transcription (STAT 1 and STAT 2). STAT1 and STAT2 are activated to form a homodimer and interact with IFN regulatory factor 9 resulting in IFN‐stimulated gene factor 3. IFN‐stimulated gene factor 3 is translocated to the nucleus to induce the genes that are controlled by IFN‐stimulated response elements, which leads to antiviral gene expression [194].
IFN‐β‐1a has been administered via intravenous, inhalation, and subcutaneous means and shows promise in improving patient outcomes for COVID‐19 when applied in the early stages of infection [198]. In a double‐blind clinical trial where 100 adults with COVID‐19 were randomized to receive either a placebo or IFN‐β‐1a via a nebulizer for 14 days, three of the placebo group died during the study, whereas no deaths occurred for the experimental group. The experimental group also experienced a faster recovery rate compared to the placebo group [199]. Although there are promising data on the efficacy of IFN‐β‐1a, late application of IFN‐β‐1a contributes little to no effect on improving patient outcomes [200]. In a randomized clinical trial, 446 COVID‐19 patients with an age range of 8–96 years were administered IFN‐β‐1a, and the early treatment group, late treatment group, and placebo had a recovery rate of 99.1%, 84.6%, and 95.1% respectively [200]. Studies have also revealed that IFN‐β‐1a may lead to an increase in acute respiratory distress, hypersensitivity, and neurological illness in some patients. However, patients did experience increased discharge rates and decreased morbidity from the IFN‐β‐1a treatment [201]. IFN‐β‐1b shares functions with IFN‐β‐1a. Studies have shown that IFN‐β‐1b is safer than IFN β‐1a, but more research is needed to determine its efficacy [198]. The emergence of robust therapeutics for COVID‐19 coupled with the potential side effects of IFN‐β‐1a has hampered the drug's potential utilization for treating COVID‐19.
Dexamethasone
Dexamethasone functions as a corticosteroid medication and has been of interest in the fight of COVID‐19 as a result of its use treating rheumatoid arthritis, asthma, different types of cancers, and systemic lupus erythematosus (Fig. 18) [202]. Corticosteroids are a class of medications that act as an immunosuppressant. The relative abundance of dexamethasone is consistent around the globe because of its status as an Essential Medication by the WHO, and this further ensures its accessibility [203].
Fig. 18.
Chemical structure of dexamethasone, a corticosteroid that acts as an immunosuppressant and reduces inflammation. Dexamethasone is used to treat rheumatoid arthritis, asthma, some cancers, and systemic lupus erythematosus and was investigated as a treatment to improve COVID‐19 patient outcomes in the early stages of the COVID‐19 pandemic [202].
The corticosteroid reacts with receptor proteins in the cytoplasm, creating a steroid‐receptor complex, which then enters the nucleus and acts by inhibiting the transcription of mRNA to downregulate target protein synthesis [204]. Dexamethasone alleviates the inflammation that can occur with COVID‐19 infection by slowing the migration of immune cells to infected areas and reducing immune cell chemotaxis (through inhibition of cytokine release) and vasodilation [205].
Clinical trials testing the use of dexamethasone dosed patients at 6 mg·day−1 for 10 days. One study conducted by the Recovery Collaborative Group based at Oxford University found that the risk of mortality in the experimental group was significantly lower compared to the control group (21.6% compared to 24.6%). They also found that outcomes improved when patients underwent mechanical ventilation (40.7% compared to 29.0%) or just non‐invasive oxygen (25.0% compared to 21.5%) [206].
Corticosteroid resistance may arise through the course of diseases that cause inflammation. Multiple proposed mechanisms of resistance include reduced steroid‐receptor complex translocation into the nucleus, competition for corticosteroid binding between glucocorticoid receptors α and β, where glucocorticoid receptor‐B inhibits glucocorticoid receptor‐A activity, and increased secretion of macrophage migration inhibitory factor [207]. These resistances pose a large problem to populations that have chronic inflammation, namely in asthmatics and smokers. However, few studies have been conducted specifically looking at resistance pertaining to the use of dexamethasone in COVID‐19 patients.
Conclusions and perspectives
As the global community has raced to develop and improve vaccines in response to the COVID‐19 pandemic, the need for antiviral therapies has become more pressing. Antiviral therapies present major benefits in the fight against the COVID‐19 pandemic, including ameliorating symptoms in mild to moderate COVID‐19 cases by providing a mechanism for preventing successful viral infection of a cell, with the potential to save thousands of lives. Successful drugs improve outlooks for both vaccinated and unvaccinated individuals, mitigating symptoms, decreasing mortality, and increasing discharge rates. Currently existing therapeutics that have been shown to be effective have a wide range of mechanisms targeting various points in the COVID‐19 maturation cycle (Table 5).
Table 5.
Summary of antiviral therapies and the category of the virus life cycle that they target. Therapies marked with a single asterisk (*) are authorized for use by the FDA in the USA under an EUA as of 31 July 2022. Therapies marked with two asterisks (**) were once approved under an EUA but are not currently authorized for treatment in the USA because of the prevalence of the Omicron variant of SARS‐CoV‐2. The therapy marked with three asterisks (***) has been fully approved by the FDA for the treatment of SARS‐CoV‐2, which supersedes the therapy's original EUA. Therapies without asterisks have not been authorized for the treatment of COVID‐19 temporarily or permanently by the FDA.
| Stage of virus maturation | Antiviral therapy targets | References |
|---|---|---|
| Viral entry into host cell | Evusheld*, REGEN‐COV**, bamlanivimab and etesevimab**, bebtelovimab*, sotrovimab**, Arbidol, nitazoxanide, chloroquine | [8, 23, 33, 44, 48, 50, 59, 63, 65, 66, 168] |
| Viral replication | Paxlovid*, Kaletra, remdesivir***, favipiravir, ribavirin, molnupiravir* | [90, 92, 93, 94, 95, 96, 119, 127, 138, 150, 153, 157] |
| Protein trafficking and post‐translational processing | Nitazoxanide, ivermectin | [168, 183, 186, 188] |
| Immune response regulation | Interferons, dexamethasone | [194, 205] |
Of the drugs mentioned in this review, molnupiravir, Paxlovid, Evusheld, sotrovimab, REGEN‐COV, BNE, and bebtelovimab have all been granted EUAs for SARS‐CoV‐2 treatment [23]. Paxlovid holds the most promise because of its higher efficacy and oral administration compared to other approved drugs. Accordingly, governmental bodies in the USA have invested heavily in this treatment, purchasing 10 million treatment courses of Paxlovid [22]. Paxlovid is highly effective because of its close imitation of the amino acid sequence found at splice sites in SARS‐CoV‐2 polyproteins, which allows it to inhibit the viral protease that processes the proteins required for viral genome replication. However, as an oral inhibitor, Paxlovid does not confer immunity against COVID‐19 to patients [90, 95, 96]. Of the monoclonal antibodies discussed, Evusheld is most promising as a result of its authorization for use as a preventative treatment against contracting COVID‐19 and experiencing severe symptomatic illness [9]. A preventative treatment would provide a greater form of defense against COVID‐19 infections because individuals would no longer have to rely on the post‐infection therapeutic techniques. Post‐infection treatment with bebtelovimab is advantageous because its effectiveness has been demonstrated on all known variants of concern (as of August 2022), although bebtelovimab has a lower efficacy rate than Paxlovid and Evusheld [51].
