

Abstract
Aim
To assess viral clearance, pharmacokinetics, tolerability and symptom evolution following ensovibep administration in symptomatic COVID‐19 outpatients.
Methods
In this open‐label, first‐in‐patient study a single dose of either 225 mg (n = 6) or 600 mg (n = 6) of ensovibep was administered intravenously in outpatients with mild‐to‐moderate COVID‐19 symptoms. Pharmacokinetic profiles were determined (90‐day period). Pharmacodynamic assessments consisted of viral load (qPCR and cultures) and symptom questionnaires. Immunogenicity against ensovibep and SARS‐CoV‐2‐neutralizing activity were determined. Safety and tolerability were assessed throughout a 13‐week follow‐up.
Results
Both doses showed similar pharmacokinetics (first‐order) with mean half‐lives of 14 (SD 5.0) and 13 days (SD 5.7) for the 225‐ and 600‐mg groups, respectively. Pharmacologically relevant serum concentrations were maintained in all subjects for at least 2 weeks postdose, regardless of possible immunogenicity against ensovibep. Viral load changes from baseline at day 15 were 5.1 (SD 0.86) and 5.3 (SD 2.2) log10 copies/mL for the 225‐ and 600‐mg doses, respectively. COVID‐19 symptom scores decreased from 10.0 (SD 4.1) and 11.3 (SD 4.0) to 1.6 (SD 3.1) and 3.3 (SD 2.4) in the first week for the 225‐ and 600‐mg groups, respectively. No anti‐SARS‐CoV‐2 neutralizing activity was present predose and all patients had SARS‐CoV‐2 antibodies at day 91. Adverse events were of mild‐to‐moderate severity, transient and self‐limiting.
Conclusion
Single‐dose intravenous administration of 225 or 600 mg of ensovibep appeared safe and well tolerated in patients with mild‐to‐moderate COVID‐19. Ensovibep showed favourable pharmacokinetics in patients and the pharmacodynamic results warrant further research in a larger phase 2/3 randomized‐controlled trail.
Keywords: COVID‐19, medication safety, pharmacodynamics, pharmacokinetics, virology
What is already known about this subject
SARS‐CoV‐2 is continuously mutating, with new variants emerging and there remains a population at risk of developing severe COVID‐19. Novel compounds are needed to expand and diversify the therapeutic arsenal for COVID‐19.
Ensovibep acts by blocking SARS‐CoV‐2‐host interaction.
Ensovibep was safe and well tolerated in healthy volunteers.
What this study adds
This study is the first to describe ensovibep pharmacokinetics, viral load dynamics and symptom reduction in COVID‐19 patients.
There was an overall decline in SARS‐CoV‐2 viral load and symptoms following ensovibep administration.
Ensovibep showed a favourable safety and pharmacokinetic profile, warranting further research in a randomized‐controlled setting.
1. INTRODUCTION
Severe acute respiratory syndrome coronavirus‐2 (SARS‐CoV‐2) has affected over half a billion people since it was first identified in December 2019. 1 Therapies targeting the receptor binding domain (RBD) of the SARS‐CoV‐2 spike glycoprotein (S)—thereby preventing virus‐host interaction via the ACE‐2 receptor—have proven successful in a clinical setting. Several monoclonal antibodies (mAbs) targeting this RBD have been shown to reduce hospitalization and death in high‐risk COVID‐19 patients with mild‐to‐moderate disease, 2 , 3 , 4 confirming the clinical benefits of early initiation of virus blocking therapy. As an RNA virus that is transmitted by millions of people worldwide, new SARS‐CoV‐2 variants will likely continue to emerge. 5 Virus susceptibility to vaccine‐induced antibodies and mAbs may be (partially) reduced in new variants. 6 It is therefore of the utmost importance to bolster the arsenal of therapeutic viral blocking agents.
Ensovibep is a recombinant multispecific designed ankyrin‐repeat protein (DARPin) molecule that was engineered to neutralize SARS‐CoV‐2 with high potency. While the mechanism of action of ensovibep—neutralization of the S protein by binding to its RBD—is comparable to (monoclonal) antibodies, there are inherent differences in the binding pattern that differentiate ensovibep from currently available antibody therapies. Ensovibep is a single molecule consisting of five DARPin modules that are covalently linked. Three DARPins bind to an overlapping epitope of the RBD, but with different antigen‐binding sequences (paratopes). This allows for cooperative binding of the tri‐specific molecule with high avidity and could limit the development of mutations under therapeutic pressure from ensovibep. 7 In addition, ensovibep contains two human serum albumin binding domains to extend its systemic half‐life.
In vitro studies confirmed the high potency neutralization by ensovibep of all SARS‐CoV‐2 variants of concern described to date. 7 Moreover, ensovibep was able to neutralize an omicron pseudovirus variant with high potency, signifying that ensovibep's neutralizing potential endures for the currently prevalent highly mutated variant of concern. 7 Studies performed in a SARS‐CoV‐2 hamster infection model showed in vivo efficacy with a significant reduction in viral load and pathogenesis after administration of ensovibep compared to placebo. 8
Recently a phase 1, randomized, placebo‐controlled, single ascending intravenous (IV) dose study was completed in healthy volunteers and showed a favourable safety and pharmacokinetic (PK) profile (dose range 3‐20 mg/kg) (manuscript in preparation).
Ensovibep is anticipated to provide benefit to COVID‐19 patients at the early stages of infection, when virus replication should be halted to limit downstream immune‐related damage and improve clinical outcomes. The pharmacological properties of ensovibep in combination with its high yield production process using an Escherichia coli fermentation‐based process could provide a needed diversification of the current treatment arsenal against COVID‐19.
In this article we present the results of a phase 2a, first‐in‐patient, IV single‐dose escalation study that assessed the viral clearance, PKs, tolerability and evolution of COVID‐19 symptoms following ensovibep administration in early symptomatic COVID‐19 patients.
