

Abstract
Background
Severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) variant of concern (VoC) Omicron (B.1.1.529) has rapidly spread around the world, presenting a new threat to global public human health. Due to the large number of mutations accumulated by SARS‐CoV‐2 Omicron, concerns have emerged over potentially reduced diagnostic accuracy of reverse‐transcription polymerase chain reaction (RT‐qPCR), the gold standard diagnostic test for diagnosing coronavirus disease 2019 (COVID‐19). Thus, we aimed to assess the impact of the currently endemic Omicron sublineages BA.4 and BA.5 on the integrity and sensitivity of RT‐qPCR assays used for coronavirus disease 2019 (COVID‐19) diagnosis via in silico analysis. We employed whole genome sequencing data and evaluated the potential for false negatives or test failure due to mismatches between primers/probes and the Omicron VoC viral genome.
Methods
In silico sensitivity of 12 RT‐qPCR tests (containing 30 primers and probe sets) developed for detection of SARS‐CoV‐2 reported by the World Health Organization (WHO) or available in the literature, was assessed for specifically detecting SARS‐CoV‐2 Omicron BA.4 and BA.5 sublineages, obtained after removing redundancy from publicly available genomes from National Center for Biotechnology Information (NCBI) and Global Initiative on Sharing Avian Influenza Data (GISAID) databases. Mismatches between amplicon regions of SARS‐CoV‐2 Omicron VoC and primers and probe sets were evaluated, and clustering analysis of corresponding amplicon sequences was carried out.
Results
From the 1164 representative SARS‐CoV‐2 Omicron VoC BA.4 sublineage genomes analyzed, a substitution in the first five nucleotides (C to T) of the amplicon's 3′‐end was observed in all samples resulting in 0% sensitivity for assays HKUnivRdRp/Hel (mismatch in reverse primer) and CoremCharite N (mismatch in both forward and reverse primers). Due to a mismatch in the forward primer's 5′‐end (3‐nucleotide substitution, GGG to AAC), the sensitivity of the ChinaCDC N assay was at 0.69%. The 10 nucleotide mismatches in the reverse primer resulted in 0.09% sensitivity for Omicron sublineage BA.4 for Thai N assay. Of the 1926 BA.5 sublineage genomes, HKUnivRdRp/Hel assay also had 0% sensitivity. A sensitivity of 3.06% was observed for the ChinaCDC N assay because of a mismatch in the forward primer's 5′‐end (3‐nucleotide substitution, GGG to AAC). Similarly, due to the 10 nucleotide mismatches in the reverse primer, the Thai N assay's sensitivity was low at 0.21% for sublineage BA.5. Further, eight assays for BA.4 sublineage retained high sensitivity (more than 97%) and 9 assays for BA.5 sublineage retained more than 99% sensitivity.
Conclusion
We observed four assays (HKUnivRdRp/Hel, ChinaCDC N, Thai N, CoremCharite N) that could potentially result in false negative results for SARS‐CoV‐2 Omicron VoCs BA.4 and BA.5 sublineages. Interestingly, CoremCharite N had 0% sensitivity for Omicron Voc BA.4 but 99.53% sensitivity for BA.5. In addition, 66.67% of the assays for BA.4 sublineage and 75% of the assays for BA.5 sublineage retained high sensitivity. Further, amplicon clustering and additional substitution analysis along with sensitivity analysis could be used for the modification and development of RT‐qPCR assays for detecting SARS‐CoV‐2 Omicron VoC sublineages.
Keywords: amplicon, BA.4 sublineage, BA.5 sublineage, COVID‐19, detection, in silico sensitivity assessment, mismatches, Omicron, primers and probes, RT‐qPCR, SARS‐CoV‐2, substitutions, variant, whole genome sequencing data
1. INTRODUCTION
The emergence of severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) B.1.1.529 lineage presents a new threat to global public human health and coronavirus disease 2019 (COVID‐19) pandemic containment efforts. 1 Due to an unprecedented number of mutations, the high potential for transmission and immune escape, both leading to rapid global spread and an increasing number of cases, the World Health Organization (WHO) labeled B.1.1.529 as Omicron Variant of Concern (VoC) and has called for immediate global action in response. 2
SARS‐CoV‐2 is a positive‐sense, single‐stranded RNA virus, with a genome of ~30 000 base pairs in length. 3 As officially recommended by the WHO 4 and International Federation of Clinical Chemistry and Laboratory Medicine (IFCC) 5 the current gold standard diagnostic assay for detecting SARS‐CoV‐2 in human specimens is a Nucleic Acid Amplification Test (NAAT); more specifically, a quantitative reverse‐transcription polymerase chain reaction (RT‐qPCR), to detect viral RNA in clinical specimens, most commonly, from nasopharyngeal or oropharyngeal swabs. RT‐qPCR employs three oligonucleotides: a forward primer, a reverse primer, and a probe, which specifically hybridize with respective sequence targets in the SARS‐CoV‐2 genome between where the forward and reverse primers recognize. 6 Subsequent amplification of the viral genome and production of a fluorescence indicator by the probe during repeated application cycles, leads to the identification and possible quantification of SARS‐CoV‐2 RNA. 6 Given the high accuracy and rapid turnaround times, NAAT are the most widely used tests by clinical laboratories for COVID‐19 diagnostics. 5 However, this accuracy is threatened by the rapid evolution of SARS‐CoV‐2, in which some RT‐qPCR assays may lose sensitivity due to mutations in the viral genome of newly evolving SARS‐CoV‐2 VoC. 6 , 7
The most likely sources of RT‐qPCR false negative results are preanalytical errors. 8 Problems can arise from specimen collection (e.g., swab), handling, transport and storage, inappropriate and/or inadequate swab quality, and/or volume. 8 Other analytical errors, such as testing carried out outside of the reasonable diagnostic window, as well as mismatches between primers/probes and viral genome due to emerging mutations, remain a threat to diagnostic accuracy in the context of novel SARS‐CoV‐2 VoC. 8 Mutations located in regions hybridizing with the 3′‐end of primers are the most sensitive, with a single mismatch in an annealing site leading to inhibition of amplification and potential reduced diagnostic accuracy and/or false negative results. 9 , 10 , 11 , 12 , 13 Mutations located in regions hybridizing with 5′‐end of primers or other annealing segments of the oligonucleotide may have a more variable impact. Nevertheless, 2–3 mismatches in such regions may result in reduced RT‐qPCR performance. 9 , 10 , 11 , 12 , 13
Given that SARS‐CoV‐2 Omicron VoC displays a high mutational profile, evaluation of all diagnostic assays (molecular and antigen immunoassays) should be urgently performed to ensure diagnostic accuracy and proper identification of COVID‐19 cases. Indeed, the United States (US) The food and Drug Administration (FDA) has already noted potential issues with several diagnostic assays in the context of the SARS‐CoV‐2 Omicron VoC. 14 With respect to RT‐qPCR, previous in silico studies evaluating less mutated variants have demonstrated several mismatches, including mutations located in oligonucleotide annealing sites, which may reduce the sensitivity for some NAAT. 7 , 15 , 16 Besides, there is a need for development of primers specific for Omicron VoC due to several mutations in the currently available primer target genes. 14
RT‐qPCR primers and probes are generally designed to hybridize with relatively conserved sequences in the SARS‐CoV‐2 RNA genome, with most assays targeting one or more of the coding sequences for Spike (S), Envelope (E), and Nucleocapsid (N) structural proteins and Open Reading Frame (ORF) ORF1ab, which encodes the viral RNA‐dependent RNA polymerase (RdRp). 5 , 6 , 7 , 15 , 16 Mutations in all of the above genes are observed in the SARS‐CoV‐2 Omicron VoC, including over 30 nonsynonymous mutations in the S, a nonsynonymous mutation in the E, multiple amino acid substitution, and a deletion in the N, and multiple nonsynonymous mutations in ORF1ab. The nonstructural protein (nsp)12, the RdRp which is a defined target for multiple antiviral drugs, such as molnupiravir 17 and remdesivir 18 is encoded by ORF1ab. An overview of the mutations found in the sublineages of SARS‐CoV‐2 Omicron VoC is provided in Table 1. 19
Table 1.