As the world is now past its second anniversary subsequent to the onset of the COVID‐19 pandemic, these antiviral therapies provide hope for a future without the fear of SARS‐CoV‐2. Research and development are paramount in the continued fight to save lives and improve future pandemic responses. More techniques should continue to be investigated, both as preventative treatments and to ameliorate symptoms after infection. Importantly, more extensive investigation of therapeutic techniques for severe COVID‐19 cases is necessary, given that such cases pose the greatest risk of life‐threatening symptoms. This review aptly summarizes the state of COVID‐19 therapeutics as they stand and provides insight into the areas that require further investigation.
Author contributions
DKB, ARG, LH, AAH, ALL, OGP, BAR, TTS and KTV reviewed the literature, wrote the manuscript, and revised the manuscript. DKB, ARG, LH, AAH, ALL, OGP, BAR, TTS, and KTV contributed equally to this manuscript. TPC carefully revised and co‐wrote the manuscript.
Conflicts of interest
OGP is currently participating in an unpaid internship at AstraZeneca as of Fall 2022 until Spring 2023. The remaining authors declare that they have no conflicts of interest.
Acknowledgements
DKB, ARG, LH, AAH, ALL, BAR, TTS, and KTV are undergraduate students who are part of a discovery‐based research lab course under the discretion of Senior Lecturer Dr Thomas Clements. OGP has been carrying out undergraduate research with Dr Clements since Spring 2019 and has been serving as a TA for this course. In addition to their work in the lab focused on elucidating novel loss of function phenotypes in zebrafish using CRISPR‐Cas9 mutagenesis, all of the authors are equally passionate about science communication. Thus, we set out to write this paper as follow‐up to the 2020 paper written by Dr Clements' students in the same course on the COVID‐19 pandemic [1]. We thank Dr James Pask, the Introductory Biology Lab Coordinator, for creating and fostering an environment that encourages us to explore these endeavors. Additionally, we are grateful for the Biological Sciences Department at Vanderbilt University for funding our experiments. We also acknowledge the use of ucsf chimerax, which was developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from NIH R01‐GM129325 and the Office of Cyber Infrastructure and Computational Biology, National Institute of Allergy and Infectious Diseases [208].
References
- 1. Atzrodt CL, Maknojia I, McCarthy RDP, Oldfield TM, Po J, Ta KTL, et al. A guide to COVID‐19: a global pandemic caused by the novel coronavirus SARS‐CoV‐2. FEBS J. 2020;287:3633–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. World Health Organization . COVID‐19 vaccine tracker and landscape. Geneva: World Health Organization; 2022. [Google Scholar]
- 3. Lee ARYB, Wong SY, Chai LYA, Lee SC, Lee MX, Muthiah MD, et al. Efficacy of covid‐19 vaccines in immunocompromised patients: systematic review and meta‐analysis. BMJ. 2022;376:e068632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lazarus JV, Wyka K, White TM, Picchio CA, Rabin K, Ratzan SC, et al. Revisiting COVID‐19 vaccine hesitancy around the world using data from 23 countries in 2021. Nat Commun. 2022;13:3801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Alanagreh L, Alzoughool F, Atoum M. The human coronavirus disease COVID‐19: its origin, characteristics, and insights into potential drugs and its mechanisms. Pathogens. 2020;9:331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. BioRender templates [cited 2022 Jul 28]. Available from: app.biorender.com/biorender‐templates
- 7. Meyerowitz EA, Richterman A, Gandhi RT, Sax PE. Transmission of SARS‐CoV‐2: a review of viral, host, and environmental factors. Ann Intern Med. 2021;174:69–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Meo S, Klonoff D, Akram J. Efficacy of chloroquine and hydroxychloroquine in the treatment of COVID‐19. Eur Rev Med Pharmalogical Sci. 2020;24:4539–47. [DOI] [PubMed] [Google Scholar]
- 9. Food and Drug Administration (US) . Fact sheet for healthcare providers: emergency use authorization for Evusheld™ (tixagevimab co‐packaged with cilgavimab). 2022. Available from: FDA.gov
- 10. Hinton D. Casirivimab and Imdevimab EUA letter of authorization. Silver Spring: FDA; 2020. [Google Scholar]
- 11. O'Shaughnessy J. Evusheld letter of authorization. Silver Spring: FDA; 2022. [Google Scholar]
- 12. Dougan M, Nirula A, Azizad M, Mocherla B, Gottlieb RL, Chen P, et al. Bamlanivimab plus Etesevimab in mild or moderate Covid‐19. N Engl J Med. 2021;385:1382–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. O'Shaughnessy J. Letter of authorization Eli Lilly bebtelovimab Emergency Use Authorization. Silver Spring: FDA; 2022. [Google Scholar]
- 14. Food and Drug Administration (US) . Fact sheet for healthcare providers Emergency Use Authorization (EUA) of sotrovimab. Silver Spring: FDA; 2021. [Google Scholar]
- 15. Alavi Darazam I, Shokouhi S, Mardani M, Pourhoseingholi MA, Rabiei MM, Hatami F, et al. Umifenovir in hospitalized moderate to severe COVID‐19 patients: a randomized clinical trial. Int Immunopharmacol. 2021;99:107969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lian N, Xie H, Lin S, Huang J, Zhao J, Lin Q. Umifenovir treatment is not associated with improved outcomes in patients with coronavirus disease 2019: a retrospective study. Clin Microbiol Infect. 2020;26:917–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yang C, Ke C, Yue D, Li W, Hu Z, Liu W, et al. Effectiveness of Arbidol for COVID‐19 prevention in health professionals. Front Public Health. 2020;8:249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhou X, Hou H, Yang L, Ding G, Wei T, Li C, et al. Arbidol is associated with increased in‐hospital mortality among 109 patients with severe COVID‐19: a multicenter, retrospective study. J Glob Health. 2021;11:05017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Khamitov RA, Loginova SI, Shchukina VN, Borisevich SV, Maksimov VA, Shuster AM. [Antiviral activity of Arbidol and its derivatives against the pathogen of severe acute respiratory syndrome in the cell cultures]. Vopr Virusol. 2008;53:9–13. [PubMed] [Google Scholar]
- 20. Cortegiani A, Ingoglia G, Ippolito M, Giarratano A, Einav S. A systematic review on the efficacy and safety of chloroquine for the treatment of COVID‐19. J Crit Care. 2020;57:279–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dong J, Zost SJ, Greaney AJ, Starr TN, Dingens AS, Chen EC, et al. Genetic and structural basis for SARS‐CoV‐2 variant neutralization by a two‐antibody cocktail. Nat Microbiol. 2021;6:1233–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dong J, Crowe JE. Crystal structure of SARS‐CoV‐2 spike RBD in complex with human monoclonal antibodies AZD8895 and AZD1061. 2022. 10.2210/pdb7L7E/pdb [DOI]