2. METHODS
2.1. Study design and patients
This was an open‐label, IV single‐dose escalation, phase 2a study conducted at the Leiden University Medical Center in nonhospitalized COVID‐19 patients. The protocol was reviewed and approved by the Medical Ethical Committee Leiden, Den Haag, Delft, the Netherlands (NL76642.058.21) and was registered at ClinicalTrials.gov (NCT04834856). The trial was conducted in accordance with the Declaration of Helsinki, Good Clinical Practice Guidelines and the principles of Dutch law on clinical experiments in humans. Written informed consent was obtained from patients prior to study‐related activities. The Dutch Municipal Healthcare Services assisted in the recruitment of individuals with positive SARS‐CoV‐2 polymerase chain reaction (PCR) tests who expressed an interest in study participation. Male and female patients were eligible if they were 18‐70 years of age with symptomatic mild‐to‐moderate COVID‐19, defined as experiencing at least one mild‐to‐moderate symptom (fever, cough, sore throat, malaise, fatigue, headache, muscle pain, gastrointestinal symptoms or shortness of breath with exertion) and a positive SARS‐CoV‐2 rapid antigen test (Panbio COVID‐19 Ag Rapid Test; Abbott Diagnostics, Jena, Germany) on the day of ensovibep administration. The main exclusion criteria were a high risk for COVID‐19‐related complications or mortality, including immunodeficiency, need for hospitalization prior to screening or anti‐SARS‐CoV‐2 treatment initiation. The protocol did not allow a prior history of SARS‐CoV‐2 infection (or vaccination), concurrent or previous use of antiviral medication (including antibodies) or convalescent plasma therapy. Vaccination was not allowed during the study until day 29.
2.2. Procedures
Patients received a single dose of either 225 mg (cohort 1) or 600 mg (cohort 2) of ensovibep administered as a 250 mL IV infusion over 60 minutes. Clinical dose and regimen projections for ensovibep were based on an integrated analysis of preclinical pharmacology results, available clinical safety, tolerability and PK results from the phase 1 dose‐escalation first‐in‐human study (NCT04870164) and PK/pharmacodynamic modelling. 9 The current study had a dose‐escalation design, meaning that the 600 mg dose was administered after the data review committee assessed the day 15 safety and tolerability data of the 225‐mg dose. Patients remained in the clinical unit for 2 hours after ensovibep administration to monitor any direct untoward effects. Nasopharyngeal swabs (viral load), blood samples (PK, immunogenicity, blood chemistry and haematology) and questionnaires (14 Common COVID‐19‐Related Symptoms and Long‐COVID Syndrome questionnaires) were obtained prior to ensovibep administration and on selected time points postdose (Figure 1).
FIGURE 1.
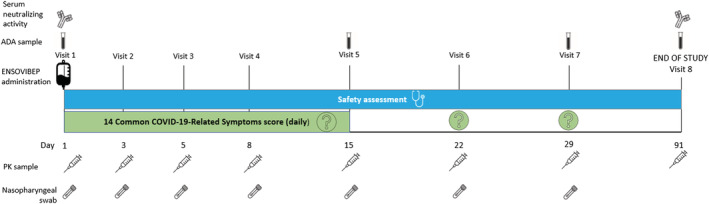
Schedule of assessment. ADA, antidrug antibodies
A validated electrochemiluminescence assay (Molecular Partners AG, Zürich Switzerland), which uses the RBD of SARS‐CoV‐2 spike glycoprotein as a capture reagent, was used to quantify free ensovibep levels in serum. The limit of quantification (LLOQ) of this assay was 0.02 μg/mL. PK profiles of ensovibep were determined for both dose levels. Descriptive PK parameters included the maximum concentration (C max), time to reach maximum concentration (T max), area under the drug serum concentration‐time curve (AUC), half‐life (t 1/2), volume of distribution (V D) and clearance (CL). As an exploratory assessment outcome, antidrug antibodies (ADAs) in human serum were measured using an electrochemiluminescence assay with acid dissociation sample pretreatment followed by neutralization (Molecular Partners AG, Zürich Switzerland). Antibodies were specifically captured via biotinylated ensovibep and detected with antihuman immunoglobuline G (IgG)/ immunoglobuline M (IgM) SulfoTag detection antibodies. The assay was validated according to the US Food and Drug Administration Guide for Industry: Immunogenicity Testing of Therapeutic Protein Products – Developing and Validating assays for Antidrug Antibody Detection. 10
Viral load was assessed by quantitative real‐time PCR (qPCR) and quantitative virus cultures at Viroclinics Biosciences BV (Rotterdam, the Netherlands). SARS‐CoV‐2 qPCR analysis was performed according to a fully validated proprietary assay that is based on the US Centers for Disease Control and Prevention N1 assay, 11 but with different dyes‐quenchers and a PCR program internally optimized by Viroclinics Biosciences BV.
Determination of infectious SARS‐CoV‐2 virus titres was performed according to a validated proprietary assay (Viroclinics Biosciences BV, Rotterdam, the Ne). Briefly, in this assay VeroE6 cells were grown to subconfluent density, after which a serial dilution of an upper respiratory sample in infection medium was added to the cells in quadruplicate and incubated for 6 days. Cells were then fixed using a formalin solution and the presence of viral plaques was detected following immunostaining with an antinucleoprotein antibody, a peroxidase conjugate and TrueBlue staining. Virus titres were calculated as median Tissue Culture Infectious Dose 50% (TCID50)/mL using the Spearman & Kärber method. 12 , 13
Whole‐virus next‐generation sequencing (NGS) for SARS‐CoV‐2 was performed from a separate aliquot of the same nasopharyngeal swab used for viral load assessment by qPCR. NGS analysis was performed at baseline and at the last positive qPCR time point above the cut‐off value of ≥4.0 log10 copies/mL, which was defined by the assay's capacity for successful amplification.