Assessment of the sensitivity of RT‐qPCR assays using SCREENED for BA.4 sublineage genomes
| Assay | Target gene | Assay name | Mismatches in the first five nucleotides of the primer's 3′ end | >10% mismatches in the annealing sites of primers and probes | False negative results* | Sensitivity (%) | ||
|---|---|---|---|---|---|---|---|---|
| No. of genomes | Substitutions in genome | No. of genome | Substitutions in genome | |||||
| 1 | N | ChinaCDC N | 1 | Rv CAGCTTGAGAGCAAAAT T TCTG | 1 | Fw AACT AACTTCTCCTGCTAGAAT | 1156 | 0.69 |
| 1 | Fw AACG AACTTCTCCTGCCAGAAT | |||||||
| 1153 | Fw AAC GAACTTCTCCTGCTAGAAT | |||||||
| 1 | ORF1ab | ChinaCDC ORF1ab | 1 | Rv TCAGCTGATGCACAATC A T | 0 | * | 1 | 99.91 |
| 2 | RdRp P1 | CoremChariteRdRp P1 | 1 | Fw GTGAAATGGTCATGTGTGGC A G | 0 | * | 1 | 99.91 |
| 2 | RdRp P2 | CoremChariteRdRp P2 | 1 | Fw GTGAAATGGTCATGTGTGGC A G | 0 | * | 1 | 99.91 |
| 2 | E | CoremCharite E | 0 | * | 0 | * | 0 | 100.00 |
| 2 | N | CoremCharite N | 14 | Fw CACATTGGCACCCG T AAT T | 0 | * | 1164 | 0.00 |
| 11 | Fw CATATTGGCACCCGCAAT T | |||||||
| 1137 | Fw CACATTGGCACCCGCAAT T | |||||||
| 1 | Rv CAAGCCTCTTCTCGTTCCT T | |||||||
| 1 | Rv CAAGCCTCTTCTCGTTC T TC | |||||||
| 3 | RdRp IP2 | Pasteur RdRp IP2 | 1 | Rv ACAACACAACAAAG A GAG | 0 | * | 1 | 99.91 |
| 3 | RdRp IP4 | Pasteur RdRp IP4 | 0 | * | 0 | * | 0 | 100.00 |
| 3 | E | Pasteur E | 0 | * | 0 | * | 0 | 100.00 |
| 4 | N‐1 | USCDC N1 | 0 | * | 0 | * | 0 | 100.00 |
| 4 | N‐2 | USCDC N2 | 34 | Fw TTACAAACATTGGCC A CAAA | 0 | * | 34 | 97.08 |
| 4 | N‐3 | USCDC N3 | 1 | Fw GGGAGCCTTGAATACACC T AAA | 0 | * | 1 | 99.91 |
| 5 | N | Japan N | 1 | Fw AAATTTTGGGGACCA T GAAC | 0 | * | 1 | 99.91 |
| 6 | ORF1b/NSP14 | HKFacMed ORF1b/nsp14 | 0 | * | 0 | * | 0 | 100.00 |
| 6 | N | HKFacMed N | 1 | Rv CATGGAAGTCACACCTT T G | 0 | * | 2 | 99.83 |
| 1 | Rv CATGGAAGTCACAC T TTCG | |||||||
| 7 | N | Thai N | 0 | * | 5 | Rv AATGG T GA GGCGC G ATCAA | 1163 | 0.09 |
| 3 | Rv G AT TC A ACTG GCAGT AACC | |||||||
| 2 | Rv GTAACCAG A ATG NGTGGGG | |||||||
| 1153 | Rv A G T AACC A GAATG GTGGGG | |||||||
| 8 | RdRp/Hel | HKUnivRdRp/Hel | 1 | Rv GACCATATCATATCAACATCACA T | 0 | * | 1164 | 0.00 |
| 1163 | Rv GACCATGTCATATCAACATCACA T | |||||||
| 8 | S | HKUniv S | 0 | * | 0 | * | 0 | 100.00 |
| 8 | N | HKUniv N | 0 | * | 0 | * | 0 | 100.00 |
| 9 | ORF1a | RoujianLu ORF1a | 0 | * | 0 | * | 0 | 100.00 |
| 10 | RdRp | Won RdRP | 0 | * | 0 | * | 0 | 100.00 |
| 10 | S | Won S | 0 | * | 0 | * | 0 | 100.00 |
| 10 | E | Won E | 0 | * | 0 | * | 0 | 100.00 |
| 10 | N | Won N | 1 | Rv CCTCATCACGTAGTCGCAA T | 0 | * | 1 | 99.91 |
| 11 | N‐1 | SigmAldr N1 | 1 | Fw GCCTCTTCTCGTTCCTCATCA T | 0 | * | 2 | 99.83 |
| 1 | Fw GCCTCTTCTCGTTCCTC G TCAC | |||||||
| 11 | N‐2 | SigmAldr N2 | 1 | Fw GCCTCTTCTCGTTCCTCATCA T | 1 | Rv CT A GAATG GGCAATGGCGG | 4 | 99.66 |
| 1 | Fw GCCTCTTCTCGTTCCTC G TCAC | |||||||
| 1 | Rv GAATGGCTGGCAATGGC A G | |||||||
| 11 | ORF1a‐3 | SigmAldr ORF1ab3 | 0 | * | 1 | Fw TGAAG A TT T A CT CA TTCGTAAG | 3 | 99.74 |
| 1 | Rv TGGT AC AACT ACACT T AAC GGTCTT | |||||||
| 1 | Pb CTTA ACG GT CTT T G GCT TGAT GACG | |||||||
| 11 | ORF1a‐4 | SigmAldr ORF1ab4 | 0 | * | 1 | Fw CA CTTA A CG GTCTT T GGC T T | 3 | 99.74 |
| 1 | Rv TGGT AC AACT ACACT T AAC GGTCTT | |||||||
| 1 | Pb CTTA ACG GT CTT T G GCT TGAT GACG | |||||||
| 11 | S‐5 | SigmAldr S5 | 0 | * | 1 | Pb T GACTAA G TCTC A TCGGCGGGCACG | 3 | 99.74 |
| 1 | Pb AGACTAA G TCTC A TCGGCGGG T ACG | |||||||
| 1 | Pb AGACTAA G TCTC A TC T GCGGGCACG | |||||||
| 11 | S‐6 | SigmAldr S6 | 0 | * | 1 | Pb T GACTAA G TCTC A TCGGCGGGCACG | 3 | 99.74 |
| 1 | Pb AGACTAA G TCTC A TCGGCGGG T ACG | |||||||
| 1 | Pb AGACTAA G TCTC A TC T GCGGGCACG | |||||||
| 12 | E | Huang E | 0 | * | 0 | * | 0 | 100.00 |
Note: Results were obtained from 1164 BA.4 sublineage genomes. The mismatches are shown in bold and underline. The forward (Fw) and reverse (Rv) primers, and probe (Pb) sequences are directed from 5′–3′. The Rv primer sequences are reverse complemented. The substitutions represent mutations in the genome sequences.“*” represents that no mismatches are obtained by SCREENED.