- 23. Bojadzic D, Alcazar O, Chen J, Chuang S‐T, Condor Capcha JM, Shehadeh LA, et al. Small‐molecule inhibitors of the coronavirus spike: ACE2 protein–protein interaction as blockers of viral attachment and entry for SARS‐CoV‐2. ACS Infect Dis. 2021;7:1519–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. AstraZeneca . AstraZeneca's antibody combination, Evusheld (tixagevimab co‐packaged with cilgavimab) authorized for use in Great Britain for pre‐exposure prophylaxis (prevention) of COVID‐19. Cambridge: AstraZeneca; 2022. [Google Scholar]
- 25. AstraZeneca . Update of AZD7442 STORM CHASER trial in post‐exposure prevention of symptomatic COVID‐19. Cambridge: AstraZeneca; 2021. [Google Scholar]
- 26. EMA . Evusheld. Amsterdam: European Medicines Agency; 2022. [Google Scholar]
- 27. Wise J. Covid‐19: evusheld is approved in UK for prophylaxis in immunocompromised people. BMJ. 2022;376:o722. [DOI] [PubMed] [Google Scholar]
- 28. Tixagevimab and Cilgavimab (Evusheld) for pre‐exposure prophylaxis of COVID‐19. JAMA. 2022;327:384–5. [DOI] [PubMed] [Google Scholar]
- 29. Case JB, Mackin S, Errico J, Chong Z, Madden EA, Guarino B, et al. Resilience of S309 and AZD7442 monoclonal antibody treatments against infection by SARS‐CoV‐2 Omicron lineage strains. bioRxiv. 2022. 10.1101/2022.03.17.484787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Weinreich DM, Sivapalasingam S, Norton T, Ali S, Gao H, Bhore R, et al. REGN‐COV2, a neutralizing antibody cocktail, in outpatients with Covid‐19. N Engl J Med. 2021;384:238–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hansen J, Baum A, Pascal KE, Russo V, Giordano S, Wloga E, et al. Studies in humanized mice and convalescent humans yield a SARS‐CoV‐2 antibody cocktail. Science. 2020;369:1010–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Franklin M, Saotome K, Romero Hernandez A, Zhou Y. Complex of SARS‐CoV‐2 receptor binding domain with the Fab fragments of two neutralizing antibodies. 2020. 10.2210/pdb6xdg/pdb [DOI]
- 33. Committee for Medicinal Products for Human Use (CHMP) . Regeneron Ireland DAC use of casirivimab and imdevimab for the treatment of COVID‐19. Amsterdam: European Medicines Agency; 2021. [Google Scholar]
- 34. Copin R, Baum A, Wloga E, Pascal KE, Giordano S, Fulton BO, et al. The monoclonal antibody combination REGEN‐COV protects against SARS‐CoV‐2 mutational escape in preclinical and human studies. Cell. 2021;184:3949–61.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Regeneron . Regeneron's COVID‐19 response efforts. Tarrytown: Regeneron; 2022. [Google Scholar]
- 36. The Medical Letter on Drugs and Therapeutics . Casirivimab and Imdevimab (REGEN‐COV) for post‐exposure prophylaxis of COVID‐19. Med Lett Drugs Ther. 2021;63:130–1. [PubMed] [Google Scholar]
- 37. Food and Drug Administration (US) . Fact sheet for patients, parents and caregivers Emergency Use Authorization (EUA) of REGEN‐COV (casirivimab and imdevimab) for coronavirus disease 2019 (COVID‐19). Silver Spring: FDA; 2022. [Google Scholar]
- 38. Updated guidelines regarding allocation of bamlanivimab/etesevimab and REGEN‐COV therapeutics states and territories can continue to order both products. U.S. Department of Health & Human Services, Public Health Emergency; 2021. [Google Scholar]
- 39. Cavazzoni P. Coronavirus (COVID‐19) update: FDA limits use of certain monoclonal antibodies to treat COVID‐19 due to the Omicron variant. Silver Spring: FDA; 2022. [Google Scholar]
- 40. Shi R, Shan C, Duan X, Chen Z, Liu P, Song J, et al. A human neutralizing antibody targets the receptor‐binding site of SARS‐CoV‐2. Nature. 2020;584:120–4. [DOI] [PubMed] [Google Scholar]
- 41. Shi R, Qi J, Wang Q, Gao F, Yan J. Molecular basis for a potent human neutralizing antibody targeting SARS‐CoV‐2 RBD. 2020. 10.2210/pdb7c01/pdb [DOI]
- 42. Jones BE, Brown‐Augsburger PL, Corbett KS, Westendorf K, Davies J, Cujec TP, et al. The neutralizing antibody, LY‐CoV555, protects against SARS‐CoV‐2 infection in nonhuman primates. Sci Transl Med. 2021;13:eabf1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hendle J, Pustilnik A, Sauder J, Coleman K, Boyles J, Dickinson C. LY‐CoV555 neutralizing antibody against SARS‐CoV‐2. 2020. 10.2210/pdb7kmg/pdb [DOI]
- 44. The Medical Letter on Drugs and Therapeutics . An EUA for Bamlanivimab and Etesevimab for COVID‐19. Med Lett Drugs Ther. 2021;63:49–50. [PubMed] [Google Scholar]
- 45. Nabel KG, Clark SA, Shankar S, Pan J, Clark LE, Yang P, et al. Structural basis for continued antibody evasion by the SARS‐CoV‐2 receptor binding domain. Science. 2021;375:eabl6251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Taylor PC, Adams AC, Hufford MM, de la Torre I, Winthrop K, Gottlieb RL. Neutralizing monoclonal antibodies for treatment of COVID‐19. Nat Rev Immunol. 2021;21:382–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Balasundaram P, Morgan‐Joseph T. Etesevimab. Treasure Island: StatPearls Publishing; 2022. [PubMed] [Google Scholar]
- 48. Westendorf K, Wang L, Žentelis S, Foster D, Vaillancourt P, Wiggin M, et al. LY‐CoV1404 (bebtelovimab) potently neutralizes SARS‐CoV‐2 variants. bioRxiv. 2022. 10.1101/2021.04.30.442182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hendle J, Pustilnik A, Sauder J, Coleman K, Boyles J, Dickinson C. LY‐CoV1404 neutralizing antibody against SARS‐CoV‐2. 2021. 10.2210/pdb7mmo/pdb [DOI]
- 50. Iketani S, Liu L, Guo Y, Liu L, Chan JF‐W, Huang Y, et al. Antibody evasion properties of SARS‐CoV‐2 Omicron sublineages. Nature. 2022;604:553–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wang Q, Yicheng G, Iketani S, Zhiteng L, Mohri H, Wang M, et al. SARS‐CoV‐2 Omicron BA.2.12.1, BA.4, and BA.5 subvariants evolved to extend antibody evasion. bioRxiv. 2022. 10.1101/2022.05.26.493517 [DOI] [Google Scholar]
- 52. Greasley SE, Noell S, Plotnikova O, Ferre R, Liu W, Bolanos B, et al. Structural basis for the in vitro efficacy of nirmatrelvir against SARS‐CoV‐2 variants. J Biol Chem. 2022;298:101972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hentzien M, Autran B, Piroth L, Yazdanpanah Y, Calmy A. A monoclonal antibody stands out against omicron subvariants: a call to action for a wider access to bebtelovimab. Lancet Infect Dis. 2022;22(9):1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Dougan M, Azizad M, Chen P, Feldman B, Frieman M, Igbinadolor A, et al. Bebtelovimab, alone or together with bamlanivimab and etesevimab, as a broadly neutralizing monoclonal antibody treatment for mild to moderate, ambulatory COVID‐19. bioRxiv. 2022. [Google Scholar]