Serum anti‐SARS‐CoV‐2 neutralizing activity and anti‐SARS‐CoV‐2 IgG antibody levels were determined prior to dosing and at the final follow‐up visit to assess the endogenous neutralizing immune response to SARS‐CoV‐2. In the virus neutralization activity assay, performed according to validated proprietary assay (Viroclinics Biosciences BV, Rotterdam, the Netherlands), a serial dilution in triplicate of a serum sample in infection medium was mixed with a fixed amount of Isolate Germany/BavPat1/2020 and incubated for 1 hour. The mixture was added to VeroE6 cells at subconfluent density and incubated for 1 hour, after which the inoculum was removed and replaced by infection medium. Cells were incubated for 16‐24 hours, then fixed using a formalin solution, and the presence of viral plaques was detected following immunostaining with an antinucleoprotein antibody, a peroxidase conjugate and TrueBlue staining. Microplaques were imaged and counted in a SX Ultimate‐V Analyzer (Cellular Technology Limited, Shaker Heights, USA). The neutralization titres were calculated from these data according to the method described by Zielinska et al. 14 Quantification of SARS‐CoV‐2 IgG antibodies was performed with a multiplex serology Meso Scale Discovery (MSD, Rockville, USA) assay (V‐PLEX SARS‐CoV‐2 Panel 2, IgG kit, K15383U). Serum samples were added in duplicates in 96‐wells assay plates coated with specific antigens. Following binding of serum antibodies to the respective antigens, antihuman IgG antibodies conjugated to MSD SULFO‐TAG were used for subsequent detection. The emitted light was measured with an MSD instrument (Meso SECTOR 600 device). Anti‐SARS‐CoV‐2 antibody levels were reported as international standard units, Binding Antibody Units (BAU)/mL.
Serum cytokines concentrations (interferon gamma [IFN‐γ], interleukins [ILs]: IL‐1β, IL‐6, IL‐8, IL‐10, and tumor necrosis facter [TNF‐α]) were determined using a multiplex electrochemiluminescent sandwich immunoassay from MSD validated in human serum by BioAgilytix Labs (Durham, NC, USA). Additional information on the bio‐analytical assays used in this trial is provided in the Supporting Information .
Safety was assessed at each follow‐up visit by assessment of treatment emergent adverse events (TEAEs), vital signs, physical examination, routine blood chemistry and haematology testing. Local tolerability at the infusion site was determined by the Visual Infusion Phlebitis (VIP) scale. 15 Patients were monitored throughout the study for adverse events of special interest, defined as infusion‐related reactions, hypersensitivity reactions and cytokine release syndrome, and serious adverse events (SAEs). Adverse events were coded according to the Medical Dictionary for Regulatory Activities (MedDRA), version 24.0.
Symptoms related to COVID‐19 were assessed daily (predose until day 15) and on days 22 and 29 using the 14 Common COVID‐19‐Related Symptoms questionnaire. 16 Symptoms were rated on either a three‐ or four‐point ordinal scale and a total symptom score was calculated as the sum individual symptoms (range 0‐40). Assessment of long‐term COVID‐19 symptoms was performed using an experimental Long‐COVID Syndrome questionnaire on days 29 and 91 ( Supporting Information ).
2.3. Statistical analysis
As this was an exploratory study, no formal power calculation was performed. Instead, a conventional (for early‐phase studies) group size per dose level was used. The study protocol included prespecified criteria to expand the cohort size to a maximum of 20 patients per dose level, in case of high interindividual PK variability or signification deviation from expected viral clearance. No formal hypothesis tests were planned nor performed. Data were summarized using descriptive statistics and graphically presented using GraphPad Prism for Windows (version 6.05, San Diego, USA). Viral load measurements <LLOQ were considered negative for the analysis. The slope of decline of viral load was estimated by the mean difference of viral load between successive study days divided by the interval (days) between measurements. Repeated measures correlations were calculated for viral load (independent variable) and the COVID‐19‐related total symptom scores to preliminary assess the relation between virus shedding and symptomatology within subjects. Noncompartmental PK analysis was performed using R 3.6.1 for Windows or newer (R Foundation for Statistical Computing/R Development Core Team, Vienna, Austria, 2019) using all PK samples collected according to protocol (until day 91).
2.4. Nomenclature
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY, and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20. 17
3. RESULTS
3.1. Patient characteristics
Between April and June 2021, 12 COVID‐19 patients who met the inclusion criteria were enrolled and received either 225 mg (n = 6) or 600 mg (n = 6) of ensovibep (Figure 2). No patients were vaccinated against COVID‐19 at baseline. Three patients received their first dose of SARS‐CoV‐2 vaccine (BNT162b2, Pfizer‐BioNTech) approximately 43, 52 and 69 days, respectively, after ensovibep administration. Baseline characteristics are described in Table 1. Median time from onset of symptoms was 5 days in both groups (total range 2‐8 days). All patients were symptomatic on baseline and median COVID‐19‐related symptom scores were similar for both dose groups.
FIGURE 2.
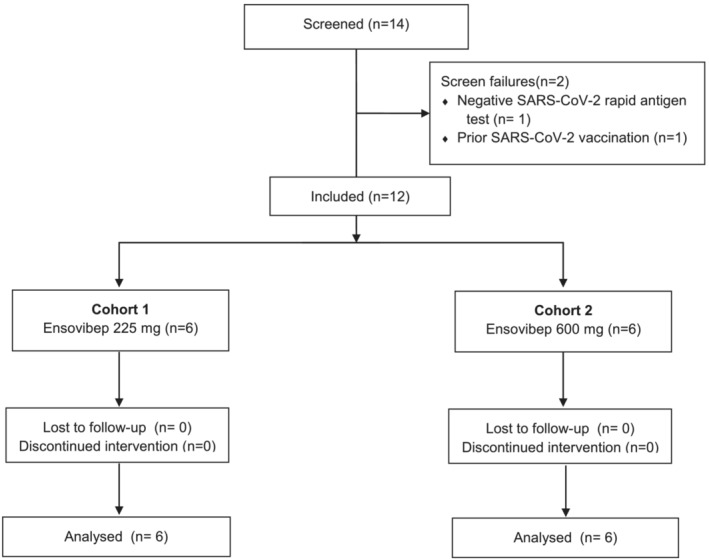
CONSORT diagram
TABLE 1.