Abbreviation: qRT‐PCR, quantitative reverse‐transcription polymerase chain reaction.
Here, we aimed to assess the potential impact of Omicron VoC on the integrity of RT‐qPCR assays currently used for COVID‐19 diagnosis and evaluate the potential for false negatives or test failure due to mismatches between primers/probes and viral genome. The primers and probes used in this study are those for which the sequences are publicly available and thus we were able to analyze them. Most of the RT‐qPCR assays used in this study are the World Health Organization (WHO) recommended publicly available RT‐qPCR tests for SARS‐CoV‐2 detection mentioned in their published technical guidance, 20 while others were identified in the literature. Ideally, the evaluation of diagnostic assays should be performed in vitro by qualified personnel at clinical and research laboratories. However, the limited access to the Omicron VoC, including the multiple sublineages, limited availability of reagents and assays due to global laboratory shortages, and lack of personnel as recently noted in a survey by the American Association of Clinical Chemistry (AACC) 21 and all primers/probes needed to perform such comprehensive evaluation has decelerated the assessment. Therefore, a bioinformatics approach, using an in silico specificity (sensitivity) evaluation, as recently demonstrated by Gand et al., 7 , 15 Khan et al., 16 and Nayar et al. 6 can be useful for initial and rapid evaluation. Thus, it is critical to take full advantage of the immense efforts of the scientific community, especially in South Africa, to generate whole genome sequencing (WGS) data that are made publicly available in databases such as National Center for Biotechnology Information (NCBI) and Global Initiative on Sharing Avian Influenza Data (GISAID).
In this study, the in silico sensitivity of 12 RT‐qPCR tests (containing 30 primers and probe sets) developed for detection of SARS‐CoV‐2 was assessed for detecting SARS‐CoV‐2 BA.4 and BA.5 Omicron VoCs sublineages as classified by Pangolin, obtained after removing redundancy from publicly available genomes from NCBI and GISAID databases. Mismatches between the amplicon regions of the SARS‐CoV‐2 Omicron VoC and primers and probe sets were evaluated and clustering analysis of corresponding amplicon sequences was carried out. Moreover, we have used an in silico approach to design primers specific for BA.4 and BA.5 sublineages, which will facilitate the development of more accurate primers.
2. METHODOLOGY
2.1. Data set
Whole genome sequencing data of the SARS‐CoV‐2 Omicron (B.1.1.529) VoCs BA.4 and BA.5 sublineage were downloaded from GISAID EpiCoV (https://www.epicov.org, 3905, and 4207 genome sequences, respectively) and NCBI virus (https://www.ncbi.nlm.nih.gov/labs/virus/, 660 and 567 genome sequences) databases on June 2, 2022. The geographical distribution of genome sequences is shown in Figure 1. To minimize the sequencing errors, the filters used for selecting data from GISAID were, “Complete” which means only complete genome sequences (>29 000 nt) were selected, and “low coverage excl,” that is, entries with more than 5% unknowns (“Ns”) were excluded. For NCBI genomes, data were selected based on filters, “SARS‐CoV‐2” as the virus (taxid: 2697049), “Homo sapiens” as the host (taxid: 9606), and “BA.4” or “BA.5” as the Pango lineage. A total of 30 primers and probes sets were evaluated, which are parts of 12 RT‐qPCR tests developed for detecting SARS‐CoV‐2.
Figure 1.

Geographical distribution of (A) 1164 BA.4 and (B) 1926 BA.5 sublineage genomes.
2.2. Selection of representative genomes by sequence identity clustering
The clustering of Omicron VoC sequences was performed to remove redundancy in the data set. All the downloaded sequences were clustered using CD‐HIT‐EST version 4.6.8 (https://github.com/weizhongli/cdhit) (developed by Weizhong Li's lab at UCSD, USA) setting sequence identity cut‐off equal to 1.0 (other parameters were left at default settings). Representative genomes of lower quality, that is, showing more than three ambiguous nucleotides (such as “N”) in the genomic regions targeted by the evaluated RT‐qPCR assays were removed. A total of 3539 clusters of sequences were obtained from CD‐HIT‐EST for SARS‐CoV‐2 Omicron BA.4 sublineage, and 3566 clusters for BA.5 sublineage. Total sequences after removing redundancy and taking high‐quality sequences were 1164 for BA.4 and 1926 for BA.5 sublineage of Omicron VoC.
2.3. Evaluation of in silico analytical sensitivity
The evaluation of the RT‐qPCR tests was done through in silico sensitivity analysis. A theoretical positive RT‐qPCR signal (the primers could successfully anneal to the genome sequence) was considered based on the criteria used by SCREENED version 1.0 (polymeraSe Chain Reaction Evaluation through largE‐scale miNing of gEnomic Data) developed by Vanneste et al., Belgium. 13 The settings used by SCREENED for a positive RT‐qPCR signal are:
-
(i)
No mismatch was observed in the initial five nucleotides of primers' 3′‐end;
-
(ii)
Total number of mismatches was not more than 10% of oligonucleotides length; and,
-
(iii)
90% or more of the oligonucleotides sequence accurately aligned with their targets. 9 , 10 , 11 , 12 , 13 , 15
The sensitivity is known as the potential of a method to detect varying targets by a positive relation. Therefore, the in silico sensitivity was calculated as the ratio of the number of genomes detected, that is, producing a positive RT‐qPCR signal to the total number of genomes analyzed.
The in silico sensitivity was evaluated as shown in Equation (1).
| (1) |
The in silico sensitivity of an assay is more qualitative than quantitative as it signifies whether the genome is detected and could relate to the diagnostic sensitivity of the assay.
2.4. Design of SARS‐CoV‐2 primers specific to Omicron VoCs BA.4 and BA.5 sublineage
The sequences representing Wuhan‐Hu‐1, Alpha, Beta, Gamma, Delta, Lambda, Mu, Omicron BA.4 and Omicron BA.5 SARS‐CoV‐2 variants of concern were aligned using MAFFT version 7 developed by Katoh et al., Japan 22 with default parameters. SARS‐CoV‐2 genome Wuhan‐Hu‐1 was considered as the reference for the alignments (GenBank accession: NC_045512). The alignments were viewed using Jalview software (version 2 by Waterhouse et al., UK). 23 The regions of the SARS‐CoV‐2 Omicron BA.4 and BA.5 sublineage genome, containing mutations compared to the reference genome, were considered for development of BA.4 and BA.5 specific primers. The characteristics of the designed primers were then assessed using Primer‐BLAST (by Ye et al., USA). 24 We selected 10 primer pairs based on the low self‐complementarity for total annealing, max 6 nucleotides, and also for annealing in the 3′ region, max 4 nucleotides.