- 55. Food and Drug Administration (US) . Fact sheet for healthcare providers: Emergency Use Authorization for bebtelovimab. Silver Spring: FDA; 2022. [Google Scholar]
- 56. Pinto D, Park Y‐J, Beltramello M, Walls AC, Tortorici MA, Bianchi S, et al. Cross‐neutralization of SARS‐CoV‐2 by a human monoclonal SARS‐CoV antibody. Nature. 2020;583:290–5. [DOI] [PubMed] [Google Scholar]
- 57. Pinto D, Park Y, Beltramello M, Walls A, Tortorici M, Bianchi S, et al. Structure of the SARS‐CoV‐2 spike glycoprotein in complex with the S309 neutralizing antibody Fab fragment. 2020. 10.2210/pdb6wps/pdb [DOI]
- 58. Cathcart AL, Havenar‐Daughton C, Lempp FA, Ma D, Schmid MA, Agostini ML, et al. The dual function monoclonal antibodies VIR‐7831 and VIR‐7832 demonstrate potent in vitro and in vivo activity against SARS‐CoV‐2. bioRxiv. 2022. 10.1101/2021.03.09.434607 [DOI] [Google Scholar]
- 59. Elesdoudy A. Sotrovimab: is it effective in early treatment of mild and moderate COVID‐19 infections? A retrospective study. Egypt J Bronchol. 2021;15:56. [Google Scholar]
- 60. Gupta A, Gonzalez‐Rojas Y, Juarez E, Crespo Casal M, Moya J, Falci DR, et al. Early treatment for Covid‐19 with SARS‐CoV‐2 neutralizing antibody sotrovimab. N Engl J Med. 2021;385:1941–50. [DOI] [PubMed] [Google Scholar]
- 61. Primary endpoint met in COMET‐TAIL phase III trial evaluating intramuscular administration of sotrovimab for early treatment of COVID‐19. Brentford: GSK; 2021. [Google Scholar]
- 62. Rockett R, Basile K, Maddocks S, Fong W, Agius JE, Johnson‐Mackinnon J, et al. Resistance mutations in SARS‐CoV‐2 delta variant after sotrovimab use. N Engl J Med. 2022;386:1477–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Blaising J, Polyak SJ, Pécheur E‐I. Arbidol as a broad‐spectrum antiviral: an update. Antiviral Res. 2014;107:84–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Boriskin SY, Leneva AI, Pecheur E‐I, Polyak JS. Arbidol: a broad‐spectrum antiviral compound that blocks viral fusion. Curr Med Chem. 2008;15:997–1005. [DOI] [PubMed] [Google Scholar]
- 65. Kadam RU, Wilson IA. Structural basis of influenza virus fusion inhibition by the antiviral drug Arbidol. Proc Natl Acad Sci USA. 2017;114:206–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Pécheur E‐I, Borisevich V, Halfmann P, Morrey JD, Smee DF, Prichard M, et al. The synthetic antiviral drug Arbidol inhibits globally prevalent pathogenic viruses. J Virol. 2016;90:3086–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Padhi AK, Seal A, Khan JM, Ahamed M, Tripathi T. Unraveling the mechanism of Arbidol binding and inhibition of SARS‐CoV‐2: insights from atomistic simulations. Eur J Pharmacol. 2021;894:173836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Vankadari N. Arbidol: a potential antiviral drug for the treatment of SARS‐CoV‐2 by blocking trimerization of the spike glycoprotein. Int J Antimicrob Agents. 2020;56:105998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Shuster A, Pechalrieu D, Jackson CB, Abegg D, Choe H, Adibekian A. Clinical antiviral drug Arbidol inhibits infection by SARS‐CoV‐2 and variants through direct binding to the spike protein. ACS Chem Biol. 2021;16:2845–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Wang X, Cao R, Zhang H, Liu J, Xu M, Hu H, et al. The anti‐influenza virus drug, Arbidol is an efficient inhibitor of SARS‐CoV‐2 in vitro. Cell Discov. 2020;6:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Nojomi M, Yassin Z, Keyvani H, Makiani MJ, Roham M, Laali A, et al. Effect of Arbidol (Umifenovir) on COVID‐19: a randomized controlled trial. BMC Infect Dis. 2020;20:954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Wang Y, Zhu L‐Q. Pharmaceutical care recommendations for antiviral treatments in children with coronavirus disease 2019. World J Pediatr. 2020;16:271–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Bahadur Gurung A, Ajmal Ali M, Elshikh MS, Aref I, Amina M, Lee J. An in silico approach unveils the potential of antiviral compounds in preclinical and clinical trials as SARS‐CoV‐2 omicron inhibitors. Saudi J Biol Sci. 2022;29:103297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Touret F, de Lamballerie X. Of chloroquine and COVID‐19. Antiviral Res. 2020;177:104762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Ibáñez S, Martínez O, Valenzuela F, Silva F, Valenzuela O. Hydroxychloroquine and chloroquine in COVID‐19: should they be used as standard therapy? Clin Rheumatol. 2020;39:2461–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Fishbane S, Hirsch JS, Nair V. Special considerations for paxlovid treatment among transplant recipients with SARS‐CoV‐2 infection. Am J Kidney Dis. 2022;79:480–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Elliott W, Chan J. Nirmatrelvir and ritonavir tablets (Paxlovid). Intern Med Alert. 2022;44:1. [Google Scholar]
- 78. National Institutes of Health (US) . Coronavirus disease 2019 (COVID‐19) treatment guidelines. Bethesda: NIH NLM; 2021. [PubMed] [Google Scholar]
- 79. O'Shaughnessy J. EUA 046 Gilead remdesivir LOA outpatients (01212022). Silver Spring: FDA; 2022. [Google Scholar]
- 80. Tsuzuki S, Hayakawa K, Doi Y, Shinozaki T, Uemura Y, Matsunaga N, et al. Effectiveness of favipiravir on nonsevere, early‐stage COVID‐19 in Japan: a large observational study using the COVID‐19 Registry Japan. Infect Dis Ther. 2022;11:1075–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Food and Drug Administration (US) . Fact sheet for healthcare providers: Emergency Use Authorization for molnupiravir. Silver Spring: FDA; 2021. [Google Scholar]