Baseline characteristics
| Cohort 1 225 mg (n = 6) | Cohort 2 600 mg (n = 6) | |
|---|---|---|
| Age, years | 23 (21‐26) | 24 (22‐44) |
| Sex, n female (%) | 2 (33) | 2 (33) |
| Race or ethnicity a , n (%) | ||
| Mixed | 0 (0) | 1 (17) |
| White | 6 (100) | 5 (83) |
| BMI | 26 (24‐30) | 25 (22‐30) |
| Days between symptom onset and dosing | 5 (2‐8) | 5 (3‐5) |
| Positive viral culture, n (%) | 4 (67) | 0 |
| Positive qPCR result, n (%) | 6 (100) | 6 (100) |
| Viral load by qPCR b , mean (SD) | 7.3 (1.0) | 6.6 (1.6) |
| COVID‐19‐related symptom score c | 10.5 (4‐15) | 11.0 (7‐18) |
Note: Data are presented as median (range) unless indicated otherwise.
Abbreviations: BMI, body mass index; qPCR, quantitative polymerase chain reaction, SD, standard deviation.
Self‐reported race or ethnicity of patients, who could choose from multiple categories.
Viral load expressed as log10 copies/mL.
Possible range of aggregated COVID‐19‐related symptom score: 0‐40.
3.2. Viral clearance
All patients had a quantifiable SARS‐CoV‐2 viral RNA load in upper respiratory tract samples determined by qPCR at baseline, collected before the administration of ensovibep (Table 1). The alpha virus variant (B.1.1.7) was detected in all patients. The mean viral load determined by qPCR was 7.3 ± 1.0 log10 copies/mL in the 225‐mg group and 6.6 ± 1.6 log10 copies/mL in the 600‐mg group at baseline. Viral RNA in nasopharyngeal swabs decreased rapidly in both dose groups during the first 2 weeks with mean changes from baseline of 5.1 and 5.3 log10 copies/mL for the 225‐ and 600‐mg doses, respectively (Figure 3). The rate of viral load decline was highest in the first week (Supporting Information Table S1), with estimated daily decreases of 0.72 and 0.67 log10 copies/mL (days 1‐3), 0.41 and 0.71 log10 copies/mL (days 3‐5) and 0.45 and 0.39 log10 copies/mL (days 5‐8) for the 225‐ and 600‐mg dose groups, respectively. The results for time to PCR negativity are summarized in Supporting Information Figure S1. No virus mutations that could potentially trigger resistance to ensovibep were identified in postdose nasopharyngeal samples. Viral load in saliva samples was lower compared to nasopharyngeal samples but showed a similar reduction over time (data not shown). Three patients in the 225‐mg group had positive viral culture results at baseline and one patient had a baseline titre equal to the LLOQ (0.75 log10 TCID50/mL) of the assay. By day 5, all viral cultures were negative. Viral cultures were negative for all analysed samples (baseline and follow‐up) in the 600‐mg group.
FIGURE 3.
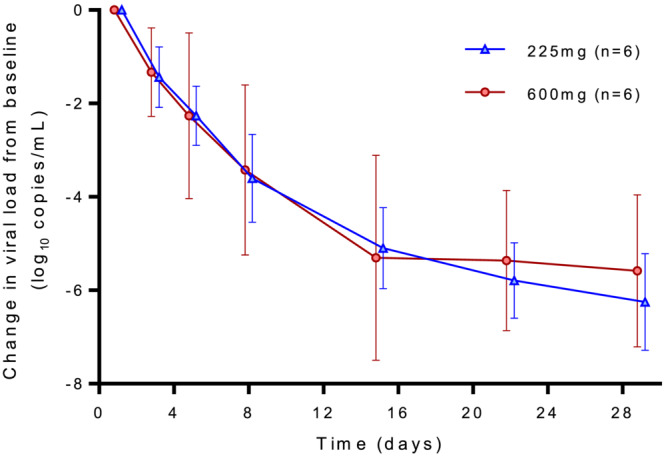
Mean change from baseline in SARS‐CoV‐2 viral load determined by qPCR on upper respiratory tract samples
3.3. Pharmacokinetics
The PK parameters are summarized in Table 2. Mean concentration‐time profiles showed log‐linear monophasic elimination of ensovibep (Figure 4). The volume of distribution (VD) was 2844 mL (SD 34.3 mL) in the 225‐mg group and 2735 mL (SD 37.2 mL) in the 600‐mg group. Drug elimination rates were similar for both doses, with mean t 1/2 of approximately 14 days (SD 5.0 days) and 13 days (SD 5.7 days) for the 225‐ and 600‐mg dose groups, respectively. Dose escalation to 600 mg resulted in a proportional increase of C max and AUC compared to 225 mg. Two individual patients (one in each cohort) showed an accelerated elimination of ensovibep at day 22 (Supporting Information Figure S2).
TABLE 2.
Summary of pharmacokinetic parameters
| Parameter | Ensovibep 225 mg | Ensovibep 600 mg | ||||
|---|---|---|---|---|---|---|
| n | Mean a | CV (%) or SD a | n | Mean a | CV (%) or SD a | |
| AUCinf (h * μg/mL) | 5 b | 37 170 | 18.1 | 6 | 100 068 | 37.9 |
| V D (mL) | 5 b | 2844 | 34.3 | 6 | 2735 | 37.2 |
| CL (mL/h) | 5 b | 6.13 | 1.02 | 6 | 6.39 | 2.81 |
| C max (μg/mL) | 6 | 88.8 | 20.3 | 6 | 233 | 19.3 |
| t 1/2 (h) | 5 b | 326 | 119 | 6 | 303 | 136 |
| T max (h) | 6 | 1.42 | 1.40‐2.70 | 6 | 2.04 | 1.37‐2.68 |
Abbreviations: CV, coefficient of variation; AUCinf, area under the concentration‐time curve from time 0 to infinity; CL, clearance; C max, maximum concentration; t ½, half‐life; T max, time to maximum concentration following start of infusion; V D, apparent volume of distribution.
C max and AUCinf are reported as geometric mean and coefficient of variation (%); CL, V D and t 1/2 are reported as arithmetic mean and standard deviation (SD); T max is reported as median (minimum − maximum).
t 1/2 could not be accurately estimated in one patient. PK parameters related to t 1/2 estimation (including V D, AUCinf and CL) are not reported for this patient.