3. RESULTS
SCREENED was used for investigating the in silico sensitivity of 30 primers and probe sets from 12 RT‐qPCR assays targeting different regions of the SARS‐CoV‐2 genome. After data preprocessing, we utilized 1164 representative genomes from BA.4 sublineage and 1926 from BA.5 sublineage SARS‐CoV‐2 Omicron VoCs genomes.
3.1. Determination of sensitivity for RT‐qPCR assays
3.1.1. For SARS‐CoV‐2 Omicron VoC BA.4 sublineage
Our results in Table 1 indicate that all of the 1164 BA.4 sublineage genomes produced a false negative (genome could not be amplified in silico by the RT‐qPCR assay) result for HKUnivRdRp/Hel and CoremCharite N assay because of substitution in the first five nucleotides of the amplicon for reverse primer's 3′ end (C to T), and thus resulting in 0% sensitivity of this test. A sensitivity of 0.69% was observed for the ChinaCDC N assay owing to a mismatch in the forward primer's 5′end (3‐nucleotide substitution, GGG to AAC), resulting in a false negative result for as many as 1153/1164 genomes. For the Thai N assay, 1163/1164 genomes could not be detected correctly, yielding 0.09% sensitivity mainly because of 10 nucleotide mismatches in the reverse primer. A sensitivity of 100% was obtained for assays CoremCharite E, Pasteur RdRp IP4, Pasteur E, USCDC N1, HKFacMed ORF1b/nsp14, HKUniv S, HKUniv N, RoujianLu ORF1a, Won RdRP, Won S, Won E, and Huang E as presented in Table 1, which means that these assays resulted in in silico amplification of all BA.4 sublineage genomes included in our analysis. All other assays showed a sensitivity of more than 99% except USCDC N2 (i.e., a sensitivity of 97.08%).
3.1.2. For SARS‐CoV‐2 Omicron BA.5 sublineage
Results in Table 3 indicate that all of the 1926 BA.5 sublineage genomes produced a false negative (genome could not be amplified in silico by the RT‐qPCR assay) result for HKUnivRdRp/Hel assay because of substitution in the first five nucleotides of the amplicon for reverse primer's 3′ end (C to T), thus resulting in 0% sensitivity of this test. A sensitivity of 3.06% was observed for the ChinaCDC N assay owing to a mismatch in the forward primer's 5′ end (3‐nucleotide substitution, GGG to AAC), resulting in a false negative result for 1862/1926 genomes. For the Thai N assay, 1890/1926 genomes could not be detected correctly, which yielded 0.21% sensitivity mainly because of 10 nucleotide mismatches in the reverse primer. A sensitivity of 100% was obtained for assays ChinaCDC ORF1ab, CoremCharite E, Pasteur E, Japan N, HKFacMed ORF1b/nsp14, HKUniv S, Won RdRP, Won S, Won E, SigmAldr N1, and Huang E as presented in Table 3, which means that these assays resulted in in silico amplification of all BA.5 sublineage genomes included in our analysis. All other assays showed a sensitivity of more than 99%.
Table 3.
Assessment of the sensitivity of RT‐qPCR assays using SCREENED for BA.5 sublineage genomes
| Assay | Target gene | Assay name | Mismatches in the first five nucleotides of the primer's 3′ end | >10% Mismatches in the annealing sites of primers and probes | False‐negative results* | Sensitivity (%) | ||
|---|---|---|---|---|---|---|---|---|
| No. of genomes | Substitutions in genome | No. of genome | Substitutions in genome | |||||
| 1 | N | ChinaCDC N | 0 | * | 1 | Fw AACT AACTTCTCCTGCTAGAAT | 1867 | 3.06 |
| 1 | Fw AACG AAATTCTCCTGCTAGAAT | |||||||
| 1 | Fw AAC GAACTTCTCCTG T TAGAAT | |||||||
| 2 | Fw AAC GAACTTCTCCT T CTAGAAT | |||||||
| 1862 | Fw AAC GAA T TTCTCCTGCTAGAAT | |||||||
| Fw AAC GAACTTCTCCTGCTAGAAT | ||||||||
| 1 | ORF1ab | ChinaCDC ORF1ab | 0 | * | 0 | * | 0 | 100.00 |
| 2 | RdRp P1 | CoremChariteRdRp P1 | 1 | Fw GTGAAATGGTCATGTGTGG A GG | 0 | * | 3 | 99.84 |
| 2 | Fw GTGAAATGGTCATGTGTGGC T G | |||||||
| 2 | RdRp P2 | CoremChariteRdRp P2 | 1 | Fw GTGAAATGGTCATGTGTGG A GG | 0 | * | 3 | 99.84 |
| 1 | Fw GTGAAATGGTCATGTGTGGC T G | |||||||
| 2 | E | CoremCharite E | 0 | * | 0 | * | 0 | 100.00 |
| 2 | N | CoremCharite N | 7 | Fw CACATTGGCACCCGCAAT T | 0 | * | 9 | 99.53 |
| 1 | Fw CACATTGGCACCCG T AATC | |||||||
| 1 | Rv CAAGCCTCTTCTCGTTCCT T | |||||||
| 3 | RdRp IP2 | Pasteur RdRp IP2 | 6 | Fw ATGAGCTTAGTCCTGT C G | 0 | * | 6 | 99.69 |
| 3 | RdRp IP4 | Pasteur RdRp IP4 | 1 | Rv CCTATATTAACCTTGAC T AG | 0 | * | 2 | 99.90 |
| 1 | Rv CCTATATTAACCTTGA T CAG | |||||||
| 3 | E | Pasteur E | 0 | * | 0 | * | 0 | 100.00 |
| 4 | N‐1 | USCDC N1 | 1 | Fw GACCCCAAAATCAGCGAAA C | 0 | * | 3 | 99.84 |
| 2 | Pb A TT CCGCATTACGTTTGGTGG G CC | |||||||
| 4 | N‐2 | USCDC N2 | 1 | Rv TTCTTCGGAATGTCGC A C | 0 | * | 2 | 99.90 |
| 1 | Rv TTCTTCGGAATGTCGCG T | |||||||
| 4 | N‐3 | USCDC N3 | 1 | Fw GGGAGCCTTGAATACACC T AAA | 1 | Fw T GGAGCC G TGAA C ACACCAAAA | 3 | 99.84 |
| 1 | Rv CAATGCTGCAATCGTG T TACA | |||||||
| 5 | N | Japan N | 0 | * | 0 | * | 0 | 100.00 |
| 6 | ORF1b/NSP14 | HKFacMed ORF1b/nsp14 | 0 | * | 0 | * | 0 | 100.00 |
| 6 | N | HKFacMed N | 1 | Fw TAATCAGACAAGGAACT T ATTA | 0 | * | 5 | 99.74 |
| 4 | Rv CATGGAAGTCACACCTT T G | |||||||
| 7 | N | Thai N | 0 | * | 1 | Rv AATGGAG GGGCGC G ATCAA | 1922 | 0.21 |