- 82. Kneller DW, Li H, Phillips G, Weiss KL, Zhang Q, Arnould MA, et al. Covalent narlaprevir‐ and boceprevir‐derived hybrid inhibitors of SARS‐CoV‐2 main protease. Nat Commun. 2022;13:2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Romano M, Ruggiero A, Squeglia F, Maga G, Berisio R. A structural view of SARS‐CoV‐2 RNA replication machinery: RNA synthesis, proofreading and final capping. Cells. 2020;9:1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Yadav R, Chaudhary JK, Jain N, Chaudhary PK, Khanra S, Dhamija P, et al. Role of structural and non‐structural proteins and therapeutic targets of SARS‐CoV‐2 for COVID‐19. Cells. 2021;10:821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Xu J, Zhao S, Teng T, Abdalla AE, Zhu W, Xie L, et al. Systematic comparison of two animal‐to‐human transmitted human coronaviruses: SARS‐CoV‐2 and SARS‐CoV. Viruses. 2020;12:244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Ullrich S, Nitsche C. The SARS‐CoV‐2 main protease as drug target. Bioorg Med Chem Lett. 2020;30:127377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Mótyán JA, Mahdi M, Hoffka G, Tőzsér J. Potential resistance of SARS‐CoV‐2 main protease (Mpro) against protease inhibitors: lessons learned from HIV‐1 protease. Int J Mol Sci. 2022;23:3507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Hartenian E, Nandakumar D, Lari A, Ly M, Tucker JM, Glaunsinger BA. The molecular virology of coronaviruses. J Biol Chem. 2020;295:12910–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Kovalevsky A, Kneller D, Coates L. Room temperature X‐ray structure of SARS‐CoV‐2 main protease (Mpro) in complex with PF‐07321332. 2021. 10.2210/pdb7si9/pdb [DOI] [PMC free article] [PubMed]
- 90. Hammond J, Leister‐Tebbe H, Gardner A, Abreu P, Bao W, Wisemandle W, et al. Oral nirmatrelvir for high‐risk, nonhospitalized adults with Covid‐19. N Engl J Med. 2022;386:1397–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Halford B. The path to paxlovid. ACS Cent Sci. 2022;8:405–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Sevrioukova IF, Poulos TL. Structure and mechanism of the complex between cytochrome P4503A4 and ritonavir. Proc Natl Acad Sci USA. 2010;107:18422–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Guy‐Alfandary S, Zhurat S, Berlin M, De Haan T, Gueta I, Shihmanter R, et al. Managing potential drug interactions of Nirmatrelvir/Ritonavir in COVID‐19 patients: a perspective from an Israeli Cross‐Sector Collaboration. Clin Pharmacol Ther. 2022. 10.1002/cpt.2610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Boras B, Jones RM, Anson BJ, Arenson D, Aschenbrenner L, Bakowski MA, et al. Discovery of a novel inhibitor of coronavirus 3CL protease for the potential treatment of COVID‐19. Chem Commun. 2021;72:8999–9154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Ramos‐Guzmán CA, Ruiz‐Pernía JJ, Tuñón I. Binding and reactivity of a nitrile oral inhibitor of SARS‐CoV‐2 main protease revealed by computational simulations. ChemRxiv. 2021. [DOI] [PubMed] [Google Scholar]
- 96. Pavan M, Bolcato G, Bassani D, Sturlese M, Moro S. Supervised molecular dynamics (SuMD) insights into the mechanism of action of SARS‐CoV‐2 main protease inhibitor PF‐07321332. J Enzyme Inhib Med Chem. 2021;36:1646–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Pepperrell T, Ellis L, Wang J, Hill A. Barriers to worldwide access for Paxlovid, a new treatment for COVID‐19. Open Forum Infect Dis. 2022;9:ofac174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Extance A. Covid‐19: what is the evidence for the antiviral Paxlovid? BMJ. 2022;377:1037. [DOI] [PubMed] [Google Scholar]
- 99. Pfizer shares top‐line results from phase 2/3 EPIC‐PEP study of PAXLOVID™ for post‐exposure prophylactic use. New York: Pfizer; 2022. [Google Scholar]
- 100. Pfizer reports additional data on PAXLOVID™ supporting upcoming new drug application submission to U.S. FDA. New York: Pfizer; 2022. [Google Scholar]
- 101. Pfizer . A phase 2/3, randomized, double‐blind, double‐dummy, placebo controlled study to evaluate the safety and efficacy of 2 regimens of orally administered PF 07321332/ritonavir in preventing symptomatic SARS‐COV‐2 infection in adult household contacts of an individual with symptomatic COVID‐19. 2022. Available from: ClinicalTrials.gov
- 102. Pfizer . An interventional efficacy and safety, phase 2/3, double‐blind, 2 arm study to investigate orally administered PF 07321332/ritonavir compared with placebo in nonhospitalized symptomatic adult participants with COVID‐19 who are at low risk of progressing to severe illness. 2022. Available from: ClinicalTrials.gov
- 103. Center for Disease Control and Prevention . COVID‐19 rebound after Paxlovid treatment. Atlanta: CDC; 2022. [Google Scholar]
- 104. Alshanqeeti S, Bhargava A. COVID‐19 rebound after Paxlovid treatment: a case series and review of literature. Cureus. 2022;14:e26239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Carlin AF, Clark AE, Chaillon A, Garretson AF, Bray W, Porrachi M, et al. Virologic and immunologic characterization of COVID‐19 recrudescence after nirmatrelvir/ritonavir treatment. Res Sq. 2022. 10.21203/rs.3.rs-1662783/v1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Kuehn BM. Rehospitalization, emergency visits after Paxlovid treatment are rare. JAMA. 2022;328:323. [DOI] [PubMed] [Google Scholar]
- 107. Ullrich S, Ekanayake KB, Otting G, Nitsche C. Main protease mutants of SARS‐CoV‐2 variants remain susceptible to nirmatrelvir. Bioorg Med Chem Lett. 2022;62:128629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Sacco MD, Hu Y, Gongora MV, Meilleur F, Kemp MT, Zhang X, et al. The P132H mutation in the main protease of Omicron SARS‐CoV‐2 decreases thermal stability without compromising catalysis or small‐molecule drug inhibition. Cell Res. 2022;32:498–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Yang KS, Leeuwon SZ, Xu S, Liu WR. Evolutionary and structural insights about potential SARS‐CoV‐2 evasion of nirmatrelvir. J Med Chem. 2022;65:8686–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Jochmans D, Liu C, Donckers K, Stoycheva A, Boland S, Stevens SK, et al. The substitutions L50F, E166A and L167F in SARS‐CoV‐2 3CLpro are selected by a protease inhibitor in vitro and confer resistance to nirmatrelvir. bioRxiv. 2022. 10.1101/2022.06.07.495116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Zhou Y, Gammeltoft KA, Ryberg LA, Pham LV, Fahnøe U, Binderup A, et al. Nirmatrelvir resistant SARS‐CoV‐2 variants with high fitness in vitro. bioRxiv. 2022. 10.1101/2022.06.06.494921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Hu Y, Lewandowski EM, Tan H, Morgan RT, Zhang X, Jacobs LMC, et al. Naturally occurring mutations of SARS‐CoV‐2 main protease confer drug resistance to nirmatrelvir. bioRxiv. 2022. 10.1101/2022.06.28.497978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Vilar S, Isom DG. One year of SARS‐CoV‐2: how much has the virus changed? Biology. 2021;10:91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Magro P, Zanella I, Pescarolo M, Castelli F, Quiros‐Roldan E. Lopinavir/ritonavir: repurposing an old drug for HIV infection in COVID‐19 treatment. Biomed J. 2021;44:43–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Choy K‐T, Wong AY‐L, Kaewpreedee P, Sia SF, Chen D, Hui KPY, et al. Remdesivir, lopinavir, emetine, and homoharringtonine inhibit SARS‐CoV‐2 replication in vitro. Antiviral Res. 2020;178:104786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Cvetkovic RS, Goa KL. Lopinavir/ritonavir: a review of its use in the management of HIV infection. Drugs. 2003;63:769–802. [DOI] [PubMed] [Google Scholar]