FIGURE 4.
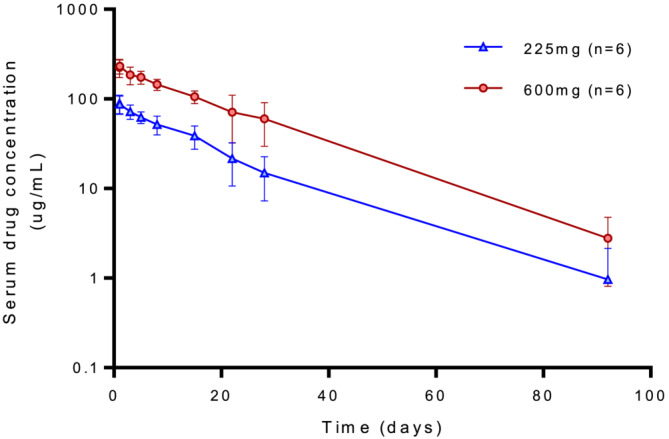
Semi‐log mean serum concentration versus time profiles of ensovibep following 225‐ and 600‐mg administration
ADAs at baseline were detected in one patient in the 225‐mg group. Treatment‐induced ADAs were detected in 5/6 (83%) patients in each dose group (time of onset, range 14‐91 days). For most patients who developed ADAs, the elimination of ensovibep remained unaffected. The two patients who showed increased elimination had a relatively early peak of ADA titres at day 29 compared to other patients.
3.4. Safety and tolerability
No SAEs, infusion site reactions, hypersensitivity, cytokine release syndrome or worsening of COVID‐19 (such as immune enhancement) were observed. At least one TEAE was reported by 4/6 (66%) patients in the 225‐mg group and 3/6 (50%) patients in the 600‐mg group (Table 3). All TEAEs were transient, resolved without intervention and were of mild‐to‐moderate severity. Out of 16 reported TEAEs, five were deemed related to the treatment and all five occurred in the 225‐mg group. These events consisted of diarrhoea (n = 2) and elevated liver tests (alanine aminotransferase [ALT], aspartate aminotransferase and bilirubin, n = 3). One of the two patients with transient liver enzyme increase had pre‐existing elevated ALT tests. Elevated liver tests were below two times the upper limit of normal.
TABLE 3.
Number of treatment emergent adverse events (number of patients, % of patients) classified by MedDRA System Organ Class and preferred term, and investigator‐assigned relationship to study drug
| Cohort 1, 225 mg (N = 6) | Cohort 2, 600 mg (N = 6) | |||
|---|---|---|---|---|
| Related to drug administration | Not related to drug administration | Related to drug administration | Not related to drug administration | |
| Ear and labyrinth disorder | … | … | … | 1 (1, 17%) |
| Ear pain | … | … | … | 1 (1, 17%) |
| Gastrointestinal disorders | 2 (2, 33%) | 1 (1, 17%) | … | … |
| Diarrhoea | 2 (2, 33%) | 1 (1, 17%) | … | … |
| General disorders and administration site conditions | … | 1 (1, 17%) | … | … |
| Alcoholic hangover | … | 1 (1, 17%) | … | … |
| Investigations | 3 (2, 33%) | … | … | … |
| Alanine aminotransferase increased | 1 (1, 17%) | … | … | … |
| Aspartate aminotransferase increased | 1 (1, 17%) | … | … | … |
| Blood bilirubin increased | 1 (1, 17%) | … | … | … |
| Metabolism and nutrition disorders | … | 1 (1, 17%) | … | … |
| Hypophosphatemia | … | 1 (1, 17%) | … | … |
| Musculoskeletal and connective tissue disorders | … | 2 (2, 33%) | … | … |
| Back pain | … | 1 (1, 17%) | … | … |
| Myalgia | … | 1 (1, 17%) | … | … |
| Nervous system disorders | … | … | … | 4 (3, 50%) |
| Headache | … | … | … | 4 (3, 50%) |
| Respiratory, thoracic, and mediastinal disorders | … | … | … | 1 (1, 17%) |
| Cough | … | … | … | 1 (1, 17%) |
3.5. Patient‐reported COVID‐19 symptoms
For both groups, an overall decrease in symptoms scores (range of total symptom score 0‐40) was observed. Total symptoms scores showed a relatively fast decline in the first week after ensovibep administration, from 10.0 and 11.3 (baseline) to 1.6 and 3.3 (day 8) in the 225‐ and 600‐mg dose groups, respectively (Supporting Information Figure S3). Mean total symptom scores were <1 in both treatment groups on day 29, indicating minimal symptomatology at the end of the observation period. Only tiredness (n = 2), myalgia (n = 1) and loss of smell/taste (n = 1) were reported by individual patients on day 29. Within patients, there was a linear correlation between viral load and total symptom score (r = 0.77, P < .0001).
Compared to their pre‐COVID status, the majority of patients reported either no change on all items or only mild worsening on one single item of the Long‐COVID Syndrome questionnaire (16 items in total). Mild fatigue compared to pre‐COVID status was reported most frequently (2/6 [33%] in the 225‐mg group and 3/6 [50%] in the 600‐mg group). On day 91, 1/6 (17%) patients in each dose group reported mild worsening of ≥2 items on the Long‐COVID Syndrome questionnaire. One patient reported an incidental severe change from the pre‐COVID status at day 91 for the domain chest pain/tightness. There were no clinical abnormalities that could explain the self‐reported complaints, but the complaints could be due to excessive exercise as the patient visited the gym frequently. The daily occupational activities of patients were not affected by long‐COVID symptoms.
3.6. Anti‐SARS‐CoV‐2 serum neutralizing activity and cytokine production
Patients had neither anti‐SARS‐CoV‐2 neutralizing activity nor anti‐SARS‐CoV‐2 antibodies at baseline in serum prior to ensovibep administration (data not shown). At day 91 (final follow‐up visit), virus neutralizing activity was detected in 3/6 (50%) patients in each dose group. All three patients who received COVID‐19 vaccinations had positive microneutralization titres at day 91. All patients had developed various levels of endogenous anti‐SARS‐CoV‐2 antibodies, with the highest values observed in vaccinated subjects. Decreases in serum levels of IFN‐γ, TNF‐α, IL‐8 and IL‐10 cytokine were observed during the study in most patients (data not shown). No apparent changes in IL‐6 and IL‐1β were observed.