| 5 | Rv GTAACCAG A ATG NGTGGGG | |||||||
| 1890 | Rv A G T AACC A GAATG GTGGGG | |||||||
| 1 | Rv AAT AACC A GAATG GTGGGG | |||||||
| 25 | Rv CGTTTG GTGGG C | |||||||
| 8 | RdRp/Hel | HKUnivRdRp/Hel | 0 | * | 1926 | 0.00 | ||
| 8 | S | HKUniv S | 0 | * | 0 | * | 0 | 100.00 |
| 8 | N | HKUniv N | 2 | Fw GCGTTCTTCGGAAT T TCG | 0 | * | 2 | 99.90 |
| 9 | ORF1a | RoujianLu ORF1a | 5 | Fw AGAAGATTGGTTAGATGATG G TAGT | 0 | * | 8 | 99.58 |
| 1 | Rv GGTTCAACCTCAATTAGAGAT A GAA | |||||||
| 1 | Rv GGTTCAACCTCAATTAGAGATG A AA | |||||||
| 1 | Rv GGTTCAACCTCAATTAGAGATGGA G | |||||||
| 10 | RdRp | Won RdRP | 0 | * | 0 | * | 0 | 100.00 |
| 10 | S | Won S | 0 | * | 0 | * | 0 | 100.00 |
| 10 | E | Won E | 0 | * | 0 | * | 0 | 100.00 |
| 10 | N | Won N | 1 | Fw CAATGCTGCAATCGTG T TAC | 0 | * | 2 | 99.90 |
| 1 | Rv CCTCATCACGTAGTCG T AAC | |||||||
| 11 | N‐1 | SigmAldr N1 | 0 | * | 0 | * | 0 | 100.00 |
| 11 | N‐2 | SigmAldr N2 | 2 | Rv GAATGGCTGGCAATG T CGG | 1 | Rv GAATGGCT A GC G A AA GC TT | 3 | 99.84 |
| 11 | ORF1a‐3 | SigmAldr ORF1ab3 | 0 | * | 1 | Fw G C CAT AGGT A C GG C GC CG ATCT | 2 | 99.95 |
| 11 | ORF1a‐4 | SigmAldr ORF1ab4 | 0 | * | 1 | Fw GG C CAT AGGT A C GG C | 2 | 99.95 |
| 11 | S‐5 | SigmAldr S5 | 0 | * | 1 | Pb A A ACTAA G TCTC A TCGGCGGG T ACG | 5 | 99.74 |
| 1 | Pb AGACTAA G TC C C A TCGGCGGGCACG | |||||||
| 1 | Pb AGACTAA G TCT AA TCGGCGGGCACGPb | |||||||
| 1 | AGACTAA G TCTC A TCGGCGGG T ACG | |||||||
| 1 | Pb AGACTAA G TCTC A TCG T CGGGCACG | |||||||
| 11 | S‐6 | SigmAldr S6 | 0 | * | 1 | Pb A A ACTAA G TCTC A TCGGCGGG T ACG | 5 | 99.74 |
| 1 | Pb AGACTAA G TC C C A TCGGCGGGCACG | |||||||
| 1 | Pb AGACTAA G TCT AA TCGGCGGGCACG | |||||||
| 1 | Pb AGACTAA G TCTC A TCGGCGGG T ACG | |||||||
| 1 | Pb AGACTAA G TCTC A TCG T CGGGCACG | |||||||
| 12 | E | Huang E | 0 | * | 0 | * | 0 | 100.00 |
Note: Results were obtained from 1926 BA.4 sublineage genomes. The mismatches are shown in bold and underline. The forward (Fw) and reverse (Rv) primers, and probe (Pb) sequences are directed from 5′–3′. The Rv primer sequences are reverse complemented. The substitutions represent mutations in the genome sequences. “*” represents that no mismatches are obtained by SCREENED.
Abbreviation: qRT‐PCR, quantitative reverse‐transcription polymerase chain reaction.
3.2. Analysis of the amplicon clusters
SCREENED also clustered the amplicon sequences from all genomes analyzed in our study targeted by the evaluated primers/probes. For each RT‐qPCR assay, genomes were clustered based on the identical amplicon sequence, where the greater number of clusters represents a higher diversity of the genomic region. The total number of clusters obtained for each assay and redistribution of genomes in the top three clusters (from largest to smallest) are shown in Tables 2 and 4 for BA.4 and BA.5 sublineages respectively. For BA.4, SigmAldr S6 showed the largest amplicon diversity with 21 clusters followed by SigmAldr N1 and N2 with 17 clusters. For BA.5, CoremCharite N andSigmAldr N2 showed the largest diversity with 25 clusters followed by SigmAldr N1 and SigmAldr S6 with 24 clusters.
Table 2.
Amplicon clusters among the SARS‐CoV‐2 Omicron BA.4 sublineage sequences amplified by the evaluated primers and probes and percentage of genomes within the largest three clusters.
| Assay | Target gene | Assay name | No. of clusters | Cluster I (%) | Cluster II (%) | Cluster III (%) |
|---|---|---|---|---|---|---|
| 1 | N | ChinaCDC N | 15 | 98.1 | 0.6 | 0.17 |
| 1 | ORF1ab | ChinaCDC ORF1ab | 9 | 98.7 | 0.43 | 0.26 |
| 2 | RdRp P1 | CoremChariteRdRp P1 | 4 | 96.9 | 2.23 | 0.77 |
| 2 | RdRp P2 | CoremChariteRdRp P2 | 4 | 96.9 | 2.23 | 0.77 |
| 2 | E | CoremCharite E | 5 | 99.23 | 0.09 | 0.09 |
| 2 | N | CoremCharite N | 10 | 97.16 | 1.2 | 0.95 |
| 3 | RdRp IP2 | Pasteur RdRp IP2 | 8 | 98.45 | 0.86 | 0.17 |
| 3 | RdRp IP4 | Pasteur RdRp IP4 | 4 | 99.74 | 0.09 | 0.09 |
| 3 | E | Pasteur E | 5 | 99.23 | 0.52 | 0.09 |
| 4 | N‐1 | USCDC N1 | 6 | 99.4 | 0.26 | 0.09 |
| 4 | N‐2 | USCDC N2 | 4 | 96.9 | 2.92 | 0.09 |
| 4 | N‐3 | USCDC N3 | 6 | 97.5 | 1.2 | 0.94 |
| 5 | N | Japan N | 13 | 95.45 | 2.92 | 0.69 |
| 6 | ORF1b/NSP14 | HKFacMed ORF1b/nsp14 | 12 | 98.19 | 0.86 | 0.17 |
| 6 | N | HKFacMed N | 9 | 96.48 | 2.92 | 0.09 |
| 7 | N | Thai N | 9 | 98.54 | 0.43 | 0.26 |
| 8 | RdRp/Hel | HKUnivRdRp/Hel | 7 | 99.31 | 0.17 | 0.17 |
| 8 | S | HKUniv S | 14 | 92.53 | 5.67 | 0.34 |
| 8 | N | HKUniv N | 6 | 98.88 | 0.69 | 0.17 |
| 9 | ORF1a | RoujianLu ORF1a | 3 | 99.83 | 0.09 | 0.09 |
| 10 | RdRp | Won RdRp | 4 | 96.9 | 2.23 | 0.77 |
| 10 | S | Won S | 2 | 99.91 | 0.09 | 0 |
| 10 | E | Won E | 5 | 99.23 | 0.51 | 0.09 |
| 10 | N | Won N | 11 | 99.14 | 0.09 | 0.09 |
| 11 | N‐1 | SigmAldr N1 | 19 | 97.42 | 0.6 | 0.34 |
| 11 | N‐2 | SigmAldr N2 | 19 | 97.42 | 0.6 | 0.34 |
| 11 | ORF1a‐3 | SigmAldr ORF1ab3 | 16 | 91.5 | 6.01 | 1.2 |
| 11 | ORF1a‐4 | SigmAldr ORF1ab4 | 17 | 91.32 | 6.01 | 1.2 |
| 11 | S‐5 | SigmAldr S5 | 15 | 93.73 | 3.6 | 0.69 |
| 11 | S‐6 | SigmAldr S6 | 21 | 92.61 | 3.6 | 0.69 |
| 12 | E | Huang E | 4 | 99.74 | 0.09 | 0.09 |
Table 4.