- 117. Sham HL, Kempf DJ, Molla A, Marsh KC, Kumar GN, Chen CM, et al. ABT‐378, a highly potent inhibitor of the human immunodeficiency virus protease. Antimicrob Agents Chemother. 1998;42:3218–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Food and Drug Administration (US) . Drug approval package: Kaletra (Lopinavir/Ritonavir) NDA #21‐226 & 21‐251. Silver Spring: FDA; 2000. [Google Scholar]
- 119. Li G, De Clercq E. Therapeutic options for the 2019 novel coronavirus (2019‐nCoV). Nat Rev Drug Discov. 2020;19:149–50. [DOI] [PubMed] [Google Scholar]
- 120. Savarino A. Expanding the frontiers of existing antiviral drugs: possible effects of HIV‐1 protease inhibitors against SARS and avian influenza. J Clin Virol. 2005;34:170–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Cao B, Wang Y, Wen D, Liu W, Wang J, Fan G, et al. A trial of lopinavir–ritonavir in adults hospitalized with severe Covid‐19. N Engl J Med. 2020;382:1787–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Horby PW, Mafham M, Bell JL, Linsell L, Staplin N, Emberson J, et al. Lopinavir–ritonavir in patients admitted to hospital with COVID‐19 (RECOVERY): a randomised, controlled, open‐label, platform trial. Lancet. 2020;396:1345–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Aguilera C, Danés I, Guillén E, Vimes A, Bosch M, Cereza G, et al. Safety of drugs used during the first wave of COVID‐19: a hospital‐registry‐based study. Diagnostics. 2022;12:1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Yin W, Mao C, Luan X, Shen D‐D, Shen Q, Su H, et al. Structural basis for inhibition of the RNA‐dependent RNA polymerase from SARS‐CoV‐2 by remdesivir. Science. 2020;368:1499–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Khan S, Attar F, Bloukh SH, Sharifi M, Nabi F, Bai Q, et al. A review on the interaction of nucleoside analogues with SARS‐CoV‐2 RNA dependent RNA polymerase. Int J Biol Macromol. 2021;181:605–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Yin W, Mao C, Luan X, Shen D, Su H, Wang X, et al. The nsp12‐nsp7‐nsp8 complex bound to the template‐primer RNA and triphosphate form of remdesivir (RTP). 2020. 10.2210/pdb7bv2/pdb [DOI]
- 127. Kokic G, Hillen HS, Tegunov D, Dienemann C, Seitz F, Schmitzova J, et al. Mechanism of SARS‐CoV‐2 polymerase stalling by remdesivir. Nat Commun. 2021;12:279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Tchesnokov EP, Feng JY, Porter DP, Götte M. Mechanism of inhibition of Ebola virus RNA‐dependent RNA polymerase by remdesivir. Viruses. 2019;11:326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Food and Drug Administration (US) . Coronavirus (COVID‐19) update: FDA approves first COVID‐19 treatment for young children. Silver Spring: FDA; 2022. [Google Scholar]
- 130. Beigel JH, Tomashek KM, Dodd LE, Mehta AK, Zingman BS, Kalil AC, et al. Remdesivir for the treatment of Covid‐19 – final report. N Engl J Med. 2020;383:1813–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Gottlieb RL, Vaca CE, Paredes R, Mera J, Webb BJ, Perez G, et al. Early remdesivir to prevent progression to severe Covid‐19 in outpatients. N Engl J Med. 2022;386:305–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. EMA . First COVID‐19 treatment recommended for EU authorisation. Amsterdam: European Medicines Agency; 2020. [Google Scholar]
- 133. Dal‐Ré R, Banzi R, Georgin‐Lavialle S, Porcher R, Sofat R, Zeitlinger M, et al. Remdesivir for COVID‐19 in Europe: will it provide value for money? Lancet Respir Med. 2021;9:127–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Lapid N. Gilead's remdesivir fails to show benefit in European trial; no fetus risk seen with first trimester vaccination. London: Reuters; 2022. [Google Scholar]
- 135. WHO Solidarity Trial Consortium . Remdesivir and three other drugs for hospitalised patients with COVID‐19: final results of the WHO Solidarity randomised trial and updated meta‐analyses. Lancet. 2022;399:1941–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Gandhi S, Klein J, Robertson AJ, Peña‐Hernández MA, Lin MJ, Roychoudhury P, et al. De novo emergence of a remdesivir resistance mutation during treatment of persistent SARS‐CoV‐2 infection in an immunocompromised patient: a case report. Nat Commun. 2022;13:1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Singh TU, Parida S, Lingaraju MC, Kesavan M, Kumar D, Singh RK. Drug repurposing approach to fight COVID‐19. Pharmacol Rep. 2020;72:1479–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Unal MA, Bitirim CV, Summak GY, Bereketoglu S, Cevher Zeytin I, Besbinar O, et al. Ribavirin shows antiviral activity against SARS‐CoV‐2 and downregulates the activity of TMPRSS2 and the expression of ACE2 in vitro. Can J Physiol Pharmacol. 2021;99:449–60. [DOI] [PubMed] [Google Scholar]
- 139. Chen F, Chan KH, Jiang Y, Kao RYT, Lu HT, Fan KW, et al. In vitro susceptibility of 10 clinical isolates of SARS coronavirus to selected antiviral compounds. J Clin Virol. 2004;31:69–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Saijo M, Morikawa S, Fukushi S, Mizutani T, Hasegawa H, Nagata N, et al. Inhibitory effect of mizoribine and ribavirin on the replication of severe acute respiratory syndrome (SARS)‐associated coronavirus. Antiviral Res. 2005;66:159–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Barnard DL, Day CW, Bailey K, Heiner M, Montgomery R, Lauridsen L, et al. Enhancement of the infectivity of SARS‐CoV in BALB/c mice by IMP dehydrogenase inhibitors, including ribavirin. Antiviral Res. 2006;71:53–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Omrani AS, Saad MM, Baig K, Bahloul A, Abdul‐Matin M, Alaidaroos AY, et al. Ribavirin and interferon alfa‐2a for severe Middle East respiratory syndrome coronavirus infection: a retrospective cohort study. Lancet Infect Dis. 2014;14:1090–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Li H, Xiong N, Li C, Gong Y, Liu L, Yang H, et al. Efficacy of ribavirin and interferon‐α therapy for hospitalized patients with COVID‐19: a multicenter, retrospective cohort study. Int J Infect Dis. 2021;104:641–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Dong L, Hu S, Gao J. Discovering drugs to treat coronavirus disease 2019 (COVID‐19). Drug Discov Ther. 2020;14:58–60. [DOI] [PubMed] [Google Scholar]