4. DISCUSSION
In this exploratory phase 2a study, ensovibep was administered for the first time to nonhospitalized symptomatic COVID‐19 patients. The time of symptom onset and high viral load, in combination with absent SARS‐CoV‐2 neutralization activity at baseline, confirmed that enrolled patients were in their initial phase of infection. All patients showed a reduction in viral load after ensovibep administration. The change from baseline of viral load by qPCR was comparable for both the 225‐ and 600‐mg doses, suggesting no dose‐dependent difference on viral clearance in this study population. Jones et al proposed a model of natural SARS‐CoV‐2 infection with a linear increase and then decline of approximately 0.17 log10 units per day after a peak of viral load was reached (estimated on approximately day 4 after onset of shedding). 18 In our study, we did not observe an initial increase in viral load, suggesting that most of the subjects surpassed their initial peak viral load. This is also expected as the study enrolled symptomatic patients and peak viral load is expected to occur 1 to 3 days before symptom onset. 18 The viral load decline in our study was relatively high in the first week following infection (3.6 log10 copies/mL in the 225‐mg group and 3.4 log10 copies/mL in the 600‐mg group on day 8). Although the viral load dynamics in this first‐in‐patient study do not permit comparison to a placebo group, the observed viral load reduction, in comparison to the model of Jones et al, suggests a potential signal that ensovibep has an effect on viral clearance in a population with low risk of COVID‐19‐related complications, consistent with the results obtained after monoclonal antibody treatments. 3 , 19 Ensovibep displayed first‐order kinetics with a long systemic half‐life in COVID‐19 patients, confirming the in vivo half‐life extension properties of its antihuman serum albumin DARPin modules in the presence of the compound's main binding target (SARS‐CoV‐2 S‐protein RBD).
Both doses showed consistent PK profiles with dose escalation from 225 to 600 mg resulting in a proportional increase of serum concentration and exposure. Due to the low variability of the PK data and consistent pharmacodynamic results, the expansion of the cohort was not needed. Noncompartmental PK analysis showed a relatively low V D of approximately 2.8 L (in the range of systemic circulation) and a long half‐life of approximately 13 days. These characteristics can be attributed to ensovibep's albumin‐binding domains. It is anticipated that ensovibep will distribute through tissues alongside albumin. Albumin (like many other proteins) distributes across the epithelial lining of the lungs, despite a low V D. 20 Monoclonal antibodies exhibit a similar V D because they are presumed to be relatively confined to the vascular space, but they still distribute to a sufficient degree to exert local effects. Moreover, several mAbs with a similar V D as ensovibep have been shown to be effective as treatment or prophylaxis for COVID‐19. 21 , 22
All patients, including the two patients with an increased elimination rate, had similar ensovibep exposures (combined with a slow elimination rate during the first 2 weeks following dosing). Neutralization of SARS‐CoV‐2 will be most important during this initial phase of infection, as prolonged viral shedding of high viral quantities is associated with poor outcomes. 23
Like other protein therapeutics, including mAbs, immunogenicity has been described previously. 24 , 25 In this study, ADA formation was observed at various timepoints in most patients, but mono‐exponential elimination appeared to remain unaffected in most patients (83%). More importantly, immunogenicity did not appear to alter ensovibep concentrations in the first 2 weeks postdose, the time interval where antiviral efficacy is anticipated to be most relevant.
Ensovibep was well tolerated in COVID‐19 patients. There were no SAEs, infusion site reactions, hypersensitivity or clinical worsening of COVID‐19 (such as immune enhancement‐like phenomena described for antibody‐based drugs and vaccines targeting coronaviruses). 26 Adverse events were of mild‐to‐moderate severity. Related TEAEs consisted of diarrhoea and transient mild liver enzyme increases, and were only observed in the low‐dose (225‐mg) group. A possible relationship of these TEAEs and ensovibep could not be ruled out based on the timing of onset. However, SARS‐CoV‐2 can cause gastrointestinal symptoms and can lead to (transient) hepatocyte injury in various degrees of severity and via various mechanisms, 27 , 28 , 29 therefore these adverse events could also be attributed to COVID‐19.
Assessment of common COVID‐19‐related symptoms indicated an overall decrease in COVID‐19 symptoms during the 29‐day follow‐up period, but the study population already had a paucity of symptoms at baseline, which makes the interpretability of results difficult. Similar to viral load decline, there were no apparent differences between the 225‐ and 600‐mg doses in the resolution of COVID‐19 symptoms. Heterogeneity of clinical outcome measures in current literature and the timing of participant inclusion in relation to symptom onset make it difficult to compare the observed symptom resolution with natural COVID‐19 disease course. A study by Bliddal et al in nonhospitalized PCR‐positive COVID‐19 patients showed a median time until cessation of symptoms of 12‐14 days, with persistence of symptoms ≥4 weeks in approximately 36% of patients. 30 In our study a subset of subjects (four out of 12 [33%]) reported symptoms at day 29 (fatigue, myalgia, smell/taste loss). These symptoms were also most prevalent in the study population of Bliddal et al. 30
At the time of the study, there was no standardized clinical case definition of long COVID. The Long‐COVID Syndrome questionnaire was used as exploratory tool to gain preliminary insights into the occurrence of long‐term post‐COVID symptoms after ensovibep administration. Case identification of long COVID according to the current World Health Organisation (WHO) definition could therefore not be made. 31 Patients reported predominantly no or only mild symptoms on day 91 compared to their pre‐COVID status and no patients reported an impact of symptoms on daily occupational functioning.