Amplicon clusters among the SARS‐CoV‐2 Omicron BA.5 sublineage sequences amplified by the evaluated primers and probes and percentage of genomes within largest three clusters.
| Assay | Target gene | Assay name | No. of clusters | Cluster I (%) | Cluster II (%) | Cluster III (%) |
|---|---|---|---|---|---|---|
| 1 | N | ChinaCDC N | 17 | 96.21 | 2.49 | 0.31 |
| 1 | ORF1ab | ChinaCDC ORF1ab | 9 | 98.7 | 0.88 | 0.1 |
| 2 | RdRp P1 | CoremChariteRdRp P1 | 7 | 96.94 | 2.23 | 0.57 |
| 2 | RdRp P2 | CoremChariteRdRp P2 | 7 | 96.94 | 2.23 | 0.57 |
| 2 | E | CoremCharite E | 4 | 99.37 | 0.67 | 0.05 |
| 2 | N | CoremCharite N | 25 | 97.46 | 0.36 | 0.31 |
| 3 | RdRp IP2 | Pasteur RdRp IP2 | 15 | 98.29 | 0.36 | 0.31 |
| 3 | RdRp IP4 | Pasteur RdRp IP4 | 8 | 99.53 | 0.1 | 0.1 |
| 3 | E | Pasteur E | 4 | 99.22 | 0.67 | 0.05 |
| 4 | N‐1 | USCDC N1 | 20 | 75.03 | 23.68 | 0.21 |
| 4 | N‐2 | USCDC N2 | 6 | 99.69 | 0.1 | 0.05 |
| 4 | N‐3 | USCDC N3 | 18 | 89.87 | 7.42 | 1.25 |
| 5 | N | Japan N | 12 | 99.12 | 0.21 | 0.16 |
| 6 | ORF1b/NSP14 | HKFacMed ORF1b/nsp14 | 13 | 98.6 | 0.31 | 0.26 |
| 6 | N | HKFacMed N | 9 | 99.38 | 0.21 | 0.1 |
| 7 | N | Thai N | 19 | 75.03 | 22.43 | 1.04 |
| 8 | RdRp/Hel | HKUnivRdRp/Hel | 11 | 98.8 | 0.31 | 0.26 |
| 8 | S | HKUniv S | 13 | 90.7 | 5.3 | 2.13 |
| 8 | N | HKUniv N | 9 | 99.06 | 0.26 | 0.2 |
| 9 | ORF1a | RoujianLu ORF1a | 10 | 99.22 | 0.26 | 0.16 |
| 10 | RdRp | Won RdRP | 8 | 96.88 | 2.23 | 0.57 |
| 10 | S | Won S | 7 | 99.64 | 0.1 | 0.05 |
| 10 | E | Won E | 4 | 99.22 | 0.67 | 0.05 |
| 10 | N | Won N | 18 | 98.23 | 0.31 | 0.26 |
| 11 | N‐1 | SigmAldr N1 | 24 | 95.53 | 2.5 | 0.21 |
| 11 | N‐2 | SigmAldr N2 | 25 | 95.48 | 2.5 | 0.21 |
| 11 | ORF1a‐3 | SigmAldr ORF1ab3 | 20 | 93.25 | 3.27 | 2.1 |
| 11 | ORF1a‐4 | SigmAldr ORF1ab4 | 22 | 93.15 | 3.27 | 2.1 |
| 11 | S‐5 | SigmAldr S5 | 15 | 97.3 | 1.5 | 0.36 |
| 11 | S‐6 | SigmAldr S6 | 24 | 96.31 | 1.5 | 0.36 |
| 12 | E | Huang E | 4 | 99.22 | 0.67 | 0.05 |
3.3. Primers specific for BA.4 and BA.5 sublineage
Table 5 represents the seven designed primers specific for BA.4 and BA.5 sublineage, two primers specific for BA.4, and one primer specific for BA.5. These primers target ORF1ab, S, E, M, N, ORF6, ORF7a, and ORF8 regions of the SARS‐CoV‐2 genome. These primers have high specificity, low self‐complementarity, and optimum GC content and Tm required for annealing of primers with the genome in vitro. Figure 2 represents the alignment of the VoCs with primer pair 4, showing the specificity towards BA.4 and BA.5 sublineage.
Table 5.
Designed primers specific to BA.4 and BA.5 sublineage genomes.
| Primer | Target | Length | Start | Stop | Tm | GC% | Self complementarity | Self 3′ complementarity | Specificity | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | fw | TACCTACTAAAAAGGCT A GT | ORF1ab | 20 | 4202 | 4222 | 50.13 | 35 | 4 | 2 | BA.4 & BA.5 |
| Rv | TAAAAAGTGTAAAAGTGC T T | 20 | 4339 | 4359 | 48.89 | 25 | 4 | 2 | |||
| 2 | fw | CCAGGAGTTTTCTGTGGTGTA | ORF1ab | 21 | 9345 | 9365 | 57.51 | 47.62 | 3 | 2 | BA.4 & BA.5 |
| rv | TAGCTGGTGGTATTGT G GCT | 20 | 9444 | 9464 | 58.71 | 50 | 4 | 2 | |||
| 3 | fw | GATCTGCACCTCTGAAGA T A | ORF1ab | 20 | 10216 | 10236 | 53.97 | 45 | 6 | 4 | BA.4 & BA.5 |
| rv | ACCAATGTGCTATGAG A C A C | 20 | 10467 | 10487 | 55.45 | 45 | 4 | 2 | |||
| 4 | fw | TAATT T CGCA C CATTTT T C G | S | 20 | 22712 | 22732 | 53.17 | 35 | 4 | 2 | BA.4 & BA.5 |
| rv | TAGAGGT A ATGAAGTCAG C C | 20 | 22811 | 22831 | 53.87 | 45 | 4 | 2 | |||
| 5 | fw | TTCAAGATGTGGTCAACCA T | S | 20 | 24427 | 24446 | 54.84 | 40 | 6 | 4 | BA.4 & BA.5 |
| rv | TCAATTTGCACTTCAGCCTC | 20 | 24565 | 24546 | 56.62 | 45 | 5 | 1 | |||
| 6 | fw | ATTCACACAATCGACGGTTC | E & M | 20 | 26180 | 26200 | 56.47 | 45 | 5 | 3 | BA.4 & BA.5 |
| rv | TTCGTTTCGGAAGAGA T AGG | 20 | 26305 | 26325 | 54.63 | 45 | 5 | 3 | |||
| 7 | fw | TAACCAGAACTCAAT C ACCC | S | 20 | 21637 | 21656 | 54.66 | 45 | 2 | 0 | BA.4 & BA.5 |
| rv | AGGTAAGAACAAGTCCTGAGT | 21 | 21752 | 21732 | 55.93 | 42.86 | 4 | 2 | |||
| 8 | fw | GGAATCTTGATTACATCATAAACCT | ORF6 & 7a | 20 | 27333 | 27353 | 55.19 | 32 | 6 | 2 | BA.4 |
| rv | GCAACCAATGGAGATT CTC T | 20 | 27417 | 27437 | 55.41 | 45 | 7 | 2 | |||
| 9 | fw | CACATTGGCACCCGCAAT T C | N | 20 | 28732 | 28751 | 60.74 | 55 | 4 | 2 | BA.4 |
| rv | TGCGACTACGTGATGAGGAA | 20 | 28873 | 28854 | 58.83 | 50 | 4 | 2 | |||
| 10 | fw | AAACTTGTCACGCCTAAA T G | ORF8 | 20 | 27905 | 27924 | 54.52 | 40 | 3 | 0 | BA.5 |
| rv | ACACGGGTCATCAACTACATA | 21 | 28038 | 28018 | 56.47 | 42.86 | 4 | 2 | |||
Note: The mismatches from other VoCs are shown in bold and underline. The forward (Fw) and reverse (Rv) primers sequences are directed from 5′–3′.