- 145. Shiraki K, Daikoku T. Favipiravir, an anti‐influenza drug against life‐threatening RNA virus infections. Pharmacol Ther. 2020;209:107512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Łagocka R, Dziedziejko V, Kłos P, Pawlik A. Favipiravir in therapy of viral infections. J Clin Med. 2021;10:273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Furuta Y, Komeno T, Nakamura T. Favipiravir (T‐705), a broad spectrum inhibitor of viral RNA polymerase. Proc Jpn Acad Ser B Phys Biol Sci. 2017;93:449–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Naydenova K, Muir KW, Wu L‐F, Zhang Z, Coscia F, Peet MJ, et al. Structure of the SARS‐CoV‐2 RNA‐dependent RNA polymerase in the presence of favipiravir‐RTP. Proc Natl Acad Sci USA. 2021;118:e2021946118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Naydenova K, Muir K, Wu L, Zhang Z, Coscia F, Peet M, et al. Nsp7‐Nsp8‐Nsp12 SARS‐CoV2 RNA‐dependent RNA polymerase in complex with template:primer dsRNA and favipiravir‐RTP. 2020. 10.2210/pdb7aap/pdb [DOI]
- 150. Agrawal U, Raju R, Udwadia ZF. Favipiravir: a new and emerging antiviral option in COVID‐19. Med J Armed Forces India. 2020;76:370–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Joshi S, Parkar J, Ansari A, Vora A, Talwar D, Tiwaskar M, et al. Role of favipiravir in the treatment of COVID‐19. Int J Infect Dis. 2021;102:501–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Chen C, Zhang Y, Huang J, Yin P, Cheng Z, Wu J, et al. Favipiravir versus Arbidol for COVID‐19: a randomized clinical trial. medRxiv. 2020. 10.1101/2020.03.17.20037432 [DOI] [Google Scholar]
- 153. Singh AK, Singh A, Singh R, Misra A. Molnupiravir in COVID‐19: a systematic review of literature. Diabetes Metab Syndr. 2021;15:102329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. O'Shaughnessy J. EUA 108 Merck molnupiravir 03232022. Silver Spring: FDA; 2022. [Google Scholar]
- 155. Kabinger F, Stiller C, Schmitzová J, Dienemann C, Kokic G, Hillen HS, et al. Mechanism of molnupiravir‐induced SARS‐CoV‐2 mutagenesis. Nat Struct Mol Biol. 2021;28:740–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Kabinger F, Stiller C, Schmitzova J, Dienemann C, Kokic G, Hillen H, et al. SARS‐CoV‐2 RdRp with Molnupiravir/NHC in the template strand base‐paired with A. 2021. 10.2210/pdb7ozu/pdb [DOI]
- 157. Imran M, Kumar Arora M, Asdaq SMB, Khan SA, Alaqel SI, Alshammari MK, et al. Discovery, development, and patent trends on molnupiravir: a prospective oral treatment for COVID‐19. Molecules. 2021;26:5795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Tian L, Pang Z, Li M, Lou F, An X, Zhu S, et al. Molnupiravir and its antiviral activity against COVID‐19. Front Immunol. 2022;13:855496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. National Institutes of Health (US) . Nitazoxanide COVID‐19 treat guidel. Bethesda: NIH; 2021. [PubMed] [Google Scholar]
- 160. Popp M, Stegemann M, Metzendorf M‐I, Gould S, Kranke P, Meybohm P, et al. Ivermectin for preventing and treating COVID‐19. Cochrane Database Syst Rev. 2021;7:CD015017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Rossignol JF. Thiazolides: a new class of antiviral drugs. Expert Opin Drug Metab Toxicol. 2009;5:667–74. [DOI] [PubMed] [Google Scholar]
- 162. Furdui C, Ragsdale SW. The role of pyruvate ferredoxin oxidoreductase in pyruvate synthesis during autotrophic growth by the Wood‐Ljungdahl pathway. J Biol Chem. 2000;275:28494–9. [DOI] [PubMed] [Google Scholar]
- 163. Stachulski AV, Pidathala C, Row EC, Sharma R, Berry NG, Iqbal M, et al. Thiazolides as novel antiviral agents. 1. Inhibition of hepatitis B virus replication. J Med Chem. 2011;54:4119–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Theodos CM, Griffiths JK, D'Onfro J, Fairfield A, Tzipori S. Efficacy of nitazoxanide against Cryptosporidium parvum in cell culture and in animal models. Antimicrob Agents Chemother. 1998;42:1959–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Fox LM, Saravolatz LD. Nitazoxanide: a new thiazolide antiparasitic agent. Clin Infect Dis. 2005;40:1173–80. [DOI] [PubMed] [Google Scholar]
- 166. Rossignol JF, La Frazia S, Chiappa L, Ciucci A, Santoro MG. Thiazolides, a new class of anti‐influenza molecules targeting viral hemagglutinin at the post‐translational level. J Biol Chem. 2009;284:29798–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Rossignol JF. Nitazoxanide, a new drug candidate for the treatment of Middle East respiratory syndrome coronavirus. J Infect Public Health. 2016;9:227–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Lokhande AS, Devarajan PV. A review on possible mechanistic insights of nitazoxanide for repurposing in COVID‐19. Eur J Pharmacol. 2021;891:173748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Calderón JM, Flores MRF, Coria LP, Garduño JCB, Figueroa JM, Contretas MJV, et al. Nitazoxanide against COVID‐19 in three explorative scenarios. J Infect Dev Ctries. 2020;14:982–6. [DOI] [PubMed] [Google Scholar]
- 170. Rocco PRM, Silva PL, Cruz FF, Melo‐Junior MAC, Tierno PFGMM, Moura MA, et al. Early use of nitazoxanide in mild COVID‐19 disease: randomised, placebo‐controlled trial. Eur Respir J. 2021;58:2003725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Broekhuysen J, Stockis A, Lins RL, De Graeve J, Rossignol JF. Nitazoxanide: pharmacokinetics and metabolism in man. Int J Clin Pharmacol Ther. 2000;38:387–94. [DOI] [PubMed] [Google Scholar]
- 172. Rossignol JF. Nitazoxanide: a first‐in‐class broad‐spectrum antiviral agent. Antiviral Res. 2014;110:94–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Hoffman PS, Sisson G, Croxen MA, Welch K, Harman WD, Cremades N, et al. Antiparasitic drug nitazoxanide inhibits the pyruvate oxidoreductases of Helicobacter pylori, selected anaerobic bacteria and parasites, and Campylobacter jejuni . Antimicrob Agents Chemother. 2007;51:868–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Haffizulla J, Hartman A, Hoppers M, Resnick H, Samudrala S, Ginocchio C, et al. Effect of nitazoxanide in adults and adolescents with acute uncomplicated influenza: a double‐blind, randomised, placebo‐controlled, phase 2b/3 trial. Lancet Infect Dis. 2014;14:609–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Jasenosky LD, Cadena C, Mire CE, Borisevich V, Haridas V, Ranjbar S, et al. The FDA‐approved oral drug nitazoxanide amplifies host antiviral responses and inhibits Ebola virus. iScience. 2019;19:1279–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Beyerstedt S, Casaro EB, Rangel ÉB. COVID‐19: angiotensin‐converting enzyme 2 (ACE2) expression and tissue susceptibility to SARS‐CoV‐2 infection. Eur J Clin Microbiol Infect Dis. 2021;40:905–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Bhutta MS, Gallo ES, Borenstein R. Multifaceted role of AMPK in viral infections. Cells. 2021;10:1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Jeon S‐M. Regulation and function of AMPK in physiology and diseases. Exp Mol Med. 2016;48:e245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Alers S, Löffler AS, Wesselborg S, Stork B. Role of AMPK‐mTOR‐Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Mol Cell Biol. 2012;32:2–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Fung TS, Liu DX. Post‐translational modifications of coronavirus proteins: roles and function. Future Virol. 2018;13:405–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Scheife RT. Protein binding: what does it mean? DICP. 1989;23:S27–31. [DOI] [PubMed] [Google Scholar]