SARS‐CoV‐2 serum neutralizing activity and endogenous antibody formation were assessed as an exploratory endpoint. All patients had detectable levels of SARS‐CoV‐2 antibodies and half of the patients had SARS‐CoV‐2 serum neutralizing activity at day 91. Because ensovibep levels were predicted to be very low at day 91, any neutralizing activity in serum was initially expected to be due to the endogenous response to SARS‐CoV‐2 infection. However, the protocol allowed for COVID‐19 vaccination after day 29. Anti‐SARS‐CoV‐2 antibody titres were highest in vaccinated patients (n = 3). These preliminary results indicate that administration of ensovibep does not prevent an endogenous immune response to SARS‐CoV‐2 antigens.
Our study had some limitations. This study was intended to assess the feasibility of IV ensovibep administration in ambulatory COVID‐19 before initiation of larger phase 2/3 randomized controlled trials, while exploring the first‐in‐patient PK and pharmacodynamic effects. The patients in this study showed clear improvement, both in symptoms and in viral clearance. However, to fully determine the clinical efficacy and effect size, a comparison with an unexposed and representative control group must be made to differentiate from a natural disease course. The sample size in this exploratory study in combination with relatively young Caucasian adults and the relatively mild disease manifestation limit the extrapolation of the results to a broader population. Lastly, COVID‐19 can cause many clinical abnormalities, which made it difficult to discriminate between disease and treatment‐related adverse effects.
In conclusion, this study provides the first clinical data of ensovibep in symptomatic, nonhospitalized, COVID‐19 patients. Single IV administration of ensovibep (225 and 600 mg) was safe and well tolerated in ambulatory COVID‐19 patients. Both explored doses had similar effects on preliminary pharmacodynamic outcome measures, such as viral load and symptoms, suggesting that low doses of ensovibep could be targeted in the future. The results of this study support the continued development of ensovibep as a potential treatment for COVID‐19 in a follow‐up randomized, double‐blind, placebo‐controlled phase 2/3 trial (NCT04828161).
COMPETING INTERESTS
This study was sponsored by Molecular Partners AG. G.T., M.S., E.F., V.S., M.Z. and R.D. are employees of Molecular Partners AG and hold shares in the company. All other authors have no competing interest to declare.
CONTRIBUTORS
M.P and J.P. wrote the manuscript. M.V. and C.B. coordinated the clinical trial under supervision of I.K. M.P., J.P., M.V. and C.B. performed the clinical research. G.H. carried the final medical responsibility. M.P., J.P., M.V., G.T., M.S., E.F., C.Z., V.S., M.Z., R.D., J.B., G.H. and I.K contributed to the conception and design of the study. D.D. was responsible for and coordinated the bioanalysis. N.B. contributed to the acquisition of data. All authors discussed the analysis and interpretation of data, provided critical feedback and contributed to the intellectual content of the manuscript.
Supporting information
FIGURE S1 Time to PCR negativity (Kaplan‐Meier curve)
FIGURE S2 Pharmacokinetic profiles of ensovibep in serum on nominal time points
FIGURE S3 Mean (SD) of total COVID‐19‐Related Symptom score
TABLE S1 Decrease of viral load following ensovibep administration per time interval and estimated slope of decline of viral load.
Long‐COVID Syndrome questionnaire
ACKNOWLEDGEMENTS
The authors would like to thank all patients for their participation in the clinical trial.
Prins MLM, van der Plas JL, Vissers MFJM, et al. Viral clearance, pharmacokinetics and tolerability of ensovibep in patients with mild to moderate COVID‐19: A phase 2a, open‐label, single‐dose escalation study. Br J Clin Pharmacol. 2022;1‐10. doi: 10.1111/bcp.15560
J.L. van der Plas and M.L.M Prins should be considered joint first author.
G.H. Groeneveld and I.M.C. Kamerling should be considered joint last author.
The authors confirm that the PI for this paper is I.M.C. Kamerling PhD. G.H. Groeneveld MD PhD is the medical responsible person and had direct clinical responsibility for the patients.
Funding Information Molecular Partners AG.
DATA AVAILABILITY STATEMENT
Research data are not shared.
REFERENCES
- 1. World Health Organization . WHO Coronavirus (COVID‐19) Dashboard. 2022. Accessed January 26, 2022. https://covid19.who.int/
- 2. Gupta A, Gonzalez‐Rojas Y, Juarez E, et al. Early treatment for Covid‐19 with SARS‐CoV‐2 neutralizing antibody sotrovimab. N Engl J Med. 2021;385(21):1941‐1950. doi: 10.1056/NEJMoa2107934 [DOI] [PubMed] [Google Scholar]
- 3. Weinreich DM, Sivapalasingam S, Norton T, et al. REGEN‐COV antibody combination and outcomes in outpatients with Covid‐19. N Engl J Med. 2021;384(3):238‐251. doi: 10.1056/NEJMoa2035002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dougan M, Nirula A, Azizad M, et al. Bamlanivimab plus etesevimab in mild or moderate Covid‐19. N Engl J Med. 2021;385(15):1382‐1392. doi: 10.1056/NEJMoa2102685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Neuzil KM. Interplay between emerging SARS‐CoV‐2 variants and pandemic control. N Engl J Med. 2021;384(20):1952‐1954. doi: 10.1056/NEJMe2103931 [DOI] [PubMed] [Google Scholar]
- 6. Chen RE, Zhang X, Case JB, et al. Resistance of SARS‐CoV‐2 variants to neutralization by monoclonal and serum‐derived polyclonal antibodies. Nat Med. 2021;27(4):717‐726. doi: 10.1038/s41591-021-01294-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rothenberger S, Hurdiss DL, Walser M, et al. 2021. Ensovibep, a novel trispecific DARPin candidate that protects against SARS‐CoV‐2 variants. bioRxiv. doi: 10.1101/2021.02.03.429164 [DOI]
- 8. Walser M, Rothenberger S, Hurdiss DL, et al. 2021. Highly potent anti‐SARS‐CoV‐2 multivalent DARPin therapeutic candidates. bioRxiv. doi: 10.1101/2020.08.25.256339 [DOI]
- 9. Knutson C, Claas A, Gaudet S, et al. Ensovibep clinical dose selection rationale to treat COVID‐19 in empathy (P‐086). 2022 ASCPT Annual Meeting abstract supplement 2022.