Figure 2.

Alignment of VoCs with primer pair 4 specific to BA.4 and BA.5 sublineage. VoC, variant of concern.
4. DISCUSSION
With an increasing number of mutations in SARS‐CoV‐2 emerging VoCs, as in B.1.1.529 (also commonly known as “Omicron”), the evaluation of current RT‐qPCR assays used for detecting SARS‐CoV‐2 is important for accurate diagnosis of acute viral infection. Evaluation of these assays in the wet laboratory is limited in this rapidly evolving SARS‐CoV‐2 Omicron VoC outbreak, because of the time constraint and lack of representative strains available for clinical laboratories, as previously noted. 25 Therefore, an in silico approach was used to evaluate the sensitivity of current RT‐qPCR assays (Table 6) using the whole genome sequencing data of SARS‐CoV‐2 Omicron VoCs BA.4 and BA.5 (particularly from the publicly available GISAID database), and employing suitable bioinformatics tools. 7 , 25 Using our in silico approach for primer design, we take into account many more sequences than traditionally done, thus enabling the design of highly accurate and flexible primers that are then tested against many sequences (unlike traditional primer design). This approach could help researchers design primers in a quick and rapid manner. From the NIHRIO COVID‐10 directory, 26 which compiles the diagnostic landscape of SARS‐CoV‐2 globally, the majority of the nucleic acid tests (NAT) that are capable of recognizing the Omicron variant use the N1, N2, and ORF1ab genes to detect the presence of the virus. Since our study demonstrated that poor sensitivity is independent of the target gene amplified (e.g., both ChinaCDC and Hong Kong University N assays amplified the N gene, but ChinaCDC N assay had poor sensitivity), it may be difficult to track each assay used in the market. This is because product information documents provided by different companies for their respective regulatory agencies do not usually include the protocol from the country or company of origin from which the target gene was determined. Hence, we recommend organizations such as the WHO issue statements to biotechnology companies manufacturing COVID‐19 NAT test kits that would ensure the accuracy and validity of their target gene protocol.
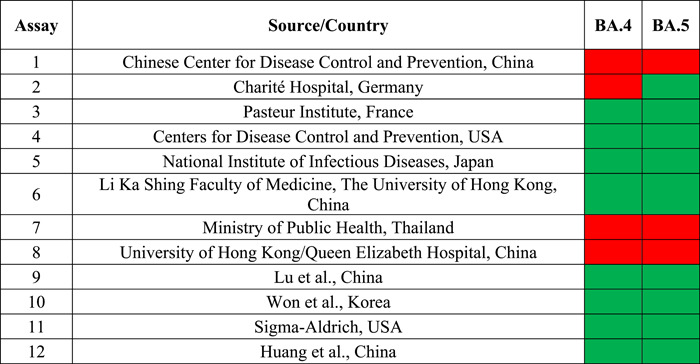
Table 6.
Assays with potentially false negative results based on in silico analysis
|
|
Note: Red indicates risk of false negatives (low sensitivity, <99% for all primers and probes in assay). Green indicates low risk of false negative (high sensitivity, ≥99% for all primers/probes in assay).
One of the limitations of the study is the lack of experimental validation. While there are disparities between in silico and in vitro experiments, experimental validation would be time‐demanding in such a manner that by the date this would be accomplished, the virus would have indeed mutated again and the work irrelevant. Hence, to screen and identify potential issues with the diagnostic performance of assays in an extremely rapid manner following the emergence of new variants, in silico analysis was performed. As such, laboratories around the world would be aware of potential issues and be enabled with the data provided in our article to explore further and design appropriate wet lab experiments.
5. CONCLUSION
In our in silico analysis evaluating 12 RT‐qPCR assays with a total of 30 primers and probes, for SARS‐CoV‐2 Omicron BA.4 and BA.5 sublineage, four assays (HKUnivRdRp/Hel, ChinaCDC N, Thai N, CoremCharite N) demonstrated potential for false negatives. Many mutations in the SARS‐CoV‐2 Omicron VoC genomes led to the low sensitivity of the included RT‐qPCR assays because the new mutated sequences do not anneal with the retained sequences of the primers for the given tests. Apart from the sensitivity analysis, the amplicon sequence clustering and design of specific primers revealed the potential new primer and probe sequences that could be used for the development of RT‐qPCR tests for detecting the SARS‐CoV‐2 Omicron VoC sublineages. As the number of SARS‐CoV‐2 Omicron VoC sequences is increasing rapidly, our analysis on a larger data set could reveal more mutations and amplicon clusters, thus providing more insights into the specificity of RT‐qPCR assays. The effect of mismatches in primers and probe sets revealed in this study on the sensitivity of RT‐qPCR assays could be further investigated in the wet laboratory for the preparation of more specific diagnostics for SARS‐CoV‐2 Omicron VoC detection. Lastly, given the number of unresolved potential issues with COVID‐19 diagnosis testing with respect to Omicron VoC, symptomatic patients, vulnerable patients, or those with high‐risk contact with infected patients, but testing negative, should be confirmed as being “true” negative by using a second assay (i.e., alternative RT‐qPCR or SARS‐CoV‐2 antigenic assays).
AUTHOR CONTRIBUTIONS
All the authors cited in the manuscript had substantial contributions to the concept and design, the execution of the work, or the analysis and interpretation of data; drafting or revising the manuscript, and have read and approved the final version of the paper. Divya Sharma: Conceptualization, Visualization, Methodology, Validation, Formal analysis, Data curation, Writing – original draft, Writing – review and editing. Kin I. Notarte: Conceptualization, Formal analysis, Writing – original draft, Writing – review and editing. Rey A. Fernandez: Data curation, Writing – original draft, Writing – review and editing. Michael Gromiha: Conceptualization, Visualization, Validation, Formal analysis, Writing – review and editing. Brandon M. Henry: Conceptualization, Visualization, Validation, Formal analysis, Writing – review and editing.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ETHICS STATEMENT
Not required.