- 182. Mullokandov E, Ahn J, Szalkiewicz A, Babayeva M. Protein binding drug‐drug interaction between warfarin and tizoxanide in human plasma. Austin J Pharmacol Ther. 2014;2:1038. [Google Scholar]
- 183. Onyeaka H, Tamasiga P, Agbara JO, Mokgwathi OA, Uwishema O. The use of Ivermectin for the treatment of COVID‐19: Panacea or enigma? Clin Epidemiol Glob Health. 2022;16:101074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Chaccour C, Hammann F, Ramón‐García S, Rabinovich NR. Ivermectin and COVID‐19: keeping rigor in times of urgency. Am J Trop Med Hyg. 2020;102:1156–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Crump A, Ōmura S. Ivermectin, ‘wonder drug’ from Japan: the human use perspective. Proc Jpn Acad Ser B Phys Biol Sci. 2011;87:13–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Wehbe Z, Wehbe M, Iratni R, Pintus G, Zaraket H, Yassine HM, et al. Repurposing ivermectin for COVID‐19: molecular aspects and therapeutic possibilities. Front Immunol. 2021;12:663586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Wagstaff KM, Sivakumaran H, Heaton SM, Harrich D, Jans DA. Ivermectin is a specific inhibitor of importin α/β‐mediated nuclear import able to inhibit replication of HIV‐1 and dengue virus. Biochem J. 2012;443:851–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Rizzo E. Ivermectin, antiviral properties and COVID‐19: a possible new mechanism of action. Naunyn Schmiedebergs Arch Pharmacol. 2020;393:1153–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Rajter JC, Sherman MS, Fatteh N, Vogel F, Sacks J, Rajter J‐J. Use of ivermectin is associated with lower mortality in hospitalized patients with coronavirus disease 2019: the ivermectin in COVID nineteen study. Chest. 2021;159:85–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Reis G, Silva EASM, Silva DCM, Thabane L, Milagres AC, Ferreira TS, et al. Effect of early treatment with ivermectin among patients with Covid‐19. N Engl J Med. 2022;386:1721–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Group AC‐19 TI and V (ACTIV)‐6 S , Naggie S. Ivermectin for treatment of mild‐to‐moderate COVID‐19 in the outpatient setting: a decentralized, placebo‐controlled, randomized, platform clinical trial. medRxiv. 2022. 10.1101/2022.06.10.22276252 [DOI] [Google Scholar]
- 192. Romark Laboratories L.C . A randomized, double‐blind, placebo controlled trial to evaluate the efficacy and safety of nitazoxanide (NTZ) for pre‐ and post exposure prophylaxis of COVID‐19 and other viral respiratory illnesses (VRI) in healthcare workers and others at increased risk of SARS‐CoV‐2 infection. 2021. Available from: ClinicalTrials.gov
- 193. Tanzi MG. FDA has authorized these therapies to manage patients with COVID‐19. Pharmacy Today. 2021;27:18–20. [Google Scholar]
- 194. Schneider WM, Chevillotte MD, Rice CM. Interferon‐stimulated genes: a complex web of host defenses. Annu Rev Immunol. 2014;32:513–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Jefferies CA. Regulating IRFs in IFN driven disease. Front Immunol. 2019;10:325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Karpusas M, Nolte M, Benton CB, Meier W, Lipscomb WN, Goelz S. The crystal structure of human interferon β at 2.2‐Å resolution. Proc Natl Acad Sci USA. 1997;94:11813–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Karpusas M, Nolte M, Lipscomb W. Human interferon‐beta crystal structure. 1997. 10.2210/pdb1au1/pdb [DOI] [PMC free article] [PubMed]
- 198. Abdolvahab MH, Moradi‐Kalbolandi S, Zarei M, Bose D, Majidzadeh‐A K, Farahmand L. Potential role of interferons in treating COVID‐19 patients. Int Immunopharmacol. 2021;90:107171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Monk PD, Marsden RJ, Tear VJ, Brookes J, Batten TN, Mankowski M, et al. Safety and efficacy of inhaled nebulised interferon beta‐1a (SNG001) for treatment of SARS‐CoV‐2 infection: a randomised, double‐blind, placebo‐controlled, phase 2 trial. Lancet Respir Med. 2021;9:196–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Wang N, Zhan Y, Zhu L, Hou Z, Liu F, Song P, et al. Retrospective multicenter cohort study shows early interferon therapy is associated with favorable clinical responses in COVID‐19 patients. Cell Host Microbe. 2020;28:455–64.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Davoudi‐Monfared E, Rahmani H, Khalili H, Hajiabdolbaghi M, Salehi M, Abbasian L, et al. A randomized clinical trial of the efficacy and safety of interferon β‐1a in treatment of severe COVID‐19. Antimicrob Agents Chemother. 2020;64:e01061‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Noreen S, Maqbool I, Madni A. Dexamethasone: therapeutic potential, risks, and future projection during COVID‐19 pandemic. Eur J Pharmacol. 2021;894:173854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. World Health Organization . WHO model list of essential medicines – 22nd list, 2021. Geneva: WHO; 2021. [Google Scholar]
- 204. Kragballe K. Topical corticosteroids: mechanisms of action. Acta Derm Venereol Suppl (Stockh). 1989;151:7–10; discussion 47–52. [PubMed] [Google Scholar]
- 205. Ahmed MH, Hassan A. Dexamethasone for the treatment of coronavirus disease (COVID‐19): a review. Sn Compr Clin Med. 2020;2:2637–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Horby P, Lim WS, Emberson J, Mafham M, Bell J, Linsell L, et al. Effect of dexamethasone in hospitalized patients with COVID‐19 – preliminary report. medRxiv. 2020. 10.1101/2020.06.22.20137273 [DOI] [Google Scholar]
- 207. Barnes PJ. Corticosteroid resistance in patients with asthma and chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2013;131:636–45. [DOI] [PubMed] [Google Scholar]
- 208. Pettersen EF, Goddard TD, Huang CC, Meng EC, Couch GS, Croll TI, et al. UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Sci. 2021;30:70–82. [DOI] [PMC free article] [PubMed] [Google Scholar]