- 10. US Food and Drug Administration . Immunogenicity Testing of Therapeutic Protein Products – Developing and Validating Assays for Anti‐Drug Antibody Detection. 2019; 25.
- 11. Lu X, Wang L, Sakthivel SK, et al. US CDC real‐time reverse transcription PCR panel for detection of severe acute respiratory syndrome coronavirus 2. Emerg Infect Dis. 2020;26(8):1654‐1665. doi: 10.3201/eid2608.201246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Spearman C. The method of “right and wrong cases” (constant stimuli) without Gauss's formula. Br J Psychol. 1908;2(3):227‐242. doi: 10.1111/j.2044-8295.1908.tb00176.x [DOI] [Google Scholar]
- 13. Kärber G. Beitrag zur kollektiven Behandlung pharmakologischer Reihenversuche. Naunyn Schmiedebergs Arch Exp Pathol Pharmakol. 1931;162(4):480‐483. doi: 10.1007/BF01863914 [DOI] [Google Scholar]
- 14. Zielinska E, Liu D, Wu HY, Quiroz J, Rappaport R, Yang DP. Development of an improved microneutralization assay for respiratory syncytial virus by automated plaque counting using imaging analysis. Virol J. 2005;2(1):84. doi: 10.1186/1743-422X-2-84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gallant P, Schultz AA. Evaluation of a visual infusion phlebitis scale for determining appropriate discontinuation of peripheral intravenous catheters. J Infus Nurs. 2006;29(6):338‐345. doi: 10.1097/00129804-200611000-00004 [DOI] [PubMed] [Google Scholar]
- 16. FDA . Assessing COVID‐19‐Related Symptoms in Outpatient Adult and Adolescent Subjects in Clinical Trials of Drugs and Biological Products for COVID‐19 Prevention or Treatment – Guidance for industry. September 2020. Accessed October 15, 2021.
- 17. Alexander SP, Kelly E, Marrion NV, et al. The Concise Guide to PHARMACOLOGY 2017/18: overview. Br J Pharmacol. 2017;174(Suppl 1):S1‐S16. doi: 10.1111/bph.13882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jones TC, Biele G, Muhlemann B, et al. Estimating infectiousness throughout SARS‐CoV‐2 infection course. Science. 2021;373(6551):eabi5273. doi: 10.1126/science.abi5273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gottlieb RL, Nirula A, Chen P, et al. Effect of bamlanivimab as monotherapy or in combination with etesevimab on viral load in patients with mild to moderate COVID‐19: a randomized clinical trial. JAMA. 2021;325(7):632‐644. doi: 10.1001/jama.2021.0202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kim KJ, Malik AB. Protein transport across the lung epithelial barrier. Am J Physiol Lung Cell Mol Physiol. 2003;284(2):L247‐L259. doi: 10.1152/ajplung.00235.2002 [DOI] [PubMed] [Google Scholar]
- 21. Fact sheet for health care providers emergency use authorization (EUA) of bamlanivimab and etesevimab. Accessed September 7, 2022. https://www.lillymedical.com/en-us/medical-information/covid-19/bamlanivimab-etesevimab
- 22. Summary of product characteristics: evusheld. Accessed September 7, 2022. https://www.ema.europa.eu/en/documents/product-information/evusheld-epar-product-information_en.pdf
- 23. Shenoy S. SARS‐CoV‐2 (COVID‐19), viral load and clinical outcomes; lessons learned one year into the pandemic: a systematic review. World J Crit Care Med. 2021;10(4):132‐150. doi: 10.5492/wjccm.v10.i4.132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tolcher AW, Sweeney CJ, Papadopoulos K, et al. Phase I and pharmacokinetic study of CT‐322 (BMS‐844203), a targeted adnectin inhibitor of VEGFR‐2 based on a domain of human fibronectin. Clin Cancer Res. 2011;17(2):363‐371. doi: 10.1158/1078-0432.CCR-10-1411 [DOI] [PubMed] [Google Scholar]
- 25. Callanan D, Kunimoto D, Maturi RK, et al. Double‐masked, randomized, phase 2 evaluation of abicipar pegol (an anti‐VEGF DARPin therapeutic) in neovascular age‐related macular degeneration. J Ocul Pharmacol Ther. 2018;34(10):700‐709. doi: 10.1089/jop.2018.0062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lee WS, Wheatley AK, Kent SJ, DeKosky BJ. Antibody‐dependent enhancement and SARS‐CoV‐2 vaccines and therapies. Nat Microbiol. 2020;5(10):1185‐1191. doi: 10.1038/s41564-020-00789-5 [DOI] [PubMed] [Google Scholar]
- 27. Bertolini A, van de Peppel IP, Bodewes F, et al. Abnormal liver function tests in patients with COVID‐19: relevance and potential pathogenesis. Hepatology. 2020;72(5):1864‐1872. doi: 10.1002/hep.31480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Silva F, Brito BB, Santos MLC, et al. COVID‐19 gastrointestinal manifestations: a systematic review. Rev Soc Bras Med Trop. 2020;53:e20200714. doi: 10.1590/0037-8682-0714-2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bzeizi K, Abdulla M, Mohammed N, Alqamish J, Jamshidi N, Broering D. Effect of COVID‐19 on liver abnormalities: a systematic review and meta‐analysis. Sci Rep. 2021;11(1):10599. doi: 10.1038/s41598-021-89513-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bliddal S, Banasik K, Pedersen OB, et al. Acute and persistent symptoms in non‐hospitalized PCR‐confirmed COVID‐19 patients. Sci Rep. 2021;11(1):13153. doi: 10.1038/s41598-021-92045-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Organization WH . A clinical case definition of post COVID‐19 condition by a Delphi consensus. 2021. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
FIGURE S1 Time to PCR negativity (Kaplan‐Meier curve)
FIGURE S2 Pharmacokinetic profiles of ensovibep in serum on nominal time points
FIGURE S3 Mean (SD) of total COVID‐19‐Related Symptom score
TABLE S1 Decrease of viral load following ensovibep administration per time interval and estimated slope of decline of viral load.
Long‐COVID Syndrome questionnaire
Data Availability Statement
Research data are not shared.