ACKNOWLEDGMENTS
We thank the Department of Biotechnology and Indian Institute of Technology Madras for computational facilities and PMRF, Ministry of Education (MHRD) to Divya Sharma.
Sharma D, Notarte KI, Fernandez RA, Lippi G, Gromiha MM, Henry BM. In silico evaluation of the impact of Omicron variant of concern sublineage BA.4 and BA.5 on the sensitivity of RT‐qPCR assays for SARS‐CoV‐2 detection using whole genome sequencing. J Med Virol. 2022;95:e28241. 10.1002/jmv.28241
Contributor Information
M. Michael Gromiha, Email: gromiha@iitm.ac.in.
Brandon M. Henry, Email: Brandon.Henry@cchmc.org.
DATA AVAILABILITY STATEMENT
The supporting data are available within the article.
REFERENCES
- 1. Lippi G, Mattiuzzi C, Henry BM. Updated picture of SARS‐CoV‐2 variants and mutations. Diagnosis (Berl). 2021;9:11‐17. 10.1515/dx-2021-0149 [DOI] [PubMed] [Google Scholar]
- 2.Classification of Omicron (B.1.1.529): SARS‐CoV‐2 Variant of Concern. Accessed December 30, 2021. https://www.who.int/news/item/26-11-2021-classification-of-omicron-(b.1.1.529)-sars-cov-2-variant-of-concern
- 3. Wu F, Zhao S, Yu B, et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579(7798):265‐269. 10.1038/s41586-020-2008-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. World Health Organization . Molecular assays to diagnose COVID‐19. Accessed December 10, 2021. https://www.who.int/docs/default-source/coronaviruse/whoinhouseassays.pdf
- 5. Bohn MK, Mancini N, Loh TP, et al. IFCC interim guidelines on molecular testing of SARS‐CoV‐2 infection. Clin Chem Lab Med. 2020;58(12):1993‐2000. 10.1515/cclm-2020-1412 [DOI] [PubMed] [Google Scholar]
- 6. Nayar G, Seabolt EE, Kunitomi M, et al. Analysis and forecasting of global real time RT‐PCR primers and probes for SARS‐CoV‐2. Sci Rep. 2021;11(1):8988. 10.1038/s41598-021-88532-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gand M, Vanneste K, Thomas I, et al. Deepening of in silico evaluation of SARS‐CoV‐2 detection RT‐qPCR assays in the context of new variants. Genes. 2021;12(4):565. 10.3390/genes12040565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lippi G, Simundic AM, Plebani M. Potential preanalytical and analytical vulnerabilities in the laboratory diagnosis of coronavirus disease 2019 (COVID‐19). Clin Chem Lab Med. 2020;58(7):1070‐1076. 10.1515/cclm-2020-0285 [DOI] [PubMed] [Google Scholar]
- 9. Kwok S, Kellogg DE, McKinney N, et al. Effects of primer‐template mismatches on the polymerase chain reaction: human immunodeficiency virus type 1 model studies. Nucleic Acids Res. 1990;18(4):999‐1005. 10.1093/nar/18.4.999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lefever S, Pattyn F, Hellemans J, Vandesompele J. Single‐nucleotide polymorphisms and other mismatches reduce performance of quantitative PCR assays. Clin Chem. 2013;59(10):1470‐1480. 10.1373/clinchem.2013.203653 [DOI] [PubMed] [Google Scholar]
- 11. Christopherson C. The effects of internal primer‐template mismatches on RT‐PCR: HIV‐1 model studies. Nucleic Acids Res. 1997;25(3):654‐658. 10.1093/nar/25.3.654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Whiley DM, Sloots TP. Sequence variation in primer targets affects the accuracy of viral quantitative PCR. J Clin Virol. 2005;34(2):104‐107. 10.1016/j.jcv.2005.02.010 [DOI] [PubMed] [Google Scholar]
- 13. Vanneste K, Garlant L, Broeders S, Van Gucht S, Roosens NH. Application of whole genome data for in silico evaluation of primers and probes routinely employed for the detection of viral species by RT‐qPCR using dengue virus as a case study. BMC Bioinformatics. 2018;19(1):312. 10.1186/s12859-018-2313-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. US Food and Drug Administration . SARS‐CoV‐2 viral mutations: impact on COVID‐19 tests. Accessed December 30, 2021. https://www.fda.gov/medical-devices/coronavirus-covid-19-and-medical-devices/sars-cov-2-viral-mutations-impact-covid-19-tests
- 15. Gand M, Vanneste K, Thomas I, et al. Use of whole genome sequencing data for a first in silico specificity evaluation of the RT‐qPCR assays used for SARS‐CoV‐2 detection. Int J Mol Sci. 2020;21(15):E5585. 10.3390/ijms21155585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Khan KA, Cheung P. Presence of mismatches between diagnostic PCR assays and coronavirus SARS‐CoV‐2 genome. Royal Soc Open Science. 2020;7(6):200636. 10.1098/rsos.200636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kabinger F, Stiller C, Schmitzová J, et al. Mechanism of molnupiravir‐induced SARS‐CoV‐2 mutagenesis. Nat Struct Mol Biol. 2021;28(9):740‐746. 10.1038/s41594-021-00651-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yin W, Mao C, Luan X, et al. Structural basis for inhibition of the RNA‐dependent RNA polymerase from SARS‐CoV‐2 by remdesivir. Science. 2020;368(6498):1499‐1504. 10.1126/science.abc1560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pango designation: Proposal to split B.1.1.529 to incorporate a newly characterised sibling lineage 361. Accessed December 23, 2021. https://github.com/cov-lineages/pango-designation/issues/361
- 20. World Health Organization . Technical guideline for the detection of SARS‐CoV‐2. Accessed December 30, 2021. https://www.who.int/docs/default-source/coronaviruse/whoinhouseassays.pdf
- 21. American Association for Clinical Chemistry . Coronavirus testing survey. Accessed December 30, 2021. https://www.aacc.org/science-and-research/covid-19-resources/aacc-covid-19-testing-survey
- 22. Katoh K, Rozewicki J, Yamada KD. MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinformatics. 2019;20(4):1160‐1166. 10.1093/bib/bbx108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Waterhouse AM, Procter JB, Martin D.M.A., Clamp M, Barton GJ. Jalview version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25(9):1189‐1191. 10.1093/bioinformatics/btp033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ye J, Coulouris G, Zaretskaya I, Cutcutache I, Rozen S, Madden TL. Primer‐BLAST: a tool to design target‐specific primers for polymerase chain reaction. BMC Bioinformatics. 2012;13:134. 10.1186/1471-2105-13-134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. US Food and Drug Administration . Guidelines for the validation of analytical methods for the detection of microbial pathogens in foods and feeds. Published online 2015. Accessed December 21, 2021. https://www.fda.gov/media/83812/download
- 26. Oyewole AO, Barrass L, Robertson EG, et al. COVID‐19 impact on diagnostic innovations: emerging trends and implications. Diagnostics. 2021;11(2):182. 10.3390/diagnostics11020182 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The supporting data are available within the article.